

Abstract
The regulatory relationship between silent information regulator 2 (SIRT2) and glucose 6‐phosphate dehydrogenase (G6PD) in clear cell renal cell carcinoma (ccRCC) is still unclear. The present study aimed to explore the function of SIRT2 and its regulatory effect on G6PD in ccRCC. The Cancer Genome Atlas data mining of SIRT2 was first analyzed. Quantitative real‐time PCR and western blot analyses were used to assess the mRNA and protein expression levels, respectively. Cell viability, colony formation, cell cycle, cell apoptosis, and TUNEL assays and EdU staining were used to investigate the roles of SIRT2 in ccRCC proliferation and apoptosis. The coimmunoprecipitation (Co‐IP) assay was used to analyze the association between SIRT2 and G6PD in ccRCC cells. Quantitative Co‐IP assay was used to detect the levels of G6PD ubiquitination and small ubiquitin‐related modifier 1 (SUMO1). An in vivo experiment was also carried out to confirm in vitro findings. The results indicated that SIRT2 promoted ccRCC proliferation and inhibited apoptosis by regulating cell cycle and apoptosis related proteins. Silent information regulator 2 interacted with G6PD, facilitated its activity through deacetylation, and increased its stability by reducing its ubiquitination and enhancing its SUMO1 modification. Silent information regulator 2 also promoted ccRCC tumor development in vivo. Taken together, the present study indicated that SIRT2 promoted ccRCC progression by increasing G6PD activity and stability, and it could be a potential new diagnostic and therapeutic target for ccRCC.
Keywords: clear cell renal cell carcinoma, deacetylation, G6PD, SIRT2, SUMO1
The present study explores the function of silent information regulator 2 (SIRT2) and its regulatory effect of on glucose 6‐phosphate dehydrogenase in clear cell renal cell carcinoma (ccRCC). It aims to provide new molecular clues for targeted treatment of patients with ccRCC. The study supports an oncogenic role for SIRT2 in ccRCC and SIRT2 might be a potential therapeutic target for ccRCC.

Abbreviations
- APC/C
anaphase‐promoting complex/cyclosome
- ccRCC
clear cell renal cell carcinoma
- CDK
cyclin‐dependent kinase
- CHX
cycloheximide
- Co‐IP
coimmunoprecipitation
- F
forward
- G6PD
glucose 6‐phosphate dehydrogenase
- KD
knockdown
- OE
overexpressed
- R
reverse
- RT‐qPCR
quantitative real‐time PCR
- RCC
renal cell carcinoma
- SIRT2
silent information regulator 2
- SUMO1
small ubiquitin‐related modifier 1
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Clear cell renal cell carcinoma, the predominant histologic subtype in RCC, has a high degree of malignancy.1, 2 However, molecular mechanisms underlying ccRCC tumorigenesis are still unclear, and new therapies targeting ccRCC are still challenging.2
Recently, accumulated evidence has revealed that SIRT2 is widely expressed in different types of human tissues.3 As one of the important proteins in posttranslational modification, SIRT2 plays a decisive role in deacetylation to regulate the activity of proteins.4, 5 A report indicated that SIRT2, as a cancer stem cell marker, is highly expressed in RCC stem‐like cells and RCC patients with higher levels of SIRT2 have poor survival.6 However, the function and underlying mechanism of SIRT2 in ccRCC are not completely delineated.
Previous studies revealed that ccRCC patients with high G6PD expression levels have poor prognosis.7 Glucose 6‐phosphate dehydrogenase facilitates ccRCC progression by modulating p‐STAT3 and the ROS‐MAPK axis pathway.8, 9 Another study confirmed that G6PD activity is significantly high in RCC tumor tissues.10, 11 Therefore, in addition to the increased expression level, posttranslation modification might also contribute to the overactivation of G6PD in ccRCC. Silent information regulator 2 can increase G6PD activity through deacetylating its lysine 403.12, 13 This suggests that SIRT2 might be important in regulating G6PD activity. However, the interplay between SIRT2 and G6PD in ccRCC at the posttranslational level has not been reported.
A report pointed out that VHL, a subunit of E3 ubiquitin ligase, induced G6PD ubiquitination, degradation and can decrease G6PD stability in a high‐glucose environment, finally resulting in podocyte injury related to diabetic kidney disease.14 Small ubiquitin‐related modifier and ubiquitin are similar in structure and bind to selected proteins, but the function of SUMOylation is opposite to that of ubiquitination and it prevents marker proteins from degradation.15, 16, 17 However, whether G6PD could be posttranslationally modified by ubiquitination or SUMOylation, as well as the underlying mechanism, in ccRCC is still unclear.
The present study explores the function of SIRT2 and its regulatory effect on G6PD in ccRCC, aiming to provide new molecular clues for targeted treatment of patients with ccRCC. Silent information regulator 2 was highly expressed in patients with ccRCC, promoted proliferation, and inhibited the apoptosis of ccRCC cells through regulating cell cycle‐ and apoptosis‐related factors. Moreover, SIRT2 facilitated G6PD activity by deacetylation and increased G6PD protein stability through reducing its ubiquitination and enhancing its SUMO1 modification at the posttranslational level. Additionally, SIRT2 could promote ccRCC development in vivo, indicating that SIRT2 might be a proto‐oncogene and potential therapeutic target for ccRCC.
2. MATERIALS AND METHODS
2.1. Clinical sample collection
All human samples were carried out in accordance with the regulations of Helsinki Declaration and approved by the Ethics Committee of Kunming Medical University. Patients were obtained from the Department of Organ Transplantation of the First Affiliated Hospital of Kunming Medical University and provided signed informed consent.
2.2. Cell culture, treatment, and transfection
ACHN (CRL‐1611; ATCC), 786‐O (CRL‐1932; ATCC), Caki‐1 (HTB‐46; ATCC), and the normal renal tubular epithelial cell line HK‐2 (CRL‐2190; ATCC) were purchased from Kunming Institute of Zoology, Chinese Academy of Sciences. These cell lines were cultured as described previously.8
The SIRT2‐specific inhibitor AGK2 (#A8231; Sigma) was dissolved in DMSO to make a 4.6 mM storage solution. The protein synthesis inhibitor CHX (#GC17198; GLPBIO) was dissolved in DMSO to prepare 100 mg/mL storage solution. Proteasome inhibitor MG132 (#GC10383; GLPBIO) powder was dissolved in DMSO to prepare 10 mg/mL storage solution. SUMOylation inhibitor 2‐D08 (#S8696; Selleck) was dissolved in DMSO to prepare 10 mM storage solution and treated with 10 μM for 24 hours.18, 19 All these stock solutions were kept in the refrigerator at −20°C.
