

Abstract
Immunotherapy with immune‐checkpoint therapy has recently been used to treat oral squamous cell carcinomas (OSCCs). However, improvements in current immunotherapy are expected because response rates are limited. Transforming growth factor‐β (TGF‐β) creates an immunosuppressive tumor microenvironment (TME) by inducing the production of regulatory T‐cells (Tregs) and cancer‐associated fibroblasts and inhibiting the function of cytotoxic T‐lymphocytes (CTLs) and natural killer cells. TGF‐β may be an important target in the development of novel cancer immunotherapies. In this study, we investigated the suppressive effect of TGF‐β on CTL function in vitro using OSCC cell lines and their specific CTLs. Moreover, TGFB1 mRNA expression and T‐cell infiltration in 25 OSCC tissues were examined by in situ hybridization and multifluorescence immunohistochemistry. We found that TGF‐β suppressed the function of antigen‐specific CTLs in the priming and effector phases in vitro. Additionally, TGF‐β inhibitor effectively restored the CTL function, and TGFB1 mRNA was primarily expressed in the tumor invasive front. Interestingly, we found a significant negative correlation between TGFB1 mRNA expression and the CD8+ T‐cell/Treg ratio and between TGFB1 mRNA expression and the Ki‐67 expression in CD8+ T‐cells, indicating that TGF‐β also suppressed the function of CTLs in situ. Our findings suggest that the regulation of TGF‐β function restores the immunosuppressive TME to active status and is important for developing new immunotherapeutic strategies, such as a combination of immune‐checkpoint inhibitors and TGF‐β inhibitors, for OSCCs.
Keywords: cytotoxic T‐cells, immunotherapy, oral squamous cell carcinoma, regulatory T‐cells, TGF‐β
We found that, TGF‐β suppressed the function of antigen‐specific CTLs in the priming and effector phases in vitro. And, we found a significant negative correlation between TGFB1 mRNA expression and the CD8+ T‐cell/Treg ratio. Inhibition of TGF‐β restores the immunosuppressive TME to active status by a mechanism different from that of immune‐checkpoint inhibitors, suggesting that the combination of both may lead to a new therapeutic strategy for OSCCs.

Abbreviations
- APC
allophycocyanin
- EBV
Epstein‐Barr virus
- Foxp3
Forkhead box protein P3
- HLA
human leucocyte antigen
- IFN
interferon
- OSCC
oral squamous cell carcinoma
- PD‐1
Programmed death receptor 1
- PD‐L1
Programmed death ligand 1
- Smad
small mothers against decapentaplegic
- TCM
central memory T‐cell
- TEM
effector memory T‐cell
- TGF‐β
transforming growth factor‐beta
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- Treg
regulatory T‐cell
1. INTRODUCTION
OSCC is the most common histological type of oral cancer. Definitive surgery is a standard treatment for patients with OSCC and involves multidisciplinary approaches with chemotherapy and/or radiotherapy.1 Recently, anti‐programmed death receptor 1 (PD‐1) immune‐checkpoint inhibitors, such as nivolumab and pembrolizumab, demonstrated significant improvement in the overall survival of patients with recurrent and/or metastatic head and neck squamous cell cancers in the CheckMate 1412 and KEYNOTE‐0483 studies, respectively, and have been approved worldwide for the treatment of OSCC. However, further investigations are needed to develop novel therapeutics such as combinational immunotherapy, as their efficacy is only seen in 10%‐20% of cases. Although studies are currently underway to identify strategies that would be synergistic with available checkpoint inhibitors, there are currently no combination immunotherapy regimens that have demonstrated definitive therapeutic efficacy.4, 5
Transforming growth factor‐beta (TGF‐β) is a well known multifunctional cytokine. It is produced by many types of cancer infiltrating cells and promotes tumor growth by inducing angiogenesis, stemness, invasion, and epithelial‐mesenchymal transition.6, 7, 8, 9 TGF‐β expression is shown to be increased in several cancer types, including oral cancers, and has been reported to correlate with cancer invasion, metastasis, and poor prognosis.10, 11, 12, 13 With the recent widespread use of immunotherapy, there is a growing interest in the effects of TGF‐β on immune cells in the tumor microenvironment (TME). In the immune system, TGF‐β induces the differentiation of regulatory T‐cells (Tregs) by promoting forkhead box protein P3 (Foxp3) expression, inducing cancer‐associated fibroblasts, and suppressing the proliferation and function of CTLs via the TGF‐β/Smad3 pathway and of natural killer cells via the inhibition of mTOR.14, 15, 16 CTLs are induced by antigen‐presenting cells that present cancer antigens to T‐cells in the priming phase and attack cancer cells in an antigen‐specific manner in the effector phase (Figure 1). As induced cancer‐specific CTLs function as direct effector cells against cancer cells, the effect of TGF‐β on CTLs needs further investigation. Recent studies have shown that TGF‐β1 promotes the creation of an immunosuppressive environment by eliminating CTLs from the TME.17, 18 Moreover, the gene set variance analysis scores of TGF‐β signaling‐associated genes were higher in melanoma patients who did not respond to anti‐PD‐1 antibody therapy.19 Therefore, inhibition of TGF‐β could be a promising cancer treatment strategy by releasing the immunosuppressive state in the TME and enhancing the anti‐tumor response. TGF‐β inhibition may also be used in combination with immune‐checkpoint inhibitors20, 21, 22; such clinical trials for advanced solid tumors, including head and neck cancers, are ongoing, and therapeutic effects have been observed.23, 24
FIGURE 1.

Illustration of in vitro experiment reproducing priming phase and effector phase. Antigen‐presenting cells present tumor antigens to T‐cells through binding of the HLA‐cancer antigen peptide complex on antigen‐presenting cells and T‐cell receptor (TCR) on T‐cells, which then induces cancer antigen‐specific CTLs in the priming phase (left upper panel). The induced cancer antigen‐specific CTLs recognize the presented cancer antigens and specifically attack cancer cells in the effector phase (right upper panel). These processes were reproduced using in vitro experiments. Induction of cancer antigen‐specific CTLs during the priming phase was reproduced in vitro using CMVpp65 antigen to induce CMVpp65 antigen‐specific CTLs from PBMCs (left lower panel). The cytotoxic activity of tumor antigen‐specific CTLs during the effector phase was reproduced in vitro by co‐culturing CMVpp65 CTLs and HSC‐3pp65 cells (right lower panel)
In our study, we focused on the effects of TGF‐β1 on the priming and effector phases of antigen‐specific CTLs in OSCCs and elucidated the role of TGF‐β in the creation of an immunosuppressive state in the TME.
2. MATERIALS AND METHODS
2.1. Cell culture
Human OSCC cell lines (HSC‐2, HSC‐3, and HSC‐4) were obtained from the Japanese Collection of Research Bioresource Cell Bank. HSC‐3pp65 cells transfected with cytomegalovirus (CMV)pp65 antigen and Td‐Tomato in HSC‐3 cells were prepared as previously described.25 The cells were maintained in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Inc South Logan) and 1% penicillin‐streptomycin (Gibco, Grand Island, NY, USA) in 75 mL flasks at 37°C in 5% CO2 humidified air. Only HSC3pp65 cells were cultured in a culture medium supplemented with 250 μg/mL G418.
2.2. Clinical samples
Twenty‐five patients with OSCC who had not received neoadjuvant chemotherapy or radiation therapy and who had undergone surgical resection at the Department of Maxillofacial Surgery, Aichi Gakuin University, were included in the study. The demineralized samples were analyzed using preoperative biopsy tissues. The clinical characteristics of the patients are presented in Table 1. Programmed cell ligand 1 (PD‐L1) expression was assessed by its positivity in cancer cells (>5% was regarded as positive). This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethical Committee of Aichi Gakuin University (approval number: 82) and Aichi Medical University (approval number: 2020‐H033).
TABLE 1.
