

Abstract
Reactive oxygen species (ROS), a class of highly bioactive molecules, have been widely studied in various types of cancers. ROS are considered to be normal byproducts of numerous cellular processes. Typically, cancer cells exhibit higher basal levels of ROS compared with normal cells as a result of an imbalance between oxidants and antioxidants. ROS have a dual role in cell metabolism: At low to moderate levels, ROS act as signal transducers to activate cell proliferation, migration, invasion, and angiogenesis. In contrast, high levels of ROS cause damage to proteins, nucleic acids, lipids, membranes, and organelles, leading to cell death. Extensive studies have revealed that anticancer therapies that manipulate ROS levels, including immunotherapies, show promising in vitro as well as in vivo results. In this review, we summarize molecular mechanisms and oncogenic functions that modulate ROS levels and are useful for the development of cancer therapeutic strategies. This review also provides insights into the future development of effective agents that regulate the redox system for cancer treatment.
Keywords: cell death, neoplasms, oxidative stress, reactive oxygen species, therapeutics
In this review, we summarize molecular mechanisms and oncogenic functions that modulate ROS levels and that are useful for the development of cancer therapeutic strategies. This review also provides insights into the future development of effective agents that regulate the redox system for cancer treatment.

1. INTRODUCTION
Worldwide, cancer ranks as a leading cause of death and a crucial barrier to increasing life expectancy. An estimated 19.3 million new cases and almost 10 million deaths have occurred from cancer in 2020.1 Despite numerous research efforts in understanding cancer and in developing anticancer strategies, cancer remains a major lethal disease in people.
Reactive oxygen species (ROS) have been extensively studied in various human diseases, including cancers. ROS are normal byproducts of a wide variety of cellular processes, including oxygen metabolism.2, 3 The term “ROS” is a collective term referring to unstable, reactive, partially reduced oxygen derivatives that include hydrogen peroxide (H2O2), superoxide anion (O2 −), hypochlorous acid (HOCl), singlet oxygen (1O2), and hydroxyl radical (•OH).4 These act as second messengers in cell signaling and are essential for various biological processes in normal and cancer cells.5 Accumulating evidence suggests that ROS show activity that is a “double‐edged sword” in cancer cells. At low to moderate levels, ROS act as signaling transducers to activate cancer cell proliferation, migration, invasion, angiogenesis, and drug resistance.2, 6, 7 In other words, adequate levels of ROS are important for cancer cell homeostasis involved in the development of cellular processes such as proliferation, differentiation, migration, and cell death. In contrast, high levels of ROS are harmful to cancer cells and ultimately lead to cell death.8
With regard to the bidirectional nature of ROS, strategies to downregulate or upregulate ROS in cancer cells appear to be promising treatments. Antioxidants are usually considered to be beneficial for both cancer prevention and treatment as they can quench ROS levels, leading to a reduction in oxidative stress. Indeed, we have shown that intracellular ROS suppression using deferasirox (DFX), an oral iron chelator, induced apoptosis in multiple myeloma (MM) cells. Mechanistically, DFX exerts anti‐MM activity via the inhibition of proline‐rich tyrosine kinase 2 phosphorylation accompanied by the reduced production of ROS.9 However, in reality, previous studies have demonstrated that antioxidants, including N‐acetyl l‐cysteine (NAC) and vitamin E, markedly increased tumor progression and cancer metastasis.10, 11
In contrast, the production of ROS is reported to be elevated in cancer cells as a result of increased metabolic rate, gene mutation, and relative hypoxia.12 Previous studies demonstrated that cancer cells adapt to a high level of ROS by activating antioxidant pathways resulting in increased ROS clearance.13 The higher endogenous ROS levels in cancer cells endow them with increased sensitivity to ROS‐inducing therapy. In fact, many chemotherapeutics are known to increase the production of ROS in cancer cells.14, 15, 16 We have previously reported that the suppression of cell growth accompanied by apoptosis was evoked through increased intracellular ROS levels in MM and colorectal cancer cells.17, 18 Therefore, the induction of ROS would be an appropriate strategy to combat cancer.
