

Abstract
Genetic engineering of innate and adaptive immune cells represents a potential solution to treat numerous immune-mediated pathologies. Current immune engineering methods to introduce nucleic acids into cells with high efficiency rely on physical mechanisms such as electroporation, viral vectors, or other chemical methods. Gene delivery using non-viral nanoparticles offers significant flexibility in biomaterial design to tune critical parameters such as nano-bio interactions, transfection efficiency, and toxicity profiles. However, their clinical utility has been limited due to complex synthetic procedures, high toxicity at increased polymer (nitrogen, N) to DNA ratios (phosphate, P) (N/P ratios), poor transfection efficiency and nanoparticle stability in the presence of serum, and short-term gene expression. Here, we describe the development of a simple, polymer-based non-viral gene delivery platform based on simple modifications of polyethylenimine (PEI) that displays potent and serum-independent transfection of innate and adaptive immune cells. Cationic acetylated PEI (Ac-PEI) was synthesized and complexed with plasmid DNA (pDNA) followed by enveloping with an anionic polyelectrolyte layer of poly(ethylene-alt-maleic acid) (PEMA) to form immunoplexes (IPs). Cellular interactions and gene expression could be precisely controlled in murine RAW 264.7 macrophages, murine DC2.4 dendritic cells, and human Jurkat T cells by altering the levels of PEMA envelopment, thus providing a strategy to engineer specific cell targeting into the IP platform. Optimally formulated IPs for immune cell transfection in the presence of serum utilized high N/P ratios to enable high stability, displayed reduced toxicity, high gene expression, and a lengthened duration of gene expression (>3 days) compared to non-enveloped controls. These results demonstrate the potential of engineered IPs to serve as simple, modular, targetable, and efficient non-viral gene delivery platform to efficiently alter gene expression within cells of the immune system.
Keywords: Gene delivery, Serum independent, Nanoparticle, Polyethylenimine, Immune cells
Graphical Abstract
Introduction
The delivery of nucleic acids to lymphocytes has the potential to improve therapeutic outcomes through correction of genetic aberrations and development of next generation cell-based therapies1. Recently, significant efforts have been focused on achieving in situ gene delivery to eliminate the need for time and labor intensive ex vivo immune cell manipulations2–3. Traditional methods for gene delivery rely heavily on physical, viral, or chemical approaches. Physical methods including electroporation, microinjection, ultrasound, or other hydrodynamic methods show high transfection efficiency yet suffer from high toxicity and are not amenable to cell-specific targeting, which hinders their potential in vivo applicability. Viral vectors are highly efficient, but the use of live viruses is limited by their potential mutagenicity and immunogenicity posing concerns for safety in clinical translation4–6. Chemical methods for gene delivery including calcium phosphate, lipids, cationic polymers, and others have the potential to overcome many of these limitations, particularly regarding toxicity and targetability in vivo.
Non-viral gene delivery has been routinely investigated for the delivery of various nucleic acid-based cargoes including plasmid DNA (pDNA), DNA oligonucleotides, mRNA, non-coding RNA species (i.e., siRNA, shRNA, lncRNA), Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated Protein 9 (CRISPR-Cas9) to alter the expression of target genes and translation of their gene products2–3, 7–13. Cationic polymers like polyethylenimine (PEI), poly(β-amino ester) (PBAE), poly(L-lysine) (PLL), polyamidoamine (PAMAM) dendrimers, and others have shown variable abilities to efficiently transfect lymphocytes. Several groups have developed approaches for high-throughput synthesis of carriers with the objective to optimize lymphocyte transfection and mitigate toxicity using cationic polymers (such as PBAE and others) or lipid nanoparticles (LNPs)9–10, 13–15. However, because of the wide variety of materials tested, it is difficult to draw direct relationships between studies based on the physicochemical properties of the materials to predict effectiveness of gene delivery.
PEI is one of the most effective off-the-shelf polymers used for gene delivery, yet it is associated with toxicity at high concentrations through cell membrane damage and apoptosis16–17. Strategies to reduce the inherent toxicity of PEI by modification of its chemical properties such as using low molecular weight PEI, functionalization of PEI (i.e., acetylation, PEGylation, etc.), and PEI encapsulation have yielded significant improvements18–20. PEI-based polyplexes are internalized via endosomal/lysosomal trafficking and escape from vesicles by the “proton-sponge” mechanism20–23. Once released into the cytosol, DNA must dissociate from PEI, transported to the nucleus for gene transcription, and then shuttled back to the cytoplasm for translation to protein. To limit the toxicity associated with PEI and to increase the ability of polyplexes to unpackage DNA in the cells, acetylation of PEI (Ac-PEI) was evaluated. Ac-PEI modified the proton buffering capacity, increased the ability for polyplexes to unpackage cargos in the cell, and reduced the toxicity associated with unmodified PEI20, 24. However, only limited effectiveness has been demonstrated for PEI and Ac-PEI to transfect adaptive immune cells25–26, potentially due to reduced proliferative capacity of resting immune cells and additional defense mechanisms within immune cells such as pattern recognition receptors.
An important challenge to overcome for efficient non-viral gene delivery is the biomolecule corona. The biomolecule corona forms due to the adsorption of serum proteins to the surface of nanoparticles, which can negatively affect the formation of specific cellular interactions as well as reduce transfection efficiency27–29. Several groups have attempted to overcome the negative contribution of serum on transfection efficiency. Olden et al. evaluated the transfection efficiency of Jurkat cells in serum-free and serum-containing media using comb- and sunflower-type poly(2-dimethylaminoethyl methacrylate) (pDMAEMA) cationic polymer architectures25. Serum-free transfection conditions of Jurkat cells using either polymer type resulted in approximately 30% and 10% gene expression, respectively. However, in serum containing medium, the efficiencies were significantly reduced to approximately 7% and 6%, respectively. Notably, the use of PEI as a control did not result in any measurable transfection in either condition. Another group focused on transfection of hard-to-transfect lymphoma/leukemia cells30. Using poly(β-amino ester) (PBAE) polymers in vitro, gene expression was found to be limited in serum-free conditions. However, pre-treatment of cells with polybrene prior to the addition of PBAE polyplexes was necessary to yield significant improvements in gene expression (up to 32%), an 8-fold increase over that mediated by Lipofectamine®. The need to pre-treat cells prior to transfection is expected to hinder the in vivo applicability of these PBAE polymers. Many of these strategies have improved upon issues related to limited endocytosis, protein expression, and low cell viability; however, the relative complexity in formulation design or treatment schedules, in addition to reduced gene expression in the presence of serum may limit their utility for in vivo applications and subsequent future development. Thus, a critical need remains to establish simple and effective methods to transfect pDNA into lymphocytes.
Polyelectrolyte enveloping of cationic polyplexes enables the surface chemistry of substrates to be modified through electrostatic absorption with the purpose of decreasing toxicity, reducing non-specific binding interactions, enabling enhanced cell targeting, and co-delivery of multiple therapeutic agents31–33. Anionic peptides have been used to coat the surface of cationic polyplexes to reduce the non-specific interactions for a targeting application using HUVECs34. Gene delivery to immune cells was achieved in vivo using PBAE polyplexes coated with poly(glutamic acid) for transfection of T cells or tumor-associated macrophages2–3. More recently, modification of nanoparticle surfaces gave rise to controlled cellular interactions and specificity to ovarian cancer, demonstrating the crucial role played by each polyelectrolyte used in the formulation35.
Here, we describe the development of immunoplexes (IPs) that consist of an inner Ac-PEI/pDNA polyplex enveloped within an anionic poly(ethylene-alt-maleic acid) (PEMA) polyelectrolyte layer for the serum-independent transfection of innate and adaptive immune cells. IPs were prepared and screened for various physical and biochemical properties and pDNA transfection efficiency was determined in RAW 264.7 macrophages, DC2.4 dendritic cells, and human Jurkat T cells. Cellular interactions of IPs were controllable through modulation of PEMA content and correlated with levels of gene expression. By incorporating pDNA into IPs at unusually high N/P ratios, IP stability in the presence of serum was greatly improved, PEMA enveloping improved pDNA unpackaging from IPs, and mitigated toxicity associated with Ac-PEI delivery at high N/P ratios. The ability to specifically control the cellular interactions of IPs highlights their potential use as a modular and targetable gene delivery platform to achieve high levels of cell-specific transfection in vivo. These results support IPs as a simple, modular, serum-independent, and highly effective non-viral gene delivery platform to efficiently transfect cells of the innate and adaptive immune system.
