

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) (eg, gefitinib) exert potent therapeutic efficacy in non–small‐cell lung cancer (NSCLC) harboring EGFR‐activating mutations. However, the resistance to EGFR TKIs limits their clinical therapeutic efficacy. TIP30, a newly identified tumor suppressor, appears to be involved in the regulation of cytoplasmic and nuclear EGFR signaling in NSCLC. Our previous study demonstrated that TIP30 regulated EGF‐dependent cyclin D1 transcription in human lung adenocarcinoma and suppressed tumorigenesis. In the present study, the involvement of TIP30 in combating gefitinib resistance in NSCLC was determined for the first time in vitro and in vivo. Gain and loss of function studies showed that overexpression of TIP30 effectively sensitized cells to gefitinib in vitro, whereas TIP30 inhibition promoted gefitinib cell resistance. Moreover, TIP30 negatively regulated the activation of the p‐AKT and p‐MEK signaling pathways in PC9/GR. Importantly, PC9/GR harbored high levels of nuclear EGFR, and overexpression of TIP30 restored irregular EGFR trafficking and degradation from early endosomes to the late endosomes, decreasing the nuclear accumulation of EGFR, which may partly or totally inhibit EGFR‐mediated induction of c‐Myc transcription. Xenographic tumors induced by overexpression of TIP30 by PC9/GR cells in nude mice were suppressed compared with their original counterparts. Overall, it was revealed that TIP30 overexpression restored gefitinib sensitivity in NSCLC cells and attenuated the cytoplasmic and nuclear EGFR signaling pathways and may be a promising biomarker in gefitinib resistance in NSCLC.
Keywords: EGFR, gefitinib resistance, non–small‐cell lung cancer (NSCLC), TIP30
This work investigated the vital role of TIP30 in gefitinib resistance of NSCLC‐involved modulation of the nuclear EGFR signaling pathway. Findings indicated that a low TIP30 level may confer gefitinib resistance and overexpression of TIP30 may predict a favorable response to gefitinib‐based chemotherapy in NSCLC.

1. INTRODUCTION
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) such as gefitinib have been demonstrated to improve progression‐free survival following unsuccessful front‐line chemotherapy in patients with advanced non–small‐cell lung cancer (NSCLC).1, 2, 3 Based on its marked clinical efficacy, in 2015, gefitinib was approved by the Food and Drug Administration (FDA) as the first‐line treatment for metastatic NSCLC in patients with activating EGFR mutations.4 All patients, however, who initially responded to EGFR TKI, developed acquired resistance to TKIs within 6‐12 mo after the initial treatment.5 Recent studies have indicated that the key mechanisms leading to acquired resistance to gefitinib involve secondary resistance mutations, such as T790 M,6 MET proto‐oncogene amplification,7 and activation of other signaling pathways.8, 9 Moreover, although third‐generation EGFR TKI has been shown to be effective in the treatment of NSCLC, the majority of patients still developed resistance and disease progression, indicating the existence of unknown mechanisms of gefitinib resistance. It has been proposed that multiple resistance mechanisms may coexist in the same cell population.10 Therefore, combining therapies with multiple targets could improve the efficacy of therapies involving EGFR TKI.
Ligand‐activated EGFR internalization and downstream signaling cascades play vital roles in cancer proliferation and progression.11 Previous studies have shown increased accumulation of EGFR in the nuclei of cancer cells following various therapies, including radiotherapy,12 chemotherapy,13 and anti‐EGFR therapies such as gefitinib and cetuximab.14, 15 The exact molecular and cellular mechanisms of resistance remain unclear; however, the involvement of nuclear EGFR in the acquired resistance of cancer cells to EGFR TKI has been confirmed. Huang et al reported that nuclear EGFR functions as a transcriptional factor to regulate the expression of breast cancer‐resistant protein (ie, BCRP/ABCG2), a plasma membrane‐bound ATP‐dependent transporter, which can expel anti‐cancer drugs from cells and induce gefitinib resistance.16 Nuclear EGFR is associated with both first‐ and third‐generation acquired resistance to TKIs. Rong et al demonstrated that membrane EGFR is translocated to the cytoplasm/nucleus, inducing first generation TKI resistance. Moreover, nuclear translocation of EGFR might also be a significant and independent factor contributing to Osimertinib, effecting third‐generation TKI resistance.17
Tat‐interacting protein (TIP30), also known as CC3 or HTATIP2, is widely expressed in numerous normal human tissues, but is downregulated in various cancers.18, 19, 20 For instance, low expression of TIP30 is associated with poor tumor differentiation and high risk of metastasis in NSCLC.21, 22 It has previously been shown that in mice deletion of TIP30 led to the spontaneous development of lung cancer. In addition, TIP30 knockdown in human lung adenocarcinoma cells has been demonstrated to delay EGFR degradation, as well as to increase nuclear localization of EGFR.23 Our previous work suggested that nuclear TIP30‐induced downregulation of cyclin D1 transcription disturbed EGFR signaling and suppressed tumorigenesis in lung adenocarcinoma.24 The above studies indicated that the functional role of TIP30 in lung adenocarcinoma is closely associated with the EGFR signaling pathway. Intriguingly, Zhu et al reported that decreased TIP30 expression contributed to chemotherapeutic resistance by regulating the AKT/glycogen synthase‐3AKT‐catenin signaling pathway in laryngeal squamous cell carcinoma.25 Therefore, we speculated that TIP30 might be involved in EGFR‐induced acquired resistance to TKI in NSCLC. In the present study, we determined that TIP30 was downregulated in gefitinib‐resistant lung cancer PC9/GR cells. Investigations involving cells and xenograft models have highlighted the essential role of TIP30 in gefitinib resistance. Mechanistically, it was found that TIP30 regulated subcellular trafficking of EGFR and transcriptionally inactivated the expression of c‐Myc, subsequently contributing to gefitinib resistance.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
The human NSCLC cell line PC9 and PC9/GR cells were provided by the Guangdong Lung Cancer Research Institute (Guangdong, China) and cultured following previously described methods.26 To maintain drug resistance, prior to conducting the experiments, PC9/GR cells were first grown in a medium containing 0.5 µM gefitinib and subsequently in a drug‐free medium for at least 1 wk. Gefitinib was diluted in DMSO at the stock concentration of 20 mM (stored at −20℃). Recombinant human epidermal growth factor (EGF) was purchased from R&D Systems. An EGFR internalization assay was conducted as described before.27 Cells were incubated in a serum‐starved medium overnight, pretreated without or with gefitinib for 60 min, followed by 50 ng/mL EGF stimulation using the designated times.
