

Summary
Due to climate change, drought has become a severe abiotic stress that affects the global production of all crops. Elucidation of the complex physiological mechanisms underlying drought tolerance in crops will support the cultivation of new drought‐tolerant crop varieties. Here, two drought‐tolerant lines, RIL70 and RIL73, and two drought‐sensitive lines, RIL44 and RIL93, from recombinant inbred lines (RIL) generated from maize drought‐tolerant line PH4CV and drought‐sensitive line F9721, were selected for a comparative RNA‐seq study. Through transcriptome analyses, we found that gene expression differences existed between drought‐tolerant and ‐sensitive lines, but also differences between the drought‐tolerant lines, RIL70 and RIL73. ZmbHLH124 in RIL73, named as ZmbHLH124 T‐ORG which origins from PH4CV and encodes a bHLH type transcription factor, was specifically up‐regulated during drought stress. In addition, we identified a substitution in ZmbHLH124 that produced an early stop codon in sensitive lines (ZmbHLH124 S‐ORG). Overexpression of ZmbHLH124 T‐ORG, but not ZmbHLH124 S‐ORG, in maize and rice enhanced plant drought tolerance and up‐regulated the expression of drought‐responsive genes. Moreover, we found that ZmbHLH124T‐ORG could directly bind the cis‐acting elements in ZmDREB2A promoter to enhance its expression. Taken together, this work identified a valuable genetic locus and provided a new strategy for breeding drought‐tolerant crops.
Keywords: drought stress, maize, recombinant inbred lines, RNA‐seq, ZmbHLH124
Introduction
Maize (Zea mays L.) is grown more than any other staple cereal with the current grain production at about 1 billion tons/year globally. The majority of maize germplasms are produced in rain‐fed regions and susceptible to drought, and maize is sensitive to drought and intolerant of water‐deficient soils because of its shallow roots. Thus, water scarcity can lead to huge reduction in maize yield, limit maize expansion outside present‐day agriculture areas, and trigger enormous social problems (Chaves and Oliveira, 2004; Fang and Xiong, 2015). Given ongoing climate crisis, the development of maize germplasm with enhanced drought tolerance and higher water use efficiency (WUE) has become an urgent goal for major breeding programs (Ribaut et al., 2009).
Previous studies in Arabidopsis and rice have elucidated multiple drought response pathways and metabolic networks. The processes include stress signal perception, signal transduction and amplification, and adjustments at the whole‐plant level. Genes encoding various transcription factor (TF) family members, such as ABA‐responsive element‐binding proteins/factors (AREBs/ABFs), dehydration‐responsive element‐binding factors (DREBs), basic leucine zipper (bZIP) proteins, zinc‐finger proteins, and NAC (NAM‐ATAF‐CUC2) TFs, play important roles as hubs of transcription networks and have been used to elicit multiple biochemical and developmental pathways to improve plant performance during drought stress (Kuromori, 2014; Hu and Xiong, 2014; Seki et al., 2007; Zhu, 2016). To date, a few studies have been conducted to reveal how maize responds to drought stress and several new key components have been discovered to modulate plant response to drought stress (Kakumanu et al., 2012; Liu et al., 2015; Thatcher et al., 2016; Zhang et al., 2014). Basic helix‐loop‐helix (bHLH) proteins are found in the three eukaryotic kingdoms (fungi, plants and animals) and constitute the second largest family of TFs in plants, but their function in drought tolerance remains poorly described (Carretero‐Paulet et al., 2010; Feller et al., 2011). Three bHLH TFs, ZmPIF3 (Gao et al., 2015), ZmPIF1 (Gao et al., 2018), and ZmPTF1 (Li et al., 2019), have been reported to be involved in drought response in maize. Currently, identifying the genetic components underlying drought tolerance is still considered to be challenging in maize owing to the complexity of genome (Jiao et al., 2017; Schnable et al., 2009) and the multi‐strategic nature of drought response. However, RNA‐seq is a powerful method for understanding transcriptomic dynamics across different tissues or conditions with little background noise, concise normalization of data sets, and relatively low cost (Hrdlickova et al., 2017; Wang et al., 2009).
In this study, we screened four lines with different drought‐responsive phenotypes isolated from a recombinant line population for RNA‐seq analysis. RIL44 and RIL93 were drought‐sensitive lines, whereas RIL70 and RIL73 were drought‐tolerant lines. After comparative transcriptomic analyses, we found that the drought tolerance mechanism of RIL73 was different from that of RIL70. According to gene expression patterns, we identified a bHLH type TF, named ZmbHLH124, involved in drought response of maize. Transgenic analysis showed that ZmbHLH124 T‐ORG overexpression in both rice and maize improved survival rates and decreased water loss rates when plants were treated with drought treatment. Moreover, ZmbHLH124T‐ORG could bind recognition sites in the promoter of ZmDREB2A, a well‐known drought response TF, and promote its expression to enhance drought tolerance in plants. Our study provided a new avenue to discover drought‐responsive genes and identified a valuable signalling protein for maize breeding.
Results
Phenotypic and physiological characteristics of RILs responding to drought stress
The contribution of natural variation to stress tolerance in plants has been observed in many plant species. To discover genetic components related to drought tolerance in maize, a population of recombinant inbred lines (RILs), together with their parental lines, the drought‐tolerant line PH4CV and the drought‐sensitive line F9721 (Figure S1), were grown to perform drought tolerance screening at the seedling stage (Min et al., 2016). Four lines with contrasting drought response phenotypes, two sensitive lines and two tolerant lines, were selected for physiological and molecular analysis to try to discover the genes involved. After one day without water (DT1), there were no obvious phenotype differences among the four lines, while after 2 days (DT2), sensitive lines exhibited growth inhibition compared to tolerant lines. This difference was particularly distinct after 5 days (DT5) when sensitive lines showed wilted and curled leaves while leaves of tolerant lines remained green and erect. As shown in Figure 1a, RIL44 and RIL93 are drought‐sensitive lines while RIL70 and RIL73 are drought‐tolerant lines.
Figure 1.
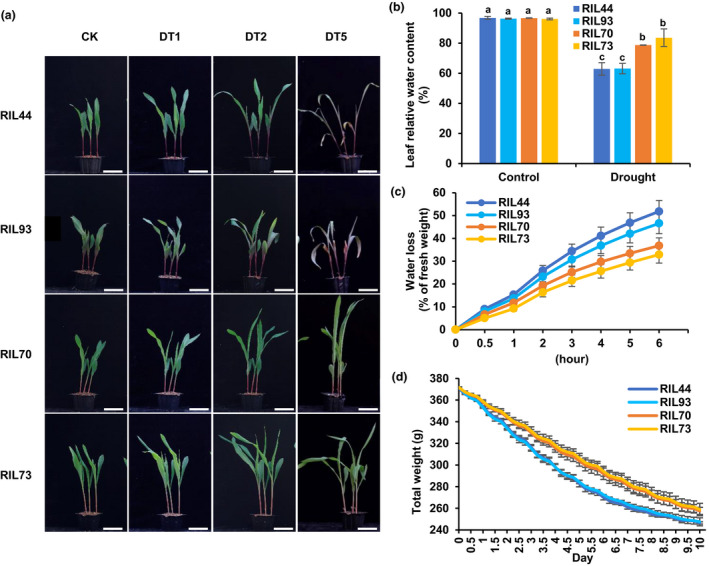
The physiological characteristics of four different lines in response to drought stress. (a) Phenotypic changes of four lines during drought treatment. CK, DT1, DT2, and DT5 refer to drought treatment for 0, 1, 2 and 5 days, respectively. Bar = 5 cm. (b) Leaf relative water content. The leaves were collected when the soil moisture content was about 86% (control) and 12% (drought). Statistical significance was determined by one‐way ANOVA Duncan’s multiple range tests (P < 0.05). (c) Water loss rates of detached leaves. (d) Water loss rates of the whole seedlings during drought treatment. Y‐axis represents the total weight of pots, soil and seedlings. Error bars, s.d., calculated from the results of at least three independent experiments.
Leaf water content is an important parameter used for determining plant drought tolerance, and guiding fertilizer application and irrigation regimens (Jin et al., 2017). Plants tolerant to drought reliably show stable water content, therefore, we measured the water retention capacity of the four lines. The leaf relative water contents (LRWC) of tolerant lines were higher than those of sensitive lines when they were grown in drought conditions with a soil moisture level of 12%, while the LRWC of four lines showed no obvious differences under well‐watered conditions with a soil moisture level of 86% (Figure 1b). The detached leaves of tolerant lines, RIL70 and RIL73, lost water slower than those of the sensitive lines, RIL44 and RIL93 (Figure 1c). To estimate water transpiration during drought treatment, we further examined the weight loss of plants grown in soil. Figure 1d showed a continuous water loss pattern of seedlings during a 12‐day period. Starting from equal weight and without additional water supplementation, RIL70 and RIL73 lost less weight than RIL44 and RIL93. These results indicated that RIL70 and RIL73 could maintain a more stable water status than RIL44 and RIL93 under water deficit.
