

Abstract
Various molecular‐targeting drugs have markedly improved the treatment of patients with breast cancer. As yet, therapies for triple‐negative breast cancer are mainly cytotoxic agents. To investigate the novel therapy for triple‐negative breast cancer, we herein examined the effects of a new combination therapy comprising a RAF/MEK inhibitor CH5126766, also known as VS‐6766, which we originally discovered, and eribulin. The combination of CH5126766 and eribulin potently inhibited cell growth in the triple‐negative breast cancer cell lines tested. The underlying mechanism in the efficacy of this combination treatment in vitro and in vivo was due to enhanced apoptosis through the suppression of survivin and Bcl‐2 family proteins. We also showed the suppressed expression of programmed cell death ligand 1 (PD‐L1) in combination therapy in vivo. We found that combination therapy with eribulin and CH5126766 for triple‐negative breast cancer inhibited cell growth by apoptosis and raised a possibility that immune responses through suppression of PD‐L1 might partially contribute to inhibition of tumor growth, indicating the potential of this combination as a novel strategy for triple‐negative breast cancer.
Keywords: apoptosis, CH5126766/VS‐6766, eribulin, PD‐L1, TNBC
We showed that combination therapy with eribulin and CH5126766 for triple‐negative breast cancer inhibited cell growth by apoptosis and raised a possibility that immune responses, through suppression of programmed death ligand 1, might partially contribute to inhibition of tumor growth, indicating the potential of this combination as a novel strategy for triple‐negative breast cancer.

Abbreviations
- EMT
epithelial‐mesenchymal transition
- IFN
interferon
- NSCLC
non‐small‐cell lung cancer
- PD‐1
programmed cell death‐1
- PD‐L1
programmed cell death‐ligand 1
- PE
phycoerythrin
- TNBC
triple‐negative breast cancer
1. INTRODUCTION
Marked advances have been achieved in the development of molecular‐targeting drugs for breast cancer; however, the established systemic therapy for recurrent TNBC is still cytotoxic chemotherapy.1 Personalized therapies, such as poly (ADP‐ribose) polymerase (PARP) inhibitors and immunotherapies, have recently been used to treat TNBC patients,2, 3, 4 and could improve the poor outcome of TNBC in the future. Due to the variety and heterogeneity of TNBC, novel effective therapies are expected.5, 6
In our research on molecular‐targeting therapy for TNBC, we previously proposed combination therapy with the novel histone deacetylase inhibitor OBP‐801 and eribulin.7 Eribulin is a well‐tolerated chemotherapeutic drug for TNBC.8 The findings indicated the potential of eribulin to suppress the MAPK pathway.7 Therefore, we examined the effects of a new combination therapy comprising eribulin and a novel RAF/MEK inhibitor for TNBC that directly suppresses the MAPK pathway.
To develop a new strategy for the treatment of TNBC, we investigated whether the RAF/MEK inhibitor CH5126766, which was derived from our established cell‐based assay9 and now also known as VS‐6766, enhanced the antiproliferative effects of eribulin in TNBC cells. The efficacy of CH5126766 against solid tumors with activated MAPK pathways has already been assessed in a phase I study (ClinicalTrials.gov NCT02407509).10 We expected combination therapy with eribulin and CH5126766 to be an effective treatment for TNBC, particularly that with activated MAPK pathways. The PI3K‐Akt signaling pathway is one of the most fundamental oncogenic pathways, in addition to the MAPK pathway, and also contributes to the regulation of cell growth and survival.11 The suppression of the MAPK pathway has been shown to activate the Akt pathway.12, 13, 14, 15 Therefore, we investigated the effects of our recommended combination therapy on the Akt pathway.
Cancer immunotherapy has been a revolution and clinically meaningful for TNBC patients.4 The PD‐1/PD‐L1 axis is one of the most important immune checkpoint signals, and the overexpression of PD‐L1 has been reported in TNBC.16 We assessed the contribution of immune responses focused on PD‐L1 expression to the efficacy of our recommended combination therapy for TNBC. Several chemotherapeutic drugs have been reported to enhance PD‐L1 expression stimulated by IFNs in TNBC.17, 18 Moreover, suppression of the MAPK pathway by the first‐in‐class MEK inhibitor trametinib was previously reported to be immunosuppressive with the upregulated expression of PD‐L1.19 The PD‐1/PD‐L1 interaction has been reported to upregulate the MAPK and PI3K/Akt pathways in TNBC.20 Therefore, the immunoreactive efficacy of eribulin in combination with CH5126766, which is expected to suppress the MAPK pathway, is of interest.
2. MATERIALS AND METHODS
2.1. Reagents
Eribulin was purchased from Eisai Co., Ltd., and CH5126766/VS‐6766 was kindly provided by Chugai Pharmaceutical Co., Ltd. Paclitaxel was purchased from Selleck Biotech Co., Ltd., and trametinib/GSK1120212 was purchased from AdipoGen Life Science. These agents were dissolved in DMSO. Recombinant human IFN‐γ was purchased from Tonbo Biosciences.
2.2. Triple‐negative breast cancer cell lines and cell culture
The human breast cancer cell lines MDA‐MB‐231, Hs‐578‐t, and BT‐549 were obtained as cell lines of NCI‐60 from the NCI Developmental Therapeutics Program. SUM159PT and SUM229PE cells were purchased from Asterand Bioscience, and murine 4T1 cells were from ATCC. MDA‐MB‐231, BT‐549, and 4T1 cells were cultured in RPMI‐1640 with 10% FBS, 2 mmol/L glutamine, 50 units/mL penicillin, and 100 µg/mL streptomycin, and Hs‐578‐t cells in DMEM supplemented with 10% FBS, 4 mmol/L glutamine, 50 units/mL penicillin, and 100 µg/mL streptomycin. SUM159PT and SUM229PE cells were maintained in Ham’s F‐12 medium supplemented with 5% FBS, 1 µg/mL hydrocortisone, and 5 µg/mL insulin. All cell cultures were incubated at 37°C in 5% CO2.