Experimental Section:
Materials:
Acetic anhydride, triethylamine, and branched polyethylenimine (PEI; MW 25 kDa) was purchased from Millipore Sigma (St. Louis, MO). Poly(ethylene-alt-maleic anhydride) (PEMA) ZeMAC™ E60 (PEMA) was received as a gift from Vertellus™ (Indianapolis, IN) was purchased from Polyscience, Inc. (Warrington, PA), NHS-Rhodamine (NHS-RHO) was purchased from Fisher Scientific (Waltham, MA). Maxiprep Endotoxin Free Kit was purchased from Qiagen (Germantown, MD). Unless otherwise noted, any additional reagents were purchased from Millipore Sigma.
Cell culture:
RAW 264.7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Millipore Sigma; St. Louis, MO) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (VWR; Radnor, PA) and 1% penicillin/streptomycin (P/S) (Invitrogen Corporation; Carlsbad, CA). DC2.4 cells were cultured in in RPMI 1640 (Millipore Sigma; St. Louis, MO) supplemented with 10% FBS, 1% P/S, and 50 μM β-mercaptoethanol. Jurkat cells were cultured in RPMI 1640 supplemented with 10% FBS and 1% P/S. Both cell lines were incubated at 37°C and 5% CO2.
Synthesis of Ac-PEI and rhodamine-conjugated PEI (Ac-PEI-RHO):
Ac-PEI with varying degrees of acetylation was synthesized using a previously reported method20. Briefly, 500 mg of PEI (0.02 mmol, 25000 g/mol) was added to 20 mL scintillation vials and dissolved in 10 mL methanol. Triethylamine (5 molar equivalents to acetic anhydride) was then added followed by acetic anhydride (Figure 1C) to achieve theoretical degrees of acetylation of 20%, 40%, and 60% of the total amines (Ac20-PEI, Ac40-PEI, and Ac60-PEI respectively). The reaction was allowed to stir overnight at room temperature and the modified PEI was purified by dialysis against water using a 12–14 kDa MWCO membrane and recovered by lyophilization. 1H-NMR was used to determine the actual percent acetylation of PEI as previously reported20. Ac-PEI polymer was dissolved in 800 μL of D2O and the 1H-NMR spectra was acquired using a 400 MHz Varian spectrometer. Fluorescently-labeled Ac-PEI was also synthesized by conjugating NHS-Rhodamine (RHO) with Ac-PEI. Briefly, 10 mg Ac20-PEI (3.51×10−4 mmol) was dissolved in methanol and then 1 mg of NHS-RHO (1.89×10−3 mmol) and 50 μL of triethylamine was added dropwise and stirred for 4 h. After the reaction, the solution was dialyzed against water with a 12–14 kDa MWCO membrane and lyophilized for further use.
Figure 1. Synthesis and characterization of acetylated PEI (Ac-PEI).
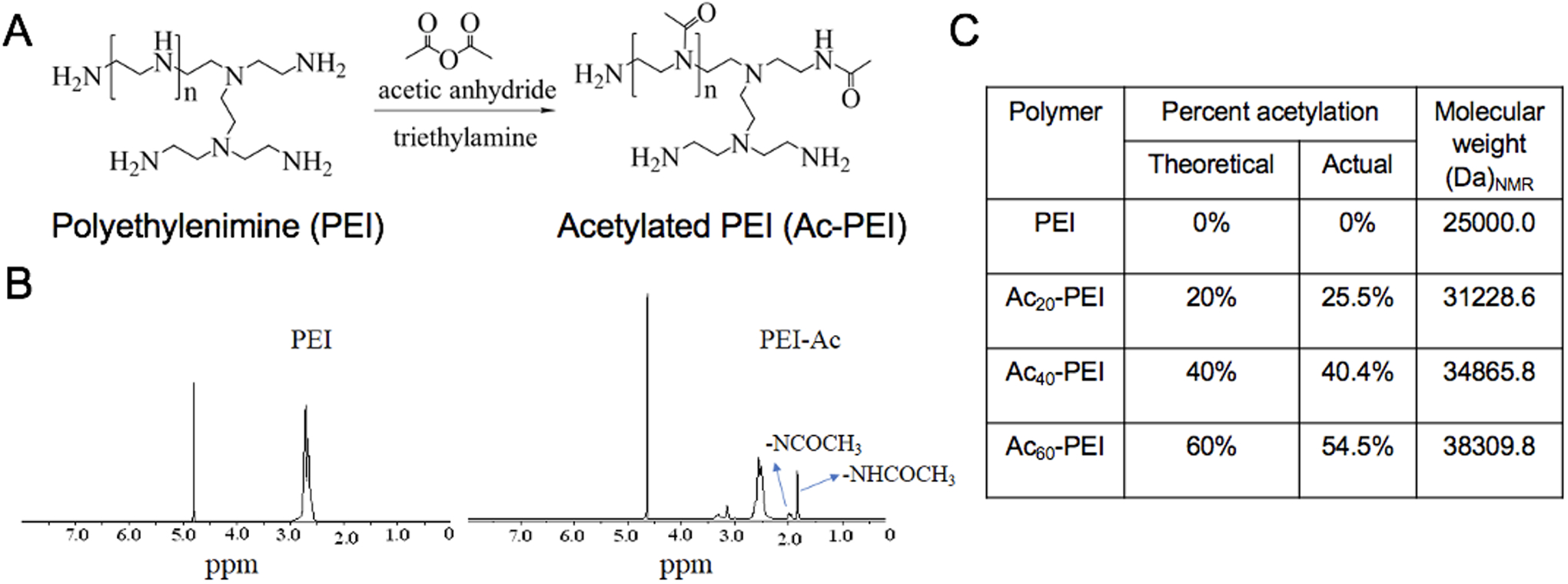
A) Synthesis scheme for acetylated PEI. B) Representative 1H-NMR spectra for PEI and Ac-PEI in D2O. The peak at 1.7–1.8 ppm is due to acetylated primary amines whereas the peak at 1.8–1.9 ppm is from acetylated secondary amines of PEI. C) Quantification of PEI degree of acetylation and molecular weight determined by 1H-NMR for various modified PEI polymers.
GFP Plasmid preparation:
DH5α E. coli competent cells (Invitrogen Corporation; Carlsbad, CA) were transformed with eGFP (GFP) pDNA (courtesy of National Center for Toxicological Research, FDA, Jefferson, AR), which encoded for kanamycin resistance. Transformed cells were expanded in an overnight liquid LB culture at 37°C under vigorous shaking, lysed, and purified using a Qiagen Maxiprep Endotoxin Free Kit. The concentration of pDNA was verified using a SpectraMax iD3 microplate reader and a SpectraDrop micro-volume microplate (Molecular Diagnostics; San Jose, CA) by measuring absorbance at 260 nm and 280 nm and agarose gel electrophoresis. pDNA was stored at -20°C until further use.
Stability of Ac20-PEI, Ac40-PEI, and Ac60-PEI polyplexes:
The binding capacity of polymers to DNA was analyzed by DNA gel electrophoresis. Fresh polyplex solutions were prepared immediately prior to each experiment. Polyplexes were prepared using Ac20-PEI, Ac40-PEI, and Ac60-PEI, at different N/P ratios ranging from 0 to 20. First, plasmid DNA was diluted in HEPES buffer (20 mM, pH 7) at a concentration 500 ng/mL. Fresh Ac-PEI was prepared at a concentration of 1 mg/mL in water. Next, the appropriate amount of Ac-PEI and plasmid DNA were mixed together in a microcentrifuge tube and incubated for 10 minutes at room temperature. 6X Loading dye (Invitrogen Corporation; Carlsbad, CA) was added to the polyplexes prior to running on a 1% agarose gel, followed by staining with ethidium bromide. pDNA was visualized with an Invitrogen™ E-Gel™ Imager System (Fisher Scientific; Waltham, MA).
Preparation of IPs:
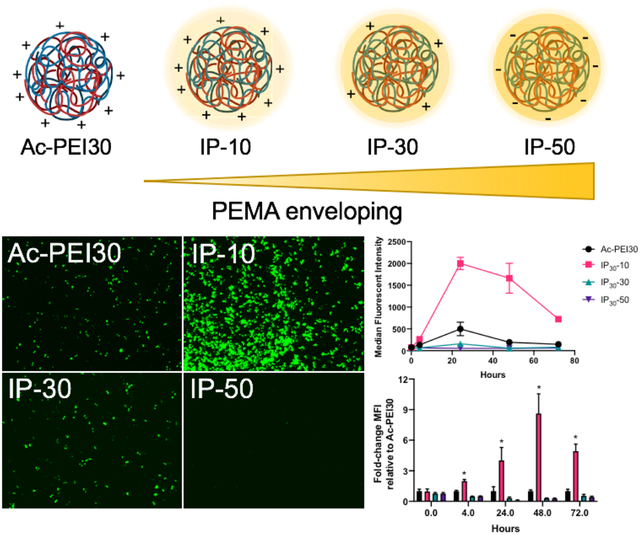
All IPs evaluated used Ac20-PEI polymers and were complexed with pDNA at N/P ratios of 7.5, 15, and 30. The formed polyplexes are referred to as Ac-PEI(N/P ratio). Polyplexes were enveloped by adding PEMA (1 mg/mL) in HEPES at various weight ratios (10 wt%, 30 wt%, and 50 wt%) with respect to Ac-PEI followed by incubation at room temperature for 10–20 minutes to form IPs. IPs are denoted as IP(N/P ratio)-(PEMA wt%). For example, IP30-10 describes an IP prepared at N/P 30 and 10 wt% PEMA.