Patient and tumor characteristics
| n | % | |
|---|---|---|
| Age | ||
| Mean (SD) | 63.8 | (18.7) |
| Median (range) | 73 | (23‐89) |
| Gender | ||
| Male | 10 | 40 |
| Female | 15 | 60 |
| Disease presentation | ||
| Primary cancer | 20 | 80 |
| Recurrent cancer | 5 | 20 |
| Tumor site | ||
| Tongue | 14 | 56 |
| Gingiva | 9 | 36 |
| Buccal | 2 | 8 |
| Floor of mouth | 0 | 0 |
| Stage | ||
| ≦Ⅱ | 23 | 92 |
| ≧Ⅲ | 2 | 8 |
| PD‐L1 expression | ||
| < 5% | 18 | 72 |
| ≧ 5% | 7 | 28 |
2.3. Induction of CMV‐CTLs and EBV‐CTLs
CMV‐CTLs (CMVpp65 antigen‐specific CTLs) and EBV‐CTLs (EBV LMP2 antigen‐specific cytotoxic CTLs) were prepared in accordance with a method reported previously.25 TGF‐β1 (10 ng/mL, Miltenyi Biotec Inc) and/or SB525334 (TGF‐β receptor I inhibitor, 1 μmol/L, Selleck Chem) were added only on day 0 during the 14‐day culture. We evaluated the effect of TGF‐β1 on antigen‐specific CTL induction on days 7 and 14. The total live cell number was calculated using the trypan blue dye inclusion method, and the cell numbers of CMV‐CTLs and EBV LMP2‐CTLs were calculated in accordance with the following formula: (total cell number) × (tetramer and CD8 double‐positive rate).
2.4. Flow cytometry
To analyze antigen‐specific CTL populations, cells were stained for 10 min at 4°C with APC‐conjugated human leucocyte antigen (HLA) HLA‐A*24:02 CMVpp65 tetramer‐QYDPVAALF or APC‐conjugated HLA‐A*02:01 CMVpp65 tetramer‐NLVPMVATV, and for EBV LMP2‐CTLs, with APC‐conjugated HLA‐A*24:02 EBV LMP2 tetramer‐TYGPVFMSL and PYLFWLAAI (MBL Co., Nagoya, Japan). Subsequently, the cells were stained with an APC‐Cy7 conjugated anti‐CD8 monoclonal antibody (mAb) (Biolegend) for 20 min at 4℃. When CTL memory subsets were evaluated, FITC‐CD45 mAb (Biolegend) and BV421 conjugated anti‐CCR7 mAb (Biolegend) were also used. For analysis of CTL proliferation, 10 μmol/L bromodeoxyuridine and 5‐bromo‐2′‐deoxyuridine (BrdU) was added to each sample and incubated for 1 h. Cells were collected and antigen‐specific CTLs were stained as described above and fixed in 4% formaldehyde for 30 min at 4°C. Subsequently, the cells were permeabilized with 1% Triton X‐100 for 30 min at 4°C. After washing, 200× diluted DNase was added, and the cells were incubated for 30 min at 37°C. They were then washed once and stained with FITC‐conjugated anti‐BrdU antibody (BioLegend) for 30 min at 4°C. Tumor necrosis factor (TNF)‐α and interferon (IFN)‐γ in the culture medium supernatant were measured using a Human Th1/Th2 Cytokine Kit II (BD Biosciences) in accordance with the manufacturer's protocol. Cells or beads were analyzed on a BD LSRFortessa flow cytometer (BD Biosciences) using the FlowJo software (Tree Star, Inc).
2.5. Measurement of CMV‐CTLs cytotoxicity with water soluble tetrazolium‐1 (WST‐1)
The viability assay was performed in accordance with a previously reported method.25 HSC‐3pp65 cells were seeded in 96‐well plates at 104 cells/well and incubated for 2 d. To measure HSC3pp65 cell viability, serial dilutions of TGF‐β1, SB525334, and serial numbers of CMV‐CTLs were added and cultured for 5 d in ALyS505N medium supplemented with 100 IU/mL of IL‐2 and 5% FBS . Then, each well was washed to remove dead cells. The remaining CMV‐CTLs and the viability of HSC‐3pp65 cells was measured by incubating in the WST‐1 Cell Proliferation Assay System (Takara Bio, Otsu, Japan). Absorbance was measured using an iMark microplate reader (Bio‐Rad Laboratories, Hercules, CA, USA) at 450 nm and 620 nm wavelengths. Cell viability was calculated using the following formula: % viability = 100 × (E − B)/(H − B), where E is the absorbance of the experimental well, H is the absorbance of the HSC3pp65 well without drugs and CMV‐CTLs, and B is the medium alone (blank).
2.6. Measurement of CMV‐CTL cytotoxicity and apoptosis of CMV‐CTLs with annexin V
HSC‐3pp65 cells were seeded into 24‐well plates at 5 × 104 cells per well and cultured for 2 d in 1 mL ALyS505N medium. The cells were co‐cultured with 105 CMV‐CTLs for 48 h in the presence or absence of TGF‐β1, TGF‐β3, and/or SB525334. The cells in each well were washed twice with 500 µL phosphate‐buffered saline and incubated with the APC‐conjugated HLA‐A*24:02 CMVpp65 tetramer‐QYDPVAALF and APC‐Cy7‐conjugated anti‐CD8 mAb for 20 min at 4℃. After washing with 500 µL of annexin buffer (10 mmol/L HEPES, 150 mmol/L NaCl, and 2 mmol/L CaCl2 [pH 7.4]) twice, the cells were incubated with FITC‐conjugated annexin V for 10 min at room temperature. Flow cytometry was performed. Annexin V positivity in cells gated on the Tomato+ population was analyzed using FlowJo version 10 software. Cytotoxicity was calculated using the following formula: % cytotoxicity = 100 × (E − S)/(100 − S), where E is annexin V positivity of the experimental well and S is annexin V positivity in the absence of CTLs (target cells were incubated with medium alone). Annexin V positivity in cells gated on the HLA‐tetramer+CD8+ population was analyzed for apoptosis of CMV‐CTLs.
2.7. Isolation of Treg from PBMC and suppression assay
eTreg cells were isolated from PBMCs of healthy donors using a CD4+CD25+ Regulatory T‐cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) in accordance with the manufacturer's recommended protocol. CMV‐CTLs from the same donor for isolation of Tregs were co‐cultured with the isolated eTreg fraction and HSC‐3pp65 at a ratio of 1:1:1. At 7 d after co‐coculture, the percentages and cell numbers of the CMVpp65‐tetramer+CD8+ fraction were measured.
2.8. Western blotting
We cultured 106 CMV‐CTLs in the presence of TGF‐β1 (10 ng/mL) or in 50% of the culture supernatant from HSC‐3 cells for 30 min. The same amount of CTL lysate was resolved on SDS‐PAGE gels and transferred to nitrocellulose membranes. After blocking with Tris‐buffered saline (TBS) with 1% skimmed milk for 1 h at room temperature, the membranes were incubated with primary antibodies. The following primary antibodies were used: Smad2/3 and phospho‐Smad2/3 (Cell Signaling Technology) at 1:1000 dilution. The sections were then washed with TBS 4 times and incubated with peroxidase polymer anti‐rabbit IgG (Vector Laboratories) at 1:1000 dilution for 30 min at room temperature. After washing with TBS 4 times, the protein signal was detected using ECL Prime western blotting Detection Reagent and imaged using an Amersham Imager 600 (GE Healthcare Systems).
2.9. TGF‐β1 analysis by ELISA
OSCC cell lines were cultured in 6‐well plates at 5 × 105 cells per well in 2 mL of the culture medium for 5 d, after which the supernatant was collected. TGF‐β1 concentration was determined using the Human TGF‐β1 Quantikine ELISA kit (R&D Systems Inc) in accordance with the manufacturer's protocol.