2.2. Antibodies
Rabbit anti‐TIP30 antibody was provided by Dr. Hua Xiao (Michigan State University, USA). ERK, p‐ERK, AKT, p‐AKT, EGFR, p‐EGFR, and STAT3 antibodies were purchased from Cell Signaling Technology. c‐Myc antibodies were obtained from Invitrogen. Anti‐STAT3 recombinant rabbit polyclonal antibody used for the chromatin immunoprecipitation (ChIP) assay was acquired from Thermo Scientific. Anti‐LAMP1 antibodies and anti‐EGFR antibodies used for immunofluorescence studies were obtained from Abcam.
2.3. Lentivirus transfection
TIP30 lentiviral plasmid LV5‐TIP30‐homo and shRNA‐containing lentiviral vectors against TIP30 were purchased from Shanghai GenePharma Co., Ltd. Virus packaging, transfection, and puromycin selection were carried out in accordance with the manufacturer’s instructions.
2.4. MTT cell viability assay
The MTT assay was conducted following the manufacturer’s protocol. Half maximal inhibitory concentrations (IC50) were calculated using GraphPad Prism 8 software.
2.5. Apoptosis assessment by Annexin V staining
An Annexin V‐APC/PI Apoptosis Detection Kit was used for the apoptosis assay in accordance with the manufacturer’s instructions. The percentages of apoptotic cells in the Annexin V+/PI− and Annexin V+/PI+ populations were determined.
2.6. Colony formation assay
Here, 35 mm dishes were coated with 0.8% agarose in complete medium (1.5 mL agarose/well), which was allowed to solidify at room temperature for 15 min. Cells (5 × 103 cells/mL) were individually mixed with 0.4% agarose and added to the bottom of agarose‐coated wells. The solutions were allowed to solidify for another 20 min. Complete medium (750 µL) was added to each well to prevent drying, and the cells were incubated for 14 d. Colonies larger than 75 μm in diameter or containing more than 50 cells were counted.
2.7. Co‐immunoprecipitation (co‐IP) and immunoblotting analyses
The preparation of total cell lysates and subcellular fractionation, co‐IP, and western blot analyses were performed using a previously described method.24 For western blots of phosphoproteins, the cells were lysed in a lysis buffer consisting of RIPA reagent, protease inhibitor cocktail used at 1:20, and phosphatase inhibitor cocktails used at 1:100.
2.8. Reverse transcription and real‐time PCR
Complementary DNA (cDNA) was synthesized from total RNA using the cDNA Reverse Transcription Kit (TaKaRa). RT‐qPCR was performed using the FastStart DNA Master SYBR Green I Kit (TaKaRa). Primer sequences are shown in supplementary data Table S1.
2.9. Chromatin immunoprecipitation assay
The ChIP assay was conducted using the Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) in accordance with the manufacturer’s recommendations. Immunoprecipitation was performed using anti‐STAT3 antibodies. Primer information is shown in Table S1.
2.10. In vivo animal model experiments
Five‐wk‐old female Balb/c‐nude mice were purchased from the Guangdong Animal Experimental Center and were fed in a specific pathogen‐free (SPF) environment with sterilized food and water; 20 mice were divided into 4 groups randomly. PC9, PC9/GR + Mock or PC9/GR + TIP30 cells (5 × 106) were injected subcutaneously into the right flank of nude mice. Tumors were monitored and measured using electronic calipers every 2 d. When the subcutaneous tumor reached a volume of approximately 50 mm3, PC9 group, PC9/GR + TIP30 group, and one of the PC9/GR + Mock groups were subjected to treatment, the other PC9/GR + Mock group was set as vehicle groups (n = 5 per group). The mice were administered saline or gefitinib (20 mg/kg/per day) orally. After 12 d consecutively of observation, all mice were anesthetized and sacrificed by cervical dislocation, and subcutaneous tumors were surgically excised and weighed. Tumor size was calculated based on the following formula: volume (mm3) = length (mm) × width (mm)2/2, where length and width indicated the longest and shortest tumor diameters, respectively.
2.11. Tissue samples and immunohistochemistry analysis
Tumor tissue was sliced and placed in 4% paraformaldehyde for posterior fixation, followed by dehydration with gradient alcohol. Xylene treatment was conducted to make the tissue transparent. Tumor sections were stained with antibodies following standard immunohistochemistry protocols. The following antibodies and dilutions were used: 1:200 anti‐c‐Myc, 1:100 anti‐EGFR, and 1:200 anti‐TIP30.
2.12. Confocal immunofluorescence
Fixed cells were permeabilized using 0.3% Triton X‐100 for 5 min, and incubated with normal goat serum for 1.5 h. The cells were incubated with anti‐EGFR (1:200) or anti‐LAMP1 (1:300) antibody overnight at 4℃. After thorough washing, the bound primary antibodies were visualized using Alexa Fluor 594 and Alexa Fluor 488 and counterstained with DAPI nucleic acid stain for cell counting. Fluorescence images were obtained using the ACAS Ultima 312 confocal laser‐scanning microscope.
2.13. Statistical analysis
Results are reported as the mean ± standard deviation (SD). SPSS software, v.13.0 was used for statistical analysis. The comparison between 2 groups was analyzed statistically using an independent sample t test, and one‐way ANOVA was used for comparisons of multiple groups. A P‐value < .05 was considered statistically significant. All experiments were conducted in triplicate.
3. RESULTS
3.1. TIP30 correlates with gefitinib resistance in NSCLC cells
As shown in Figure 1A, the MTT assay confirmed the acquired resistance of the PC9/GR clones toward gefitinib. PC9/GR2 cells exhibited a higher IC50 (9.01 ± 0.32 μM) than PC9/GR1 (1.328 ± 0.15 μM) and PC9 cells (0.15 ± 0.19 μM), P < .01. Furthermore, it was found that PC9/GR cells exhibited lower expression levels of TIP30 compared with PC9 cells (P < .01, Figure 1B). To determine the functional role of TIP30 in PC9/GR, PC9/GR2 cells were selected for subsequent investigation. TIP30 was overexpressed using a lentivirus encoding TIP30 cDNA (LV‐TIP30). Compared with the mock transfection group (Mock) and negative control (NC) group, the cells in the PC9/GR + TIP30 group stably overexpressed TIP30 at both at mRNA and protein levels (P < .001, Figure 1C). We also performed TIP30 knockdown in PC9 cells. Silencing efficiency is shown in Figure 1D. The MTT assay (Figure 1E) revealed that overexpression of TIP30 evidently reversed the sensitivity of PC9/GR2 cells to gefitinib (IC50 value, PC9/GR2 + TIP30 vs PC9/GR2 + Mock, 1.468 ± 0.172 μM vs 9.01 ± 0.32 μM, P < .001); conversely, TIP30 knockdown promoted gefitinib resistance of PC9 cells (IC50 value, PC9 + TIP30‐SH2 vs PC9 + shRNA‐Con, 0.262 ± 0.119 μM vs 3.679 ± 0.789 μM, P < .01). As shown in Figure 1F, the percentage of apoptotic cells significantly increased in PC9/GR2 cells that overexpressed TIP30 compared with PC9/GR2 + Mock cells with or without gefitinib, respectively, and TIP30 knockdown in PC9 cells inhibited apoptosis relative to that in PC9 + shRNA‐Con cells with or without gefitinib. Moreover, colony formation assays revealed that either gefitinib treatment or TIP30 overexpression decreased the number of colony formations in PC9/GR cells. Gefitinib treatment combined with or without TIP30 knockdown showed a similar trend in PC9 cells. However, gefitinib seemed to be more effective than TIP30 in regulating colony formation in both cells (Figure 1G). Overall, the data suggested that TIP30 might re‐sensitize gefitinib‐resistant cells to gefitinib in vitro.
