

Abstract
Evi1 is a transcription factor essential for the development as well as progression of acute myeloid leukemia (AML) and high Evi1 AML is associated with extremely poor clinical outcome. Since targeting metabolic vulnerability is the emerging therapeutic strategy of cancer, we herein investigated a novel therapeutic target of Evi1 by analyzing transcriptomic, epigenetic, and metabolomic profiling of mouse high Evi1 leukemia cells. We revealed that Evi1 overexpression and Evi1‐driven leukemic transformation upregulate transcription of gluconeogenesis enzyme Fbp1 and other pentose phosphate enzymes with interaction between Evi1 and the enhancer region of these genes. Metabolome analysis using Evi1‐overexpressing leukemia cells uncovered pentose phosphate pathway upregulation by Evi1 overexpression. Suppression of Fbp1 as well as pentose phosphate pathway enzymes by shRNA‐mediated knockdown selectively decreased Evi1‐driven leukemogenesis in vitro. Moreover, pharmacological or shRNA‐mediated Fbp1 inhibition in secondarily transplanted Evi1‐overexpressing leukemia mouse significantly decreased leukemia cell burden. Collectively, targeting FBP1 is a promising therapeutic strategy of high Evi1 AML.
Keywords: acute myeloid leukemia, gluconeogenesis, oncogene, pentose phosphate pathway, transcription factors
The activated pentose phosphate pathway enhances progression of Evi1‐overexpressing leukemia. Fbp1 and other glucose metabolism‐related genes cooperatively upregulate pentose phosphate pathway activity.

Abbreviations
- EVI1
ecotropic virus integration site 1
- F1,6P
fructose 1,6‐bisphosphate
- F6P
fructose 6‐phosphate
- FBP1
fructose‐1,6‐bisphosphatase 1
- G6P
glucose‐6‐phosphate
- GA3P
glyceraldehyde 3‐phosphate
- GATA2
GATA binding protein 2
- Hk3
hexokinase 3
- HSC
hematopoietic stem cell
- IDH1
isocitrate dehydrogenase 1
- IDH2
isocitrate dehydrogenase 2
- JUN
jun proto‐oncogene
- MECOM
myelodysplasia syndrome 1 and ecotropic virus integration site 1 complex locus
- OXPHOS
oxidative phosphorylation
- 3‐PG
3‐phosphoglyceric acid
- Pgam1
phosphoglycerate mutase 1
- Pgd
phosphogluconate dehydrogenase
- R5P
Ribose 5‐phosphate
- Rpia
ribose 5‐phosphate isomerase A
- SPI1
spleen focus forming virus proviral integration oncogene
- Taldo1
transaldolase 1
1. INTRODUCTION
Acute myeloid leukemia (AML) is a hematological malignancy characterized by differentiation arrest and clonal expansion of immature hematopoietic progenitor or stem cells mainly in bone marrow and peripheral blood.1 Its prognosis is highly heterogeneous depending on its molecular profiles.2, 3 A number of genetic abnormalities and accompanying transcriptomic characteristics are related to poor prognostic AML, which is resistant to current standard chemotherapy. Deregulated expression of ecotropic viral integration site 1 (Evi1) occurs in approximately 10% of AML and is associated with extremely poor prognosis.4, 5, 6, 7 Evi1 is encoded by the MECOM gene located on human chromosome 3q26 and the commonly found chromosomal alteration t(3:3)(q21:q26)/inv(3)(q21q26) causes GATA2 enhancer‐mediated Evi1 overexpression in AML.8, 9 Evi1 is a nuclear transcription factor that is indispensable for physiological functions of normal hematopoietic stem cells (HSCs), and it also contributes to leukemogenesis by multiple mechanisms such as suppressing transforming growth factor‐β signaling, inhibiting c‐Jun N‐terminal kinase, activating activator protein‐1, upregulating mammalian target of rapamycin signaling pathway through phosphatase and tensin homologue deleted from chromosome 10 repression, and inducing PU.1 mediated myeloid skewing through Spi1 upregulation.10, 11, 12, 13, 14, 15, 16, 17 The indispensability of Evi1 in the hematopoietic system makes it difficult to target Evi1 as a therapy of Evi1high AML. Although many transcriptional targets of Evi1 have been found, novel specific therapies of this AML subtype have not been established so far.18, 19, 20, 21
Recent investigations identified that cancer cells have cancer‐type specific metabolic vulnerabilities as a result of dysregulated transcriptomic alteration. Isocitrate dehydrogenase 1/2(IDH1/2) mutations in AML lead to the aberrant generation of oncometabolite R‐2HG, which causes aberrant epigenetic modification resulting in transcriptional upregulation of oncogenes.22, 23, 24 IDH1/2 inhibition decreases R‐2HG and ameliorates its oncogenic phenotypes. MLL‐rearranged leukemia cells are dependent on glycolysis in its leukemia initiation.25 In this subtype of AML, inhibition of glycolytic enzymes effectively decreases leukemia cells. Metabolome and transcriptome analysis of Evi1‐overexpressing mouse hematopoietic cells revealed that Evi1 induces metabolic vulnerability due to dependence on creatine kinase pathway and glutaminolysis.26, 27 Although these studies demonstrated novel therapeutic possibilities of Evi1high AML, the metabolic features of Evi1high AML were not fully understood. Since targeting the cancer‐dependent metabolic pathway is the emerging therapeutic strategy, identifying novel metabolic features is still of great interest.
Herein we sought to clarify specific molecular features of AML with high Evi1 expression by using an Evi1‐overexpressing leukemia mouse model that we had previously established.16 RNA‐sequencing (RNA‐seq) analysis of Evi1‐overexpressing pre‐ and post‐leukemia cells revealed transcriptional deregulation of Fbp1 and multiple pentose phosphate pathway enzymes. Furthermore, we showed that Evi1 overexpression‐mediated metabolic alteration in which the pentose phosphate pathway and oxidative phosphorylation are upregulated become crucial metabolism pathways in Evi1‐overexpessing cells. Taken together, these results provide a potential therapeutic target of AML with high Evi1 expression.
2. MATERIALS AND METHODS
2.1. Subjects
Studies using human subjects were done in accordance with the ethical guidelines for medical and health research involving human subjects, which were developed by the Japanese Ministry of Health, Labour and Welfare and the Japanese Ministry of Economy, Trade and Industry, and enforced on 22 December 2014. This study was approved by the ethical committee of the University of Tokyo. Written informed consent was obtained from all patients whose samples were collected after the guidelines were enforced in accordance with the Declaration of Helsinki. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Tokyo.
2.2. Plasmids
pMYs‐human Evi‐1‐internal ribosome entry site (IRES)‐green fluorescent protein (GFP) vector was produced by cloning human Flag‐tagged Evi1 cDNA into the multiple cloning site of pMYs‐IRES‐GFP. To obtain short hairpin RNA (shRNA) for silencing the target Fbp1, G6pd, Pgd, and Rpia genes, oligonucleotides were inserted into the RNAi‐Ready pSIREN‐RetroQ‐DsRed Vector or RNAi‐Ready pSIREN‐puro Vector (Clontech). Target sequences of shRNAs were selected by using Clontech RNAi Target Sequence Selector and are described separately (Table S1). Luciferase shRNA Annealed Oligonucleotide (Clontech) was used as a control.
2.3. Retrovirus production and transduction
To produce retroviruses, Plat‐E packaging cells were transiently transfected with 12 µg of each retrovirus vector mixed with 96 µL of PEI and 500 µL of 150 mmol/L NaCl, followed by incubation at 37℃. Culture medium was replaced 12 hours after transfection. The retrovirus‐containing supernatant was collected 24 hours after medium change, filtered through 0.45 µm membrane, and added to the culture plate coated with RetroNectin (Takara Bio). The culture plate was centrifuged at 2000 × g, 37℃ for 4 hours and supernatant was discarded. Cells were seeded onto the virus‐binding plate and infected with retroviruses for 48 hours.
2.4. Isolation of murine bone marrow cells
C57BL/6 mice were sacrificed by cervical dislocation. Femora, tibiae, and ilia were excised, cleaned of attached mouse tissue, and stored on ice in PBS. After crushing these bones in PBS, bone marrow cells in supernatant were collected through 70 µm EASY strainerTM (Greiner Bio One), centrifuged at 250 x g for 5 minutes, resuspended in 4 mL of cold PBS, and carefully layered onto Histopaque‐1077 (Sigma‐Aldrich) in a 15 mL conical centrifuge tube. After centrifugation at 400 × g for 30 minutes at room temperature, a opaque interface containing mononuclear cells was added to 10 mL of PBS, and centrifuged at 250 × g for 5 minutes.