ACHN and 786‐O cells were infected with SIRT2 OE lentivirus or control lentivirus to construct a cell line with stable overexpression of SIRT2. 786‐O and Caki‐1 cells were infected with SIRT2 KD lentivirus (SIRT2 KD 1, 5′‐TAAGCTGGATGAAAGAGAA‐3′; SIRT2 KD 2, 5′‐CAACCATCTGTCACTACTT‐3′; and SIRT2 KD 3, 5′‐CATCTTTGAGATCAGCTAT‐3′) or nonsilencer (5′‐TTCTCCGAACGTGTCACGT‐3′) to establish stable SIRT2 KD cell lines. These viruses were purchased from GeneChem. Puromycin (0.5 μg/mL) was used for stable cells screening after 72 hours.
2.3. Western blot analysis
All cells and xenograft mouse model tissues were lysed using RIPA lysis buffer (#R0010; Solarbio) containing protease inhibitors (#P0100; Solarbio). Western blot analysis was carried out as described previously.20
The Abs used in the study included anti‐G6PD Ab (ab133525; Abcam), anti‐SIRT2 Ab (ab51023; Abcam), anti‐CDK4 Ab (11026‐1‐AP; Proteintech), anti‐cyclin D1 Ab (60186‐1‐Ig; Proteintech), anti‐CDK2 Ab (10122‐1‐AP; Proteintech), anti‐cyclin E1 Ab (11554‐1‐AP; Proteintech), anti‐Bax Ab (50599‐2‐Ig; Proteintech), anti‐Bcl2 Ab (12789‐1‐AP; Proteintech), anti‐GAPDH Ab (bs‐2188R; Bioss), anti‐β‐actin Ab (bs‐0061R; Bioss), anti‐rabbit IgG (111‐035‐003; Jackson ImmunoResearch Laboratories), and anti‐mouse IgG Ab (115‐035‐003; Jackson ImmunoResearch Laboratories).
2.4. Quantitative real‐time PCR analysis
Total RNA was isolated using TRIzol Reagent (#9109; Takara) following the manufacturer’s protocol. Real‐time PCR was carried out using SYBR Green Master (ROX) (#04913914001; Roche) following the manufacturer’s protocol. The primers used were as follows: SIRT2 F, 5′‐CCTTCTACACATCACACTGCGTC‐3′ and SIRT2 R, 5′‐TTCACACTTGGGCGTCACCT‐3′; CDK4 F, 5′‐TCAGCCAGCTTGACTGTTCCA‐3′ and CDK4 R, 5′‐GCCTAGATTTCCTTCATGCCA‐3′; cyclin D1 F, 5′‐GCTGCGAAGTGGAAACCATC‐3′ and cyclin D1 R, 5′‐CCTCCTTCTGCACACATTTGAA‐3′; CDK2 F, 5′‐CCAGGAGTTACTTCTATGCCTGA‐3′ and CDK2 R, 5′‐TTCATCCAGGGGAGGTACAAC‐3′; cyclin E1 F, 5′‐ACTCAACGTGCAAGCCTCG‐3′ and cyclin E1 R, 5′‐GCTCAAGAAAGTGCTGATCCC‐3′; Bax F, 5′‐AGACACTCGCTCAGCTTCTTG‐3′ and Bax R, 5′‐CTTTTGCTTCAGGGTTTCATC‐3′; Bcl2 F, 5′‐GTGCCTGCTTTTAGGAGACCGA‐3′ and Bcl2 R, 5′‐GAGACCACACTGCCCTGTTGATC‐3′; and U6 F, 5′‐CTCGCTTCGGCAGCACA −3′ and U6 R, 5′‐AACGCTTCACGAATTTGCGT‐3′.
2.5. Cell viability, colony formation, and cell cycle assays and EdU staining
An MTS cell proliferation assay kit (#CTB169; Promega) was used for cell viability assay following the manufacturer’s protocol. The colony formation assay was carried out based on a previous report.21 The EdU staining was carried out using a BeyoClick EdU Cell Proliferation Kit with Alexa Fluor 594 (#C00788S; Beyotime). The images were acquired with a fluorescence microscope (#DM4 B, ×400; Leica). The cell cycle was assessed using a Cell Cycle Analysis Kit (#FXP0211; 4A Biotech), following the manufacturer’s protocols. A PARTEC CyFlow Space flow cytometer was used for cell cycle detection.
2.6. Glucose 6‐phosphate dehydrogenase activity analysis
Glucose 6‐phosphate dehydrogenase activity was analyzed using a G6PD assay kit (#GMS70013.1; Genmed) and carried out as described previously.8
2.7. Apoptosis detection
Cell apoptosis was measured by flow cytometry using an apoptosis detection kit (#AD11; Dojindo). A PARTEC CyFlow Space flow cytometer was used for the analysis.
A one‐step TUNEL apoptosis assay Kit (#C1090; Beyotime) was used for the TUNEL assay and a fluorescence microscope (#DM4 B, ×400; Leica) was used for the observation.
2.8. Coimmunoprecipitation assay
Cells were lysed using weak lysate (#P0013D; Beyotime) containing a protease inhibitor cocktail (#04906845001; Roche). The Co‐IP analysis was carried out as described previously using the corresponding Ab.16 The Abs used in this study included anti‐SIRT2 Ab (ab51023; Abcam), anti‐G6PD Ab (sc‐373886; Santa Cruz Biotechnology), anti‐acetyl lysine Ab (ab190479; Abcam), anti‐ubiquitin Ab (ab7780; Abcam), anti‐ubiquitin Ab (#3933; Cell Signaling Technology), anti‐ubiquitin Ab (#3933; Cell Signaling Technology), and anti‐SUMO1 Ab (#4930; Cell Signaling Technology).
2.9. Animal experiments
A total of 12 6‐week‐old female BALB/c nude mice were purchased from the Department of Experimental Animals, Kunming Medical University. The injection and calculation methods are described in a previous article.8 The animal experiments were approved by the Institutional Animal Care and Use Committee of Kunming Medical University.