FIGURE 1.
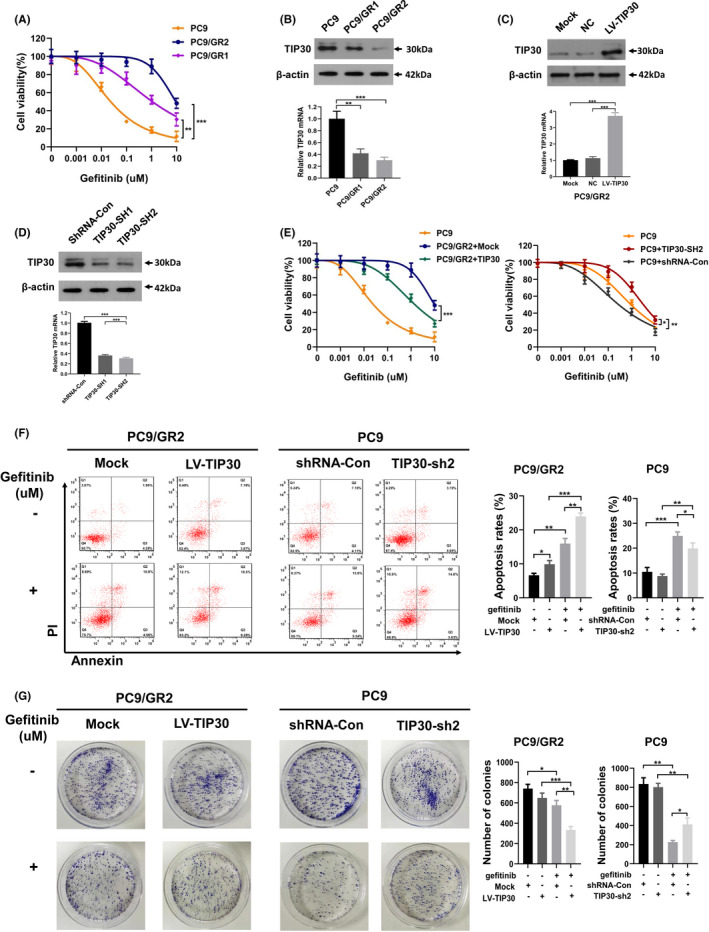
Overexpression/downregulation of TIP30 increases/decreases the sensitivity of PC9/GR and PC9 cells to gefitinib. A, The growth inhibitory effects of gefitinib on PC9 and two different gefitinib‐resistant clones of PC9/GR cells were determined by MTT assays. B, C, The mRNA and protein levels of TIP30 expressed in PC9, PC9/GR, PC9/GR2 + TIP30, Mock, and negative control cell lines. D, Western blotting and RT‐qPCR analysis of the TIP30 levels in PC9 + shTIP30 and shRNA‐Con cells. E, Gefitinib sensitivities of PC9, PC9/GR2 + Mock, PC9/GR2 + TIP30, PC9 + TIP30‐SH2, and PC9 + shRNA‐Con cells were evaluated using the MTT assay. F, Transfected PC9 cells or PC9/GR2 cells were treated with 0.1 μM gefitinib or 5 μM gefitinib for 24 h, respectively, followed by flow cytometry to evaluate the apoptotic rate. The same concentration of DMSO was used as the control. G, Colony formation assay for transfected PC9/GR2 and transfected PC9 cells under the stimulation of 5 μM or 0.1 μM gefitinib, respectively.* P < .05; ** P < .01; *** P < .001
3.2. Specific overexpression of TIP30 decreased activation of p‐AKT, p‐ERK, and p‐EGFR in PC9/GR cells
Previous studies revealed persistent AKT and ERK phosphorylation in NSCLC cells exhibiting acquired gefitinib resistance.28 In the present study, we used western blotting to evaluate the role of TIP30 in the whole protein and phosphorylated protein expression of the EGFR signaling pathway. As demonstrated in Figure 2A, the expression of p‐EGFR, p‐AKT, and p‐ERK in PC9 cells was dose‐dependently attenuated following gefitinib treatment. Conversely, a minor decrease in the expression levels of p‐EGFR, p‐ERK, and p‐AKT was observed for PC9/GR cells (Figure 2A). We subsequently examined whether TIP30 was involved in gefitinib resistance by regulating tyrosine phosphorylation in the EGFR signaling pathway. Compared with PC9/GR + Mock cells, an obvious decrease in the levels of p‐AKT, p‐EGFR, and p‐ERK in response to a gradual increase in gefitinib concentrations was detected for PC9/GR + TIP30 cells (Figure 2B). These results indicated the presence of persistently activated EGFR signaling cascades in PC9/GR cells. Moreover, the activation of this signaling cascade was partly blocked by TIP30.
FIGURE 2.