2.5. Quantitative real‐time PCR
Total RNA was isolated by using a NucleoSpin kit (Takara Bio) and the cDNA was synthesized using ReverTra Ace (Toyobo). Quantitative real‐time PCR (qPCR) was performed by using THUNDERBIRD qPCR Mix (Toyobo), with a LightCycler 480 System (Roche Applied Science). All procedures were performed according to the manufacturer's instructions. The results were normalized to the expression levels of 18s rRNA. PCR primers used for qPCR were described separately (Table S2).
2.6. Flow cytometry
Cell sorting and analysis were performed by using FACS AriaIII (BD Biosciences) and FACS AriaII (BD Biosciences). The data were analyzed using FACSDiva software (BD Biosciences) and FlowJo software (FlowJo LLC). To stain cells, 0.5‐1 µL of each antibody was added to 100 µL of isolated mononucleated cell suspended in PBS with 3% FCS and incubated for 30 minutes on ice. Antibodies are listed in Table S3. To detect apoptotic cells, 0.5 µL of 20 mg/ml 4ʹ,6‐diamidino‐2‐phenylindole (DAPI) (Sigma‐Aldrich) was added to each sample.
2.7. Evi1‐overexpressing leukemia mouse model
Lineage marker negative (KSL) cells were precultured from one day before transduction in α‐MEM (FUJIFILM Wako Pure Chemical Corporation) supplemented with 20% FCS, 1% penicillin‐streptomycin, 40 ng/mL SCF, 10 ng/mL IL‐3, 20 ng/mL IL‐6, 20 ng/mL TPO, and 20 ng/mL Flt3‐ligand and subjected to retroviral transduction with pMYs‐Evi1‐IRES‐GFP or pMYs‐mock‐IRES‐GFP vector. The infected cells were injected through the tail vein into sublethally irradiated (5.25 Gy) syngeneic recipient mice. After primary recipients developed leukemia, recipient mice were sacrificed and mononuclear cells of bone marrow and spleen were cryopreserved. For secondary bone marrow transplantation, approximately 5 × 104 leukemia cells were intravenously injected to sublethally irradiated (5.25 Gy) syngeneic recipient mice.
2.8. shRNA‐mediated knockdown and colony forming cell assay
Retroviral transduction was performed as described in the section above. When pSIREN‐RetroQ‐puro vector was used these cells were further cultured in medium containing 1.5 µg/mL puromycin for 24 hours. Puromycin‐resistant cells were used for colony‐forming cell assays. When pSIREN‐RetroQ‐DsRed vector was used, sorted DsRed positive cells were used for colony‐forming cell assays. Sorted cells (2000 cells) in methylcellulose Methocult GF M3434 medium (Stem Cell Technologies) supplemented with 1% penicillin‐streptomycin and 1.5 µg/mL puromycin if pSIREN‐RetroQ‐puro vector was used and were plated in duplicate in 35 mm dishes and incubated at 37℃, 5% CO2. The number of colonies was scored after 7 days and 2000 cells recovered from the cultured plates were replated every 7 days.
2.9. shRNA‐mediated knockdown of Fbp1 in Evi1 leukemia cells
Evi1‐overexpressing leukemia mice bone marrow GFP‐positive cells were sorted and precultured from 1 day before transduction in α‐MEM supplemented with 20% FCS, 1% penicillin‐streptomycin, 40 ng/mL SCF, 10 ng/mL IL‐3, 20 ng/mL IL‐6, 20 ng/mL TPO, and 20 ng/mL Flt3‐ligand and subjected to retroviral infection. After 48 hours of culture on a retrovirus‐coated plate, sorted cells were used for bone marrow transplantation assays.
2.10. Fbp1 inhibitor administration in vitro and in vivo
In the colony‐forming cell assay, CAY18860 (Cayman Chemical) dissolved in DMSO was added to M3434 (Stem Cell Technologies). CAY18860 dissolved in peanut oil (Sigma‐Aldrich) with 5% DMSO (Sigma‐Aldrich) was intraperitoneally administrated to Evi1 leukemia mice daily (100 µg/mouse) from the 21st day to the 27th day after bone marrow transplantation.
2.11. RNA‐sequencing analysis
Total RNA was isolated from GFPpos, lineageneg, c‐kitpos Evi1‐overexpressing or the control GFPpos, lineageneg, c‐kitpos bone marrow mononuclear cells by using a NucleoSpin kit (Takara Bio). The quality of the RNA samples (RNA Integrity Number >8) was validated using a Agilent 2100 Bioanalyzer (Agilent Technology). For RNA library preparation, a NEB Next Ultra RNA Library Prep Kit (New England Biolabs) was used. RNA library samples were sequenced according to the manufacturer's protocol using an Illumina Hiseq 2000 sequencer (Illumina). The sequence data obtained were analyzed using CLC Genomics Workbench software (Qiagen) and R software (http://www.R‐project.org).
2.12. Chromatin immunoprecipitation and sequencing
Detailed protocols for chromatin immunoprecipitation assays are presented in the supplemental Methods. Briefly, immunoprecipitated DNA fragments were diluted and quantified by next‐generation sequencing performed by Illumina Hiseq 2500. The antibody used in chromatin immunoprecipitation (ChIP) assays was monoclonal Anti‐FLAG M2 antibody (F3165; Sigma‐Aldrich). The data obtained were aligned using Bowtie2 and visualized using an Integrative Genomics Viewer.28, 29 Calling ChIP‐seq peaks was done using Macs2.30 Motif analysis was conducted using HOMER v4.31
2.13. Metabolome analysis
Metabolite extraction was performed following the Human Metabolome Technologies (HMT) manual. Briefly, ME‐1 cells were washed and treated with methanol. After centrifugation, supernatant was filtered through a Millipore 5‐kDa cutoff filter (Ultrafree MC‐PLHCC, HMT). The filtrate was centrifugally concentrated and re‐suspended in Milli‐Q water for metabolome analysis Metabolome analysis was conducted by the C‐SCOPE package of HMT using capillary electrophoresis time‐of‐flight mass spectrometry for cation analysis and CE‐tandem mass spectrometry for anion analysis based on the methods described previously.32, 33 Peaks were extracted using MasterHands (Keio University, Japan)34 and Mass Hunter Quantitative Analysis B.04.00 (Agilent Technologies) to obtain information for peaks. The detected peak area of each metabolite was normalized to that of the internal standard. For more details, see supplemental Methods.
2.14. Extracellular metabolic flux analysis
A cell mito stress test and a glycolysis stress test were performed with a Seahorse XFe24 Analyzer (Agilent Technologies) following the manufacturer's instructions. For the cell mito stress test, the oxygen consumption rate was measured. For the glycolysis stress test, the extracellular acidification rate was measured. For more information, see supplemental Methods.
2.15. Data sharing statement
RNA‐seq data are available through Gene Expression Omunibus (accession no. GSE 147736). ChIP‐seq data are available through Sequence Read Archive (accession no. PRJNA615776).