Hydrodynamic size and zeta potential measurement:
Fresh polyplex and IPs solutions were prepared with 2 μg of GFP plasmid in HEPES buffer. 1 mL of each sample was added into a disposable cuvette and the size was measured using a Zetasizer Nano ZS (Malvern Instruments Inc.). 15 runs were performed in triplicate for each sample. Subsequently, samples were transferred to a folded capillary cell (Malvern) and zeta-potential measurements were performed in triplicate for each sample using the Zetasizer Nano ZS (Malvern).
Stability of IPs in presence of PEMA and serum:
The stability of IPs is critical to ensure pDNA can be efficiently delivered to immune cells of interest in vivo without destabilization. The stability of IP30-10, 30, 50 was assessed by DNA gel electrophoresis as described above in buffer or after incubating with 55% fetal bovine serum (FBS) in PBS, a physiologically-relevant serum concentration, for 30–60 minutes29.
In vitro transfection of RAW 264.7 macrophages, DC2.4 dendritic cells, and human Jurkat T cells :
Polyplexes and corresponding IPs were prepared at N/P 30 containing 2 μg of pDNA encoding GFP (Ac-PEI30). RAW 264.7, DC2.4, or Jurkat cells were treated for 4 h in serum-containing or serum-free media followed by washing to remove excess complex prior to overnight incubation in complete media. RAW 264.7 and DC2.4 cells are adherent and excess complex was easily removed by washing with PBS and replaced with complete DMEM or RPMI1640, respectively. In contrast, Jurkat cells are suspension cells and were washed by first transferring to a microcentrifuge tube followed by centrifugation at 500 × g for 5 min to separate from polyplexes and IPs. The supernatant was removed, and the cell pellet was dispersed in a fresh complete media. Rhodamine and GFP expression was visualized using a Revolve fluorescence microscope (ECHO, San Diego, CA) at 24 h post-transfection. The cellular interactions and uptake were determined using polyplex and IPs prepared with Ac20-PEI-RHO.
Flow cytometry:
RAW 264.7, DC2.4, or Jurkat cells were cultured in a 24-well plate at a density 2×105 cells/ well for 24 h. Next, cells were transfected with different formulations of IPs in both serum-free and serum-containing media for 4 h. Subsequently, cells were washed and incubated for another 24 h in fresh serum-containing media. After 24 h, cells were collected using a cell scraper followed by centrifugation at 500 × g for 5 minutes (RAW 264.7 and DC2.4 cells are adherent) or by centrifugation at 500 × g for 5 minutes (Jurkat cells are in suspension) and resuspended in fresh flow cytometry buffer. For analysis of live cells only, 4′,6-diamidino-2-phenylindole dilactate (DAPI) was used as an exclusion dye to determine cell viability. Data was collected using an LSR II (Becton-Dickinson, San Jose, CA) flow cytometer and analyzed by FCS Express 7 (De Novo Software, Pasadena, CA) and transfection efficiency was measured as the percentage of live cells which were GFP+ compared to un-transfected controls.
In vitro persistence of GFP expression:
Raw 264.7 cells were grown in a 24-well plate at a density 2×105 cells/well for 24 h in complete DMEM. After that, cells were transfected with Ac-PEI30, IP30-10, IP30-30 and IP30-50 for 4 h in serum-containing medium. The cells were then washed and incubated with fresh DMEM for 4 h, 24 h, 48 h, and 72 h. The percentage of live cells which were GFP+ compared to controls was determined using flow cytometry.
MTS assay for assessing cytotoxicity:
The cytotoxicity of IPs was evaluated using an MTS assay (Abcam; Cambridge, MA). RAW 264.7 and Jurkat cells were cultured in a 24-well plate at a density 2×105 cells/well overnight prior to treatment with PEI variants [Ac-PEI7.5, Ac-PEI15, Ac-PEI30, PEI30] or IPs [IP30-10, IP30-30 and IP30-50] for 4 h in both serum-containing and serum-free media. Following the incubation, cells were washed and further incubated with fresh media for 24 h. Next, 50 μL of MTS solution was added in each well and incubated for another 3 h. The optical density (O.D.) of the solution was measured using a SpectraMax iD3 microplate reader at 570 nm. The percentage of cell viability was measured as the ratio of O.D. at 570 nm to no treatment control. Each treatment was replicated for a total of three times.
Results and Discussion
Synthesis and characterization of Ac-PEI polyplexes:
Ac-PEI was synthesized by reacting 25 kDa branched PEI with various amounts of acetic anhydride to yield three variations of Ac-PEI (Figure 1A). The primary and secondary amine groups of PEI react with acetic anhydride to form secondary and tertiary amides, respectively. 1H-NMR was used to determine the total percent of acetylation, where the peak at 1.7–1.75 ppm signifies the acetylation of primary amine groups and the peak at 1.8–1.85 ppm depicts the acetylation of secondary amine groups of PEI (Figure 1B and Figure S1)20. The actual percentage of acetylation for Ac20-PEI, Ac40-PEI, and Ac60-PEI was measured to be 25.5%, 40.4% and 54.5%, respectively (Figure 1C).
Polyplexes were formed at various N/P ratios to assess the effect of percentage of acetylation of Ac-PEI to form stable polyplexes with pDNA using DNA gel electrophoresis (Figure 2A and B). Ac20-PEI formed more stable complexes than Ac40-PEI and Ac60-PEI, which was consistent with previous reports that polyplexes prepared at higher percentages of acetylation were less able to condense pDNA20, 24. Further, the degree of acetylation correlated with ability of Ac20-PEI, Ac40-PEI, and Ac60-PEI to transfect RAW 264.7 cells in vitro (Figure 2C). Transfection is largely dependent on two factors, endosomal buffering capacity of the polyplexes, which aids in endosomal escape, and dissociation/unpackaging of the polyplexes intracellularly to release pDNA inside the cells20, 24. Acetylation of PEI can enhance the dissociation of the polyplexes inside cells but also decreases the endosomal escape efficiency24. A lower percentage of acetylation with Ac20-PEI enables retention of strong endosomal buffering capacity, whereas the polyplexes prepared with high levels of acetylation are not as effective24. Furthermore, acetylation of PEI also enhances the lipophilicity of PEI which can lead to enhancement of transfection36. Due to the higher stability and transfection efficiency, Ac20-PEI (referred to as Ac-PEI) was used in all subsequent experiments.
Figure 2. Ac-PEI polyplex formation and characterization.
