

Abstract
Yes‐associated protein 1 (YAP1) and its paralogue PDZ‐binding motif (TAZ) play pivotal roles in cell proliferation, migration, and invasion, and abnormal activation of these TEAD transcriptional coactivators is found in diverse cancers in humans and mice. Targeting YAP1/TAZ signaling is thus a promising therapeutic avenue but, to date, few selective YAP1/TAZ inhibitors have been effective against cancer cells either in vitro or in vivo. We screened chemical libraries for potent YAP1/TAZ inhibitors using a highly sensitive luciferase reporter system to monitor YAP1/TAZ‐TEAD transcriptional activity in cells. Among 29 049 low‐molecular‐weight compounds screened, we obtained nine hits, and the four of these that were the most effective shared a core structure with the natural product alantolactone (ALT). We also tested 16 other structural derivatives of ALT and found that natural ALT was the most efficient at increasing ROS‐induced LATS kinase activities and thus YAP1/TAZ phosphorylation. Phosphorylated YAP1/TAZ proteins were subject to nuclear exclusion and proteosomic degradation such that the growth of ALT‐treated tumor cells was inhibited both in vitro and in vivo. Our data show for the first time that ALT can be used to target the ROS‐YAP pathway driving tumor cell growth and so could be a potent anticancer drug.
Keywords: alantolactone, hippo‐YAP pathway, ROS, TAZ, YAP1
YAP1 and its paralogue TAZ play pivotal roles in cell proliferation, migration, and invasion, and abnormal activation of these TEAD transcriptional coactivators is found in diverse cancers in humans and mice. We screened chemical libraries for potent YAP1/TAZ inhibitors using a highly sensitive luciferase reporter system to monitor YAP1/TAZ‐TEAD transcriptional activity in cells. We found that alantolactone primarily targets the ROS‐YAP pathway driving tumor cell growth and so could be a potent anticancer drug.

Abbreviations
- β2MG
β2‐microglobulin
- ALT
alantolactone
- CHX
cycloheximide
- Cpd
compound
- Ct
cycle threshold
- DKO
double knockout
- EGFR
epidermal growth factor receptor
- F. Luc
firefly luciferase
- H2DCFDA
2′,7′‐dichlorodihydrofluorescein diacetate
- IHC
immunohistochemistry
- IKK
IκB kinase
- LATS
large tumor suppressor kinase
- MEF
murine embryonic fibroblast
- MOB1
MOB kinase activator
- MPTP
mitochondrial permeability transition pore
- MST
mammalian STE‐20 like kinase
- NAC
N‐acetylcysteine
- NFκB
nuclear factor‐κB
- PCNA
proliferating cell nuclear antigen
- PFA
paraformaldehyde
- R. Luc
Renilla luciferase
- ROS
reactive oxygen species
- SVFC
stromal vascular fraction cell
- TAZ
PDZ‐binding motif
- TEAD
TEA domain transcription factor
- Tmx
tamoxifen
- TrxR
thioredoxin reductase
- YAP1
Yes‐associated protein 1
1. INTRODUCTION
Cancer is one of the most lethal diseases globally, and the Hippo‐YAP pathway is one of the most important cell signaling pathways involved in tumor onset and development.1 High levels of YAP1/TAZ activation are observed in most human cancers, and engineered activation of YAP1 in mice results in various malignancies, notably the immediate onset of squamous cell carcinoma and hepatocellular carcinoma in mice.2, 3, 4, 5 Because YAP1/TAZ activation enhances the self‐renewal, proliferation, migration, and invasion of most human cancer cell types,1 Hippo‐YAP signaling is a promising therapeutic target. However, there are few potent selective YAP1/TAZ inhibitors that have been found to be effective in suppressing cancer cells either in vitro or in vivo.6
Both YAP1 and TAZ are paralogous coactivators of the TEADs, which regulate numerous target genes involved in cell growth.7 In the unphosphorylated state, YAP1 and TAZ act in the nuclei of most cell types to positively regulate proliferation. Negative control of YAP1/TAZ is mediated by the Hippo pathway, which is triggered in response to signaling associated with cell polarity, cell‐cell contact, mechanical tension, and other stresses, as well as certain soluble factors. The Hippo pathway has four core components: MST, LATS, the salvador family WW domain‐containing protein 1 adaptor protein (SAV1), and MOB1 adaptor protein. YAP1/TAZ are usually suppressed through efficient phosphorylation mediated by LATS, which confines them to the cytoplasm. Phospho‐YAP1/TAZ are then subject to E3‐ubiquitin ligase SCFβ‐TRCP‐mediated ubiquitination and proteasomal degradation.8 This strict regulation of YAP1/TAZ prevails in normal cells, but if YAP1 and/or TAZ become continuously activated, the cells grow abnormally and can become transformed.
Low levels of ROS are required for many normal cellular functions, including proliferation, differentiation, and gene expression. When ROS production becomes elevated as a consequence of increased metabolism, gene mutation, and/or relative hypoxia, these excess ROS are usually neutralized by an increase in the activities of enzymatic and nonenzymatic antioxidant pathways.9 Should this quenching be insufficient and moderate ROS accumulate, DNA mutation and malignant transformation could occur. If ROS levels then become very high, the programmed cell death of the abnormal cells is often triggered.9 Thus, the development of therapeutic strategies to increase excess ROS in incipient cancer cells is now in progress.9, 10 A particularly important example is the thioredoxin antioxidant system, which is often upregulated in malignant cells.11, 12 Ablation of TrxR in mice leads to increased ROS production and a delay in tumor progression and metastasis.13, 14 Conversely, overexpression of thioredoxin inhibits cancer cell apoptosis and promotes tumor angiogenesis.15, 16 Chemical inhibition of the thioredoxin system has thus become a therapeutic goal.
Alantolactone is a sesquiterpene lactone that is isolated from the roots of Inula helenium L. It was discovered more than 80 years ago and is a traditional Chinese herbal medicine with potent anti‐inflammatory, antiparasite, and antitumor activities in vivo and in vitro.17, 18 However, the exact mechanism underlying ALT’s anticancer activity remains unclear. Alantolactone binds to and inhibits the enzymatic activity of TrxR, leading to ROS accumulation.19 Alantolactone also reportedly inhibits STATs,20 IKK‐NFκB,21, 22 and others,18 but the relative contributions of these signaling pathways to ALT’s antitumor activity have yet to be clearly defined. In addition, the relationship between ALT and the Hippo‐YAP1 signaling pathway is totally unknown. Here, we present the results of an in vitro screening of small‐molecule chemical compound libraries using a sensitive luciferase reporter assay system to detect inhibitors of YAP1/TAZ signaling. We identified ALT as a powerful natural agent that triggers the accumulation of lethal ROS levels in malignant cells. These ROS induce YAP1/TAZ degradation, leading to suppression of tumor cell growth both in vitro and in vivo. Alantolactone is thus a good candidate for a novel anticancer therapy based on YAP1/TAZ inhibition.
2. MATERIALS AND METHODS
2.1. Chemicals
Alantolactone was purchased from BLDpharm and dissolved in DMSO. Tamoxifen was purchased from Toronto Research Chemicals and dissolved in ethanol. N‐acetylcysteine was purchased from Wako Chemical. We purchased H2DCFDA from Invitrogen and it was dissolved in DMSO. Auranofin was purchased from FUJIFILM Wako Pure Chemical Corporation and dissolved in DMSO.
2.2. Cell culture
H1299‐Luc cells5 were cultured in RPMI‐1640 (Nacalai) supplemented with 10% FBS (Hyclone). Human MDA‐MB‐231 (ATCC) and KKU‐M213 (JCRB Cell Bank) cells were cultured in DMEM (Wako) supplemented with 10% FBS. Primary MEFs were isolated from E16 embryos of Rosa26‐CreERT; Yap1flox/flox ; Tazflox/flox mice using a standard procedure.23 These MEFs were cultured in DMEM supplemented with 10% FBS and passaged more than 40 times to generate the spontaneously immortalized iYap1/Taz DKO MEF line. For the induction of Yap1 and Taz gene deletion in cultured iYap1/Taz DKO MEFs, the cells were treated with 1 μmol/L Tmx for 7 days. The frequency of Yap1 and Taz deletion in iYap1/Taz DKO MEFs was determined by genotyping PCR using the following primers: Yap1flox, 5′‐aatccattgaggagactgat‐3′/5′‐gtgatccttgagtgtcacgt; Yap1Δ, 5′‐gcccaaacatacccacgtaat‐3′/5′‐gtgatccttgagtgtcacgt‐3′; Tazflox, 5′‐aagcagtttccacttcatgaaac‐3′/5′‐agtcaagaggggcaaagttgtga‐3′; and TazΔ, 5′‐aagcagtttccacttcatgaaac‐3′/5′‐caccctctgtggcgtccggctctt‐3′.