3. RESULTS
3.1. Whole transcriptome analysis identified Fbp1 as a highly upregulated gene in Evi1‐overexpressing cells
To determine the transcriptional consequences of Evi1 overexpression and the resultant leukemic transformation in vivo, we compared gene expression profiles among mouse bone marrow cells with or without ectopic expression of Evi1 and bone marrow leukemia cells transformed by Evi1 overexpression by RNA‐seq analysis. Bone marrow KSL cells derived from wild‐type mice were retrovirally transduced with Evi1‐GFP or GFP and were intravenously transplanted into sublethally irradiated mice. As previously reported by Yoshimi et al, recipient mice developed acute myeloid leukemia with high blast cell proliferation and marked splenomegaly at approximately 6 months after bone marrow transplantation in this model (Figure 1A).16 Although frequencies of GFP positive peripheral blood white blood cells of both groups at 4 weeks after transplantation showed no significant difference, the number of bone marrow GFP positive cells in mice transplanted with Evi1‐overexpressing cells was significantly increased at 4 weeks after transplantation (Figure 1B,C). To avoid the bias affecting RNA‐seq results due to the difference in the fraction of hematopoietic cells, we examined frequencies of hematopoietic stem cell fractions in these mice. The frequencies of long‐term hematopoietic stem cells (LT‐HSC, defined as CD150+ CD48‐ c‐kitpos Sca1pos Linneg(KSL)), short‐term hematopoietic stem cells (ST‐HSC, CD150+ CD48+ KSL), and multipotent progenitor cells (MPP, CD150‐ CD48+ KSL) in KSL cells and the frequencies of megakaryocyte‐erythroid progenitor cells (MEP, CD34‐ CD16/32‐ Linneg c‐kitpos Sca1neg), committed myeloid progenitor cells (CMP, CD34+ CD16/32‐ Linneg c‐kitpos Sca1neg), and granulocyte‐macrophage progenitor cells (GMP, CD34‐ CD16/32+ Linneg c‐kitpos Sca1neg) in Linnegc‐kitposSca1neg cells of GFP positive bone marrow cells isolated from transplanted mice at 4 weeks after transplantation were not significantly different between Evi1‐GFP and GFP overexpressing groups (Figure 1D,E).35, 36 Therefore, the GFP‐positive, lineage‐negative, c‐kit‐positive (GFPpos Linneg c‐kitpos) bone marrow cells were sorted and subjected to RNA‐seq analysis (Figure 2A).
FIGURE 1.
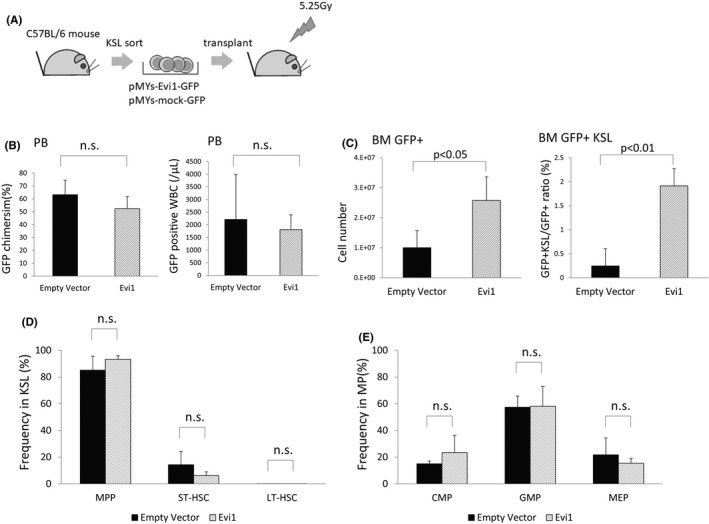
Evi1‐overexpressing leukemia mouse showed increased bone marrow GFP positive cells at 4 weeks after transplantation. (A) Schematic representation of generation of Evi1‐overexpressing leukemia mouse model. Bone marrow KSL cells collected from C57BL/6 mice were retrovirally transduced with Evi1‐GFP or GFP and were intravenously transplanted into sublethally (5.25 Gy) irradiated syngeneic mice for Evi1‐overexpressing mouse generation. (B) Chimerism (left) and number (right) of GFP positive cells in peripheral blood at 4 weeks after transplantation. (n = 3, unpaired t test) (C) Number of GFP positive cells in bone marrow at 4 weeks after transplantation (left). Frequency of KSL cells in GFP positive cells in bone marrow at 4 weeks after transplantation (right) (n = 3 unpaired t test) (D), (E) Frequency of LT‐HSC, ST‐HSC, and MPP in KSL, CMP, GMP, and MEP in myeloid progenitor (MP) cells (Linnegc‐kitposSca1neg) in GFP positive bone marrow cells collected from mice transplanted with Evi1‐GFP or GFP overexpressing cells. Error bars indicate SD (n = 3, unpaired t test)
FIGURE 2.
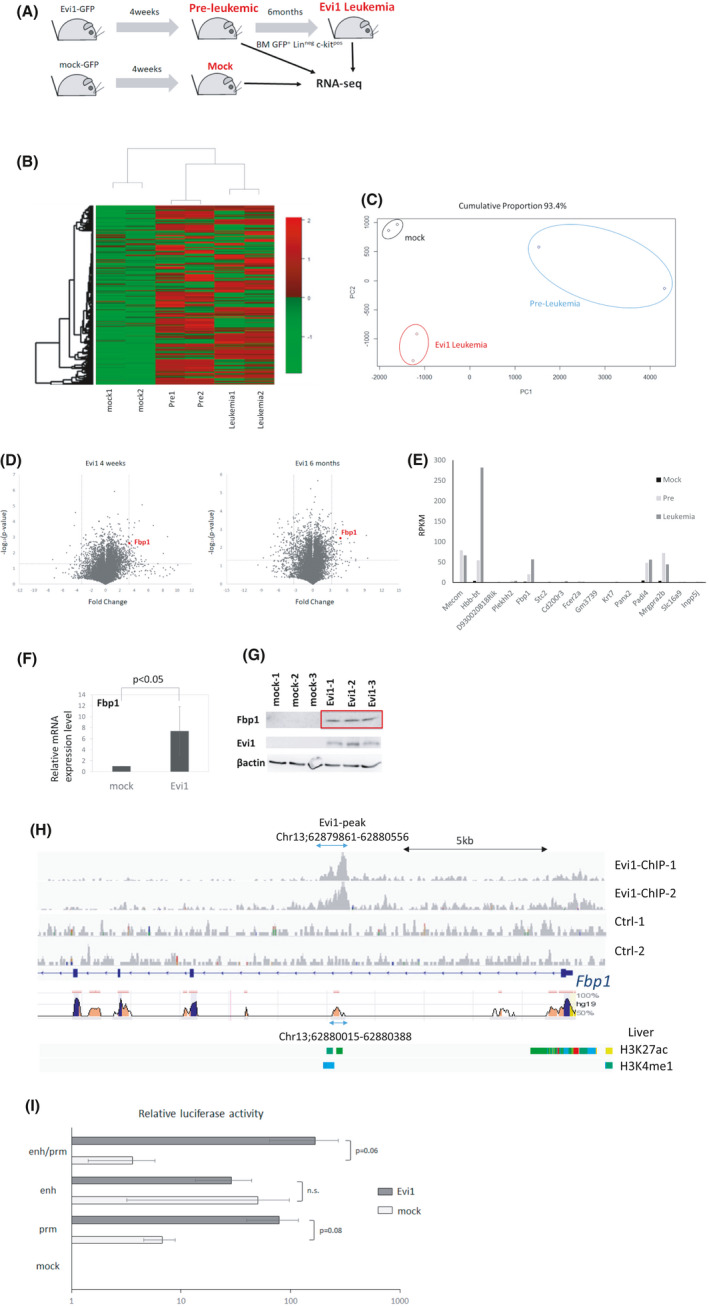
Evi1 upregulates Fbp1 expression. (A) Schematic representation of sample preparation for RNA‐seq analysis. Evi1‐GFP or GFP‐transduced cells were transplanted to sublethally irradiated mice. GFPpos, Linneg, c‐kitpos bone marrow cells were isolated at 4 weeks (pre‐leukemic phase) and 6 months (leukemic phase) after transplantation and subjected to RNA‐seq analysis (n = 2 in each experiment). (B) Unsupervised hierarchical clustering of differentially expressed genes among the three types of the cells. (C) Principle component analysis of RNA‐seq result. (D) Volcano plot comparing mRNA expression of Evi1‐overexpressing bone marrow LK cells at 4 weeks to control cells (left) or leukemia cells at 6 months to control cells (right). Fbp1 is highlighted as a red dot. (E) RPKM of 15 genes highly upregulated in Evi1‐overexpressing bone marrow cells at 4 weeks and at 6 months (F) Relative mRNA expression of Fbp1 in KSL cells retrovirally transduced with Evi1. Error bars indicate SD (n = 3, unpaired t test). (G) Western blot analysis of Fbp1 and EVI1 of whole cell lysates from mouse LK cells transduced with mock or EVI1‐FLAG retroviral vector. (H) EVI1‐FLAG ChIP‐seq signals in Fbp1 locus in mouse Evi1‐overexpressing leukemia cells. Regions conserved between mouse and human are shown at the bottom. (I) Luciferase activity of Fbp1 promoter and/or enhancer with or without Evi1 overexpression in 293T cells. Values are normalized to values using 293T cells transfected with empty luciferase vector (n = 3 in each, unpaired t test). prm, promoter; enh, enhancer; prm/enh, promoter and enhancer; emp, empty
Each group formed a distinct cluster in primary component analysis and unbiased hierarchical clustering (Figure 2B,C). Overall, 634 and 641 genes were significantly upregulated (RPKM ratio >2 and P < .05) in Evi1‐overexpressing mouse bone marrow GFPpos Linneg c‐kitpos cells at 4 weeks and 6 months after each transplantation (Tables S4 and S5). To explore the critical genes in leukemia progression, we further focused on highly (RPKM ratio >10) upregulated 15 genes both 4 weeks and 6 months after transplantation. Among these 15 genes, Hbb‐bt, Fbp1, Padi4, and Mrgpra2b showed high expression in the Evi1‐overexpressing group (Figure 2D,E). Since metabolic alteration in malignant cells was found to be important for carcinogenesis and survival of transformed cells, we focused on the fructose bisphosphatase 1 (Fbp1) gene, a rate‐limiting enzyme of gluconeogenesis that conversely plays a negative role in glycolysis.
We next investigated whether Fbp1 transcription is directly regulated by Evi1. Consistent with the RNA‐seq results, Fbp1 mRNA and protein levels rapidly increased in Evi1‐transduced mouse bone marrow Linneg c‐kitpos cells within 48 hours after transduction (Figure 2F,G). We assessed enrichment of Evi1 protein in the promoter and enhancer region of Fbp1 by performing ChIP‐seq on Evi1 overexpressing mouse leukemia cells. Overall, approximately 8600 peaks were identified by Evi1 ChIP‐seq analysis. EVI1 protein was enriched in the first intron region of Fbp1 gene locus (Figure 2H). According to ENCODE Candidate Cis‐Regulatory Elements, this region is classified as a distal enhancer‐like signature region.37 Using rVISTA 2.0, we confirmed that this region is conserved across mouse and human38 (Figure 2H). We next sought whether this enhancer region is related to active enhancer histone marks such as H3K4me1 and H3K27ac. Using ChIP‐Atlas, we found this enhancer region is enriched in H3K4me1 and H3K27ac in mouse liver cells, which highly express Fbp1 (Figure 2H).39 Since HOMER motif analysis did not find EVI1 binding motifs in the identified region, other DNA binding proteins may act as a scaffold and play a critical role in upregulation of Fbp1. Next we sought to determine whether EVI1 directly upregulates FBP1 using luciferase assay. We measured the luciferase activity of 293T cells transfected with luciferase reporter vector and Evi1 expression vector. Evi1 overexpression increased luciferase activity in a Fbp1‐promoter‐dependent manner in this assay. Although Evi1 did not bind Fbp1 promoter region in our mouse Evi1‐overexpressing leukemia cells, indirect upregulating mechanisms by Evi1 may be present. On the other hand, the Fbp1 enhancer sequence did not increase luciferase activity by Evi1 overexpression (Figure 2I). This is partly explained by the fact that the Evi1 binding domain was not included in the Fbp1 enhancer region and in 293T cells the unknown cofactor essential for Evi1 binding to enhancer region may not be expressed enough to bind Evi1 to this enhancer region. These results indicate that Evi1 binds this enhancer region and subsequently upregulates Fbp1 in Evi1‐overexpressing cells.
3.2. Inhibition of FBP1 suppresses acute myeloid leukemia driven by Evi1
In some cancers, such as breast, pancreatic and non‐small‐cell lung carcinoma, silencing of Fbp1 is caused by various mechanisms and related to proliferation, chemoresistance, and poor prognosis by increasing glycolytic flux.40, 41, 42, 43, 44 However, the role of Fbp1 in the context of leukemia and the in vivo tumorigenic or tumor promoting potential of Fbp1 remains poorly understood. We investigated whether enhanced Fbp1 expression mediated by Evi1 overexpression contributes to Evi1‐overexpressing cell proliferation by analyzing the colony‐forming cell capacity of Evi1‐overexpressing KSL cells with or without FBP1 inhibition. shRNA‐mediated silencing of Fbp1 significantly suppressed the colony‐forming cell capacity of mouse bone marrow KSL cells transformed by Evi1 (Figure 3A). In contrast to this, the colony number of normal KSL cells was not reduced by Fbp1‐targeting shRNA (Figure 3B). These data suggest that Fbp1 has a critical role in proliferation of Evi1‐overexpressing cells but not in normal hematopoietic stem cells.
FIGURE 3.
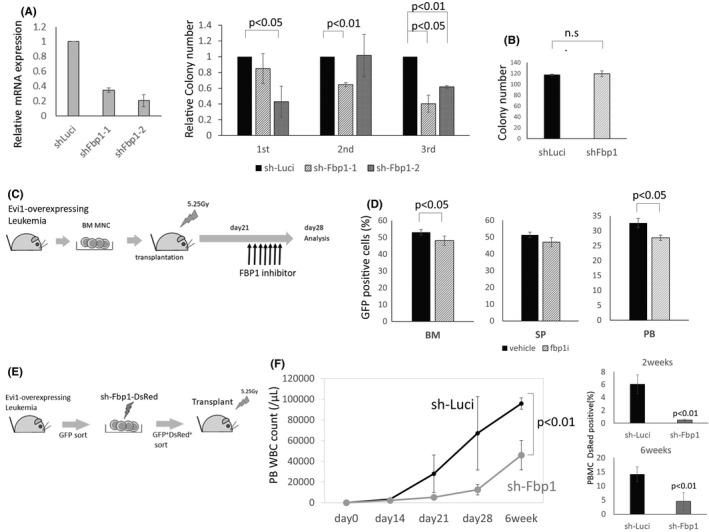
Fbp1 inhibition suppresses Evi1‐overexpressing leukemia in vitro and in vivo. (A) Relative mRNA expression of Fbp1 of Ba/F3 cell lines transduced with Fbp1 knockdown vector. Ba/F3 was transduced with shFbp1. After 24 hours of puromycin selection, GFP‐positive cells are sorted and subjected to qPCR (n = 2 each). Error bars indicate SD. Left: Relative colony numbers of KSL cells transduced with Evi1‐GFP and sh‐Fbp1compared with the control shRNA‐transduced cells (n = 3 in each experiment, unpaired t test). Middle: Colony numbers of Linneg c‐kitpos cells with or without Fbp1 knockdown (n = 3 in each experiment, unpaired t test). Right: (B) Schematic representation of Fbp1 inhibitor administration in vivo. Leukemia cells isolated from the bone marrow of Evi1 leukemia mice were intravenously transplanted into sublethally irradiated normal C57BL/6 mice (day 0). An FBP1 inhibitor CAY18860 was intraperitoneally injected daily into the leukemic mice on days 21‐27. (C) On day 28, the mice were sacrificed and analyzed for the frequency of GFP‐positive cells in the bone marrow (BM), spleen (SP), and peripheral blood (PB) (n = 4 in each experiment, unpaired t test). (D) Schematic representation of shRNA‐mediated Fbp1 knockdown of Evi1‐leukemia mice. Bone marrow GFP‐positive cells were isolated from the Evi1‐overexpressing leukemia cells, transduced with sh‐Fbp1‐DsRed or sh‐Luci‐DsRed (control). DsRed positive GFP positive cells were sorted and transplanted into sublethally irradiated normal C57BL/6 mice. (F) Peripheral blood white blood cell counts of the transplanted mice at the indicated time points (n = 4 in each group, unpaired t test). Left: Frequency of DsRed‐positive cells within the peripheral blood mononuclear cells is shown (n = 4 in each group, unpaired t test). (right)
To further explore the therapeutic potential of targeting Fbp1 in Evi1high AML, we investigated the effect of Fbp1 inhibition on Evi1‐overexpressing leukemia in vivo. Leukemia cells isolated from bone marrow of Evi1 leukemia mice were intravenously transplanted into sublethally irradiated secondary recipient mice. These mice were treated with daily intraperitoneal injection of vehicle or FBP1 inhibitor CAY18860 from day 21 through day 27 after bone marrow transplantation (Figure 3C).45, 46 Although all of the secondary recipient mice developed leukemia, pharmacological inhibition of Fbp1 significantly decreased leukemia cell burden in bone marrow and peripheral blood (Figure 3D). To interrogate the effect of more specific Fbp1 inhibition on Evi1‐overexpressing leukemia, we analyzed secondarily transplanted Evi1‐overexpressing leukemia mice with or without shRNA mediated Fbp1 knockdown (Figure 3E). Strikingly, shRNA‐mediated Fbp1 knockdown significantly delayed leukemia progression (Figure 3F). Collectively, these data clearly show that Fbp1 promotes Evi1‐overexpressing leukemia in vivo.