2.3. Cell viability assay
Cell growth was evaluated using CCK‐8 (Dojindo), as previously reported.7, 21
2.4. Analyses of cell cycle and apoptosis
The nuclei of cells treated with agents were stained with propidium iodide. The DNA content in stained nuclei was analyzed by FACSCalibur (Becton Dickinson) as previously reported.7, 21
2.5. Colony formation assay
Cells were seeded on 6‐well plates. After incubation for 24 hours, cells were treated with each agent, and cultured for 7 days. Colonies were fixed in 10% formalin and stained in crystal violet. The area of stained colonies was quantified using ImageJ (US NIH).
2.6. Western blot analysis
Cells treated with agents were lysed with RIPA buffer, and western blotting was carried out as previously reported.7, 21 In the molecular analysis of tumor samples in vivo, single‐cell suspensions were prepared using the gentleMACS Octo Dissociator (Miltenyi Biotec). After tumors had been transformed to single cells, the cells obtained were treated as described above. Primary Abs were rabbit anti‐survivin (R&D Systems), anti‐Bcl‐2 and PD‐L1 (Abcam), anti‐Bcl‐xL, phosphorylated ERK and Akt, and anti‐total ERK and Akt (Cell Signaling Technology), and mouse anti‐β‐actin (Sigma‐Aldrich) Abs.
2.7. Quantitative RT‐PCR
Total cellular RNA extraction from cells and cDNA synthesis were undertaken as previously reported.7 A quantitative RT‐PCR analysis was carried out using QuantStudio3 (Applied Biosystems) with the cDNA and TaqMan probes (Thermo Fisher Scientific) to PD‐L1 (Hs00204257_m1) and GAPDH (Hs02758991_g1).
2.8. Detection of PD‐L1 (CD274) expression on cell surfaces
Cells treated with agents were harvested with or without the induction of IFN‐γ, washed with ice‐cold PBS, and resuspended in PBS with 1% BSA. Phycoerythrin‐conjugated anti‐human CD274 (B7‐H1) mAb (eBioscience; Thermo Fisher Scientific) and the PE‐conjugated IgG isotype control (eBioscience; Thermo Fisher Scientific) were then added. After incubation on ice for 30 minutes, cells were washed in ice‐cold PBS and analyzed by FACSCalibur.
2.9. Caspase assay
To investigate caspase activity during apoptosis, the Caspase‐Glo 3/7 assay (Promega) was used according to the guided protocol. Luminescence was recorded using SpectraMax iD3 (Molecular Devices) 60 minutes after the addition of Caspase‐Glo 3/7 reagent to the cells treated with agents.
2.10. In vivo
Murine breast cancer 4T1 cells were subcutaneously inoculated at 1 × 105 cells per mouse into female 6‐week‐old BALB/c mice (Shimizu Laboratory Supplies). Tumor sizes were measured using a caliper twice a week. Tumor volumes were calculated using the following formula: 1/2 × (length) × (width)2. The drug injection was started when the tumor volume reached approximately 40 mm3. Mice were randomly assigned to each group (n = 5 per group). The treatments in each group were vehicle, 0.8 mg/kg eribulin, 0.4 mg/kg CH5126766, and both drugs. Eribulin was dissolved in saline and injected into the tail vein. Mice were injected twice a week for 3 weeks with eribulin. CH5126766 was dissolved in 20% hydroxypropyl‐β‐cyclodextrin/saline. Mice were treated with CH5126766 p.o. five times a week for 3 weeks. We observed mice for more 3 weeks after termination of treatments to study their survival. When tumors were resected 6 days after the initiation of the treatment for molecular analysis, mice were killed. All animal procedures were carried out under the approval of the institutional guidelines of Kyoto Prefectural University of Medicine. Data are represented as the mean ± SD. Statistical analyses of body weight and tumor volume were carried out using a two‐way ANOVA by GraphPad Prism version 8.4., and samples were considered to be significantly different at P < .0001.
2.11. Combination index
Combination index values were analyzed using CalcuSyn software (Biosoft) as previously reported.7 Synergism is defined as more than the expected additive effect with combination index less than 1.0.
2.12. Statistical analysis
Data in vitro are represented as the mean ± SD in triplicate. Samples were considered to be significantly different at P < .05. Statistical analyses were undertaken using multiple Bonferroni comparisons.
3. RESULTS
3.1. CH5126766 inhibits growth of TNBC cells with cell cycle arrest and induction of apoptosis
We evaluated the effects of the novel RAF/MEK inhibitor CH5126766 in several TNBC cell lines. We used human MDA‐MB‐231, SUM159PT, SUM229PE, Hs‐578‐t, and BT‐549 cells, in which we investigated the activated MAPK pathway (Figure S1). CH5126766 inhibited TNBC cell growth as shown in Figure 1A; however, its effects at high concentrations were not strong. We then examined these inhibitory effects on the cell cycle and apoptosis in several TNBC cells. The results showed that CH5126766 induced apparent G1‐phase arrest with a decrease in the S‐phase population in MDA‐MB‐231 and SUM159PT cells, and also caused slight G2/M‐phase arrest (Figures 1B and S2). Moreover, CH5126766 increased the percentage of sub‐G1 cells (Figure 1C). As shown in Figure 1C, the dependence of the inhibition of growth on apoptosis differed among the cell lines examined, and apoptosis was partially dependent on the caspase pathway in MDA‐MB‐231 cells (Figure S3). Therefore, additional agents appear to be necessary for the development of more effective treatments for TNBC.