2.10. Multifluorescence immunohistochemistry (MF‐IHC)
Formalin‐fixed, paraffin‐embedded OSCC sections were incubated with Universal HIER antigen retrieval reagent (Abcam, Cambridge, UK) for 10 min at 110°C in an autoclave. After cooling to 60°C, the cells were washed with 10 mmol/L Tris‐HCl (pH 7.4) buffered saline for 5 min and stained with primary antibody for 60 min at room temperature. The following primary antibodies were used: CD3 (clone M4622, Spring Biosciences), CD8 (clone 1A5, Biogenex, Fremont, CA, USA), Foxp3 (clone 236A/E7, Abcam), Ki‐67 (clone MIB‐1, DAKO, Denmark A/S), and cytokeratin (Biogenex). After washing twice for 5 min each, the sections were reacted with peroxidase polymer anti‐mouse IgG and anti‐rabbit IgG (Vector Laboratories) for 30 min at room temperature. After washing twice for 5 min each, the signals in the sections were visualized with tyramide signal amplification and the OPAL system (PerkinElmer, Waltham, MA, USA). The sections were heated with 10 mmol/L sodium citrate buffer solution (pH 6.0) for 10 min at 95°C to remove the binding antibodies before progressing to the next primary antibody staining. The staining method for the second and subsequent colors was the same as described above. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI).
2.11. TGFB1 RNA‐in situ hybridization (ISH)
TGFB1 mRNA in formalin‐fixed, paraffin‐embedded OSCC tissue sections was detected in accordance with the manufacturer's protocol by RNA‐ISH using an RNAscope assay (Advanced Cell Diagnostics). The following probes were used: Hs‐TGFB1‐CDS, Hs‐POLR2A, and DapB (Advanced Cell Diagnostics). Hs‐POLR2A was used as a positive control and DapB was used as a negative control. Chromogenic detection was performed using 3,3′‐diaminobenzidine tetrahydrochloride (DAB), and the sections were counterstained with hematoxylin.
2.12. Image analysis
Slides were scanned at low resolution with a Vectra system (PerkinElmer), and regions of interest (ROIs) in the invasive front of the tumor were chosen using Phenochart software (PerkinElmer). Adjacent normal epithelium was defined as the epithelial edge of the resected tissue, which was diagnosed as negative for tumor margins. At least 3 ROIs were captured with a ×20 magnification objective for MF‐IHC and a ×40 magnification objective for RNA‐ISH. Then, the reconstructed multispectral images were obtained, and the number of marker‐positive cells or dot signals was counted using the Inform software (PerkinElmer). For MF‐IHC analysis, ROIs were separated into tumor nest regions and tumor stroma regions using cytokeratin staining, and cells were segmented and phenotyped by software identification. The identification tools were trained by staining a lymph node sample. CD8+ T‐cell: CD3+CD8+Foxp3− cell; CD4+ T‐cell: CD3+CD8− Foxp3− cells; and Treg: CD3+CD8−Foxp3+ cells (Figure S1). For RNA‐ISH analysis, the ROIs were separated into tumor nest regions and tumor stroma regions by manual selection, and the number of dot signals was counted by software identification. Then, cell or dot density was calculated using the following formula: (cell counts or dot signal counts)/(tissue area [megapixels]).
2.13. Statistical analysis
Numerical data are presented as the mean (±SE) of independent experiments. Differences between 2 groups were examined using Student t test. Mann‐Whitney U test and paired t test were performed to determine the significance of the RNA‐ISH analysis. Correlation between RNA‐ISH analysis data and MF‐IHC analysis data was determined using the Spearman rank correlation coefficient. Statistical analyses were performed using R Statistical Software (version 3.6.3; Foundation for Statistical Computing, Vienna, Austria). A P‐value < .05 was considered statistically significant.
3. RESULTS
3.1. TGF‐β1 inhibited the induction of antigen‐specific CTLs from PBMCs
First, we reproduced the immune responses of tumor antigen‐specific CTLs in the priming phase by in vitro induction of CMV or EBV‐specific CTL by the stimulation of PBMCs from healthy donors with T‐cell epitope peptides (Figure 1, left). Flow cytometric analysis revealed that stimulation of PBMCs from donor 1 (HLA‐A24) with CMVpp65 peptide increased the frequency of CMV‐CTLs (CD8 and A24 CMVpp65 tetramer double‐positive cells) from <0.01% on day 0 (data not shown) to 45.4% on day 14 (Figure 2A, control). TGF‐β1 inhibited the peptide‐stimulated increase in CMV‐CTL frequency to 8.8% on day 14 (Figure 2A, plot T), and inhibition of TGF‐β signaling by SB525334 suppressed the inhibition. The frequency recovered was from 8.8% to 51.1% on day 14 (Figure 2A, plot T+S). Peptide stimulation increased the number of CMV‐CTLs from 120 on day 0 to 8.6 × 105 cells on day 14, which expanded approximately 7200‐fold (Figure 2B, ●). When the peptide‐stimulated cells were cultured with TGF‐β1, the number of CMV‐CTLs decreased to 0.7 × 105 cells on day 14, showing that TGF‐β1 inhibited peptide‐stimulated proliferation by approximately 1/12 (Figure 2B, ▲). SB525334 eliminated the inhibition and the cell number of CMV‐CTLs on day 14 recovered from 0.7 × 105 to 1.3 × 106 cells (Figure 2B,◆). In the wells cultured with peptide and SB525334, the frequency and cell number of CMV‐CTLs were lower than those of peptide alone. This may be because SB525334 increases the number of tetramer negative non‐specific CD8+ T‐cells. Similar results were obtained when the donor was changed to an HLA‐A2 positive individual (Figure 2C, D), and the antigen was changed to EBV (Figure 2E, F). These results suggested that TGF‐β1 inhibits the induction of antigen‐specific CTLs in cancer. Memory subsets of CMV‐CTLs were evaluated on days 0 and 7 (Figure 3). In total, 50% of CMV‐CTLs were central memory T‐cells (TCM; CCR7+CD45RA−) and 32.1% were effector memory T‐cells (TEM; CCR7−CD45RA−) on day 0 (Figure 3A). On day 7, the percentage of the TCM fraction of CMV‐CTLs in the well without TGF‐β1 decreased to 18.6% and that of the TEM fraction increased to 57.1% (Figure 3A), indicating that the differentiation of CMV‐CTLs was advanced. In contrast, the TCM percentage of CMV‐CTLs increased to 82.5% in the wells with TGF‐β and, conversely, that of TEM decreased to 10.0% (Figure 3A), indicating that the differentiation of CMV‐CTLs was inhibited. Additionally, in the well with TGF‐β1 and TGF‐β inhibitor (SB325334), the percentages of TCM and TEM in CMV‐CTL approximated those of the well without TGF‐β1 (Figure 3A). Figure 3B, C shows the average and SE of TCM and TEM under each condition. These results suggested that TGF‐β1 inhibited CTL differentiation from TCM to TEM.
FIGURE 2.
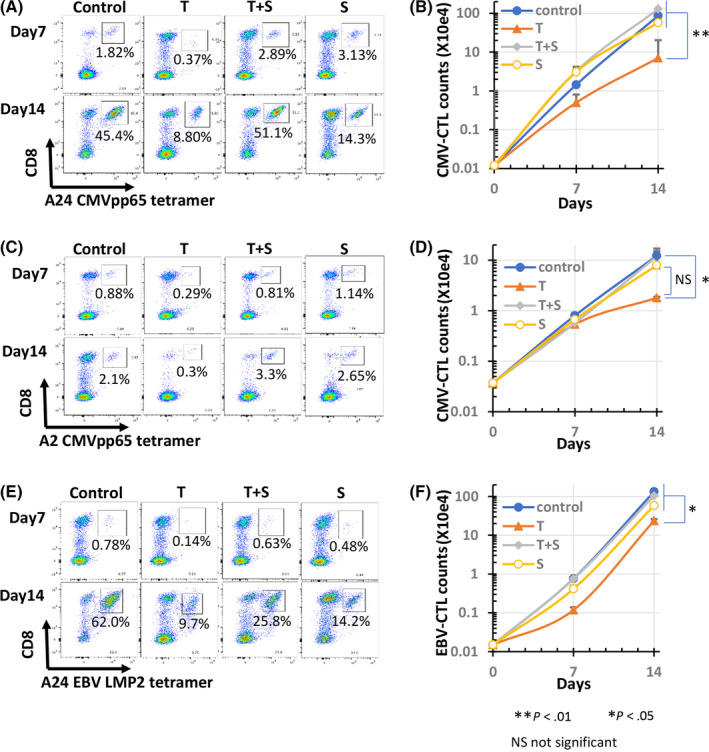
Effect of TGF‐β1 on antigen‐specific CTL induction from PBMCs. In total, 106 PBMCs isolated from 2 healthy donors (SS, ST) were co‐cultured with an antigen peptide (1 μmol/L) in the presence of SB525334 (1 μmol/L) and/or TGF‐β1 (10 ng/mL). On days 7 and 14, the cells were stained with HLA‐tetramer and anti‐CD8 and analyzed using flow cytometry. The number of HLA‐tetramer+CD8+ cells was counted. Cytograms of the induced CMVpp65‐CTLs from donor SS and donor ST (A, C) and the induced EBV LMP2‐CTLs from donor SS (E) are shown. Cell number transition of the induced CMVpp65‐CTLs from donor SS and donor ST (B, D), and the induced EBV LMP2‐CTLs from donor SS (F) are shown. Control: treated with peptides only; T: induction with peptides in the presence of TGF‐β1; T+S: induction with peptides in the presence of TGF‐β1 and SB525334
FIGURE 3.