Differential analysis of chromosome regions constructed by bin map through RNA‐seq
The different drought response phenotypes of the four RILs could be due to the rearrangement and combination of parental genomes. To better understand RIL genetic diversity and transcriptome changes during drought treatment, RNA‐seq was performed to facilitate the comparative analysis. Leaves of the four RILs sampling at four‐time points (CK, DT1, DT2, and DT5) during drought treatment were harvested and the total RNAs were extracted for RNA‐seq. Two biological replicates per RIL at each time point were used for sequencing and reducing experimental errors. After measuring the gene expression level correlation among replicates, the Pearson correlation coefficient’s square was at least above 0.96 (R 2 > 0.96) between two replicates of each sample (Figure S2), indicating that expression patterns of each sample between two replicates were significantly similar and the data was suitable and reliable for subsequent studies.
First, SNPs of each RIL were identified from RNA‐seq data by using maize B73 RefGen_v3 as the reference genome. Then, the four RIL genotypes (Table S1) were called according to the physical positions of SNPs. Because of the lack of detailed parental genome information, we could not specify the genetic sources of individual SNPs, that is to say, at least two genotypes were needed to distinguish genetic backgrounds and find regions involved in stress response. Thus, we combined four genotypes into one file for comparative analysis. In order to find the differences in response to drought stress among RILs, or the uniqueness of drought response of each RIL, we constructed bin maps (Figure S3) of RIL44, RIL93, RIL70, and RIL73 without parental genotypes by using a modified sliding window method (Huang et al., 2009) to discover each RIL’s specific chromosome regions after excluding the common regions shared by any of the other three RILs in our study. Bin maps could be homozygous or heterozygous depending on the types of SNPs in the sliding windows. Finally, 15 regions of the four types were detected in total by comparing these bin maps (Table 1), including the specific region of each RIL, common regions between two RILs, or among three RILs, and common regions of all four RILs.
Table 1.
Gene numbers in different chromosome regions among four RILs
| Region no. | Region type | DEG number in these regions | Total gene number in these regions |
|---|---|---|---|
| 1 | RIL44 specific regions different from other three RILs | 162 | 7057 |
| 2 | RIL93 specific regions different from other three RILs | 684 | 5161 |
| 3 | RIL70 specific regions different from other three RILs | 89 | 6540 |
| 4 | RIL73 specific regions different from other three RILs | 488 | 8923 |
| 5 | common regions between RIL44 and RIL93 different from RIL70 or RIL73 background | 903 | 6951 |
| 6 | common regions between RIL44 and RIL70 different from RIL73 or RIL93 background | 168 | 5946 |
| 7 | common regions between RIL44 and RIL73 different from RIL70 or RIL93 background | 973 | 14 492 |
| 8 | common regions between RIL93 and RIL70 different from RIL73 or RIL93 background | 2286 | 18 476 |
| 9 | common regions between RIL70 and RIL73 different from RIL44 or RIL93 background | 381 | 6065 |
| 10 | common regions between RIL73 and RIL93 different from RIL44 or RIL70 background | 1292 | 8933 |
| 11 | common regions among RIL70, RIL44 and RIL93 different from RIL73 background | 662 | 4852 |
| 12 | common regions among RIL70, RIL44 and RIL73 different from RIL93 background | 313 | 3909 |
| 13 | common regions among RIL70, RIL73 and RIL93 different from RIL44 background | 548 | 3806 |
| 14 | common regions among RIL44, RIL73 and RIL93 different from RIL70 background | 528 | 3432 |
| 15 | common regions among four RILs | 42 | 188 |
DEG, Differentially Expressed Gene.
Analysis of differentially expressed TFs in RIL73 specific regions
To discover genes related to drought tolerance in maize, we followed with interest in specific regions in the drought‐tolerant lines RIL70 and RIL73. We found that RIL73 had a higher capacity for leaf water retention than RIL70 (Figure 1b,c); therefore, we speculated that there could be regions specific in RIL73 for this phenotype. Therefore, genes in specific regions of RIL73 (red boxes in Figure S3) were targets for further analysis.
Among the 488 differentially‐expressed genes (DEGs) in specific regions of RIL73 (Table 1), 31 DEGs were found to have DNA binding activity and/or TF activity based on gene annotations (Table S2). According to gene expression patterns, they were divided into two clusters: one cluster was repressed genes and the other was induced genes under drought stress (Figure 2a). After searching the detailed information of homologous genes of these 31 TFs, eight genes with exhibiting upregulation by drought treatment, GRMZM2G041761, GRMZM2G042895, GRMZM2G081782, GRMZM2G091044, GRMZM2G125775, GRMZM2G132550, GRMZM2G155217, and GRMZM5G801627 annotated as stress response, drought tolerance, or unknown, were selected to validate the accuracy and reproducibility of RNA‐seq data by quantitative real‐time PCR (qRT‐PCR). The result showed that the expression of these genes was up‐regulated by drought stress (Figure 2b). In addition, among the four unknown function genes, GRMZM2G041761, GRMZM2G042895, GRMZM2G091044, and GRMZM2G132550, only GRMZM2G132550 from RIL73 had the same genetic background as PH4CV, and the genetic backgrounds of the other three genes were the same as that of drought‐sensitive line F9721. Thus, we chose GRMZM2G132550 as the candidate gene for drought analysis.
Figure 2.
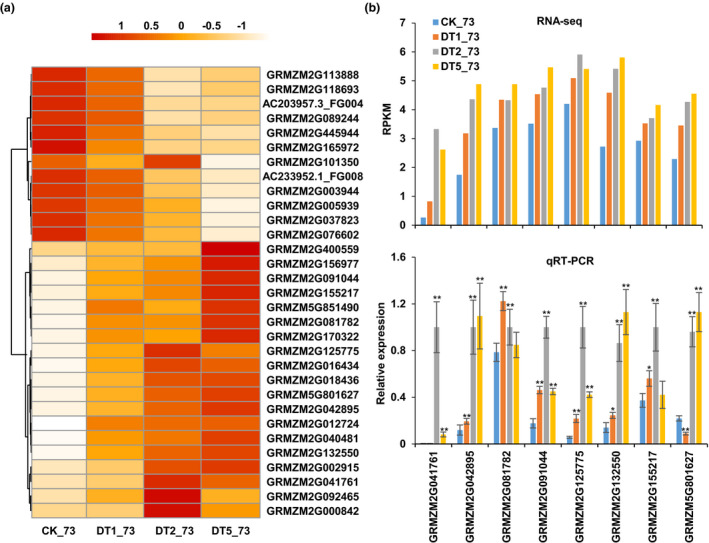
Differentially expressed transcription factors in specific regions from RIL73. (a) Heat map of transcription factors with differential expression in specific regions. Colour was determined by Z score. (b) Validation of RNA‐seq results by qRT‐PCR. The top half is the result of RNA‐seq, the bottom half is the result of qRT‐PCR. RPKM: Reads Per Kilobase per Million mapped reads. Error bars, s.d., calculated from the results of at least three independent experiments. Statistical significance compared with CK was determined by a two‐sided t‐test: *P < 0.05, **P < 0.01.
ZmbHLH124 is involved in drought tolerance in plants
GRMZM2G132550 encodes a basic helix‐loop‐helix (bHLH) type TF referred to as ZmbHLH124 in MaizeGDB (https://www.maizegdb.org/). Thus, we designated this gene as such and will heretofore refer to it in this fashion. We amplified and sequenced the genomic sequence of ZmbHLH124 from two parental lines, ZmbHLH124 T‐ORG originally from the drought‐tolerant line PH4CV (T‐ORG) and ZmbHLH124 S‐ORG from the drought‐sensitive line F9721 (S‐ORG). In comparison to ZmbHLH124 T‐ORG, two main variants, indel +936 (5 bp) and indel +1458 (647 bp), were discovered downstream from the transcript start site in ZmbHLH124 S‐ORG (Figure 3a). Indel +936 was a short fragment substitution, in ZmbHLH124 T‐ORG, the nucleotide sequences from +936 to +949 were 5′‐CGAGGCTCTCAAGA‐3′, while in the ZmbHLH124 S‐ORG, sequences from +936 to +949 were 5′‐TATATGCTAAGGATTATAT‐3′, which resulted in an immediate TAA translation stop codon gained in ZmbHLH124 S‐ORG. Indel +1458 was a 647 bp deletion in the fourth intron of ZmbHLH124 S‐ORG, and adapted as a marker to screen near‐isogenic lines (NIL). From the structural changes caused by DNA substitution, we speculated that ZmbHLH124 S‐ORG might lose its function because of the premature stop codon which might disrupt the dimerization domain of the predicted bHLH TF (Figure S4). To verify whether ZmbHLH124 T‐ORG can complement the lost function of ZmbHLH124 S‐ORG in maize line S‐ORG, we introgressed the ZmbHLH124 T‐ORG into the S‐ORG by generating NIL‐ZmbHLH124 T‐ORG through five generations of successive backcrossing of the F1 plants (RIL73 × S‐ORG). For each generation, the ZmbHLH124‐heterozygous plants were selected and backcrossed with S‐ORG. After two generations of self‐pollination, we obtained BC5F3 plants carrying the homozygous‐tolerant ZmbHLH124 T‐ORG allele (Figure S5). Evaluations of drought tolerance confirmed that the NIL‐ZmbHLH124 T‐ORG plants were more tolerant than S‐ORG seedlings (Figure 3b,c). These results indicated that the intact ZmbHLH124 T‐ORG contributed to drought tolerance in maize germplasm T‐ORG.