FIGURE 1.
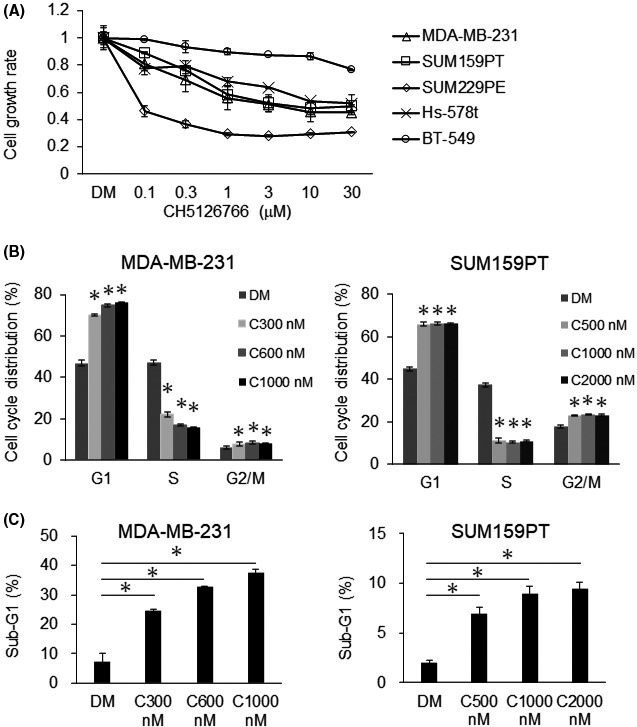
CH5126766 inhibits the growth of triple‐negative breast cancer (TNBC) cells with cell cycle arrest and induction of apoptosis. A, Several TNBC cells were treated with the indicated concentrations of CH5126766 for 72 h. The inhibition of cell growth was evaluated using CCK‐8. The cell growth rate of each treatment was calculated by comparisons with DMSO (DM) samples. B, Cell cycle populations treated with CH5126766 were analyzed by flow cytometry. Cell cycle populations at the indicated concentrations of CH5126766 were compared with DMSO samples. MDA‐MB‐231 and SUM159PT cells were treated for 24 h. C, Apoptotic cells were analyzed as the sub‐G1 population by flow cytometry. Sub‐G1 populations with the CH5126766 treatment were compared with DMSO samples. MDA‐MB‐231 cells were treated with DMSO (DM) or the indicated concentrations of CH5126766 (C300‐C1000) for 72 h, as well as SUM159PT cells (C500‐C2000) for 24 h. Columns, mean values of triplicate data; bars, SD. *P < .05
3.2. Combination treatment with eribulin and CH5126766 inhibits growth of TNBC cells
We previously examined the inhibition of cell growth by eribulin in several human TNBC cell lines. The findings showed that eribulin inhibited cell growth in TNBC cell lines in a dose‐dependent manner, and downregulated the MAPK pathway.7 We herein investigated whether CH5126766, which directly suppresses the MAPK pathway, enhanced the efficacy of eribulin against TNBC cells. As shown in Figure 2A, the growth of MDA‐MB‐231 and SUM159PT cells was synergistically and more strongly inhibited by the combination of these agents than by either agent alone (Figure S4). As the combined effects in SUM159PT cells were not as strong as those in MDA‐MB‐231 cells, we also undertook the colony formation assay using SUM159PT cells. In a long incubation, combined effects more potently inhibited the growth of SUM159PT cells (Figure 2B,C). We also compared our recommended treatment with the combination of other microtubule inhibitor paclitaxel and MEK inhibitor trametinib. As shown in Figure S5, cotreatment of paclitaxel plus CH5126766 and eribulin plus trametinib showed synergistic effects in MDA‐MB‐231 cells, but not in SUM159PT cells. However, cotreatment of paclitaxel plus trametinib did not show synergistic effects in MDA‐MB‐231 or SUM159PT cells.
FIGURE 2.
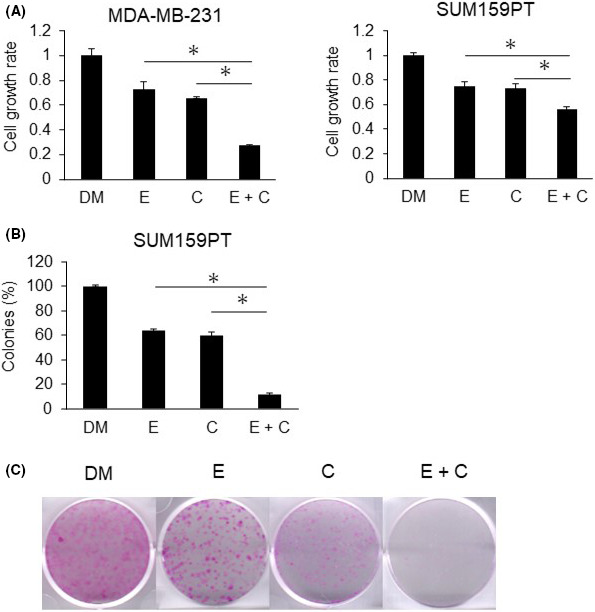
Combination of eribulin and CH5126766 inhibits the growth of triple‐negative breast cancer cells. MDA‐MB‐231 cells were treated with 0.5 nmol/L eribulin and 300 nmol/L CH5126766, while SUM159PT cells were treated with 3.5 nmol/L eribulin and 500 nmol/L CH5126766. A, Cells were treated with DMSO (DM) or eribulin (E) and/or CH5126766 (C) for 72 h. The inhibition of cell growth was evaluated using CCK‐8. The cell growth rate of each treatment was calculated by comparisons with DMSO samples. B, Efficacy of the combined treatment in SUM159PT cells was investigated using a colony formation assay. Cells were treated with DMSO or eribulin and/or CH5126766. Duration of drug exposure was 144 h. The colony area was assessed by ImageJ. A, B, Columns, mean values of triplicate data; bars, SD. *P < .05. C, Representative images of colonies in the assessment of B, are shown
3.3. Combination treatment with eribulin and CH5126766 induces apoptosis with suppression of survivin and Bcl‐2 family proteins
We examined the effects of combination treatment on the cell cycle and apoptosis because eribulin inhibits the growth of TNBC cells through G2/M‐phase arrest and induction of apoptosis.7, 22, 23 As a result, apoptosis was more potent with the combination of eribulin and CH5126766 than with either agent alone (Figure 3A), but the enhancement of cell cycle arrest was not observed to be more potent (Figure S6). To clarify the mechanisms underlying the enhancement of apoptosis by the combined treatment, we investigated the expression of various apoptotic‐related proteins. We initially assessed the expression of survivin, which is known to be upregulated by eribulin at the protein and mRNA levels.7 CH5126766 markedly suppressed eribulin‐induced increases in the expression of survivin (Figure 3B). We also investigated the expression of Bcl‐2 family proteins, which are major caspase‐dependent antiapoptotic proteins. The combination treatment suppressed Bcl‐2 and Bcl‐xL in MDA‐MB‐231 cells, but not in SUM159PT cells (Figure 3C).
FIGURE 3.
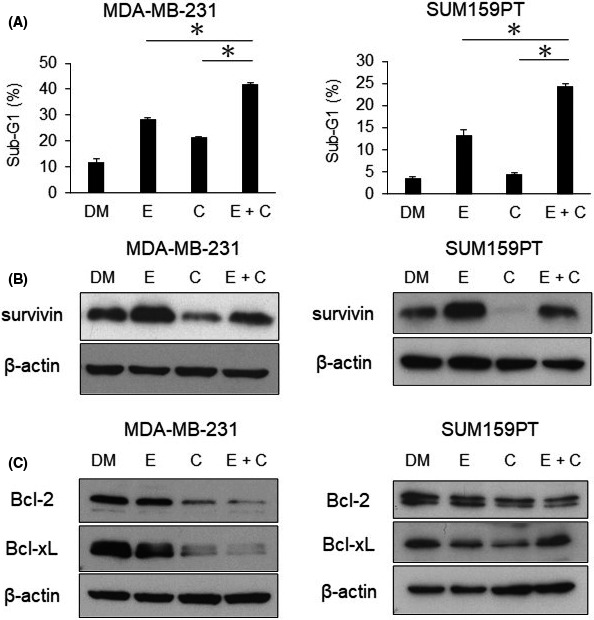
Combination treatment with eribulin and CH5126766 enhances apoptosis in triple‐negative breast cancer (TNBC). Cells were treated with DMSO (DM) or eribulin (E) and/or CH5126766 (C) for 48 h. MDA‐MB‐231 cells were treated with 0.5 nmol/L eribulin and 300 nmol/L CH5126766; SUM159PT cells were treated with 3.5 nmol/L eribulin and 500 nmol/L CH5126766. A, TNBC cells were treated with each drug for 72 h. The sub‐G1 population was analyzed by flow cytometry. The sub‐G1 population with combination treatment was compared with that of single treatment. Columns, mean values of triplicate data; bars, SD. *P < .05. B, Western blot for the evaluation of survivin expression. Cells were harvested after 48 h of the drug treatment. C, Western blot for the investigation of Bcl‐2 family protein expression. The time course was the same as that in (B)
3.4. Eribulin suppresses activation of the Akt pathway induced by CH5126766 and combination treatment decreases PD‐L1 expression
Activation of the Akt pathway represents a serious obstacle to the expanded use of MEK inhibitors for clinical cases. Therefore, we investigated the effects of the combination of eribulin and CH5126766 on the Akt and MAPK pathways in TNBC cells. The MAPK pathway was more strongly suppressed in MDA‐MB‐231 cells by the combination of eribulin and CH5126766 than by either agent alone, whereas its effects were similar to those by CH5126766 alone in SUM159PT cells (Figure 4A). However, eribulin suppressed the Akt pathway activated by CH5126766 in both cell types (Figure 4A). These results suggest that eribulin makes a significant contribution to the inhibitory effects on Akt pathway activation due to resistance to CH5126766.
FIGURE 4.
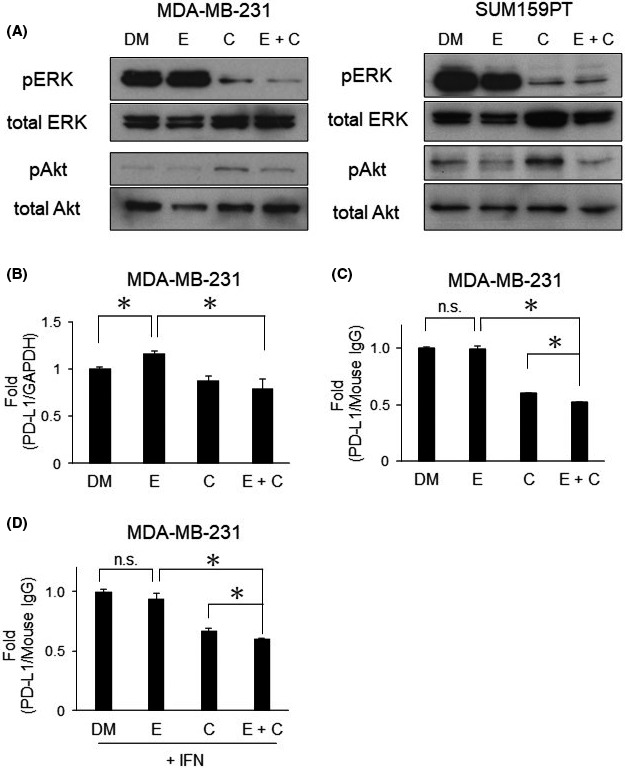
Eribulin suppresses the activated Akt pathway and the combination decreases programmed cell death‐ligand 1 (PD‐L1) expression in triple‐negative breast cancer cells. A, Regulation of the MAPK and Akt pathways was evaluated by western blotting. The samples in these figures were the same as those in Figure 3C. B, PD‐L1 expression at the mRNA level in MDA‐MB‐231 cells analyzed by quantitative RT‐PCR. C, PD‐L1 expression on the surface of MDA‐MB‐231 cells was analyzed by FACSCalibur. D, PD‐L1 expression on the surface of MDA‐MB‐231 cells treated with IFN‐γ (20 ng/mL) was analyzed by FACSCalibur. B‐D, Columns, mean values of triplicate data; bars, SD. *P < .05. MDA‐MB‐231 cells were treated with 0.5 nmol/L eribulin and 300 nmol/L CH5126766; SUM159PT cells were treated with 3.5 nmol/L eribulin and 500 nmol/L CH5126766. Cells were harvested after 48 h of the drug treatment. C, CH5126766; DM, DMSO; E, eribulin
We then investigated the local immunoreaction because of previous reports concerning immunosuppression in TNBC induced by chemotherapy17 as well as trametinib.19 We examined the expression of PD‐L1, a crucial immune‐related biomarker. The expression of PD‐L1 at the mRNA level was slightly upregulated by eribulin in MDA‐MB‐231 cells (Figure 4B), but was not upregulated at the protein level in the cell surface analysis of TNBC cells (Figures 4C,D and S7). We continued the analysis using stimulation with IFN‐γ. Gamma‐interferon is a cytokine that is secreted from activated T cells and is involved in oncoimmune responses with the upregulation of PD‐L1.24 Eribulin in combination with CH5126766 decreased PD‐L1 expression levels in TNBC cells regardless of the stimulation with IFN‐γ (Figures 4D and S7). We further evaluated the enhancement of apoptosis and immune responses by this combination therapy using an immunocompetent model.
3.5. Effects of combination therapy with eribulin and CH5126766 in a 4T1 syngeneic mouse model
We assessed the effects of combination therapy in vivo using murine 4T1 cells, which are highly metastatic TNBC cells. We initially confirmed the combined effects of eribulin and CH5126766 on 4T1 cells in vitro. We assessed the inhibitory effects of eribulin alone, CH5126766 alone (Figure S8), and also their combination on 4T1 cell growth (Figure 5A). As shown in Figure 5B, this combination was effective in 4T1 cells due to the enhancement of apoptosis, which was suggested to be induced by the suppression of Bcl‐2 and survivin (Figure 5C,D). Moreover, the combination of eribulin and CH5126766 more potently suppressed the MAPK pathway than either agent alone, and also overcame the resistance of the Akt pathway in 4T1 cells (Figure 5D), similar to human TNBC cells (Figure 4A). After the effects of the combined treatment had been confirmed in 4T1 cells in vitro, we undertook in vivo experiments. Cells were subcutaneously inoculated into female 6‐week‐old BALB/c mice. Mice assigned to each group were treated with vehicle, intravenous eribulin, oral CH5126766, or a combination of intravenous eribulin and oral CH5126766. The weights of mice did not significantly decrease during these therapies (Figure 6A). Tumor growth curves per treatment showed that combination therapy exerted stronger inhibitory effects on tumor growth than either agent alone (Figure 6B). Overall survival also improved in the group that received combination therapy (Figure 6C). We then undertook molecular and oncoimmunologic analyses of tumors in this model. Six days after the start of treatment, tumors were excised. Based on in vitro results, we investigated the expression levels of survivin, Bcl‐2 family proteins, and PD‐L1 in tumors. As shown in Figure 6D, the expression levels of survivin, Bcl‐2, Bcl‐xL, and PD‐L1 were reduced more by the combination of eribulin and CH5126766 than by either agent alone.
FIGURE 5.
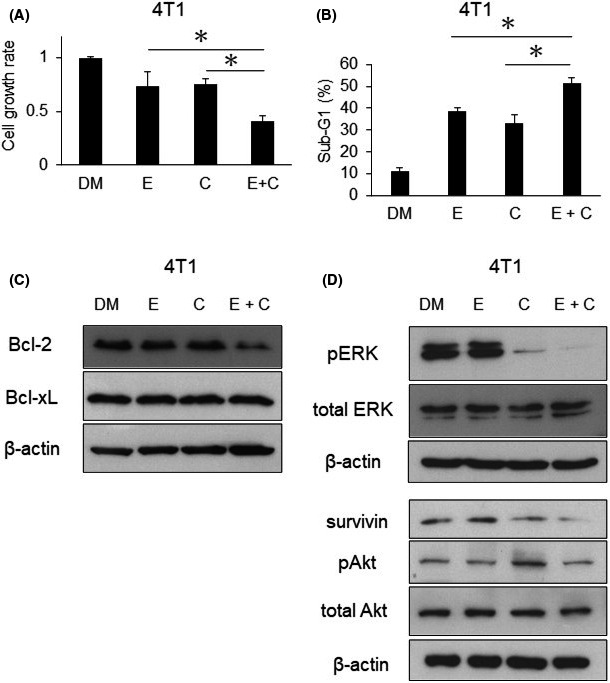
Eribulin enhances the efficacy of CH5126766 with apoptosis and overcomes resistance in 4T1 cells. A, Murine 4T1 cells were treated with DMSO (DM) or 20 nmol/L eribulin (E) and/or 300 nmol/L CH5126766 (C) for 48 h. Inhibition of cell growth was evaluated using CCK‐8. The cell growth rate of each treatment was calculated by comparisons with the DMSO samples. B, The sub‐G1 population of 4T1 cells treated with each drug was analyzed by FACSCalibur. The sub‐G1 population of combination treatment was compared with that of single treatment. C, Expression of Bcl‐2 family proteins was evaluated by western blotting. D, Expression of survivin and the regulation of the MAPK and Akt pathways were evaluated by western blotting. A, B, Columns, mean values of triplicate data; bars, SD. *P < .05. B‐D, Concentrations of drugs and the time course were the same as those in (A)
FIGURE 6.
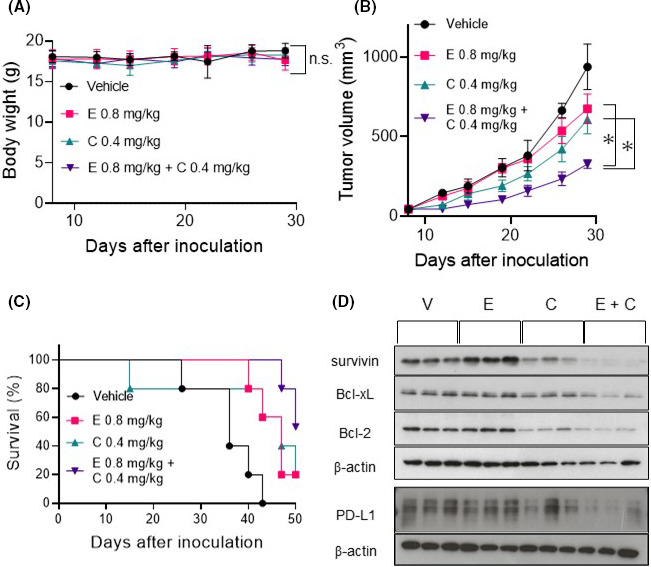
Combination therapy with eribulin and CH5126766 in the 4T1 syngeneic model of triple‐negative breast cancer. Mice assigned to each group were treated with vehicle (V), 0.8 mg/kg eribulin (E), 0.4 mg/kg CH5126766 (C), and both drugs. Eribulin was dissolved in saline and injected into the tail vein. Mice were injected with eribulin twice a week for 3 wk (on days 9, 13, 16, 20, 23, and 27). CH5126766 was dissolved in 20% hydroxypropyl‐β‐cyclodextrin/saline. Mice were given CH5126766 orally 5 d a week for 3 wk (on days 9‐13, 16‐20, and 23‐27). A, Points, means of weights measured twice a week (error bars, SD), in comparisons with the control (vehicle). B, Points, means of tumor volumes measured twice a week (error bars, SD). C, Overall survival. The observation duration was 50 d after inoculation. D, Molecular mechanism underlying the inhibition of tumor growth was investigated by western blotting. PD‐L1, programmed cell death‐ligand 1 A, B, Significance of differences in comparisons between two groups was examined using two‐way ANOVA by GraphPad Prism. Combination therapy was compared with monotherapy. *P < .0001
4. DISCUSSION
In the present study, we recommended combination therapy with eribulin and CH5126766 as a treatment strategy for TNBC. The growth of TNBC cells was inhibited by this combination therapy (Figures 2, 5A, and 6B) through the enhancement of apoptosis with the suppression of survivin and Bcl‐2 family proteins (Figures 3, 5B‐D, and 6D). We also showed that our recommended therapy with eribulin overcame the activation of the Akt pathway as feedback from the suppression of the MAPK pathway (Figures 4A and 5D). This is the first study to report a treatment with eribulin and a RAF/MEK inhibitor in TNBC. According to other combination effects, shown in Figure S5, the results in MDA‐MB‐231 cells showed that the synergistic effects of the combination were not specific on CH5126766 or eribulin. However, no other combinations showed synergistic effects in SUM159PT cells (Figure S5). These results suggest that the combination of eribulin and CH5126766 appears to be an efficient treatment for TNBC. CH5126766 has been believed to be effective against malignancies with activated MAPK pathway.10 However, interestingly in the current study, the activity of the MAPK pathway in TNBC cells was not associated with the efficacy of CH5126766 (Figure S1). Therefore, the activity of the MAPK pathway is not a useful biomarker in our combination therapy.
Eribulin, developed in Japan, has recently been used worldwide, not only in Japan, the US, and Europe, but also in other Asian countries. The major frequent adverse effect is neutropenia, which is able to be managed mostly by changing to a biweekly regimen for breast cancer.25, 26 As eribulin is acceptable for the majority of breast cancer patients, it is used in monotherapy and combination therapy.27 In the present study, we showed the efficacy of the combination of eribulin and CH5126766 for TNBC, which might be due to the induction of apoptosis and the suppression of PD‐L1 expression in tumor (Figure 6). Immunotherapy has recently become an essential treatment for various types of carcinomas. Immune checkpoint blockade toward the PD‐1/PD‐L1 axis is an important strategy for the evasion of immunosuppressive responses.28 Immune checkpoint antagonists have significant efficacy when used as single agents as well as in combination therapy with chemotherapeutic drugs. The effectiveness of the combination of chemotherapy and immunotherapy for NSCLC and TNBC was reported in the CheckMate‐816 trial (ClinicalTrials.gov NCT02998528) and Impassion 130 trial.4 Based on the mechanisms underlying chemotherapy‐induced immunomodulation,29 eribulin is a candidate chemotherapeutic agent in combination therapy for TNBC with antagonists of immune checkpoint signaling because it does not always require steroids and is a well‐tolerated drug, which is particularly suitable for TNBC.8, 30
Epithelial‐mesenchymal transition is a key process in cancer metastasis. It has been shown to contribute to immunosuppression in breast cancer.31 Eribulin is a microtubule dynamic inhibitor that has been shown to reverse EMT.32, 33 Various effects of eribulin on the tumor microenvironment, including the reversal of EMT, have been reported.34, 35, 36 However, limited information is currently available on oncoimmune responses by eribulin.37, 38 In studies from the same research group, the expression of PD‐L1 was downregulated in breast carcinoma samples obtained from patients treated with eribulin. As shown in Figure 6D, the expression of PD‐L1 in murine TNBC 4T1 tumors was not suppressed by eribulin. 4T1 cells easily become metastatic and appear to have an immunosuppressive mesenchymal phenotype31; therefore, the expression of PD‐L1 in 4T1 cells might only be weakly regulated by eribulin. Considering previous findings on the upregulation of PD‐L1 by other chemotherapeutic drugs in the various types of carcinomas,17, 39, 40 further studies are needed to clarify the oncoimmune response induced by eribulin and the combination with CH5126766.
The suppression of the MAPK pathway is important for the treatment of cancer. We originally discovered two potent molecular‐targeting agents, the RAF/MEK inhibitor CH5126766 and MEK inhibitor trametinib, from cell‐based screening assays in collaborations with pharmaceutical companies. Trametinib is already in widespread use today, while CH5126766 is under clinical development.10 CH5126766 is superior to trametinib for the suppression of CRAF activation as a result of feedback.41, 42, 43 In addition, we investigated oncoimmune responses induced by this therapy with a focus on the expression of PD‐L1. CH5126766 significantly downregulated the expression of PD‐L1 in TNBC cells (Figures 4B‐D and S7). The suppression of PD‐L1 expression by the combination shown in murine TNBC 4T1 tumors suggests the possibility of enhanced invasion of activated T lymphocytes in the tumor microenvironment (Figure 6D). The results raise a possibility that immune responses might partially contribute to the efficacy of this combination therapy. Trametinib has been reported to induce expression of PD‐L1 in TNBC cells in vitro and in vivo,19 whereas another MEK inhibitor, selumetinib, has been reported to downregulate the expression of PD‐L1 in NSCLC cells,44 which is consistent with our results on CH5126766. These differences appear to be dependent on the phenotype, such as epithelial or mesenchymal carcinomas.31 Moreover, selumetinib was previously shown to prevent lung metastasis through the reversal of EMT in TNBC.45 If suppression of the MAPK pathway induces the reversal of EMT, our recommended combination therapy with eribulin, which potentiates EMT reversal,32, 33 and CH5126766 could have significant efficacy against metastasis in TNBC. Based on EMT and immunosuppression in the tumor microenvironment of TNBC,31 the efficacy of our recommended combination therapy might be partially caused by immune responses related to the reversal of EMT.
DISCLOSURE
T. Sakai reports receiving patent fees from Chugai Pharmaceutical Co., Ltd. No potential conflicts of interest were disclosed by the other authors.
Supporting information
Fig S1‐S8
ACKNOWLEDGMENTS
We thank Medical English Service for English language editing.
Ono H, Horinaka M, Sukeno M, et al. Novel RAF/MEK inhibitor CH5126766/VS‐6766 has efficacy in combination with eribulin for the treatment of triple‐negative breast cancer. Cancer Sci. 2021;112:4166–4175. 10.1111/cas.15071
REFERENCES
- 1.Cortes J, O'Shaughnessy J, Loesch D, et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open‐label randomised study. Lancet. 2011;377:914‐923. [DOI] [PubMed] [Google Scholar]
- 2.Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patient with a germline BRCA mutation. N Engl J Med. 2017;377:523‐533. [DOI] [PubMed] [Google Scholar]
- 3.Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid P, Adam S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379:2108‐2121. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple‐negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple‐negative breast cancer‐‐deciphering the heterogeneity. Clin Cancer Res. 2014;20:782‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ono H, Sowa Y, Horinaka M, et al. The histone deacetylase inhibitor OBP‐801 and eribulin synergistically inhibit the growth of triple‐negative breast cancer cells with the suppression of survivin, Bcl‐xL and the MAPK pathway. Breast Cancer Res Treat. 2018;171:43‐52. [DOI] [PubMed] [Google Scholar]
- 8.Twelves C, Cortes J, Vahdat L, et al. Efficacy of eribulin in women with metastatic breast cancer: a pooled analysis of two phase 3 studies. Breast Cancer Res Treat. 2014;148:553‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai T, Sowa Y. Molecular‐targeting therapies against quantitative abnormalities in gene expression with malignant tumors. Cancer Sci. 2017;108:570‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo C, Chénard‐Poirier M, Roda D, et al. Intermittent schedules of the oral RAF‐MEK inhibitor CH5126766/VS‐6766 in patients with RAS/RAF‐mutant solid tumors and multiple myeloma: a single‐centre, open‐label, phase 1 dose‐escalation and basket dose‐expansion study. Lancet Oncol. 2020;21:1478‐1488. [DOI] [PubMed] [Google Scholar]
- 11.Cantley LC. The phosphoinositide 3‐kinase pathway. Science. 2002;296(5573):1655‐1657. [DOI] [PubMed] [Google Scholar]
- 12.Mendoza MC, Er EE, Blenis J. The Ras‐ERK and PI3K‐mTOR pathways: cross‐talk and compensation. Trends Biochem Sci. 2011;36:320‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halilovic E, She QB, Ye Q, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010;70:6804‐6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dry JR, Pavey S, Pratilas CA, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010;70:2264‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jing J, Greshock J, Holbrook JD, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11:720‐729. [DOI] [PubMed] [Google Scholar]
- 16.Mittendorf EA, Philips AV, Meric‐Bernstam F, et al. PD‐L1 expression in triple‐negative breast cancer. Cancer Immunol Res. 2014;2:361‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samanta D, Park Y, Ni X, et al. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple‐negative breast cancer cells. Proc Natl Acad Sci. 2018;115:E1239‐E1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brockwell NK, Owen KL, Zanker D, et al. Neoadjuvant interferons: critical for effective PD‐1 based immunotherapy in TNBC. Cancer Immunol Res. 2017;5:871‐884. [DOI] [PubMed] [Google Scholar]
