

Abstract
The activation of cannabinoid CB1 receptors (CB1R) by Δ9-tetrahydrocannabinol (THC), the main component of Cannabis sativa, induces analgesia. CB1R activation, however, also causes cognitive impairment via the serotonin 5HT2A receptor (5HT2AR), a component of a CB1R–5HT2AR heteromer, posing a serious drawback for cannabinoid therapeutic use. We have shown that peptides reproducing CB1R transmembrane (TM) helices 5 and 6, fused to a cell-penetrating sequence (CPP), can alter the structure of the CB1R–5HT2AR heteromer and avert THC cognitive impairment while preserving analgesia. Here, we report the optimization of these prototypes into drug-like leads by (i) shortening the TM5, TM6, and CPP sequences, without losing the ability to disturb the CB1R–5HT2AR heteromer, and (ii) extensive sequence remodeling to achieve protease resistance and blood–brain barrier penetration. Our efforts have culminated in the identification of an ideal candidate for cannabis-based pain management, an orally active 16-residue peptide preserving THC-induced analgesia.
Introduction
About 20% of the population suffers from chronic pain that seriously affects personal and professional life, highlighting the urgent need for analgesics that effectively alleviate pain with minimal side effects.1 Opioids, thus far the mainstay in severe pain management, are under intense debate for their poor safety profiles and high potential for abuse, as reflected in the opioid abuse disorder epidemic claiming about 145 deaths/day in the recent years in the USA.2,3 On the other hand, patients with minor pain manifestations are usually dealt with nonsteroidal anti-inflammatories (NSAIDs) that not only fail quite often to achieve adequate relief but also have serious side effects (e.g., gastrointestinal and renal).4 Thus, there is a need to fill the therapeutic gap between opioids and NSAIDs under multiple moderate pain conditions or in situations where opioids are ineffective, such as severe neuropathic pain.5
The analgesic potential of cannabinoid compounds has long been recognized.6−8 The activation of cannabinoid CB1 receptors (CB1R) by agonists, such as Δ9-tetrahydrocannabinol (THC), the main psychoactive component of Cannabis sativa, elicits therapeutically relevant responses for the treatment of migraine,9,10 rheumatoid arthritis,10 osteoarthritis,11 neuropathic,12 or cancer-related pain,13 among many others. Unfortunately, CB1R activation is also linked to side effects, such as memory impairment, with major consequences in cannabis users.8,14−17 These adverse effects pose a serious hurdle to the therapeutic use of cannabinoids; hence, the possibility to minimize their impact would constitute a major breakthrough.
Behavioral studies in mice lacking the serotonin 5HT2A receptor (5HT2AR) revealed a remarkable dissociation between the beneficial antinociceptive effects of THC and its detrimental amnesic properties.18 As CB1R and 5HT2AR are G protein-coupled receptors (GPCRs), a class of proteins frequently forming oligomers in cells,19 we provided evidence that the underlying molecular basis for that observation was the formation of CB1R–5HT2AR heteromers.18 Both receptors are functionally active in specific brain regions; the heteromer shows specific biochemical signatures distinct from those of the protomers, in artificial cell lines as well as native tissues; very importantly, while the cognitive impairment caused by THC is due to the CB1R–5HT2AR heteromer, the antinociceptive effects result solely from CB1R activation.18 The arrangement of CB1R and 5HT2AR protomers in the heteromer is via transmembrane (TM) helices 5 and 618 as studied by bimolecular fluorescence complementation (BiFC) experiments with synthetic peptides TM5-Tat and Tat-TM6, where TM5 and 6 helices are fused to cell-penetrating peptide (CPP) GRKKRRQRRR, that is, HIV Tat(48–57),20 respectively, at N- or C-termini, ensuring TM native alignment relative to the membrane.
The formation of a stable four-helix bundle between these TM5 and 6 helices of both protomers, as described for the dimeric crystal of the μ-opioid receptor,21 was suggested to be involved in the allosteric interactions between protomers and in the mechanisms behind the specific signaling signatures of the heteromer.18,22 Moreover, interfering TM5-Tat and Tat-TM6 peptides were able to alter the structure of the heteromer in vivo, leading to a selective abrogation of memory impairments caused by the exposure to THC.18 From this, it was logical to assume that disturbing the formation of the CB1R–5HT2AR heteromer might be an effective strategy for harnessing the therapeutic potential of THC while avoiding its side effects. TM5-Tat and Tat-TM6 peptides, however, were ungainly therapeutic leads with poor prospects related to their large size, low solubility, and inability to cross the blood–brain barrier (BBB). We now report on a comprehensive optimization effort leading from those prototypes to a candidate with vastly improved, drug-like features (Table 1) including small size, proteolytic stability, BBB crossing ability, and, remarkably, oral activity, thereby achieving the desired effects, both in vitro and in vivo.
Table 1. From TM5/6 Prototypes to a Drug-Like Finalist Peptidea.
| peptide | sequenceb | residues | |
|---|---|---|---|
| prototypes18 | TM5-Tat | ETYLMFWIGVTSVLLLFIVYAYMYILWGRKKRRQRRR | 37 |
| Tat-TM6 | GRKKRRQRRRKTLVLILVVLIICWGPLLAIMVYDVF | 36 | |
| optimized candidate | wliymyayvaGilkrw | 16 | |
A detailed list of all peptides used in the various rounds of the optimization process is given in Tables S1–S4.
Upper and lower case denote l- and d-residues, respectively.
Results
Shorter Peptides Mimicking CB1R TM Helices 5 and 6 and Altering CB1R–5HT2AR Heteromers
The design of these peptides required the identification of the amino acid residues in TM5-Tat and Tat-TM6 that contribute more significantly to the binding to 5HT2AR. This was studied by several replicas of unbiased 1 μs molecular dynamic (MD) simulations of TM5-Tat and Tat-TM6 in complex with 5HT2AR (see Figure 1a–c and Experimental Section). The simulations show that the intracellular part of the peptides remains more stable than the extracellular one (more noticeable for Tat-TM6) due to specific interactions with 5HT2AR (mainly the V19-Y20-Y22-M23-L26-W27 stretch in TM5-Tat or the K11-V14-L17-V18 stretch in Tat-TM6, Figure 1d,e).
Figure 1.

(a–e) MD simulations of 5HT2AR in complex with TM5-Tat and Tat-TM6 peptides. The previously published computational model of the CB1R–5HT2AR heteromer18,20 (panel a) was used as the starting point for MD simulations (see Experimental Section) of a membrane-embedded (yellow spheres) 5HT2AR (green ribbons) in complex with peptides where the TM5 (blue ribbon, panel b) or TM6 (red ribbon, panel c) sequences are fused to HIV-Tat(48–57) (orange). These TM5-Tat and Tat-TM6 peptides (Table 1) were stable in the three replicas of 1 μs unbiased MD, as shown by root-mean-square deviations (Figure S1). Panels (d,e) show detailed views of the interactions between TM5-Tat and Tat-TM6, respectively, with TM helices 5 and 6 of 5HT2AR. Snapshots along the trajectories are shown as transparent ribbons, whereas representative structures are solid ribbons. Side chain residues involved in stable interactions of TM5-Tat and Tat-TM6 with 5HT2AR (Ballesteros–Weinstein notation) during the MD simulations are labeled. Interaction frequency maps for frequencies above 50% in at least one of the three replicas (r1, r2, and r3) are depicted. TM5-Tat and Tat-TM6 residues involved in stable interactions with two or more 5HT2AR residues are highlighted in bold.