Primary murine SVFCs were isolated from 8‐week‐old Rosa26‐CreERT; Lats1flox/flox ; Lats2flox/flox (iLats1/2 DKO) and Rosa26‐CreERT; Mob1aflox/flox ; Mob1b−/− (iMob1a/1b DKO) mice as described previously24 but with a minor modification. Briefly, inguinal fat tissue was minced and digested in 0.1% collagenase (FUJIFILM Wako Pure Chemical Corporation) at 37°C for 20‐40 minutes. After filtration and centrifugation at 140 g for 3 minutes, the pellet representing the stromal vascular fraction was recovered. Immortalized SVFCs were established by culturing the resuspended SVFC pellet in DMEM supplemented with 10% FBS for 20‐30 passages. For the induction of Lats1 and Lats2 gene deletion in cultured iLats1/2 DKO SVFCs, and Mob1a and Mob1b gene deletion in cultured iMob1a/1b DKO SVFCs, the cells were treated with 1 μmol/L Tmx for 7 days.
2.3. Reporter assay for YAP1/TAZ‐TEAD transcriptional activity
H1299‐Luc cells are a reporter cell line designed to monitor YAP1/TAZ‐TEAD transcriptional activity.5 H1299‐Luc cells were seeded onto 384‐well plates (1.3 × 103 cells/well) or 96‐well plates (5 × 103 cells/well). After incubation for 12‐16 hours, the cells were treated for 24 hours with individual chemical compounds from the screening libraries. Reporter luciferase activities (F. Luc and R. Luc) were determined using the Dual‐Glo Luciferase assay kit (Promega) according to the manufacturer’s instructions. Luminescence was detected using an Enspire multimode plate reader (PerkinElmer).
2.4. Chemical library screening
The above YAP1/TAZ‐TEAD reporter assay was used to undertake an initial screening of 29 049 compounds in a 384‐well format, and a secondary screening of 147 compounds in a 96‐well format. The screened compounds were derived from the NPDepo chemical library (19 449 compounds; RIKEN) plus the Drug Discovery Initiative (DDI) chemical library (9600 compounds; University of Tokyo). The structural derivatives of the hit compounds were selected by 2D and 3D ligand‐based similarity searches, as previously described.25 The concentration of each screened compound was 20 μmol/L.
2.5. Treatment in vitro with ALT or Auranofin
Cells were seeded at 7.6 × 103 cells/cm2 (eg 1.6 × 105 cells per 6‐cm dish) and cultured for 18‐26 hours prior to the addition of ALT or auranofin to 10 μmol/L (or as otherwise indicated). The vehicle control was DMSO (0.1% or as otherwise indicated) for each experiment. After incubation for 1‐24 hours, cells were subjected to immunoblotting, quantitative RT‐PCR, and/or a ROS production assay as described below. For ALT treatment of iYap1/Taz DKO MEFs, cells were left untreated or pretreated with Tmx as described above (1 μmol/L for 7 days), and seeded at 1 × 104 cells/well (96‐well plate). After incubation for 18 hours, cells were treated with 10 μmol/L ALT or 0.1% DMSO for 24 hours. Cells were then subjected to the MTT proliferation assay as described below.
For ALT treatment of iLats1/2 DKO and iMob1a/1b DKO SVFCs, cells were left untreated or pretreated with Tmx as described above (1 μmol/L for 7 days), and seeded at 6.7 × 104 cells/well (6‐well plate). After incubation for 18 hours, cells were treated with 10 μmol/L ALT or 0.1% DMSO for 6 hours, then subjected to immunoblotting as described below.
2.6. Immunoblotting
Cells were washed in PBS, dissolved in 60‐100 μL solubilizing solution (9M urea, 2% NP40), and subjected to immunoblotting as previously described.3 Primary Abs recognizing the following proteins were used: YAP1 (D8H1X), TAZ (v386), phospho‐(p)‐YAP1 (Ser127), p‐YAP1 (Ser397), p‐TAZ (S89), LATS1, and p‐LATS1 (Ser909) (all from Cell Signaling Technology), actin (Sigma), or GAPDH (Wako Chemical). Horseradish peroxidase‐conjugated anti‐rabbit Ab (Cell Signaling Technology) was used as the secondary Ab. Band intensities were determined by Fujifilm Multi Gauge software or the ImageJ program. The YAP1 and TAZ protein levels were normalized to actin or GAPDH, as indicated. Phosphorylated YAP1, p‐TAZ, and p‐LATS1 were normalized to total YAP1, TAZ, and LATS1 levels, respectively.
2.7. siRNA transfection
The siRNA targeting of YAP1 and TAZ gene expression was carried out using the following specific siRNA oligonucleotides: YAP1, 5′‐GACAUCUUCUGGUCAGAGA‐3′; TAZ, 5′‐AGAGGUACUUCCUCAAUCA‐3′; and Scr (control), 5′‐ACUACUGAGUGACAGUAGA‐3′. H1299‐Luc, MDA‐MB‐231, or KKU‐M213 cells were transfected for 2 days with these siRNA oligonucleotides using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Transfected cells were then subjected to quantitative RT‐PCR and/or the MTT proliferation assay as described below.
2.8. Quantitative RT‐PCR
Total RNA was extracted using RNAiso (Takara) according to the manufacturer’s protocol and reverse‐transcribed using the Transcriptor First Strand cDNA Synthesis Kit (Roche). Polymerase chain reaction amplifications were undertaken using the StepOne real‐time PCR system (Applied Biosystems). Primer sequences used for quantitative PCR were as follows: β2MG (internal control), 5′‐agatgagtatgcctgccgtg‐3′/5′‐tcatccaatccaaatgcggc‐3′; YAP1, 5′‐tcggcttcaggtcctcttc‐3′/5′‐ctggagcactctgactgattc‐3′; WWTR1 (TAZ), 5′‐catggcagtatcccagccaa‐3′/5′‐ctggattctctgaagccgca‐3′; CTGF, 5′‐tgtgtgacgagcccaagga‐3′/5′‐tctgggccaaacgtgtcttc‐3′; CYR61, 5′‐cgccttgtgaaagaaacccg‐3′/5′‐ggttcgggggatttcttggt‐3′; and ANKRD1, 5′‐aggaactggtcactggaaagaag‐3′/5′‐cacagggtgggctagaagtg‐3′.
The Ct values for each gene amplification were normalized by subtracting the Ct value calculated for β2MG. Normalized gene expression values were deemed to represent the relative quantity of mRNA.
2.9. Nuclear localization of YAP1
H1299‐Luc cells (2 × 104) were seeded onto a 13‐mm coverslip and incubated for 12‐16 hours to allow cell attachment to approximately 30% confluence. Attached cells were treated with 10 μmol/L ALT or 0.1% DMSO for 3 hours. Immunofluorescent assays to detect total YAP1 and nuclearly localized YAP1 were carried out as described previously.2 The intensities of three points of nuclear YAP1 staining and three points of cytoplasmic YAP1 staining were measured in each cell by ImageJ software, and the ratio of nuclear / cytoplasmic YAP1 staining intensity was calculated. At least 30 cells were evaluated for each culture. Experiments were repeated three times.
2.10. Cell proliferation assay
Cells (1 × 103 cells/well) were seeded onto a 96‐well plate and incubated for 18‐26 hours. Cells were treated with 10 μmol/L ALT or 0.1% DMSO for 24 hours, followed by incubation with 0.5 mg/mL MTT for 1 hour. After removal of the medium from each well, cells containing dye were dissolved in 150 μL/well solubilizing solution (4 mmol/L HCl, 0.1% NP40 in isopropanol), and absorbance at 570 nm was measured.
2.11. Cellular ROS measurement
Cells (5 × 104 cells/well) were seeded onto a 6‐well plate and incubated for 12‐16 hours. Cells were pretreated (or not) with 2 mmol/L NAC for 1 hour, then treated with 10 μmol/L ALT or 0.1% DMSO for 4 hours for KKU‐M213 cells, or for 12‐13 hours for MDA‐MB‐231 cells. Treated cells were then incubated with 25 μmol/L H2DCFDA for 0.5 hour at 37°C to detect ROS. Cells were trypsinized and fluorescence intensity was analyzed using a FACSCalibur (Becton Dickinson) instrument.
2.12. In vivo effects of ALT
The in vivo effects of ALT on the initiation of tongue cancer were evaluated using tgMob1DKO mice.2 To delete the floxed Mob1a gene, Tmx (10 mg/mL) was applied daily directly to tongues of 6‐week‐old male Rosa26‐CreERT; Mob1aflox/flox ; Mob1−/− mice (C57BL/6 background) for 5 days by brush, as described previously.2 Alantolactone (20 mg/kg, in 50 μL DMSO) or vehicle (50 μL DMSO) was given daily for 13‐17 days, starting at 3 days before Tmx application. For NAC treatment, NAC‐containing water (2 g/L in drinking water) was given starting at 5 days before Tmx application. The NAC‐containing water was prepared daily and water intake was monitored. Ten or 14 days after the initial Tmx treatment, tongue tumor tissues were fixed in 4% PFA in PBS and subjected to H&E staining and/or IHC analysis as described below.
For tumor xenograft studies, MDA‐MB‐231 cells (5 × 106) were subcutaneously injected into the right flanks of 5‐week‐old female BALB/c nude mice. At 10 days post‐injection, the animals were given ALT (5 mg/kg in 100 μL DMSO) or vehicle (100 μL DMSO) every 2 days over 15 days. Tumor dimensions were measured using a caliper and tumor volumes calculated according to the following formula: length × width2 × π/6. After 15 days of treatment, mice were killed and tumors were removed, weighed, and fixed in 4% PFA for IHC analysis.
2.13. Tissue immunostaining
Mouse tissues fixed in 4% paraformaldehyde in PBS were embedded in paraffin and sectioned (5 μm). For IHC, fixed sections were incubated overnight at 4°C with primary Abs recognizing: YAP1 (WH0010413M1; Sigma), TAZ (E9J5A; Cell Signaling Technology), or PCNA (610 664, BD). Anti‐rabbit/mouse HRP (Dako) and anti‐rabbit/mouse IgG conjugated with Alexa Fluor 488 dye (Molecular Probes) were used for DAB staining and immunofluorescence detection, respectively. Slides were counter‐stained with Mayer’s hematoxylin (Muto) or DAPI (Dojindo) before mounting using PermaFluor (Thermo Fisher Scientific). For staining quantification, at least 200 cells/sample/mouse were counted. Intensities of nuclear YAP1 and TAZ staining were measured using the ImageJ program with the IHC profiler plug‐in as described.26 At least 300 nuclei were counted for each image.
3. RESULTS
3.1. Identification of new chemical inhibitors of YAP1/TAZ
Because inhibition of the Hippo pathway might represent a novel approach to anticancer therapy, we devised a two‐phase screening strategy to identify YAP1 inhibitors within libraries of small‐molecule chemical compounds (Figure 1A). To detect inhibition, we took advantage of our previously established H1299 cell‐based F. Luc assay system (H1299‐Luc), which sensitively monitors YAP1/TAZ‐TEAD transcriptional activity.5 Among the 29 049 low‐molecular‐weight chemicals examined in our first phase of screening, 251 compounds were positive hits (inhibited F. Luc by >50% and had a [F. Luc − R. Luc] / F. Luc ratio of >0.5] (Figure 1B). In our secondary screen, after excluding 32 compounds with structures unsuitable for drug candidates, we treated H1299 cells with each of the 147 (out of 219) primary hit compounds that were commercially available and in stock, and then analyzed cell extracts by immunoblot and RT‐PCR. Among these 147 compounds, nine were positive as secondary hits, defined as showing more than 75% reduction in YAP1 protein and more than 80% reduction in CTGF and CYR61 mRNAs. Among these nine hits, four compounds (Cpd A‐D) shared a core structure with the natural product ALT and more effectively reduced YAP1 protein and inhibited YAP1‐dependent transcriptional activity compared to the other five secondary hit compounds. We then carried out F. Luc inhibition assays of ALT as well as Cpd A‐D and 16 other structural derivatives of ALT (Figure S1) that had been selected by 2D and 3D ligand‐based similarity searches undertaken as previously described.25 As shown in Figure 1C, ALT itself was the most effective inhibitor, demonstrating the lowest IC50 value (2.35 ± 0.10 μmol/L) in the YAP1/TAZ reporter luciferase assay. Also shown in Figure 1C is the dose‐dependent inhibition of YAP1/TAZ reporter activity by ALT and the top four ALT derivatives (Cpd A‐D). Immunoblotting confirmed that YAP1/TAZ protein expression in H1299‐Luc cells was dramatically decreased after ALT (10 μmol/L) exposure for 24 hours (Figure 1D). These data suggested that ALT was potentially a powerful inhibitor of Hippo pathway activity.
FIGURE 1.
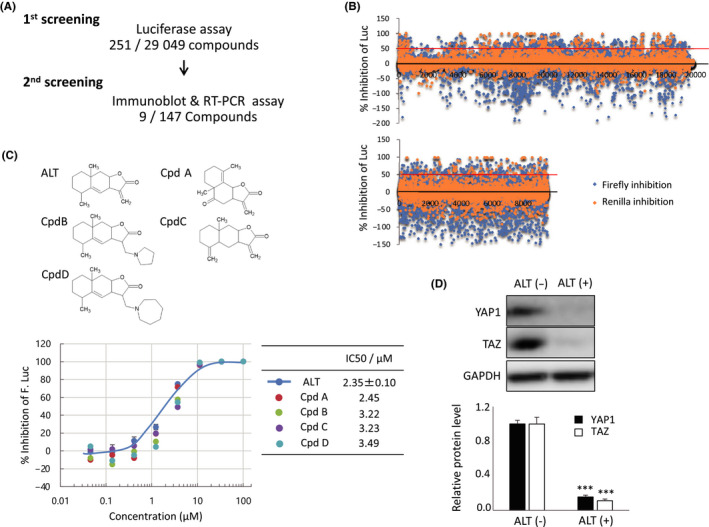
Screening of chemical compound libraries identifies alantolactone (ALT) as a potent YAP1/TAZ inhibitor. A, Illustration of screening workflow and summary of hits selected from the NPDepo chemical library (first screening) and Drug Discovery Initiative (DDI) chemical library (second screening). B, Dot plot representations of high‐throughput screening results for the NPDepo chemical library (upper panel) and DDI chemical library (lower panel). Data are the percentage of inhibition of luciferase activity exerted by each compound relative to DMSO (control; 0% inhibition). Firefly luciferase (Luc) inhibition (blue dots) and Renilla Luc inhibition (orange dots) values are individually plotted. C, Upper panel: Molecular structures of ALT and selected ALT derivatives (compound [Cpd] A‐D). Lower left panel: YAP1/TAZ reporter assay of H1299‐Luc cells that were treated with ALT or Cpd A‐D at the indicated concentrations for 24 h. DMSO (0.2%) was used as a vehicle control. Data are mean ± SEM of three independent experiments. Lower right panel: IC50 values for the compounds in the lower left panel. D, Upper panel: Immunoblot to detect total YAP1 and TAZ proteins in H1299‐Luc cells treated with/without 10 μmol/L ALT for 24 h. GAPDH, loading control. Lower panel: Quantification of data in the upper panel. Data are mean ± SEM of three independent experiments. ***P < .001
3.2. Alantolactone increases YAP1/TAZ phosphorylation and decreases YAP1/TAZ protein levels, especially in the nucleus
The phosphorylation of both YAP1 and TAZ is mainly executed by LATS kinases, which are core components of Hippo signaling. The LATS‐induced phosphorylation of YAP1 S127 or TAZ S89 leads to their binding of 14‐3‐3 protein, followed by nuclear exclusion.27, 28 Furthermore, LATS‐induced phosphorylation of YAP1 S397 or TAZ S311 causes these proteins to bind to SCFβ‐TRCP, triggering YAP1/TAZ ubiquitination and proteasomal degradation.8, 29 We therefore speculated that ALT’s inhibitory effect might be due to an ability to enhance the phosphorylation, nuclear exclusion, and degradation of YAP1/TAZ protein. In accordance with our hypothesis, ALT‐treated H1299‐Luc cells showed decreased total amounts of YAP1/TAZ proteins and increased phosphorylation of YAP1 S127, YAP1 S397, and TAZ S89 in a time‐dependent manner (Figure 2A). Immunohistochemical analysis confirmed that YAP1 appeared to be localized in the nucleus in control DMSO‐treated H1299‐Luc cells cultured at low cell density (Figure 2B, left panel). In contrast, ALT treatment for 3 hours led to significant exclusion of YAP1 from the nucleus (Figure 2B, right panel).
FIGURE 2.
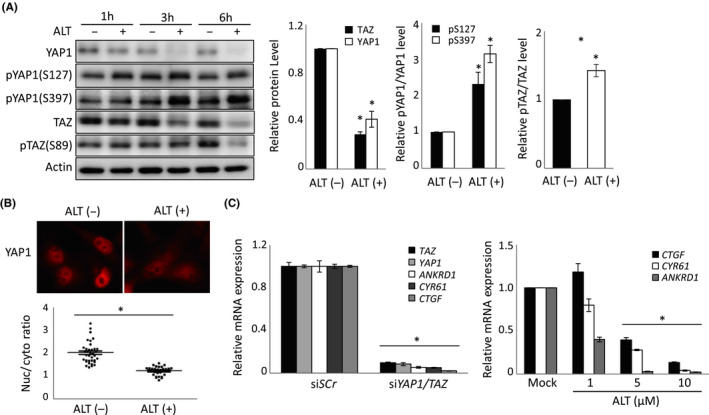
Alantolactone (ALT) decreases YAP1/TAZ protein levels, nuclear localization, and YAP1/TAZ‐dependent transcriptional activity. A, Left panel: Immunoblot to detect the indicated total and phosphorylated YAP1 and TAZ proteins in H1299‐Luc cells that were treated with/without 10 μmol/L ALT for the indicated times. Actin, loading control. Right panels: Quantification of data in left panel. Data are mean ± SEM of three independent experiments. B, Upper panel: Immunofluorescent detection of nuclear localization of YAP1 in H1299‐Luc cells treated for 3 h with DMSO (ALT (−); control) or 10 μmol/L ALT (ALT (+)). Lower panel: Quantification of data in upper panel. Data are presented as the ratio of nuclear (Nuc) to cytoplasmic (cyto) YAP1 in the indicated individual cultures. Results are representative of three independent experiments. C, Left panel: Quantitative RT‐PCR analysis of YAP1/TAZ and their indicated target genes in H1299‐Luc cells treated for 72 h with either siScr (control) or siYAP1/TAZ. Right panel: Quantitative RT‐PCR analysis of indicated genes in H1299‐Luc cells treated for 10 h with indicated concentrations of ALT. Mock, DMSO. For both panels, data are mean ± SEM of three independent experiments. *P < .05
Usually, YAP1/TAZ associate with TEAD transcriptional factors to regulate the expression of target genes such as CTGF, CYR61, and ANKRD1.30 To establish that these targets were mainly regulated by YAP1/TAZ in H1299‐Luc cells, we treated these cells with siYAP1/TAZ and observed dramatic decreases in the mRNA levels of all of these target genes (Figure 2C, left panel). We then treated H1299‐Luc cells with ALT for 10 hours and noted a similar significant decrease in CTGF, CYR61, and ANKRD1 mRNAs in a dose‐dependent manner (Figure 2C, right panel). Collectively, these data indicate that ALT increases YAP1/TAZ phosphorylation and nuclear exclusion, and thereby decreases both YAP1/TAZ protein levels and the expression of their target genes.
3.3. Alantolactone increases YAP1/TAZ protein degradation
We next investigated ALT’s role in YAP1 and TAZ protein degradation. When we pretreated H1299‐Luc cells with the protein translation inhibitor CHX, and then treated these cells with ALT for 1‐6 hours, we observed marked reductions in YAP1 and TAZ proteins in cells treated with ALT plus CHX compared to those treated with CHX alone (Figure 3A). It is of note that YAP1/TAZ mRNA levels in H1299‐Luc cells were not significantly altered after ALT treatment for 3 hours (Figure 3B). The enhanced degradation of YAP1/TAZ proteins observed in response to ALT treatment was blunted by the proteasome inhibitor MG132 (Figure 3C), suggesting that ALT promotes the degradation of the YAP1 and TAZ proteins through the canonical polyubiquitination‐proteasome system.
FIGURE 3.
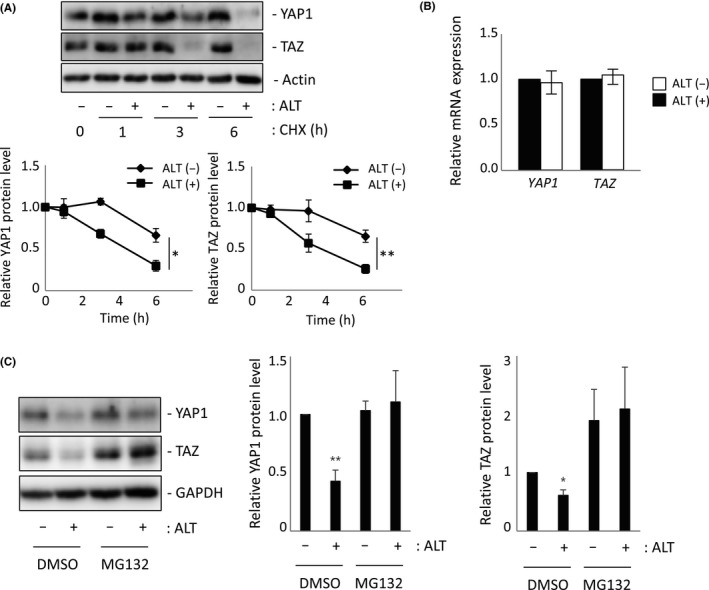
Alantolactone (ALT) enhances YAP1/TAZ degradation. A, Upper panel: Immunoblot to detect total YAP1 and TAZ proteins in H1299‐Luc cells treated with ALT (10 μmol/L; ALT+) or DMSO (0.1%; ALT−) in combination with 100 μg/mL cycloheximide (CHX) for indicated times. Actin, loading control. Lower panel: Quantification of data in upper panel. Results are mean ± SEM of three independent experiments. B, Quantitative RT‐PCR analysis of YAP1 and TAZ mRNA levels in H1299‐Luc cells treated with 10 μmol/L ALT (ALT (+)) or 0.1% DMSO (ALT (−)) for 1 h. Data are mean ± SEM of three independent experiments. C, Left panel: Immunoblot to detect total YAP1 and TAZ proteins in H1299‐Luc cells treated with ALT (10 μmol/L; ALT+) in combination with MG132 (10 μmol/L) or DMSO (0.1%) for 6 h. GAPDH, loading control. Right panel: Quantification of data in left panel. Results are mean ± SEM of three independent experiments. *P < .05, **P < .01
3.4. Alantolactone inhibits cell growth in a YAP1/TAZ‐dependent manner
To explore in depth the effects of ALT on cell growth, we took advantage of a Tmx‐inducible Yap1/Taz DKO MEF clone (iYap1/Taz DKO MEF). These cells arose by spontaneous immortalization of MEFs that were isolated from Rosa26‐CreERT; Yap1flox/flox ; Tazflox/flox mice cultured without Tmx. When iYap1/Taz DKO MEFs were treated with Tmx for 7 days, YAP1 and TAZ were deleted at both the DNA (Figure 4A, upper panel) and protein (Figure 4A, lower panel) levels. When we treated iYap1/Taz DKO MEFs with ALT for 24 hours (after 7 days pre‐incubation with or without Tmx), we found significant ALT‐mediated cell growth inhibition in the absence of Tmx treatment, but no significant effect of ALT on Tmx‐treated iYap1/Taz DKO MEFs (Figure 4B). Thus, ALT inhibits the growth of MEFs in a YAP1/TAZ‐dependent manner.
FIGURE 4.
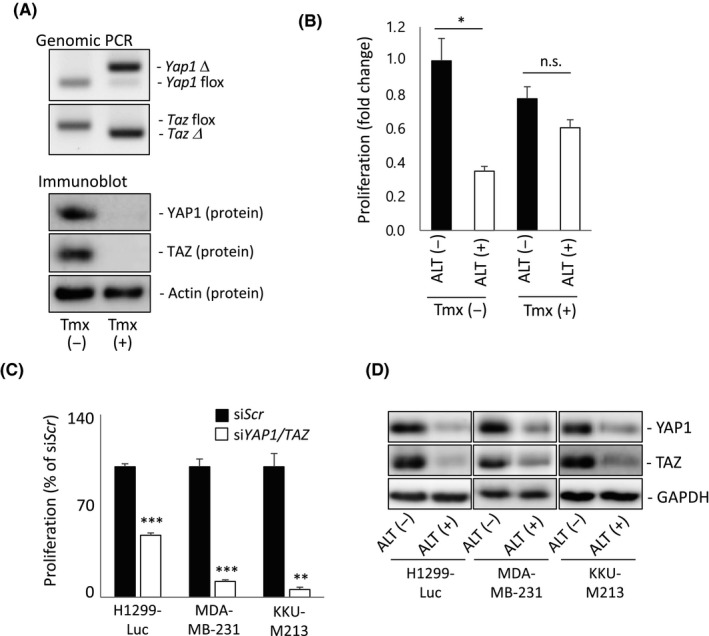
Alantolactone (ALT)‐induced growth inhibition is dependent on YAP1/TAZ. A, Genotyping genomic PCR (upper panels) and immunoblot to detect YAP1 and TAZ proteins (lower panels) in iYap1/Taz DKO murine embryonic fibroblasts that were either left untreated (Tmx (−)) or treated with tamoxifen (Tmx (+)) to delete YAP1/TAZ. Results are representative of three independent experiments. B, MTT assay of proliferation of the cells in (A). Data are expressed as fold change relative to Tmx (−) / ALT (−) control and are mean ± SEM of three independent experiments. *P < .05. n.s., not significant. C, MTT assay of proliferation of indicated cell lines after transfection with siYAP1/TAZ or siScr (control) and incubation for 48 h. Data are mean ± SEM of three independent experiments. **P < .01, ***P < .001. D, Immunoblot to detect total YAP1 and TAZ proteins in indicated cell lines treated with 10 μmol/L ALT (ALT (+)) or 0.1% DMSO (ALT (−)) for either 8 h (for H1299‐Luc and KKU‐M213) or 12 h (for MDA‐MB‐231). GAPDH, loading control. Data are representative of at least three independent experiments
We next replicated these experiments in H1299‐Luc cells as well as in two other human cancer cell lines, namely MDA‐MB‐231 breast cancer cells31 and KKU‐M213 cholangiocarcinoma cells,5 whose growth in both cases is strongly dependent on the Hippo‐YAP1 pathway. First, we used an siRNA approach to show that the growth of MDA‐MB‐231 and KKU‐M213 cells is more dependent on YAP1/TAZ than is the growth of H1299‐Luc cells (Figures 4C and S2). However, YAP1 and TAZ protein levels in all three of these human cell lines were equally decreased by 8‐12 hours after initiation of ALT treatment (Figure 4D). We therefore selected the more YAP1/TAZ‐dependent MDA‐MB‐231 and KKU‐M213 cell lines for a series of biological assays.
3.5. Effect of ALT on YAP1/TAZ activity and cell growth due to enhanced ROS accumulation
As noted above, ALT directly inhibits TrxR and so promotes ROS accumulation.19 We confirmed the presence of significant ROS levels in both KKU‐M213 and MDA‐MB‐231 cells after ALT treatment (Figure 5A). Interestingly, we also observed increased p‐LATS1 and decreased YAP1/TAZ protein levels in ALT‐treated KKU‐M213 cells (Figure 5B), as well as decreased F. Luc reporter activity in ALT‐treated H1299‐Luc cells (Figure 5C). Most importantly, the proliferation of both KKU‐M213 and MDA‐MB‐231 cells was inhibited by ALT in a manner that was almost completely rescued by pretreatment with NAC, a ROS scavenger (Figure 5D). These results indicate that the effects of ALT, including the observed reduction in YAP1/TAZ proteins and inhibition of cell growth, are mediated mainly by enhanced ROS production.
FIGURE 5.
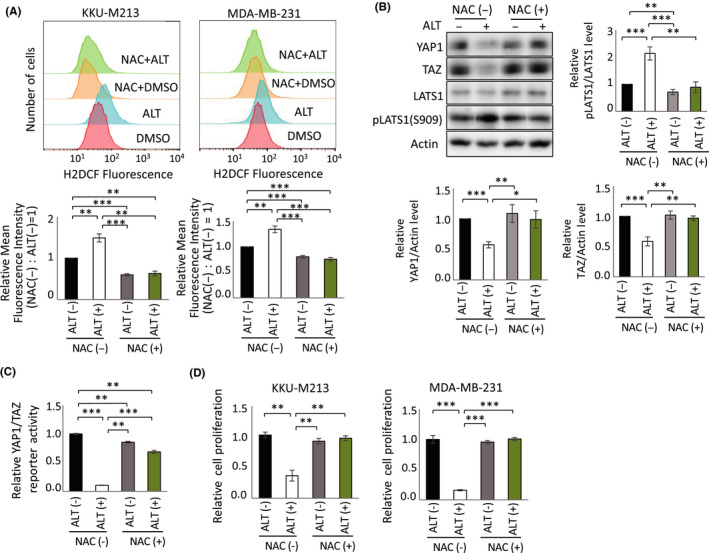
Alantolactone (ALT) suppresses YAP1/TAZ activity through reactive oxygen species (ROS) generation. A, Upper panels: Representative FlowJo histograms of flow cytometric assays to detect 2′,7′‐dichlorodihydrofluorescein diacetate (H2DCFDA) staining of cellular ROS in KKU‐M213 (left) and MDA‐MB‐231 (right) cells pretreated with/without 2 mmol/L N‐acetylcysteine (NAC) for 1 h, followed by treatment with 10 μmol/L ALT or 0.1% DMSO for 4 h (KKU‐M213 cells) or 13 h (MDA‐MB‐231 cells). Lower panels: Quantification of relative fluorescence intensity data in upper panels. Data are mean ± SEM of three or four experiments. B, Upper left panel: Immunoblot to detect indicated total and phosphorylated proteins in KKU‐M213 cells pretreated with/without NAC for 1 h, followed by treatment with 10 μmol/L ALT or 0.1% DMSO for 6 h. Upper right and lower panels: Quantification of relative levels of phosphorylated LATS, YAP1, and TAZ from data in upper left panel. Results are mean ± SEM of at least four independent experiments. C, Quantification of relative YAP1/TAZ reporter activities in H1299‐Luc cells pretreated with/without 0.5 mmol/L NAC for 1 h, followed by treatment with 10 μmol/L ALT or 0.1% DMSO for 24 h. Data are mean ± SEM of three independent experiments. D, Quantification of relative cell proliferation as determined by MTT assay at 24 h posttreatment of KKU‐M213 or MDA‐MB‐231 cells pretreated with/without 2 mmol/L NAC for 1 h, followed by treatment with 10 μmol/L ALT or 0.1% DMSO for 24 h. Data are mean ± SEM of three independent experiments. *P < .05, **P < .01, ***P < .001
Elevated ROS are known to activate MST1 kinase,32 which then activates LATS1/2 and MOB1A/B, the adaptor protein essential for LATS1/2 activation. In turn, LATS1/2 inactivates YAP1/TAZ. To explore whether ALT’s action was solely dependent on LATS1/2 and/or Hippo signaling, we investigated the effects of ALT treatment on our already‐established lines of immortalized SVFCs bearing Tmx‐inducible conditional deletion mutations of LATS1/2 or MOB1A/B (unpublished data, 2020 and 2021). After treating MOB1A/B‐deficient or LATS1/2‐deficient immortalized SVFCs with ALT, we found that the ALT‐induced reduction in YAP1/TAZ proteins was less than in ALT‐treated WT SVFCs, with LATS1/2 depletion showing a greater degree of ALT‐induced YAP1/TAZ inactivation than MOB1A/B depletion (Figure S3). These data suggest that ALT’s effects are largely dependent on LATS1/2 and partially dependent on MOB1A/B, indicating that ALT’s prime target for inhibition of the Hippo‐YAP1/TAZ pathway is the LATS kinases.
To confirm that the ALT‐mediated inhibition we observed was indeed due to targeting of the TrxR system, we analyzed the effects of another TrxR inhibitor, auranofin,33 on YAP1/TAZ proteins in KKU‐M213 cells. Just like ALT, auranofin reduced YAP1/TAZ proteins in these cells, and the effect was again almost completely prevented by pretreatment with NAC (Figure S4). Thus, it appears to be a common property of TrxR inhibitors that they can inactivate YAP1/TAZ by inducing the intracellular accumulation of large amounts of ROS.
3.6. Alantolactone inhibits YAP1‐dependent tumor development in vivo
We recently reported that YAP1 activation causes very rapid oral squamous carcinoma onset in Tmx‐inducible tgMob1 DKO mice.2 To investigate the effect of ALT‐mediated YAP1 inhibition on these mutants, we treated them with ALT (or DMSO) for 3 days before applying Tmx ointment to the tongue. When these animals were killed at 10‐14 days post‐Tmx, we found that ALT treatment blocked both the excessive cell proliferation (Figure 6A, upper right panel) and YAP1 activation (Figure 6B, upper right panel) observed in Tmx‐treated mice without ALT. Treatment with NAC in vivo significantly reduced the repressive effect of ALT on cell proliferation (Figure 6A, lower right panel) and YAP1 activation (Figure 6B, lower right panel) in tongue tissues of Tmx‐treated tgMob1 DKO mice, reflecting our in vitro cell line results (Figure 5B‐D). Macroscopically, the mucosal irregularity in control mutant tongues had clearly improved after ALT treatment (Figure 6C, left panels). Histological examination revealed that the mean thickness of the epithelial layer (Figure 6C, middle panel), and the mean percentage of the dysplastic cell area (Figure 6C, right panel), were reduced after ALT treatment.
FIGURE 6.
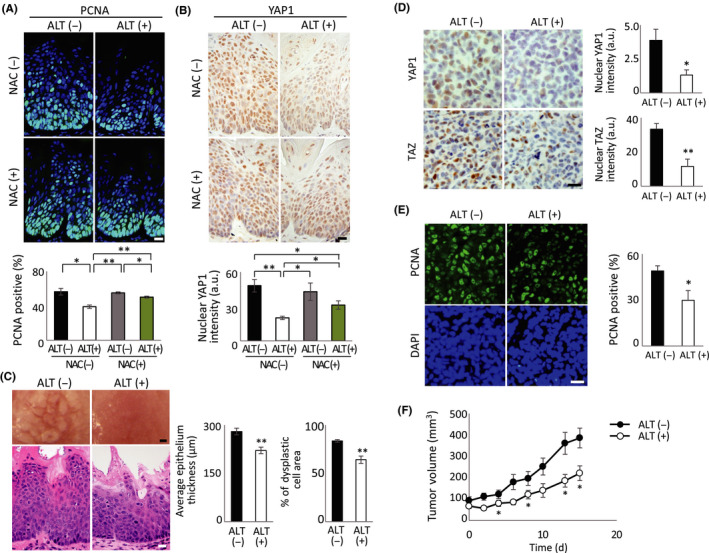
Alantolactone (ALT) reduces tumor growth in a YAP1‐dependent tongue cancer model and a xenograft tumor model. A, B, tgMob1 double knockout (DKO) mice received daily injection of 20 mg/kg ALT i.p. or vehicle control (50 μL DMSO) for a total of 13 d starting at 3 d before tamoxifen (Tmx) application to the tongue. N‐acetylcysteine (NAC) treatment was started at 5 d before Tmx application. Mice were killed at 10 d post‐Tmx. Upper panels: Mouse tongue sections were analyzed for (A) proliferation by proliferating cell nuclear antigen (PCNA) staining and (B) YAP1 protein expression by immunostaining. Mouse numbers were: ALT (−) NAC (−), n = 4; ALT (−) NAC (+), n = 3; ALT (+) NAC (−), n = 4; and ALT (+) NAC (+), n = 5. Scale bar = 20 μm. Lower left panel: Quantification of percentages of PCNA‐positive cells in tongue sections in upper left panel. Data are mean ± SEM. Lower right panel: Quantification of relative intensity of nuclear YAP1 staining in cells in upper right panel. Data are mean ± SEM. a.u., arbitrary units. C, Left panels: Macroscopic views (top) and H&E‐stained sections (bottom) of tongues of tgMob1 DKO mice that received daily injection of 20 mg/kg ALT i.p. (n = 10) or vehicle control (50 μL DMSO; n = 8) for a total of 17 d starting at 3 d before Tmx application. Tongue tissues were sectioned after 2 wk of treatment with ALT or DMSO. Scale bar = 200 μm (top) and 20 μm (bottom). Middle and right panels: Quantification of thickness of epithelial layer (middle) and area of dysplastic cells in this layer expressed as a percentage (right; n = 10 different sites/sample). Data are mean ± SEM. *P < .05. D, MDA‐MB‐231 cells were xenografted s.c. into nude mice. After 10 d, mice were injected with 5 mg/kg ALT i.p. (n = 9) or vehicle control (100 μL DMSO; n = 10) for 15 d, after which xenografted tumor tissues were sectioned. Left panels: Immunostaining to detect nuclear YAP1 and TAZ in xenografted tumor tissues. Scale bar = 10 μm. Right panels: Quantification of intensities of nuclear YAP1 and TAZ staining in left panels. Data are mean ± SEM. E, Left panels: PCNA (top) and DAPI (bottom) staining of xenografted tumor tissues in (D). Scale bar = 10 μm. Right panels: Quantification of percentage of PCNA‐positive cells in tissues in left panels. Data are mean ± SEM. F, Quantification of xenograft tumor volumes in ALT‐treated (ALT (+), n = 9) and DMSO‐treated (ALT (−), n = 10) mice in (D). Tumor volumes were measured every second day. Data are mean ± SEM. *P < .05, **P < .01.
Finally, we investigated whether ALT treatment could inhibit not only tumor initiation but also progression in vivo. We xenografted MDA‐MB‐231 cells into the subcutaneous region in nude mice and intraperitoneally injected these animals with ALT or DMSO. We found that ALT treatment in vivo blocked YAP1/TAZ activation (Figure 6D) and PCNA positivity (Figure 6E) in xenografted tumor tissues, and suppressed the ability of these MDA‐MB‐231‐derived tumors to expand in vivo (Figure 6F). Thus, both tumor initiation and progression can be blocked by ALT treatment in vivo, and these effects are at least partly due to inhibition of YAP1/TAZ by ROS accumulation.
4. DISCUSSION
Reactive oxygen species take on the properties of a double‐edged sword in carcinogenesis.10, 34 When mitochondrial ROS are continuously moderate, mutagenesis could increase to drive malignant transformation of normal cells. These ROS also play critical roles in cell signaling pathways that can stabilize factors supporting cancer initiation and progression,35 and/or inactivate tumor‐suppressive phosphatases such as PTEN.36 These observations have led to attempts to use antioxidants to neutralize ROS levels and thereby prevent or reduce cancer cell growth. Unfortunately, massive multicenter clinical trials of antioxidant treatment of cancer patients showed not only no benefit but also a striking increase in tumor incidence.37, 38 Similar analyses in mouse cancer models confirmed that antioxidant supplementation was protumorigenic rather than antitumorigenic.39 For example, antioxidant treatment of melanoma‐bearing mice strongly enhanced metastasis in these animals.40 Thus, removal of ROS can clearly augment cancer cell growth.
In contrast, excessive oxidative stress can induce a cell death pathway that involves the activation of the MPTP.41, 42 After a cell transforms, it often sustains mutations and undergoes metabolic changes that can increase ROS. If these ROS accumulate to very high levels within a tumor cell, it could suffer growth arrest or MPTP‐mediated death, blocking cancer progression. In particular, cells that hyperproliferate vigorously enough that they are forced from their natural “niches” become starved for essential growth factors, altering their oxygen metabolism such that lethal levels of ROS are generated.43 Many tumor cells increase their production of antioxidants to combat these high ROS and stave off death,43, 44 and so strategies that suppress the production of these antioxidants have been used to reduce cancer initiation, progression, and/or metastasis.45 Much effort is now being devoted to finding antioxidant inhibitors that increase ROS‐induced tumor cell killing without unduly damaging normal cells.9, 10 Our work points to a novel therapeutic avenue whereby ALT‐mediated ROS production can suppress the growth of tumor cells featuring activated YAP1/TAZ.
An important but complex relationship has recently been identified between ROS and the Hippo pathway. These studies have revealed that Hippo signaling can act both upstream and downstream of ROS, and can either promote ROS‐triggered cell death or the scavenging of ROS in a variety of species. For example, in mammals, YAP1 can transcriptionally coactivate FOXO1, which upregulates antioxidant genes.46 In Drosophila, overexpression of the YAP1 ortholog Yki enhances the transcription of genes such as mitochondria assembly regulatory factor (Marf) and opa1‐like (opa1), resulting in mitochondrial fusion and decreased ROS levels.47 In the human renal cell carcinoma cell line SN12C, shRNA‐mediated inhibition of YAP1/TAZ increases ROS generation and apoptosis.48 These findings imply that upstream YAP1/TAZ activation reduces ROS, favoring tumor cell survival.
Reactive oxygen species can also act on the Hippo pathway. For example, in Drosophila showing Ras activation, mitochondrial function is disrupted and moderate ROS accumulate that inactivate Hippo signaling, leading to Yki activation that drives tumor progression.49 Similarly, in mice, moderate ROS can inactivate the tumor suppressor PTEN,50 which could lead to YAP1/TAZ activation.51, 52, 53, 54, 55
Previous work has shown that the TrxR inhibitor curcumin induces the accumulation of high levels of ROS, leading to the phosphorylation and activation of MST1.32, 56, 57, 58, 59, 60, 61 The MST1‐mediated phosphorylation of YAP1 inactivates it, leaving a cancer cell vulnerable to mechanisms of defense against oxidative stress. We have shown here that two other TrxR inhibitors, ALT and auranofin, induce reductions in YAP1/TAZ proteins, and that the effects of these agents can be almost completely rescued by pretreatment with NAC (Figure S4). Thus, antioxidant inhibitors that induce sustained high levels of ROS, and that continuously and negatively control YAP1 activation, are the best candidates for novel anticancer drugs.
We found that YAP1/TAZ inactivation due to ALT‐induced ROS was largely dependent on LATS kinase, supporting the notion that ALT‐induced ROS play a critical tumor‐suppressive role through the LATS‐YAP/TAZ pathway. In contrast, deficiency for MOB1 (the adaptor protein supporting LATS kinase activation) had a milder suppressive effect on ALT action than did LATS deficiency, presumably due to the incomplete repression of LATS kinase activity caused by the MOB1 deficiency. For these reasons, ALT was still able to inactivate YAP1/TAZ to some degree in MOB1‐deficient cells.
Many signaling molecules are reportedly targeted by ALT. In addition to TrxR,19 ALT inhibits STATs20 and IKK‐NFκB.21, 22 Because continuous accumulation of high amounts of ROS depresses JAK2/Src‐STAT3 signaling,62, 63 as well as IKK‐NFκB activity in the late phase post‐stimulation,64, 65 the observed ALT‐induced inhibition could be due to excessive ROS accumulation. In our study, as ALT‐induced YAP1 inhibition and the resulting reduction in tumor cell growth were substantially rescued by NAC treatment, these effects of ALT are most likely due to increased ROS. These findings imply that the most important molecular target of ALT is TrxR.
To summarize, we screened approximately 30 000 chemical compounds and identified ALT, a TrxR inhibitor, as promoting excessive ROS accumulation that strongly inhibits YAP1/TAZ activation in tumor cells, thus blocking cancer initiation and progression in vivo. Cancer cells are characterized by increased aerobic glycolysis and high ROS levels that are counteracted by elevations in antioxidant defense mechanisms. Thus, targeting antioxidants so as to generate lethal levels of intracellular ROS could be a promising approach for a new form of cancer therapy. Auranofin (Ridaura; Prometheus) is a gold‐based compound that functions as a TrxR inhibitor and is used in the clinic as an antirheumatic agent. Human head and neck squamous cell carcinoma cells that were treated in vitro and in vivo with auranofin in combination with buthionine sulfoximine (an inhibitor of the antioxidant glutathione) showed increased sensitivity to inhibitors of EGFR signaling.66 Notably, the master regulator of the EGFR pathway is the YAP1 pathway.2 Thus, it might be worth exploring whether patients whose cancers show excessive YAP1/TAZ activity respond favorably to treatment with auranofin, ALT, and/or related antioxidant inhibitors.
DISCLOSURES
K. Nakatani and S. Hagiwara are employees of FUJIFILM Corporation. A. Suzuki received collaborative research grants from FUJIFILM Corporation.
Supporting information
Fig S1‐S4
ACKNOWLEDGMENTS
We are grateful for funding provided by the Japanese Agency for Medical Research and Development (P‐CREATE [AMED] grant JP21cm0106114h0006), the Japanese Society for the Promotion of Science (grants 21H04806 and 20H04905), and the Nanken‐Kyoten (grant no. 2021‐02), TMDU. The high‐throughput first screening of the chemical libraries was performed by Takeshi Sonoda, RIKEN CSRS and the technical support team of P‐CREATE.
Nakatani K, Maehama T, Nishio M, et al. Alantolactone is a natural product that potently inhibits YAP1/TAZ through promotion of reactive oxygen species accumulation. Cancer Sci. 2021;112:4303–4316. 10.1111/cas.15079
Tomohiko Maehama, Miki Nishio and Junji Otani contributed equally as second authors.
Funding information
Nanken‐Kyoten, TMDU, Grant/Award Number: 2021‐02; Japan Society for the Promotion of Science, Grant/Award Number: 21H04806, 20H04905; Japan Agency for Medical Research and Development, Grant/Award Number: JP21cm0106114h0006.
Contributor Information
Tomohiko Maehama, Email: tmaehama@med.kobe-u.ac.jp.
Akira Suzuki, Email: suzuki@med.kobe-u.ac.jp.
REFERENCES
- 1.Nishio M, Maehama T, Goto H, et al. Hippo vs. crab: tissue‐specific functions of the mammalian hippo pathway. Genes Cells. 2017;22:6‐31. [DOI] [PubMed] [Google Scholar]
- 2.Omori H, Nishio M, Masuda M, et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci Adv. 2020;6:eaay3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishio M, To Y, Maehama T, et al. Endogenous YAP1 activation drives immediate onset of cervical carcinoma in situ in mice. Cancer Sci. 2020;111(10):3576‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maehama T, Nishio M, Otani J, Mak TW, Suzuki A. The role of hippo‐YAP signaling in squamous cell carcinomas. Cancer Sci. 2021;112(1):51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishio M, Sugimachi K, Goto H, et al. Dysregulated YAP1/TAZ and TGF‐beta signaling mediate hepatocarcinogenesis in Mob1a/1b‐deficient mice. Proc Natl Acad Sci USA. 2016;113:E71‐E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatani K, Maehama T, Nishio M, et al. Targeting the hippo signalling pathway for cancer treatment. J Biochem. 2017;161:237‐244. [DOI] [PubMed] [Google Scholar]
- 7.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev. 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev. 2010;24:72‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931‐947. [DOI] [PubMed] [Google Scholar]
- 11.Lincoln DT, Ali Emadi EM, Tonissen KF, Clarke FM. The thioredoxin‐thioredoxin reductase system: over‐expression in human cancer. Anticancer Res. 2003;23:2425‐2433. [PubMed] [Google Scholar]
- 12.Berggren M, Gallegos A, Gasdaska JR, Gasdaska PY, Warneke J, Powis G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996;16:3459‐3466. [PubMed] [Google Scholar]
- 13.Hellfritsch J, Kirsch J, Schneider M, et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor‐derived angiogenesis. Antioxid Redox Signal. 2015;22:938‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo MH, Xu XM, Carlson BA, Gladyshev VN, Hatfield DL. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J Biol Chem. 2006;281:13005‐13008. [DOI] [PubMed] [Google Scholar]
- 15.Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin‐1 (Trx‐1) increases hypoxia‐inducible factor 1alpha protein expression: Trx‐1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62:5089‐5095. [PubMed] [Google Scholar]
- 16.Baker A, Payne CM, Briehl MM, Powis G. Thioredoxin, a gene found overexpressed in human cancer, inhibits apoptosis in vitro and in vivo. Cancer Res. 1997;57:5162‐5167. [PubMed] [Google Scholar]
- 17.Tavares WR, Seca AML, Inula L. Secondary metabolites against oxidative stress‐related human diseases. Antioxidants (Basel). 2019;8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babaei G, Gholizadeh‐Ghaleh Aziz S, Rajabi Bazl M, Khadem Ansari MH. A comprehensive review of anticancer mechanisms of action of alantolactone. Biomed Pharmacother. 2021;136:111231. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Li Y, Duan D, Yao J, Gao K, Fang J. Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress‐mediated apoptosis of HeLa cells. Biochem Pharmacol. 2016;102:34‐44. [DOI] [PubMed] [Google Scholar]
- 20.Chun J, Li RJ, Cheng MS, Kim YS. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA‐MB‐231 cells. Cancer Lett. 2015;357:393‐403. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Yu Z, Wang C, et al. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKbeta kinase activity and interrupting NF‐kappaB/COX‐2‐mediated signaling cascades. J Exp Clin Cancer Res. 2017;36:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei W, Huang H, Zhao S, et al. Alantolactone induces apoptosis in chronic myelogenous leukemia sensitive or resistant to imatinib through NF‐kappaB inhibition and Bcr/Abl protein deletion. Apoptosis. 2013;18:1060‐1070. [DOI] [PubMed] [Google Scholar]
- 23.Nagy A, Gertsenstein M, Vintersten K, Behringer R.. Manipulating the Mouse Embryo: A Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press; 2003:371‐373. [Google Scholar]
- 24.Matsumoto T, Kano K, Kondo D, et al. Mature adipocyte‐derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol. 2008;215:210‐222. [DOI] [PubMed] [Google Scholar]
- 25.Nara T, Nakagawa Y, Tsuganezawa K, et al. The ubiquinone synthesis pathway is a promising drug target for chagas disease. PLoS One. 2021;16:e0243855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varghese F, Bukhari AB, Malhotra R, De A. IHC profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9:e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747‐2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lei QY, Zhang H, Zhao B, et al. TAZ promotes cell proliferation and epithelial‐mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426‐2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu CY, Zha ZY, Zhou X, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}‐TrCP E3 ligase. J Biol Chem. 2010;285:37159‐37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Xu X, Maglic D, et al. Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 2018;25(1304–1317):e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han H, Yang B, Nakaoka HJ, et al. Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene. 2018;37:6414‐6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu T, Ji J, Guo YL. MST1 activation by curcumin mediates JNK activation, Foxo3a nuclear translocation and apoptosis in melanoma cells. Biochem Biophys Res Commun. 2013;441:53‐58. [DOI] [PubMed] [Google Scholar]
- 33.Omata Y, Folan M, Shaw M, et al. Sublethal concentrations of diverse gold compounds inhibit mammalian cytosolic thioredoxin reductase (TrxR1). Toxicol In Vitro. 2006;20:882‐890. [DOI] [PubMed] [Google Scholar]
- 34.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an achilles' heel? Nat Rev Cancer. 2014;14:709‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao P, Zhang H, Dinavahi R, et al. HIF‐dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336‐20342. [DOI] [PubMed] [Google Scholar]
- 37.Klein EA, Thompson IM Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT). JAMA. 2011;306:1549‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177‐178. [DOI] [PubMed] [Google Scholar]
- 39.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215. [DOI] [PubMed] [Google Scholar]
- 40.Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re308. [DOI] [PubMed] [Google Scholar]
- 41.Loor G, Kondapalli J, Iwase H, et al. Mitochondrial oxidant stress triggers cell death in simulated ischemia‐reperfusion. Biochim Biophys Acta. 2011;1813:1382‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schriewer JM, Peek CB, Bass J, Schumacker PT. ROS‐mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia‐reperfusion. J Am Heart Assoc. 2013;2:e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schafer ZT, Grassian AR, Song L, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris IS, Treloar AE, Inoue S, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211‐222. [DOI] [PubMed] [Google Scholar]
- 46.Shao D, Zhai P, Del Re DP, et al. A functional interaction between hippo‐YAP signalling and FoxO1 mediates the oxidative stress response. Nat Commun. 2014;5:3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagaraj R, Gururaja‐Rao S, Jones KT, et al. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev. 2012;26:2027‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White SM, Avantaggiati ML, Nemazanyy I, et al. YAP/TAZ inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2‐deficient tumor cells. Dev Cell. 2019;49(425–443):e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, Igaki T. Mitochondrial defect drives non‐autonomous tumour progression through hippo signalling in Drosophila. Nature. 2012;490:547‐551. [DOI] [PubMed] [Google Scholar]
- 50.Kwon J, Lee SR, Yang KS, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA. 2004;101:16419‐16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixit D, Ghildiyal R, Anto NP, Sen E. Chaetocin‐induced ROS‐mediated apoptosis involves ATM‐YAP1 axis and JNK‐dependent inhibition of glucose metabolism. Cell Death Dis. 2014;5:e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Wang C, Tao Z, et al. Curcumin derivative WZ35 inhibits tumor cell growth via ROS‐YAP‐JNK signaling pathway in breast cancer. J Exp Clin Cancer Res. 2019;38:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gandhirajan RK, Jain M, Walla B, et al. Cysteine S‐Glutathionylation promotes stability and activation of the hippo downstream effector transcriptional co‐activator with PDZ‐binding motif (TAZ). J Biol Chem. 2016;291:11596‐11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho Y, Park MJ, Kim K, et al. Reactive oxygen species‐induced activation of Yes‐associated protein‐1 through the c‐Myc pathway is a therapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2020;26:6599‐6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tung JN, Lin PL, Wang YC, Wu DW, Chen CY, Lee H. PD‐L1 confers resistance to EGFR mutation‐independent tyrosine kinase inhibitors in non‐small cell lung cancer via upregulation of YAP1 expression. Oncotarget. 2018;9:4637‐4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen T, Zhao L, Chen S, et al. The curcumin analogue WZ35 affects glycolysis inhibition of gastric cancer cells through ROS‐YAP‐JNK pathway. Food Chem Toxicol. 2020;137:111131. [DOI] [PubMed] [Google Scholar]
- 57.Kakeya H, Onose R, Osada H. Caspase‐mediated activation of a 36‐kDa myelin basic protein kinase during anticancer drug‐induced apoptosis. Cancer Res. 1998;58:4888‐4894. [PubMed] [Google Scholar]
- 58.Shi L, Liang F, Zheng J, et al. Melatonin regulates apoptosis and autophagy Via ROS‐MST1 pathway in subarachnoid hemorrhage. Front Mol Neurosci. 2018;11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST‐FOXO signaling pathway mediates oxidative‐stress responses and extends life span. Cell. 2006;125:987‐1001. [DOI] [PubMed] [Google Scholar]
- 60.Kim YH, Choi J, Yang MJ, et al. A MST1‐FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat Commun. 2019;10:838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu H, Xiao Y, Zhang S, et al. The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep. 2013;3:1663‐1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miao Z, Yu F, Ren Y, Yang J. d, l‐Sulforaphane induces ROS‐dependent apoptosis in human gliomablastoma cells by inactivating STAT3 signaling pathway. Int J Mol Sci. 2017;18:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: role of antioxidative nutraceuticals. Cancer Lett. 2017;387:95‐105. [DOI] [PubMed] [Google Scholar]
- 64.Xie J, Shaikh ZA. Cadmium‐induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor‐kappa B activity. Toxicol Sci. 2006;91:299‐308. [DOI] [PubMed] [Google Scholar]
- 65.Nakajima S, Kitamura M. Bidirectional regulation of NF‐kappaB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162‐174. [DOI] [PubMed] [Google Scholar]
- 66.Sobhakumari A, Love‐Homan L, Fletcher EV, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7:e48175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4