In this review, we initially focus on the molecular mechanisms and oncogenic functions of ROS, and subsequently discuss several potential therapeutic strategies that modulate ROS levels in cancer cells.
2. MOLECULAR MECHANISM OF ROS PRODUCTION IN CANCER CELLS
2.1. Generation of ROS
ROS can be generated by multiple endogenous and exogenous factors.19 Mitochondria are known to produce significant amounts of endogenous ROS that contribute to intracellular oxidative stress.20 At an ultrastructural level, mitochondria have a four‐layer structure consisting of an outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane, and matrix. The generation of ROS mainly occurs in the electron transport chain on the inner mitochondrial membrane during oxidative phosphorylation, a process that creates adenosine triphosphate (ATP) from oxygen and simple sugars.21 Five protein complexes contribute to the process: NADH:ubiquinone reductase (complex I), succinate:ubiquinone reductase (complex II), ubiquinol:cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and F1F0‐ATP synthase (complex V).22 The 2 major sites for ROS generation are complexes I and III where large changes occur in the potential energy of electrons related to the reduction of oxygen.23 The leakage of electrons at complexes I and III leads to the generation of superoxide. Once generated, superoxide is quickly dismutated to hydrogen peroxide by superoxide dismutase 1 (SOD1) in the intermembrane space or by SOD2 in the matrix. Both superoxide and hydrogen peroxide in this process are recognized as ROS that are generated in mitochondria.21
Transition metals such as iron can also generate ROS non‐enzymatically via a Fenton reaction.24 The Fenton reaction involves Fe2+ reacting with hydrogen peroxide to yield a hydroxy radical, which can cause damage to DNA and other biomolecules.25
In addition, multiple external factors induce exogenous ROS, including air pollutants, tobacco smoke, radiation, foods, and drugs.26 For instance, tobacco smoke contains more than 4000 chemicals, including superoxide and hydroxyl radicals.27 Ionizing radiation can also generate hydroxyl radicals either directly by oxidation of water or indirectly by the formation of partial secondary ROS.28Collectively, various intracellular and extracellular cues stimulate ROS formation in cancer cells (Figure 1).
FIGURE 1.
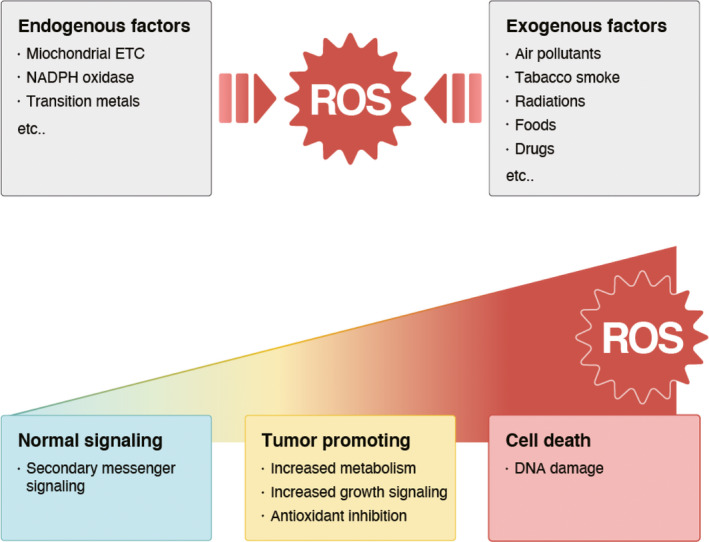
Generation of reactive oxygen species (ROS) and their effects. Reactive oxygen species can be generated by multiple endogenous and exogenous factors, which, in turn, lead to various biological consequences. Low levels of ROS act as intracellular second messengers. Moderate levels of ROS are beneficial to cancer cells because they can increase cancer metabolism and growth signaling, and inhibit antioxidants, which contribute to oncogenesis. Conversely, high levels of ROS can lead to cell death induced by DNA damage