2.10. Statistical analysis
Data of the SIRT2 expression profile in ccRCC and control tissues were downloaded from TCGA and analyzed using the Mann‐Whitney U test. For animal experiments, the significance of tumor volumes in different groups was determined using mixed ANOVA. For other statistical analyses, unpaired or paired‐sample Student’s t test (two groups) and one‐way ANOVA (multiple groups) were used. All results were expressed as mean ± SD. A P value of less than 0.05 indicated a statistically significant difference.
3. RESULTS
3.1. High expression of SIR2 in ccRCC
Some studies have shown that SIRT2 is highly expressed in certain types of cancers,20, 22, 23 whereas it is slightly expressed in other cancers.4, 24, 25 However, the expression pattern of SIRT2 in ccRCC tumorigenesis remains unknown. We explored the TCGA datasets and found that SIRT2 mRNA levels were significantly high in ccRCC tissues (Figure 1A). We collected 20 ccRCC specimens and relevant adjacent normal tissues and analyzed the mRNA and protein expression levels of SIRT2 by RT‐qPCR and western blot analyses, respectively. The results showed that both the mRNA and protein expressions are significantly higher in ccRCC tissues compared to normal tissues (Figure 1B,C). Likewise, the mRNA and protein expression levels of SIRT2 were higher in ACHN, Caki‐1, and 786‐O cells compared to those HK2 cells (Figure 1D,E). Taken together, these findings indicated that SIRT2 was highly expressed in ccRCC and might be involved in ccRCC tumorigenesis.
FIGURE 1.
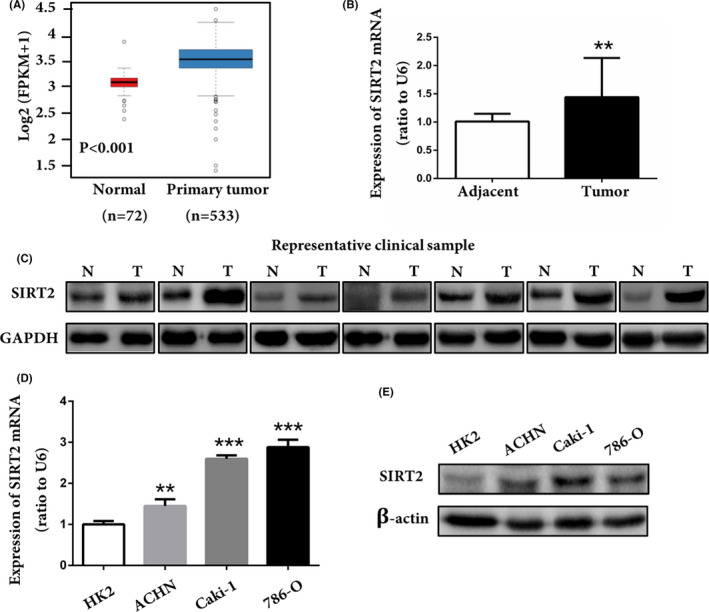
Silent information regulator 2 (SIRT2) was overexpressed in clear cell renal cell carcinoma (ccRCC). A, Transcriptome data of ccRCC samples (n = 533) and normal renal tissue (n = 72) were downloaded from The Cancer Genome Atlas database, and the expression profile of SIRT2 was analyzed. P < .001 vs normal tissue (Mann‐Whitney U test). B, C, mRNA and protein expression of SIRT2 in ccRCC (T) and adjacent tissues (N) from different patients. **P < .01 vs adjacent tissues (paired‐sample Student’s t test). D, E, Expression of SIRT2 in normal renal tubular epithelial cells (HK2) and ccRCC cell lines (ACHN, Caki‐1, and 786‐O) was detected by (D) quantitative real‐time PCR and (E) western blot analysis. Data represent three independent experiments, each carried out in triplicate. **P < .01, ***P < .001 vs HK2 cells (one‐way ANOVA)
3.2. Silent information regulator 2 promoted the proliferation of ccRCC cells in vitro
First, we checked whether the cells were successfully transfected not. The results from RT‐qPCR and western blot analyses showed that the transfected cell lines were successfully established and SIRT2 KD cells with SIRT2 KD 1 sequences were selected for further experiments (Figure S1). Next, we evaluated the function of SIRT2 in ccRCC cell proliferation by different analyses. The cell viability assay showed that ccRCC cells were inhibited in a dose‐dependent manner after AGK2 treatment (Figure 2A). In contrast, SIRT2 OE cells displayed an increase in the proliferation rate (Figure 2B). Colony formation assay and EdU staining analysis also showed similar results (Figure 2C,D). The results of cell cycle analysis revealed that, in SIRT2 OE cells, the proportion of cells in the G0/G1 phase was low, whereas it was high in the S and G2/M phases (Figure 2E, top panel). The opposite results were found in SIRT2 KD cells (Figure 2E, bottom panel). These results supported that SIRT2 accelerated cell cycle progression and promoted ccRCC proliferation. Finally, we undertook a mechanistic evaluation and found that cell cycle proteins, including CDK4, cyclin D1, CDK2, and cyclin E1, were highly expressed in SIRT2 OE cells, whereas they were restrained in SIRT2 KD cells (Figure 3). Taken together, these results suggested that SIRT2 can promote ccRCC cells proliferation by upregulating cell cycle proteins.
FIGURE 2.
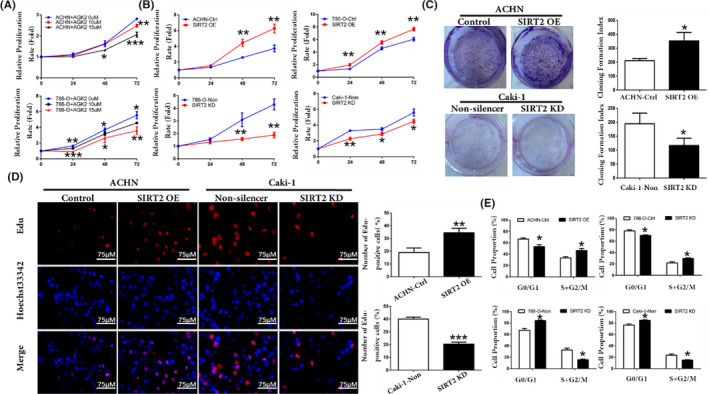
Silent information regulator 2 (SIRT2) promoted clear cell renal cell carcinoma cell proliferation and affected cell cycle phase distribution. A, ACHN and 786‐O cells were treated with SIRT2 specific inhibitor AGK2 (10 or 15 μM), and the cell proliferation rate was detected by MTS assay. B, Relative proliferation rate of ACHN, 786‐O, and Caki‐1 stably transfected cells with SIRT2 overexpression (OE) or knockdown (KD) and the number of relevant control cells were determined using MTS assay at indicated hours after seeding. C, Colony formation assay and quantification assessment were used to determine the proliferation rate of stably transfected ACHN and Caki‐1 cells. D, EdU staining was undertaken in stably transfected ACHN and Caki‐1 cells, and the number of EdU‐positive cells was recorded. E, Cycle distributions of stably transfected ACHN, 786‐O, and Caki‐1 cells were determined by propidium iodide staining and flow cytometry analysis. Data are means ± SD from three independent experiments, each carried out in triplicate. *P < .05, **P < .01, ***P < .001 vs corresponding controls (unpaired sample Student’s t test). Ctrl, control
FIGURE 3.
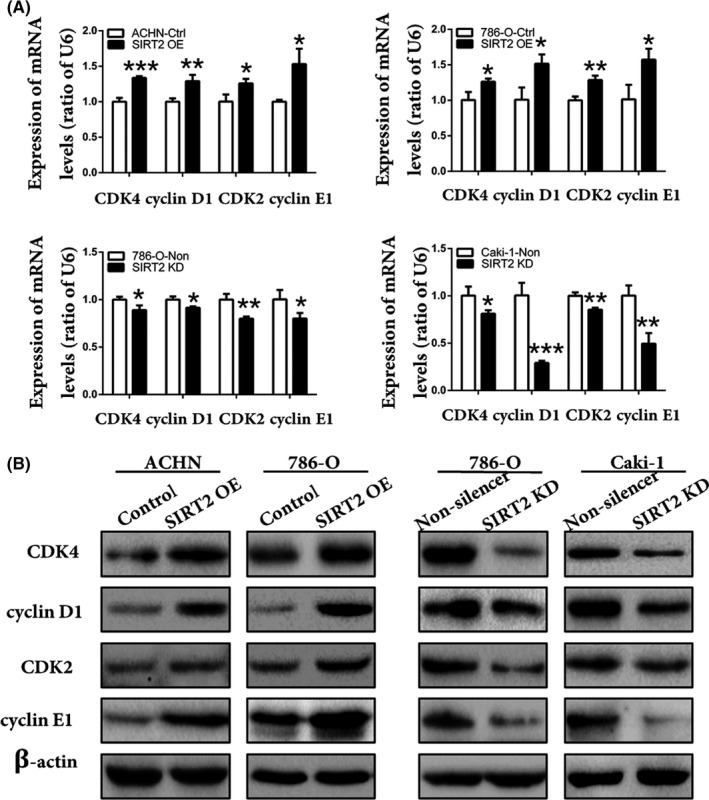
Silent information regulator 2 (SIRT2) positively regulated the expression of cyclin‐dependent kinase (CDK)4, cyclin D1, CDK2, and cyclin E1 in clear cell renal cell carcinoma. A, B, Expression of CDK4, cyclin D1, CDK2, and cyclin E1 in stably transfected ACHN, 786‐O, and Caki‐1 cells was analyzed by (A) quantitative real‐time PCR and (B) western blot analysis. Bars represent means ± SD from three independent experiments, each carried out in triplicate. *P < .05, **P < .01, ***P < .001 vs corresponding controls (unpaired‐sample Student t test). Ctrl, control; KD, knockdown; OE, overexpression
3.3. Silent information regulator 2 inhibited apoptosis of ccRCC cells in vitro
The overexpression of SIRT2 facilitates ccRCC cell proliferation. Hence, this study further explored the effects of SIRT2 on ccRCC cell apoptosis. Flow cytometry and double staining analyses showed that the apoptotic rate of SIRT2 OE cells was low, whereas it was higher in SIRT2 KD cells (Figure 4A). In addition, the TUNEL assay showed that the number of TUNEL‐positive cells increased with SIRT2 knockdown (Figure 4C). Moreover, the western blotting analysis revealed that, when SIRT2 was overexpressed in cells, the expression level of proapoptotic protein Bax decreased whereas the expression level of antiapoptotic protein Bcl2 increased (Figure 5). Taken together, these results suggested that SIRT2 can inhibit ccRCC cells apoptosis by regulating the expression of apoptosis‐related factors Bax and Bcl2.
FIGURE 4.
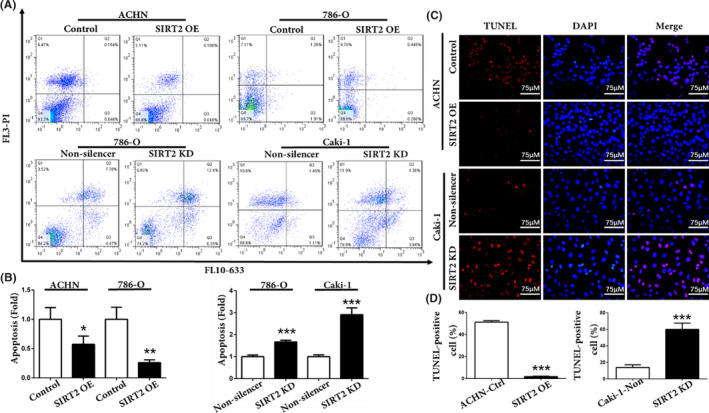
Silent information regulator 2 (SIRT2) inhibited clear cell renal cell carcinoma cell apoptosis. A, B, Apoptosis of stably transfected ACHN, 786‐O, and Caki‐1 cells was (A) measured by flow cytometry and (B) statistically analyzed. C, D, TUNEL assays were used to (C) observe the apoptotic cells and (D) record the number of TUNEL‐positive cells. Bars represent the means ± SD. Data represent three independent experiments, each carried out in triplicate. *P < .05, **P < .01, ***P < .001 vs corresponding controls (unpaired‐sample Student’s t test). KD, knockdown; OE, overexpression
FIGURE 5.
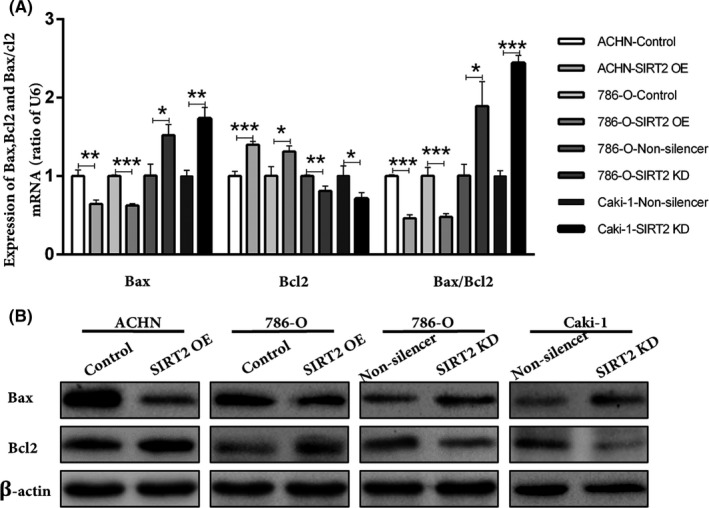
Silent information regulator 2 (SIRT2) regulated the expression of apoptosis‐related factors in clear cell renal cell carcinoma cells. A, B, Expression of Bax and Bcl2 in stably transfected ACHN, 786‐O, Caki‐1, and relevant control cells were detected by (A) quantitative real‐time PCR and (B) western blot analysis. Bars represent means ± SD from three independent experiments, each carried out in triplicate. *P < .05, **P < .01 vs corresponding controls (unpaired‐sample Student’s t test). KD, knockdown; OE, overexpression
3.4. Silent information regulator 2 enhanced G6PD activity through deacetylation
As the aforementioned results showed that SIRT2 played an oncogenic function in ccRCC, we continued our experiments by investigating the possible underlying mechanism. We first analyzed the G6PD activity after treating cells with AGK2 and found that it significantly decreased dose‐dependently (Figure S2). In addition, the results showed an increase of G6PD activity in SIRT2 OE cells and a decrease in SIRT2 KD cells (Figure 6A). However, as shown in Figure 6B,C, the expression of G6PD was not modified following SIRT2 overexpression or knockdown. Our results indicated that SIRT2 might promote G6PD activity through facilitating the formation of its homodimer in ccRCC, as reported in previous studies.12, 13, 26 To elucidate the mechanism, we analyzed the expression of G6PD monomers and dimers after transfections. As shown in Figure 6D, G6PD dimer formation was significantly increased with SIRT2 overexpression while G6PD monomer formation showed obviously opposite results. These data indicated that SIRT2 enhances G6PD activity by promoting its active dimer formation in ccRCC.
FIGURE 6.
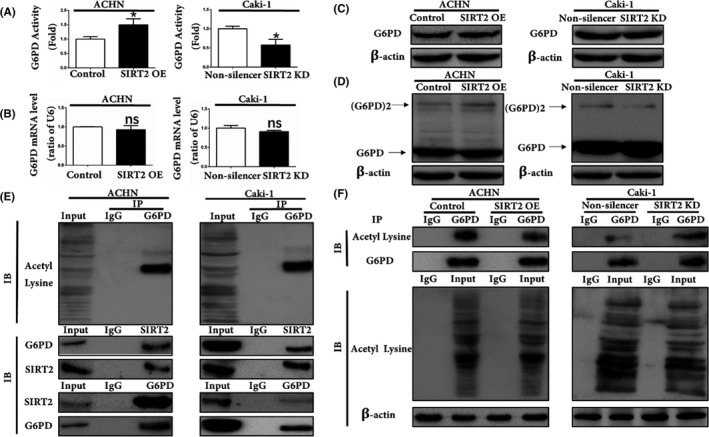
Silent information regulator 2 (SIRT2) enhanced glucose 6‐phosphate dehydrogenase (G6PD) activity by deacetylation in clear cell renal cell carcinoma. A, G6PD activity was measured in stably transfected ACHN and Caki‐1 cells. **P < .01 vs corresponding controls (unpaired‐sample Student’s t test). B, C, mRNA and total protein expression levels of G6PD in SIRT2‐overexpressing (OE) ACHN cells and SIRT2‐knockdown (KD) Caki‐1 cells were detected by (B) quantitative real‐time PCR and (C) western blot analysis. D, G6PD dimer and monomer expression levels in ACHN SIRT2 OE and Caki‐1 KD cells were analyzed using western blot assays with glutaraldehyde cross‐linking. E, Acetylation level of G6PD and interactions between SIRT2 and G6PD were analyzed by coimmunoprecipitation (Co‐IP) assays in ACHN and Caki‐1 cells. F, G6PD acetylation levels were determined by quantitative Co‐IP analysis in stably transfected ACHN and Caki‐1 cells. Data represent three independent experiments, each carried out in triplicate. IB, immunoblot
Subsequently, Co‐IP analysis was undertaken to detect the acetylation status of G6PD and the interaction between SIRT2 and G6PD. The results indicated that G6PD was acetylated in ccRCC cells (Figure 6D, top panel) and that SIRT2 and G6PD interact with each other to form a protein‐protein complex in ccRCC cells (Figure 6E, bottom panel). The quantitative Co‐IP analysis showed that the G6PD acetylation level was remarkably low in SIRT2 OE cells (Figure 6F). Taken together, these results indicated that SIRT2 formed a protein‐protein complex with G6PD and deacetylated it in order to promote its active dimer formation and enhance its activity in ccRCC.
3.5. Silent information regulator 2 stabilized G6PD by affecting the balance between its ubiquitination and SUMOylation
In addition to transcriptional regulation, the amount of protein in a cell is also regulated by degradation. The protein half‐life of G6PD was first analyzed using CHX to inhibit the protein synthesis. The G6PD protein level was detected at indicated time points by western blot analysis. The results showed that the overexpression of SIRT2 leads to an increase in G6PD protein half‐life, whereas SIRT2 knockdown caused an opposite result (Figure 7A,B). However, when cells were treated with CHX combined with the proteasome inhibitor MG132, the changes in G6PD protein half‐life were diminished (Figure 7A,B, bottom panel). These results indicated that SIRT2 overexpression favored G6PD protein stability in ccRCC, while SIRT2 knockdown promoted G6PD degradation probably through the ubiquitin proteasome pathway. As shown in Figure 7C, the G6PD ubiquitination level was reduced by SIRT2 overexpression, whereas it was increased following SIRT2 knockdown. Furthermore, SIRT2 overexpression or knockdown had no effect on the whole ubiquitination level in ccRCC cells.