Based on these predictions,18 peptides VYAYMYILW-Tat and Tat-KTLVLILVV (1 and 3, Table S1), respectively, mimicking the key interactions of TM5-Tat and Tat-TM6, were designed. Their ability to alter CB1R–5HT2AR heteromer formation was first tested by BiFC assays,18,23,24 in HEK-293T cells transfected with receptors fused to two complementary halves of YFP (5HT2AR-cYFP and CB1R-nYFP). In this assay, 1 and 3 caused a decrease in fluorescence complementation comparable to the TM5-Tat and Tat-TM6 prototypes, in contrast to peptide TM7-Tat, used as a negative control (Figure 2a). Moreover, the biochemical properties of the heteromer differ from those of the protomers.18,23,24 In CB1R–5HT2AR, in addition to a Gq to Gi switch in 5HT2AR coupling preference, antagonist binding to one of the receptors blocks signaling of the interacting receptor (known as cross-antagonism), and costimulation with both agonists does not produce an additive effect (known as negative cross-talk).23,24 Thus, we next determined signaling after receptor activation by measuring cAMP levels [decrease in forskolin (FK)-induced cAMP as the result of adenylate cyclase inhibition by Gi] or the increase in pERK in the ERK1/2 phosphorylation pathway (Figure 2d,g). Cells stimulated with FK and treated with the CB1R agonist WIN 55,212-2 (WIN) or 5HT2AR agonist 2,5-dimethoxy-4-iodoamphetamine (DOI) showed reduced cAMP production in all groups, as expected for Gi-coupled receptors. Neither TM5-Tat, Tat-TM6, peptides 1 or 3, nor TM7-Tat (negative control) influenced G protein coupling preferences (5HT2AR remains Gi-coupled in the presence of interfering peptides). The CB1R antagonist rimonabant (RIM) blocked the decrease in FK-induced cAMP or the increase in pERK triggered by the 5HT2AR agonist DOI, and the 5HT2AR antagonist MDL 100,907 (MDL) also blocked the decrease in cAMP and increase in pERK induced by the CB1R agonist WIN (bidirectional cross-antagonism). Notably, peptides 1 and 3 eliminated this bidirectional cross-antagonism as efficiently as TM5-Tat and Tat-TM6 peptides, an effect that was not observed with TM7-Tat (negative control). Coadministration of WIN and DOI agonists, in the absence of peptides, does not lead to a further statistically significant decrease in cAMP or increase in pERK (negative cross-talk). However, in the presence of 1 and 3, as well as TM5-Tat and Tat-TM6 (but not TM7-Tat), the simultaneous addition of both agonists results in a significant decrease in cAMP or an increase in pERK (absence of negative cross-talk). In sum, interfering peptides 1 and 3 successfully blocked key biochemical signatures (cross-antagonism and negative cross-talk) of the CB1R–5HT2AR heteromer as efficiently as TM5-Tat and Tat-TM6.
Figure 2.

(a–c) BiFC analysis of the effect of designed peptides on CB1R–5HT2AR heteromerization. Fluorescence (530 nm) of HEK-293T cells transfected with 5HT2AR-cYFP and CB1R-nYFP treated with a vehicle (no peptide) or 4 μM peptide for 4 h. Values are mean ± SEM of n = 10–30; TM5-Tat and Tat-TM6 are positive controls, whereas TM7-Tat is a negative control; ns (no significant), *(p < 0.05), **(p < 0.01), and ***(p < 0.001) represent significantly different (two-tailed) values (one-way ANOVA followed by Tukey’s multiple comparison tests). (d–i) Effect of designed peptides on the decrease in FK-induced cAMP (d–f) or increase in ERK1/2 phosphorylation. (g–i) Transfected cells preincubated with a vehicle (no peptide) or with 5HT2AR (5HT2ARago, DOI, 100 nM) or CB1R (CB1Rago, WIN, 100 nM) agonists or with 5HT2AR (5HT2ARanta, MDL, 300 nM) or CB1R (CB1Ranta, RIM, 1 μM) antagonists and combinations thereof, in the presence or absence of 0.5 μM FK. Values in panels (d–f) are mean ± SEM of n = 3–6 of FK-treated cells. Quantification of phosphorylated ERK-1/2 was determined by the α-screen bead-based technology. Values in panels (g–i), expressed as a percentage of basal (nontreated cells), are mean ± SEM of n = 4–9. Two-way ANOVA followed by Tukey’s multiple comparison tests was used to analyze the data (Table S5). Cartoons depict the designed peptides (cylinders with color codes as in Figure 1) fused to different CPPs (lines).
We assumed that the segments of peptides TM5-Tat and Tat-TM6 replicating CB1R TM helices 5 and 6 are membrane-embedded as in the receptor, whereas Tat(48–57) is in the intracellular space (Figure 1b,c). Peptides 2 and 4 (Table S1), lacking the Tat(48–57) sequence, had no effect on decreasing fluorescence complementation (Figure 2a) or in signaling (Figure 2d–g), underscoring the essential role of the CPP motif.
HIV-Tat Replacement by Shorter CPPs
Inspired by recent studies,25−28 a second set of analogues were devised (Table S2) in which four alternatives to 10-residue Tat(48–57) were explored: (i) 7-residue PepH3 (AGILKRW), a reported BBB shuttle25 (peptides 5 and 9); (ii) 6-residue SKSKSK26 (6 and 10); (iii) 5-residue RLRWR27 (7 and 11), and (iv) 3-residue RKR28 (8 and 12). The last two CPP motifs (7, 8, 11, and 12) were not as efficient as Tat(48–57) in decreasing fluorescence complementation, as measured in BiFC assays (Figure 2b), neither could 8 or 12 abolish the cross-antagonism and negative cross-talk signatures of the CB1R–5HT2AR heteromer (Figure 2e,h). In contrast, peptides 5/9 and 6/10 were as effective as peptides 1/3 in fluorescence complementation or signaling (Figure 2b,e,h) and were thus selected for further optimization.
CPP Stereochemistry and Its Significance
Replacement of natural l-by d-amino acids results in CPP sequences with improved cell entry and metabolic stability.29,30 We thus switched the CPP motifs of peptides 5, 6, 9, and 10 to their enantiomeric d-versions, while preserving the L configuration for the cell membrane-embedded sections replicating CB1R TM5 or 6 (peptides 13–16, Table S3). Trypsin incubation over 24 h confirmed our expectations of improved proteolytic stability in the 13–16 set, in contrast to poor survival for all-l counterparts (5, 6, 9, and 10) (Figure S2a). More relevantly, 13–16 maintained the CB1R–5HT2AR heteromer-disturbing ability (Figure 2c) and blocking of cross-antagonism and negative cross-talk (Figure 2f,i), as somehow expected from the stereochemical nonmodification of the membrane-embedded TM segments.
BBB Translocation
In our earlier report,18 tacitly assuming the inability of TM5-Tat and Tat-TM6 to traverse the BBB, these peptides were administered by intracerebroventricular (ICV) infusion, an invasive route of little interest for human therapeutic application. However, for a peptide drug to be a successful candidate, BBB-crossing capability is mandatory. Therefore, to explore the BBB-crossing features of optimized peptides 13–16, their 5(6)-carboxyfluorescein-labeled versions (see Experimental Section) were tested for translocation in an in vitro BBB model using mouse b.End3 cells.30,31 Fluorescence intensity readings at the basolateral chamber of the transwell device showed that 13 and 15, with the TM-altering sequence fused to d-CPP motif aGilkrw, translocated more efficiently than 14 and 16, where the TM-altering sequence was fused to the sksksk vector (Figure S2b). The high in vitro BBB integrity observed after 24 h incubation ruled out paracellular transport (Figure S2c). In parallel, cytotoxicity studies on b.End3 cells were conducted to discard potential toxic effects modifying barrier permeability, that is, artifactual peptide ability to cross the BBB (Figure S2d).
Applying Configurational Switch and Sequence Reversal for Last-Stage Lead Optimization
Although considerable progress had been achieved in terms of BBB-crossing, in the CPP-enabled constructs delivering short TM payloads to the CB1R–5HT2AR heteromer, the essential modifying segments (i.e., short TM5: VYAYMYILW and short TM6: KTLVLILVV) were still made up of natural l-amino acids, susceptible to endopeptidases such as chymotrypsin, thermolysin, or pepsin, targeting preferentially hydrophobic and/or aromatic residues (Ile, Leu, Val, Ala, Met, Trp, and Tyr) whose abundance in those TM sequences was an important concern. To boost protease resistance, both in body fluids and within the target cell, we developed two last-stage candidates (peptides 17–18, Table S4) where the optimal BBB-permeable shuttle (i.e., aGilkrw) was fused to the retro-enantio (r/e) versions of the TM5- and TM6-altering sequences (i.e., short TM5 and short TM6 composed of d-amino acids in the reversed sequence). In such an arrangement (Figure 3), if the all-l sequence is displayed in the conventional N- to C-terminal fashion but the r/e analogue is laid out in the opposite (C- to N-terminal) sense by way of a 180° rotation in the plane, the side chains adopt similar orientations,32−35 although the amide bonds show inverted (CONH vs NHCO) directions. In summary, peptides 17–18, recapitulating the various rounds of structural elaboration so far, met a priori all desired requirements, including (i) the same TM5/TM6 side chain orientation than previous CB1R–5HT2AR-modifying analogues, (ii) high proteolytic resistance as expected from an all-d composition,36 and (iii) ability to traverse the BBB.30
Figure 3.