2.2. Redox homeostasis
Excessive concentrations of ROS result in cell‐cycle arrest and apoptosis. To prevent excessive intracellular ROS, cancer cells respond to oxidative stress by inducing the transcription of antioxidant enzymes.12 The transcription factor, nuclear erythroid 2‐related factor (NRF2), is a pivotal regulator of antioxidant responses in cancer cells.29 NRF2 is activated and overexpressed in cancer to promote cancer cell survival; the cellular antioxidant system is largely regulated by NRF2 and its associated genes.30 Under normal conditions, the expression and activity of NRF2 is tightly degraded by kelch‐like ECH‐associated protein 1 (KEAP1). However, under oxidative conditions, NRF2 dissociates from KEAP1 and translocates to the nucleus to bind and activate the antioxidant response element (ARE) in various target genes.31 ARE regulates downstream antioxidant enzymes, including NAD(P)H quinone dehydrogenase 1, heme oxygenase 1, thioredoxin reductase 1, superoxide dismutase, glutathione peroxidase, and catalase.32 Therefore, cancer cells defend themselves from excessive ROS. Specifically, we have previously reported that the six‐transmembrane epithelial antigen of the prostate1 (STEAP1), which was identified as a cell surface protein and works as an intracellular transporter, is overexpressed in colorectal cancer cells compared with normal counterparts. Knockdown analysis of STEAP1 using small interfering (si)RNA revealed that it regulates ROS levels by manipulating NRF2 and downstream genes in colorectal cancer cells (Figure 2).18 Therefore, as in previous reports, NRF2 plays a vital role in defending cancer cells from excessive ROS.
FIGURE 2.
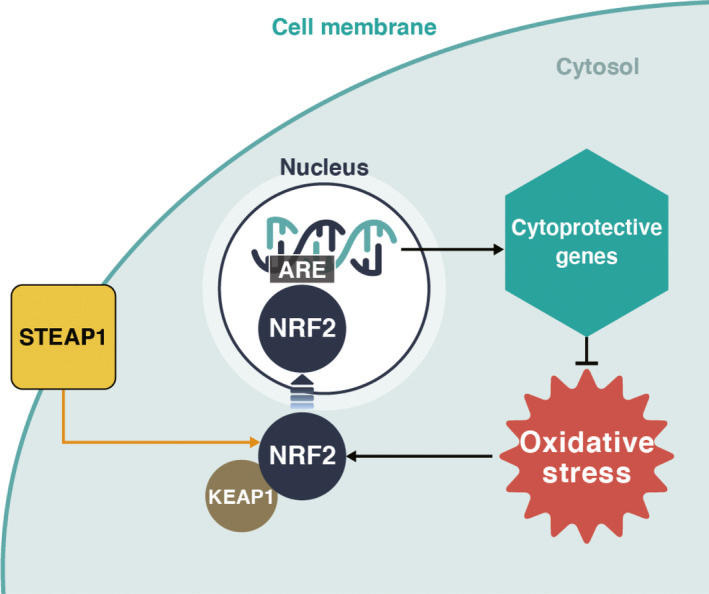
Schematic representation of STEAP1‐NRF2 pathway. Under nonoxidative conditions, nuclear erythroid 2‐related factor (NRF2) is located in the cytoplasm, adjacent to kelch‐like ECH‐associated protein 1 (KEAP1). Oxidative stress causes the dissociation of NRF2 from KEAP1. NRF2 enters the nucleus and activates several cytoprotective genes for protection against oxidative stress. Our previous report suggested that six‐transmembrane epithelial antigen of the prostate 1 (STEAP1) plays an important role in upregulating this pathway. ARE, antioxidant response element
3. ONCOGENIC FUNCTIONS OF ROS
ROS can directly induce oxidative DNA damage. Such damage consists of DNA double‐stranded breaks and the formation of mutagenic 8‐oxo‐7‐hydro‐2′‐deoxyguanosine (8‐oxodG). In particular, 8‐oxodG is a major cause of spontaneous mutagenesis, because it induces guanine to thymine transversion through its capacity to pair with both cytosine and adenine.33, 34, 35Therefore, the accumulation of 8‐oxodG in cellular genomes leads to carcinogenesis.
