

The discovery of medical therapies to reduce morbidity and mortality related to coronavirus disease (COVID-19) remains an area of intense investigation. Beyond supportive care (e.g., oxygen supplementation, vasopressors), a broad spectrum of therapeutics has been investigated, including antivirals, immunomodulators, monoclonal antibodies to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and antithrombotic agents. However, a limited understanding of downstream molecular drivers that cause clinical sequelae in severe COVID-19 has hindered progress in identifying effective therapies that prevent adverse clinical outcomes. In contrast, there has been greater success in preventing viral transmission with the availability of multiple effective vaccines. Despite the increasing availability of vaccines for COVID-19, the United Nations estimates that widespread global immunization will not be complete until 2024. As evidenced by the recent catastrophic COVID-19 surge in India, there remains an urgent need to identify novel therapies to mitigate the severity of COVID-19 infection.
It is now well-documented that COVID-19 affects nearly every organ system in the body, extending beyond the expected pulmonary manifestations from a respiratory virus (e.g., pneumonia, acute respiratory distress syndrome) (1). Whether the clinical complications observed with COVID-19 infection are due to direct or indirect viral toxicity remains a question of significant debate. The available histopathological evidence has identified intracellular SARS-CoV-2 in the lungs, heart, kidney, brain, and hematological organs; however, uncertainty remains as to whether the pathophysiology of the observed multiorgan injury in COVID-19 is largely driven by systemic activation of the immune system in response to sepsis (like any other cause of distributive shock) or is mechanistically specific to SARS-CoV-2 (2). One unique histopathologic finding that has emerged as a prominent feature in COVID-19 infection is the presence of venous and arterial thrombi in small and large vessels, in addition to more common cardiovascular complications of severe illness such as myocardial injury, heart failure, and stroke (3, 4). In aggregate, this constellation of pathologic findings suggests a central mechanistic role of endothelial dysfunction in COVID-19 (Figure 1). These findings are further supported by clinical data documenting high levels of PAI-1, a key marker of endothelial dysfunction, in patients with severe COVID-19 (5). However, to date, studies examining the underlying molecular mechanisms linking endothelial dysfunction and the plasminogen system in COVID-19 remain limited.
Figure 1.
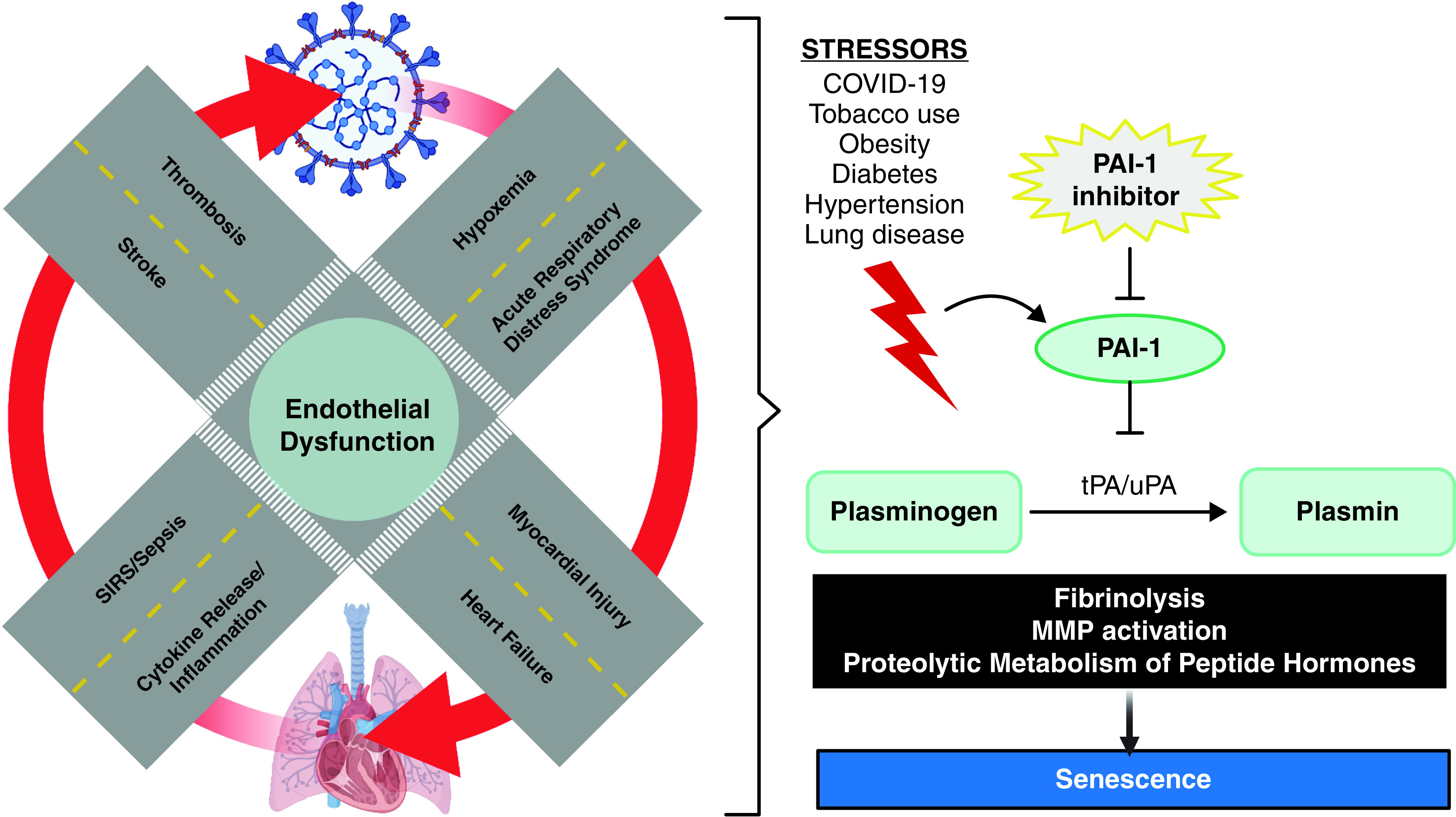
Central role of endothelial dysfunction and PAI-1 at the intersection of pulmonary and cardiovascular complications in COVID-19 infection. COVID-19 = coronavirus disease; MMP = matrix metalloproteinases; SIRS = systemic inflammatory response syndrome; tPA = tissue plasminogen activator; uPA = urokinase plasminogen activator.
In this issue of the Journal, Han and Pandey (pp. 300–308) describe an elegant set of in vitro experiments to investigate cellular mechanisms underlying SARS-CoV-2–triggered endothelial dysfunction in human pulmonary microvascular endothelial cells (HPMECs) (6). The central finding of their work is the robust release of PAI-1 by HPMECs in response to the uptake of recombinant SARS-CoV-2 spike protein (S1). They identify two upstream drivers of PAI-1 expression in HPMECs exposed to SARS-CoV-2-S1: 1) KLF2, a transcription factor, and 2) ZPMSTE24, a zinc metalloproteinase. The first factor, KLF2, has been previously identified as an important regulator of thrombin-mediated endothelial activation whereby KLF2 overexpression is known to reduce the secretion of PAI-1 (7). To confirm this pathway in endothelial injury due to SARS-CoV-2-S1, the authors demonstrate the protective role of bortezomib, a proteasomal inhibitor, via KLF2-mediated inhibition of PAI-1. The second pathway implicated by the authors in COVID-19–related endothelial dysfunction, ZPMSTE24, is especially intriguing. Prior literature has demonstrated significant upregulation of PAI-1 with declining ZPMSTE24 function in aging (8). The authors build on these findings by demonstrating increased PAI-1 expression in response to injury (cigarette smoke) and with an in vitro model of aging (prelamin A endothelial cells). In addition, the authors identify the role of ZPMSTE24 in regulating the expression of ACE-2 and viral entry of SARS-CoV-2-S1. Taken together, these findings build on the emerging evidence regarding the central role of endothelial dysfunction in COVID-19 and support PAI-1 as a potential mechanistic link between known risk factors (e.g., tobacco use, age) and clinical manifestations of COVID-19 infection.
PAI-1, which is encoded by the SERPINE1 gene, was first discovered ∼4 decades ago as the principal inhibitor of tissue and urokinase plasminogen activators. Although PAI-1 was originally discovered for its role in the fibrinolytic system, a substantial body of preclinical and clinical data have now defined a broader biological role of PAI-1 as a marker and mediator of senescence and aging (9). At the cellular level, PAI-1 is part of a set of factors termed the senescence-associated secretory phenotype, which additionally includes factors that are known to upregulate expression of PAI-1 (ILs, transforming growth factor-β, TNF-α) (10, 11). In addition to the robust available evidence identifying high levels of PAI-1 in studies of both chronological and stress-induced cellular aging (e.g., doxorubicin, nicotinamide adenosine dinucleotide–dependent deacetylase sirtuin-1 [SIRT-1] inhibitor, sirtinol), a pivotal 2006 study by Kortlever and colleagues first defined PAI-1 as a promoter of cellular senescence (12). Specifically, the authors demonstrated that p53-deficient fibroblasts overexpressed PAI-1 and underwent replicative senescence. Conversely, PAI-1–deficient fibroblasts (and p53-deficient fibroblasts) were resistant to senescence. This work was subsequently extended with studies in a murine model of accelerated aging (klotho) that demonstrated that genetic deficiency or pharmacologic inhibition of PAI-1 delayed progression of aging, prevented telomere shortening, and prolonged life span (13).
