

Abstract
Kita‐kyushu lung cancer antigen 1 (KK‐LC‐1) is a kind of cancer‐testis antigen with anti‐tumor potential for clinical application. As a class of small‐molecule antigen conjugate, tumor‐targeting peptides have broad application prospects in gastric cancer diagnosis, imaging, and biological treatment. Here, we screened specific cyclic nonapeptides from a phage‐display library. The targeting peptide with the best affinity was selected and further verified in ex vivo tissue sections. Finally, enrichment of targeting peptides in tumor tissues was observed in vivo, and the dynamic biodistribution process was also observed with micro‐positron emission tomography (micro‐PET)/computed tomography (CT) imaging. Studies showed that the specific cyclic nonapeptide had a high binding capacity for KK‐LC‐1 protein. It has a strong affinity and specificity for KK‐LC‐1‐expressing positive tumor cells. Targeting peptides were significantly enriched at tumor sites in vivo, with very low normal tissue background. These findings demonstrated that the KK‐LC‐1 targeting peptide has high clinical potential.
Keywords: gastric cancer, KK‐LC‐1, molecular probe, targeted therapy, targeting peptide
The first publicly reported Kita‐kyushu lung cancer antigen 1 targeting peptide. The peptides showed excellent specificity in PET/CT.

1. INTRODUCTION
Every year there are nearly 1 million new cases of gastric cancer (GC) reported worldwide, making it the third leading cause of cancer‐related deaths and prompting the World Health Organization to declare it a public health concern.1 Although the incidence of GC has been substantially declining during the past decades, it remains a high incidence in East Asia.2 Kita‐kyushu lung cancer antigen 1 (KK‐LC‐1) is a novel tumor target with great therapeutic potential.3 The KK‐LC‐1 antigen was originally found in human lung cancer cells but was soon found to be expressed at high levels in breast cancer and GC.4, 5, 6 KK‐LC‐1 is a type of cancer‐testis antigen (CTA) that is not be expressed in most normal tissues. Differences between normal and tumor cells are key points of molecular targeted therapy.7 Small‐molecule targeting peptide engineering is a recent area of interest. Compared with antibodies, small‐molecule peptides have better tumor permeability, better immunogenicity, and pharmacokinetic parameters.8 It is worth noting that small‐molecule targeting peptides still have high affinity and selectivity for tumor antigens. These tumor‐specific drugs without the structure of immunoglobulins are also called new‐generation antibody‐like drugs.9
Phage‐display technology is an extremely powerful library construction and screening technology.10 It can efficiently construct a high‐capacity peptide library that exceeds traditional technology and at a low cost.11 By modifying the bacteriophage gene, a specific random amino acid fragment can be expressed on the surface of the bacteriophage capsid protein.12 A phage‐display peptide library is comprised of a heterogeneous mixture of billions of phage clones. Through multiple rounds of biological panning for specific antigens, the targeting peptides can be quickly and efficiently separated.13
In our research, we conducted multiple rounds of screening for CTA KK‐LC‐1 through a circular 9‐peptides phage‐display library, and obtained peptide sequences with advantageous enrichment. The ability to target KK‐LC‐1 was evaluated in vitro and in vivo (Figure 1). In the dynamic distribution study in vivo, the target peptide still showed specific binding specificity to KK‐LC‐1‐positive cancer cells and have no binding in other normal organs. This is the first KK‐LC‐1 targeting peptide reported. The results showed that this targeting peptide had excellent affinity, specificity, and excellent drug‐forming potential, and could be useful for labeling and clinical imaging of specific tumor cells in the future.
FIGURE 1.

Selection and verification of targeting peptides from phage‐display library. A, Construction of recombinant phages. By modifying phage DNA, the designed random peptide (red) was displayed on the minor coat protein III. B, Composition of the phage‐display library. A tube of recombinant phages with random foreign DNA fragments constitutes a library. C, Schematic representation of target‐coated surface. The target protein (KK‐LC‐1) was coated onto the Petri dish surface and incubated with the phage library. D, Workflow of phage‐display‐based screening in vitro. After 3 rounds of screening, target‐bound phages were enriched and isolated, while negative phages were discarded. E, Sequencing of selected peptides, and evaluation of affinity and specificity. The positive target‐bound phages were isolated and sequenced. Verification of binding capacity and specificity was performed by a variety of methods
2. MATERIALS AND METHODS
2.1. Phage library, screening, and phage enrichment
The PhD‐C7C™ Phage‐Display Peptide Libraries Kit was purchased from New England Biolabs. The C7C system is a loop‐constrained heptapeptide library, whose randomized segment is flanked by a pair of cysteine residues.14, 15 The titer of the library was 2 × 1013 pfu/mL, and the complexity was 1.2 × 109 individual clones.