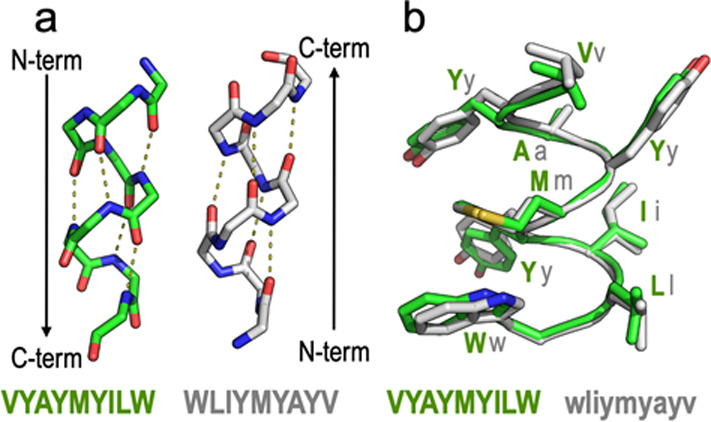
(a) Retro modification of a short TM5 peptide (VYAYMYILW) where the peptide sequence has been synthesized in the reverse order (WLIYMYAYV). (b) When the enantio modification (d-amino acids) is additionally applied (wliymyayv), the orientation of the side chains is similar to that of the original peptide.
When 17 and 18 were submitted to the same screens as previous candidates (Figure 4), our expectations were fulfilled. The ability to interfere the CB1R–5HT2AR heterodimer structure and signaling (Figure 4a,b) was preserved, serum degradation was successfully averted (Figure S3a), and despite the extensive structural modifications undergone, BBB-crossing properties were also maintained (Figures 4c,d, S3b). This last feature was further confirmed by examining internalization of both 17 and 18 in bEnd.3 cells by confocal microscopy (Figure 4e).
Figure 4.
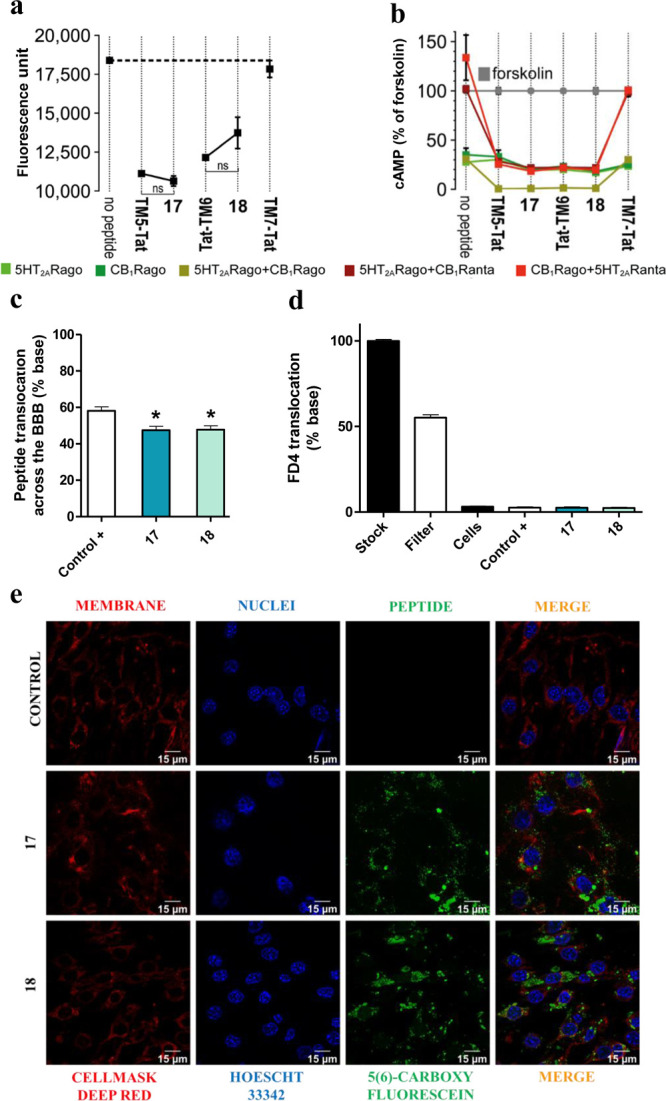
(a) BiFC analysis of the effect of peptides 17 and 18 on CB1R–5HT2AR heteromerization. Fluorescence (530 nm) of HEK-293T cells transfected with 5HT2AR-cYFP and CB1R-nYFP treated with a vehicle or peptide. Values are mean ± SEM of n = 10–30. TM5-Tat and Tat-TM6 are positive controls, whereas TM7-Tat is a negative control; ns (no significant), (one-way ANOVA followed by Tukey’s multiple comparison tests). (b) Effect of peptides 17 and 18 on cAMP production. Transfected cells preincubated with a vehicle (no peptide) or with 5HT2AR (5HT2ARago, DOI, 100 nM) or CB1R (CB1Rago, WIN, 100 nM) agonists or with 5HT2AR (5HT2ARanta, MDL, 300 nM) or CB1R (CB1Ranta, RIM, 1 μM) antagonists and combinations thereof, in the presence of FK. Values are mean ± SEM of n = 3–6 of FK-treated cells. Two-way ANOVA followed by Tukey’s multiple comparison tests was used to analyze the data (Table S5). (c) In vitro translocation of peptides 17 and 18. Translocation (%) across the transwell BBB model quantified as fluorescence in the basolateral chamber after 24 h. A trans-BBB peptide was used as the positive control. Values are mean ± SEM of n = 3–6. *p < 0.05 vs positive control (one-way ANOVA followed by Dunnett post hoc tests). (d) BBB integrity to peptides 17 and 18. Measured as permeability of fluorescent FD4 dextran upon peptide exposure. Values are mean ± SEM of n = 3–6. No statistical significance difference was observed between samples (one-way ANOVA followed by Tukey’s multiple comparison test). (e) Confocal microscopy. Images showing peptide internalization into bEnd.3 cells.
In Vivo Validation of Finalist Peptides 17 and 18
The promising in vitro data obtained for analogues 17 and 18 encouraged us to validate their in vivo efficacy in mice. Given their BBB-crossing features, we evaluated the efficacy of the peptides, intravenously (IV) administered, by examining the preservation of THC analgesic effects (hot-plate test) and the disruption of its amnesic effects (novel object recognition paradigm) (see Experimental Section for a detailed description of both assays).
The in vitro-based expectations for 17 and 18 were partially fulfilled. Thus, the analgesic effects induced by THC (10 mg/kg by intraperitoneal route, IP) were preserved when coadministered with either peptide (20 mg/kg, IV) (Figure 5a). In contrast, the memory impairment produced by THC (3 mg/kg, IP) was suppressed only when coadministered with 17 (20 mg/kg, IV), but amnesic effects remained unaffected when THC was coadministered with 18 (20 mg/kg, IV) (Figure 5b).
Figure 5.

Prevention of THC-induced amnesic effects by peptide 17, while maintaining THC analgesic responses. (a) Analgesic effects of THC (10 mg/kg, IP) in the mouse hot-plate test were preserved after pretreatment with peptides 17 and 18 (both 20 mg/kg, IV). (b) Amnesic effects of THC (3 mg/kg, IP) observed in the mouse novel object recognition test were abrogated by pretreatment with 17 (20 mg/kg, IV) but not with 18 (20 mg/kg, IV). Data are presented as mean ± SEM (n = 10–12). *p < 0.05 vs vehicle + vehicle (Fisher LSD test).
Therefore, peptide 17 was chosen to evaluate the effects following oral administration (OR), the suitable route for therapeutic purposes. THC (10 mg/kg, IP) analgesic effects remained unaffected by coadministration with 5, 10, and 20 mg/kg (OR) doses of 17 (Figure 6a), whereas amnesic effects (3 mg/kg THC, IP) were reverted by the same doses of 17 given by gavage (Figure 6b), demonstrating the effectiveness of the peptide by the most desirable route of administration.
Figure 6.