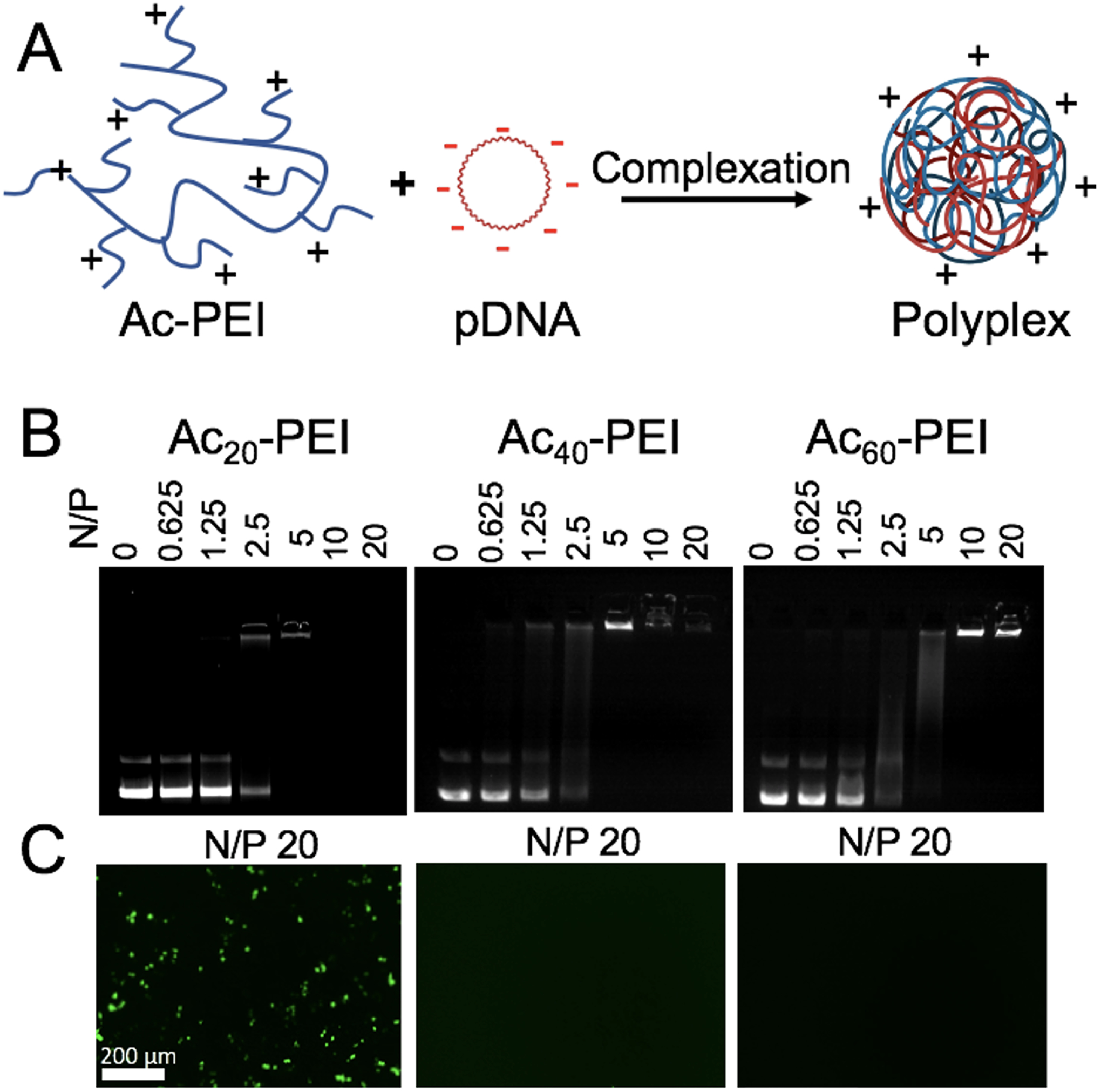
A) Schematic representation of polyplex formation using acetylated PEI (Ac-PEI) and plasmid DNA (pDNA). B) Agarose gel electrophoresis showing complex formation of various Ac-PEI polymers with GFP plasmid at different N/P ratios. C) GFP expression in RAW 264.7 macrophages using AC-PEI/GFP polyplexes prepared at N/P ratio 20. Ac20-PEI shows higher GFP signals compared to Ac40-PEI and Ac60-PEI. Representative images of at least n=3 experiments.
Overcoming serum-induced reductions in transfection efficiency in RAW 264.7 and Jurkat cells through modulation of the N/P ratio:
Immune cells, especially suspension cells, are notoriously difficulty to transfect and commercially available transfection reagents have only shown limited success25, 30. Numerous barriers must be overcome to enable effective DNA delivery to cells37. Accordingly, a balance must be achieved between DNA protection, DNA release, and toxicity to efficiently induce gene expression38. Polyplexes prepared at high N/P ratios exhibit improved serum stability yet suffer from a reduced ability to release DNA intracellularly, increased non-specific cellular interactions, and high toxicity.
We assessed the impact of N/P ratio of Ac20-PEI (Ac-PEI7.5, Ac-PEI15, Ac-PEI30) on the serum-dependent transfection of RAW 264.7 (adherent) and Jurkat (suspension) cells. The experimental setup as well as representative flow cytometry data are shown in Figure S2. The impact of serum on transfection efficiency was more prominent in RAW 264.7 cells at low N/P ratios (Figure 3A), whereas higher N/P ratios showed enhanced transfection efficiencies in serum-containing medium and decreased in serum-free medium. The differences between serum and serum-free were less apparent for Jurkat cells, where increasing the N/P ratio from Ac-PEI7.5 to Ac-PEI30 resulted in enhanced transfection (Figure 3B). Higher N/P ratios in the presence of serum are likely necessary due to the ability of free polymer to interact with serum proteins and assist with intracellular trafficking39–40. Interestingly, the %GFP+ cells was similar for Ac-PEI30 and PEI30 in RAW 264.7 cells in serum-containing medium, the MFI for Ac-PEI30 was 96% higher than non-acetylated PEI30 controls (Figure 3C). An opposite trend was observed for Jurkat cells where the %GFP+ was significantly higher for Ac-PEI30 versus PEI30, but the MFI of Ac-PEI30 was only increased by 25% (Figure 3D). Critically, not only did Ac-PEI30 show significantly improved GFP expression compared to PEI30 overall, the improvement in cell viability of Ac-PEI30 compared to PEI30 was dramatic where a 70% increase in viability was noted for RAW 264.7 cells and a 47% increase was found for Jurkat cells (Figure 3E and F). Our results are in contrast to a previous study that found unmodified PEI could not increase the transfection efficiency in Jurkat cells likely because of its inability to unpackage the genetic payload at such high N/P ratios25. The improved GFP expression and overall cell viability of Ac-PEI30 in serum-containing medium (greater than 75%) compared to other polyplexes examined, provided our rationale to utilize Ac-PEI30 in our subsequent experiments.
Figure 3. Serum-dependent transfection efficiency and toxicity of Ac-PEI polyplexes.
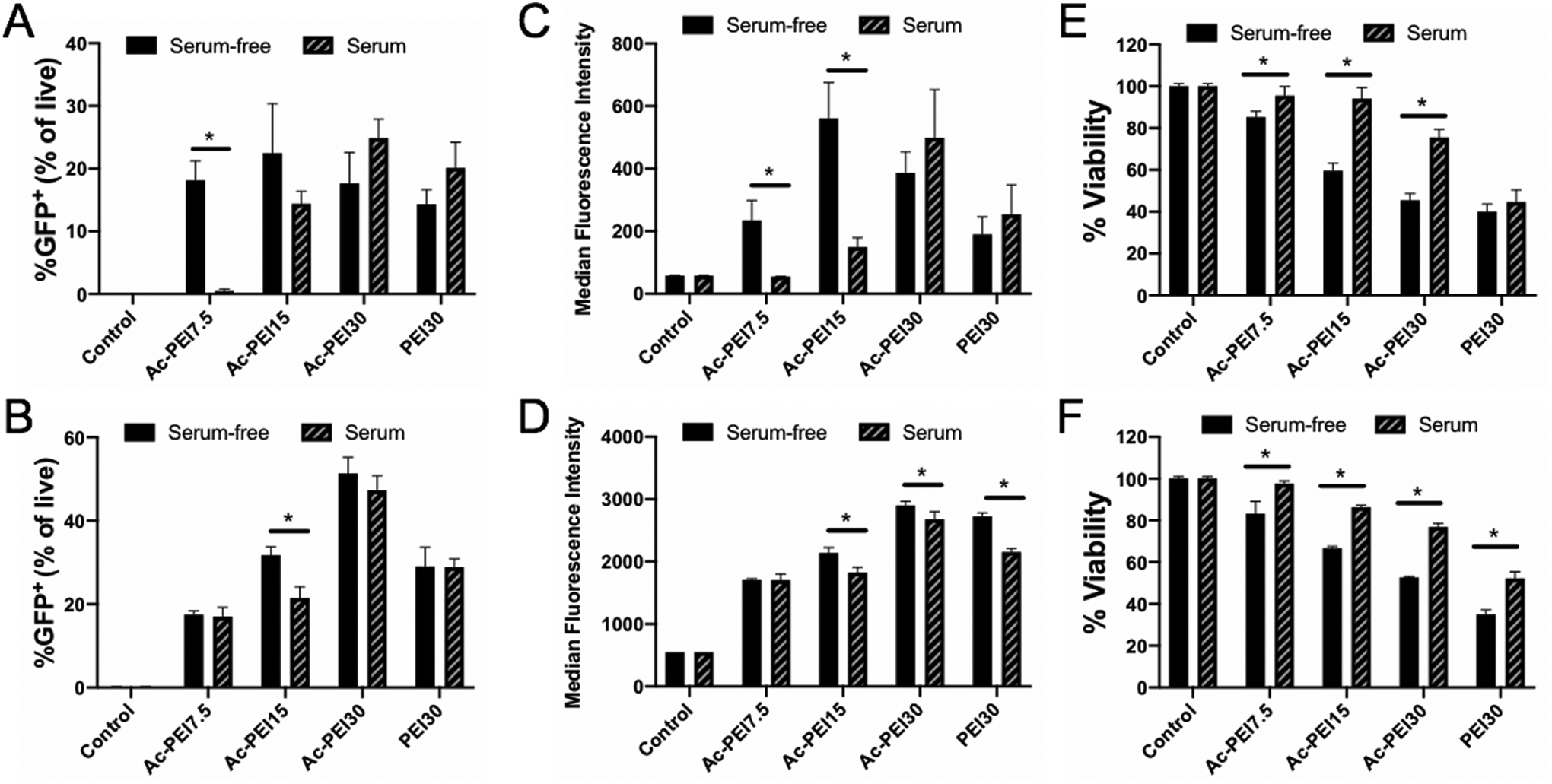
Transfection efficiency of Ac20-PEI (Ac-PEI) polyplexes at various N/P ratios in RAW 264.7 (A) and Jurkat cells (B) determined by flow cytometry. Median Fluorescence Intensity of GFP expression for RAW 264.7 (C) and Jurkat cells (D). Cell viability determined by MTS assay for RAW 264.7 (E) and Jurkat cells (F). Cells were incubated in serum-free or serum-containing medium for 4 hrs with polyplexes, washed, and incubated for 24 hrs prior to analysis. Data are representative of n=3 experiments. Statistical differences between groups were determined by performing a one-way ANOVA and Tukey’s post hoc test (p < 0.05). Error bars represent SD.