Figure 3.
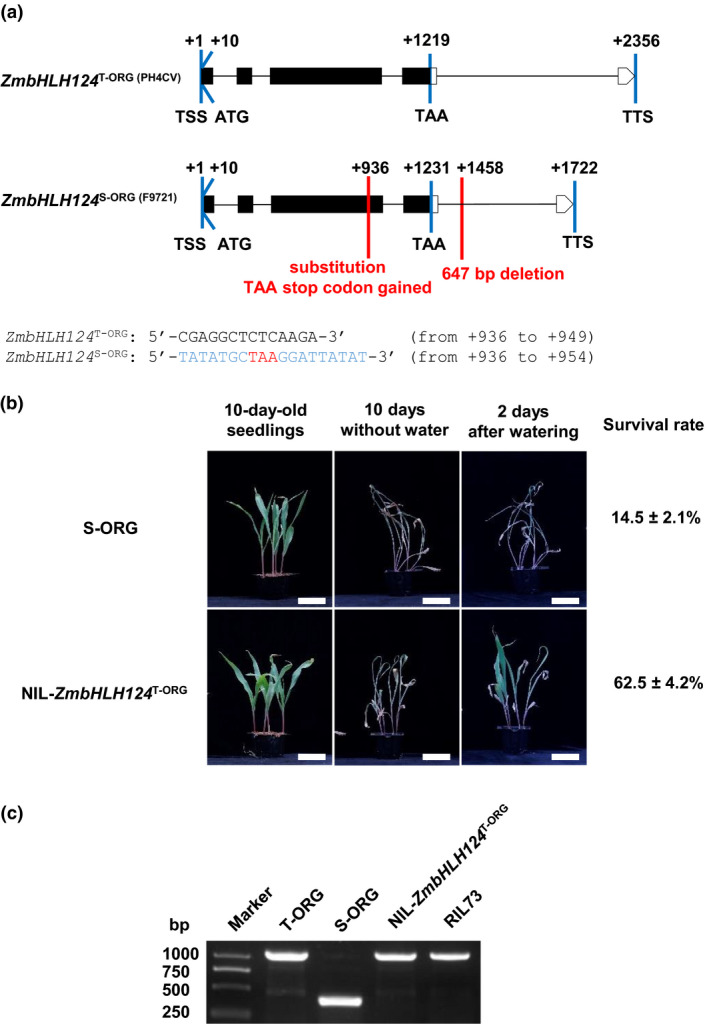
ZmbHLH124 is involved in drought response in maize seedlings. (a) Schematic diagram of ZmbHLH124 transcript regions. Black boxes represent exons, straight lines represent introns and white frames represent 5′ or 3′UTR. The highlight red lines and annotations are main differences which distinguish ZmbHLH124 S‐ORG from ZmbHLH124 T‐ORG. The Indel +936 that causes premature termination of translation are labelled. TSS: transcript start site; TTS: transcript terminal site. (b) Comparison of drought response of parental line S‐ORG and NIL‐ZmbHLH124 T‐ORG. Survival rates were calculated from the results of at least three independent experiments, n ≥ 12. Bar = 5 cm. (c) The molecular marker (647 bp deletion) selection of segregating NILs carrying a homozygous allele of ZmbHLH124 T‐ORG.
Given that drought stress up‐regulated ZmbHLH124 expression, we generated both transgenic rice and maize, overexpressing the cDNAs of ZmbHLH124 from both T‐ORG and S‐ORG lines to verify whether ZmbHLH124 T‐ORG could generally improve monocot drought tolerance. For the transgenic maize, the transcript levels of the transgenes were verified by qRT‐PCR (Figure 4a). We confirmed the full‐length mRNA and protein using semi‐qPCR and western blot assays (Figure S6). Then, two independent transgenic overexpression lines of each genotype were selected for further analysis. The detached leaves of ZmbHLH124 T‐ORG transgenic plants lost water much slower than those of transgene‐free sibling plants (WT) or ZmbHLH124 S‐ORG overexpression (OE) plants (Figure 4b). But no significant differences were found in stomatal density and aperture among the transgenic lines and the WT (Figure S7). Upon drought treatment, more than 85% of ZmbHLH124 T‐ORG transgenic maize plants survived; whereas, the survival rates of WT and ZmbHLH124 S‐ORG transgenic plants were approximately 38.9% and below 50%, while no notable changes were observed in the transgenic maize compared to WT under normal growth conditions (Figure 4c), indicating that ZmbHLH124 T‐ORG OE improved drought tolerance in maize.
Figure 4.
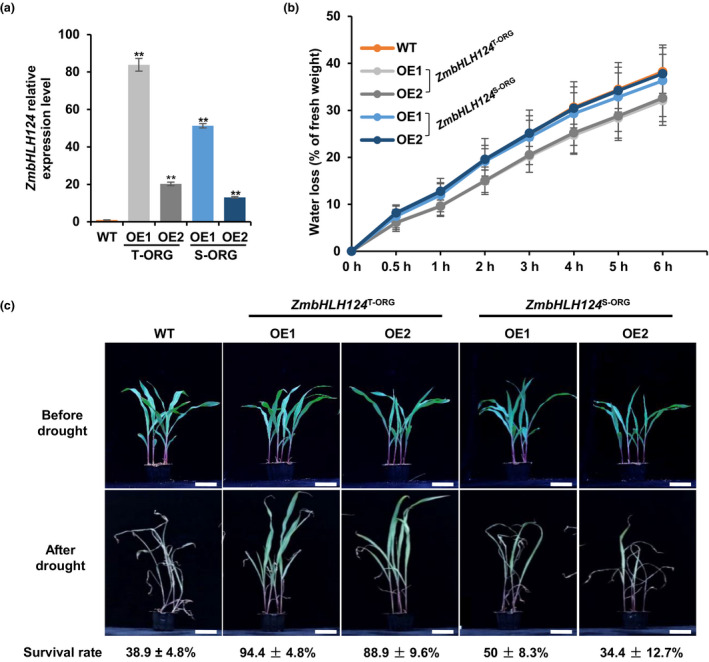
Phenotypes of ZmbHLH124 transgenic maize upon drought treatment. (a) Relative expression levels of ZmbHLH124 detected by qRT‐PCR in maize grown under normal conditions. The data were based on three independent biological replicates. Error bars, s.d.. Statistical significance compared with WT was determined by a two‐sided t‐test: *P < 0.05, **P < 0.01. (b) Water loss rates of detached leaves. Error bars, s.d., calculated from the results of at least three independent experiments. (c) Drought treatment of transgenic maize as compared to WT in different time points. Survival rates were calculated from the results of at least three independent experiments, n ≥ 12. Bar = 5 cm.
Similarly, improved drought tolerance was also observed in transgenic rice transformed with ZmbHLH124 driven by the ZmUbiquitin1 promoter. The transcript levels of transgenes were verified by qRT‐PCR (Figure S8a), and the phenotypes of two independent ZmbHLH124 transgenic lines with detected high transgene expression per parental source were determined. In comparison to WT and ZmbHLH124 S‐ORG OE plants, the ZmbHLH124 T‐ORG OE rice displayed significantly enhanced drought tolerance, while remarkable morphological changes were not observed under standard growth conditions for all transgenic lines. The survival rates of WT plants and ZmbHLH124 S‐ORG OE plants were around 11% and below 20%, respectively, more than 45% of ZmbHLH124 T‐ORG OE plants were alive in the parallel water‐withholding experiments (Figure S8b), suggesting that ZmbHLH124 T‐ORG OE could improve rice drought tolerance. Collectively, these data suggested that the single intact gene ZmbHLH124 T‐ORG could improve drought tolerance in monocot plants including rice and maize.
ZmbHLH124 activates ZmDREB2A expression by directly binding to its promoter
ZmbHLH124 encodes a bHLH type TF and it is known that such a TF can bind bHLH recognition core sequences (5′‐CANNTG‐3′) named E‐box in the promoter of downstream genes to activate or repress their transcriptions (Nakata et al., 2013; Song et al., 2013; Tian et al., 2019). Thus, some downstream genes should be regulated in response to the expression of ZmbHLH124. In order to discover the target genes, transcript levels of several drought‐inducible candidate genes of maize homologs, such as DREB2A, ERD1, GolS3, RAB18, and ABF2 (Mao et al., 2015; Zhu et al., 2020) containing copies of bHLH recognition core sequence in their promoters, were detected in the transgenic maize lines with or without exposure to drought treatment. Our qRT‐PCR results showed that two days after drought treatment, ZmRAB18 was not induced by drought stress and the transcript levels of ZmDREB2A, ZmERD1, and ZmGolS3 in both ZmbHLH124 T‐ORG transgenic maize lines were induced to higher levels than those of WT or ZmbHLH124 S‐ORG transgenic maize lines. It had been reported that ERD1 induction required the concerted action of ZFHD1 and NAC TFs (Tran et al., 2007), and GolS3‐functioned downstream of DREB2A (Qin et al., 2007; Sakuma et al., 2006a). ZmABF2 was also up‐regulated in maize upon drought treatment, but its transcript levels in ZmbHLH124 T‐ORG transgenic maize lines were not significantly higher than those of WT or ZmbHLH124 S‐ORG transgenic maize lines (Figure 5).
Figure 5.
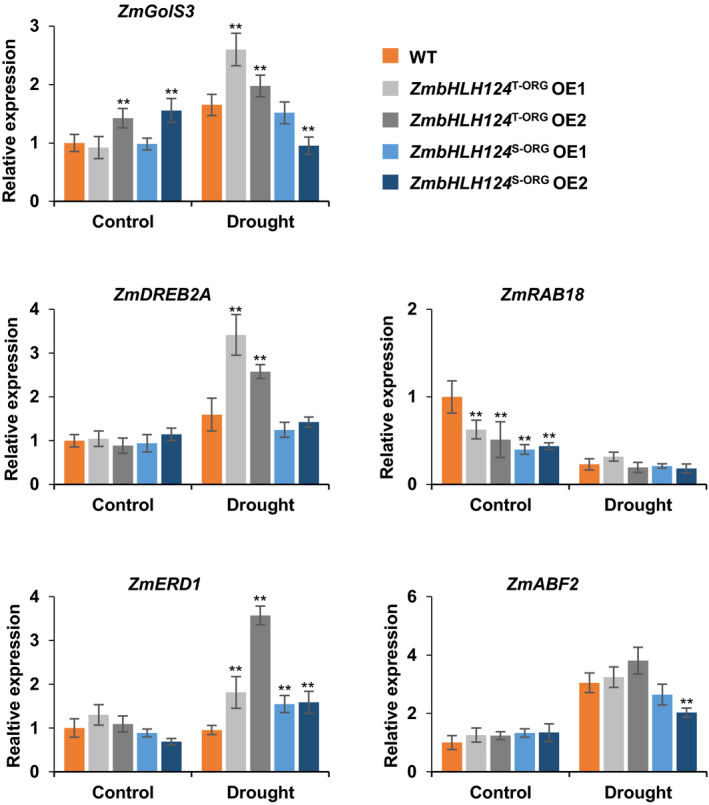
Relative expression levels of candidate drought‐inducible genes detected by qRT‐PCR in maize under control and drought conditions. The data were based on three independent biological replicates. Error bars are s.d.. Statistical significance compared with WT was determined by a two‐sided t‐test: *P < 0.05, **P < 0.01.
A previous study demonstrated that ZmDREB2A functioned as a transcriptional activator and played an important role in plant stress tolerance and that overexpression of ZmDREB2A resulted in improved drought tolerance in Arabidopsis (Qin et al., 2007). To further confirm whether ZmbHLH124 could up‐regulate the expression of ZmDREB2A in vivo, a transient expression system was employed to investigate its transcription activation effect (Shu et al., 2013). We constructed the luciferase (LUC) reporter plasmids driven by ZmDREB2A promoter, pZmDREB2A:LUC, and the effector plasmids ZmbHLH124 T‐ORG and ZmbHLH124 S‐ORG driven by the CaMV 35S promoter, respectively. When the pZmDREB2A:LUC construct combined with ZmbHLH124 T‐ORG was co‐transformed in Nicotiana benthamiana, strong LUC activity was detected. However, when pZmDREB2A:LUC was combined with an equal amount of effector ZmbHLH124 S‐ORG, the LUC activity was particularly weak (Figure 6a). This result indicated that ZmbHLH124 T‐ORG indeed promoted ZmDREB2A expression in vivo.
Figure 6.
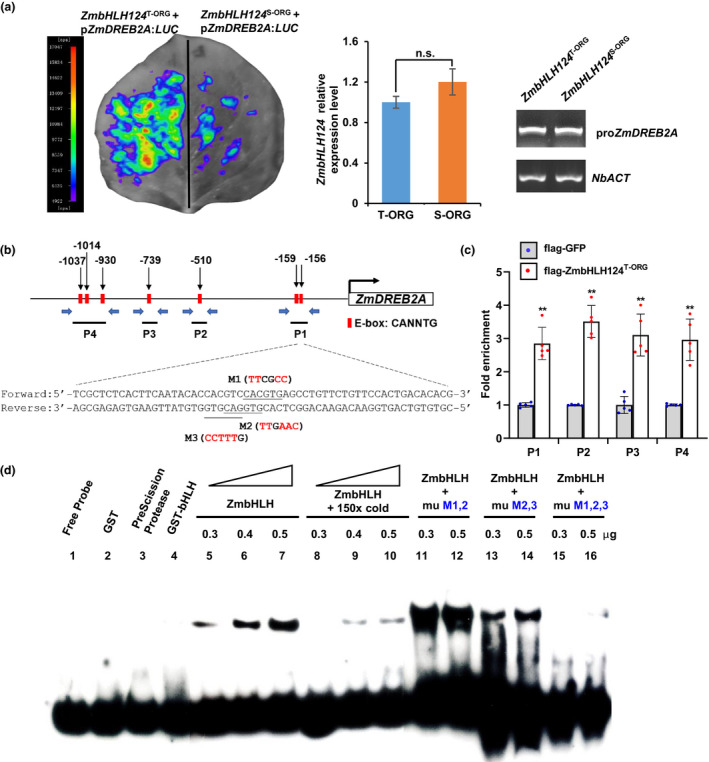
ZmbHLH124 directly activates expression of ZmDREB2A. (a) Transactivation assay in tobacco leaf by transient expression. Left: firefly luciferase activity detection; middle: the relative expression levels of ZmbHLH124; right: PCR‐DNA amount of promoters to represent the equal plasmid DNA in agro‐infiltration were applied between parallel experiments, and NbACT was employed as the internal control in the DNA analysis. The data were based on three independent biological replicates. (b) Schematic diagram of the ZmDREB2A promoter. Red boxes indicate the putative E‐box motifs. (c) ChIP‐qPCR identified significant enrichment in the fragment containing the E‐box motif in the ZmDREB2A promoter. The fragments used in ChIP‐qPCR are indicated in (b). Fold enrichments were calculated relative to input. (d) ZmbHLH124T‐ORG directly bound to the promoter of ZmDREB2A in vitro. Lane 1–4: control; lane 5–10: protein and DNA interaction; lane 11–16: protein and mutated DNA interaction. Mutations of binding sites are labelled in (b). mu: mutation. Error bars, s.d., calculated from the results of at least three independent experiments. Statistical significance was determined by a two‐tailed t‐test: **P < 0.01; n.s.: not significant.
To check whether ZmbHLH124 T‐ORG directly binds to the ZmDREB2A promoter in vivo and in vitro, chromatin immunoprecipitation (ChIP)‐qPCR assay and electrophoretic mobility shift assay (EMSA) were performed to determine the interaction between ZmbHLH124 T‐ORG and the ZmDREB2A promoter. First, we identified seven putative E‐box motifs within 1.2 kb upstream of the ZmDREB2A transcription start site, then four fragments (P1–4) were selected to be amplified by qPCR using specific primer pairs that spanned the E‐box motifs (Figure 6b). The ChIP‐qPCR assays showed that flag‐ZmbHLH124T‐ORG had a 2‐to‐4‐fold enrichment of P1‐4 regions compared with flag‐GFP (Figure 6c), suggesting that ZmDREB2A was a direct target of ZmbHLH124T‐ORG in vivo. Next, based on the ChIP‐qPCR results, EMSAs were employed to confirm further the interaction between ZmbHLH124T‐ORG and ZmDREB2A promoter in vitro. The recombinant GST‐ZmbHLH124T‐ORG fusion protein was expressed and purified from E. coli, one fragment of P1 region was chosen as a probe for this assay. In addition, the fusion protein was digested by a protease to release the bHLH protein from the GST tag before performing EMSA assays (Figure 6d, lane 2–4). The results revealed that the mobility of the probe was significantly delayed in the presence of ZmbHLH124T‐ORG (Figure 6d, lane 1, 5–7). Further, the cold‐competitor probes (excess of unlabelled fragments) were sufficient to compete for the ZmbHLH124T‐ORG binding activity (Figure 6d, lane 8–10). Moreover, in addition to the two E‐box motifs, there is another binding motif called non‐E‐box motif (5′‐NACGTG‐3′) (Li et al., 2006; Toledo‐Ortiz et al., 2003) in the probe (Figure 6b), so we had mutated three bHLH binding sites in different combinations to test TF binding. Only when all motifs were mutated did the binding activity of ZmbHLH124T‐ORG disappear (Figure 6d, lane 11–16). The EMSA results demonstrated that ZmbHLH124T‐ORG directly bound to the ZmDREB2A promoter. Taken together, these data suggested the ZmbHLH124 T‐ORG could directly bind to the ZmDREB2A promoter to promote its expression to improve plant drought tolerance.
Discussion
Drought resistance is a complex trait due to the simultaneous occurrence of other stresses and the multiple stress response strategies in plants (Hu and Xiong, 2014; Takeda and Matsuoka, 2008). In past decades, hundreds of genes in different pathways had been identified with the purpose of understanding how plants adapt to drought stress, thus providing genetic materials and targets for crop improvement. However, the detailed molecular and biochemical mechanisms underlying drought tolerance remain largely unclear (Basu et al., 2016; Deikman et al., 2012; Fang and Xiong, 2015). In the present study, four individuals from a panel of RILs, drought‐sensitive lines RIL44 and RIL93 and drought‐tolerant lines RIL70 and RIL73, were used based on the survival rates of maize seedlings upon drought treatment. RIL70 and RIL73 had higher water use efficiency (WUE) than RIL44 and RIL93 (Figure 1), and those lines could be used for identification of drought response‐related genes. Genetic components with respect to drought response within parental lines must have been rearranged among segregated RIL individuals. Due to loci homozygosity established by repeated self‐pollination, RILs can serve as powerful tools for genetic analysis and for scoring invasive phenotypes in a variety of situations (Broman, 2005; Candela and Hake, 2008). Additionally, factors such as overdominance, epistasis, and hybrid vigour contribute to transgression in RILs but are not influential as in F2 populations (deVicente and Tanksley, 1993). Therefore, it is relatively easy and straightforward to study drought tolerance in maize.
The strategy for identifying novel genetic loci determining maize drought tolerance described here is based on the subtraction of similar regions shared by any of the remaining three RILs from RIL73 by means of bin map construction to locate specific regions of RIL73 that might confer drought tolerance. This is an exploratory study using available materials and data to mine valuable genetic components. Here we use the SNPs identified from transcriptome data to construct bin maps, mainly taking into account the maize genome size and complexity. Bin construction can connect scattered SNPs into fragments for distinguishing the chromosomal composition of different RILs in a parent‐independent way. Such treatment can not only reduce the errors caused by sequencing, but also increase the accuracy of subtractive analysis. Moreover, subtractive analysis is applied to locate specific regions in transgressive individual, like RIL73, in which alleles that are involved in drought tolerance with similar or additive effects are very likely to converge. In addition, based on our observation that RIL73 had stronger water retention capacity than RIL70, the DEGs from specific regions (No.4 in Table 1 and red boxes in Figure S3) of RIL73 are likely unique and worth further analysis in our study. Our work provides a useful trait research method for the study of species with complex and large genome, and even without reference genome information.
ZmbHLH124 was discovered from a specific region of RIL73, and its expression continued to be up‐regulated during drought stress (Figure 2). ZmbHLH124 T‐ORG not only complemented the function of ZmbHLH124 S‐ORG relevant to drought tolerance by introgression into the S‐ORG background but also improved drought tolerance through its overexpression in maize and rice. Therefore, despite of its similar function between different crops, ZmbHLH124 T‐ORG must play a key role in the drought response signalling pathway by regulating known and conserved target genes involved in drought response. The bHLH type TFs target promoters of downstream genes, including other TFs. DREB2A is an important and versatile regulator in stress response, a few studies have reported that its homologs can mediate the expression of genes which respond to certain abiotic stresses in plants, with the role of DREB2A in drought response being most studied (Mallikarjuna et al., 2011; Mizoi et al., 2013; Qin et al., 2007; Sakuma et al., 2006a; Sakuma et al., 2006b; Zhang et al., 2013). Similar to other plant species, ZmDREB2A may be a candidate gene targeted by ZmbHLH124. Based on tobacco leaf transient expression assays, ChIP‐qPCR and EMSA assays, ZmbHLH124T‐ORG directly binds to the recognition sites in the promoter of ZmDREB2A to activate its expression, then ZmDREB2A regulates expression of water stress‐inducible genes to protect maize from drought stress, suggesting that ZmbHLH124 acts upstream of ZmDREB2A in the osmotic‐stress signalling pathway. The downstream genes induced by DREB2A have been revealed to be a set of genes that encode late embryogenesis abundant proteins, which can reduce water loss rate as hydration buffers during stress, stabilize membrane structures, protect proteins and organelles, and sequestrate metal ions and reactive oxygen species (Banerjee and Roychoudhury, 2016; Battaglia et al., 2008; Hand et al., 2011; Yu et al., 2018). Perhaps that’s why ZmbHLH124 has little to do with stomatal movement. Given that DREB2A is finely tuned at the transcriptional, posttranscriptional, and posttranslational level (Guerra et al., 2015; Kim et al., 2011; Morimoto et al., 2013; Morimoto et al., 2017; Qin et al., 2008), what we are concerned with is whether or how ZmbHLH124 interacts or cooperates with transcriptional regulators such as maize histone acetyltransferases, histone deacetylases (Zhao et al., 2014), and the GRF7 (Kim et al., 2012) homolog of maize to regulate the expression of ZmDREB2A tightly under normal and stress conditions.
In conclusion, through transgressive segregation in drought tolerance, four lines with different degrees of drought tolerance were identified from a maize RIL population and were used to perform RNA‐seq. Through SNP identification and comparative transcriptomic analyses of drought‐sensitive and ‐tolerant lines, we identified a specific gene locus that encodes a bHLH type TF, ZmbHLH124, which can activate the transcription of the drought‐related ZmDREB2A gene to contribute to drought tolerance in drought‐tolerant maize. Whether ZmbHLH124 can improve drought tolerance during reproductive stages, which is very important for final crop yield and grain production, needs further study.
Experimental procedures
Plant materials and growth conditions
The maize (Zea mays L.) lines PH4CV, F9721, RIL44, RIL93, RIL70, RIL73, and C01 (wild type from China National Seed Group Co Ltd. used for transgenic lines construction) were used in this study. Maize seeds were directly planted in the soil and germinated in a temperature‐controlled greenhouse with natural light (day, 16 h, 26–28 °C; night, 8 h, 18–20 °C). Rice (Oryza sativa L.) ecotype Nipponbare was used for this study. After being soaked with 3% H2O2 at 28 °C for 24 h, the rice seeds were transferred to the water and germinated in a growth chamber (28 °C with dark condition), then were moved to the greenhouse.
Water loss assays
For the drought treatment, 10‐day‐old maize seedlings and 4‐week‐old rice seedlings grown in soil under normal conditions in a greenhouse were used. After water had been withheld for 10–14 days, the phenotypic responses to drought stress were recorded. Three independent experiments were performed, with more than three replicate pots for each line in each experiment.
For the LRWC assay, the detached leaves were collected from maize plants grown in soil under normal conditions for ˜1 month, or under drought conditions when the moisture content was ˜12%. The detached leaves of maize plants were weighed (fresh weight, FW) immediately, then were placed in distilled water for 8 h or overnight and weighed again (saturated weight, SW). The water‐saturated leaves were dried to a constant weight (dry weight, DW) in a drying oven (105 °C for 30 min, then 70 °C for 12 h). Three biological replicates were assessed for each line. RWC = (FW − DW)/(SW − DW) × 100%.
For the water loss rate assay of detached leaves, the leaves of maize plants grown under normal conditions for 10 days were detached and immediately weighed, then placed in a culture chamber (26 °C, 50–55% relative humidity) and weighed at designated time intervals. Three biological replicates were assessed for each line. The percentage of loss of fresh weight was calculated on the basis of the initial weight of the leaves.
For the water transpiration assay, the maize plants had been grown in a culture chamber with conditions of 26 °C, 50–55% relative humidity for 2 weeks before measurement. Water loss of maize plants grown in soil was measured by weighing each pot with an electronic balance every 30 min for 15 days; the data were automatically recorded by a software that was connected to the balance (DroughtSpotter, Phenospex, Heerlen, The Netherlands). The experiments were repeated three times.
RNA extraction, sequencing, and DEG analysis
Total RNA was isolated from the leaf tissues at different periods by a Total Plant RNA Extraction Kit (Karroten, Beijing, China). Sequencing libraries were generated using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB). Poly(A)‐selected mRNA was sequenced on an Illumina HiSeq 2000 platform. RNA‐seq clean reads were aligned to the B73 version 3.0 reference genome by TopHat version 2.0.9 (Trapnell et al., 2009) allowing two segment mismatches. HTSeq version 0.6.1 (Anders et al., 2015) was used to count the read number of genes and the Reads Per‐Kilobase of the exon model per Million mapped reads (RPKM) was calculated based on the reads count and length of the respective gene. Differential expression analysis was performed by the DESeq R package (Anders, 2010). Genes with fold change ≥ 2 and a false discovery rate (FDR) adjusted P < 0.05 were selected as DEGs. Gene ontology (GO) analysis was performed by agriGO version 2.0 (Tian et al., 2017).
SNP calling and line specific region comparison
RNA‐seq clean reads of each sample were aligned to the reference genome by STAR version 2.6.0 (Dobin et al., 2013). Bam files from the same lines at different periods were merged and underwent SNP calling via the Genome Analysis Toolkit (GATK, version 4.1.3) (McKenna et al., 2010). The SNPs with minimum depth ≥ 5 were selected for further analysis. The modified sliding window approach for bin map construction (Huang et al., 2009) was used to identify the line‐specific regions among RILs. Contiguous SNPs within 1 kb were classified into a window. The ratio of specific SNPs inside a window was calculated. The windows with more than 65% specific SNPs were regarded as specific windows. Contiguous specific windows were combined into a specific region.
Quantitative real‐time PCR assays
Total RNA was isolated from the leaf tissues at different periods by an Ultrapure RNA Kit with DNaseI (CWBIO, Beijing, China) and was subjected to reverse transcription using a FastKing RT kit with gDNase (TIANGEN, Beijing, China). qRT‐PCR was performed using 2 × Talent qPCR PreMix (TIANGEN, Beijing, China) and the CFX96 real‐time system (Bio‐Rad, California, USA). Transcript levels were quantified at the logarithmic phase based on the expression of the housekeeping genes ZmEF1A (Lin et al., 2014) and OsACTIN (Shang et al., 2016) as internal controls. Three biological replicates were performed per line. Primer sequences for qRT‐PCR are shown in Table S3.
Transformation vectors and generation of transgenic plants
Transgenic plants carrying constitutively expressing ZmbHLH124 T‐ORG and ZmbHLH124 S‐ORG were generated. To obtain the Ubi‐ZmbHLH124 overexpression vector, an 888 bp ZmbHLH124 T‐ORG CDS fragment and an 899 bp ZmbHLH124 S‐ORG fragment were amplified by PCR and then cloned into the vector pCAMBIA‐2300‐Ubi‐OCS for rice transformation and the pZZ000026‐Ubi‐OCS for maize transformation, in which ZmbHLH124 was driven by the ZmUbiquitin1 (GRMZM2G409726) promoter. Transformation of maize was performed by China National Seed Group Co. Ltd., and the transformation of rice was performed by Wuhan BioRun Biotechnology Co. Ltd. For more detailed phenotypic analysis, two independent T2 transgenic positive lines were used.
To detect full‐length mRNA of ZmbHLH124 in transgenic plants or WT seedlings, total RNA isolation and reverse transcription were performed as described above, and gene‐specific primers were used to amplify ZmbHLH124 gene listed in Table S3 and the ZmEF1A gene as a control.
To detect ZmbHLH124 protein, leaves of transgenic plant or WT seedlings were collected and homogenized in liquid nitrogen. Three‐fold (v/w) of native extraction buffer (50 mm Tris‐MES pH 8.0, 0.5 m sucrose, 1 mm MgCl2, 10 mm EDTA, 1 mm DTT, 1 mm PMSF, protease inhibitor cocktail complete mini tablets [Roche, Basel, Switzerland] and 50 μm MG132 [Tocris Bioscience, Missouri, USA]) was added to the powder. The homogenized solutions were centrifuged at 13,523 g for 10 min at 4 °C. Finally, the supernatants with SDS loading buffer were boiled for 10 min, separated by SDS‐PAGE and detected by anti‐ZmbHLH124 antibody. Polyclonal antibodies of ZmbHLH124 were raised in rat.
Transient expression in Nicotiana benthamiana
Agrobacterium‐mediated transient expression in N. benthamiana was carried out according to a previously described protocol (Liu et al., 2010). The 2329 bp‐long genomic fragment of ZmbHLH124 T‐ORG and the 1798 bp‐long genomic fragment of ZmbHLH124 S‐ORG were amplified separately from genomic DNA and then cloned into pCAMBIA1300‐221, in which ZmbHLH124 variants were driven by the CaMV 35S promoter. The 1616 bp‐long fragment for the native ZmDREB2A promoter (pZmDREB2A) was amplified from genomic DNA, and cloned into pENTR using the pENTR Directional TOPO cloning kit (Invitrogen, Waltham, MA, USA). Then, pZmDREB2A was fused with the luciferase reporter gene LUC through gateway reactions into the plant binary vector pGWB35 (Nakagawa et al., 2007) to generate the reporter construct pZmDREB2A:LUC. The effector constructs were p35S:ZmbHLH124 T‐ORG and p35S:ZmbHLH124 S‐ORG. LUC images were captured using a low‐light cooled charge‐coupled device imaging apparatus (NightOWL II LB983 with indiGO software). For the gene expression quantification, an Agrobacterium strain carrying a construct encoding RFP was used as an internal control.
Chromatin immunoprecipitation and quantitative PCR assays
To generate flag‐tag fusion protein of GFP and ZmbHLH124T‐ORG, the fragment containing CaMV 35S promoter‐flag‐NOS terminator was amplified and cloned into the pBlueScript II SK(+) vector forming p35S:flag vector, then the full‐length CDS of GFP and ZmbHLH124 T‐ORG were amplified and sub‐cloned into p35S:flag vector forming the flag‐tagged fusion plasmids (flag‐GFP and flag‐ZmbHLH124 T‐ORG). The flag‐tagged fusion plasmids were transformed into the protoplasts collected from the leaves of 10‐day‐old seedlings of inbred line B73 to perform the chromatin immunoprecipitation (ChIP) assays. The ChIP assays were conducted following a reported protocol with minor modifications (Tu et al., 2020). Sonicated chromatin fragments were immunoprecipitated using anti‐flag (MBL, M185‐10). The IP chromatins were detected by performing qPCR using specific primers. The formula as following was used to calculate IP%: IP% = 100 × 2 (−ΔCt [normalized ChIP]), in which ΔCycle threshold ΔCt = Ct [ChIP] − (Ct [Input] − log2 (Input Dilution Factor)) and Input Dilution Factor = (fraction of the input chromatin saved)−1 (Tian et al., 2019). The fold enrichment was calculated against the flag control samples. The primers used in ChIP‐qPCR are listed in Table S3.
Electrophoretic mobility shift assay
EMSAs were performed with the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher, Waltham, MA, USA) according to a previously published protocol (Wei et al., 2015). The coding sequence of ZmbHLH124 T‐ORG was inserted into the BamHI/SalI sites of the pGEX‐6P‐1 backbone vector which contained a GST tag. The ZmbHLH124T‐ORG fusion proteins were expressed at 28 °C for 3–4 h in the Rosetta Escherichia coli strain and purified, then GST tag was excised from the fusion protein by PreScission™ (Beyotime, Shanghai, China) Protease digestion. The single‐stranded oligonucleotide fragments of probes were synthesized and then, the double‐stranded DNAs were obtained through heating oligonucleotides at 90–95 °C in annealing solution (10 mm Tris‐HCl pH 7.5, 1 mm EDTA, 100 mm NaCl) for 5 min, then slowly cooled to room temperature. Investigation of the interaction between ZmbHLH124T‐ORG protein and the corresponding probes was carried out according to the protocol provided with the LightShift™ Chemiluminescent EMSA Kit (Thermo Fisher, Waltham, MA USA). The primer sequences for constructing the GST‐ZmbHLH124T‐ORG vector and the probes for the EMSA are given in Table S3.
Stomatal density and aperture assays
For the stomatal density and aperture assays, the same regions of the second leaf from seedlings were floated on the stomatal opening buffer (20 mm KCl, 5 mm MES‐Tris and 1 mm CaCl2) (Wu et al., 2019; Xiang et al., 2021) for at least 1 h in the dark to ensure that most of the stomata had become fully closed. Then, after the leaf pieces maintained under light for 2 h, half of the samples were transferred to the opening buffer with 500 mm mannitol and incubated for an additional 2 h. Finally, the samples were immediately fixed by liquid nitrogen and coated with gold particles. The pictures were obtained using a scanning electron microscopy (S‐3000N; Hitachi, Tokyo, Japan), and the numbers of stomata in randomly chosen fields were counted. The width and length of the aperture for each stomatal pore were measured with ImageJ software (NIH, Bethesda, Maryland, USA).
Accession numbers
ZmUbiquitin1:GRMZM2G409726, other genes and their accession numbers used in this study from MaizeGDB and Phytozome are shown in Table S3.
Conflict of interest
The authors declare no competing financial interests.
Author contributions
S.W., Y.W., and Q.X. designed the research. S.W., R.X., X.S., F.G., and H.W. performed the experiments and analysed the data. S.W. and C.C. carried out the transcriptomic data analyses. Y.W., R.X. and H.C. advised on the experiments. S.W., Y.W., and Q.X. wrote the manuscript.
Supporting information
Figure S1 Different drought phenotypes between PH4CV and F9721.
Figure S2 Pearson correlations between different samples.
Figure S3 Comparative analysis of chromosome regions between RIL73 and other three RILs.
Figure S4 Comparison of two ZmbHLH124 proteins from different genetic background.
Figure S5 Schematic diagram of NIL development.
Figure S6 Expression level analyses of maize transgenic lines.
Figure S7 Effects of ZmbHLH124 overexpression on stomatal movement.
Figure S8 Phenotypes of ZmbHLH124 transgenic rice plants upon drought treatment.
Table S1 Genotypes of four RILs.
Table S2 Annotations of differentially expressed transcription factors in RIL73 specific regions.
Table S3 Primers used in this study.
Acknowledgements
We thank Dr. Feng Qin from China Agricultural University for valuable suggestion and help in this work and Mr. Tao Yun for technical help. We thank Mr. Yanbao Tian from the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, for his assistance in scanning electron microscopy observation. This work was supported by the National Key R&D Program of China (2016YFD0100605), and Transgenic Research Projects (2016ZX08009003‐002 and 2018ZX08009‐11B‐002).
Wei, S. , Xia, R. , Chen, C. , Shang, X. , Ge, F. , Wei, H. , Chen, H. , Wu, Y. and Xie, Q. (2021) ZmbHLH124 identified in maize recombinant inbred lines contributes to drought tolerance in crops. Plant Biotechnol. J., 10.1111/pbi.13637
Contributor Information
Yaorong Wu, Email: yrwu@genetics.ac.cn.
Qi Xie, Email: qxie@genetics.ac.cn.
Data availability statement
The raw sequence data reported in this paper have been deposited in the National Center for Biotechnology Information Sequence Read Archive (accession no. PRJNA662679).
References
- Anders, S. (2010) Analysing RNA‐Seq data with the DESeq package. Mol. Biol. 43, 1–17. [Google Scholar]
- Anders, S. , Pyl, P.T. and Huber, W. (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, A. and Roychoudhury, A. (2016) Group II late embryogenesis abundant (LEA) proteins: structural and functional aspects in plant abiotic stress. Plant Growth Regul, 79, 1–17. [Google Scholar]
- Basu, S. , Ramegowda, V. , Kumar, A. and Pereira, A. (2016) Plant adaptation to drought stress. F1000Res. 5, 1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia, M. , Olvera‐Carrillo, Y. , Garciarrubio, A. , Campos, F. and Covarrubias, A.A. (2008) The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 148(1), 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K.W. (2005) The genomes of recombinant inbred lines. Genetics, 169(2), 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candela, H. and Hake, S. (2008) The art and design of genetic screens: maize. Nat. Rev. Genet. 9, 192–203. [DOI] [PubMed] [Google Scholar]
- Carretero‐Paulet, L. , Galstyan, A. , Roig‐Villanova, I. , Martinez‐Garcia, J.F. , Bilbao‐Castro, J.R. and Robertson, D.L. (2010) Genome‐wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 153(3), 1398–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves, M.M. and Oliveira, M.M. (2004) Mechanisms underlying plant resilience to water deficits: prospects for water‐saving agriculture. J. Exp. Bot. 55(407), 2365–2384. [DOI] [PubMed] [Google Scholar]
- Deikman, J. , Petracek, M. and Heard, J.E. (2012) Drought tolerance through biotechnology: improving translation from the laboratory to farmers' fields. Curr. Opin. Biotechnol. 23, 243–250. [DOI] [PubMed] [Google Scholar]
- deVicente, M.C. and Tanksley, S.D. (1993) QTL analysis of transgressive segregation in an interspecific tomato cross. Genetics, 134(2), 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Y. and Xiong, L. (2015) General mechanisms of drought response and their application in drought resistance improvement in plants. Cell. Mol. Life Sci. 72(4), 673–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, A. , Machemer, K. , Braun, E.L. and Grotewold, E. (2011) Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66(1), 94–116. [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Jiang, W. , Dai, Y. , Xiao, N. , Zhang, C. , Li, H. , Lu, Y. et al. (2015) A maize phytochrome‐interacting factor 3 improves drought and salt stress tolerance in rice. Plant Mol. Biol. 87(4‐5), 413–428. [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Wu, M. , Zhang, M. , Jiang, W. , Ren, X. , Liang, E. , Zhang, D. et al. (2018) A maize phytochrome‐interacting factors protein ZmPIF1 enhances drought tolerance by inducing stomatal closure and improves grain yield in Oryza sativa . Plant Biotechnol. J. 16(7), 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra, D. , Crosatti, C. , Khoshro, H.H. , Mastrangelo, A.M. , Mica, E. and Mazzucotelli, E. (2015) Post‐transcriptional and post‐translational regulations of drought and heat response in plants: a spider's web of mechanisms. Front. Plant Sci. 6(57), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand, S.C. , Menze, M.A. , Toner, M. , Boswell, L. and Moore, D. (2011) LEA proteins during water stress: not just for plants anymore. Annu. Rev. Physiol. 73, 115–134. [DOI] [PubMed] [Google Scholar]
- Hrdlickova, R. , Toloue, M. and Tian, B. (2017) RNA‐Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA, 8, e1364.(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. and Xiong, L. (2014) Genetic engineering and breeding of drought‐resistant crops. Annu. Rev. Plant Biol. 65(1), 715–741. [DOI] [PubMed] [Google Scholar]
- Huang, X. , Feng, Q. , Qian, Q. , Zhao, Q. , Wang, L. , Wang, A. , Guan, J. et al. (2009) High‐throughput genotyping by whole‐genome resequencing. Genome Res. 19(6), 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Peluso, P. , Shi, J. , Liang, T. , Stitzer, M.C. , Wang, B. , Campbell, M.S. et al. (2017) Improved maize reference genome with single‐molecule technologies. Nature, 546(7659), 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X. , Shi, C. , Yu, C.Y. , Yamada, T. and Sacks, E.J. (2017) Determination of leaf water content by visible and near‐infrared spectrometry and multivariate calibration in miscanthus. Front. Plant Sci. 8(721), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumanu, A. , Ambavaram, M.M. , Klumas, C. , Krishnan, A. , Batlang, U. , Myers, E. , Grene, R. et al. (2012) Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA‐Seq. Plant Physiol. 160(2), 846–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.S. , Mizoi, J. , Yoshida, T. , Fujita, Y. , Nakajima, J. , Ohori, T. , Todaka, D. et al. (2011) An ABRE promoter sequence is involved in osmotic stress‐responsive expression of the DREB2A gene, which encodes a transcription factor regulating drought‐inducible genes in Arabidopsis. Plant Cell Physiol. 52(12), 2136–2146. [DOI] [PubMed] [Google Scholar]
- Kim, J.S. , Mizoi, J. , Kidokoro, S. , Maruyama, K. , Nakajima, J. , Nakashima, K. , Mitsuda, N. et al. (2012) Arabidopsis growth‐regulating factor7 functions as a transcriptional repressor of abscisic acid‐ and osmotic stress‐responsive genes, including DREB2A. Plant Cell, 24(8), 3393–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromori, T. , Mizoi, J. , Umezawa, T. , Yamaguchi‐Shinozaki, K. & Shinozaki, K. (2014) Howell, S.H. , Drought Stress Signaling Network. The Plant Sciences. Molecular Biology. New York: Springer. 383–411. [Google Scholar]
- Li, X. , Duan, X. , Jiang, H. , Sun, Y. , Tang, Y. , Yuan, Z. , Guo, J. et al. (2006) Genome‐wide analysis of basic/helix‐loop‐helix transcription factor family in rice and Arabidopsis. Plant Physiol. 141(4), 1167–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Liu, C. , Zhang, Y. , Wang, B. , Ran, Q. and Zhang, J. (2019) The bHLH family member ZmPTF1 regulates drought tolerance in maize by promoting root development and abscisic acid synthesis. J. Exp. Bot. 70(19), 5471–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Zhang, C. , Lan, H. , Gao, S. , Liu, H. , Liu, J. , Cao, M. et al. (2014) Validation of potential reference genes for qPCR in maize across abiotic stresses, hormone treatments, and tissue types. PLoS One, 9, e95445.(5), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Zhang, Y. , Tang, S. , Zhao, Q. , Zhang, Z. , Zhang, H. , Dong, L. et al. (2010) An efficient system to detect protein ubiquitination by agroinfiltration in Nicotiana benthamiana . Plant J. 61(5), 893–903. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Zhou, M. , Gao, Z. , Ren, W. , Yang, F. , He, H. and Zhao, J. (2015) RNA‐Seq analysis reveals MAPKKK family members related to drought tolerance in maize. PLoS One, 10, e0143128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallikarjuna, G. , Mallikarjuna, K. , Reddy, M.K. and Kaul, T. (2011) Expression of OsDREB2A transcription factor confers enhanced dehydration and salt stress tolerance in rice (Oryza sativa L.). Biotechnol. Lett. 33, 1689–1697. [DOI] [PubMed] [Google Scholar]