3.3. Evi1 upregulates pentose phosphate pathway‐related genes
Since Fbp1 is a gluconeogenesis enzyme, we hypothesized that glucose metabolism alteration mediated by high Fbp1 expression resulted in progression of Evi1‐overexpressing leukemia. To characterize the effect of Evi1 overexpression on the glucose metabolism pathway, we further analyzed the RNA‐seq results of mouse Evi1‐overexpressing bone marrow cells. Intriguingly, hexokinase 3 (Hk3), which catalyzes the first step of the glycolysis, was upregulated in the high Evi1 group whereas phosphoglycerate mutase 1 (Pgam1), an enzyme located in the latter step of glycolysis, was downregulated (Figure 4A). It is conceivable that these expression changes of glycolysis enzymes result in accumulation of the intermediate metabolites of glycolysis such as glucose 6‐phosphate (G6P), fructose 6‐phosphate (F6P), fructose 1,6‐bis phosphate (F1,6P), glyceraldehyde 3‐phosphate (GA3P), and 3‐phosphoglyceric acid (3‐PG). Considering high Fbp1 expression, the balance of these metabolites would be inclined towards G6P and F6P. Since G6P is the first substrate of the pentose phosphate pathway, we speculated that the pentose phosphate pathway is upregulated by the relative accumulation of G6P in Evi1‐overexpressing cells. Consistent with this idea, RNA‐seq results showed upregulation of pentose phosphate pathway enzymes such as glucose 6‐phosphate dehydrogenase (G6pdx), phosphogluconate dehydrogenase (Pgd), and ribose 5‐phosphate isomerase (Rpia) in Evi1‐overexpressing cells (Figure 4A). Moreover, gene set enrichment analysis showed relative enrichment of pentose phosphate pathway genes in Evi1‐overexpressing cells (Figure 4B). Interestingly, ChIP‐seq analysis revealed that EVI1 also binds to the enhancer and promoter region of other glycolysis enzymes such as Hk3 and Pgam1 as well as pentose phosphate pathway enzymes such as Pgd, Rpia, and Taldo1, all of which were differently expressed in Evi1‐overexpressing mouse bone marrow cells (Figure 4C‐G). These regions are mostly conserved between human and mouse. Homer de novo motif analysis of these EVI1‐binding regions did not show the EVI1 binding motif, which indicates other cofactors may contribute to DNA binding of EVI1 in these regions. Collectively, these expression changes of glucose metabolic enzymes possibly mediated by the interaction of EVI1 and DNA enhancer regions orchestrate glucose metabolism alteration, glycolysis to pentose phosphate pathway, which generates pentose and then ribose‐5‐phosphates, resulting in enhanced nucleotide synthesis, which is essential for rapidly dividing cells.
FIGURE 4.
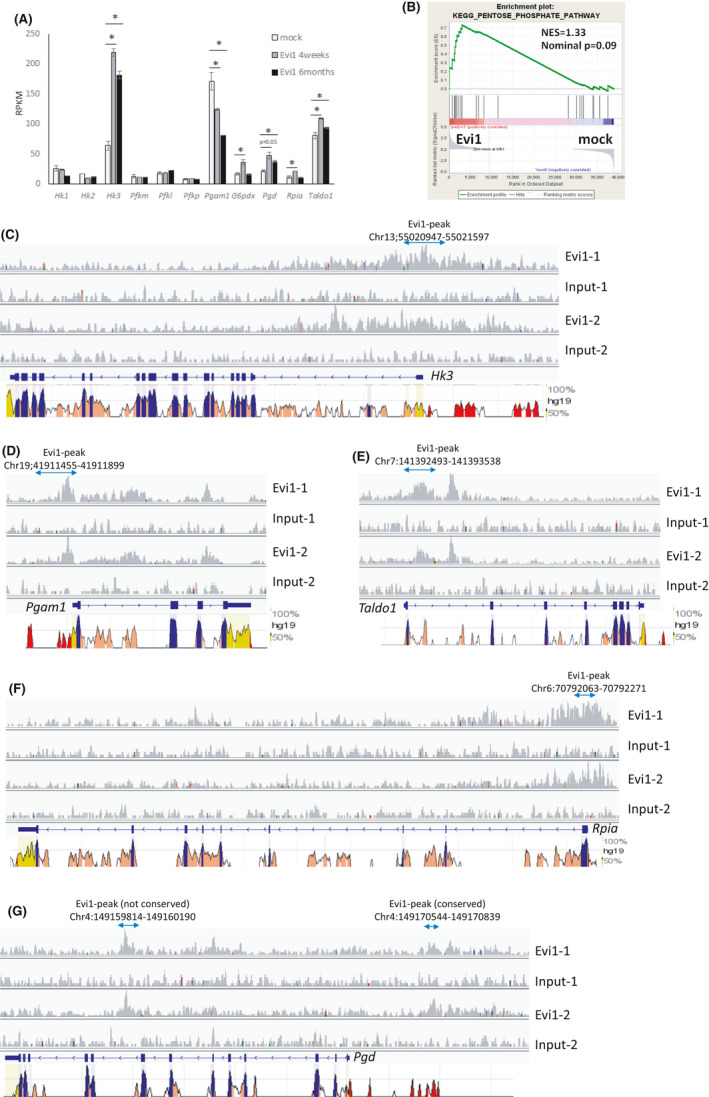
Evi1 modulates expression of glycolysis and pentose phosphate pathway enzymes. (A) Re‐analysis of RNA‐seq result. mRNA expression of glycolysis and pentose phosphate pathway enzymes of GFPpos Linneg c‐kitpos bone marrow cells from Evi1‐overexpressing and control mice. (B) Gene set enrichment analysis using RNA‐seq data of GFPpos Linneg c‐kitpos bone marrow cells collected from the transplanted mice with Evi1‐GFP overexpressing or GFP overexpressing KSL cells at 4 weeks after transplantation. Gene set name KEGG_PENTOSE_PHOSPHATE_PATHWAY was used in this analysis. (C)‐(G) Gene tracks of EVI1‐FLAG ChIP‐seq signals (rpm) at (C) Hk3, (D) Pgam1, (E) Taldo1, (F) Rpia, and (G) Pgd in mouse Evi1‐overexpressing leukemia cells. Regions conserved between mouse and human are shown at the bottom
3.4. Metabolome analysis revealed Evi1 overexpression upregulates the pentose phosphate pathway
To further investigate whether high Evi1 upregulates the pentose phosphate pathway and de novo nucleotide synthesis, we analyzed the metabolome profiles of the AML cell line with or without Evi1 overexpression using capillary electrophoresis combined with mass spectrometry. Since metabolic consequences caused by high Fbp1 expression are of interest, we have to adopt cell lines which originally express Evi1 and Fbp1 at the lowest level possible and express high Fbp1 under Evi1 overexpression. We tested some AML cell lines and employed ME‐1 cells in which FBP1 is upregulated by EVI1 overexpression to perform metabolome analysis (Figure 5A). The glucose‐6‐phosphate (G6P)/ribulose‐5‐phosphate (R5P) ratio is a parameter inversely correlated to pentose phosphate pathway activity.47 Although many metabolites related to glycolysis and pentose phosphate pathway are not significantly different between the two groups, the G6P/R5P ratio was significantly decreased in Evi1‐transduced ME‐1 cells (Figure 5B,C and Table S6). Moreover, amounts of nucleic acid such as adenosine monophosphate (AMP) and guanosine monophosphate (GMP) were significantly increased in Evi1‐overexpressing cells (Figure 5C).
FIGURE 5.
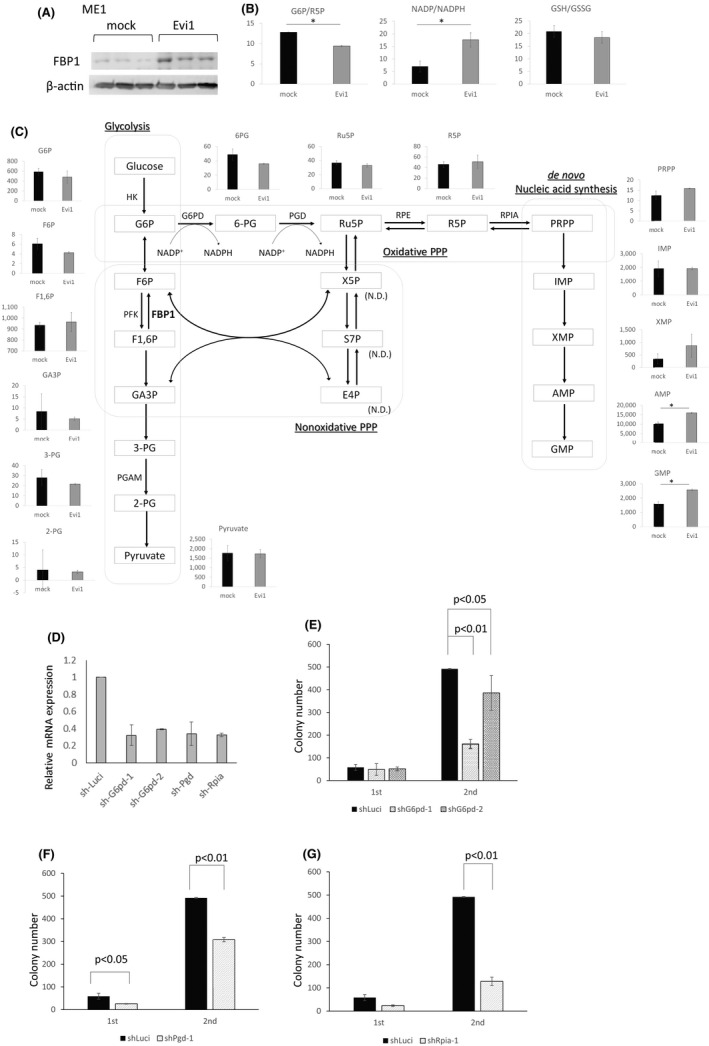
Metabolome analysis revealed Evi1‐overexpressing cells activate the pentose phosphate pathway and de novo nucleotides synthesis. (A) Western blot analysis of whole cell lysates from Evi1‐GFP or GFP‐overexpressing ME‐1. (B), (C) Schematic representation of glycolysis and pentose phosphate pathway and de novo ribonucleotide synthetic pathway. Each graph shows normalized metabolite amount or ratio of average of metabolite amounts of ME‐1 cell with (gray) or without (red) Evi1‐overexpression (unpaired t test). (D) Relative mRNA expression of g6pd, pgd, and rpia of Ba/F3 cell lines transduced with knockdown vectors targeting each gene. GFP‐positive cells are sorted and subjected to qPCR. mRNA expression was normalized to mRNA expression of cells transduced with sh‐Luci (n = 2 each). Error bars indicate SD. (E)‐(F) Colony numbers of KSL cells transduced with Evi1‐GFP and (E) shG6pd, (F) shPgd, or (G) shRpia vectors compared with the control shRNA‐transduced cells (n = 3 in each experiment, unpaired t test)
To examine whether pentose phosphate pathway inhibition is the metabolic vulnerability of Evi1‐overexpressing cells, we analyzed the colony‐forming cell capacity of Evi1‐overexpressing KSL cells with or without knockdown of each pentose phosphate pathway enzyme. Strikingly, knockdown of each enzyme significantly reduced the colony‐forming cell capacity of Evi1‐overexpressing cells (Figure 5D‐G). These results suggest that the activated pentose phosphate pathway mediated by deregulated Evi1 expression plays a critical role in leukemogenesis of Evi1high AML.
3.5. EVI1 upregulates oxidative phosphorylation as an alternative ATP source in Evi1‐overexpressing cells
When considering adenosine triphosphate (ATP) generation in high Evi1 cells, Evi1 overexpressing cells may not utilize glycolysis efficiently due to high Fbp1 expression, counteracting glycolysis. Since another main source of ATP is the citric acid cycle combined with oxidative phosphorylation (OXPHOS), we hypothesized that Evi1high leukemia cells are more dependent on OXPHOS in ATP generation. Interestingly, Evi1‐transduced mouse bone marrow Linneg c‐kitpos cells showed a significantly higher oxygen consumption rate (OCR), which means higher OXPHOS activity, whereas Evi1‐transduced cells showed a lower extracellular acidification rate (ECAR), meaning relatively less glycolysis activity (Figure 6A,B). Fbp1‐transduced mouse Linneg c‐kitpos cells also showed higher OCR (Figure 6C). Although Fbp1 itself does not seem to have any direct activity modulating the citric acid cycle or OXPHOS, this result is partly explained by the fact that the activity of OXPHOS is strongly controlled by the intracellular balance between ATP and ADP. OXPHOS inhibitor metformin suppressed the colony‐forming cell capacity of Fbp1‐overexpressing mouse bone marrow c‐kitpos cells compared with control c‐kitpos cells, which indicates that Fbp1‐overexpressing cells are more dependent on OXPHOS than normal hematopoietic cells (Figure 6D). Moreover, metformin also decreased the colony‐forming cell capacity of Evi1‐overexpressing mouse Linneg c‐kitpos cells compared with control Linneg c‐kitpos cells and Linneg c‐kitpos cells transduced with MLL‐ENL, which is known as a genetic mutation related to glycolysis dependency (Figure 6E,F). Since high Fbp1 causes OXPHOS dependency, metformin treatment combined with shFbp1 did not show a combinatory effect on Evi1‐overexpressing Linneg c‐kitpos cells (Figure 6G). These data collectively suggest that Evi1high leukemia cells harbor high OXPHOS activity and are dependent on OXPHOS.
FIGURE 6.
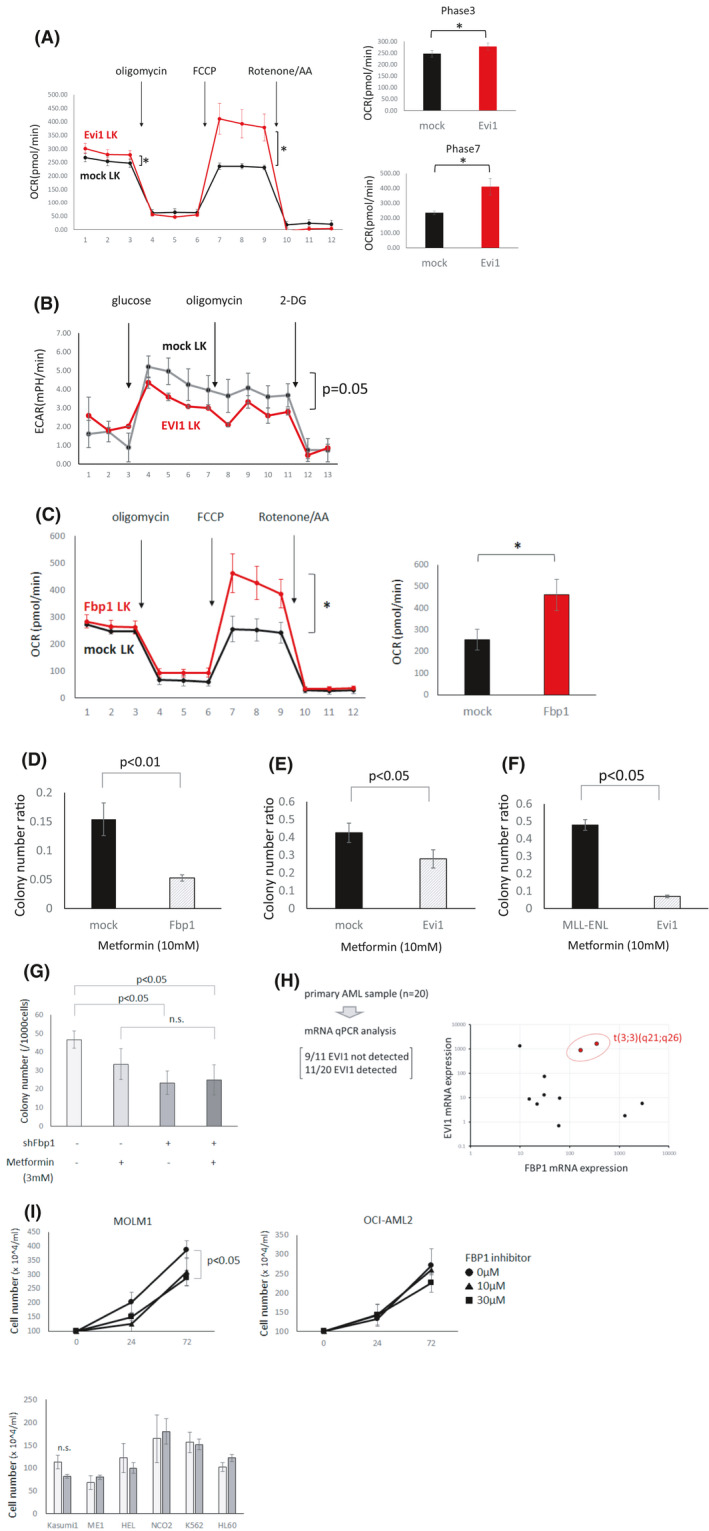
Evi1 overexpression induces OXPHOS activation and OXPHOS dependency. (A) Oxygen consumption rate (OCR) of mouse bone marrow LK cells with (red) or without (black) retroviral Evi1 overexpression (left). In each phase OCR was shown in bar graph (right) (n = 6 each, unpaired t test). (B) Extracellular acidification rate (ECAR) of mouse bone marrow LK cells with (red) or without (black) retroviral Evi1 overexpression (n = 6 each, unpaired t test). (C) Oxygen consumption rate (OCR) in mouse bone marrow LK cells with (red) or without (black) retroviral Fbp1 overexpression (left). Representative phase shown in a bar graph (right) (n = 6 each, unpaired t test). (D) Ratio of colony number of mouse Linneg bone marrow cells transduced with GFP or Fbp1‐GFP treated by 10 mmol/L metformin normalized to vehicle treatment (n = 3 in each, unpaired t test). (E) Ratio of colony number of mouse Linneg c‐kitpos bone marrow cells transduced with GFP or Evi1‐GFP treated by 10 mmol/L metformin normalized to vehicle treatment (n = 3 in each, unpaired t test). (F) Ratio of colony number of mouse Linneg c‐kitpos bone marrow cells transduced with MLL‐ENL or Evi1 treated by 10 mmol/L metformin normalized to vehicle treatment (n = 3 each group, unpaired t test). (G) Colony number of Evi1‐overexpressing LK cells. Combinatory effect by shFbp1 and metformin was tested (n = 3 in each, unpaired t test). (H) mRNA expression of Fbp1 and Evi1 in AML cells derived from AML patients. Twenty samples were subjected to analysis and in 11 samples Evi1 was detected and is plotted on the right panel. (I) Cell proliferation of MOLM1 and other human cell lines. Cell numbers of MOLM1(upper left), OCI‐AML(upper right), and other cell lines (lower) (n = 3 in each, unpaired t test)
3.6. FBP1 is highly transcribed in a part of 3q26 related leukemia and some other subtypes of AML