Enveloping polyplexes with PEMA (immunoplex (IP) formation) and physicochemical characterization:
Polyplexes prepared at higher N/P ratios using Ac-PEI30 provided higher transfection efficiencies an improved toxicity profiles in serum-containing medium compared to PEI30 (Figure 3). We hypothesized that by enveloping Ac-PEI30 polyplexes with the anionic polyelectrolyte PEMA, that we could further improve the toxicity profile at high N/P ratios and enhance the levels of gene expression by engineering improved mechanisms of endosomal escape into Ac-PEI30 through incorporation of a pH-dependent endosomal destabilization mechanism41. The polyplexes were enveloped with PEMA by first preparing Ac-PEI30 polyplexes at N/P 30 followed by the addition of PEMA at various weight ratios [10wt% (IP30-10), 30wt% (IP30-30), and 50wt% (IP30-50)] relative to Ac-PEI30 (Figure 4A). The hydrodynamic diameter of Ac-PEI30 polyplexes was approximately 150 nm and +28 mV zeta potential (Figure 4B and C). As increased amounts of PEMA were incorporated, the surface charge of the IPs was decreased and IP30-50 was -17 mV. The size increased with increasing percentage of PEMA. As the surface charge of IPs reached close to neutral (as in IP30-30), aggregation was observed due to the loss of electrostatic repulsion between polyplexes. Further increases in PEMA such as IP30-50 recovered the size distribution. A similar trend with size and zeta potential was observed for IPs prepared at N/P 7.5 and 15 (Table S1).
Figure 4. Preparation, characterization, and stability assessment of immunoplexes (IPs).
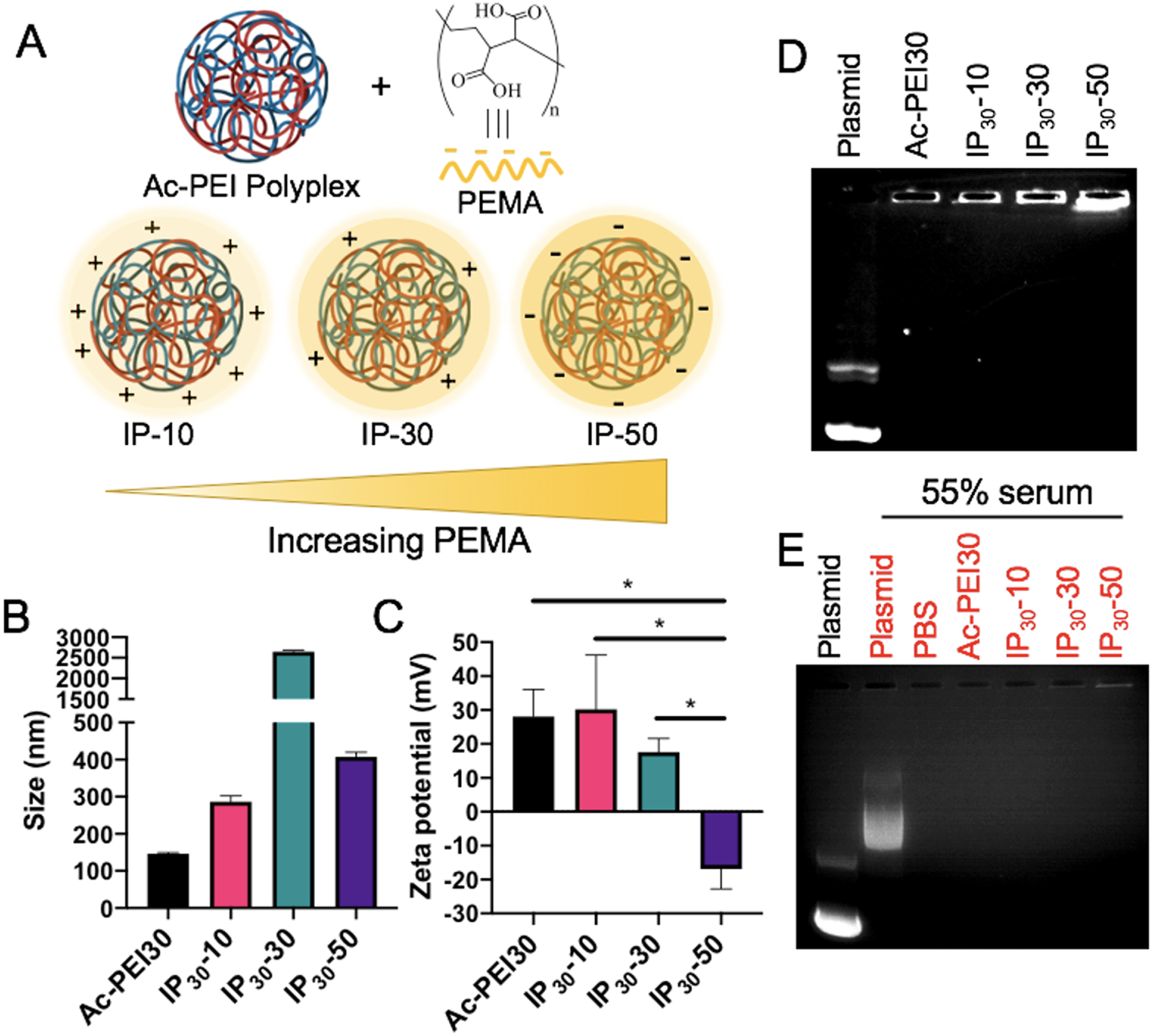
A) Schematic representation of enveloping Ac-PEI/GFP polyplexes with various wt.% of PEMA to form IPs. B, C) Hydrodynamic size and zeta potential of IPs, respectively. Agarose gel electrophoresis to assess the stability of Ac-PEI/GFP polyplexes following PEMA enveloping at N/P 30 in the absence (D) or presence (E) of physiologically-relevant concentration serum (55% v/v). IPs are stable after enveloping with PEMA and in presence of serum as no DNA band is observed. PEMA (poly(ethylene-alt-maleic acid)). n=3 for each experiment. Statistical differences between groups were determined by performing a one-way ANOVA and Tukey’s post hoc test (p < 0.05). All bars in panel B are significantly different from each other. Error bars represent SD.
Stability of IPs in the presence of PEMA and physiologically-relevant serum concentrations:
Engineering polyplexes with high stability is critical to ensure their genetic cargo is delivered appropriately to cell types of interest, particularly in vivo. The enveloping of cationic polyplexes with anionic polyelectrolyte coatings has the potential to reduce toxicity and minimize non-specific cellular interactions. However, enveloping could induce destabilization of the polyplexes through competition with negatively-charged DNA resulting in its subsequent release. To determine the effect of PEMA enveloping on IP stability, agarose gel electrophoresis was performed. Figure 4D shows that at up to 50wt% of PEMA, IPs maintained stable complexes, however IPs became unstable as the PEMA concentration was further increased (data not shown).
The effect of serum on IP stability was also confirmed by first formulating IPs followed by incubation in PBS containing 55% FBS, a physiologically-relevant serum concentration29, 42. This experiment was performed to address two potential effects that may lead to reduced performance of IPs in vivo: 1) the formation of a biomolecule corona on the surface of polyplexes may induce IP destabilization through competition with negatively-charged DNA through the adsorption of serum proteins to the surface of the particles27 and 2) FBS/serum contains nucleases that can degrade pDNA if not sufficiently protected by the polyplex42. Figure 4E shows the stability of IPs was not affected by serum as indicated by a lack of DNA bands detected in the wells containing IPs. This result correlated with previous studies that have shown incubation of PEI-based polyplexes in high concentrations of FBS (greater than 50%) can maintain the stability of plasmid against degradation for at least 3 days42. Interestingly, the control well containing a mixture of serum and pDNA was able to partially condense the DNA through interactions between the negatively-charged DNA and positively-charged species present in the serum. These results demonstrated that the formulated polyplexes display beneficial stability during formulation and under in vivo-relevant conditions, which may allow for their successful in vivo application.
Controlled transfection of immune cells using IPs in serum-containing media:
We hypothesized that PEMA enveloping of Ac-PEI/pDNA polyplexes would increase transfection efficiency of polyplexes prepared at high N/P ratios through improved unpackaging of DNA cargo and enhanced endosomal escape, while improving cell viability due to shielding of positive charges caused by the polyplexes. Enveloping of polyplexes with anionic polymers also has the potential to minimize the formation of non-specific interactions and enable cell-specific targeting to be engineered into the platform, thus improving its potential for in vivo applications. The transfection efficiency of rhodamine (RHO)-labeled IPs with variable coating with PEMA was assessed in vitro using RAW 264.7 and Jurkat cells. The uptake and transfection of IPs using RAW 264.7 and Jurkat cells in serum-containing medium is shown in Figures S3A and B, respectively. The uptake of IPs and the GFP protein expression decreased as a function of PEMA percentage in both cell types suggesting that PEMA enveloping reduced non-specific cellular interactions. Colocalization of RHO-labeled IPs and GFP signals were strongly visualized for RAW 264.7 cells and to a lesser extent for Jurkat cells due to difficulties associated with live cell imaging of suspension cells43–44.
Flow cytometry was used to quantify differences in transfection efficiency for IPs in serum-containing or serum-free medium. The impact of PEMA enveloping on transfection efficiency and toxicity of IPs in RAW 264.7 and Jurkat cells is shown in Figure 5. Transfection of RAW 264.7 cells (Figure 5A) was less effective compared to Jurkat cells (Figure 5B) in serum-free conditions, where all IPs tested resulted in less than 10% of cells GFP+. Interestingly, a 225% enhancement was measured in RAW 264.7 cells in serum-containing medium for IP30-10 compared to Ac-PEI30. However, this difference was not observed for Jurkat cells, where IP30-10 was not significantly different from Ac-PEI30 at similar conditions. The ability for IPs to transfect either cell type efficiently was lost using IP30-30 and IP30-50. A potential reason for the loss in transfection efficiency for IP30-30 is the increased size of the complex due to aggregation in this condition (Figure 4B). The reduced transfection efficiency of IP30-50 was attributed to a highly negative surface charge that reduced nano-bio interactions as supported by Figure S3. The MFI for GFP expression was assessed for RAW 264.7 and Jurkat cells in Figures 5C and D, respectively. Notably, in accordance with the %GFP+ enhancement for IP30-10 in RAW 264.7 cells, a 400% increase in MFI was measured over Ac-PEI30. The MFI for Jurkat cells followed a similar trend as %GFP+. For both cell types, the viability was increased with increasing percentages of PEMA enveloping and in the presence of serum (greater than 10% PEMA) (Figures 5E and F). A similar trend for %GFP+ enhancement and MFI for DC2.4 cells was observed as Jurkat cells except that Ac-PEI30 treatment resulted in greater expression than IP30-10 (Figure S4). These results demonstrated that IPs prepared at high N/P ratios significantly increased the transfection efficiency of RAW 264.7 cells and retained similar effectiveness of Ac-PEI30 for Jurkat and DC2.4 cells. Importantly, the reduced non-specific cellular interactions for IPs such as IP30-50 may offer an avenue for further development of the IP platform for specific cell targeting through incorporation of targeting ligands2, 45.
Figure 5. Serum- and PEMA enveloping-dependent transfection and toxicity of IPs.
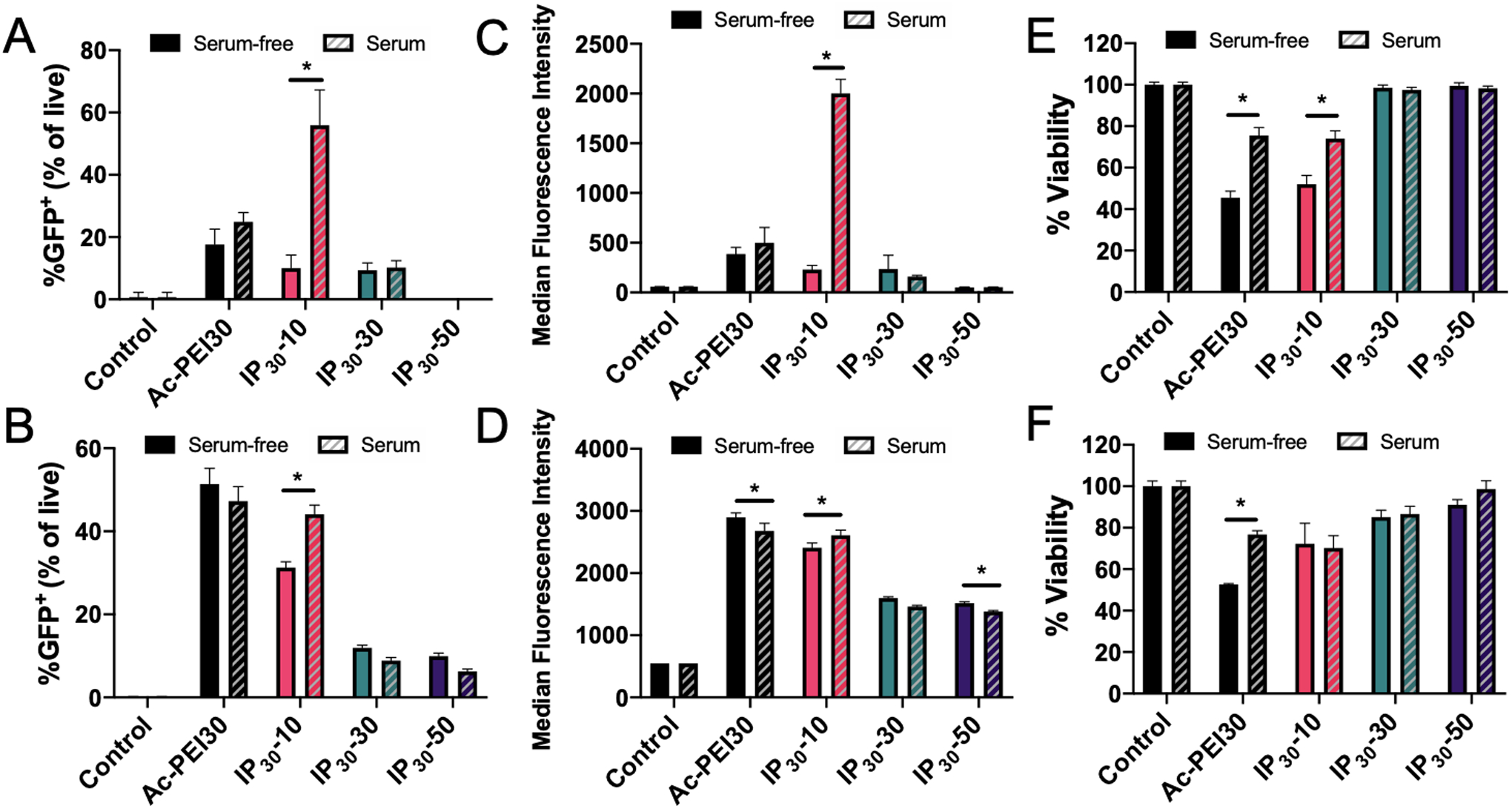
Transfection efficiency of IPs prepared with various wt.% of PEMA (10% to 50%) determined by flow cytometry in RAW 264.7 (A) and Jurkat cells (B). Median Fluorescence Intensity of GFP expression for RAW 264.7 (C) and Jurkat cells (D). Cell viability determined by MTS assay for RAW 264.7 (E) and Jurkat cells (F). Cells were incubated in serum-free or serum-containing medium for 4 hrs with polyplexes, washed, and incubated for 24 hrs prior to analysis. Data are representative of n=3 experiments. Statistical differences between groups were determined by performing a one-way ANOVA and Tukey’s post hoc test (p < 0.05). Error bars represent SD.
Controlling GFP expression in vitro:
To further understand the persistence of GFP expression and the role that PEMA enveloping of polyplexes plays in this process, we evaluated GFP expression using a combination of fluorescence microscopy and flow cytometry at 5 timepoints using RAW 264.7 cells. Figures 6A and 6B show the significant enhancements in GFP expression in cells from 24 – 72 h in serum-containing medium. Figure 6C shows the fold-change MFI relative to Ac-PEI30 for each of the IPs evaluated. The most significant enhancements were found for IP30-10 at all timepoints longer than 4 h (2-fold). The enhancements further increased to 4-fold at 24 h and 8-fold at 48 h before slightly decreasing to 5-fold at 72 h. A noteworthy finding from this study was that a significant increase in GFP expression after 4 h was measured for IP30-10 indicating that the PEMA enveloping potentially enhanced the unpackaging of plasmid DNA from Ac-PEI. This enhancement in unpackaging is likely due to three factors: 1) Ac-PEI has a greater capacity to release DNA due to its acetylation20, 24; 2) Ac-PEI has a lower pKa than PEI, which affects endosomal buffering capacity; and 3) PEMA improves the dissociation of DNA from Ac-PEI by dissociating from the surface of polyplexes at lysosomal pH (4.5–5) (PEMA: pKa1 = 4.16; pKa2 = 5.61)46 that leads to endosomal membrane destabilization and improves cytosolic delivery of DNA41. Therefore, the type of anionic polyelectrolyte enveloping the polyplex potentially plays a significant role in controlling the protein expression in cells which could be very important design criteria to leverage in for future IP designs.
Figure 6. Persistence of GFP expression in RAW 264.7 macrophages.
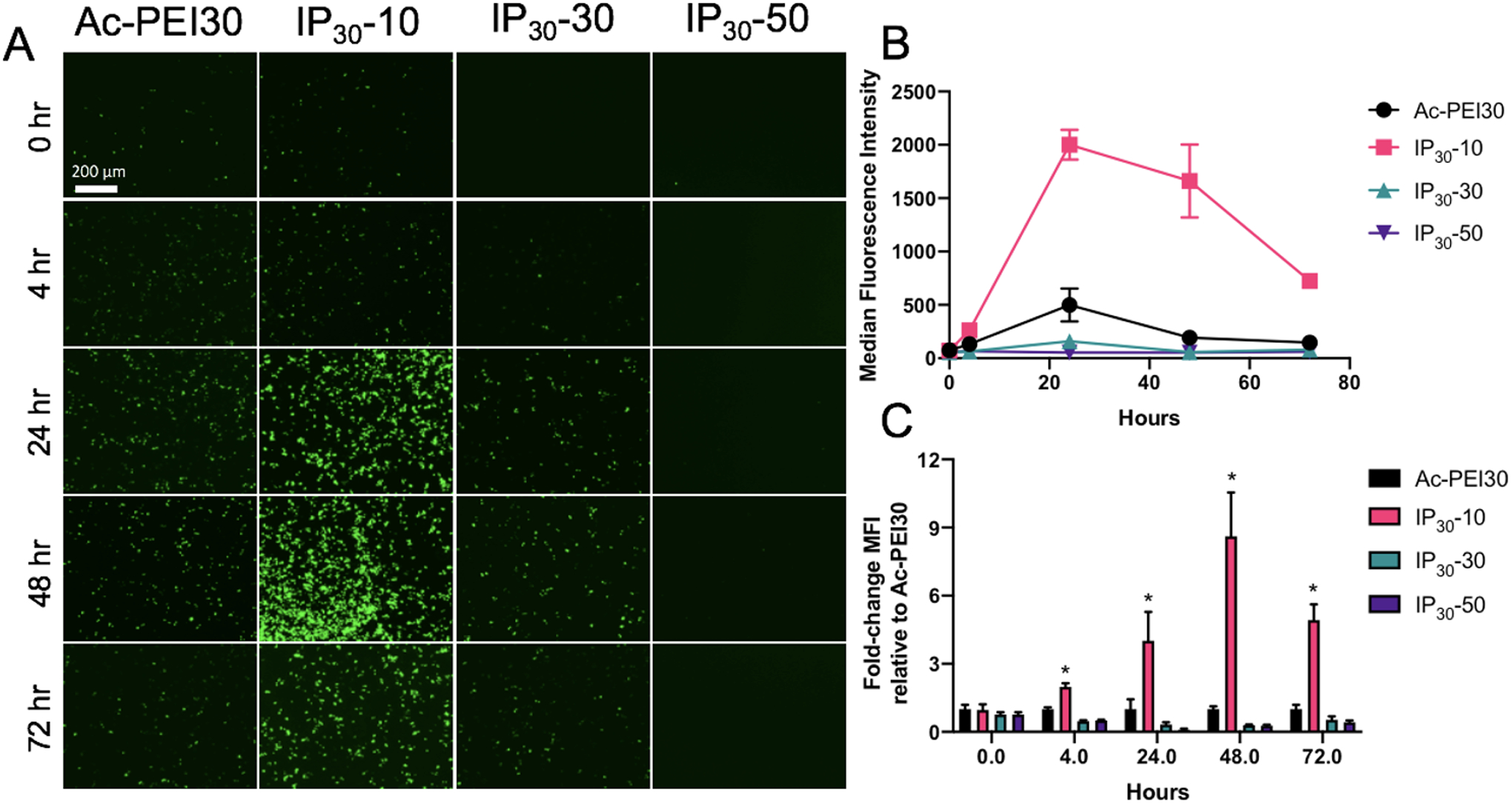
A) Fluorescence micrographs of RAW 264.7 macrophages transfected in serum-containing medium with various IPs enveloped with various wt.% of PEMA. Images are representative of at least n=3 experiments. Cells were incubated in serum-containing medium for 4 hrs with IPs, excess IPs were washed away, and images were acquired at various time points. B) Transfection efficiency of IPs, 24 hr post cell treatment. C) Fold-change enhancements in transfection efficiency relative to Ac-PEI30 for IPs. IP30-10 displayed significantly longer and higher levels of transfection compared to Ac-PEI30 polyplexes. Data are representative of n=3 experiments and as mean +/- SD.
Conclusions
Engineering of immune cells through delivery of nucleic acids has the widespread potential to correct or functionally reprogram genetic aberrations present in diseases or as therapeutics in the case of cancer, autoimmune diseases, or others47. The presence of serum has been demonstrated as a hindrance to achieve high transfection efficiencies and in situ applications of gene delivery will require delivery platforms to be effective in complex medium such as blood. Here, we developed a simple and effective, serum-independent platform technology, immunoplexes (IPs), consisting of Ac-PEI enveloped with an anionic polyelectrolyte (PEMA) to enhance the transfection of plasmid DNA into innate and adaptive immune cells. We identified through modulation of the N/P ratio that the negative impact of serum in lymphocyte transfection could be overcome and that the high N/P ratio used for the formulation of IPs was critical to ensure serum stability. Furthermore, the incorporation of PEMA enabled controllable cellular interactions and transfection efficiencies to be engineered, improved IP unpackaging, and showed a beneficial toxicity profile. The optimized IP30-10 formulation resulted in an increased duration of gene expression compared to other Ac-PEI30 and other IP variants with a maximum 8-fold enhancement in gene expression over 3 days. Finally, we foresee that formulations such as IP30-50, which showed limited cellular interactions, transfection efficiency, and low toxicity will be useful when developed further for in situ targeted gene delivery applications as PEMA can be functionalized with targeting ligands directed toward cell types of interest as similarly achieved using other anionic polymers2, 48. Our future studies aim to address these potential factors and applications to improve the efficiency of IPs as a non-viral gene delivery platform for targeted modulation of immune-mediated diseases.
Supplementary Material
Acknowledgements
This work was supported by startup funds by the University of Maryland School of Pharmacy, the New Investigator Award from the American Association of Colleges of Pharmacy (AACP), and the University of Maryland Baltimore Institute for Clinical and Translational Research (ICTR) Accelerated Translational Incubator Pilot Grant (NIH #1UL1TR003098) awarded to R.M.P. Additional support was provided by the NIAID-NIH Signaling Pathways in Innate Immunity Training Program (NIH #T32AI095190) and NHLBI-NIH Interdisciplinary Training Program in Cardiovascular Disease (NIH #T32HL007698) to support J.J.M.L. The authors declare no competing conflicts of interest.
Footnotes
Supporting information
The following files are available free of charge at www.pubs.acs.org.
Figure S1. 1H-NMR of various PEI polymers used in this study.
Figure S2. Experimental setup for transfections.
Figure S3. Controlled cellular interactions and transfection efficiency of IPs.
Figure S4. Serum- and PEMA enveloping-dependent transfection of IPs in DC2.4 cells.
Table S1. Size and Zeta potentials of various IP formulations prepared in this study.
References
- (1).Pearson RM; Casey LM; Hughes KR; Miller SD; Shea LD In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance. Adv Drug Deliv Rev 2017, 114, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Smith TT; Stephan SB; Moffett HF; McKnight LE; Ji W; Reiman D; Bonagofski E; Wohlfahrt ME; Pillai SPS; Stephan MT In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol 2017, 12, 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang F; Parayath NN; Ene CI; Stephan SB; Koehne AL; Coon ME; Holland EC; Stephan MT Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun 2019, 10, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Colella P; Ronzitti G; Mingozzi F Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol Ther Methods Clin Dev 2018, 8, 87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shirley JL; de Jong YP; Terhorst C; Herzog RW Immune Responses to Viral Gene Therapy Vectors. Mol Ther 2020, 28, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hacein-Bey-Abina S; Hauer J; Lim A; Picard C; Wang GP; Berry CC; Martinache C; Rieux-Laucat F; Latour S; Belohradsky BH; Leiva L; Sorensen R; Debré M; Casanova JL; Blanche S; Durandy A; Bushman FD; Fischer A; Cavazzana-Calvo M Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. New England Journal of Medicine 2010, 363, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wan T; Chen Y; Pan Q; Xu X; Kang Y; Gao X; Huang F; Wu C; Ping Y Genome editing of mutant KRAS through supramolecular polymer-mediated delivery of Cas9 ribonucleoprotein for colorectal cancer therapy. J Control Release 2020, 322, 236–247. [DOI] [PubMed] [Google Scholar]
- (8).Moradian H; Roch T; Lendlein A; Gossen M mRNA Transfection-Induced Activation of Primary Human Monocytes and Macrophages: Dependence on Carrier System and Nucleotide Modification. Sci Rep 2020, 10, 4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hou X; Zhang X; Zhao W; Zeng C; Deng B; McComb DW; Du S; Zhang C; Li W; Dong Y Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nature Nanotechnology 2020, 15, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Billingsley MM; Singh N; Ravikumar P; Zhang R; June CH; Mitchell MJ Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett 2020, 20, 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ryu N; Kim MA; Park D; Lee B; Kim YR; Kim KH; Baek JI; Kim WJ; Lee KY; Kim UK Effective PEI-mediated delivery of CRISPR-Cas9 complex for targeted gene therapy. Nanomedicine 2018, 14, 2095–2102. [DOI] [PubMed] [Google Scholar]
- (12).McKinlay CJ; Benner NL; Haabeth OA; Waymouth RM; Wender PA Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci U S A 2018, 115, E5859–E5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Whitehead KA; Dorkin JR; Vegas AJ; Chang PH; Veiseh O; Matthews J; Fenton OS; Zhang Y; Olejnik KT; Yesilyurt V; Chen D; Barros S; Klebanov B; Novobrantseva T; Langer R; Anderson DG Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat Commun 2014, 5, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bishop CJ; Kozielski KL; Green JJ Exploring the role of polymer structure on intracellular nucleic acid delivery via polymeric nanoparticles. Journal of Controlled Release 2015, 219, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Patel S; Ashwanikumar N; Robinson E; Xia Y; Mihai C; Griffith JP 3rd; Hou S; Esposito AA; Ketova T; Welsher K; Joyal JL; Almarsson O; Sahay G Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun 2020, 11, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Moghimi SM; Symonds P; Murray JC; Hunter AC; Debska G; Szewczyk A A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther 2005, 11, 990–5. [DOI] [PubMed] [Google Scholar]
- (17).Choi YJ; Kang SJ; Kim YJ; Lim YB; Chung HW Comparative studies on the genotoxicity and cytotoxicity of polymeric gene carriers polyethylenimine (PEI) and polyamidoamine (PAMAM) dendrimer in Jurkat T-cells. Drug Chem Toxicol 2010, 33, 357–66. [DOI] [PubMed] [Google Scholar]
- (18).Sunoqrot S; Bae JW; Jin S-E; M. Pearson R; Liu Y; Hong S Kinetically Controlled Cellular Interactions of Polymer−Polymer and Polymer−Liposome Nanohybrid Systems. Bioconjugate Chemistry 2011, 22, 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fitzsimmons REB; Uludağ H Specific effects of PEGylation on gene delivery efficacy of polyethylenimine: Interplay between PEG substitution and N/P ratio. Acta Biomaterialia 2012, 8, 3941–3955. [DOI] [PubMed] [Google Scholar]
- (20).Forrest ML; Meister GE; Koerber JT; Pack DW Partial acetylation of polyethylenimine enhances in vitro gene delivery. Pharm Res 2004, 21, 365–71. [DOI] [PubMed] [Google Scholar]
- (21).Kichler A; Leborgne C; Coeytaux E; Danos O Polyethylenimine-mediated gene delivery: a mechanistic study. J Gene Med 2001, 3, 135–44. [DOI] [PubMed] [Google Scholar]
- (22).Godbey WT; Wu KK; Mikos AG Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci U S A 1999, 96, 5177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Forrest ML; Pack DW On the kinetics of polyplex endocytic trafficking: implications for gene delivery vector design. Mol Ther 2002, 6, 57–66. [DOI] [PubMed] [Google Scholar]
- (24).Gabrielson NP; Pack DW Acetylation of Polyethylenimine Enhances Gene Delivery via Weakened Polymer/DNA Interactions. Biomacromolecules 2006, 7, 2427–2435. [DOI] [PubMed] [Google Scholar]
- (25).Olden BR; Cheng Y; Yu JL; Pun SH Cationic polymers for non-viral gene delivery to human T cells. J Control Release 2018, 282, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kircheis R; Kichler A; Wallner G; Kursa M; Ogris M; Felzmann T; Buchberger M; Wagner E Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther 1997, 4, 409–18. [DOI] [PubMed] [Google Scholar]
- (27).Pearson RM; Juettner V; Hong S Biomolecular Corona on Nanoparticles: A Survey of Recent Literature and its Implications in Targeted Drug Delivery. Front Chem 2014, 2, doi: 10.3389/fchem.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Al-Dosari MS; Gao X Nonviral gene delivery: principle, limitations, and recent progress. AAPS J 2009, 11, 671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Salvati A; Pitek AS; Monopoli MP; Prapainop K; Bombelli FB; Hristov DR; Kelly PM; Åberg C; Mahon E; Dawson KA Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nature Nanotechnology 2013, 8, 137–143. [DOI] [PubMed] [Google Scholar]
- (30).Zhao N; Qi J; Zeng Z; Parekh P; Chang CC; Tung CH; Zu Y Transfecting the hard-to-transfect lymphoma/leukemia cells using a simple cationic polymer nanocomplex. J Control Release 2012, 159, 104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bishop CJ; Liu AL; Lee DS; Murdock RJ; Green JJ Layer-by-layer inorganic/polymeric nanoparticles for kinetically controlled multigene delivery. J Biomed Mater Res A 2016, 104, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Shmueli RB; Anderson DG; Green JJ Electrostatic surface modifications to improve gene delivery. Expert Opinion on Drug Delivery 2010, 7, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hammond PT Building biomedical materials layer-by-layer. Materials Today 2012, 15, 196–206. [Google Scholar]
- (34).Green JJ; Chiu E; Leshchiner ES; Shi J; Langer R; Anderson DG Electrostatic Ligand Coatings of Nanoparticles Enable Ligand-Specific Gene Delivery to Human Primary Cells. Nano Letters 2007, 7, 874–879. [DOI] [PubMed] [Google Scholar]
- (35).Correa S; Boehnke N; Barberio AE; Deiss-Yehiely E; Shi A; Oberlton B; Smith SG; Zervantonakis I; Dreaden EC; Hammond PT Tuning Nanoparticle Interactions with Ovarian Cancer through Layer-by-Layer Modification of Surface Chemistry. ACS Nano 2020, 14, 2224–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Thomas M; Klibanov AM Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc Natl Acad Sci U S A 2002, 99, 14640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pearson RM; Hsu H.-j.; Bugno J; Hong S Understanding nano-bio interactions to improve nanocarriers for drug delivery. MRS Bulletin 2014, 39, 227–237. [Google Scholar]
- (38).Grigsby CL; Leong KW Balancing protection and release of DNA: tools to address a bottleneck of non-viral gene delivery. J R Soc Interface 2010, 7Suppl 1, S67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cai J; Yue Y; Wang Y; Jin Z; Jin F; Wu C Quantitative study of effects of free cationic chains on gene transfection in different intracellular stages. J Control Release 2016, 238, 71–79. [DOI] [PubMed] [Google Scholar]
- (40).Boeckle S; von Gersdorff K; van der Piepen S; Culmsee C; Wagner E; Ogris M Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J Gene Med 2004, 6, 1102–11. [DOI] [PubMed] [Google Scholar]
- (41).Evans BC; Fletcher RB; Kilchrist KV; Dailing EA; Mukalel AJ; Colazo JM; Oliver M; Cheung-Flynn J; Brophy CM; Tierney JW; Isenberg JS; Hankenson KD; Ghimire K; Lander C; