

Abstract
Breast cancer is the most common cancer among women. Glycoprotein non–metastatic melanoma protein B (GPNMB), a type I transmembrane protein that is highly expressed in many cancers, including breast cancer, has been shown to be a prognostic factor. We previously reported that GPNMB overexpression confers tumorigenic potential, as evidenced by invasive tumor growth in vivo, sphere formation, and cellular migration and invasion to non–tumorigenic mammary epithelial cells. In this study, we focused on the serine (S) residue in the intracellular domain of GPNMB (S530 in human isoform b and S546 in mouse), which is predicted to be a phosphorylation site. To investigate the roles of this serine residue, we made an antibody specific for S530‐phosphorylated human GPNMB and a point mutant in which S530 is replaced by an alanine (A) residue, GPNMB(SA). Established GPNMB(SA) overexpressing cells showed a significant reduction in sphere formation in vitro and tumor growth in vivo as a result of decreased stemness‐related gene expression compared to that in GPNMB(WT)‐expressing cells. In addition, GPNMB(SA) impaired GPNMB‐mediated cellular migration. Furthermore, we found that tyrosine kinase receptor signaling triggered by epidermal growth factor or fibroblast growth factor 2 induces the serine phosphorylation of GPNMB through activation of downstream oncoproteins RAS and RAF.
Keywords: breast cancer, cancer stem cell, GPNMB, sphere formation, tumorigenesis
In this study, we revealed that GPNMB serine phosphorylation is induced by growth factor signaling through activation of downstream oncoproteins RAS and RAF. Furthermore, we found that the serine residue is critical for tumorigenic potential, as indicated by sphere formation in vitro, tumor formation in vivo, and the enhancement of stemness‐related gene expression. In addition, we showed that the serine residue is important for EMT induction and cellular migration mediated by GPNMB.

1. INTRODUCTION
Breast cancer, in which malignant tumors occur in breast epithelial tissue, is the most common cancer among adult women worldwide. The global incidence of breast cancer has been on the rise since the late 1970s.1, 2, 3
Glycoprotein non–metastatic melanoma protein B (GPNMB), a type I transmembrane protein, is highly expressed in various cancers, including breast cancer. A high level of GPNMB expression is reported to be associated with poorer prognosis. There is increasing evidence that GPNMB is involved in cancer initiation and progression; thus, it is seen as a promising therapeutic target.4, 5, 6, 7, 8
We previously demonstrated that GPNMB induces tumorigenic abilities in breast epithelial cells by overexpressing GPNMB.9, 10 GPNMB‐expressing cells show epithelial‐mesenchymal transition (EMT)‐like phenotypes, such as the cadherin switch, increased cellular migration and invasion, and anchorage‐independent growth potential.9, 10, 11, 12, 13 Furthermore, we showed that cell surface localized GPNMB is mainly found in the dormant breast cancer cells and induces stem‐like properties through the tyrosine residue in its hemi–immunoreceptor tyrosine‐based activation motif (ITAM).14, 15
Another phosphorylation site, serine (S) 530, in the intracellular domain of human GPNMB isoform b, has been predicted based on data in the eukaryotic linear motif (ELM) database.16, 17 Therefore, in the present study, we focused on S530 in humans and the corresponding site in mouse GPNMB, S546, to clarify the involvement of the serine site in GPNMB‐induced tumorigenic potential. Accordingly, we made a point mutant of the phosphorylation site of GPNMB by replacing the serine with an alanine (A) residue (ie, GPNMB[SA]). Cells overexpressing GPNMB(SA) significantly reduced tumorigenic growth in terms of sphere formation in vitro and tumor growth in vivo, stemness‐related gene expression, and cellular migration. Furthermore, we found that growth factors such as epidermal growth factor (EGF) and fibroblast growth factor (FGF)‐2, which are supplemented in the sphere culture medium, induce the phosphorylation of S530 through the activation of the oncoproteins RAS and RAF.
2. MATERIALS AND METHODS
2.1. Cells and cell culture
For the present study, 293T, BT‐474, Hs578T, MCF7, MDA‐MB‐157, MDA‐MB‐231, and NMuMG cells were obtained from the ATCC. These cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Gibco‐Thermo Fisher), penicillin G (100 U/mL), and streptomycin sulfate (0.1 mg/mL, Wako). Insulin (10 μg/mL, Wako) was supplemented only for Hs578T cells. Puromycin (1 μg/mL, Sigma‐Aldrich) was used to select MCF7 and NMuMG cells showing stable expression of GPNMB(WT) or GPNMB(SA).9
2.2. DNA construction and transfection
The genes pCAGIP‐mouse GPNMB‐FLAG/HA and pcDNA3‐HA‐rat ERK2 (L73P/S151D) have been described previously.9, 18 Human GPNMB was amplified by PCR, and human GPNMB(S530A) and mouse GPNMB(S546A) mutants were constructed by PCR‐based mutagenesis using the following primers: for humans (Forward) 5′‐AAAGGCCTGGCTGTCTTTCTC‐3′, (Reverse) 5′‐GAGAAAGACAGCCAGGCCTTT‐3′; for mice (Forward) 5′‐AGGGCAAGGGCCTGGCTGTTCTCCTCAGTC‐3′, (Reverse) 5′‐GACTGAGGAGAACAGCCAGGCCCTTGCCCT‐3′. The obtained DNA was cloned into a pCAGIP‐FLAG/HA‐expressing vector. The expression constructs encoding human BRAF have been described previously.19 The BRAF(V600E) mutant was constructed by PCR‐based mutagenesis using the following primers: (Forward) 5′‐GGCACCAGAAGAGATCAGAATGCAA‐3′, (Reverse) 5′‐TTGCATTCTGATCTCTTCTGGTGCC‐3′. The expression constructs encoding rat MEK1 (S218D/S222D), which have been described previously,19 were cloned into a pcDNA3‐HA‐expressing vector. Mouse HRAS(G12V) mutant was constructed by PCR‐based mutagenesis using the following primers: (Forward) 5′‐ACAGAATACAAGCTTGTGGTGGTGGGCGCTGTAGGC‐3′, (Reverse) 5′‐TCAGGACAGCACACATTTGC‐3′. The obtained DNA was cloned into a pCAGIP‐HA‐expressing vector. These constructs were transfected into cells using PEI Max (Polysciences Asia‐Pacific). To establish stably expressing cell lines from MCF7 or NMuMG, cells were transfected with the plasmid using Lipofectamine3000 (Invitrogen) as described previously.9
2.3. Antibody establishment
We used a custom P‐site rabbit antibody service (YenZym Antibodies LLC) to establish the anti–pGPNMB(S530) antibody. CVRSKGL‐pS‐VFLNRAK peptides, which were conjugated with KLH, were injected into a rabbit. Antibodies that recognize pGPNMB(S530) were purified from rabbit sera by purification against the phosphorylated peptide and affinity absorption against the non–phosphorylated peptide.