Screening procedures were performed in accordance with the manufacturer's protocol, with some modifications. The solution of 100 µg/mL of KK‐LC‐1 target protein (Chinapeptide) was prepared in PBS buffer and was added into a sterile polystyrene Petri dish overnight at 4℃ with gentle agitation. The dish was blocked with 5% BSA to reduce non‐specific hydrophobic binding. After washing with PBST solution (1% Tween‐20 contained) 6 times, 1.5 mL of the phage‐display peptide library with 2 × 1012 pfu was added into the dish and incubated for 120 min at room temperature. Then the dish was slapped face down on a clean paper towel and washed immediately. This operation was repeated 10 times. Washing times were increased in different rounds of screening. The eluate solution (0.2 mol/L glycine‐HCl pH 2.2 and 1 mg/mL BSA) was added to the dish and incubated for 20 min at 4℃. After incubation, the phages were neutralized with 0.5 mL Tris‐HCl solution (pH 9.1).
Phage was titrated on tetracycline agar plates using a plaque‐forming assay, and amplified for the next rounds of panning. After 3 rounds of reiterative biopanning were performed, the selected phages were enriched sufficiently (Figure 1).
2.2. Binding enrichment assay and DNA sequencing of selected phage clones
Based on the titrate of the output phage solution, the enrichment rate was calculated as output pfu/input pfu, and the top‐3‐enrichment phage clones were collected and amplified for binding enrichment assay.
The sterile polystyrene Petri dish and the target protein were prepared as mentioned previously. Three individual phage solutions were added into the dish and incubated as mentioned previously. The unselected original phages were used as the control group. Phages were titrated using a plaque‐forming assay on tetracycline agar plates. The average enrichment rate was calculated as output pfu/input pfu.
Single‐stranded DNA (ssDNA) from the positive phages was prepared in accordance with a standard protocol. The ssDNA samples were sent to Shanghai Sangon Corp. for sequencing. DNA sequences of the positive phage clones were analyzed using the SnapGene program. The primer used for sequencing was 5′‐CCC TCA TAG TTA GCG TAA CG‐3′.
2.3. Cell lines, western blotting, and phage‐targeting ELISA
The human GC cell lines NUGC‐4 and AGS were obtained from China Pharmaceutical University. NUGC‐4 cells were grown in RPMI 1640 supplemented with 15% (v/v) fetal bovine serum (Gibco) at 37℃. AGS cells were grown in RPMI 1640 supplemented with 10% (v/v) FBS (Gibco) at 37℃.
NUGC‐4 knock down cells (NUGC‐4 KD) and AGS overexpression cells (AGS OE) were prepared by GenePharma Corp. for in vitro and in vivo assay. Small interfering RNA (siRNA) targeting KK‐LC‐1 mRNA was used to knock down endogenous KK‐LC‐1, and a random siRNA was used as the control (KD control). Lentivirus vectors overexpressing KK‐LC‐1 were used to prepare AGS overexpression cells, and blank vectors were used for the control (OE control).
Western blotting (WB) was used to identify KK‐LC‐1 expression in 2 cell lines. Cells were scraped off with a sterile cell spatula, and were heated with Loading Buffer (Beyotime) in boiling water for 5 min. The supernatant was obtained for WB. Proteins were then separated using 15% SDS‐PAGE, after that, proteins were transferred from SDS‐PAGE gels onto a PVDF membrane (Merck Millipore). The PVDF membrane was incubated overnight with mouse anti‐KK‐LC‐1 primary antibody (ab219971, Abcam). After washing the membrane 4 times, it was incubated with HRP‐conjugated goat anti‐mouse secondary antibody (Beyotime). Finally, the bands were detected using a chemiluminescent substrate kit (Biosharp) in Tanon‐5200 system (Tanon).
The target protein solutions (10 μg/mL) were coated overnight in a 96‐well polystyrene microtiter plates. They were washed with PBST, and blocked with blocking solution (Beyotime) immediately. The plates were slapped face down on a clean paper towel, and washed 6 times. The phage solutions and unselected original phages (control) (phages c. 1012 pfu) were added in specific wells and making a 4‐fold dilution in every single row, then they were incubated at room temperature for 2 h. Next, they were incubated with HRP‐conjugated anti‐M13 secondary antibody (11973‐MM05T‐H, Sino Biological) for 1 h. The tetramethylbenzidine substrate solution (TransGen) was added into the wells and the reaction lasted for c. 5 min. The reaction was finally stopped with 2 mol/L hydrochloric acid. The absorbance at 450 nm was detected using an automated ELISA reader (Tecan).
2.4. Affinity determination
The selected peptides were synthesized by Chinapeptide Corp. and purified using HPLC. The affinity determination was operated by surface plasmon resonance (SPR). The ligand and the analytes were synthesized individually. The ligand was dissolved in PBS (0.5 mg/mL), and the analytes were dissolved in Dimethyl sulfoxide (DMSO; 50 mg/mL). All the samples were sent to Genscript Corp. for SPR assay. The monoclonal antibody (mAb) used in the research was Abcam ab219971 and its affinity determination was measured using ELISA.16
2.5. Epitopes prediction of selected peptides
The CABS‐dock online sever was used to predict the possible residue‐residue contacts between the nonapeptide and KK‐LC‐1.17 The 3D structure of KK‐LC‐1 was predicted using trRosetta.18 The top 5 predicted models were chosen to determine the contacting cluster (residues closer than 3.5 Å in the peptide‐receptor complex were selected).
2.6. Anti‐tumor cytotoxicity assay
UNGC‐4 cells were grown in 96‐well polystyrene microtiter plates overnight. Then they were incubated with 2/20/200 μmol/L 1131 or CG7C peptide for 2 h at 37℃. Then 10 μL CCK‐8 solution (Vazyme) was added into each well and incubated for 2 h at 37℃. No peptide was added in the blank group. The absorbance of each well at 450 nm was detected with an automated ELISA reader (Tecan).
2.7. Peptide labeling and immunofluorescence
The selected peptides were synthesized and labeled with rhodamine B (RhoB) at the N‐terminus by Chinapeptide Corp. and purified using HPLC. In addition, a CG7C peptide (sequence 5′‐CGGGGGGGC‐3′) was synthesized, labeled, and purified in the same way as the selected peptides. The CG7C peptide was considered a negative control in this research.
UNGC‐4 and AGS cells were grown in 96‐well polystyrene microtiter plates overnight and fixed with 4% paraformaldehyde at room temperature for 20 min. Then they were washed with 0.3% Triton‐X100/PBS at room temperature. Samples were blocked in 1% BSA (w/v) in PBS for 60 min at 37℃. After washing with PBS, they were incubated with 20 μmol/L RhoB‐labeled peptides for 2 h and washed with PBS 3 times. This step was followed by nuclear staining with 4,6‐diamidino‐2‐phenylindole (DAPI). A microscope (Life Technologies) was used to visualize the cells in 96‐well polystyrene microtiter plates. In the competitive inhibition assay, firstly anti‐KK‐LC‐1 antibody (ab219971, Abcam) was incubated for 120 min at 37℃ as primary antibody, secondly, the FITC‐labeled goat anti‐mouse IgG (A0568, Beyotime) was incubated for 120 min at 37℃ as secondary antibody, thirdly, fluorescent peptide was incubated to bind competitively to NUGC‐4 cells.
Sections of GC tissues and normal tissues were collected from Nanjing Drum Tower Hospital. Tissues were obtained after surgery, washed twice with chilled PBS, and immediately embedded in an optimal cutting temperature medium. Samples were sent to Servicebio Corp. for immunohistochemistry (IHC) and immunofluorescence ex vivo. A microscope (Life Technologies) was used to feature the confrontation of tissues (Figure 5A).
FIGURE 5.

Peptide 1131 targeting to human tumor sections. A, Schematic illustration of ex vivo peptide targeting to human tumor and normal tissue sections. B, Sections of GC tissues and normal tissues stained with H&E (HC) and by IHC. 1131 DAPI, staining of the nucleus (blue). 1131 Merge, binding of RhoB‐1131 to sections (red). CG7C DAPI, also staining of the nucleus (blue). CG7C Merge, binding of RhoB‐CG7C to sections (red). Immunofluorescence signals (RhoB, red) were only observed on gastric adenocarcinoma and normal testis sections. Scale bar, 400 μm. C, Staining of normal tissue chip. HE, H&E staining. 1131 Merge, binding of RhoB‐1131 to sections (red). Blue is DAPI staining. CG7C Merge, binding of RhoB‐CG7C to sections (red). Blue is DAPI staining. 1131 showed no binding to normal tissue samples above. HE scale bar, 300 μm. Immunofluorescence scale bar, 400 μm
A 90‐well chip containing multiple samples from human tissues was used to verify the specificity of fluorescent peptides (Outdo; Figure 5A). The operations of incubation and nuclear staining were the same as mentioned previously.
2.8. Xenogeneic mouse models
The Ethics Committee of Drum Tower Hospital approved all experiments in this study. All mice procedures were carried out in compliance with guidelines set by the Animal Care Committee at Nanjing Drum Tower Hospital. All experimenters fully considered the principles of animal protection when feeding and caring for animals. No abuse or waste of animal resources occurred. All experiments were based on the principle of animal ethics. All efforts were made to minimize the number of animals used and their suffering.
Mice were randomized based on age and weight. For the subcutaneous tumor model, 3‐wk‐old male BALB/C‐nu/nu nude mice were injected subcutaneously with 106 cells (cells suspended in 100 μL normal saline; Figure 6A).
FIGURE 6.

Peptide 1131 shows tumor specificity in xenogeneic mouse models. A, Schematic illustration of xenogeneic mouse models and treatment. KD, knocked down; OE, overexpression. B, Cryosections of mice tumors and vital organs. Blue is DAPI staining. Red is the RhoB‐peptide fluorescence signal. Fluorescent peptide binding signals (red) were observed only in NUGC‐4, NUGC‐4 KD, KD control, and AGS OE model tumor samples. ns, normal saline. All cryosection images were DAPI and RhoB merged. Scale bar, 400 μm. C, Schematic illustration of mouse models and the treatment in micro‐PET/CT assay. D, Representative micro‐PET/CT images of 68Ga‐DOTA‐1131 in subcutaneous xenograft models. The block group was pre‐injected with non‐labeled 1131 as a blocking agent. KD, knocked down; OE, overexpression. Red arrows indicate the positions of tumors. Scale bar, 5 cm. E, Radioactivity uptake and biodistribution histogram of tumor and vital organs. NUGC‐4 model group data were used for ROI analysis. Data were analyzed using ImageJ software v,1.52. **P < .01
In the cryosection biodistribution study in vivo, 1 h after the tail vein injection of RhoB‐1131, mice were anesthetized and sacrificed. The tumors and vital organs (brain, heart, lung, liver, stomach, intestine, kidney, and bladder) were obtained by dissection for cryosection. Peptide CG7C and normal saline were used as the control. The KK‐LC‐1 negative cell line AGS was considered a negative control model. Tumors and vital organs were taken and sent to Servicebio Corp. for cryosection and immunofluorescence (peptide used with 20 μmol/L). The cryosections were observed using a fluorescence microscope (Life Technologies).
2.9. Micro‐positron emission tomography/computed tomography (micro‐PET/CT)
Here, 1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid (DOTA) was prepared to modify the peptide as an N‐terminal fusion (Figure S3). To construct 68Ga‐DOTA‐1131, the DOTA‐1131 peptide complex was synthesized and radiolabeled with 68Ga via a robust method within 15 min (Genscript).
The DOTA‐peptide was dissolve in 0.25 mol/L sodium acetate buffer. 68Ga was eluted from a 68Ge/68Ga generator (ITG Corp.) with 4 mL of 0.05 mol/L HCl. The 68Ga eluent was mixed with DOTA‐peptide solution (v/v = 4:1), and heated at 95℃ for 10 min. After cooling down, the reaction mixture was pushed through the C18 cartridge (Sep‐Pak C18 Plus Light Cartridge) and the eluent was collected in the waste tube. The product that remained on the C18 cartridge was eluted with 60% ethanol. The ethanol concentration in the product was no more than 10% before use. Radioactivity was calculated using the RM‐905a system (Yida).
Mice in this study were divided into 2 groups (Figure 6C). The block group was injected with 100 μL normal peptide solution by tail vein, and 60 min later both groups mice were injected with 100 μL 68Ga‐DOTA‐peptide solution by tail vein. The total radioactivity of each mouse injected was not more than 0.5 mCi. All mice were anesthetized by isoflurane inhalation before micro‐PET/CT imaging. Four mice at a time were imaged using micro‐PET/CT (Inveon). Imaging was performed at 30, 60, and 120 min after the mice were anesthetized. The energy window ranged from 384 to 638 keV. PET scans were acquired for 10 min in list mode followed by CT scans for 5 min. Micro‐PET/CT images were analyzed using Inveon Research Workplace software. Region of interest (ROI) was used for the analysis of biodistribution.19 The percentage of injected dose per gram of tissue (% ID/g) was calculated in this study.
2.10. Statistical analysis
GraphPad Prism 7.0 (GraphPad Software, Inc.) was used for statistical analysis of the data. The significance of the difference from the respective controls for each experimental test condition was assessed using the Student t test for each paired experiment. Analysis of variance (one‐way ANOVA) was used to evaluate significance of the difference in the anti‐tumor cytotoxicity assay. Quantification of fluorescence intensity was determined using ImageJ 1.52 software (National Institutes of Health).
3. RESULTS
3.1. The top‐3‐enrichment phage clones were identified
Three selection rounds were performed on KK‐LC‐1 recombinant protein (Figure 1), and the output/input ratio of phages (phage enrichment rate) was used to evaluate the phage recovery efficiency.20 In the last round of screening, phage enrichment was significantly improved. Finally, 3 of the most advantageous phage clones (1131/0931/0431) were selected for sequencing and subsequent verification (Figure 2A).
FIGURE 2.
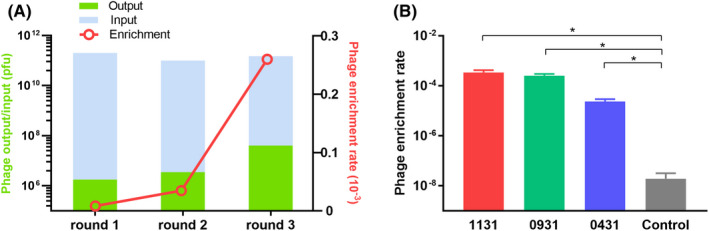
The enrichment of the top‐3 phage clones. A, The input and output of each round of screening. The phage enrichment rate was calculated as output number/input number. B, Histogram of the binding enrichment assay. The original library was used as a control. Compared with the control, 1131, 0931, and 0431 showed a significantly higher phage enrichment rate, indicating the effectiveness of screening. *P < .05
For preliminary confirmation of the binding capacity of 3 selected phage clones, the top‐3‐enrichment phage clones (1131/0931/0431) were collected and amplified for binding enrichment assay. The original phage library was used as a control group in this assay. Compared with the control, phage clones 1131, 0931, and 0431 were shown to have a significantly higher phage recovery rate (Figure 2B), indicating the effectiveness of screening. Therefore, the 3 phage clones were considered positive clones.
3.2. NUGC‐4 cells showed KK‐LC‐1 positive in WB
NUGC‐4 is a cell line derived from gastric signet ring cell carcinoma, while AGS is derived from gastric adenocarcinoma. High expression of KK‐LC‐1 was observed in NUGC‐4 cells as predicted band size 13 kDa, whereas no KK‐LC‐1 was detected in the AGS cells (Figure 3A). We therefore took NUGC‐4 and AGS cell lines to be high KK‐LC‐1 expressing and non‐KK‐LC‐1 expressing controls, respectively.
FIGURE 3.
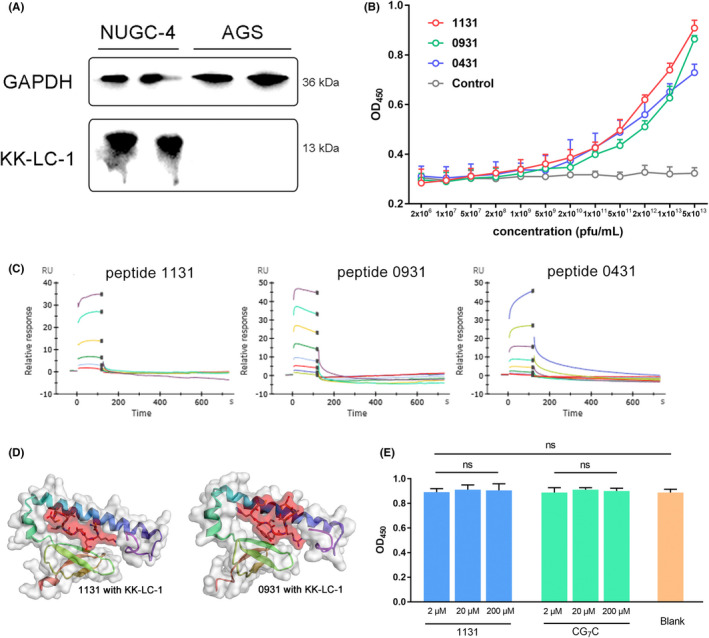
Peptides showed affinity at the molecular level. A, KK‐LC‐1 expression in NUGC‐4 and AGS cells. The NUGC‐4 cell lysate showed a band at 13 kDa, while the AGS cell lysate showed no band. GAPDH was used as an internal reference. B, Phage‐targeting ELISA. The original phage library was used as a control. The affinity of phage clones 1131, 0931, and 0431 was greater than the control when the phage titer was 5 × 109 pfu/mL or higher. In particular, phage clone 1131 showed the best affinity. C, Time‐response curve of BIAcore SPR analysis. The affinities of peptides 1131, 0931 and 0431 were measured respectively. D, Structure of peptides binding with KK‐LC‐1. Red sticks with surface indicate peptides and ribbon cartoon with a gray surface indicate KK‐LC‐1. E, Cytotoxicity experiments of 1131 and CG7C. Peptide 1131 showed no cytotoxicity to NUGC‐4 cells in 2, 20 and 200 μmol/L compared with CG7C control and blank. ns, not significant
3.3. Peptides showed high affinity and epitopes were predicted preliminarily
Phage‐targeting ELISA was performed to estimate the affinity of selected phage clones with KK‐LC‐1 protein. As shown (Figure 3B), the selected phage clones 1131, 0931, and 0431 had a higher affinity with the increase in phage titer. The original phage library was used as a control group in this assay. The affinity of phage clones 1131, 0931, and 0431 was significantly greater than that of the control when the phage titer was 5 × 109 pfu/mL or higher, which indicated that the binding capacity of selected phage clones with KK‐LC‐1 protein was sequence‐dependent. In particular, phage clone 1131 showed the best affinity among all the 3 selected phage clones.
To further estimate the binding of selected peptides with KK‐LC‐1 protein, the affinity of peptides to KK‐LC‐1 was subsequently characterized by SPR assay. Sequences from phage clones 1131, 0931, and 0431 were synthesized and named peptides 1131, 0931, and 0431 respectively. The affinity estimated from SPR measurements was consistent with the phage‐targeting ELISA assay. In accordance with the SPR measurements (Figure 3C), peptide 1131 had a KD of 19.5 μmol/L, peptide 0931 had a KD of 13.1 μmol/L, and peptide 0431 had a KD of 28.2 μmol/L. Also, the affinity KD of mAb (Abcam ab219971) was measured as 2.38 nmol/L (Figure S1). We determined peptide 1131 and 0931 as the first‐class sequences for subsequent validation due to their greater affinity.
The 3D structure of peptides binding with KK‐LC‐1 were modeled (Figure 3D). The most possible KK‐LC‐1 epitope residues of 1131 interaction were 27L, 39Y, 41L, 45I, 49F, 59I, 63L, 87P. The most possible KK‐LC‐1 epitope residues of 0931 interaction were 25S, 27L, 39Y, 41L, 49F, 56Q, 59I, 63L, 70L (Figure S2). From Abcam official information, the epitope of mAb (ab219971) has already been identified STALALVRPSSSGLI (extracellular domain positions 14‐28).
3.4. Peptides showed affinity and specificity to human cancer cells in vitro
RhoB‐1131 and RhoB‐0931 showed abundant binding to NUGC‐4 cells, while in contrast there was no binding to AGS cells (Figure 4A). Also, RhoB‐1131 showed a better binding effect than RhoB‐0931. After quantification of fluorescence intensity, the mean ± standard error of the mean (SEM) was shown in the bar graph (Figure 4C). RhoB‐1131 had a better mean fluorescence intensity (MFI) than RhoB‐0931, while RhoB‐CG7C exhibited a small positive MFI with NUGC‐4 cells.
FIGURE 4.

Fluorescent peptides 1131 and 0931 showed specific targeting to NUGC‐4 cells. A, Fixed NUGC‐4 and AGS cells were incubated with fluorescent‐labeled peptides. CG7C was considered a negative control. DAPI, staining of the nucleus (blue); 1131, binding of fluorescent peptides to cells (red, RhoB). Compared with the control, strong fluorescence signals were observed in the 1131 and 0931 groups. Scale bar, 100 μm. B, Competitive inhibition assay. Fixed cells were incubated with mAb (Abcam ab219971) and fluorescent peptides. mAb, binding fluorescence of Abcam ab219971 (green). 1131, a competitive inhibition against mAb. Scale bar, 100 μm. C, Fluorescence intensity histogram of (A). MFI was analyzed using ImageJ software v.1.52. ****P < .0001. D, Fluorescence intensity histogram of (B). MFI was analyzed using ImageJ software v.1.52. *P < .05, **P < .01
In the mAb inhibition assay as shown (Figure 4B), the KK‐LC‐1 antibody (ab219971, Abcam) bound to NUGC‐4 cells with a FITC fluorescence signal. However, RhoB‐1131 and RhoB‐0931 also showed binding with a RhoB fluorescence signal, which indicates the binding capacity of fluorescent peptides. The MFI of the green and red fluorescence signals was calculated, and the SEM values are shown in the bar graph (Figure 4D). Binding of RhoB‐1131 to NUGC‐4 was found to be stronger than for RhoB‐0931. These findings demonstrated that peptide 1131 has specific binding to NUGC‐4. Therefore we determined peptide 1131 as the best sequence in our research and retained peptide 1131 for subsequent validation.
In the anti‐tumor cytotoxicity assay (Figure 3E), peptide 1131 was incubated with NUGC‐4 for 2 h but no significant difference was found in a concentration gradient. Also no significant difference was found compared with the control and blank. Peptide 1131 alone could have no anti‐tumor cytotoxicity but only tumor binding capacity.
3.5. Peptide 1131 showed affinity and specificity to human cancer sections ex vivo
To further verify the affinity and specificity of peptide 1131 ex vivo, sections of GC tissues and normal tissues were collected and stained with H&E. Compared with the CG7C control, peptide 1131 had significant immunofluorescence binding (red) to KK‐LC‐1‐positive gastric adenocarcinoma, while it showed no binding to most normal tissue samples (Figure 5B). This confirmed the specificity of peptide 1131 ex vivo. The targeting peptide had significant binding to the tumor but showed no off‐target effect on normal tissues. Also, binding to normal testis was performed. As a member of CTA, KK‐LC‐1 is richly expresses in testis tissues. RhoB‐1131 showed specifically binding to normal testis (Figure 5B).
Furthermore, a 90‐well chip containing multiple samples from human tissues was used to verify the specificity of fluorescent peptides (Figure 5C). After incubation with peptide 1131, the samples showed no binding signal. These findings demonstrated the affinity and specificity of peptide 1131, as it did not bind to human non‐tumor tissues but selectively bound to tumor tissues ex vivo.
3.6. RhoB‐labeled 1131 peptide specificity accumulated in xenogeneic mouse models
To investigate the biodistribution of peptide 1131 in vivo, xenogeneic mouse models were established (Figure 6A). As shown in Figure 6B, fluorescence signals (RhoB, red) were detected distinctly in NUGC‐4 tumors, but no signal was present in normal organs. In addition, no signal was detected in the CG7C group and the normal saline group. Also, the NUGC‐4 KD tumor showed a weak fluorescence signal compared with the KD control. The AGS OE tumor showed a strong fluorescence signal compared with OE control. These results demonstrated that peptide 1131 was greatly enriched in KK‐LC‐1‐positive tumors in vivo and no off‐target effect was detected.
3.7. Isotope‐labeled 1131 peptide showed high radioactivity uptake in xenogeneic mouse models
The radiolabeling efficiency of 68Ga‐DOTA‐1131 was over 70% with no further HPLC purification. As shown in Figure 6D, high focal accumulation was clearly visualized at tumor site at all three‐time points from 30 to 120 min in the non‐blocked group. In addition, no signal was visualized at tumor site in the blocked group. Compared with other models, NUGC‐4 KD model tumors had weaker radioactivity uptake. Also, some accumulation in kidneys and bladders demonstrated the major renal‐urinary clearance of the radiotracer. Analysis of radiotracer accumulated in ROI is shown in Figure 6E, the radioactivity uptake of tumor site and vital organs (muscle, brain, heart, lung, liver, digestive tract, and kidney) was calculated as %ID/g. The tumor sites showed peak radioactivity uptake and other organs, except the kidney, showed low radioactivity uptake. These findings demonstrated that the specific radioactivity uptake of KK‐LC‐1 positive tumors was dependent on peptide 1131.
4. DISCUSSION
In this study, we screened several candidate peptides from a phage library. Its affinity was confirmed by ELISA and SPR. In cellular immunofluorescence, the targeting peptide showed good tumor cell affinity. Validation in patient tumors and tissue sections also showed excellent specificity. In vivo, the peptides could be effectively enriched in the tumor with little systemic non‐specific binding. The enrichment process of target peptides in vivo was reproduced by micro‐PET/CT imaging. The radioactivity uptake of nuclide‐labeled peptides at the tumor site was obvious and higher than the background, which suggested the high specific uptake of peptides by tumor.
Targeting peptides have a variety of unique active advantages, such as good tumor penetration, high affinity, and stable structure. The KK‐LC‐1 antigen‐specific peptide obtained in this study is the first publicly reported KK‐LC‐1 small‐molecule conjugate. However, the analysis and modification of molecular structure may help to further improve its affinity and activity.21 Also, pharmacokinetic parameters can be further improved by adding modified groups, therefore prolonging plasma half‐life and improving clinical application value.22 Peptides showed competitive binding with mAb, epitope prediction indicated that there was a partial epitope overlap, and that could interfere with mAb binding. The anti‐tumor cytotoxicity assay indicated that peptide 1131 used alone could have no anti‐tumor cytotoxicity. Peptides were designed to target the tumor but simply binding with KK‐LC‐1 could not inhibit tumor growth. Peptide‐drug conjugate (PDC) could be a good way for peptide application.
In conclusion, we screened KK‐LC‐1 specific targeting binding peptides based on the phage‐display library, and a series of in vivo and in vitro experiments verified its excellent affinity and specificity. Targeting peptides showed excellent imaging, therapeutic potential, and smaller organ side effects. The PET/CT imaging reagents or coupling drugs based on the targeting peptide may have extremely high clinical application value. GC diagnostic reagents, imaging reagents, or therapeutic drugs based on targeting peptides may bring practical clinical benefits to more patients with GC in the near future.