To evaluate the potential relevance of these findings to the clinical setting, we analyzed gene expression data of bone marrow AML cells derived from 20 evaluable patients. We compared Evi1 and Fbp1 mRNA levels by real‐time PCR. Although Evi1 and Fbp1 did not show significant correlation as a whole, leukemia cells derived from AML with inv(3)(q21q26) chromosomal abnormality showed high Evi1 and high Fbp1 mRNA expression (Figure 6H). On the other hand, in the BeatAML dataset a part of inv(3)(q21q26) leukemia and some other subtypes of leukemia showed higher Fbp1 expression, which indicates that Evi1 and other factors may coordinately cause high Fbp1 upregulation and that high Fbp1 may also play an important role in other subtypes of leukemia.48To clarify whether high EVI1 human AML cells are susceptible to FBP1 inhibition, we compared cell proliferation of AML cell lines with or without FBP1 inhibitor. FBP1 inhibitor decreased the proliferation of the inv(3) MOLM1 cell line whereas proliferation of no other tested cell lines was suppressed (Figure 6I). These results indicate that cell proliferation of a certain type of high EVI1 human AML cells was also inhibited by FBP1 inhibition.
4. DISCUSSION
Here we demonstrated that Fbp1 is a critical metabolic regulator of Evi1‐overexpressing leukemia cells by balancing the pentose phosphate pathway and glycolysis, and that Fbp1, as well as pentose phosphate pathway enzymes, is a vulnerability of Evi1‐overexpressing leukemia cells. Previous reports revealed the pentose phosphate pathway contributes to the survival and chemotherapy resistance of some cancer cells as well as AML cells by the synthesis of ribonucleotides.49, 50, 51, 52, 53, 54 Consistent with this idea, upregulated ribonucleotide synthesis was clearly induced by Evi1 transduction in the human AML cell line. Since the pentose phosphate pathway is also known as a main NADPH source, which contributes to maintenance of intracellular homeostasis as a scavenger of reactive oxygen species and a cofactor essential for fatty acid synthesis, pentose phosphate pathway activation is usually believed to result in a reduction of the NADP/NADPH ratio.49, 55 However, in Evi1‐overexpressing cells we found the NADP/NADPH ratio was increased (Figure 5B). This result indicates Evi1‐overexpressing cells show excess consumption of NADPH compared to cells without Evi1 overexpression. This hypothesis may be partly supported by the fact that upregulated OXPHOS by Evi1 overexpression causes cellular oxidative stress, which results in excess consumption of NADPH. For example, IDH mutant cells which consume much NADPH for 2‐hydroxyglutarate synthesis showed high pentose phosphate pathway flux and a high NADP/NADPH ratio.56 Collectively, Evi1‐overexpressing cells may consume excess NADPH coupled with the upregulated pentose phosphate pathway.
Fbp1 has been reported as a tumor suppressor gene in other solid cancers.40, 41, 42, 43, 44, 57 In this context, high Fbp1 expression decreases glycolysis flux indispensable for quick ATP supply in the low‐oxygen environment to which many highly proliferative cancers are exposed and suppression of Fbp1 function is related to cell proliferation, chemoresistance, and metastasis.40, 42 On the contrary, in Evi1‐overexpressing cells Fbp1 acts as an oncogene which promotes the pentose phosphate pathway. Since which aspect of Fbp1 emerges may vary according to the cellular context such as types of cancer, dependent oncogenic pathway, and tumor microenvironment, the determinant factor of the Fbp1 function needs to be elucidated. On the other hand, this bifacial feature of Fbp1 makes it attractive to target Fbp1 as a therapeutic target. We showed that shRNA‐mediated knockdown of Fbp1 and pharmacological Fbp1 inhibition had little effect on the colony formation ability of mouse KSL cells in this paper (Figures 2B and 3A). Similarly, previous reports showed that the function of normal hematopoietic stem cells derived from human cord blood was totally undamaged by Fbp1 inhibition.58 These results strongly indicate that Fbp1 inhibition does not compromise the normal hematopoietic system and is an ideal therapeutic target of Evi1‐overexpressing leukemia.
Recent analyses have revealed that AML with high OXPHOS activity is related to chemotherapy resistance.59, 60 Although the mechanisms of this chemotherapy resistance have not yet been discovered, upregulation of OXPHOS by Evi1 overexpression is consistent with the fact that Evi1high AML clinically shows extremely high resistance to the conventional chemotherapy. We showed that Fbp1 overexpression alone could partially induce OXPHOS activation to a lesser extent than Evi1 overexpression. This OXPHOS upregulation may be indirectly caused by an ATP shortage related to downregulation of glycolysis. These data collectively suggest that Fbp1 inhibition can ameliorate resistance to chemotherapy in Evi1‐overexpressing leukemia by downregulating OXPHOS.
In summary, our study demonstrated that the activated pentose phosphate pathway through transcriptional upregulation of Fbp1 as well as catalyzing enzymes is crucial for progression of Evi1‐driven leukemia. Since inhibition of Fbp1 did not compromise normal hematopoiesis, targeting the enzyme can be a promising therapeutic approach for Evi1high AML.
DISCLOSURE
The authors declare no competing financial interests.
Supporting information
Supplementary Material
Tables S1‐S6
ACKNOWLEDGMENTS
We thank T. Kitamura for Plat‐E and Plat‐A packaging cells, and H. Nakauchi for pGCDNsam‐IRES‐GFP retroviral vector. This work was supported by JSPS KAKENHI Grant Number JP18H04052.