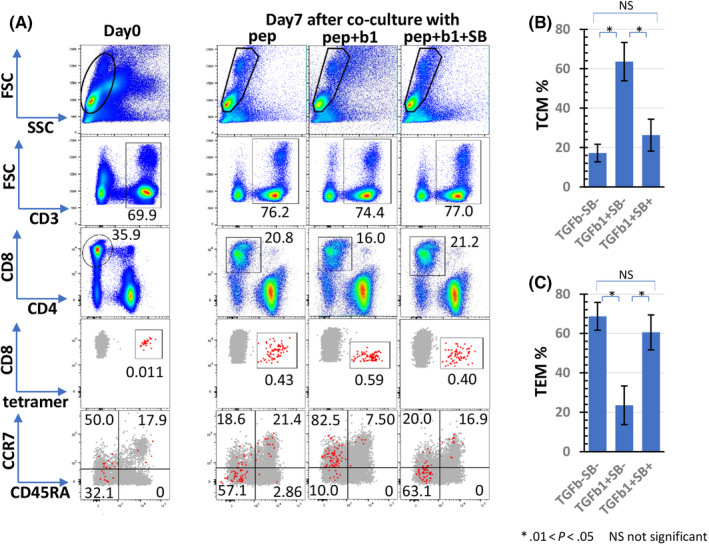
Effect of TGF‐β1 on differentiation of antigen‐specific CTLs. In total, 106 PBMCs isolated from 2 healthy donors (SS) were co‐cultured with a CMVpp65 peptide (1 μmol/L) in the presence of SB525334 (1 μmol/L) and/or TGF‐β1 (10 ng/mL) in triplicate. Cells were stained with tetramer and anti‐CD8 and analyzed using flow cytometry on day 0 and day 7. Tetramer+CD8+ cells were separated by CD45RA and CCR7, and percentages of central memory T‐cells (TCM CD45RA−CCR7+) and effector memory T‐cells (TEM, CD45RA−CCR7−) were measured. Representative cytograms are shown in panel (A). Red large dots indicate tetramer+CD8+ population and brown large dots indicate antigen non‐specific CD8+ T‐cells. Panels (B) and (C) show the average and SE of TCM and TEM in each condition. b1, TGF‐β1; pep, CMVpp65 peptide; SB, SB525334
3.2. TGF‐β1 inhibited cytotoxicity and proliferation of antigen‐specific CTLs in effector phase
Next, we examined the effect of TGF‐β on the effector phase. Tumor antigen‐specific cytotoxic activity was reproduced in vitro by co‐culturing CMVpp65‐CTL and HSC‐3pp65 (Figure 1, right). HSC‐3pp65 was co‐cultured with CMVpp65‐CTL at each E/T ratio (effector: CMVpp65‐CTL, target: HSC‐3pp65) for 5 d. The viability of HSC‐3pp65 was decreased in an E/T ratio‐dependent manner. In the control assay, the viability decreased from 70% at an E/T ratio of 0.125 to 10% at an E/T ratio of 1.0 in the control assay (Figure 4A, ●). TGF‐β1 increased the viability of all E/T ratios (Figure 4A, 〇), indicating that TGF‐β1 suppresses CMV‐CTL cytotoxicity. The addition of SB525334 inhibited cytotoxicity in a dose‐dependent manner and enhanced cytotoxicity compared with the control (Figure 4A, ▲, □, ◇). Furthermore, Figure 4B showed that cytotoxicity against HSC‐3pp65 was enhanced by SB525234, even in the absence of external addition of TGF‐β1. We additionally performed an annexin V assay (Figure 4C) to confirm the direct cytotoxicity of CMV‐CTLs (Figure 4D). Cytotoxicity in the wells without TGF‐β was 77.1%, which decreased to 42.2% and 39.4% in the wells with TGF‐β1 or TGF‐β3, respectively. In the wells with TGF‐β and SB525344, the cytotoxicity was close to that of the wells without TGF‐β. Therefore, it was confirmed that TGF‐β also inhibited antigen‐specific cytotoxic activity of CMV‐CTLs in the annexin V assay.
FIGURE 4.
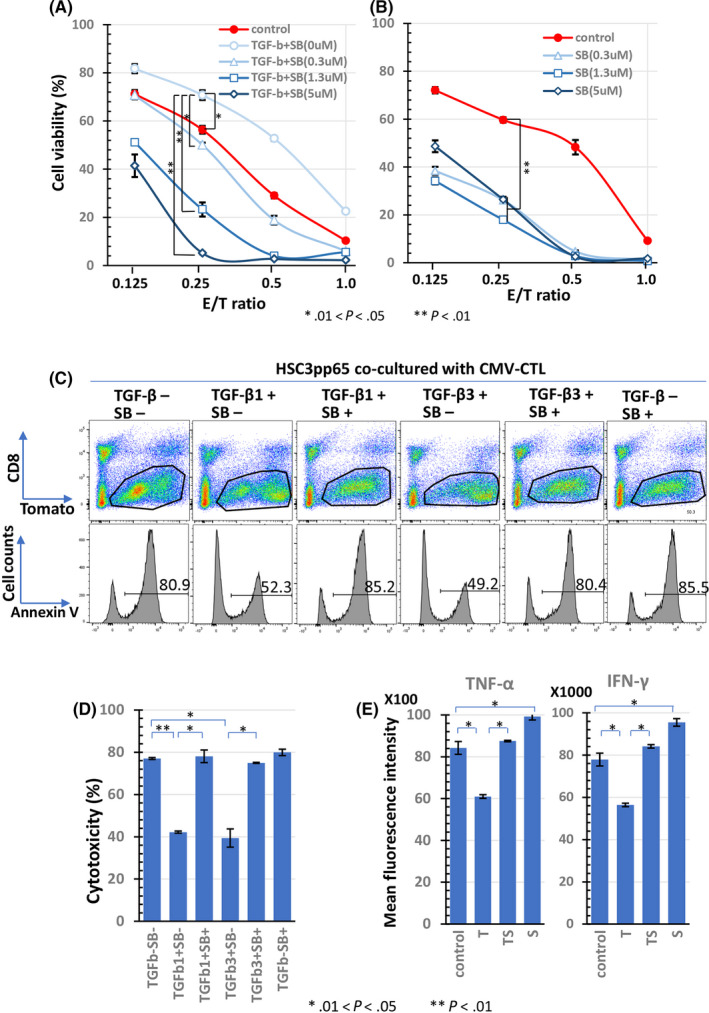
Effect of TGF‐β on cytotoxicity of antigen‐specific CTLs. CMV‐CTLs induced from PBMCs of donor SS were co‐cultured with HSC‐3pp65 in serial E/T ratio for 5 d. HSC‐3pp65 viability was measured using the WST‐1 assay. Cells were treated with SB525234 at the indicated doses in the presence of 10 ng/mL TGF‐β1 (A) or in the absence of TGF‐β1 (B). The mean fluorescence intensity (MFI) of IFN‐γ and TNF‐α concentrations in the supernatant of the CMV‐CTLs co‐cultured with HSC‐3pp65 at an E/T ratio of 1 for 24 h in the presence of SB525334 (1 μmol/L) and/or TGF‐β1 (10 ng/mL) are shown in (C). At 2 d after co‐culture of the CMV‐CTLs and HSC‐3pp65 with TGF‐β1 (10 ng/mL) or TGF‐β3 (10 ng/mL) in the presence or absence of SB525334(1 μmol/L), an annexin V assay was performed. Cells were separated by CD8 and Tomato, the percentage of annexin V+ cells in the Tomato+ gated cells was measured (D), and the cytotoxicity was calculated for each condition (E). The numbers of histograms in panel (D) indicate the percentages of annexin+ cells. SB, SB525334
We measured the relative concentrations of TNF‐α and IFN‐γ in the supernatant of CMVpp65‐CTLs that were co‐cultured with HSC‐3pp65 for 24 h at an E/T ratio of 1 (Figure 4E). The relative concentrations are indicated as mean fluorescence intensity. The relative concentrations of TNF‐α and IFN‐γ in the supernatant of the control were 8400 and 78 000, respectively, which decreased to 6000 and 56 000, respectively, by adding TGF‐β1. These concentrations were recovered by SB525334 to 8800 and 84 000, respectively. This indicated that TGF‐β1 suppressed the production of TNF‐α and IFN‐γ by CTLs, which were stimulated with the target cells, HSC‐3pp65. Interestingly, the concentrations were higher in the wells with SB525334 alone than in the control wells.