DISCLOSURE
The authors declare no potential conflicts of interest.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
{kind=link}
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by grants from the National Natural Science Foundation of China (No. 81930080), and the Program of Jiangsu Provincial Key Medical Center (No. YXZXB2016002). The funding sources had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Yu X, Yan J, Chen X, et al. Identification of a peptide binding to cancer antigen Kita‐kyushu lung cancer antigen 1 from a phage‐display library. Cancer Sci. 2021;112:4335–4345. 10.1111/cas.15109
Xiaoxiao Yu and Jiayao Yan contributed equally to this work.
REFERENCES
- 1.Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635‐648. [DOI] [PubMed] [Google Scholar]
- 2.Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomark Prev. 2014;23(5):700‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukuyama T, Hanagiri T, Takenoyama N, et al. Identification of a new cancer/germline gene, KK‐LC‐1, encoding an antigen recognized by autologous CTL induced on human lung adenocarcinoma. Can Res. 2006;66(9):4922‐4928. [DOI] [PubMed] [Google Scholar]
- 4.Shida A, Futawatari N, Fukuyama T, et al. Frequent high expression of Kita‐Kyushu lung cancer antigen‐1 (KK‐LC‐1) in gastric cancer. Anticancer Res. 2015;35(6):3575‐3579. [PubMed] [Google Scholar]
- 5.Kondo Y, Fukuyama T, Yamamura R, et al. Detection of KK‐LC‐1 protein, a cancer/testis antigen, in patients with breast cancer. Anticancer Res. 2018;38(10):5923‐5928. [DOI] [PubMed] [Google Scholar]
- 6.Cohen AS, Khalil FK, Welsh EA, et al. Cell‐surface marker discovery for lung cancer. Oncotarget. 2017;8(69):113373‐113402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chau CH, Steeg PS, Figg WD. Antibody‐drug conjugates for cancer. Lancet. 2019;394(10200):793‐804. [DOI] [PubMed] [Google Scholar]
- 8.Li YI, Moysey R, Molloy PE, et al. Directed evolution of human T‐cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23(3):349‐354. [DOI] [PubMed] [Google Scholar]
- 9.Ma L, Wang C, He Z, Cheng B, Zheng L, Huang K. Peptide‐drug conjugate: a novel drug design approach. Curr Med Chem. 2017;24(31):3373‐3396. [DOI] [PubMed] [Google Scholar]
- 10.Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discovery Today. 2015;20(1):122‐128. [DOI] [PubMed] [Google Scholar]
- 11.Saw PE, Song EW. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell. 2019;10(11):787‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frenzel A, Schirrmann T, Hust M. Phage display‐derived human antibodies in clinical development and therapy. Mabs‐Austin. 2016;8(7):1177‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheehan J, Marasco WA. Phage and yeast display. Microbiology Spectrum. 2015;3(1):AID‐0028‐2014. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen KM, Kyneb MH, Alstrup AKO, et al. (68)Ga‐labeled phage‐display selected peptides as tracers for positron emission tomography imaging of Staphylococcus aureus biofilm‐associated infections: selection, radiolabelling and preliminary biological evaluation. Nucl Med Biol. 2016;43(10):593‐605. [DOI] [PubMed] [Google Scholar]
- 15.Altmann A, Sauter M, Roesch S, et al. Identification of a novel ITGalphavbeta6‐binding peptide using protein separation and phage display. Clin Cancer Res. 2017;23(15):4170‐4180. [DOI] [PubMed] [Google Scholar]
- 16.Eble JA. Titration ELISA as a method to determine the dissociation constant of receptor ligand interaction. J Vis Exp. 2018;132:57334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaszczyk M, Ciemny MP, Kolinski A, Kurcinski M, Kmiecik S. Protein‐peptide docking using CABS‐dock and contact information. Brief Bioinform. 2019;20(6):2299‐2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci USA. 2020;117(3):1496‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng GK, Ye JC, Zhang WG, et al. Integrin alpha6 targeted positron emission tomography imaging of hepatocellular carcinoma in mouse models. J Controlled Release. 2019;310:11‐21. [DOI] [PubMed] [Google Scholar]
- 20.Hou L, Zhu D, Liang YU, et al. Identification of a specific peptide binding to colon cancer cells from a phage‐displayed peptide library. Br J Cancer. 2018;118(1):79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai W, Shukla RS, Qin B, Li B, Cheng K. Development of a peptide‐drug conjugate for prostate cancer therapy. Mol Pharm. 2011;8(3):901‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou LI, Kong W‐P, Chuang G‐Y, et al. Preclinical development of a fusion peptide conjugate as an HIV vaccine immunogen. Sci Rep. 2020;10(1):3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material