As mentioned earlier in Section 1, 2, iron is one of the major sources of ROS production, and is increasingly recognized as an important initiator and mediator of cell death in a variety of organisms and pathological situations through the regulation of ROS.36 Of note, iron‐induced oxidative stress has been shown to be a risk factor for the development of numerous cancers.37 Our group has investigated the clinical impact of excess iron‐induced ROS in cancers. We therefore focus on the association between iron‐induced ROS and carcinogenesis in this section.
We have previously revealed that a close link exists between oxidative DNA damage induced by hepatic iron overload and hepatocarcinogenesis in patients with chronic hepatitis C (CHC). With a reduction in therapeutic iron, elevated hepatic ROS, evaluated by immunostaining 8‐oxodG, significantly decreased to almost normal levels with a concomitant improvement of hepatitis, followed by a lowered risk of the development of hepatocellular carcinoma (HCC) in patients with CHC.38, 39 Additionally, we found that feeding iron with a high‐fat high‐cholesterol diet increased the incidence of HCC accompanied by the upregulation of ROS in mutY DNA glycosylase (MUTYH), an enzyme that repairs oxidative DNA damage, in a deficiency non‐alcoholic steatohepatitis (NASH) mouse model. Markedly, the development of HCC in this NASH model could be suppressed by the administration of NAC.40 Collectively, ROS induced by excess hepatic iron is associated with hepatocarcinogenesis in both CHC and NASH (Figure 3).
FIGURE 3.

Iron overload leads to ROS and 8‐oxodG. Reactive oxygen species (ROS) can be induced by iron and contribute to the formation of mutagenic 8‐oxo‐7‐hydro‐2′‐deoxyguanosine (8‐oxodG), which leads to hepatocarcinogenesis (HCC) in patients with chronic hepatitis C (CHC)/non‐alcoholic steatohepatitis (NASH), and leukemic transformation in myelodysplastic syndrome (MDS). Therapeutic iron reduction contributes to inhibiting both hepatocarcinogenesis in CHC/NASH and leukemic transformation in MDS
Apart from hepatocarcinogenesis, we have also reported that iron overload‐induced ROS and 8‐oxodG levels in peripheral blood mononuclear cells (PBMC) of patients with myelodysplastic syndrome (MDS) were higher than those in healthy volunteers. MDS is a disease that is characterized by dysplasia and a high risk of leukemic transformation.41 We conducted iron chelation therapy (ICT) using DFX in patients with MDS and showed that 8‐oxodG levels in PBMC dramatically decreased 3 mo after DFX administration. Recently, a randomized study and meta‐analysis clarified that patients who were assigned to an ICT group showed longer survival, with low risk of progression to leukemia, compared with a non‐ICT group.42, 43 Therefore, we concluded that excess iron contributed to oxidative DNA damage in patients with MDS and that ICT improved survival by inhibiting leukemic change through mitigating oxidative DNA damage (Figure 3).
4. CELL DEATH INDUCED BY ROS
4.1. ROS and apoptosis
Excess cellular levels of ROS cause damage to proteins, nucleic acids, lipids, membranes, and organelles, which can lead to activation of cell death processes such as apoptosis.44 Mitochondria play an important role in initiating apoptosis and are considered to be both a source and target of ROS. High levels of mitochondrial ROS can initiate intrinsic apoptosis leading to the release of cytochrome c into the cytosol from the mitochondrial intermembrane space.45 In the cytosol, cytochrome c engages the apoptotic protease activating factor‐1 (APAF1), followed by the formation of an apoptosome, which activates caspase‐9. Caspase‐9, a key player in the intrinsic pathway, then activates effector caspases, such as caspase‐3, ‐6, and ‐7, resulting in cleavage of cellular proteins and cell death by apoptosis.46 Links between ROS and the extrinsic pathway of apoptosis also exist. ROS can activate transmembrane death receptors, including Fas, tumor necrosis factor‐related apoptosis inducing ligand (TRAIL‐R1/2), and tumor necrosis factor receptor 1. Activation of transmembrane death receptors recruits the adaptor proteins, Fas‐associated protein with death domain (FADD), and procaspase‐8 and ‐10, to the cytoplasmic surface to form death‐inducing signaling complexes (DISCs), subsequently triggering caspase‐8 and ‐10 activation that can directly activate effector caspases and trigger apoptosis. Caspase‐8 and ‐10 also cleave Bid to produce truncated Bid (tBid), which translocates to mitochondria, blocks the anti‐apoptotic activity of Bcl‐2 and Bcl‐XL, and activates Bax and Bak. This leads to activation of the mitochondrial pathway of apoptosis.44 In summary, ROS promote both intrinsic and extrinsic pathways of apoptosis in cancer cells (Figure 4A).
FIGURE 4.

Schematic representation of cell death induced by ROS. A, Reactive oxygen species (ROS) can lead to activation of apoptosis. High levels of mitochondrial ROS can release cytochrome c into the cytosol from the mitochondrial intermembrane space. In the cytosol, cytochrome c engages apoptotic protease activating factor‐1 (APAF1) and activates caspase‐9. Furthermore, as the extrinsic pathway, ROS can activate transmembrane death receptors, including Fas, tumor necrosis factor‐related apoptosis inducing ligand (TRAIL‐R1/2), Fas‐associated protein with death domain (FADD), and procaspase‐8 and ‐10 at the cytoplasmic surface to form death‐inducing signaling complexes (DISCs), subsequently triggering caspase‐8 and ‐10 activation, and apoptosis. Caspase‐8 and ‐10 also cleave Bid to produce truncated (t)Bid, which translocates to mitochondria, blocks Bcl‐2 and Bcl‐XL, and activates Bax and Bak. B, ROS can regulate autophagy induction in cells. Increased ROS leads to oxidation and inactivation of autophagy‐related (ATG)4. Inactivation of ATG4 results in promoting lipidation of ATG8, an essential step in autophagy. ROS also directly activate adenosine monophosphate (AMP)‐activated protein kinase (AMPK), upstream of mammalian target of rapamycin (mTOR), to suppress its phosphorylation, resulting in the induction of autophagy
4.2. ROS and autophagy
Autophagy is a self‐digestion process aimed at recycling cellular components and damaged organelles in response to diverse conditions of stress.47 In cancer cells, autophagy plays dual roles in tumor promotion and suppression.48 The tight interaction between ROS and autophagy is reflected in 2 ways: the induction of autophagy by oxidative stress and the reduction of ROS by autophagy.49 Induction of autophagy following nutrient starvation requires the production of hydrogen peroxide that oxidizes autophagy‐related (ATG)4. Of ATG proteins, ATG4 is the sole protease that regulates autophagy by processing and deconjugating ATG8.50 The oxidization modification mainly inactivates the delipidation activity of ATG4 leading to increased formation of light chain 3‐associated autophagosomes.51, 52 In addition to the above, which is regarded as a direct mechanism, an indirect induction of autophagy by ROS can also occur. Adenosine monophosphate (AMP)‐activated protein kinase (AMPK), which can suppress the activity of the mammalian target of rapamycin (mTOR), is activated by ROS and leads to the induction of autophagy.53 In summary, a close relationship exists between ROS and autophagy in cancer cells (Figure 4B).
5. ROS IN CANCER TREATMENT
5.1. ROS and cancer chemotherapy
Chemotherapy has been widely used to treat cancer patients in a clinical setting. Most chemotherapeutic agents produce ROS, and many can alter redox homeostasis in cancer cells.4 Anthracyclines (doxorubicin, epirubicin, and daunorubicin), alkylating agents, platinum coordination complexes (cisplatin, carboplatin, and oxaliplatin), and camptothecins (topotecan and irinotecan) are the major drugs that increase ROS in cancer cells.54 The generation of mitochondrial ROS and inhibition of the cellular antioxidant system are 2 major reasons for the elevation of ROS in response to chemotherapeutic agents.4 For example, cisplatin, which is one of the most effective and widely used chemotherapeutic agents for various cancers, induces mitochondria‐dependent ROS that contributes to cell death via the formation of nuclear DNA damage. Cisplatin‐related ROS generation occurs as a result of a direct effect on mitochondrial DNA, resulting in the impairment of the synthesis of electron transport chain proteins.55
ROS also play an important role in multidrug resistance. Such resistance is one of the major reasons for the failure of chemotherapy in cancer treatment.56 P‐glycoprotein (P‐gp) and other related transporter‐based efflux pumps in the plasma membrane are strongly associated with multidrug resistance in cancer cells.57 P‐gp which is encoded by the MDR1/ABCB1 gene, is a member of the large ATP‐binding cassette family of proteins and acts as a barrier to the uptake of xenobiotics, including chemotherapeutic agents.58 As mentioned previously, NRF2 is activated and overexpressed in cancer cells as a protective mechanism against excessive ROS MDR1/ABCB1 is an NRF2 target gene that contributes to the increased expression of P‐gp and multidrug resistance in cancer cells.59, 60 In short, increased ROS levels in response to chemotherapy are crucial for damaging cancer cells and also play a vital role in multidrug resistance.
5.2. ROS in cancer immunity
In this section, we focus on impacts of CD4+Foxp3+ regulatory T (Treg) cells and dendritic cells (DCs) in anti‐tumor immune responses as both cell types play essential roles in the tumor microenvironment (TME).
Growing evidence has shown that Treg cells are recruited into the TME and behave as powerful immunosuppressors.61, 62 It is well documented that the TME, comprised of many different cell types, such as cancer cells, cancer‐associated fibroblasts and numerous immune cells, creates a field with a high concentration of ROS.63 In the TME, the high ROS level is one of the major mechanisms responsible for refractoriness to immunotherapies, including immune checkpoint blockades.64, 65 Tumor‐infiltrating Treg cells undergo apoptosis in the high ROS concentration existing within the TME. Of note, tumor‐infiltrating apoptotic Treg cells, which are highly vulnerable to ROS due to their weak NRF2‐associated antioxidant system, subvert programmed death‐ligand 1–blockade‐mediated anti‐tumor T cell immunity by converting ATP to adenosine. Intriguingly, the immunosuppressive effect of apoptotic Treg cells is more powerful compared with live Treg cells.65 In line with this scenario, the suppression of ROS in the TME can lead to ameliorating immunotherapies for cancer. Indeed, a novel nano‐scavenger anchoring on the extracellular matrix relieved suppressive immunogenic cell death via the elimination of ROS.66
Aside from Treg cells, ROS are also critical metabolic regulators in DC‐mediated and cytotoxic T cell‐mediated anti‐tumor immunity.67, 68 In terms of DCs, ROS play an essential role in the initiation of a stimulator of interferon (STING)‐induced DC anti‐tumor response in a SUMO‐specific protease 3 (SENP3)‐dependent fashion.69 Mechanistically, ROS induces SENP3 accumulation in DCs, followed by the promotion of a SENP3‐interferon inducible (IFI)204 interaction and IFI204 deSUMOylation, which elicits STING‐mediated anti‐tumor activities. Therefore, ROS drive the anti‐tumor immune responses of DCs, unlike Treg cells. Conversely, ROS can blunt anti‐tumor immunity through endoplasmic reticulum (ER) stress‐XBP1 signaling in DCs.70
Overall, the relationship between ROS and cancer immunity is still elusive, underlining the continuing need to explore this to improve the efficacies of immunotherapies.
6. CONCLUSION
Convincing evidence suggests that ROS are virtually indispensable in understanding the pathophysiology of cancer. Initially, ROS were thought to be an evil for cancer cells. However, increasing reports have highlighted the vital roles of ROS in cancer cell survival. In contrast, high levels of ROS can be lethal for cancer cells. Markedly, ROS also act as a double‐edged sword in cancer immunity. Even though these distinct paradoxes make this difficult, targeting ROS manipulations may offer a new approach to cancer therapy.
DISCLOSURE
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This work was supported in part by the Japan Society for the Promotion of Science (grants C/19K08397, C/16K07178 to KT). We are grateful to M. Nakatsugawa (Tokyo Medical University Hachioji Medical Center) for critical discussions on cancer immunity.
Nakamura H, Takada K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021;112:3945–3952. 10.1111/cas.15068
REFERENCES
- 1.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;10:1‐41. [DOI] [PubMed] [Google Scholar]
- 2.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85‐95. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Villani RM, Wang H, et al. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res. 2018;37:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chio IIC, Tuveson DA. ROS in Cancer: the burning question. Trends Mol Med. 2017;23:411‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol Res. 2015;100:170‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931‐947. [DOI] [PubMed] [Google Scholar]
- 9.Kamihara Y, Takada K, Sato T, et al. The iron chelator deferasirox induces apoptosis by targeting oncogenic Pyk2/β‐catenin signaling in human multiple myeloma. Oncotarget. 2016;7:64330‐64341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6(221):221ra15. [DOI] [PubMed] [Google Scholar]
- 11.Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schafer ZT, Grassian AR, Song L, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeAtley SM, Aksenov MY, Aksenova MV, et al. Antioxidants protect against reactive oxygen species associated with adriamycin‐treated cardiomyocytes. Cancer Lett. 1999;136:41‐46. [DOI] [PubMed] [Google Scholar]
- 15.Siomek A, Tujakowski J, Gackowski D, et al. Severe oxidatively damaged DNA after cisplatin treatment of cancer patients. Int J Cancer. 2006;119:2228‐2230. [DOI] [PubMed] [Google Scholar]
- 16.Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ. Catalytic organometallic anticancer complexes. Proc Natl Acad Sci USA. 2008;105:11628‐11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arihara Y, Takada K, Kamihara Y, et al. Small molecule CP‐31398 induces reactive oxygen species‐dependent apoptosis in human multiple myeloma. Oncotarget. 2017;8:65889‐65899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura H, Takada K, Arihara Y, et al. Six‐transmembrane epithelial antigen of the prostate 1 protects against increased oxidative stress via a nuclear erythroid 2‐related factor pathway in colorectal cancer. Cancer Gene Ther. 2019;26:313‐322. [DOI] [PubMed] [Google Scholar]
- 19.Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144‐164. [DOI] [PubMed] [Google Scholar]
- 20.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schägger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I‐V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem. 2001;276:37861‐37867. [DOI] [PubMed] [Google Scholar]
- 23.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483‐495. [DOI] [PubMed] [Google Scholar]
- 24.Beyersmann D, Hartwig A. Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch Toxicol. 2008;82:493‐512. [DOI] [PubMed] [Google Scholar]
- 25.Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal. 2014;20:1917‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Toorn M, Rezayat D, Kauffman HF, et al. Lipid‐soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L109‐L114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27‐33. [DOI] [PubMed] [Google Scholar]
- 29.Motohashi H, Yamamoto M. Nrf2‐Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549‐557. [DOI] [PubMed] [Google Scholar]
- 30.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1‐Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error‐prone repair. Cancer Lett. 2008;270:1‐9. [DOI] [PubMed] [Google Scholar]
- 34.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation‐damaged base 8‐oxodG. Nature. 1991;349:431‐434. [DOI] [PubMed] [Google Scholar]
- 35.Oka S, Nakabeppu Y. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011;102:677‐682. [DOI] [PubMed] [Google Scholar]
- 36.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9‐17. [DOI] [PubMed] [Google Scholar]
- 37.Toyokuni S. Oxidative stress as an iceberg in carcinogenesis and cancer biology. Arch Biochem Biophys. 2016;595:46‐49. [DOI] [PubMed] [Google Scholar]
- 38.Kato J, Kobune M, Nakamura T, et al. Normalization of elevated hepatic 8‐hydroxy‐2'‐deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001;61:8697‐8702. [PubMed] [Google Scholar]
- 39.Kato J, Miyanishi K, Kobune M, et al. Long‐term phlebotomy with low‐iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J Gastroenterol. 2007;42:830‐836. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto H, Miyanishi K, Tanaka S, et al. MUTYH is associated with hepatocarcinogenesis in a non‐alcoholic steatohepatitis mouse model. Sci Rep. 2021;11:3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kikuchi S, Kobune M, Iyama S, et al. Improvement of iron‐mediated oxidative DNA damage in patients with transfusion‐dependent myelodysplastic syndrome by treatment with deferasirox. Free Radic Biol Med. 2012;53:643‐648. [DOI] [PubMed] [Google Scholar]
- 42.Angelucci E, Li J, Greenberg P, et al. Iron chelation in transfusion‐dependent patients with low‐ to intermediate‐1‐risk myelodysplastic syndromes: a randomized trial. Ann Intern Med. 2020;172:513‐522. [DOI] [PubMed] [Google Scholar]
- 43.Liu H, Yang N, Meng S, Zhang Y, Zhang H, Zhang W. Iron chelation therapy for myelodysplastic syndrome: a systematic review and meta‐analysis. Clin Exp Med. 2020;20:1‐9. [DOI] [PubMed] [Google Scholar]
- 44.Redza‐Dutordoir M, Averill‐Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977‐2992. [DOI] [PubMed] [Google Scholar]
- 45.Simon HU, Haj‐Yehia A, Levi‐Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415‐418. [DOI] [PubMed] [Google Scholar]
- 46.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532‐542. [DOI] [PubMed] [Google Scholar]
- 47.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yun CW, Lee SH. The roles of autophagy in cancer. Int J Mol Sci. 2018;19:3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35:615‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maruyama T, Noda NN. Autophagy‐regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot (Tokyo). 2017;71:72‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherz‐Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poillet‐Perez L, Despouy G, Delage‐Mourroux R, Boyer‐Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emerling BM, Weinberg F, Snyder C, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009;46:1386‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conklin KA. Chemotherapy‐associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther. 2004;3:294‐300. [DOI] [PubMed] [Google Scholar]
- 55.Marullo R, Werner E, Degtyareva N, et al. Cisplatin induces a mitochondrial‐ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One. 2013;8:e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szakács G, Paterson JK, Ludwig JA, Booth‐Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219‐234. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Gao Z, Liu X, et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat Commun. 2018;9:562. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Ambudkar SV, Kimchi‐Sarfaty C, Sauna ZE, Gottesman MM. P‐glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468‐7485. [DOI] [PubMed] [Google Scholar]
- 59.Jeddi F, Soozangar N, Sadeghi MR, et al. Nrf2 overexpression is associated with P‐glycoprotein upregulation in gastric cancer. Biomed Pharmacother. 2018;97:286‐292. [DOI] [PubMed] [Google Scholar]
- 60.Sadeghi MR, Jeddi F, Soozangar N, et al. Nrf2/P‐glycoprotein axis is associated with clinicopathological characteristics in colorectal cancer. Biomed Pharmacother. 2018;104:458‐464. [DOI] [PubMed] [Google Scholar]
- 61.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942‐949. [DOI] [PubMed] [Google Scholar]
- 62.Sakaguchi S. Naturally arising Foxp3‐expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non‐self. Nat Immunol. 2005;6:345‐352. [DOI] [PubMed] [Google Scholar]
- 63.Weinberg F, Ramnath N, Nagrath D. Reactive oxygen species in the tumor microenvironment: an overview. Cancers (Basel). 2019;11:1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, et al. ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maj T, Wang W, Crespo J, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD‐L1‐blockade resistance in tumor. Nat Immunol. 2017;18:1332‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deng H, Yang W, Zhou Z, et al. Targeted scavenging of extracellular ROS relieves suppressive immunogenic cell death. Nat Commun. 2020;11:4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gehrke N, Mertens C, Zillinger T, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING‐dependent immune sensing. Immunity. 2013;39:482‐495. [DOI] [PubMed] [Google Scholar]
- 68.Chamoto K, Chowdhury PS, Kumar A, et al. Mitochondrial activation chemicals synergize with surface receptor PD‐1 blockade for T cell‐dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114:761‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu Z, Teng XL, Zhang T, et al. SENP3 senses oxidative stress to facilitate STING‐dependent dendritic cell antitumor function. Mol Cell. 2021;81:940‐952. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Azad N, Kongkaneramit L, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down‐regulation. J Immunol. 2008;180:3072‐3080. [DOI] [PMC free article] [PubMed] [Google Scholar]