We then examined the effect of TGF‐β1 on the proliferation of CMV‐CTLs in the effector phase. In total, 5 × 104 CMV‐CTLs were co‐cultured with HSC‐3pp65 at an E/T ratio of 1, and the cell number was counted on day 7. The cell number of CMVpp65‐CTLs increased to 4.5 × 105 cells on day 7 in the control well (Figure 5A, ●). In contrast, a slight increase in the cell number to 1.0 × 105 was seen in the well containing TGF‐β1 (Figure 5A, ▲). SB525334 recovered the number of cells inhibited by TGF‐β1 to 3.5 × 105 cells (Figure 5A, ◆). These results indicated that TGF‐β1 inhibited the proliferation of CMV‐CTLs. Interestingly, the cell number was higher in the wells containing SB525334 than in the control wells on day 7 (Figure 5A, 〇). In addition, BrdU incorporation into CMV‐CTL on day 2 after stimulation with HSC‐3pp65 was examined (Figure 5B, C). Flow cytometric analysis demonstrated that BrdU positivity in TGF‐β1‐ or TGF‐β3‐containing wells was 30.2% and 31.8%, respectively, which was lower than that in the control well (39.3%) (Figure 5C). BrdU positivity in the well containing TGF‐β1 and SB525334 or in the well containing TGF‐β3 and SB525334 was almost equal to that in the control well (Figure 5C). The percentage of BrdU incorporation in CMV‐CTLs was 7.52% before antigen stimulation (data not shown). The average and SE of BrdU incorporation in each condition are shown in Figure 5B. These results suggested that TGF‐β1 and β3 attenuate CTL cell cycle. In addition, induction of apoptosis in CMV‐CTLs by TGF‐β after stimulation with HSC‐3pp65 was investigated (Figure 6). In the well without TGF‐β, the percentage of annexin V‐positive cells among CMV‐CTLs was 18.8%, which increased to 31.7% or 33.6% in the well with TGF‐β1 or TGF‐β3, respectively (Figure 6A). In addition, in the well with TGF‐β1 or TGF‐β3 in the presence of SB525334, the percentages were similar to those in the well without TGF‐β (Figure 6A). The percentage of HLA‐A24‐tetramer+CD8+ cells decreased by TGF‐β1 or TGF‐β3 and was restored by SB525334, although the difference was not statistically significant (Figure 6A, C). The average and SE for the percentage of annexin V‐positive cells in the CMV‐CTL and percentage of HLA‐A24‐tetramer+CD8+ cells in each condition are shown in Figure 6B and Figure 6C, respectively. These results suggested that TGF‐β1 and TGF‐β3 induce CTL apoptosis. We also examined the direct effect of SB525334 on the proliferation of target cells (HSC3pp65) and induction of apoptosis using WST‐1, BrdU incorporation, and annexin V assay, respectively. Neither inhibition of proliferation nor induction of apoptosis was observed (Figure S2).
FIGURE 5.

Effect of TGF‐β on proliferation of antigen‐specific CTLs. The cell number transition of the CMV‐CTLs co‐cultured in triplicate with HSC‐3pp65 at an E/T ratio of 1 for 7 d in the presence of SB525334 (1 μmol/L) and/or TGF‐β1 (10 ng/mL) are shown in (A). The percentage of BrdU incorporated into CMV‐CTLs after co‐culture in duplicate with HSC‐3pp65 for 2 d at an E/T ratio of 1 in the presence of SB525334 (1 μmol/L), and/or TGF‐β1 (10 ng/mL) or TGF‐β3 (10 ng/mL) was analyzed by flow cytometry (C), and is indicated as a bar graph (B). SB, SB525334
FIGURE 6.
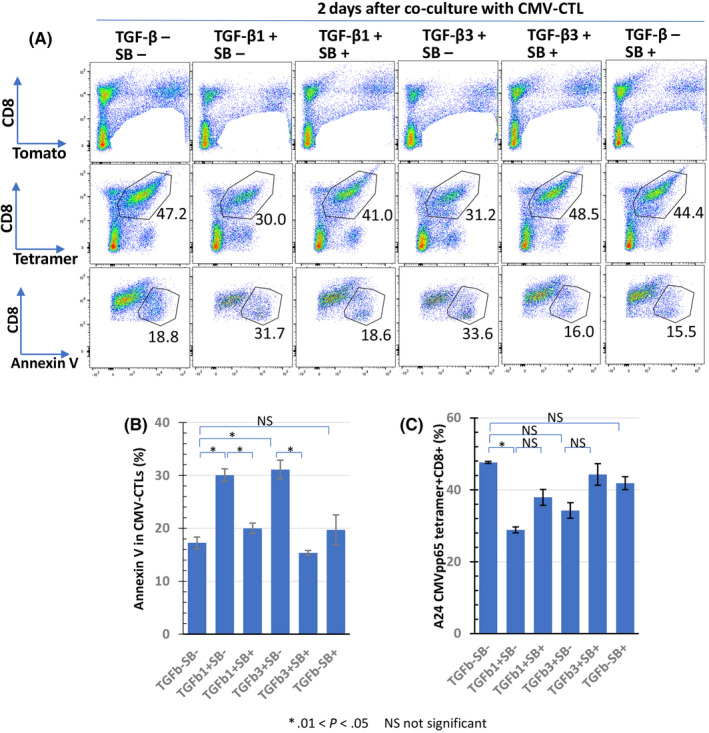
Effect of TGF‐β on apoptosis induction of antigen‐specific CTLs. CMV‐CTLs were co‐cultured with HSC‐3pp65 in duplicate in the presence of SB525334 (1 μmol/L), TGF‐β1 (10 ng/mL), or TGF‐β3 (10 ng/mL) for 2 d. Cells that excluded the CD8‐Tomato+ fraction were separated by CD8 and tetramers. Percentages of annexin V+ cells in the CD8+tetramer+ fraction were measured using flow cytometry (A); these are presented as a bar graph (B). Panel (C) shows the percentages of the CD8+tetramer+ fraction in each culture condition
Collectively, these results suggested that TGF‐β attenuates CTL proliferation through several mechanisms such as suppression of differentiation, DNA synthesis, and apoptosis induction.
Furthermore, we examined the effect of Tregs on CMV‐CTL proliferation and explored whether CTL proliferation was restored by SB525334. Cytograms of isolated Tregs and conventional T‐cells (Tconvs, CD4+CD25−) are shown in Figure S3A. The percentage of CD8+tetramer+ cells and the number of CMV‐CTLs before co‐culture were 36.8% and 3 × 104, respectively (Figure S3B, C). At 7 d after co‐cultured with Tconvs and HSC3pp65 in the absence of SB525334, the number of CMV‐CTLs increased to 3.3 × 105 and, in the well co‐cultured with Treg and HSC3pp65, the increase in the number of CMV‐CTLs was limited to 1.2 × 105, but increased to 2.4 × 105 in the presence of SB525334 (Figure S3C). Although these data were obtained from a single culture because the number of isolated Tregs was limited, TGF‐β may be considered to play an important role in the inhibition of T‐cell function by Tregs.