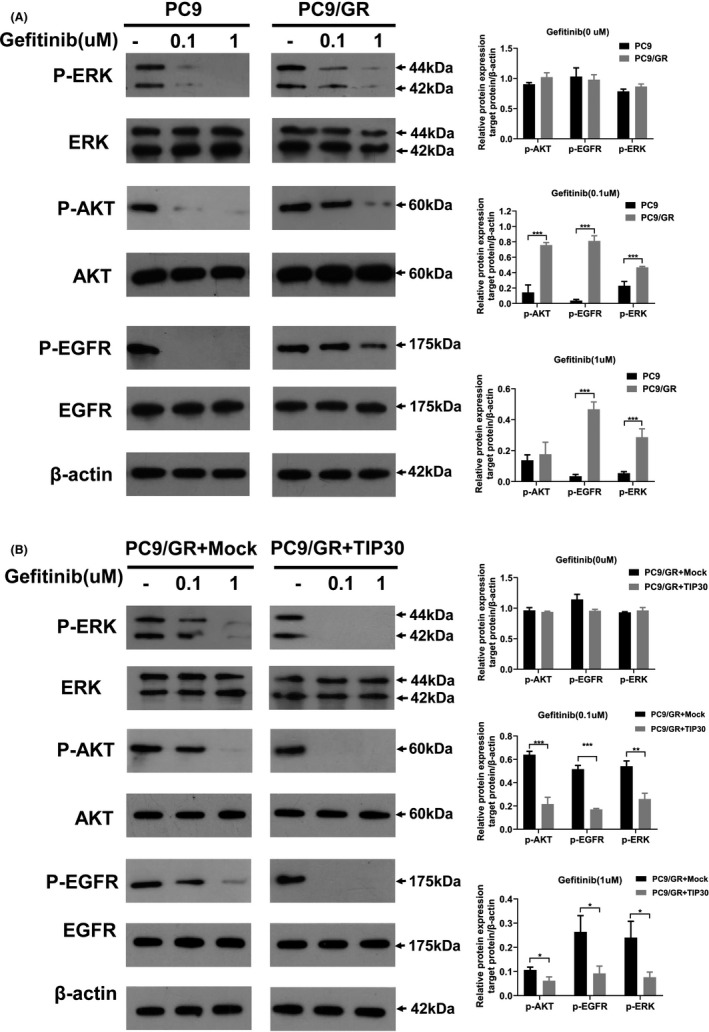
Cytotoxic effects of gefitinib on PC9, PC9/GR, and PC9/GR + TIP30 cells. A, PC9 and PC9/GR cells were pretreated with different concentrations of gefitinib (0‐1 μM) for 24 h. Expression of p‐AKT, total AKT, p‐EGFR, total EGFR, p‐ERK, and total ERK in the cell lysates was evaluated by western blotting. B, PC9/GR + TIP30 and PC9/GR + Mock cells were pretreated with different concentrations of gefitinib (0‐1 μM) for 24 h. Expression of p‐AKT, total AKT, p‐EGFR, total EGFR, p‐ERK, and total ERK was investigated by western blotting. * P < .05; *** P < .001
3.3. TIP30 increases EGFR degradation in late endosomes/lysosomes in PC9/GR cells, which is also increased by gefitinib treatment
To compare the intracellular distribution of EGFR between the gefitinib‐resistant PC9/GR cells and PC9/GR + TIP30 cells following EGF stimulation for 30 min, the cells were labeled with EGFR antibodies and DAPI, revealing the nuclei. For PC9/GR + Mock cells, EGFR (red) partly localized within small vesicular structures distributed throughout the cytoplasm and cell membrane. Additionally, some punctate signals were clearly detected in the nucleus (white arrows, Figure 3A). The expression of TIP30 resulted in a significant decrease in the nuclear EGFR signal in PC9/GR cells. Furthermore, western blotting also confirmed the decreased protein expression of nuclear EGFR in PC9/GR + TIP30 cells (P < .01, Figure 3B). These results suggested that TIP30 might contribute to the degradation of EGFR in the cytoplasm of PC9/GR + TIP30 cells.
FIGURE 3.
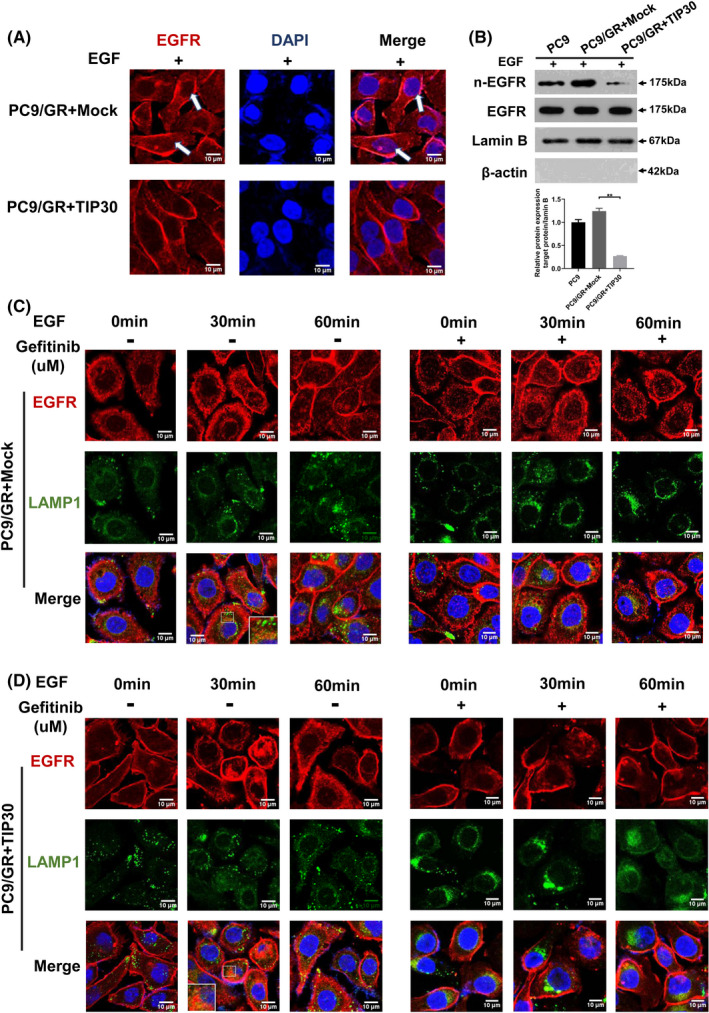
TIP30 modulates lysosome‐mediated EGFR degradation and nuclear localization upon EGF treatment in gefitinib‐resistant NSCLC cells. A, Confocal microscopy analysis of EGFR localization was carried out in PC9/GR + Mock and PC9/GR + TIP30 cells. B, Western blot analysis of EGFR expression in the nuclear fraction and total fraction of PC9, PC9/GR + Mock, and PC9/GR + TIP30 cells following EGF treatment for 30 min. C, D, Fluorescence microscopy analysis of the EGFR localization in PC9/GR + Mock and PC9/GR+TIP30 cells pre‐incubated with or without 5 μM gefitinib at 37°C, following stimulation of EGF for 0 min, 30 min, or 60 min. The quantitative analysis of EGFR and LAMP1 co‐lozalizations was shown in Figure S1. * P < .05; ** P < .01;*** P < .001. (Scale bars, 10 μm)
Furthermore, we attempted to monitor the consequences of TIP30 expression on the degradation of EGFR in PC9/GR cells. Lysosomes/endosomes were marked with lysosomal associated membrane protein 1 (LAMP1) to exhibit green fluorescence. As shown in Figure 3C (left), following EGF treatment, nuclear EGFR in PC9/GR + Mock cells increased in a time‐dependent manner, which was evident by the formation of red puncta. The staining between EGFR and LAMP1 in PC9/GR + Mock cells overlapped after EGF treatment, suggesting that some nuclear EGFR degraded in aggregated late endosomes following stimulation with EGF. Notably, EGF treatment for 30 min resulted in an increase in EGFR in the LAMP1‐labeled late‐endosome/lysosome compartments in PC9/GR + TIP30 cells (Figure 3D, left). Furthermore, decreased accumulation of internalized EGFR was observed in the late endosomes of PC9/GR + TIP30 cells after EGF treatment for 60 min. Negligible EGFR was detected in the nucleus. These data support the hypothesis that overexpression of TIP30 increases the degradation of EGFR in late endosomes/lysosomes and decreases EGFR nuclear localization in PC9/GR cells.