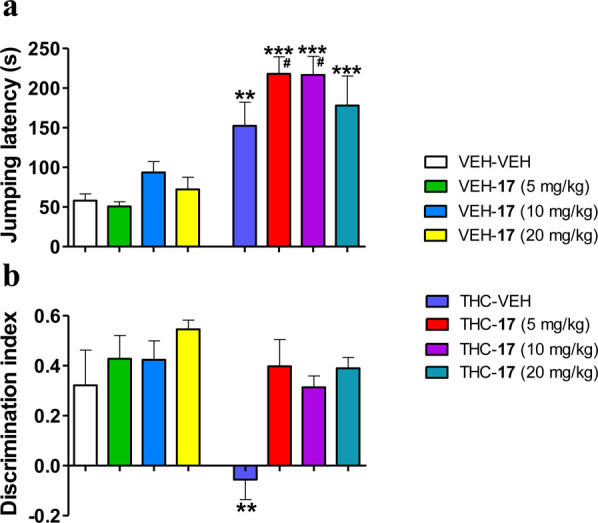
Prevention of THC-induced amnesic effects by peptide 17 administered orally, while maintaining THC analgesic responses. (a) Analgesic effects of THC (10 mg/kg, IP) revealed in the mouse hot-plate test were preserved after pretreatment with 17 (5, 10, and 20 mg/kg, OR). (b) Amnesic effects of THC (3 mg/kg, IP) observed in the mouse novel object recognition test were abrogated by pretreatment with 17 (5, 10, and 20 mg/kg, OR). Data are presented as mean ± SEM (n = 5–8). **p < 0.01, ***p < 0.001 vs vehicle + vehicle. #p < 0.05 vs vehicle + THC (Fisher LSD test).
Non-immunogenicity Confirmed
After showing that peptide 17 epitomized our wish list for an optimal Cannabis-complementing drug candidate, a final concern was its possible immunogenicity, despite expectations that a small 16-residue peptide would most likely go undetected by the immune system. To this end, we immunized mice using a repeated schedule with a high dosage (100 μg of 17 on 5 consecutive days, and a boost at day 15). As shown in Figure 7, repeated administration of 17 in the free form did not evoke any detectable immune response. Antibodies to 17 could only be eventually raised by immunization with the keyhole limpet hemocyanin (KLH)-conjugated version, a positive control of immunogenicity that is, on the other hand, irrelevant with regard to administration of 17 as a therapeutic agent.
Figure 7.

(a) Immunogenicity assessment. The procedure was devised to mimic a daily chronic pain therapy. Mice were treated for 5 consecutive days and boosted at day 15 with peptide 17 in phosphate-buffered saline (PBS) (group 1, 100 μg/dose), peptide 17-KLH conjugate in PBS (group 2, positive control, 30 μg), or PBS (group 3, negative control). The lower dose of the 17-KLH conjugate, compared to 17 alone, was chosen to prevent potential protein toxicity in the mice. Serum samples were collected on days 0, 9, and 19 and animals were euthanized on day 19. (b) ELISA of humoral response. Peptide-specific antibody response in sera collected at the indicated days. Each point depicts mean antibody titers ± SD. The dotted line indicates the detection limit. Free (nonconjugated) 17 induces no significant response; immunogenicity is observed only upon conjugation to a large-size carrier protein (KLH, MW ∼ 5 MDa) and boosting.
In sum, our research has led to an orally active, proteolytically stable, and nonimmunogenic short peptide that constitutes an optimal candidate for combined administration with THC, taking advantage of its analgesic effects but avoiding its associated cognitive impairment.
Discussion
CB1R is the most-abundant GPCR in the mammalian brain and the main target of THC.37−40 While originally thought to exist exclusively as a monomer,41 strong evidence has accumulated indicating its ability to form homo- and heterodimer complexes42−48 with new pharmacological and functional properties.49 Of particular relevance in this regard is the formation of the CB1R–5HT2AR heteromer, as previously revealed by (i) in vitro proximity-based biophysical techniques, (ii) specific biochemical signatures of heteromers distinct from those of the protomers, (iii) in situ proximity ligation assay to detect protein–protein interactions in native tissues, (iv) membrane-permeable peptides altering heteromeric interfaces, and (v) mice expressing heteromerization-deficient receptors, all of them established criteria for the existence of CB1R–5HT2AR heteromers in vivo.50 This CB1R–5HT2AR heteromer, therefore, is not simply due to receptor overexpression, but the consequence of an explicit molecular mechanism of THC to dissociate beneficial antinociceptive response and detrimental amnesic effects,50 hence defining it as a novel therapeutic target.
The development of heteromer-specific drugs is more challenging than traditional drug-discovery programs, where efforts are focused on compounds binding on orthosteric sites of monomeric GPCRs of endogenous ligands.51,52 One of the most widespread options to target heteromers is designing bivalent ligands, that is, single chemical entities capable of interacting simultaneously with the orthosteric sites of a (homo/hetero) dimer via two pharmacophore units covalently linked by an appropriate spacer.53,54 We have developed here a new approach in which the ligand/peptide binds at the TM dimerization interface of the heteromer. This rests on the assumption of allosteric binding sites at the extrahelical part of GPCRs, facing the membrane bilayer. The numerous recent structures of GPCRs bound to allosteric modulators have confirmed the presence of these sites for families A and B.55 Related to the GPCRs of this manuscript, the negative allosteric modulator ORG27569 of CB1R binds to an extrahelical site, mostly interacting with TM4.56
Protein–protein interfaces in general, and GPCR interfaces in particular, consist of large and flat contact areas with no well-defined pockets.57 Here, we have taken advantage of our previous studies in which peptides with the TM5 and TM6 sequences of CB1R (TM5-Tat and Tat-TM6), fused to a HIV-Tat CPP vector, can bind 5HT2AR and modify the quaternary structure of the CB1R–5HT2AR heteromer.18 MD simulations of TM5-Tat and Tat-TM6 in complex with 5HT2AR have allowed to identify these “hotspots” near the intracellular part.
The proteolytic instability of TM5-Tat or Tat-TM6 and their inability to penetrate the BBB complicate any prospect of systemic administration. With this in mind, we have set out to develop strategies aimed at improving the druggability of the TM5-Tat and Tat-TM6 prototypes by (i) downsizing the length of the interfering peptides to only include the essential identified “hotspots”, (ii) enabling CNS delivery via a BBB-permeating CPP shuttle, (iii) ensuring proteolytic robustness, hence bioavailability, by judicious structural manipulation, including the switch to enantio and retro-enantio versions of CPP and interfering motifs, respectively. To this end, we have used in silico tools and peptide medicinal chemistry criteria through a series of design/optimization rounds and a variety of in vitro and in vivo screens that have eventually allowed to identify peptide wliymyayvaGilkrw (17) as a highly effective, orally available candidate with encouraging therapeutic prospects.
Conclusions
Pain is a highly relevant pharmacological area with still significant unmet needs. It has been suggested that the therapeutic gap between benign but rather feeble NSAID painkillers and effective but high-risk opioids could be ideally bridged by cannabinoids, although their important undesirable side effects represent a major limitation. In addition, new markets are emerging, also aimed at using cannabis-derived products with other (i.e., non pain-related) purposes, and again entraining significant risks associated to these side effects. Our work has resulted in a novel tool to minimize the most prominent of these adverse outcomes, namely cognitive impairment. We specifically propose a new approach based on the administration of a CB1R cannabinoid agonist (e.g., THC) in combination with a CB1R–5HT2AR altering agent. This compound is a 16-residue peptide (17: wliymyayvaGilkrw) that provides a convincing proof of concept that appropriately modified peptides constitute valid therapeutic candidates for treating pain with cannabinoids minimizing their side effects. This novel pharmacological approach could also be of interest for attenuating the side effects associated to the use of cannabis derivatives for other purposes.
Experimental Section
Computational Models
The CB1R–5-HT2AR heteromer (Figure 1a) was built from the TM5/6 dimeric interface observed in the crystal structure of the μ-opioid receptor (PDB id 4DKL),21 using the crystal structure of CB1R (5TGZ)58 and a 5HT2B-based (4IB4)59 homology model of 5HT2AR as previously proposed.18,20 The molecular 3D-coordinates corresponding to CB1R TM helices 5 and 6 were extracted and fused with the HIV-Tat(48–58) motif. The resulting TM5-Tat and Tat-TM6 peptides were capped at the N- and C-termini with acetyl and carboxamide groups and energy-minimized using the Molecular Operating Environment (MOE) software (Chemical Computing Group Inc., Montreal, Quebec, Canada). Molecular complexes of 5HT2AR with TM5-Tat and Tat-TM6 were embedded in a pre-equilibrated lipid bilayer box containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), water molecules (TIP3P), and monoatomic Na+ and Cl– ions (0.2 M) (Figure 1b,c). The assignment of ionization states and hydrogens at physiological pH for the selected structures was conducted with the Protonate3D method as implemented in MOE. Molecular systems were subject to 1000 cycles of energy minimization, followed by 20 ns of gradual relaxation of positional restraints (corresponding to 100, 50, 25, and 10 kJ mol–1 nm–2) at protein backbone coordinates before the production phase in order to hydrate the receptor cavities and allow lipids to pack around the protein. The AMBER99SB force field as implemented in GROMACS and Berger parameters for POPC lipids were used for the MD simulations. After equilibration, three replicas of 1 μs of unrestrained MD simulation were performed at a constant temperature of 300 K and a time step of 2.0 fs. Lennard-Jones interactions were computed using a cutoff of 10 Å, and the electrostatic interactions were treated using PME with the same real-space cutoff under periodic boundary conditions. MD simulations were performed using GROMACS 2019.
Peptide Synthesis, Analysis, and Purification
Peptides were assembled in a Prelude instrument (Protein Technologies, Tucson, AZ) running optimized Fmoc synthesis protocols as described before.60 For labeled peptides, the incorporation of 5(6)-carboxyfluorescein to the carboxy-terminal end was performed by adding an additional Fmoc-Lys(Mtt)-OH residue to the sequence, the Mtt group selectively removable on-resin with 1% trifluoroacetic acid, after which the fluorescent dye was coupled via its carboxyl function to the Lys free ε-NH2 group. Final deprotection and cleavage were performed with a CF3COOH/H2O/3,6-dioxa-1,8-octanedithiol/triisopropylsilane (94:2.5:2.5:1 v/v) cocktail for 90 min. Peptide analysis and purification were performed as previously detailed.60 Fractions of satisfactory purity (>90%) by analytical high-performance liquid chromatography (HPLC) were pooled, lyophilized, and analyzed for identity by HPLC–MS (Supporting Information, section 3). For immunogenicity studies, conjugation to bovine serum albumin (BSA) or KLH was carried out with a peptide containing an extra C-terminal Cys residue. Both carrier proteins were preactivated with m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS). After Sephadex G25 gel filtration, purified KLH-MBS or BSA-MBS was added to the peptide (r.t., 5 h, pH 7.0). The unreacted peptide was removed by dialysis and the peptide–protein conjugates were lyophilized and quantified by the AccQTag method.
Enzyme Digestion
Peptide samples, incubated with trypsin, were collected and analyzed as before.60 For human serum digestion, peptide [1000 μM, 2% dimethyl sulfoxide (DMSO) in filtered H2O] was added to human serum (Sigma, St Louis, Missouri) in a 1:1 ratio and incubated at 37 °C with gentle shaking. At 0 min and 24 h, aliquots were taken and proteolysis was stopped with CH3CN (80% in filtered H2O), chilled at 0 °C for 15 min to precipitate serum proteins, and centrifuged at 13,000 rpm at 4 °C for 10 min. The supernatant was collected and analyzed by analytical RP-HPLC and LC–MS as described before,60 with a 0–95% linear gradient of CH3CN over 15 min. All experiments are performed in duplicate and data were fitted with GraphPad Prism.
In Vitro Biochemical and Molecular Assays
Expression Vectors, HEK-293T Cell Culture, and Transient Transfection
Sequences encoding YFP Venus protein amino acid residues 1–155 and 156–238 were subcloned in the pcDNA3.1 vector to obtain the YFP Venus hemitruncated proteins. The human cDNAs for 5HT2AR and CB1R, cloned into the pcDNA3.1, were amplified and subcloned as described18 to give 5HT2AR-cYFP and CB1R-nYFP. Human embryonic kidney (HEK-293T) cells obtained from ATCC were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 2 mM l-glutamine, 100 μg/mL sodium pyruvate, 100 U/mL penicillin/streptomycin, MEM nonessential amino acid solution (1/100), and 5% (v/v) heat-inactivated fetal bovine serum (FBS) (all supplements from Invitrogen, Paisley, Scotland, UK). Cells were maintained at 37 °C in an atmosphere of 5% CO2. HEK-293T cells were transfected with the corresponding fusion protein cDNA by the polyethylenimine (Sigma) method, as described before.18
BiFC Assay
HEK-293T cells, after 48 h transient cotransfection with the cDNA encoding for 5HT2AR, fused to c-YFP, and CB1R fused to n-YFP (4 μg of cDNA for each construct), were treated or not with the indicated peptides (4 μM) for 4 h at 37 °C. To quantify protein-reconstituted YFP Venus expression, cells (20 μg protein) were distributed in 96-well microplates (black plates with a transparent bottom, Porvair, King’s Lynn, UK), and emission fluorescence at 530 nm was read in a Fluo Star Optima fluorimeter (BMG Labtechnologies, Offenburg, Germany) equipped with a high-energy Xe flash lamp, using a 10 nm bandwidth excitation filter at 400 nm reading. Protein fluorescence expression was determined as fluorescence of the sample minus the fluorescence of cells not expressing the fusion proteins (basal). Cells expressing 5HT2AR-cVenus and nVenus or CB1R-nVenus and cVenus showed similar fluorescence levels to nontransfected cells.
cAMP Production and ERK-1/2 Phosphorylation Assays
For cAMP production, homogeneous time-resolved fluorescence energy transfer (HTRF) assays were performed as previously described.18 Cells (1000 cells/well) growing in medium containing 50 μM zardeverine were pretreated with the CB1R antagonist, RIM (1 μM, RIM), and the 5-HT2AR antagonist, MDL 100,907 (300 nM, MDL), or the corresponding vehicle in white ProxiPlate 384-well microplates (PerkinElmer, Waltham, Massachusetts, US) at 25 °C for 20 min and stimulated with the CB1 agonist, WIN 55,212-2 (100 nM, WIN) and the 5HT2A agonist, DOI (100 nM, DOI) for 15 min before adding 0.5 μM FK, or vehicle and incubated for an additional 15 min period. Fluorescence at 665 nm was analyzed on a PHERAstar Flagship microplate reader equipped with an HTRF optical module (BMG Lab technologies, Offenburg, Germany).
For ERK-1/2 phosphorylation assay, HEK-293 cells (30.000 cells/well) seeded in 96-well poly-d-lysine-coated plates (Sigma-Aldrich, Madrid, Spain) were pretreated at 25 °C for 15 min with RIM (1 μM, RIM) and MDL 100,907 (300 nM, MDL) or the corresponding vehicle and stimulated for an additional 7 min with the WIN 55,212-2 (100 nM, WIN) and DOI (100 nM, DOI). Phosphorylation was determined in white ProxiPlate 384-well microplates (PerkinElmer Life Sciences) by the α-screen bead-based technology using the amplified luminescent proximity homogeneous assay kit (PerkinElmer Life Sciences) and using the Enspire multimode plate reader (PerkinElmer Life Sciences). Phosphorylation is expressed in arbitrary units, ALPHAcounts, as measured by light emission at 520–620 nm of the acceptor beads.
bEnd.3 Cell Culture and Viability Assay
bEnd.3 brain endothelial cells (ATCC CRL-2299TM) were grown as a monolayer in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin solution (Gibco, USA). Cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37 °C (MCO-18AIC (UV), Sanyo, Japan), with daily medium replacement.
The cytotoxicity of peptides toward bEnd.3 cells seeded into a 96-well plate (Corning, USA) at 10,000 cells/100 μL/well was tested using CellTiter-Blue cell viability assay.31 IC50 values were determined with GraphPad Prism using a log(inhibitor) vs normalized response. Experiments were performed on different days using independently grown cell cultures.
In Vitro BBB Translocation and Integrity Studies
The translocation capabilities of 5(6)-carboxyfluorescein-labeled peptides were determined using a protocol previously described.30,31 After the experiment, the integrity of the endothelial barrier was checked by measuring the permeability of a 4 kDa fluorescently labeled dextran (FD4) as previously described.25 Experiments were carried out on different days using independently grown cell cultures.
Confocal Microscopy
The internalization of 5(6)-carboxyfluorescein-labeled peptides was evaluated using confocal microscopy as previously described.31 The acquisition was done on a confocal point-scanning Zeiss LSM 880 microscope (Carl Zeiss, Germany) equipped with an alpha Plan-Apochromat X 63 oil immersion objective (1.40 numerical aperture). A diode 405–30 laser was used to excite Hoechst 33342 (Sigma-Aldrich, Spain) and CellMask Deep Red plasma membrane stain (Thermo Fisher, USA). The 488 nm line from an Ar laser was used to excite labeled peptides. In the normal confocal mode, X 0.6 zoom images were recorded at 1024 × 1024 resolution. ZEN software was used for image acquisition and Fiji software for image processing.
In Vivo Behavior and Pain Response Assays
Memory Impairment Measurements
On day 1 (Habituation), mice were habituated for 9 min to the V-maze in which the task was performed. The following day (Training), mice were placed in the V-maze for 9 min, two identical objects were presented and the time that the mice spent exploring each object was recorded. Mice were again placed in the maze 24 h later for 9 min (Test), one of the familiar objects was replaced with a novel object and the total time spent exploring each of the two objects (novel and familiar) was recorded. Object exploration was defined as the orientation of the nose to the object at less than 2 cm. A discrimination index was calculated as the difference between the time spent exploring the novel or familiar object divided by the total time exploring the two objects. A higher discrimination index is considered to reflect greater memory retention for the familiar object.
Peptide or vehicle were administered by IV (20 mg/kg) or oral route (5, 10, and 20 mg/kg) immediately after the training session. Thirty min after peptide administration, mice received an IP injection of vehicle (5% v/v of ethanol, 5% v/v of Cremophor-EL, and 90% v/v saline) or 3 mg/kg of THC dissolved in vehicle.
Hot-Plate Test
THC-induced analgesia was measured using a hot-plate meter (Hot/Cold Plate Test, Bioseb, USA) 60 min after IP injection of either THC dissolved in IP vehicle or vehicle alone. Peptide or vehicle were administered by IV (20 mg/kg) or oral route (5, 10 and 20 mg/kg) 30 min before THC. The plate was heated to 52 ± 0.5 °C and the time (in s) until mice showed a jumping response on the plate was recorded. A cutoff time of 240 s was set to prevent tissue damage.
Mice Immunization, Bleedings, and Serum Analysis by ELISA
Experiments were performed at the ICTS “NANBIOSIS” Custom Antibody Service (CAbS, CIBER-BBN, IQAC-CSIC). The immune response to 17 was assessed in outbred Swiss ICR (CD-1) mice strain Hsd:ICR (CD-1) (Envigo RMS, The Netherlands), following the immunization schedule in Figure 7. Animals were housed under standard conditions in the CID-CSIC animal facility. Animal experimental procedures were conducted in accordance with protocols approved by CSIC Committee on Ethics of Animal Experiments and Biosafety, as well as the Spanish National Committee on Ethics and Animal Welfare. Briefly, a first group of 10 female mice received IV doses of 17 (200 μL, 20 mg/kg, PBS + 2% DMSO), a second group of 5 mice received IV doses of 17-KLH conjugate (200 μL, 30 μg) and a third group of 5 mice received IVdoses of PBS (200 μL, negative control). The 17-KLH conjugate, administered with Freund’s adjuvant, was also used to generate polyclonal antibody (positive control) (data not shown). Serum samples were collected by tail vein bleeds on days 0, 9, and 19. The animals were humanely sacrificed on day 19. The peptide-specific humoral response was determined by indirect ELISA. Microtiter plates (Nunc) were coated with the 17-BSA conjugate (1 μg/mL in coating buffer, 100 μL/well) overnight at 4 °C. The next day, plates were washed and antiserum samples (diluted in PBST, serial dilutions starting at 1/100, 100 μL/well) were added and incubated for 30 min at r.t.. The plates were again washed and a solution of anti-IgG–HRP (1/4000 in 10 mM PBST) was added to the wells (100 μL/well) and incubated for 30 min at r.t.. After another wash, the tetramethylbenzidine substrate solution was added (100 μL/well) to plates. Color development was stopped with 4 N H2SO4 (50 μL/well) after 30 min at r.t., and absorbance read at 450 nm. Titers in a log10 scale were expressed as the reciprocal of the last dilution giving the absorbance recorded in the control wells (serum at day 0) plus 2 SD.
Statistical Analysis
Experimental data were managed and analyzed with GraphPad Prism software version 9 (San Diego, CA, USA) or IBM SPSS Statistics version 27.0 (IBM Corp., NY, USA). P-values lower than 0.05 were considered statistically significant.
Acknowledgments
The authors thank Dr. Núria Pascual from Custom Antibody Service (CAbS, CIBER-BBN, IQAC-CSIC) for help with immunizations.
Glossary
Abbreviations
- BiFC
bimolecular fluorescence complementation
- BBB
blood–brain barrier
- CB1R
cannabinoid receptor 1
- CNS
central nervous system
- CPP
cell-penetrating peptide
- DOI
2,5-dimethoxy-4-iodoamphetamine
- FK
forskolin
- GPCR
G-protein-coupled receptor
- ICV
intracerebroventricular
- IP
intraperitoneal
- IV
intravenous
- KLH
keyhole limpet hemocyanin
- MD
molecular dynamics
- MDL
MDL 100,907
- NSAID
nonsteroidal anti-inflammatory drug
- r/e
retro-enantio
- 5HT2AR
serotonin (5-hydroxytryptamine, 5-HT) receptor 2A
- THC
tetrahydrocannabinol
- TM
transmembrane
- WIN
WIN 55,212-2
- YFP
yellow fluorescent protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00484.
Design and synthesis of TMs 5 and 6 short analogues; HIV-Tat replacement by shorter CPPs; HIV-Tat replacement by shorter CPPs; applying CPP stereochemistry change; applying configurational switch and sequence reversal for last-stage lead optimization; statistical analyses used in signaling experiments; stabilities of the peptide-receptor complexes; effect of CPP stereochemistry on trypsin digestion; HPLC analysis of serum-incubated peptides 17 and 18; analytical data; and molecular formula strings (PDF)
Crystal structure of the μ-opioid receptor (PDB)
Molecular formula strings (CSV)
Author Contributions
M.G. and E.M. contributed equally. V.C., L.P., R.M., and D.A. are cosenior authors.
Rhodes Pharmaceuticals (L.P., Coventry, R.I., USA; 2016–2017), Marie Skłodowska-Curie Research and Innovation Staff Exchange (call H2020-MSCA-RISE-2014, 2015–2019), the Spanish Ministry of Economy and Innovation with FEDER funds (SAF2017-87629-R, PID2019-109240RB-I00), and the Fundació Bancària La Caixa (CaixaImpulse 2018 call; 2018–2020).
The authors declare no competing financial interest.
Notes
Authors will release the atomic coordinates upon article publication.
This paper was published on the Web on April 23, 2021. Additional corrections were received post ASAP, and the corrected version was reposted on April 23, 2021.
Supplementary Material
References
- Dahlhamer J.; Lucas J.; Zelaya C.; Nahin R.; Mackey S.; DeBar L.; Kerns R.; Von Korff M.; Porter L.; Helmick C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults - United States, 2016. Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. 10.15585/mmwr.mm6736a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl L.; Seth P.; Kariisa M.; Wilson N.; Baldwin G. Drug and Opioid-Involved Overdose Deaths—United States, 2013-2017. Morb. Mortal. Wkly. Rep. 2018, 67, 1419–1427. 10.15585/mmwr.mm675152e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye A. D.; Jones M. R.; Kaye A. M.; Ripoll J. G.; Galan V.; Beakley B. D.; Calixto F.; Bolden J. L.; Urman R. D.; Manchikanti L. Prescription Opioid Abuse in Chronic Pain: An Updated Review of Opioid Abuse Predictors and Strategies to Curb Opioid Abuse: Part 1. Pain Physician 2017, 2, S93–S109. 10.36076/ppj.2017.s109. [DOI] [PubMed] [Google Scholar]
- Harirforoosh S.; Asghar W.; Jamali F. Adverse Effects of Nonsteroidal Antiinflammatory Drugs: An Update of Gastrointestinal, Cardiovascular and Renal Complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. 10.18433/j3vw2f. [DOI] [PubMed] [Google Scholar]
- Carter G. T.; Duong V.; Ho S.; Ngo K. C.; Greer C. L.; Weeks D. L. Side Effects of Commonly Prescribed Analgesic Medications. Phys. Med. Rehabil. Clin. N. Am. 2014, 25, 457–470. 10.1016/j.pmr.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Walker J. M.; Hohmann A. G. Cannabinoid Mechanisms of Pain Suppression. Handb. Exp. Pharmacol. 2005, 168, 509–554. 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- Valjent E.; Mitchell J. M.; Besson M.-J.; Caboche J.; Maldonado R. Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br. J. Pharmacol. 2002, 135, 564–578. 10.1038/sj.bjp.0704479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fonseca F. R.; Rubio P.; Menzaghi F.; Merlo-Pich E.; Rivier J.; Koob G. F.; Navarro M. Corticotropin-Releasing Factor (CRF) Antagonist [D-Phe12,Nle21,38,C Alpha MeLeu37]CRF Attenuates the Acute Actions of the Highly Potent Cannabinoid Receptor Agonist HU-210 on Defensive-Withdrawal Behavior in Rats. J. Pharmacol. Exp. Ther. 1996, 276, 56–64. [PubMed] [Google Scholar]
- Kandasamy R.; Dawson C. T.; Craft R. M.; Morgan M. M. Anti-Migraine Effect of (9)-Tetrahydrocannabinol in the Female Rat. Eur. J. Pharmacol. 2018, 818, 271–277. 10.1016/j.ejphar.2017.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron E. P.; Lucas P.; Eades J.; Hogue O. Patterns of Medicinal Cannabis Use, Strain Analysis, and Substitution Effect among Patients with Migraine, Headache, Arthritis, and Chronic Pain in a Medicinal Cannabis Cohort. J. Headache Pain 2018, 19, 37. 10.1186/s10194-018-0862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Porta C.; Bura S. A.; Llorente-Onaindia J.; Pastor A.; Navarrete F.; García-Gutiérrez M. S.; De la Torre R.; Manzanares J.; Monfort J.; Maldonado R. Role of the Endocannabinoid System in the Emotional Manifestations of Osteoarthritis Pain. Pain 2015, 156, 2001–2012. 10.1097/j.pain.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R.; Baños J. E.; Cabañero D. The Endocannabinoid System and Neuropathic Pain. Pain 2016, 157, S23–S32. 10.1097/j.pain.0000000000000428. [DOI] [PubMed] [Google Scholar]
- Donovan K. A.; Oberoi-Jassal R.; Chang Y. D.; Rajasekhara S.; Haas M. F.; Randich A. L.; Portman D. G. Cannabis Use in Young Adult Cancer Patients. J. Adolesc. Young Adult Oncol. 2020, 9, 30–35. 10.1089/jayao.2019.0039. [DOI] [PubMed] [Google Scholar]
- Maldonado R.; Berrendero F.; Ozaita A.; Robledo P. Neurochemical Basis of Cannabis Addiction. Neuroscience 2011, 181, 1–17. 10.1016/j.neuroscience.2011.02.035. [DOI] [PubMed] [Google Scholar]
- Moreira F. A.; Lutz B. The Endocannabinoid System: Emotion, Learning and Addiction. Addict. Biol. 2008, 13, 196–212. 10.1111/j.1369-1600.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- Castellano C.; Rossi-Arnaud C.; Cestari V.; Costanzi M. Cannabinoids and Memory; Animal Studies. Curr. Drug Targets: CNS Neurol. Disord. 2003, 2, 389–402. 10.2174/1568007033482670. [DOI] [PubMed] [Google Scholar]
- Puighermanal E.; Busquets-Garcia A.; Maldonado R.; Ozaita A. Cellular and Intracellular Mechanisms Involved in the Cognitive Impairment of Cannabinoids. Philos. Trans. R. Soc., B 2012, 367, 3254–3263. 10.1098/rstb.2011.0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñals X.; Moreno E.; Lanfumey L.; Cordomí A.; Pastor A.; de La Torre R.; Gasperini P.; Navarro G.; Howell L. A.; Pardo L.; Lluís C.; Canela E. I.; McCormick P. J.; Maldonado R.; Robledo P. Cognitive Impairment Induced by Delta9-Tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194 10.1371/journal.pbio.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S.; Casadó V.; Devi L. A.; Filizola M.; Jockers R.; Lohse M. J.; Milligan G.; Pin J.-P.; Guitart X. G Protein-Coupled Receptor Oligomerization Revisited: Functional and Pharmacological Perspectives. Pharmacol. Rev. 2014, 66, 413–434. 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze S. R.; Ho A.; Vocero-Akbani A.; Dowdy S. F. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572. 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S.; Ciruela F.; Casadó V.; Pardo L.. Oligomerization of G protein-coupled receptors: Still doubted?. In Oligomerization in Health and Disease: From Enzymes to G Protein-Coupled Receptors; Giraldo J., Ciruela F., Eds.; Academic Press, 2020; Vol. 169, pp 297–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinart D.; Moreno E.; Galindo L.; Cuenca-Royo A.; Barrera-Conde M.; Pérez E. J.; Fernández-Avilés C.; Correll C. U.; Canela E. I.; Casadó V.; Cordomi A.; Pardo L.; de la Torre R.; Pérez V.; Robledo P. Altered Signaling in CB1R-5-HT2AR Heteromers in Olfactory Neuroepithelium Cells of Schizophrenia Patients Is Modulated by Cannabis Use. Schizophr. Bull. 2020, 46, 1547–1557. 10.1093/schbul/sbaa038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo L.; Moreno E.; López-Armenta F.; Guinart D.; Cuenca-Royo A.; Izquierdo-Serra M.; Xicota L.; Fernandez C.; Menoyo E.; Fernández-Fernández J. M.; Benítez-King G.; Canela E. I.; Casadó V.; Pérez V.; de la Torre R.; Robledo P. Cannabis Users Show Enhanced Expression of CB1-5HT2A Receptor Heteromers in Olfactory Neuroepithelium Cells. Mol. Neurobiol. 2018, 55, 6347–6361. 10.1007/s12035-017-0833-7. [DOI] [PubMed] [Google Scholar]
- Neves V.; Aires-da-Silva F.; Morais M.; Gano L.; Ribeiro E.; Pinto A.; Aguiar S.; Gaspar D.; Fernandes C.; Correia J. D. G.; Castanho M. A. R. B. Novel Peptides Derived from Dengue Virus Capsid Protein Translocate Reversibly the Blood-Brain Barrier through a Receptor-Free Mechanism. ACS Chem. Biol. 2017, 12, 1257–1268. 10.1021/acschembio.7b00087. [DOI] [PubMed] [Google Scholar]
- Jastrzebska B.; Chen Y.; Orban T.; Jin H.; Hofmann L.; Palczewski K. Disruption of Rhodopsin Dimerization with Synthetic Peptides Targeting an Interaction Interface. J. Biol. Chem. 2015, 290, 25728–25744. 10.1074/jbc.m115.662684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. F.; Xu X.; Fan X.; Zhang C.; Wei Q.; Wang X.; Guo W.; Xing W.; Yu J.; Yan J.-L.; Liang H.-P. A Cell-penetrating Peptide Suppresses Inflammation by Inhibiting NF-κB Signaling. Mol. Ther. 2011, 19, 1849–1857. 10.1038/mt.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto-Escuela D. O.; Romero-Fernandez W.; Mudó G.; Pérez-Alea M.; Ciruela F.; Tarakanov A. O.; Narvaez M.; Di Liberto V.; Agnati L. F.; Belluardo N.; Fuxe K. Fibroblast Growth Factor Receptor 1- 5-Hydroxytryptamine 1A Heteroreceptor Complexes and Their Enhancement of Hippocampal Plasticity. Biol. Psychiatry 2012, 71, 84–91. 10.1016/j.biopsych.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Najjar K.; Erazo-Oliveras A.; Brock D. J.; Wang T.-Y.; Pellois J.-P. An L- to d-Amino Acid Conversion in an Endosomolytic Analog of the Cell-Penetrating Peptide TAT Influences Proteolytic Stability, Endocytic Uptake, and Endosomal Escape. J. Biol. Chem. 2017, 292, 847–861. 10.1074/jbc.m116.759837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaco M.; Valle J.; da Silva R.; Correia J. D. G.; Castanho M. A. R. B.; Andreu D.; Neves V. DPepH3, an Improved Peptide Shuttle for Receptor-Independent Transport across the Blood-Brain Barrier. Curr. Pharm. Des. 2020, 26, 1495–1506. 10.2174/1381612826666200213094556. [DOI] [PubMed] [Google Scholar]