3.3. Smad2/3 phosphorylation in CMV‐CTLs by TGF‐β1 and culture supernatant from OSCC cells
In total, 5 × 105 OSCC cells (HSC‐2, HSC‐3, and HSC‐4) were cultured in 2 mL of medium for 5 d, and the concentration of TGF‐β1 in the supernatant was measured. TGF‐β1 concentrations of HSC‐2, HSC‐3, and HSC‐4 were 2.2, 1.7, and 4.2 ng/mL, respectively (Figure 8A). Western blot analysis demonstrated Smad2/3 phosphorylation in CMV‐CTLs by TGF‐β1 and culture supernatant from HSC‐3 cells (Figure 7B). The enhancement of cytotoxic activity and cytokine production by the addition of SB525334 alone without TGF‐β1 (Figure 4B, C) may be due to the effect of TGF‐β1 produced by HSC‐3pp65. These results indicate that OSCC cells release TGF‐β1 and inhibit the cytotoxic activity of CTLs by suppressing cytokine production and proliferation in the effector phase.
FIGURE 8.
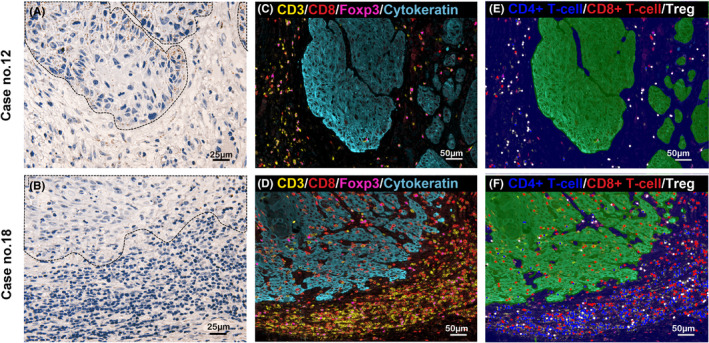
Representative images of RNA‐ISH of TGF‐β1 and MF‐IHC of immune cells. Representative cases of high (case 12; A) and low (case 18; B) expression of TGFB1 mRNA in the tumor nests are shown, respectively (×400 magnification). The area surrounded by dotted lines indicates the tumor nest. Tiny brown dots indicate TGFB1 mRNA expression. The distribution of T‐cells by MF‐IHC of the same area as (A) and (B) are shown in (C) and (E), and (D) and (F), respectively (×200 magnification). The MF‐IHC is indicated by red circles for CD8+ T‐cells, blue circles for CD4+ T‐cells, and white circles for Tregs (E, F)
FIGURE 7.
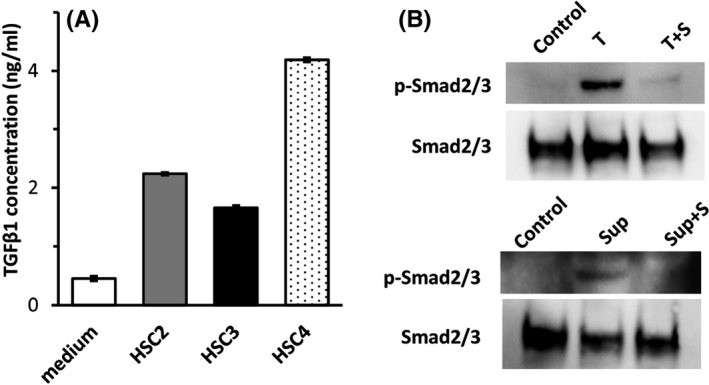
Production of TGF‐β1 from OSCC cells and Smad2/3 phosphorylation in CMV‐CTLs by TGF‐β1 or culture supernatant of HSC‐3. TGF‐β1 concentration in supernatants from OSCC cell line is shown in panel (A). Smad2/3 phosphorylation in CMV‐CTLs by TGF‐β1 or culture supernatant of HSC‐3 in panel (B). S, SB525334; sup, supernatant; T, TGF‐β1
3.4. Relationship between expression and distribution of TGFB1 mRNA with T‐cell infiltration in OSCC tissues
Representative images of TGFB1 mRNA expression by RNA‐ISH and T‐cell infiltration by MF‐IHC are shown in Figure 8. TGFB1 mRNA expression in cases 12 and 18 are shown as examples of high expression and low expression, respectively (Figure 8A, B). TGFB1 mRNA tended to be mainly expressed in the invasive front of cancer cells, where its expression was higher than that in the stroma (Figure 9A). In this study, we could not identify the type of cells that expressed TGFB1 in the stroma. TGFB1 density was higher in cancer nests than in adjacent normal epithelial cells (Figure 9B).
FIGURE 9.
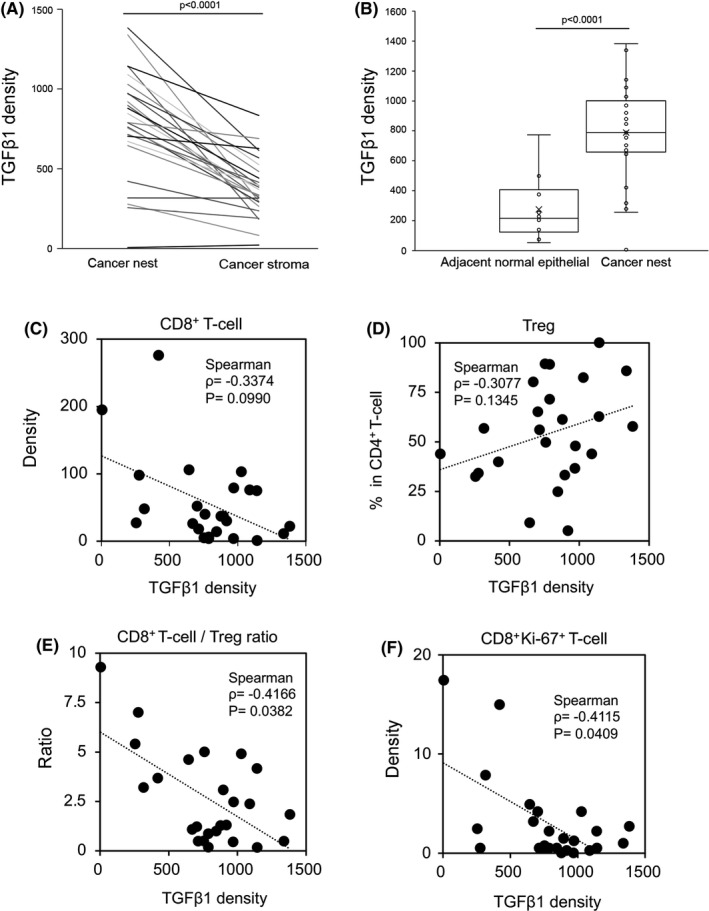
Comparison of TGFB1 mRNA distribution within OSCC tissues and correlation of TGFB1 mRNA expression with T‐cell infiltration. Comparison of TGF‐β1 density in the cancer nests and cancer stroma in the same tissue (A). Comparison of TGF‐β1 density in adjacent normal epithelium and cancer nests (B). Correlation between TGF‐β1 density and CD8+ T‐cell density (C), TGF‐β1 density and Ki‐67+CD8+ density (D), TGF‐β1 density and the percentage of Tregs in CD4+ T‐cells (E), and TGF‐β1 density and CD8+ T‐cell/Treg ratio (F)
Next, T‐cell infiltration was compared with TGFB1 mRNA expression. A representative image of MF‐IHC staining of the T‐cell subpopulation is shown in Figure S1. TGFB1 mRNA expression was negatively and positively correlated with infiltration of CD8+ T‐cells and Tregs, respectively, however the correlations were not statistically significant (Figure 9C, D). Interestingly, we found a statistically significant negative correlation between TGFB1 expression and the CD8+ T‐cell/Treg ratio (P = .038; Figure 9E). Cases 12 and 18 are representative of these trends. In case 12, which expressed high TGFB1 mRNA, very few CD8+ T‐cells infiltrated the cancer nest (Figure 8E). In case 18, which expressed low TGFB1 mRNA, a high number of CD8+ T‐cells infiltrated both the cancer nest and stroma, and the CD8+ T‐cell/Treg ratio was very high, especially in the cancer nest (Figure 8F). Furthermore, we analyzed Ki‐67 expression in CD8+ T‐cells. CD8+Ki‐67+ T‐cells were distributed along the periphery of the cancer nest, and a statistically significant negative correlation was observed between CD8+Ki‐67+ T‐cell number and TGF‐β expression (P = .041; Figure 9F). These observations are representatively shown in cases 12 and 18 (Figure S4). A higher expression of Ki‐67 in CD8+ T‐cells was observed in case 18 compared with in case 12.