In our experiments, after gefitinib treatment and EGF stimulation, the endocytosis of EGFR was significantly suppressed in both PC9/GR + TIP30 and PC9/GR + Mock cells (Figure 3C, right and 3D, right). Most of the EGFR was located on the plasma membrane, exhibiting red fluorescence. The suppressive effect of gefitinib on the endocytosis of p‐EGFR in PC9/GR + TIP30 cells was much stronger than that in PC9/GR + Mock cells.
3.4. TIP30 negatively regulates c‐Myc transcription in PC9/GR cells
Nuclear EGFR functions as a transcription cofactor rather than a DNA‐binding transcription factor to mediate its downstream gene transcription. Jaganathan et al reported that EGFR, Src, and STAT3 formed a heteromeric complex and upregulated the transcription of c‐Myc in pancreatic cancer cells.29 As TIP30 was identified as the co‐transcription factor regulating the c‐Myc transcription in mammary glands,30 we speculated that TIP30 might also act as a co‐transcription factor of STAT3‐mediated c‐Myc transcription in PC9/GR cells. RT‐qPCR was performed to detect whether TIP30 had a direct role in regulating nuclear EGFR‐mediated transcription of common downstream genes, such as cyclin D1, iNOS, and c‐Myc. As demonstrated by the data shown in Figure 4A,B as well as supplementary data in Figures S2 and S3, c‐Myc mRNA and protein levels were negatively associated with TIP30 expression in PC9 or PC9/GR cells (P < .001).
FIGURE 4.
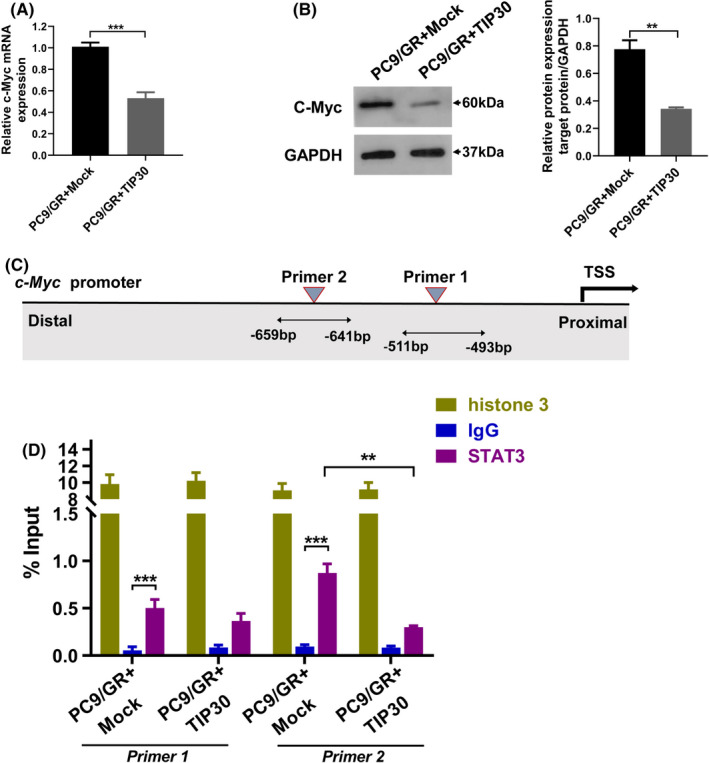
TIP30 negatively regulates c‐Myc transcription in PC9/GR cells. A, B, The c‐Myc mRNA and protein expressions were analyzed after 96 h of transduction of Mock or TIP30 in PC9/GR cells. C, Schematic of the STAT3‐binding sites in the c‐Myc promoter. D, ChIP analysis of STAT3 binding to the c‐Myc promoter in PC9/GR + Mock and PC9/GR + TIP30 cells. The lysates were analyzed using the anti‐histone 3 antibody as the positive control, and anti‐IgG was used as the negative control. ** P < .01, *** P < .001
Based on the putative binding sites for STAT3 in the promoter region of c‐Myc determined using online MatInspector software (v.8.1, Genomatix, http://www.genomatix.de/), we designed 2 pairs of primers for ChIP analysis that annealed to these putative binding sites (Figure 4C). ChIP analysis demonstrated that STAT3 bound directly to the c‐Myc promoter in both PC9/GR and PC9/GR + TIP30 cells. Both binding sites of STAT3 were statistically significant compared with IgG, and enrichment of STAT3 in binding site 2 (−659 to −641 bp) was higher than in binding site 1 (−511 to −493 bp) (P < .001). Furthermore, overexpression of TIP30 resulted in reduced binding of STAT3 to the c‐Myc promoters, indicating the dependency of STAT3 binding on TIP30 (P < .01, Primer 2) (Figure 4D).
3.5. The formation of subcutaneous tumors induced by PC9/GR cells is suppressed by overexpression of TIP30
To provide further in vivo evidence regarding the role of TIP30 in reversing gefitinib resistance in NSCLC, the investigated tumor bearing mice were randomly divided into 4 groups for treatment with vehicle or gefitinib at 3‐d intervals (Figure 5A). Mice injected with PC9 cells and treated with gefitinib were used as a negative control, and the PC9/GR + Mock group was the positive control. Figure 5A shows the representative tumor burdens induced by each group. Compared with PC9/GR + gefitinib or PC9/GR + Mock groups, the tumor burdens normally induced by PC9/GR cells overexpressing TIP30 were mostly inhibited by gefitinib. Intriguingly, there was no obvious difference in the tumor volume induced by PC9/GR + Mock cells with or without gefitinib treatment.
FIGURE 5.
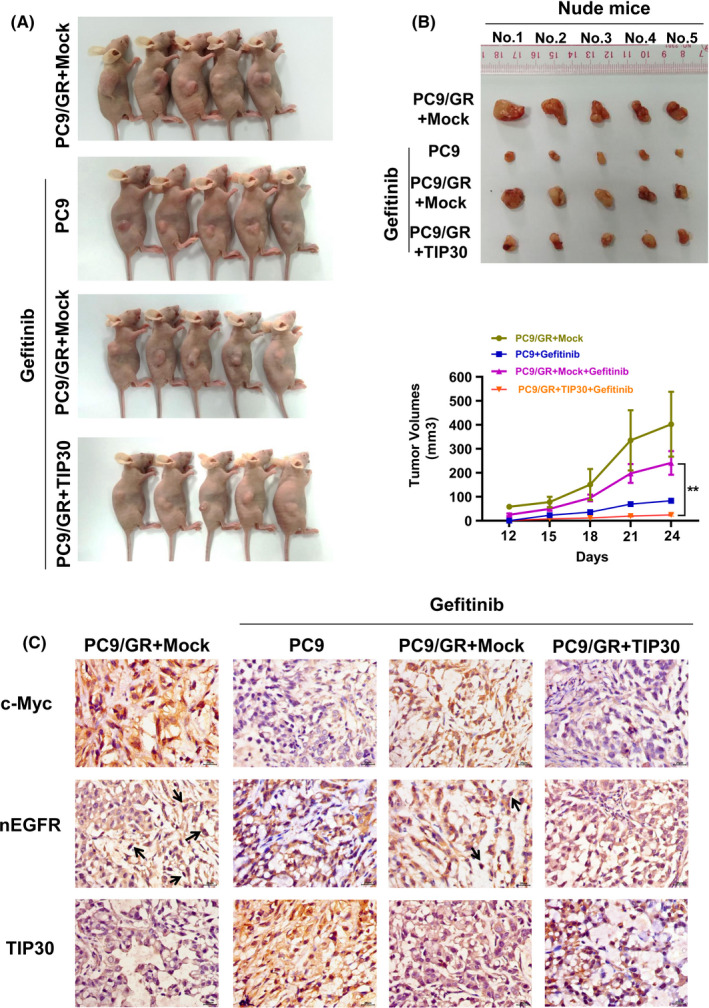
Overexpression of TIP30 attenuates growth of PC9/GR xenograft tumors in nude mice cells. A, Subcutaneous tumors were generated in nude mice injected with PC9 and PC9/GR cells that overexpressed TIP30 or carried a Mock vector. Representative images of the xenograft tumors dissected after treatment with gefitinib or vehicle for 12 d. B, Representative tumor burdens in the 4 investigated groups. In vivo tumor growth curves for the 4 groups were plotted and compared using Student t test. C, Representative immunohistochemical staining of c‐Myc, nuclear EGFR, and TIP30 in xenograft tumors. ** P < .01; n = 5 for each group
Furthermore, 4 groups of xenograft nude mice tumors were randomly selected and immunohistochemistry analysis was conducted to determine whether c‐Myc or nuclear EGFR expression was affected by TIP30. As shown in Figure 5C, high expression of c‐Myc and nuclear EGFR (black arrows) and low expression of TIP30 was observed in tumors induced by PC9/GR + Mock cells. Conversely, decreased c‐Myc and EGFR expression was detected in tumors induced by PC9/GR + TIP30 cells. Moreover, we noted that the expression of TIP30 in PC9/GR cells was markedly weaker than that in PC9 cells following gefitinib treatment, and was consistent with the results of the conducted in vitro experiments. These outcomes also implied that TIP30 increased the sensitivity to gefitinib by inactivating nuclear EGFR c‐Myc signaling pathway in NSCLC.
4. DISCUSSION
TIP30 has been shown to be a tumor suppressor, which plays important roles in tumor suppression, anti‐angiogenesis, and metastasis. Although the functions of TIP30 in the progression and metastasis of NSCLC have been well characterized, the relationship of TIP30 with gefitinib resistance in NSCLC has not been previously determined. In the present study, PC9 and PC9/GR cells were used as models to investigate the function of TIP30 in the acquired resistance to gefitinib in NSCLC. We identified that the expression of TIP30 in gefitinib‐resistant PC9/GR cells was naturally downregulated compared with that in the wild‐type PC9 cells. A possible explanation for this could be that continuous exposure to gefitinib downregulated the expression of TIP30, resulting in the formation of a gefitinib‐resistant phenotype of PC9 cells. These findings led us to explore the biological function as well as the underlying mechanism of TIP30 in gefitinib resistance in NSCLC in more detail. Overexpression of TIP30 sensitized PC9/GR cells to gefitinib; this was shown by changes in IC50 values and the in vitro colony formation efficiency. This led to apoptosis in NSCLC cells. Rescue experiments showed that TIP30 knockdown reversed these effects in PC9 cells. All these results highlighted the essential role of TIP30 in gefitinib resistance, and suggested that TIP30 may be a good biomarker of EGFR TKI resistance in NSCLC.
Dysregulation of the EGFR signaling pathway is associated with TKI resistance of EGFR‐mutated lung adenocarcinomas.31, 32 Therefore, understanding the roles of TIP30 in regulating divergent cytoplasmic EGFR signaling and the nuclear EGFR pathway, which was correlated with TKI resistance of EGFR‐mutated lung adenocarcinomas, is of particular importance. We noted that gefitinib attenuated phosphorylation of ERK, AKT, and EGFR in the wild‐type PC‐9 cells; however, persistently activated phosphorylation of ERK, AKT, and EGFR was detected in PC9/GR cells upon gefitinib treatment. This result is in agreement with previously reported outcomes, indicating persistent activation of the MAPK/ERK cascades in gefitinib‐resistant cells.33 This sustained EGFR signaling activation may be associated with the T790M mutation of PC9/GR cells (Table S2). In contrast, TIP30 overexpression inactivated this protein phosphorylation, indicating that the role of TIP30 in gefitinib resistance in NSCLC cells with T790M may involve the ability to inhibit EGFR downstream signals.
Upon EGF stimulation, EGFR was endocytosed through clathrin‐dependent or clathrin‐independent mechanisms, followed by endosomal recycling back to the plasma membrane, lysosomal degradation, or altered EGFR distribution to the nuclei or mitochondria (Figure 6).34, 35 Previous studies have reported that EGFR endocytic homeostasis was disturbed after competent EGF treatment in gefitinib‐resistant cell lines, leading to aggregation in early endosomes and delayed degradation.36 Consistent with earlier reports, we found that internalized EGFR aggregated in the nucleus, but not in late lysosomes of PC9/GR cells after rapid stimulation of EGF. However, large amounts of EGFR co‐localized with LAMP1‐positive late endosomes/lysosomes in PC9/GR cells that overexpressed TIP30 after 30 min of EGF stimulation. These results showed that overexpression of TIP30 significantly accelerated the degradation of EGFR in PC9/GR cells, resulting in loss of recycled EGFR to the plasma membrane and irregular EGFR trafficking to the nuclei. Decreased recycling of EGFR back to the plasma membrane provided an alternative explanation to the above observations that ERK, AKT, and EGFR phosphorylation were inhibited in PC9/GR + TIP30 cells. A previous study has reported that TIP30 facilitated the localization of Rab5a and V‐ATPases in endosomes leading to EGFR trapping in endocytic vesicles.37 This study also provided a clue to further studies involving the mechanism of TIP30‐regulated endocytic trafficking in gefitinib resistance cells. It is also noteworthy that internalization and accumulation of EGFR in late lysosomes were mostly impaired after gefitinib treatment in PC9/GR + TIP30 cells, whereas, this effect was limited in PC9/GR cells. Therefore, we speculate that TIP30 overexpression may restore the inhibition of kinase‐dependent EGFR internalization by gefitinib in PC9/GR cells. However, further investigation is required. Collectively, we propose that TIP30 activation may play a positive role in regulating the cytoplasmic EGFR signaling pathway through multiple endocytic pathways. This effect may therefore be disturbed in drug‐resistant cells that lack TIP30, in favor of nuclear EGFR translocation.
FIGURE 6.
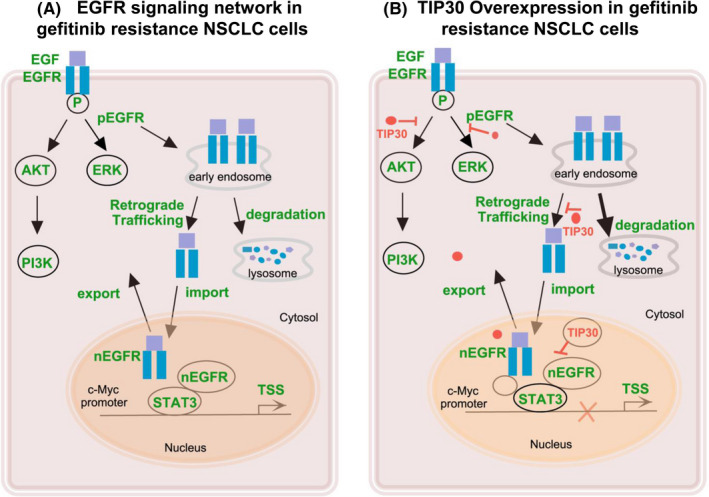
Schematic diagram of this study. A, EGFR signaling network in gefitinib resistance in NSCLC cells. B, TIP30 overexpression in gefitinib resistance NSCLC cells
In the present study, ChIP assay data showed that overexpression of TIP30 decreased the occupancy of STAT3 in the c‐Myc promoter in PC9/GR cells. We speculated that there may be several reasons. First, overexpression of TIP30 increased EGFR endocytic degradation and decreased EGFR nuclear localization. Decreased amounts of transcription cofactor may contribute to transcriptional depression. Second, TIP30 overexpression may decrease IL‐6‐induced nuclear STAT3 expression in PC9/GR cells,38, 39 although this hypothesis needs further confirmation. Third, TIP30 functions as a transcription inhibitor, negatively regulating c‐Myc transcription. Hence, based on the ChIP assay and co‐IP results (Figure S4), we found that STAT3 and EGFR formed a transcriptional complex. Nevertheless, the interaction of TIP30 with STAT3 or EGFR was not determined by the co‐IP assay (negative data were not shown). A possible explanation was that TIP30 may be located inside the transcription complex, and was therefore inaccessible to the antiserum used in the co‐IP assay. The transcriptional complex of EGFR, TIP30, and STAT3 regulated c‐Myc gene transcription, consequently inducing activation of some downstream genes, and was closely correlated with cell cycle DNA synthesis and eventually caused genomic instability in some cancers.40, 41 All these changes increased the expression of several genes and uncontrolled cell proliferation, which increased the risk of acquiring secondary mutations that contributed to tumor development.42 Therefore, PC9/GR cells, which naturally express low TIP30, maintained the activation of nuclear EGFR signaling, to send proliferative and oncogenesis signals, and avoid the traditional kinase‐dependent signaling pathway, which is inhibited by TKI. These results implied that TIP30 conferred gefitinib resistance by repressing the nuclear EGFR‐c‐Myc signaling pathway in NSCLC. Moreover, the present in vivo results were consistent with the conducted cell experiments and further confirmed our hypothesis.
In conclusion, we characterized TIP30, a tumor suppressor involved in gefitinib resistance in NSCLC cells by regulating cytoplasmic and nuclear EGFR signaling. Although the current technique of restoring the tumor suppressor gene in cancer therapy is limited, our study still showed that this specific TIP30 overexpression treatment may be a potential therapeutic approach to improve outcomes in patients with EGFR‐mutated NSCLC undergoing gefitinib therapy. Also, TIP30 may serve as a prognostic biomarker for NSCLC harboring gefitinib resistance.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Fig S1‐S4
Table S1
Table S2
ACKNOWLEDGMENTS
This study was funded by the National Natural Science Foundation for Young Scientists of China (Grant No. 81501958). We thank Dr. Xiao Hua and Dr. Aimin Li for generously sharing the anti‐TIP30 antibody. We also thank Dr. JinXin Liu for sharing PC9 and PC9/GR cell lines and for the guidance regarding the tissue culture.
Shuai S, Liao X, Wang H, et al. TIP30 overcomes gefitinib resistance by regulating cytoplasmic and nuclear EGFR signaling in non–small‐cell lung cancer. Cancer Sci. 2021;112:4139–4150. 10.1111/cas.15000
Contributor Information
Shuai Shuai, Email: shining726@hotmail.com.
Senming Wang, Email: wsenming@126.com.