- Mao, H. , Wang, H. , Liu, S. , Li, Z. , Yang, X. , Yan, J. , Li, J. et al. (2015) A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 6(8326), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20(9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, H. , Chen, C. , Wei, S. , Shang, X. , Sun, M. , Xia, R. , Liu, X. et al. (2016) Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 7(1080), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoi, J. , Ohori, T. , Moriwaki, T. , Kidokoro, S. , Todaka, D. , Maruyama, K. , Kusakabe, K. et al. (2013) GmDREB2A;2, a canonical DEHYDRATION‐RESPONSIVE ELEMENT‐BINDING PROTEIN2‐type transcription factor in soybean, is posttranslationally regulated and mediates dehydration‐responsive element‐dependent gene expression. Plant Physiol. 161(1), 346–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, K. , Mizoi, J. , Qin, F. , Kim, J.S. , Sato, H. , Osakabe, Y. , Shinozaki, K. et al. (2013) Stabilization of Arabidopsis DREB2A is required but not sufficient for the induction of target genes under conditions of stress. PLoS One, 8, e80457.(12), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, K. , Ohama, N. , Kidokoro, S. , Mizoi, J. , Takahashi, F. , Todaka, D. , Mogami, J. et al. (2017) BPM‐CUL3 E3 ligase modulates thermotolerance by facilitating negative regulatory domain‐mediated degradation of DREB2A in Arabidopsis. Proc. Natl Acad. Sci. USA, 114, E8528–E8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T. , Kurose, T. , Hino, T. , Tanaka, K. , Kawamukai, M. , Niwa, Y. , Toyooka, K. et al. (2007) Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 104(1), 34–41. [DOI] [PubMed] [Google Scholar]
- Nakata, M. , Mitsuda, N. , Herde, M. , Koo, A.J. , Moreno, J.E. , Suzuki, K. , Howe, G.A. et al. (2013) A bHLH‐type transcription factor, ABA‐INDUCIBLE BHLH‐TYPE TRANSCRIPTION FACTOR/JA‐ASSOCIATED MYC2‐LIKE1, acts as a repressor to negatively regulate jasmonate signaling in arabidopsis. Plant Cell, 25(5), 1641–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, F. , Kakimoto, M. , Sakuma, Y. , Maruyama, K. , Osakabe, Y. , Tran, L.S. , Shinozaki, K. et al. (2007) Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 50(1), 54–69. [DOI] [PubMed] [Google Scholar]
- Qin, F. , Sakuma, Y. , Tran, L.S. , Maruyama, K. , Kidokoro, S. , Fujita, Y. , Fujita, M. et al. (2008) Arabidopsis DREB2A‐interacting proteins function as RING E3 ligases and negatively regulate plant drought stress‐responsive gene expression. Plant Cell, 20(6), 1693–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribaut, J.M. , Betran, J. , Monneveux, P. and Setter, T. (2009) Drought tolerance in maize. In Handbook of Maize: Its Biology ( Bennetzen, J.L. and Hake, S.C. , eds), pp. 311–344. New York: Springer. [Google Scholar]
- Sakuma, Y. , Maruyama, K. , Osakabe, Y. , Qin, F. , Seki, M. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2006a) Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought‐responsive gene expression. Plant Cell, 18(5), 1292–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma, Y. , Maruyama, K. , Qin, F. , Osakabe, Y. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2006b) Dual function of an Arabidopsis transcription factor DREB2A in water‐stress‐responsive and heat‐stress‐responsive gene expression. Proc. Natl. Acad. Sci. USA, 103(49), 18822–18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable, P.S. , Ware, D. , Fulton, R.S. , Stein, J.C. , Wei, F. , Pasternak, S. , Liang, C. et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326(5956), 1112–1115. [DOI] [PubMed] [Google Scholar]
- Seki, M. , Umezawa, T. , Urano, K. and Shinozaki, K. (2007) Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 10(3), 296–302. [DOI] [PubMed] [Google Scholar]
- Shang, X.L. , Xie, R.R. , Tian, H. , Wang, Q.L. and Guo, F.Q. (2016) Putative zeatin O‐glucosyltransferase OscZOG1 regulates root and shoot development and formation of agronomic traits in rice. J. Integr. Plant Biol. 58(7), 627–641. [DOI] [PubMed] [Google Scholar]
- Shu, K. , Zhang, H. , Wang, S. , Chen, M. , Wu, Y. , Tang, S. , Liu, C. et al. (2013) ABI4 regulates primary seed dormancy by regulating the biogenesis of abscisic acid and gibberellins in Arabidopsis. PLoS Genet. 9, e1003577.(6), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, S. , Qi, T. , Fan, M. , Zhang, X. , Gao, H. , Huang, H. , Wu, D. et al. (2013) The bHLH subgroup IIId factors negatively regulate jasmonate‐mediated plant defense and development. PLoS Genet. 9, e1003653.(7), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, S. and Matsuoka, M. (2008) Genetic approaches to crop improvement: responding to environmental and population changes. Nat. Rev. Genet. 9, 444–457. [DOI] [PubMed] [Google Scholar]
- Thatcher, S.R. , Danilevskaya, O.N. , Meng, X. , Beatty, M. , Zastrow‐Hayes, G. , Harris, C. , Van Allen, B. et al. (2016) Genome‐wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 170(1), 586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, J. , Wang, C. , Xia, J. , Wu, L. , Xu, G. , Wu, W. , Li, D. et al. (2019) Teosinte ligule allele narrows plant architecture and enhances high‐density maize yields. Science, 365(6454), 658–664. [DOI] [PubMed] [Google Scholar]
- Tian, T. , Liu, Y. , Yan, H. , You, Q. , Yi, X. , Du, Z. , Xu, W. et al. (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 45(W1), W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo‐Ortiz, G. , Huq, E. and Quail, P.H. (2003) The Arabidopsis basic/helix‐loop‐helix transcription factor family. Plant Cell, 15(8), 1749–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, L.S. , Nakashima, K. , Sakuma, Y. , Osakabe, Y. , Qin, F. , Simpson, S.D. , Maruyama, K. et al. (2007) Co‐expression of the stress‐inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J. 49(1), 46–63. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25(9), 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, X. , Mejia‐Guerra, M.K. , Valdes Franco, J.A. , Tzeng, D. , Chu, P.Y. , Shen, W. , Wei, Y. et al. (2020) Reconstructing the maize leaf regulatory network using ChIP‐seq data of 104 transcription factors. Nat. Commun. 11(5089), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Gerstein, M. and Snyder, M. (2009) RNA‐Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, W. , Zhang, Y.Q. , Tao, J.J. , Chen, H.W. , Li, Q.T. , Zhang, W.K. , Ma, B. et al. (2015) The Alfin‐like homeodomain finger protein AL5 suppresses multiple negative factors to confer abiotic stress tolerance in Arabidopsis. Plant J. 81(6), 871–883. [DOI] [PubMed] [Google Scholar]
- Wu, Q. , Wang, M. , Shen, J. , Chen, D. , Zheng, Y. and Zhang, W. (2019) ZmOST1 mediates abscisic acid regulation of guard cell ion channels and drought stress responses. J. Integr. Plant Biol. 61(4), 478–491. [DOI] [PubMed] [Google Scholar]
- Xiang, Y. , Sun, X. , Bian, X. , Wei, T. , Han, T. , Yan, J. and Zhang, A. (2021) The transcription factor ZmNAC49 reduces stomatal density and improves drought tolerance in maize. J. Exp. Bot. 72(4), 1399–1410. [DOI] [PubMed] [Google Scholar]
- Yu, Z. , Wang, X. and Zhang, L. (2018) Structural and functional dynamics of dehydrins: a plant protector protein under abiotic stress. Int. J. Mol. Sci. 19(3420), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Han, Z. , Guo, Q. , Liu, Y. , Zheng, Y. , Wu, F. and Jin, W. (2014) Identification of maize long non‐coding RNAs responsive to drought stress. PLoS One, 9, e98958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.X. , Tang, Y.J. , Ma, Q.B. , Yang, C.Y. , Mu, Y.H. , Suo, H.C. , Luo, L.H. et al. (2013) OsDREB2A, a rice transcription factor, significantly affects salt tolerance in transgenic soybean. PLoS One, 8, e83011.(12), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Wang, P. , Yan, S. , Gao, F. , Li, H. , Hou, H. , Zhang, Q. et al. (2014) Promoter‐associated histone acetylation is involved in the osmotic stress‐induced transcriptional regulation of the maize ZmDREB2A gene. Physiol. Plant 151(4), 459–467. [DOI] [PubMed] [Google Scholar]
- Zhu, D. , Chang, Y. , Pei, T. , Zhang, X. , Liu, L. , Li, Y. , Zhuang, J. et al. (2020) MAPK‐like protein 1 positively regulates maize seedling drought sensitivity by suppressing ABA biosynthesis. Plant J. 102(4), 747–760. [DOI] [PubMed] [Google Scholar]
- Zhu, J.K. (2016) Abiotic stress signaling and responses in plants. Cell, 167(2), 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Different drought phenotypes between PH4CV and F9721.
Figure S2 Pearson correlations between different samples.
Figure S3 Comparative analysis of chromosome regions between RIL73 and other three RILs.
Figure S4 Comparison of two ZmbHLH124 proteins from different genetic background.
Figure S5 Schematic diagram of NIL development.
Figure S6 Expression level analyses of maize transgenic lines.
Figure S7 Effects of ZmbHLH124 overexpression on stomatal movement.
Figure S8 Phenotypes of ZmbHLH124 transgenic rice plants upon drought treatment.
Table S1 Genotypes of four RILs.
Table S2 Annotations of differentially expressed transcription factors in RIL73 specific regions.
Table S3 Primers used in this study.
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the National Center for Biotechnology Information Sequence Read Archive (accession no. PRJNA662679).