There was no significant correlation between TGFB1 density and clinical characteristics (Table S1), although interesting immunological findings were found.
4. DISCUSSION
Dori et al reported that, in a mouse model, TGF‐β directly inhibits the effector function of CTLs by downregulating granzyme, perforin, and IFN‐γ.26 Similarly, chimeric antigen receptor T‐cell therapy studies have shown that TGF‐β1 inhibits the production of perforin and anti‐tumor cytokines such as IFN‐γ, IL‐2, and TNF‐α, and that TGF signaling inhibition improves chimeric antigen receptor T‐cell proliferation and survival in vitro.27 Therefore, it is well known that TGF‐β suppresses T‐cell function. However, to the best of our knowledge, this report is the first to show the suppression of antigen‐specific response of CTLs by TGF‐β in humans. As the tumor mutation burden in OSCCs is relatively high28 and human papillomavirus is related to the pathogenesis of OSCCs,29 targeting tumor‐specific antigens (TSAs) such as neoantigens and virus antigens is considered reasonable for immunotherapy of OSCCs. Although our in vitro system is artificial in that it uses CMVpp65 antigen instead of TSAs, T‐cell receptor (TCR) affinity of CMV‐CTLs may be considered to be as high as that of CTLs recognizing TSAs. Therefore, our data indicated that TGF‐β1 suppresses the function of TSA‐specific human CTL. This study showed that TGF‐β1 inhibited the induction of antigen‐specific CTLs from PBMCs by epitope peptide stimulation in vitro. Inhibition of differentiation is considered one of the mechanisms for inhibiting CTL induction. We also found that TGF‐β1 or TGF‐β3 inhibited the responses of the induced CTLs to secondary stimulation with the target cells (HSC‐3pp65) by reducing proliferation, DNA replication, cytokine production, and cytotoxicity. Therefore, we suggest that TGF‐β attenuates anti‐tumor immunity by extensively suppressing CTL functions both in the priming and effector phases.
It is important to clarify the distribution of TGF‐β in cancer tissues to understand the immunosuppressive TME. Although immunohistochemical investigations of TGF‐β in numerous cancer types have been reported,13, 30, 31, 32 the results show discrepancies in the staining patterns. Moreover, after researching 6 previous studies, we found no consensus on the distribution of TGF‐β, at least in head and neck squamous cell cancer and OSCC tissues.10, 11, 33, 34, 35, 36 The lack of standardized anti‐TGF‐β antibodies for IHC and the difficulties found for immunohistochemical detection of cytokines are considered to be the reasons for the lack of consensus on the distribution of TGF‐β. Although we tested several commercially available anti‐TGF‐β1 antibodies, they could not detect recombinant TGF‐β1 by western blotting; IHC staining of the tissues by these antibodies could not be erased by absorption with recombinant TGF‐β1 (data not shown). Therefore, we evaluated TGFB1 mRNA expression in OSCC tissues using RNAscope, a novel RNA‐ISH assay without immunohistochemical detection of TGF‐β1. We found that TGFB1 mRNA was mainly located in the invasive front of OSCC cells; this may suppress the infiltration of CTL into the cancer nests. In contrast, TGFB1 expression in normal epithelium was very low compared with that in cancer cells, consistent with previous reports.10, 11 OSCC cells were thought to acquire TGF‐β1 production ability by oncogenesis and that it is crucial for immune escape. Although it was difficult to specify the cell types that express TGF‐β1 in the stroma area using RNAscope, these cell types were thought to be Tregs, M2 macrophages, myeloid‐derived suppressor cells, and cancer‐associated fibroblasts, as previously reported. Our results showed that cancer cells expressed more TGFB1 mRNA than the cells in the stroma, which suggests that cancer cells, especially in the invasive front, are the major source of TGF‐β1 in OSCC and contribute to creating an immunosuppressive TME. However, Tregs may be considered as one of the important sources of TGF‐β in the tissues because we observed a strong infiltration of Tregs in OSCC; TGF‐β was shown to suppress CMV‐CTL proliferation by Tregs. Further investigations are required to elucidate the role of local expression of TGF‐β and its functions in OSCC tissues.
Although a statistically significant correlation was not observed between CD8+ T‐cell infiltration into nest and TGFB1 mRNA expression, the expression of Ki‐67 in CD8+ T‐cells was inversely correlated with TGFB1 mRNA expression, which indicated that TGF‐β1 inhibited CTL proliferation in the tissues. Moreover, TGFB1 mRNA expression in the nest was inversely correlated with CD8+ T‐cell/Treg ratio. TGF‐β has been reported to inhibit intratumoral invasion of CTLs.17, 18 Recent reports have shown that TGF‐β eliminates CTLs by suppressing CXCR3 expression in CD8+ T‐cells.37 Additionally, a low CD8+ T‐cell/Treg ratio has been reported as a poor prognostic marker in cancers38, 39 These previous reports support our results and collectively suggest that TGF‐β plays a vital role in creating an immunosuppressive microenvironment in OSCC tissues.
Therefore, inhibition of TGF‐β function is expected to be a novel immunotherapy for OSCC. Interestingly, a study using a mouse model of squamous cell carcinoma showed that the combination of TGF‐β inhibition and anti‐PD‐1 antibody increased the CD8+ T‐cell/Treg ratio and had a greater effect than utilizing anti‐PD‐1 antibody alone.20 The CD8+ T‐cell/Treg ratio within the TME in pancreatic cancer has been shown to be increased by the combined use of anti‐TGF‐β antibody and vaccine therapy compared with vaccine therapy alone.40 Therefore, TGF‐β inhibition may improve the tumor immune microenvironment by increasing the CD8+ T‐cell/Treg ratio, which is highly useful in combinational immunotherapy.
Our results from in vitro experiments and in situ immunological analyses collectively suggest that regulation of TGF‐β is important for the development of OSCC immunotherapy. TGF‐β is known to have both proliferative and inhibitory effects on tumors.41, 42 Therefore, none of the several types of TGF‐β inhibitors, including monoclonal antibodies, antisense oligonucleotides, TGF‐β‐related vaccines, and receptor kinase inhibitors, have been approved for cancer treatment despite being tested in clinical trials over the last 15 y.43 The current development direction utilizing TGF‐β inhibition involves selecting the appropriate therapeutic combination for TGF‐β inhibition and biomarkers for identifying the patient subpopulation who might benefit from TGF‐β inhibition.44, 45 The combined utilization of anti‐PD‐L1 antibody and TGF‐β inhibition is a potent option, because increasing TGF‐β signaling in the TME correlates with poor overall survival and increased resistance to the inhibition of PD‐1/PD‐L1. The bifunctional drug Bintrafusp alfa (M7824), composed of an IgG1 monoclonal antibody targeting PD‐L1 fused with the extracellular domains of 2 TGF‐β receptor II molecules, can block both signaling pathways.23 Therefore, Bintrafusp alfa might be suitable for treating OSCC because of the expression pattern of TGF‐β1 and PD‐L1 in OSCC cancer cells.24 Our findings strongly suggest that the expression of TGF‐β1 and PD‐L1 in cancer cells and the CD8+ T‐cell/Treg ratio could be useful biomarkers for such therapies.