REFERENCES
- 1.Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein‐tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395‐411. Epub 2018/12/01. [DOI] [PubMed] [Google Scholar]
- 2.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735‐742. Epub 2011/07/26. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi K, Hagiwara K. Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC). Targeted Oncol. 2013;8(1):27‐33. Epub 2013/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazandjian D, Blumenthal GM, Yuan W, He K, Keegan P, Pazdur R. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation‐positive non‐small cell lung cancer. Clin Cancer Res. 2016;22(6):1307‐1312. Epub 2016/03/17. [DOI] [PubMed] [Google Scholar]
- 5.Chong CR, Jänne PA. The quest to overcome resistance to EGFR‐targeted therapies in cancer. Nat Med. 2013;19(11):1389‐1400. Epub 2013/11/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onitsuka T, Uramoto H, Nose N, et al. Acquired resistance to gefitinib: the contribution of mechanisms other than the T790M, MET, and HGF status. Lung Cancer. 2010;68(2):198‐203. Epub 2009/07/11. [DOI] [PubMed] [Google Scholar]
- 7.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039‐1043. Epub 2007/04/28. [DOI] [PubMed] [Google Scholar]
- 8.Xiao J, Wang F, Lu H, et al. Targeting the COX2/MET/TOPK signaling axis induces apoptosis in gefitinib‐resistant NSCLC cells. Cell Death Dis. 2019;10(10):777. Epub 2019/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu CF, Chang YW, Kuo KT, et al. NF‐kappaB‐driven suppression of FOXO3a contributes to EGFR mutation‐independent gefitinib resistance. Proc Natl Acad Sci U S A. 2016;113(18):E2526‐E2535. Epub 2016/04/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug‐resistance mechanisms can emerge from drug‐tolerant cancer persister cells. Nat Commun. 2016;7:10690. Epub 2016/02/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. British J Cancer. 2007;96(Suppl):R16‐20. Epub 2007/03/30. [PubMed] [Google Scholar]
- 12.Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011;71(3):1103‐1114. Epub 2011/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res. 2009;1(3):249‐258. Epub 2009/12/04. [PMC free article] [PubMed] [Google Scholar]
- 14.Dittmann KH, Mayer C, Ohneseit PA, et al. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA‐repair: a COX‐2 independent mechanism. Int J Radiat Oncol Biol Phys. 2008;70(1):203‐212. Epub 2007/11/13. [DOI] [PubMed] [Google Scholar]
- 15.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28(43):3801‐3813. Epub 2009/08/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang WC, Chen YJ, Li LY, et al. Nuclear translocation of epidermal growth factor receptor by Akt‐dependent phosphorylation enhances breast cancer‐resistant protein expression in gefitinib‐resistant cells. J Biol Chem. 2011;286(23):20558‐20568. Epub 2011/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rong X, Liang Y, Han Q, et al. Molecular mechanisms of tyrosine kinase inhibitor resistance induced by Membranous/Cytoplasmic/Nuclear Translocation of Epidermal Growth Factor Receptor. J Thorac Oncol. 2019;14(10):1766‐1783. Epub 2019/06/23. [DOI] [PubMed] [Google Scholar]
- 18.Lee LW, Zhang DH, Lee KT, Koay ES, Hewitt RE. CC3/TIP30 expression was strongly associated with HER‐2/NEU status in breast cancer. Ann Acad Med Singap. 2004;33(5 Suppl):S30‐S32. Epub 2005/01/18. [PubMed] [Google Scholar]
- 19.Zhao J, Zhang X, Shi M, et al. TIP30 inhibits growth of HCC cell lines and inhibits HCC xenografts in mice in combination with 5‐FU. Hepatology. 2006;44(1):205‐215. Epub 2006/06/27. [DOI] [PubMed] [Google Scholar]
- 20.Lu B, Ma Y, Wu G, et al. Methylation of Tip30 promoter is associated with poor prognosis in human hepatocellular carcinoma. Clin Cancer Res. 2008;14(22):7405‐7412. Epub 2008/11/18. [DOI] [PubMed] [Google Scholar]
- 21.Tong X, Li K, Luo Z, et al. Decreased TIP30 expression promotes tumor metastasis in lung cancer. Am J Pathol. 2009;174(5):1931‐1939. Epub 2009/04/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CJ, Chou PA, Huang MS, Liu YP. Low TIP30 protein expression is associated with a high risk of metastasis and poor prognosis for Non‐Small‐Cell lung cancer. J Clin Med. 2019;8(1):83. Epub 2019/01/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li A, Zhang C, Gao S, et al. TIP30 loss enhances cytoplasmic and nuclear EGFR signaling and promotes lung adenocarcinogenesis in mice. Oncogene. 2013;32(18):2273‐2281, 81e 1‐12. Epub 2012/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuai S, Yan X, Zhang J, et al. TIP30 nuclear translocation negatively regulates EGF‐dependent cyclin D1 transcription in human lung adenocarcinoma. Cancer Lett. 2014;354(1):200‐209. Epub 2014/08/20. [DOI] [PubMed] [Google Scholar]
- 25.Zhu M, Yin F, Yang L, et al. Contribution of TIP30 to chemoresistance in laryngeal carcinoma. Cell Death Dis. 2014;5:e1468. Epub 2014/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu M, Yuan Y, Pan YY, Zhang Y. Combined gefitinib and pemetrexed overcome the acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small cell lung cancer. Mol Med Rep. 2014;10(2):931‐938. Epub 2014/05/21. [DOI] [PubMed] [Google Scholar]
- 27.Xia W, Wei Y, Du Y, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcino. 2009;48(7):610‐617. Epub 2008/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi M, Tian Y, Li W, et al. ERK inhibition represses gefitinib resistance in non‐small cell lung cancer cells. Oncotarget. 2018;9(15):12020‐12034. Epub 2018/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS ONE. 2011;6(5):e19605. Epub 2011/05/17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Jiang C, Ito M, Piening V, Bruck K, Roeder RG, Xiao H. TIP30 interacts with an estrogen receptor alpha‐interacting coactivator CIA and regulates c‐myc transcription. J Biol Chem. 2004;279(26):27781‐27789. Epub 2004/04/10. [DOI] [PubMed] [Google Scholar]
- 31.Haber DA, Bell DW, Sordella R, et al. Molecular targeted therapy of lung cancer: EGFR mutations and response to EGFR inhibitors. Cold Spring Harb Symp Quant Biol. 2005;70:419‐426. Epub 2006/07/28. [DOI] [PubMed] [Google Scholar]
- 32.Camp ER, Summy J, Bauer TW, Liu W, Gallick GE, Ellis LM. Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor. Clin Cancer Res. 2005;11(1):397‐405. Epub 2005/01/27. [PubMed] [Google Scholar]
- 33.Huang MH, Lee JH, Chang YJ, et al. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol. 2013;7(1):112‐120. Epub 2012/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19(11):679‐696. Epub 2018/09/09. [DOI] [PubMed] [Google Scholar]
- 35.Bakker J, Spits M, Neefjes J, Berlin I. The EGFR odyssey ‐ from activation to destruction in space and time. J Cell Sci. 2017;130(24):4087‐4096. Epub 2017/11/29. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura Y, Yoshioka K, Bereczky B, Itoh K. Evidence for efficient phosphorylation of EGFR and rapid endocytosis of phosphorylated EGFR via the early/late endocytic pathway in a gefitinib‐sensitive non‐small cell lung cancer cell line. Mol Cancer. 2008;7:42. Epub 2008/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C, Li A, Zhang X, Xiao H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J Biol Chem. 2011;286(11):9373‐9381. Epub 2011/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barré B, Vigneron A, Perkins N, Roninson IB, Gamelin E, Coqueret O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol Med. 2007;13(1):4‐11. Epub 2006/11/23. [DOI] [PubMed] [Google Scholar]
- 39.Cao W, Liu Y, Zhang R, et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL‐6/JAK1/STAT3 signal pathway in Gefitinib‐resistant lung cancer cells. Sci Rep. 2015;5:8477. Epub 2015/07/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuzyk A, Mai S. c‐MYC‐induced genomic instability. Cold Spring Harb Perspect Med. 2014;4(4):a014373. Epub 2014/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prochownik EV. c‐Myc: linking transformation and genomic instability. Curr Mol Med. 2008;8(6):446‐458. Epub 2008/09/11. [DOI] [PubMed] [Google Scholar]
- 42.Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging roles of C‐Myc in cancer stem cell‐related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int J Mol Sci. 2019;20(9):2340. Epub 2019/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1
Table S2