FIGURE 7.
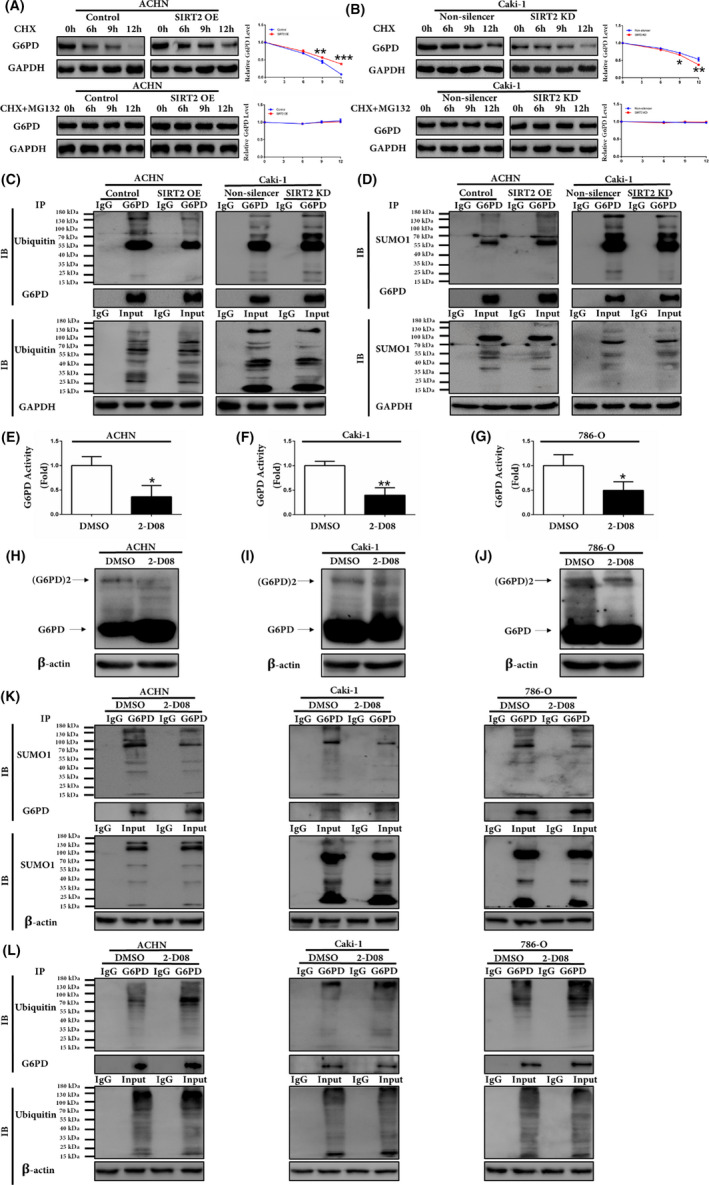
Silent information regulator 2 (SIRT2) increased glucose 6‐phosphate dehydrogenase (G6PD) protein stability by reducing G6PD ubiquitination and enhancing G6PD small ubiquitin‐related modifier 1 (SUMO1) modification. A, B, G6PD protein expression levels were analyzed by western blot analysis in stably transfected ACHN and Caki‐1 cells following treatment with 100 μg/mL cycloheximide (CHX) with or without 10 μg/mL MG132 for 0, 6, 9, 12 h. C, Expression levels of G6PD ubiquitination were analyzed by quantitative coimmunoprecipitation (Co‐IP) assays in stably transfected ACHN and Caki‐1 cells. D, Expression levels of G6PD SUMO1 modification were analyzed by quantitative Co‐IP assays in stably transfected ACHN and Caki‐1 cells. E‐G, G6PD activity was measured in ACHN, Caki‐1, and 786‐O cells with DMSO or 2‐D08. H‐J, G6PD dimer and monomer expression level in ACHN, Caki‐1, and 786‐O cells with DMSO or 2‐D08 were analyzed using western blot assays with glutaraldehyde cross‐linking. K, L, Expression levels of G6PD SUMO1 modification and ubiquitination were analyzed by quantitative Co‐IP assays in ACHN, Caki‐1, and 786‐O cells with DMSO or 2‐D08. Data represent three independent experiments, each carried out in triplicate. *P < .05, **P < .01, ***P < .001 vs corresponding controls (unpaired‐sample Student’s t test). IB, immunoblot
SUMOylation is structurally similar to ubiquitylation but exerts completely opposite functions in mediating protein stability.15 As shown in Figure 7D, SIRT2 overexpression could efficiently stimulate SUMO1 modification of G6PD. Moreover, total protein expression was not modified by SUMO1 in stably transfected SIRT2 cells. Figure 7E‐G shows that G6PD activity decreased significantly after treatment with SUMOylation inhibitor 2‐D08. At the same time, G6PD dimer formation decreased with 2‐D08 while G6PD monomer formation showed obviously opposite results (Figure 7H‐J). As shown in Figure 7K,L, the expression of G6PD SUMO1 significantly reduced after adding 2‐D08 in three ccRCC cell lines while the G6PD ubiquitination level was clearly increased. Taken together, these results indicated that SIRT2 could promote G6PD protein stability by reducing G6PD ubiquitination and enhancing its SUMO1 modification.
3.6. Silent information regulator 2 promoted ccRCC progression in vivo
The aforementioned results showed that SIRT2 overexpression contributes to ccRCC proliferation following G6PD overactivation in vitro. Subsequently, we investigated whether SIRT2 could promote ccRCC progression in vivo, by using nude mouse xenografted models (Figure 8A). We found that tumor growth was significantly faster with SIRT2 overexpression, but slower with SIRT2 knockdown (Figure 8B). Additionally, tumor weight significantly increased with SIRT2 overexpression (Figure 8C). The G6PD activity was also increased under in vivo conditions when SIRT2 was overexpressed (Figure 8D). Moreover, as shown in Figure 8E,F, no significant changes in G6PD expression were observed while SIRT2 was overexpressed. These results obtained under in vivo conditions confirmed the oncogenic function of SIRT2 in ccRCC.
FIGURE 8.
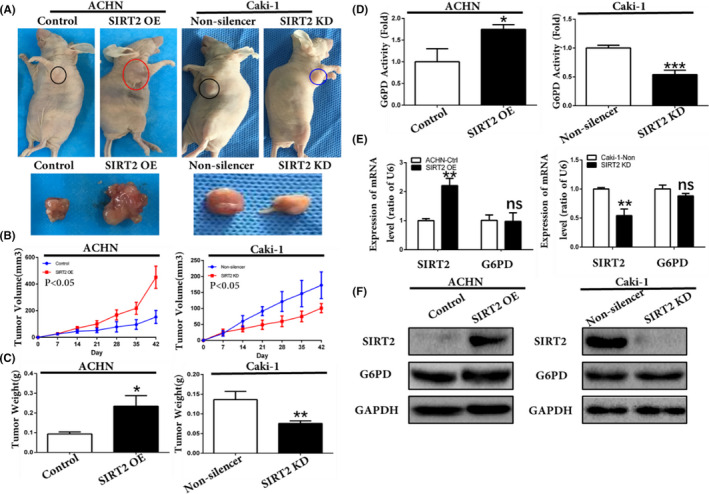
Silent information regulator 2 (SIRT2) promoted clear cell renal cell carcinoma development in vivo. A, Stably transfected ACHN, Caki‐1, and relevant control cells (1 × 107) were injected subcutaneously into the right oxter flank of BALB/c nude mice. Representative images of tumor‐bearing mice (top panel) and the tumors isolated from each group (bottom panel) on day 42. B, Tumor size was monitored on indicated days after injection. P < .05 vs corresponding controls (mixed ANOVA). C, Tumor weights of each group were measured after the mice were killed and samples were harvested. *P < .05, **P < .01 vs control or nonsilencer (unpaired‐sample Student’s t test). D, Glucose 6‐phosphate dehydrogenase (G6PD) activities of tumors in each group were measured. *P < .05, ***P < .001 vs control or nonsilencer (unpaired‐sample Student’s t test). E, F, SIRT2 and G6PD expression levels in tumors in each group were analyzed by (E) quantitative real‐time PCR and (F) western blot analysis. Bars represent means ± SD from three independent experiments, each carried out in triplicate. **P < .01. ns, P > .05 vs corresponding controls (unpaired‐sample Student’s t test). Ctrl, control; KD, knockdown; OE, overexpression
4. DISCUSSION
Previous studies indicated that SIRT2 plays contradictory roles in different types of human cancers.27 A report suggested that SIRT2 deacetylated CDH1 and CDC20 to enhance the activation of APC/C ligase complex in order to prevent the development of breast cancer.24 Conversely, in hepatocellular carcinoma, SIRT2 activated β‐catenin to promote epithelial‐mesenchymal transition.23 However, the role of SIRT2 in ccRCC is not clearly established. In this study, we explored the expression pattern of SIRT2 in ccRCC and unraveled its function in ccRCC tumorigenesis. We found that SIRT2 was highly expressed and played an oncogenic role in ccRCC. Our finding was consistent with that of previous groups.1
Previous studies showed that SIRT2 is highly expressed in glioma and promotes glioma cell proliferation;26 SIRT2 could also inhibit apoptosis in cholangiocarcinoma cells and promote tumor cell metabolic reprogramming.21 Our results confirmed that the overexpression of SIRT2 promoted ccRCC cell proliferation and attenuated cell apoptosis through regulating cell cycle‐ and apoptosis‐related factors. In addition, SIRT2 could also facilitate ccRCC development in vivo. These results provided strong evidence that SIRT2 might be a tumor promoter of ccRCC.
Glucose 6‐phosphate dehydrogenase plays a key role in various cancers, including ccRCC.8 It is a highly conserved protein with invariant regulatory sites. In fact, Wang et al discovered that two acetylated lysines (K171 and K403) are conserved sites of G6PD. They proved that K403 acetylation impairs the formation of dimeric G6PD and thus inhibits its activity.12 However, this is not irreversible. Studies showed that SIRT2 deacetylates G6PD, leading to its activation. It was reported in leukemia and glioma, where SIRT2 plays an oncogenic role.13, 26 Moreover, we and others have previously found that G6PD shows high activity in ccRCC.8, 10, 11 Taking this evidence into account, we hypothesized that there might be a relation between SIRT2 overexpression and the aberrant activation of G6PD in ccRCC. As expected, our results indicated that SIRT2 interacts with G6PD in ccRCC. As a result, the acetylation level of G6PD diminished and the enzyme activity was enhanced. Knowing that G6PD activity in ccRCC can be regulated at the transcriptional level, as revealed in previous studies,7, 8 we evaluated the mRNA and protein expressions levels of G6PD following silencing and overexpression of SIRT2. Our results displayed no obvious changes in the G6PD expression after modification of SIRT2 expression. This clearly shows that the SIRT2‐mediated G6PD activation in ccRCC occurs at the posttranslational level.
Ubiquitination and SUMOylation are important processes that regulate protein stability. For instance, a recent study showed that high glucose‐induced ubiquitination of G6PD hampered its stability.14 Thus, after finding that SIRT2 overexpression favored G6PD stability and increased its half‐life, we investigated the underlying mechanism. Giving that SUMOylation produces an opposite effect to that of ubiquitination, we hypothesized that SIRT2 overexpression might restore G6PD stability by engaging SUMOylation. Indeed, our results revealed that SIRT2 overexpression leads to SUMO1 modification of G6PD, and thereby induces its stability. In addition, G6PD activity and stability were significantly reduced after treatment with SUMO1 inhibitor 2‐D08 in ccRCC cells. These data provided novel clues for the regulation of G6PD activity in ccRCC. That is, SIRT2 promotes G6PD dimer formation through deacetylation. This is accompanied by the maintenance of G6PD stability, which is mediated by SIRT2 through the inhibition of ubiquitination and instigation of SUMOylation of G6PD. Therefore, the present study showed that G6PD activity can be regulated at the posttranslational level in ccRCC to contribute to disease progression.
In summary, this study showed that SIRT2 was aberrantly overexpressed in ccRCC and contributed to disease progression. Functionally, SIRT2 promoted ccRCC proliferation and inhibited apoptosis in vitro, and promoted tumor development in vivo. Silent information regulator 2 induced ccRCC progression by enhancing G6PD activity through its deacetylation, ubiquitination inhibition, and SUMOylation. Nonetheless, the regulatory sites of G6PD implicated in the SIRT2‐mediated ubiquitination and SUMOylation in ccRCC might be the subject of future investigations. Taken together, the findings of the present study supported the oncogenic role of SIRT2 in ccRCC and proposed novel therapeutic ways for ccRCC treatment.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
Fig S1‐S2
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Nos. 81960462, 81760455, 31960200, 31660246, and 31960145) and Yunnan province applied research funds (2018FE468(‐001), 2018FB120, and 2019FB091).
Ni Y, Yang Z, Agbana YL, et al. Silent information regulator 2 promotes clear cell renal cell carcinoma progression through deacetylation and small ubiquitin‐related modifier 1 modification of glucose 6‐phosphate dehydrogenase. Cancer Sci. 2021;112:4075–4086. 10.1111/cas.15085
Ni and Yang are equal contributors.
Contributor Information
Qiao Zhang, Email: zhangqiao200824@126.com.
Yingmin Kuang, Email: yingmin1512@aliyun.com.
Yuechun Zhu, Email: zhuyuechun20091119@163.com.
REFERENCES
- 1.Shuch B, Amin A, Armstrong AJ, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67(1):85‐97. [DOI] [PubMed] [Google Scholar]
- 2.Peng XS, Yang JP, Qiang YY, et al. PTPN3 Inhibits the Growth and Metastasis of Clear Cell Renal Cell Carcinoma via Inhibition of PI3K/AKT Signaling. Mol Cancer Res. 2020;18(6):903‐912. [DOI] [PubMed] [Google Scholar]
- 3.Wang T, Xu Z, Lu Y, et al. Recent Progress on the Discovery of Sirt2 Inhibitors for the Treatment of Various Cancers. Curr Top Med Chem. 2019;19(12):1051‐1058. [DOI] [PubMed] [Google Scholar]
- 4.Zhu H, Hu Y, Zeng C, et al. The SIRT2‐mediated deacetylation of AKR1C1 is required for suppressing its pro‐metastasis function in Non‐Small Cell Lung Cancer. Theranostics. 2020;10(5):2188‐2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou W, Ni TK, Wronski A, et al. The SIRT2 Deacetylase Stabilizes Slug to Control Malignancy of Basal‐like Breast Cancer. Cell Rep. 2016;17(5):1302‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei R, He D, Zhang X. Role of SIRT2 in Regulation of Stemness of Cancer Stem‐Like Cells in Renal Cell Carcinoma. Cell Physiol Biochem. 2018;49(6):2348‐2357. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Yi X, Yang Z, et al. Overexpression of G6PD Represents a Potential Prognostic Factor in Clear Cell Renal Cell Carcinoma. J Cancer. 2017;8(4):665‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q, Yang Z, Han Q, et al. G6PD promotes renal cell carcinoma proliferation through positive feedback regulation of p‐STAT3. Oncotarget. 2017;8(65):109043‐109060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Han Q, Yang Z, et al. G6PD facilitates clear cell renal cell carcinoma invasion by enhancing MMP2 expression through ROS‐MAPK axis pathway. Int J Oncol. 2020;57(1):197‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langbein S, Frederiks WM, zur Hausen A, et al. Metastasis is promoted by a bioenergetic switch: new targets for progressive renal cell cancer. Int J Cancer. 2008;122(11):2422‐2428. [DOI] [PubMed] [Google Scholar]
- 11.Frederiks WM, Bosch KS, Hoeben KA, van Marle J, Langbein S. Renal cell carcinoma and oxidative stress: The lack of peroxisomes. Acta Histochem. 2010;112(4):364‐371. [DOI] [PubMed] [Google Scholar]
- 12.Wang YP, Zhou LS, Zhao YZ, et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33(12):1304‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu SN, Wang TS, Li X, Wang YP. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci Rep. 2016;6:32734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Hu J, Yan L, et al. High glucose‐induced ubiquitination of G6PD leads to the injury of podocytes. FASEB J. 2019;33(5):6296‐6310. [DOI] [PubMed] [Google Scholar]
- 15.Melchior F. SUMO–nonclassical ubiquitin. Annu Rev Cell Dev Biol. 2000;16:591‐626. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Chen Y, Wang S, et al. PIASxα ligase enhances SUMO1 modification of PTEN protein as a SUMO E3 ligase. J Biol Chem. 2014;289(6):3217‐3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Floris A, Mazarei M, Yang X, et al. SUMOylation Protects FASN Against Proteasomal Degradation in Breast Cancer Cells Treated with Grape Leaf Extract. Biomolecules. 2020;10(4):529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou P, Chen X, Li M, et al. 2–D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem Biophys Res Comm. 2019;513(4):1063‐1069. [DOI] [PubMed] [Google Scholar]
- 19.Lorente M, García‐Casas A, Salvador N, et al. Inhibiting SUMO1‐mediated SUMOylation induces autophagy‐mediated cancer cell death and reduces tumour cell invasion via RAC1. J Cell Sci. 2019;132(20):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Zhang M, Dorfman RG, et al. SIRT2 Promotes the Migration and Invasion of Gastric Cancer through RAS/ERK/JNK/MMP‐9 Pathway by Increasing PEPCK1‐Related Metabolism. Neoplasia (New York, NY). 2018;20(7):745‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu L, Wang L, Zhou L, et al. The SIRT2/cMYC Pathway Inhibits Peroxidation‐Related Apoptosis In Cholangiocarcinoma Through Metabolic Reprogramming. Neoplasia (New York, NY). 2019;21(5):429‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funato K, Hayashi T, Echizen K, et al. SIRT2‐mediated inactivation of p73 is required for glioblastoma tumorigenicity. EMBO Rep. 2018;19(11):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Chan AW, To KF, et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase‐3β/β‐catenin signaling. Hepatology. 2013;57(6):2287‐2298. [DOI] [PubMed] [Google Scholar]
- 24.Kim HS, Vassilopoulos A, Wang RH, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20(4):487‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang LL, Zhan L, Jin YD, et al. SIRT2 mediated antitumor effects of shikonin on metastatic colorectal cancer. Eur J Pharmacol. 2017;797:1‐8. [DOI] [PubMed] [Google Scholar]
- 26.Ye H, Huang H, Cao F, Chen M, Zheng X, Zhan R. HSPB1 Enhances SIRT2‐Mediated G6PD Activation and Promotes Glioma Cell Proliferation. PLoS One. 2016;11(10):e0164285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55:100961. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2