2.4. Quantitative real‐time PCR
Total RNA was isolated using ISOGEN II (Nippon Gene). Real‐Time PCR was performed using GeneAce SYBR quantitative real‐time PCR (qPCR) mix α Low ROX (Nippon Gene) and the ABI7500 Fast Sequence Detection system (Applied Biosystems). All samples were run in triplicate in each experiment. Primers are listed in Table S1.
2.5. SDS‐PAGE and immunoblot analysis
The protein samples were subjected to SDS‐PAGE. The samples were then electrotransferred to PVDF membranes (Millipore) and subjected to immunoblot analysis. The following primary antibodies were used: E‐cadherin (610 181, BD Transduction, 1:2500), fibronectin (sc‐8422, Santa Cruz Biotechnology, 1:200), GPNMB (AF2550, R&D Systems, 1:2000), pGPNMB(S530) (YenZym Antibodies LLC, 1:70), HA epitope tag (3F10, Roche Applied Sciences, 1:1000), and β‐actin (010‐27841, Sigma‐Aldrich, 1:5000). The reacted antibodies were detected with HRP‐conjugated anti–mouse IgG, anti–rabbit IgG, anti–rat IgG (GE Healthcare, 1:10 000), or anti–goat IgG (Dako, 1:10 000) secondary antibodies and visualized on an AE‐9300H Ez‐Capture MG chemiluminescence imaging system (ATTO).
2.6. Protein phosphatase treatment
293T cells were transfected with pCAGIP‐human GPNMB(WT)‐FLAG/HA and cell lysates were immunoprecipitated with anti–FLAG (M2, Sigma‐Aldrich). Samples were then treated with 0 or 400 U of lambda protein phosphatase (New England Biolabs) at 37°C for 30 minutes in the manufacturer’s recommended reaction buffer.
2.7. Immunofluorescence staining
The cells were cultured on glass coverslips and then fixed in 4% formaldehyde, after which we used 0.3% Triton X‐100 for permeabilization and 1% BSA in PBS for blocking. Antibodies against GPNMB (AF2550, R&D Systems, 1:200) and E‐cadherin (610181, BD Transduction, 1:200) were used as primary antibodies. The cells were then incubated with Alexa 488‐labeled anti–goat IgG or Alexa 488–labeled anti–mouse IgG (Molecular Probes, Invitrogen, 1:500). Propidium iodide (PI) solution was used for nuclei staining. We used a laser scanning microscope (model TCS SP8, Leica Microsystems) for detection and imaging.
2.8. Transwell migration
Thirty thousand cells were seeded into a Transwell chamber (8‐μm pore) (Corning). After 16 hours, the cells were fixed with 3.7% formaldehyde and stained with 0.5% crystal violet. Four high power fields of the lower surface of each Transwell membrane were photographed, and the migrated cells were counted.
2.9. Sphere formation
Five thousand cells were cultured in DMEM/F12 medium (Sigma‐Aldrich) supplemented with B‐27 (20 μL/mL, Invitrogen), EGF (20 ng/mL, Sigma‐Aldrich), and FGF‐2 (20 ng/mL, Wako) in each ultra‐low attachment culture plate (6‐well; Corning). The number of the spheres was counted on day 7.
2.10. Tumor formation
Five million cells were injected subcutaneously into 6‐week‐old female BALB/cAJcl‐nu/nu mice (CLEA Japan). Mice were killed at 8 weeks after injection, and the tumor grafts were obtained. The tumor volumes were approximated using the following formula: volume = 0.5 × a × b2. The a and b are the lengths of the major and minor axes, respectively. All the animal experiments were performed with the approval of the Animal Ethics Committee of the University of Tsukuba and in accordance with the university’s animal experiment guidelines and the provisions of the 1995 Declaration of Helsinki.
2.11. Immunohistochemical staining
The paraffin‐embedded tissue sections were deparaffinized in xylene, rehydrated in ethanol, and immersed in citrate‐NaOH buffer (10 mmol L−1 sodium citrate, pH 6.0) at 121°C for 20 minutes. After retrieval of antigenicity, the nonspecific antibody reaction was blocked in blocking solution (Perkin Elmer Life Science), and the samples were incubated with antibodies against E‐cadherin (610181, BD Biosciences, 1:100) or Ki‐67 (ab15580, Abcam, 1:500). After the sections had been washed, the reacted antibodies were detected using the Dako EnVision + System/HRP (DAB) (DakoCytomation).
2.12. Liquid chromatography‐mass spectrometry
The cell lysates were immunoprecipitated by anti–FLAG and the resulting precipitate samples were subjected to SDS‐PAGE for purification. Samples were stained with Coomassie Brilliant Blue and extracted from the gel. After decolorization of the gel, acetonitrile was added, and the gel was sonicated until it turned white and was dry. Each sample was reduced in 20 mmol L−1 DTT and 50 mmol L−1 ammonium bicarbonate at 50°C, after which they were alkylated in 50 mmol L−1 iodoacetamide and 50 mmol L−1 ammonium bicarbonate at room temperature in the dark. After the sample was washed, acetonitrile was added, and the sample was dehydrated by sonication. The samples were then digested by MS‐grade trypsin (20 ng/µL trypsin in 50 mmol L−1 ammonium bicarbonate) and washed in 50 mmol L−1 ammonium bicarbonate at 37°C overnight. The samples were dehydrated in elution solution (5% formic acid, 40% acetonitrile). This mixture was filtered through using a 0.45‐µm filter and then condensed to approximately 20 µL using a centrifugal concentrator. The resulting peptides were analyzed using a nanoACQUITY ultrahigh performance liquid chromatography system (nanoUPLC; Waters) equipped with a BEH130 nanoACQUITY C18 column (100‐mm long, 75‐µm i.d., 1.7‐µm particle diameter), which was kept at 35°C. Mobile phase A (water containing 0.1% (v/v) formic acid) and mobile phase B (acetonitrile containing 0.1% (v/v) formic acid) were mixed in a linear fashion using a gradient program. The flow rate was 0.3 µL/min, and the mobile phase composition was as follows: initially at 3% B, which was held for 1 minute, then linearly increased over 74 minutes to 40% B, which was maintained for 4 minutes, then linearly increased over 1 minute to 95% B, which was maintained for 5 minutes, then linearly decreased over 1 minute to 3% B. The total runtime, including conditioning the column at the initial conditions, was 100 minutes. The eluted peptides were transferred to the nano‐electrospray source of a Synapt high‐definition Q‐TOF mass spectrometer (Waters) through a Teflon capillary union and a precut PicoTip (Waters). The system was controlled, and the mass spectral data was analyzed using MassLynx version 4.1 software (Waters). The mass spectrometer was set at electrospray ionization with a capillary voltage of 3.5 kV and a sampling cone voltage of 40 V. A low collision energy (6 eV) was used to generate intact peptide precursor ions, and an elevated collision energy (stepped from 15 to 30 eV) was used to generate peptide product ions. The source temperature was 100°C, and the detector was operated in positive ion mode. Data were collected in centroid mode, and the m/z range was 50‐1990. All analyses were acquired using an independent reference. Glu‐1‐fibrinopeptide B (m/z 785.8426), which was used as an external mass calibrant, was infused through the nanoLockSpray ion source and sampled every 10 s. Biopharmlynx version 1.2 software (Waters) was used to perform the database searches.20
2.13. Statistical analysis
Quantitative data were expressed as means ± SD. The statistical analyses were performed using one‐way or two‐way ANOVA with Tukey’s multiple comparison test and GraphPad Prism 7 software (GraphPad). Probability values at P <.05 were considered significant.
3. RESULTS
3.1. Serine phosphorylation of glycoprotein non–metastatic melanoma protein B and construction of a phosphorylation site mutant
In the present study, we investigated the role of S530 in the intracellular domain of human GPNMB isoform b in GPNMB‐induced tumorigenic abilities. This serine residue is predicted to be phosphorylated, according to data from the ELM database,17 and is conserved in both humans and mice (Figure 1A). To examine the phosphorylation of S530, we established an anti–S530 phosphorylated GPNMB (pGPNMB) antibody. This antibody could recognize GPNMB when it is transiently expressed in 293T cells. In contrast, the lambda protein phosphatase treatment reduced the antibody binding (Figure 1B). Furthermore, we confirmed the phosphorylation of S530 by ULPC‐MS analysis using the purified human GPNMB protein that is expressed in 293T cells (Figure 1C and Table 1). To investigate the impact of S530, we constructed a point mutant, GPNMB(SA). The antibody bound to GPNMB(SA) to a much lesser degree than it did to wild‐type (WT) GPNMB when they were expressed in 293T cells (Figure 1D). GPNMB(SA) overexpressing cell lines were established from a human breast cancer cell line, MCF7 (Figure 1E), and localization of GPNMB(SA) is comparable with that of GPNMB(WT) (Figure S1A). In addition, we made a mouse GPNMB(SA) mutant, which was stably expressed in the mouse mammary gland epithelial cell line, NMuMG (Figure 1F). The localization pattern was not affected by point mutation, similarly with MCF7‐GPNMB(SA) cells (Figure S1B).
FIGURE 1.

Detection of serine phosphorylation of glycoprotein non–metastatic melanoma protein B (GPNMB) and construction of a phosphorylation site mutant. A, Serine residue (S) marked with red square is conserved among species. B, 293T cells were transfected with human GPNMB(WT) and the lysates were immunoprecipitated and treated with or without lambda protein phosphatase. Immunoblotting for pGPNMB(S530) and total GPNMB was performed. C, The purified human GPNMB(WT) protein derived from transiently expressed 293T cells was digested by trypsin and analyzed using the nanoUPLC‐MS system. MSE data are shown in Table 1. D, 293T cells were transfected with human GPNMB(WT) or GPNMB(SA) mutant. Immunoblotting for pGPNMB(S530) and total GPNMB was performed. β‐actin was used as a loading control. E, Stably expressing cell lines were established from MCF7 cells. MCF7‐mock (m), MCF7‐GPNMB(WT) (clones 1 and 2, indicated as G1 and G2, respectively), and MCF7‐GPNMB(SA) (clones 17 and 19, indicated as SA17 and SA19, respectively). Immunoblotting for pGPNMB(S530) and total GPNMB was performed. β‐actin was used as a loading control. F, Stably expressing cell lines were established from NMuMG cells. NMuMG‐mock (m), NMuMG‐GPNMB(WT) (clones 3 and 8 indicated as G3 and G8), and NMuMG‐GPNMB(SA) (clones 8 and 10 indicated as SA8 and SA10). Immunoblotting for GPNMB was performed. β‐actin was used as a loading control
TABLE 1.
MSE data for phosphorylated S peptides in human GPNMB. The purified glycoprotein non–metastatic melanoma protein B (GPNMB) protein was digested by trypsin and analyzed by nanoUPLC‐MS system
| Position | Assignment | Calculated mass (Da) | Observed mass (Da) | Modifiers |
|---|---|---|---|---|
| 528‐537 | y10* | 1184.6193 | 1184.6010 | Phosphoryl S (1) |
| a2 | 143.1184 | 143.1147 | ||
| y4 | 488.2945 | 488.2970 |
3.2. GPNMB(SA) mutants impair sphere formation and tumor growth
The tumorigenic potential of GPNMB(SA) was assessed in terms of sphere formation in vitro and tumor growth in vivo. We performed sphere forming assays to investigate the impact of serine residues (S530 and S546 in human and mouse GPNMB, respectively). As shown in Figure 2A, human GPNMB(SA)‐expressing MCF7 cell lines showed significantly lower sphere‐forming ability than those of MCF7‐GPNMB(WT) cell lines. Similar results were obtained in mouse GPNMB(SA)‐expressing NMuMG cell lines (Figure 2B). To confirm the importance of serine phosphorylation of GPNMB, we constructed a point mutant to mimic serine phosphorylation by replacing the serine residue with an aspartic acid (D) residue and named this point mutant GPNMB(SD). We established stably expressing cell lines from MCF7 and NMuMG cells (Figure S2A, B). The sphere‐forming ability of GPNMB(SD)‐expressing cells was comparable with those of GPNMB(WT)‐expressing cells in the presence of EGF or FGF‐2 and significantly higher than those of GPNMB(SA)‐expressing cells in both MCF7 and NMuMG cell lines (Figure S2C, D). Moreover, GPNMB(SA) expression significantly reduced the size of tumors after we subcutaneously injected NMuMG‐mock, NMuMG‐GPNMB(WT), and NMuMG‐GPNMB(SA) cells into immunodeficient mice (Figure 2C‐E). The tumors derived from NMuMG‐GPNMB(WT) cells did not show any tubular structures, whereas those of NMuMG‐mock or NMuMG‐GPNMB(SA) cells formed differentiated tubular structures (Figure 2F). We then examined the proliferation marker, Ki‐67. Ratios of Ki‐67‐positive cells were higher in the tumor of NMuMG‐GPNMB(WT) cells than those of NMuMG‐mock or NMuMG‐GPNMB(SA) cells (Figure 2G), indicating that GPNMB(SA) expression reduces cellular proliferation in vivo. In addition, we examined the expression of the adherence molecule, E‐cadherin, given that we had previously reported that the tumors of NMuMG‐GPNMB(WT) cells have no E‐cadherin expression.9, 10 Interestingly, GPNMB(SA) expression failed to decrease E‐cadherin expression in the tumors (Figure 2H). These results indicate that the serine phosphorylation of GPNMB plays an important role in tumorigenic growth, as evidenced in sphere formation in vitro and tumor growth in vivo induced by GPNMB. In contrast, neither GPNMB(WT) nor GPNMB(SA) affected the monolayer cell proliferation of MCF7 cells and NMuMG cells (Figure S3A, B).
FIGURE 2.
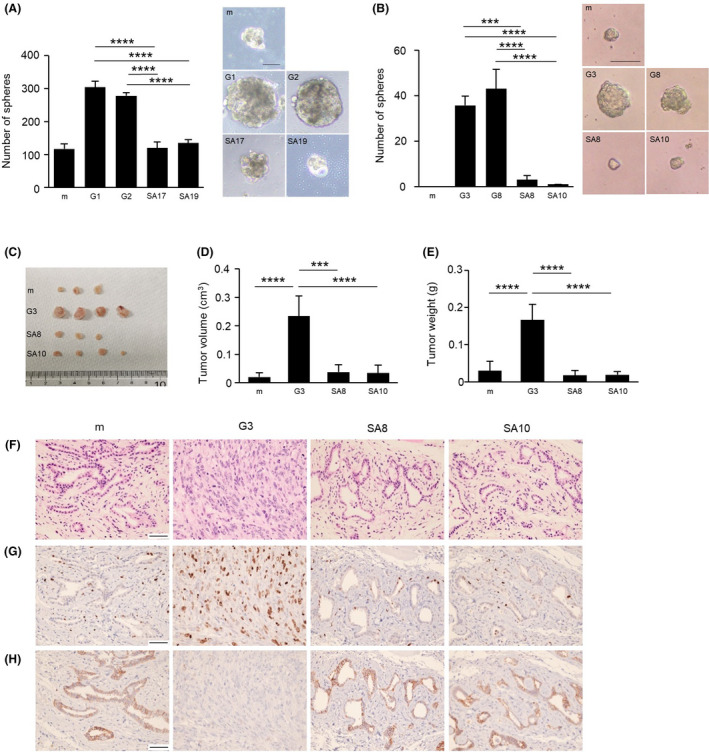
Impact of serine residue in sphere formation and tumor growth ability of glycoprotein non–metastatic melanoma protein B (GPNMB). A, Sphere formation of MCF7‐mock, MCF7‐GPNMB(WT) (G1 and G2), and MCF7‐GPNMB(SA) (SA17 and SA19) cells. Only spheres with diameter greater than 100 μm were counted. Data are means ± SD; n = 3 replicates, representative of three independent experiments. ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. Scale bar, 100 μm. B, Sphere formation of NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells. Only spheres with diameter greater than 50 μm were counted. Data are means ± SD; n = 3 replicates, representative of three independent experiments. ***P < .001, ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. Scale bar, 100 μm. C‐E, Tumor growth of NMuMG‐mock, NMuMG‐GPNMB(WT) (G3), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells was examined by subcutaneous injection into immunodeficient mice. Data are presented as mean ± SD; n = 4. ***P < .001, ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. F‐H, Histological analysis of xenograft tumors using different types of staining: H&E staining (F), immunostaining with anti–Ki‐67 antibody (G), and immunostaining with anti–E‐cadherin antibody (H). Scale bar, 50 μm. GPNMB, glycoprotein non–metastatic melanoma protein B
3.3. GPNMB(SA) mutant decreases expression levels of stemness‐related genes
In our previous study, we reported that 3D sphere‐cultured cells show higher mRNA expression levels of stemness‐related genes.14 Here, we cultured MCF7‐mock, MCF7‐GPNMB(WT), and MCF7‐GPNMB(SA) cells under 2D monolayer or 3D sphere culture condition to compare the mRNA expression levels of stemness‐related genes, such as SOX2, NANOG, OCT4, and CD44. The expression level of each of three of these genes (SOX2, OCT4, and CD44) was enhanced in the 3D‐cultured spheres of MCF7‐GPNMB(WT) cells compared to those in the 2D monolayer cultured cells. The exception was NANOG, which was highly induced, even in MCF7‐mock cells. By contrast, MCF7‐GPNMB(SA) cells had lower expression levels for stemness‐related genes, even in 3D cultured spheres (Figure 3A‐D). Similar results were obtained in NMuMG‐GPNMB(WT) and NMuMG‐GPNMB(SA) cells (Figure 3E‐H). These results indicate that the serine residue in the intracellular domain of GPNMB is essential for the induction of stemness‐related genes in the 3D‐cultured spheres. Taken together, our current results suggest that the GPNMB‐induced tumorigenic potential is dependent on the serine phosphorylation of GPNMB, without which the GPNMB cannot induce cancer stem‐like properties.
FIGURE 3.
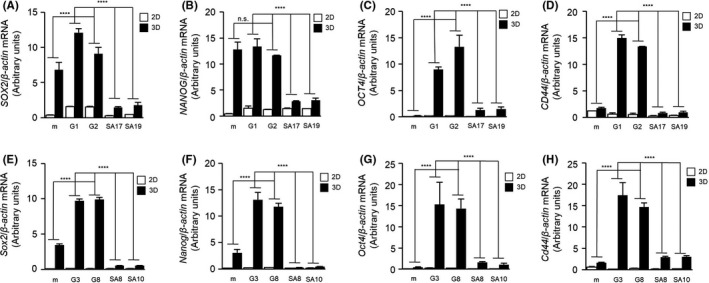
Expression levels of stemness‐related genes in 2D‐cultured or 3D‐cultured cells. A‐H, mRNA expression levels of SOX2 (A, E), NANOG (B, F), OCT4 (C, G), and CD44 (D, H) in 2D monolayer cultured (2D) or 3D sphere cultured (3D) cells of MCF7‐mock, MCF7‐GPNMB(WT) (G1 and G2), MCF7‐GPNMB(SA) (SA17 and SA19) (A‐D), NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and NMuMG‐GPNMB(SA) (SA8 and SA10) (E‐H) were analyzed by quantitative PCR and normalized to those levels of β‐actin. Data are presented as mean ± SD; n = 3. n.s., not significant, ****P < .0001, two‐way ANOVA with Tukey’s multiple comparison test. Data are representative of three independent replicates. GPNMB, glycoprotein non–metastatic melanoma protein B
3.4. GPNMB(SA) fails to induce epithelial‐mesenchymal transition in NMuMG cells
NMuMG cells are frequently used to examine the EMT, and we have previously shown that GPNMB(WT) induces EMT phenotypic changes in NMuMG cells.9 In addition, as shown in Figure 2H, the remaining expression of E‐cadherin in the tumors is derived from NMuMG‐GPNMB(SA) cells. Therefore, we evaluated the EMT phenotypic changes, such as downregulation of the epithelial marker E‐cadherin, upregulation of the mesenchymal marker fibronectin, and cellular migration in NMuMG‐GPNMB(SA) cell lines. GPNMB(SA)‐expressing cells failed to suppress both mRNA and protein expression levels of E‐cadherin and enhance fibronectin (Figure 4A‐D), while GPNMB(SD) suppressed E‐cadherin protein expression in NMuMG cells (Figure S2B). It seems that GPNMB(SA) even enhances the expression of E‐cadherin compared to that of NMuMG‐mock cells (Figure 4A, C, D). Furthermore, the result of the Transwell migration assay showed that GPNMB(SA) impairs cellular migration in contrast to GPNMB(WT) in NMuMG and MCF7 cells (Figure 4E and S4). In addition, the mRNA expression levels of EMT‐related transcription factor genes, such as Snail, Slug, and Zeb1, in NMuMG‐GPNMB(SA) cells were lower than levels in NMuMG‐GPNMB(WT) cells (Figure 4F‐H). These results indicate that the serine residue in GPNMB is important to induce EMT in NMuMG cells.
FIGURE 4.
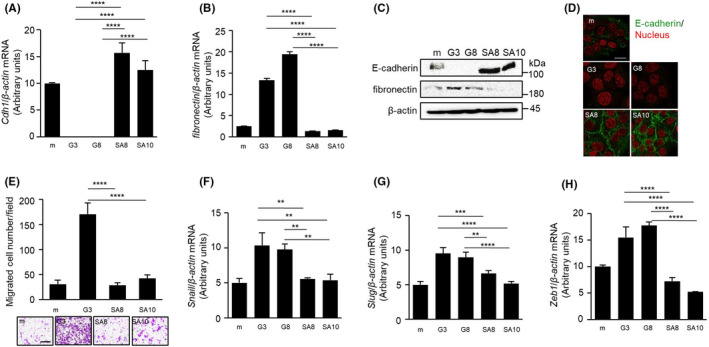
Importance of the serine residue in epithelial‐mesenchymal transition induction and cell migration of glycoprotein non–metastatic melanoma protein B (GPNMB) protein. A,B, mRNA expression levels of Cdh1 (A) and fibronectin (B), in NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells were measured by quantitative PCR (qPCR) and normalized to those levels of β‐actin. Data are presented as mean ± SD; n = 3. ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. C, Immunoblotting for E‐cadherin and fibronectin in NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and GPNMB(SA) (SA8 and SA10) cells was performed. β‐actin was used as a loading control. D, Immunofluorescence staining of E‐cadherin (green) in NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells. Nuclei were counterstained with propidium iodide (red). Scale bar, 20 μm. E, Migration of NMuMG‐mock, NMuMG‐GPNMB(WT) (G3), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells was examined by Transwell migration assay. Data are presented as mean ± SD, n = 3 replicates, representative of three independent experiments. ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. Scale bar, 100 μm. F‐H, mRNA expression levels of Snail (F), Slug (G), and Zeb1 (H) in NMuMG‐mock, NMuMG‐GPNMB(WT) (G3 and G8), and NMuMG‐GPNMB(SA) (SA8 and SA10) cells were analyzed by qPCR and normalized to those levels of β‐actin. Data are presented as mean ± SD; n = 3. **P < .01, ***P < .001, ****P < .0001, one‐way ANOVA with Tukey’s multiple comparison test. Data are representative of three independent replicates
3.5. Growth factor signaling induces the serine phosphorylation of glycoprotein non–metastatic melanoma protein B
To investigate the mechanism of S530 phosphorylation, we focused on growth factors, namely EGF and FGF‐2, because these growth factors are supplemented in the sphere culture medium and GPNMB(WT) showed comparable induction of sphere‐forming ability to that of GPNMB(SD) mutation; however GPNMB(SA) mutation significantly attenuated sphere formation in both MCF7 and NMuMG cell lines (Figure 2A, B and S2C, D). 293T cells were transfected with human GPNMB(WT) and treated with EGF or FGF‐2 for the indicated time periods. The level of pGPNMB(S530) increased gradually depending on the treatment time (Figure 5A, B). In contrast, the phosphorylation level of GPNMB(SA) was not enhanced by FGF‐2 and, in the case of a FGFR downstream molecule, ERK1/2, phosphorylation was observed (Figure S5). As shown in Figure 5C and D, phosphorylation and total protein levels of endogenous GPNMB in the human breast cancer cell line, MDA‐MB‐231, were slightly increased by EGF and FGF‐2 treatment. These results suggest that receptor tyrosine kinase (RTK) signaling is involved in the serine phosphorylation of GPNMB. Therefore, we co–transfected the downstream oncoproteins RAS, RAF, MEK1, and ERK2 and their constitutive active forms with GPNMB(WT) in 293T cells and investigated which molecule is responsible for GPNMB(S530) phosphorylation. We found that constitutive active forms of RAS(G12V) and RAF(V600E) further enhanced the pGPNMB(S530) levels, although expression of GPNMB(WT) alone yields GPNMB(S530) phosphorylation (Figure 5E). Furthermore, both phosphorylation and total protein levels of endogenous GPNMB were slightly increased when MDA‐MB‐231, Hs578T, MDA‐MB‐157, and BT‐474 cells were transiently transfected with RAS(G12V) (Figure 5F). Although further investigations are required to understand the mechanism, total protein levels of GPNMB are increased by RAS activation or the phosphorylated GPNMB protein is more stable than the non–phosphorylated GPNMB protein. These results suggest that RTK signaling induces serine phosphorylation of GPNMB, and its downstream molecules, RAS, and RAF, are responsible for this process.
FIGURE 5.
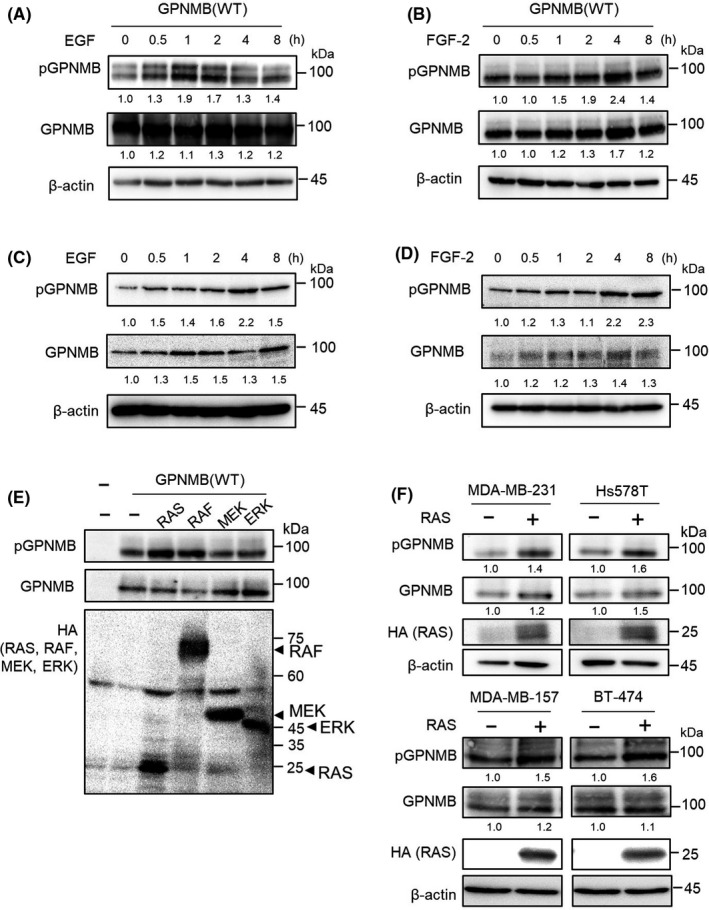
Effect of tyrosine kinase receptor signaling on the serine phosphorylation of glycoprotein non–metastatic melanoma protein B (GPNMB) protein. A,B, 293T cells were transfected with human GPNMB(WT) and treated with 10 ng/mL of EGF (A) or 10 ng/mL of FGF‐2 (B), for the indicated time periods. Immunoblotting for pGPNMB(S530) and total GPNMB was performed. β‐actin was used as a loading control. Intensities of the bands are quantified by Image J and normalized to those intensities of β‐actin. C,D, MDA‐MB‐231 cells were treated with 10 ng/mL of EGF (C) or 10 ng/mL of FGF‐2 (D) for the indicated time periods. Immunoblotting for pGPNMB(S530) and total GPNMB was performed. β‐actin was used as a loading control. Intensities of the bands were quantified by Image J and normalized to those intensities of β‐actin. E, 293T cells were transfected with HA‐tagged RAS (G12V), BRAF (V600E), MEK1 (S218D/S222D), and ERK2 (L73P/S151D). Immunoblotting for pGPNMB (S530), total GPNMB, and HA was performed. F, MDA‐MB‐231, Hs578T, MDA‐MB‐157, and BT‐474 cells were transfected with HA‐RAS (G12V). Immunoblotting for pGPNMB(S530), total GPNMB, and HA was performed. β‐actin was used as a loading control. Intensities of the bands are quantified by Image J and normalized to those intensities of β‐actin
4. DISCUSSION
In this study, we demonstrated that the serine residue in the intracellular domain of GPNMB is critical for tumorigenic potential, as indicated by sphere formation in vitro, tumor formation in vivo, and the enhancement of stemness‐related gene expression (Figures 2, 3 and S2). Furthermore, we showed that the serine residue is important for EMT induction and cellular migration mediated by GPNMB (Figure 4 and S4). In addition, we found that this serine phosphorylation is induced by activation of the RTK signaling upon EGF and FGF‐2 stimulation through the activation of downstream molecules; namely, RAS and RAF (Figure 5 and S5).
Protein phosphorylation, which occurs mainly on serine, threonine, and tyrosine residues, is one of the post‐translational modifications of proteins and affects the activity of the protein. We previously showed that the tyrosine residue in the intracellular domain of GPNMB is important for its tumorigenic activity.9, 14, 15 Moreover, Lin et al demonstrated the clinical significance of this tyrosine residue in breast cancer patients. It has been reported that the ELM database predicts several functional motifs in the intracellular domain of GPNMB, including one phosphorylation site in GPNMB.17 However, reliable experimental evidence of serine phosphorylation of GPNMB is lacking, and the present study is the first to report that S530 in human GPNMB isoform b and S546 in mouse GPNMB are phosphorylated and affect tumorigenic potential.
RTK signaling is activated upon ligand stimulation, and intracellular signaling pathways become active and regulate multiple cellular processes, such as cell survival, proliferation, and cellular migration and invasion.21, 22, 23 We focused on EGF and FGF‐2, which are supplemented in the sphere culture medium, and showed that EGF and FGF‐2 stimulation enhances pGPNMB(S530). The data shown indicate that EGF and FGF‐2 induce pGPNMB(S530) through activation of RAS and RAF but not MEK1 and ERK2 (Figure 5). These findings suggest that EGF and FGF‐2 stimulate the serine phosphorylation of GPNMB in sphere culture condition and are associated with the transcription of stemness‐related genes, such as SOX2, NANOG, OCT4, and CD44 (Figure 3). We know that one of the signaling pathways downstream of RTK, RAS/RAF/MEK/ERK, which is considered a classic MAPK pathway, regulates cell proliferation, is involved in stemness regulation and tumor growth, and also regulates drug resistance in cancers24, 25, 26, 27. Although RAS activation caused by genetic mutations is frequently found in several cancers, including lung, colorectal, and pancreatic cancers, as well as melanoma, the RAS mutation rate in breast cancers is not high. The frequency of mutations in RAS family proteins is less than 8% in breast cancer patients.28, 29, 30 In this study, we used MDA‐MB‐231 cells with KRAS and BRAF mutations (Figure 5C, D, F) and Hs578T with HRAS mutation (Figure 5F).31, 32, 33, 34 pGPNMB(S530) is present without any stimuli; however, we found further enhancement of phosphorylation levels and total GPNMB when we treated cells with EGF or FGF‐2 or overexpressed HRAS(G12V) in MDA‐MB‐231 and Hs578T cells. Furthermore, pGPNMB(S530) was induced by exogenous HRAS(G12V) overexpression in MDA‐MB‐157 and BT‐474 cells with wild‐type endogenous RAS and RAF (Figure 5F). These results suggest that RTK signaling through RAS activation induces GPNMB(S530) phosphorylation in breast cancer cell lines. Previously, Rose et al (2017) demonstrated that inhibition of the MAPK signaling pathway increases GPNMB protein expression in several melanoma cell lines with NRAS or BRAF mutations because BRAF and MEK inhibitors increase the expression of the microphthalmia‐associated transcription factor (MITF), resulting in enhancement of GPNMB expression.35 Together with our current results, the MAPK signaling pathway might be involved in the regulation of both GPNMB protein amounts and phosphorylation levels in different types of cancer.
The phosphorylation site of phosphorylase kinase (PHK) has been identified as (R/K) XXS (V/I) XX, where X is any residue phosphorylation site and is found in both human and mouse GPNMB (Figure 1A). PHK is a first characterized kinase, which regulates glycogen phosphorylase to release glucose‐1‐phosphate from glycogen and is a substrate of cAMP‐dependent protein kinase (protein kinase A or PKA).36, 37 One of its regulatory subunits, the β subunit (PHKB), has been reported to promote cell survival of colorectal cancer cells due to the regulation of glycogen metabolism and has been shown to be a prognostic factor in colorectal cancer patients.38, 39 In hepatocellular carcinoma, in contrast, PHKB inhibits cellular proliferation and tumor growth in vivo through suppression of AKT and STAT3 signaling pathways.40 The role of PHK in breast cancer has not been reported on, but it would be interesting to examine the involvement of PHK in GPNMB serine phosphorylation and tumorigenesis. Furthermore, we identified phosphorylated S519 in human GPNMB isoform b through UPLC‐MSE analysis (Figure S6 and Table S2). Therefore, it would be interesting to examine the effect of this S519 on GPNMB tumorigenic potential.
Protein phosphorylation is involved in many biological processes and is highly activated, especially in cancer cells.41, 42, 43 Therefore, forms of protein kinase are the most important therapeutic targets in cancer treatment; many FDA‐approved drugs have been developed on this basis, such as, imatinib, crizotinib, erlotinib, gefitinib, and vemurafenib.37, 44 However, resistance to those drugs is among the biggest problems to solve.37, 45 The potential of GPNMB as a therapeutic target has been assessed and demonstrated in several types of cancers, including breast cancer by using glembatumumab vedotin, an antibody against human GPNMB conjugated with an anticancer drug (monomethyl auristatin E).6, 7, 8 In addition, based on the findings in the present study, we believe that blockage of the S530 of GPNMB, which is downstream of RTK signaling, is a potential therapeutic strategy, instead of targeting protein kinase itself. Because GPNMB demonstrates essential tumorigenic function in growth‐arrested cancer cells, there may be less risk of generating drug‐resistant mutations.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Fig S1‐S6
Table S1‐S2
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by JSPS KAKENHI Grant Numbers JP17K14981, JP19K07658 (to YO), JP18H02676, and JP20K20597 (to MK).
Wang C, Okita Y, Zheng L, et al. Glycoprotein non–metastatic melanoma protein B functions with growth factor signaling to induce tumorigenesis through its serine phosphorylation. Cancer Sci. 2021;112:4187–4197. 10.1111/cas.15090
REFERENCES
- 1.Bray F, McCarron P, Parkin DM. The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 2004;6:229‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52‐62. [DOI] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 4.Zhou LT, Liu FY, Li Y, Peng YM, Liu YH, Li J. Gpnmb/osteoactivin, an attractive target in cancer immunotherapy. Neoplasma. 2012;59:1‐5. [DOI] [PubMed] [Google Scholar]
- 5.Maric G, Rose AA, Annis MG, Siegel PM. Glycoprotein non–metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco Targets Ther. 2013;6:839‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rose AAN, Biondini M, Curiel R, Siegel PM. Targeting GPNMB with glembatumumab vedotin: Current developments and future opportunities for the treatment of cancer. Pharmacol Ther. 2017;179:127‐141. [DOI] [PubMed] [Google Scholar]
- 7.Taya M, Hammes SR. Glycoprotein non–metastatic melanoma protein B (GPNMB) and cancer: a novel potential therapeutic target. Steroids. 2018;133:102‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tray N, Adams S, Esteva FJ. Antibody‐drug conjugates in triple negative breast cancer. Future Oncol. 2018;14:2651‐2661. [DOI] [PubMed] [Google Scholar]
- 9.Okita Y, Kimura M, Xie R, et al. The transcription factor MAFK induces EMT and malignant progression of triple‐negative breast cancer cells through its target GPNMB. Sci Signal. 2017;10:eaak9397. 10.1126/scisignal.aak9397 [DOI] [PubMed] [Google Scholar]
- 10.Xie R, Okita Y, Ichikawa Y, et al. Role of the kringle‐like domain in glycoprotein NMB for its tumorigenic potential. Cancer Sci. 2019;110:2237‐2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128‐134. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21:341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. 2021;21:325‐338. 10.1038/s41568-021-00332-6 [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Okita Y, Watanabe Y, et al. Glycoprotein nmb is exposed on the surface of dormant breast cancer cells and induces stem cell‐like properties. Cancer Res. 2018;78:6424‐6435. [DOI] [PubMed] [Google Scholar]
- 15.Okita Y, Chen C, Kato M. Cell‐surface GPNMB and induction of stemness. Oncotarget. 2018;9:37289‐37290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puntervoll P, Linding R, Gemund C, et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31:3625‐3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selim AA. Osteoactivin bioinformatic analysis: prediction of novel functions, structural features, and modes of action. Med Sci Monit. 2009;15:MT19‐33. [PubMed] [Google Scholar]
- 18.Asada S, Daitoku H, Matsuzaki H, et al. Mitogen‐activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519‐527. [DOI] [PubMed] [Google Scholar]
- 19.Hu J, Stites EC, Yu H, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinkai Y, Ding Y, Miura T, Kumagai Y. Aggregation of β‐crystallin through covalent binding to 1,2‐naphthoquinone is rescued by α‐crystallin chaperone. J Toxicol Sci. 2020;45:37‐43. [DOI] [PubMed] [Google Scholar]
- 21.Lin A, Li C, Xing Z, et al. The LINK‐A lncRNA activates normoxic HIF1α signalling in triple‐negative breast cancer. Nat Cell Biol. 2016;18:213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211‐225. [DOI] [PubMed] [Google Scholar]
- 23.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riverso M, Montagnani V, Stecca B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene. 2017;36:3322‐3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan LH, Zhou L, Ng KY, et al. PRMT6 regulates RAS/RAF binding and MEK/ERK‐mediated cancer stemness activities in hepatocellular carcinoma through CRAF methylation. Cell Rep. 2018;25:e698. [DOI] [PubMed] [Google Scholar]
- 27.Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 2020;9:198. 10.3390/cells9010198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira CB, Leal MF, de Souza CR, et al. Prognostic and predictive significance of MYC and KRAS alterations in breast cancer from women treated with neoadjuvant chemotherapy. PLoS ONE. 2013;8:e60576. 10.1371/journal.pone.0060576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraus MH, Yuasa Y, Aaronson SA. A position 12‐activated H‐ras oncogene in all HS578T mammary carcinosarcoma cells but not normal mammary cells of the same patient. Proc Natl Acad Sci USA. 1984;81:5384‐5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozma SC, Bogaard ME, Buser K, et al. The human c‐Kirsten ras gene is activated by a novel mutation in codon 13 in the breast carcinoma cell line MDA‐MB231. Nucleic Acids Res. 1987;15:5963‐5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949‐954. [DOI] [PubMed] [Google Scholar]
- 34.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple‐negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose AAN, Annis MG, Frederick DT, et al. MAPK pathway inhibitors sensitize BRAF‐mutant melanoma to an antibody‐drug conjugate targeting GPNMB. Clin Cancer Res. 2016;22:6088‐6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor SS, Yang J, Wu J, Haste NM, Radzio‐Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259‐269. [DOI] [PubMed] [Google Scholar]
- 37.Roskoski R Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1‐23. [DOI] [PubMed] [Google Scholar]
- 38.Terashima M, Fujita Y, Togashi Y, et al. KIAA1199 interacts with glycogen phosphorylase kinase β‐subunit (PHKB) to promote glycogen breakdown and cancer cell survival. Oncotarget. 2014;5:7040‐7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Shen W, Liu CY, et al. Phosphorylase kinase β affects colorectal cancer cell growth and represents a novel prognostic biomarker. J Cancer Res Clin Oncol. 2017;143:971‐980. [DOI] [PubMed] [Google Scholar]
- 40.Yang W, Zhang C, Li Y, et al. Phosphorylase kinase β represents a novel prognostic biomarker and inhibits malignant phenotypes of liver cancer cell. Int J Biol Sci. 2019;15:2596‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams‐Ashman HG, Kennedy EP. Oxidative phosphorylation catalyzed by cytoplasmic particles isolated from malignant tissues. Cancer Res. 1952;12:415‐421. [PubMed] [Google Scholar]
- 42.Kennedy EP, Smith SW. The isolation of radioactive phosphoserine from phosphoprotein of the Ehrlich ascites tumor. J Biol Chem. 1954;207:153‐163. [PubMed] [Google Scholar]
- 43.Singh V, Ram M, Kumar R, Prasad R, Roy BK, Singh KK. Phosphorylation: implications in cancer. Protein J. 2017;36:1‐6. [DOI] [PubMed] [Google Scholar]
- 44.Roskoski R Jr. Properties of FDA‐approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res. 2020;152:104609. 10.1016/j.phrs.2019.104609 [DOI] [PubMed] [Google Scholar]
- 45.Barouch‐Bentov R, Sauer K. Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs. 2011;20:153‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1‐S2
Supplementary Material