Mizuno H, Koya J, Masamoto Y, Kagoya Y, Kurokawa M. Evi1 upregulates Fbp1 and supports progression of acute myeloid leukemia through pentose phosphate pathway activation. Cancer Sci. 2021;112:4112–4126. 10.1111/cas.15098
REFERENCES
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136‐1152. [DOI] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rollig C, Bornhauser M, Thiede C, et al. Long‐term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol. 2011;29:2758‐2765. [DOI] [PubMed] [Google Scholar]
- 4.van Waalwijk B, van Doorn‐Khosrovani S, Erpelinck C, et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101:837‐845. [DOI] [PubMed] [Google Scholar]
- 5.Groschel S, Lugthart S, Schlenk RF, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28:2101‐2107. [DOI] [PubMed] [Google Scholar]
- 6.Lugthart S, van Drunen E, van Norden Y, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111:4329‐4337. [DOI] [PubMed] [Google Scholar]
- 7.Qin YZ, Zhao T, Zhu HH, et al. High EVI1 expression predicts poor outcomes in adult acute myeloid leukemia patients with intermediate cytogenetic risk receiving chemotherapy. Med Sci Monit. 2018;24:758‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamazaki H, Suzuki M, Otsuki A, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25:415‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369‐381. [DOI] [PubMed] [Google Scholar]
- 10.Ayoub E, Wilson MP, McGrath KE, et al. EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat Commun. 2018;9:4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goyama S, Yamamoto G, Shimabe M, et al. Evi‐1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207‐220. [DOI] [PubMed] [Google Scholar]
- 12.Katayama S, Suzuki M, Yamaoka A, et al. GATA2 haploinsufficiency accelerates EVI1‐driven leukemogenesis. Blood. 2017;130:908‐919. [DOI] [PubMed] [Google Scholar]
- 13.Kurokawa M, Mitani K, Yamagata T, et al. The evi‐1 oncoprotein inhibits c‐Jun N‐terminal kinase and prevents stress‐induced cell death. Embo j. 2000;19:2958‐2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kustikova OS, Schwarzer A, Stahlhut M, et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 2013;27:1127‐1138. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka T, Nishida J, Mitani K, Ogawa S, Yazaki Y, Hirai H. Evi‐1 raises AP‐1 activity and stimulates c‐fos promoter transactivation with dependence on the second zinc finger domain. J Biol Chem. 1994;269:24020‐24026. [PubMed] [Google Scholar]
- 16.Yoshimi A, Goyama S, Watanabe‐Okochi N, et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood. 2011;117:3617‐3628. [DOI] [PubMed] [Google Scholar]
- 17.Kataoka K, Sato T, Yoshimi A, et al. Evi1 is essential for hematopoietic stem cell self‐renewal, and its expression marks hematopoietic cells with long‐term multilineage repopulating activity. J Exp Med. 2011;208:2403‐2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glass C, Wuertzer C, Cui X, et al. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS One. 2013;8:e67134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamakawa N, Kaneda K, Saito Y, Ichihara E, Morishita K. The increased expression of integrin alpha6 (ITGA6) enhances drug resistance in EVI1(high) leukemia. PLoS One. 2012;7:e30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez‐Benito M, Conchillo A, Garcia MA, et al. EVI1 controls proliferation in acute myeloid leukaemia through modulation of miR‐1‐2. Br J Cancer. 2010;103:1292‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rommer A, Steinmetz B, Herbst F, et al. EVI1 inhibits apoptosis induced by antileukemic drugs via upregulation of CDKN1A/p21/WAF in human myeloid cells. PLoS One. 2013;8:e56308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montalban‐Bravo G, DiNardo CD. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018;14:979‐993. [DOI] [PubMed] [Google Scholar]
- 24.Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730‐741. [DOI] [PubMed] [Google Scholar]
- 25.Wang YH, Israelsen WJ, Lee D, et al. Cell‐state‐specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fenouille N, Bassil CF, Ben‐Sahra I, et al. The creatine kinase pathway is a metabolic vulnerability in EVI1‐positive acute myeloid leukemia. Nat Med. 2017;23:301‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito Y, Sawa D, Kinoshita M, et al. EVI1 triggers metabolic reprogramming associated with leukemogenesis and increases sensitivity to L‐asparaginase. Haematologica. 2019;105(8):2118‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods. 2012;9:357‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohashi Y, Hirayama A, Ishikawa T, et al. Depiction of metabolome changes in histidine‐starved Escherichia coli by CE‐TOFMS. Mol Biosyst. 2008;4:135‐147. [DOI] [PubMed] [Google Scholar]
- 33.Ooga T, Sato H, Nagashima A, et al. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol Biosyst. 2011;7:1217‐1223. [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry‐based saliva metabolomics identified oral, breast and pancreatic cancer‐specific profiles. Metabolomics. 2010;6:78‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75:14‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109‐1121. [DOI] [PubMed] [Google Scholar]
- 37.Moore JE, Purcaro MJ, Pratt HE, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oki S, Ohta T, Shioi G, et al. ChIP‐Atlas: a data‐mining suite powered by full integration of public ChIP‐seq data. EMBO Rep. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong C, Yuan T, Wu Y, et al. Loss of FBP1 by Snail‐mediated repression provides metabolic advantages in basal‐like breast cancer. Cancer Cell. 2013;23:316‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirata H, Sugimachi K, Komatsu H, et al. Decreased expression of fructose‐1,6‐bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Res. 2016;76:3265‐3276. [DOI] [PubMed] [Google Scholar]
- 42.Li B, Qiu B, Lee DS, et al. Fructose‐1,6‐bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Q, Wei P, Wu J, et al. The FOXC1/FBP1 signaling axis promotes colorectal cancer proliferation by enhancing the Warburg effect. Oncogene. 2019;38:483‐496. [DOI] [PubMed] [Google Scholar]
- 44.Chen LY, Cheng CS, Qu C, et al. CBX3 promotes proliferation and regulates glycolysis via suppressing FBP1 in pancreatic cancer. Biochem Biophys Res Commun. 2018;500:691‐697. [DOI] [PubMed] [Google Scholar]
- 45.von Geldern TW, Lai C, Gum RJ, et al. Benzoxazole benzenesulfonamides are novel allosteric inhibitors of fructose‐1,6‐bisphosphatase with a distinct binding mode. Bioorg Med Chem Lett. 2006;16:1811‐1815. [DOI] [PubMed] [Google Scholar]
- 46.Lai C, Gum RJ, Daly M, et al. Benzoxazole benzenesulfonamides as allosteric inhibitors of fructose‐1,6‐bisphosphatase. Bioorg Med Chem Lett. 2006;16:1807‐1810. [DOI] [PubMed] [Google Scholar]
- 47.Sato E, Mori T, Mishima E, et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci Rep. 2016;6:36618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu S, Wang H, Li Y, et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 2018;78:4549‐4562. [DOI] [PubMed] [Google Scholar]
- 50.Cho ES, Cha YH, Kim HS, Kim NH, Yook JI. The pentose phosphate pathway as a potential target for cancer therapy. Biomol Ther (Seoul). 2018;26:29‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poulain L, Sujobert P, Zylbersztejn F, et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia. 2017;31:2326‐2335. [DOI] [PubMed] [Google Scholar]
- 52.Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014;5:592‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santana‐Codina N, Roeth AA, Zhang Y, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 2018;9:4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF‐1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71‐87.e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verma N, Pink M, Boland S, Rettenmeier AW, Schmitz‐Spanke S. Benzo[a]pyrene‐induced metabolic shift from glycolysis to pentose phosphate pathway in the human bladder cancer cell line RT4. Sci Rep. 2017;7:9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gelman SJ, Naser F, Mahieu NG, et al. Consumption of NADPH for 2‐HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 2018;22:512‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu GM, Zhang YM. Targeting FBPase is an emerging novel approach for cancer therapy. Cancer Cell Int. 2018;18:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR‐gamma signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med. 2018;24:360‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farge T, Saland E, de Toni F, et al. Chemotherapy‐resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yucel B, Sonmez M. Repression of oxidative phosphorylation sensitizes leukemia cell lines to cytarabine. Hematology. 2018;23:330‐336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Tables S1‐S6