This study had some limitations. First, RNA‐ISH was used to evaluate TGF‐β1 expression in OSCC cells. Because TGF‐β1 protein binds to the extracellular matrix after production, RNA‐ISH may not accurately reflect TGF‐β1 protein localization. Therefore, immunohistochemical detection using anti‐TGF‐β1 antibodies is necessary. We plan to evaluate TGF‐β1 protein distribution using immunohistochemistry in the future. Second, this was a retrospective study with a small sample size in which the data were analyzed using RNA‐ISH and MF‐IHC. Future studies with larger sample size are warranted.
In conclusion, we found that TGF‐β suppressed antigen‐specific CTL functions in both the priming and effector phases. Furthermore, TGF‐β generates suppressive TMEs by decreasing the CD8+ T‐cell/Treg ratio. These results indicate that TGF‐β inhibition restores immunosuppressive TME via a mechanism different from that of immune‐checkpoint inhibitors, suggesting that the combination of these inhibitors may lead to a new therapeutic strategy for OSCCs.
DISCLOSURE
RU received research funding from Chugai Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., and Kyowa Kirin Co., Ltd. TT received remuneration from Chugai Pharmaceutical Co., Ltd. and AstraZeneca plc as advisors. The other authors declare no conflicts of interest.
Supporting information
Fig S1‐4
Table S1
ACKNOWLEDGMENTS
We thank Ms. Hiromi Tsuchida and Ms. Akiko Shimada for their excellent technical assistance and Ms. Kyoko Okumura for her excellent secretarial assistance. We thank Mr. Makoto Naruse and Ms. Natsumi Kodama (Aichi Medical University, Institute of Comprehensive Medical Research, Division of Advanced Research Promotion) for the maintenance of the BD LSRFortessa flow cytometer and Amersham Imager 600. We would like to thank Editage (www.editage.com) for English language editing.
Kondo Y, Suzuki S, Takahara T, et al. Improving function of cytotoxic T‐lymphocytes by transforming growth factor‐β inhibitor in oral squamous cell carcinoma. Cancer Sci. 2021;112:4037–4049. 10.1111/cas.15081
REFERENCES
- 1.Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burtness B, Harrington KJ, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE‐048): a randomised, open‐label, phase 3 study. The Lancet. 2019;394:1915‐1928. [DOI] [PubMed] [Google Scholar]
- 4.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer 2017;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meric‐Bernstam F, Larkin J, Tabernero J, Bonini C. Enhancing anti‐tumour efficacy with immunotherapy combinations. The Lancet. 2021;397:1010‐1022. [DOI] [PubMed] [Google Scholar]
- 6.Massagué J. TGFβ in Cancer. Cell. 2008;134:215‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neuzillet C, Tijeras‐Raballand A, Cohen R, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22‐31. [DOI] [PubMed] [Google Scholar]
- 8.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M. Intracellular and extracellular TGF‐β signaling in cancer: some recent topics. Front Med. 2018;12:387‐411. [DOI] [PubMed] [Google Scholar]
- 10.Chen MF, Wang WH, Lin PY, Lee KD, Chen WC. Significance of the TGF‐β1/IL‐6 axis in oral cancer. Clin Sci. 2012;122:459‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu SL, Reh D, Li AG, et al. Overexpression of transforming growth factor beta 1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405‐4410. [DOI] [PubMed] [Google Scholar]
- 12.Calon A, Lonardo E, Berenguer‐Llergo A, et al. Stromal gene expression defines poor‐prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320‐329. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Huang J, Ren X, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr‐1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahmani A, Delisle JS. TGF‐β in T cell biology: implications for cancer immunotherapy. Cancers. 2018;10:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngiow SF, Young A. Re‐education of the tumor microenvironment with targeted therapies and immunotherapies. Front Immunol. 2020;11:1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Y, Souza‐Fonseca‐Guimaraes F, Bald T, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18:1004‐1015. [DOI] [PubMed] [Google Scholar]
- 17.Tauriello DVF, Palomo‐Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538‐543. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell. 2016;165:35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dodagatta‐Marri E, Meyer DS, Reeves MQ, et al. α‐PD‐1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α‐TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer 2019;7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lan Y, Zhang D, Xu C, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD‐L1 and TGF‐β. Sci Transl Med. 2018;10:eaan5488. [DOI] [PubMed] [Google Scholar]
- 22.Ravi R, Noonan KA, Pham V, et al. Bifunctional immune checkpoint‐targeted antibody‐ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat Commun. 2018;9:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strauss J, Heery CR, Schlom J, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD‐L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. 2018;24:1287‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho BC, Daste A, Ravaud A, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF‐β and PD‐L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer. 2020;8:e000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishio‐Nagai M, Suzuki S, Yoshikawa K, Ueda R, Kazaoka Y. Adoptive immunotherapy combined with FP treatment for head and neck cancer: an in vitro study. Int J Oncol. 2017;51:1471‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas DA, Massagué J. TGF‐β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369‐380. [DOI] [PubMed] [Google Scholar]
- 27.Stüber T, Monjezi R, Wallstabe L, et al. Inhibition of TGF‐β‐receptor signaling augments the antitumor function of ROR1‐specific CAR T‐cells against triple‐negative breast cancer. J Immunother Cancer. 2020;8:e000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;3(92):709‐720. [DOI] [PubMed] [Google Scholar]
- 30.Aoyagi Y, Oda T, Kinoshita T, et al. Overexpression of TGF‐β by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br J Cancer. 2004;91:1316‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeh HW, Hsu EC, Lee SS, et al. PSPC1 mediates TGF‐β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol. 2018;20:479‐491. [DOI] [PubMed] [Google Scholar]
- 32.Marwitz S, Depner S, Dvornikov D, et al. Downregulation of the TGFβ Pseudoreceptor BAMBI in Non‐Small Cell Lung Cancer Enhances TGFβ Signaling and Invasion. Cancer Res. 2016;76:3785‐3801. [DOI] [PubMed] [Google Scholar]
- 33.Rosenthal E, McCrory A, Talbert M, Young G, Murphy‐Ullrich J, Gladson C. Elevated expression of TGF‐β1 in head and neck cancer‐associated fibroblasts. Mol Carcinog. 2004;40:116‐121. [DOI] [PubMed] [Google Scholar]
- 34.Kim N, Ryu H, Kim S, et al. CXCR7 promotes migration and invasion in head and neck squamous cell carcinoma by upregulating TGF‐β1/Smad2/3 signaling. Sci Rep. 2019;9:18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dasgupta S, Bhattacharya‐Chatterjee M, O’Malley BW, Chatterjee SK. Tumor metastasis in an orthotopic murine model of head and neck cancer: Possible role of TGF‐beta 1 secreted by the tumor cells. J Cell Biochem. 2006;97:1036‐1051. [DOI] [PubMed] [Google Scholar]
- 36.Guo M, Mu Y, Yu D, et al. Comparison of the expression of TGF‐β1, E‐cadherin, N‐cadherin, TP53, RB1CC1 and HIF‐1α in oral squamous cell carcinoma and lymph node metastases of humans and mice. Oncol Lett. 2017;15:1639‐1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunderson AJ, Yamazaki T, McCarty K, et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat Commun. 2020;11:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci. 2005;102:18538‐18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jordanova ES, Gorter A, Ayachi O, et al. Human leukocyte antigen class I, MHC class I chain‐related molecule A, and CD8+/regulatory T‐cell ratio: which variable determines survival of cervical cancer patients? Clinical Cancer Res. 2008;14:2028‐2035. [DOI] [PubMed] [Google Scholar]
- 40.Soares KC, Rucki AA, Kim V, et al. TGF‐β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget. 2015;6:43005‐43015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415‐424. [DOI] [PubMed] [Google Scholar]
- 42.Roberts AB, Wakefield LM. The two faces of transforming growth factor in carcinogenesis. Proc Natl Acad Sci. 2003;100:8621‐8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol. 2020;31:1336‐1349. [DOI] [PubMed] [Google Scholar]
- 44.Liu S, Ren J, ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Sig Transduct Target Ther. 2021;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Y, Agarwal R, Dituri F, et al. NGS‐based transcriptome profiling reveals biomarkers for companion diagnostics of the TGF‐β receptor blocker galunisertib in HCC. Cell Death Dis. 2017;8:e2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐4
Table S1