- 19.Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor‐infiltrating lymphocytes in triple‐negative breast cancer: therapeutic cooperation between MEK and PD‐1/PD‐L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Chen S, Yuan W, et al. PD‐1/PD‐L1 interaction up‐regulates MDR1/P‐gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. 2017;8:99901‐99912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono H, Iizumi Y, Goi W, Sowa Y, Taguchi T, Sakai T. Ribosomal protein S3 regulates XIAP expression independently of the NF‐κB pathway in breast cancer cells. Oncol Rep. 2017;38:3205‐3210. [DOI] [PubMed] [Google Scholar]
- 22.Owusu‐Brackett N, Evans KW, Akcakanat A, et al. TAK228 enhances antitumor activity of eribulin in triple negative breast cancer. Oncotarget. 2019;10:5011‐5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao SS, Stoehr J, Dokic D, et al. Synergistic eribulin and CDK inhibition for the treatment of triple negative breast cancer. Oncotarget. 2017;8:83925‐83939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity, and cancer immunoediting. Nat Rev Immunol. 2006;6:836‐848. [DOI] [PubMed] [Google Scholar]
- 25.Ohtani S, Nakayama T, Yoshinami T, et al. Bi‐weekly eribulin therapy for metastatic breast cancer: a multicenter phase II prospective study (JUST‐STUDY). Breast Cancer. 2018;25:438‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith J 2nd, Irwin A, Jensen L, et al. Phase II study of eribulin mesylate administered biweekly in patients with human epidermal growth factor receptor‐2 negative metastatic breast cancer. Clin Breast Cancer. 2020;20:160‐167. [DOI] [PubMed] [Google Scholar]
- 27.Araki K, Fukada I, Yanagi H, et al. First report of eribulin in combination with pertuzumab and trastuzumab for advanced HER2‐positive breast cancer. Breast. 2017;35:78‐84. [DOI] [PubMed] [Google Scholar]
- 28.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci. 2002;99:122293‐122297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3(5):436‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawamura T, Kasai H, Fermanelli V, et al. Pharmacodynamic analysis of eribulin safety in breast cancer patients using real‐world postmarketing surveillance data. Cancer Sci. 2018;109:2822‐2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dongre A, Rashidian M, Reinhardt F, et al. Epithelial‐to‐mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017;77:3982‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida T, Ozawa Y, Kimura T, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial‐mesenchymal transition (EMT) to mesenchymal‐epithelial transition (MET) states. Br J Cancer. 2014;110:1497‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terashima M, Sakai K, Togashi Y, et al. Synergistic antitumor effects of S‐1 with eribulin in vitro and in vivo for triple‐negative breast cancer cell lines. Springerplus. 2014;8:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funahashi Y, Okamoto K, Adachi Y, et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014;105:1334‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueda S, Saeki T, Takeuchi H, et al. In vivo imaging of eribulin‐induced reoxygenation in advanced breast cancer patients: a comparison to bevacizumab. Br J Cancer. 2016;114:1212‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kashiwagi S, Asano Y, Goto W, et al. Mesenchymal‐epithelial transition and tumor vascular remodeling in eribulin chemotherapy for breast cancer. Anticancer Res. 2018;38:401‐410. [DOI] [PubMed] [Google Scholar]
- 37.Goto W, Kashiwagi S, Asano Y, et al. Eribulin promotes antitumor immune responses in patients with locally advanced or metastatic breast cancer. Anticancer Res. 2018;38:2929‐2938. [DOI] [PubMed] [Google Scholar]
- 38.Kashiwagi S, Asano Y, Goto W, et al. Use of tumor‐infiltrating lymphocytes (TILs) to predict the treatment response to eribulin chemotherapy in breast cancer. PLoS One. 2017;12:e0170634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng J, Hamanishi J, Matsumura N, et al. Chemotherapy induces programmed cell death‐ligand 1 overexpression via the Nuclear Factor‐κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015;75:5034‐5045. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Deng Z, Wang H, Ma W, Zhou C, Zhang S. Repeated cycles of 5‐fluorouracil chemotherapy impaired anti‐tumor functions of cytotoxic T cells in a CT26 tumor‐bearing mouse model. BMC Immunol. 2016;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishii N, Harada N, Joseph EW, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050‐4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wada M, Horinaka M, Yamazaki T, Katoh N, Sakai T. The dual RAF/MEK inhibitor CH5126766/RO5126766 may be a potential therapy for RAS‐mutated tumor cells. PLoS One. 2014;9:e113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lito P, Saborowski A, Yue J, et al. Disruption of CRAF‐mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stutvoet TS, Kol A, de Veies EG, et al. MAPK pathway activity plays a key role in PD‐L1 expression of lung adenocarcinoma cells. J Pathol. 2019;249:52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartholomeusz C, Xie X, Pitner MK, et al. MEK inhibitor selumetinib (AZD6244; ARRY‐142886) prevents lung metastasis in a triple‐negative breast cancer xenograft model. Mol Cancer Ther. 2015;14:2773‐2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8