- Cavaco M.; Pérez-Peinado C.; Valle J.; Silva R. D. M.; Correia J. D. G.; Andreu D.; Castanho M. A. R. B.; Neves V. To What Extent Do Fluorophores Bias the Biological Activity of Peptides? A Practical Approach Using Membrane-Active Peptides as Models. Front. Bioeng. Biotechnol. 2020, 8, 552035. 10.3389/fbioe.2020.552035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B.; Juvvadi P.; Andreu D.; Ubach J.; Boman A.; Boman H. G. Retro and Retroenantio Analogs of Cecropin-Melittin Hybrids. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 3449–3453. 10.1073/pnas.92.8.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard G.; Benkirane N.; Zeder-Lutz G.; van Regenmortel M. H.; Briand J. P.; Muller S. Antigenic Mimicry of Natural L-Peptides with Retro-Inverso-Peptidomimetics. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 9765–9769. 10.1073/pnas.91.21.9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prades R.; Oller-Salvia B.; Schwarzmaier S. M.; Selva J.; Moros M.; Balbi M.; Grazú V.; de La Fuente J. M.; Egea G.; Plesnila N.; Teixidó M.; Giralt E. Applying the Retro-Enantio Approach to Obtain a Peptide Capable of Overcoming the Blood-Brain Barrier. Angew. Chem., Int. Ed. 2015, 54, 3967–3972. 10.1002/anie.201411408. [DOI] [PubMed] [Google Scholar]
- Vaissière A.; Aldrian G.; Konate K.; Lindberg M. F.; Jourdan C.; Telmar A.; Seisel Q.; Fernandez F.; Viguier V.; Genevois C.; Couillaud F.; Boisguerin P.; Deshayes S. A Retro-Inverso Cell-Penetrating Peptide for SiRNA Delivery. J. Nanobiotechnol. 2017, 15, 34. 10.1186/s12951-017-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almiñana N.; Alsina M. A.; Ortiz A.; Reig F. Comparative Physicochemical Study of SIKVAV Peptide and Its Retro and Retro-Enantio Analogues. Colloids Surf., A 2004, 249, 19–24. 10.1016/j.colsurfa.2004.08.041. [DOI] [Google Scholar]
- Howlett A. C.; Bidaut-Russell M.; Devane W. A.; Melvin L. S.; Johnson M. R.; Herkenham M. The Cannabinoid Receptor: Biochemical, Anatomical and Behavioral Characterization. Trends Neurosci. 1990, 13, 420–423. 10.1016/0166-2236(90)90124-s. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. Cannabinoid Pharmacology: The First 66 Years. Br. J. Pharmacol. 2006, 147, S163–S171. 10.1038/sj.bjp.0706406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y.; Mechoulam R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. 10.1021/ja01062a046. [DOI] [Google Scholar]
- Manzanares J.; Julian M.; Carrascosa A. Role of the Cannabinoid System in Pain Control and Therapeutic Implications for the Management of Acute and Chronic Pain Episodes. Curr. Neuropharmacol. 2006, 4, 239–257. 10.2174/157015906778019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda L. A.; Lolait S. J.; Brownstein M. J.; Young A. C.; Bonner T. I. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned CDNA. Nature 1990, 346, 561–564. 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Hudson B. D.; Hébert T. E.; Kelly M. E. Physical and Functional Interaction between CB1 Cannabinoid Receptors and Beta2-Adrenoceptors. Br. J. Pharmacol. 2010, 160, 627–642. 10.1111/j.1476-5381.2010.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher A. M.; Laprairie R. B.; Toguri J. T.; Kelly M. E. M.; Denovan-Wright E. M. Bidirectional Allosteric Interactions between Cannabinoid Receptor 1 (CB1) and Dopamine Receptor 2 Long (D2L) Heterotetramers. Eur. J. Pharmacol. 2017, 813, 66–83. 10.1016/j.ejphar.2017.07.034. [DOI] [PubMed] [Google Scholar]
- Rios C.; Gomes I.; Devi L. A. μ Opioid and CB1 Cannabinoid Receptor Interactions: Reciprocal Inhibition of Receptor Signaling and Neuritogenesis. Br. J. Pharmacol. 2006, 148, 387–395. 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores A.; Maldonado R.; Berrendero F. Cannabinoid-Hypocretin Cross-Talk in the Central Nervous System: What We Know so Far. Front. Neurosci. 2013, 7, 256. 10.3389/fnins.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid Receptor Homo- and Heterodimerization. Life Sci. 2005, 77, 1667–1673. 10.1016/j.lfs.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Navarro G.; Carriba P.; Gandí J.; Ciruela F.; Casadó V.; Cortés A.; Mallol J.; Canela E. I.; Lluis C.; Franco R.; Gandí J.; Ciruela F.; Casadó V.; Cortés A.; Mallol J.; Canela E. I.; Lluis C.; Franco R. Detection of Heteromers Formed by Cannabinoid CB1, Dopamine D2, and Adenosine A2AG-Protein-Coupled Receptors by Combining Bimolecular Fluorescence Complementation and Bioluminescence Energy Transfer. Sci. World J. 2008, 8, 1088–1097. 10.1100/tsw.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfalvi A.; Moreno E.; Cordomí A.; Cai N.-S.; Fernández-Dueñas V.; Ferreira S. G.; Guixà-González R.; Sánchez-Soto M.; Yano H.; Casadó-Anguera V.; Cunha R. A.; Sebastião A. M.; Ciruela F.; Pardo L.; Casadó V.; Ferré S. Control of Glutamate Release by Complexes of Adenosine and Cannabinoid Receptors. BMC Biol. 2020, 18, 9. 10.1186/s12915-020-0739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadó-Anguera V.; Cortés A.; Casadó V.; Moreno E. Targeting the Receptor-Based Interactome of the Dopamine D1 Receptor: Looking for Heteromer-Selective Drugs. Expert Opin. Drug Discovery 2019, 14, 1297–1312. 10.1080/17460441.2019.1664469. [DOI] [PubMed] [Google Scholar]
- Gomes I.; Ayoub M. A.; Fujita W.; Jaeger W. C.; Pfleger K. D. G.; Devi L. A. G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 403–425. 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Stevens R. C.; Roth B. L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Benito L.; Henry A.; Matsoukas M.-T.; Lopez L.; Pulido D.; Royo M.; Cordomí A.; Tresadern G.; Pardo L. The Size Matters? A Computational Tool to Design Bivalent Ligands. Bioinformatics 2018, 34, 3857–3863. 10.1093/bioinformatics/bty422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido D.; Casadó-Anguera V.; Pérez-Benito L.; Moreno E.; Cordomí A.; López L.; Cortés A.; Ferré S.; Pardo L.; Casadó V.; Royo M. Design of a True Bivalent Ligand with Picomolar Binding Affinity for a G Protein-Coupled Receptor Homodimer. J. Med. Chem. 2018, 61, 9335–9346. 10.1021/acs.jmedchem.8b01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Benito L.; Llinas Del Torrent C.; Pardo L.; Tresadern G. The Computational Modeling of Allosteric Modulation of Metabotropic Glutamate Receptors. Adv. Pharmacol. 2020, 88, 1–33. 10.1016/bs.apha.2020.02.004. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Yan W.; Chapman K.; Ramesh K.; Ferrell A. J.; Yin J.; Wang X.; Xu Q.; Rosenbaum D. M. Structure of an Allosteric Modulator Bound to the CB1 Cannabinoid Receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. 10.1038/s41589-019-0387-2. [DOI] [PubMed] [Google Scholar]
- Filizola M. Increasingly Accurate Dynamic Molecular Models of G-Protein Coupled Receptor Oligomers: Panacea or Pandora’s Box for Novel Drug Discovery?. Life Sci. 2010, 86, 590–597. 10.1016/j.lfs.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Pu M.; Qu L.; Han G. W.; Wu Y.; Zhao S.; Shui W.; Li S.; Korde A.; Laprairie R. B.; Stahl E. L.; Ho J.-H.; Zvonok N.; Zhou H.; Kufareva I.; Wu B.; Zhao Q.; Hanson M. A.; Bohn L. M.; Makriyannis A.; Stevens R. C.; Liu Z.-J. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X.-P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo M.; Navarro G.; Franco R.; Andreu D. A2A Receptor Homodimer-Disrupting Sequence Efficiently Delivered by a Protease-Resistant, Cyclic CPP Vector. Int. J. Mol. Sci. 2019, 20, 4937. 10.3390/ijms20194937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.