The clinical relevance of these preclinical data is supported by emerging studies in humans. Specifically, we recently completed deep phenotyping studies in an Old Order Amish community, the only known kindred in the world that harbors a rare loss-of-function variant in the SERPINE1 gene leading to lifelong deficiency of PAI-1, which offers an unprecedented opportunity to study the biology of aging and metabolic disease in humans (14). We found that heterozygosity for the null SERPINE1 variant was associated with lower PAI-1 levels, greater insulin sensitivity, and longer life span across several generations. These data help connect the well-established and fundamental role of metabolism with aging and build on studies demonstrating the contributions of insulin and insulin-like growth factor in senescence across a variety of experimental model organisms (Caenorhabditis elegans, Drosophila, and rodents). We and others have additionally demonstrated the broader relevance of PAI-1 in the general population by documenting the association of circulating PAI-1 levels with cardiometabolic diseases in large, community-based cohort studies, such as the Coronary Artery Risk Development in Young Adults study, the Multi-Ethnic Study of Atherosclerosis study, and the Framingham Heart Study (9, 15).
The findings by Han and Pandey, in addition to the growing clinical evidence that older patients and those with preexisting cardiometabolic diseases (who are more likely to have higher baseline PAI-1 levels) are at a heightened risk for severe infection, support the biological plausibility of targeted PAI-1 inhibition in patients with COVID-19. Orally active small molecule inhibitors of PAI-1 are readily available, and one such phase II trial—Study To antagOnize Plasminogen Activator Inhibitor-1 in Severe COVID-19 (STOP Severe COVID-19)—is already underway in the United States examining the safety and efficacy of a novel PAI-1 inhibitor (TM5614; www.clinicaltrials.gov NCT 04634799). In contrast to studies investigating more generalized antithrombotic strategies with therapeutic anticoagulation (e.g., low-molecular-weight heparin), targeted inhibition of PAI-1 may hold greater promise as a novel therapeutic option to improve outcomes beyond thrombosis, given the available evidence supporting the multifaceted role of PAI-1 in COVID-19 morbidity.
Footnotes
Originally Published in Press as DOI: 10.1165/rcmb.2021-0208ED on June 3, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanley B, Naresh KN, Roufosse C, Nicholson AG, Weir J, Cooke GS, et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet Microbe. 2020;1:e245–e253. doi: 10.1016/S2666-5247(20)30115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434. doi: 10.1016/j.eclinm.2020.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lombardi CM, Carubelli V, Iorio A, Inciardi RM, Bellasi A, Canale C, et al. Association of troponin levels with mortality in italian patients hospitalized with coronavirus disease 2019: results of a multicenter study. JAMA Cardiol. 2020;5:1274–1280. doi: 10.1001/jamacardio.2020.3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma tissue plasminogen activator and plasminogen activator inhibitor–1 in hospitalized COVID-19 patients. Sci Rep. 2021;11:1580. doi: 10.1038/s41598-020-80010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han M, Pandey D.ZMPSTE24 regulates SARS-CoV-2 spike protein–enhanced expression of endothelial PAI-1 Am J Respir Cell Mol Biol 202165300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin Z, Hamik A, Jain R, Kumar A, Jain MK. Kruppel-like factor 2 inhibits protease activated receptor-1 expression and thrombin-mediated endothelial activation. Arterioscler Thromb Vasc Biol. 2006;26:1185–1189. doi: 10.1161/01.ATV.0000215638.53414.99. [DOI] [PubMed] [Google Scholar]
- 8. Infante A, Rodríguez CI. Secretome analysis of in vitro aged human mesenchymal stem cells reveals IGFBP7 as a putative factor for promoting osteogenesis. Sci Rep. 2018;8:4632. doi: 10.1038/s41598-018-22855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. 2017;37:1446–1452. doi: 10.1161/ATVBAHA.117.309451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khan SS, Singer BD, Vaughan DE. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell. 2017;16:624–633. doi: 10.1111/acel.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rana T, Jiang C, Liu G, Miyata T, Antony V, Thannickal VJ, et al. PAI-1 regulation of TGF-β1-induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am J Respir Cell Mol Biol. 2020;62:319–330. doi: 10.1165/rcmb.2019-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales-Nebreda L, et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA. 2014;111:7090–7095. doi: 10.1073/pnas.1321942111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan SS, Shah SJ, Klyachko E, Baldridge AS, Eren M, Place AT, et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv. 2017;3:eaao1617. doi: 10.1126/sciadv.aao1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campbell PT, VanWagner LB, Colangelo LA, Lewis CE, Henkel A, Ajmera VH, et al. Association between plasminogen activator inhibitor-1 in young adulthood and nonalcoholic fatty liver disease in midlife: CARDIA. Liver Int. 2020;40:1111–1120. doi: 10.1111/liv.14417. [DOI] [PMC free article] [PubMed] [Google Scholar]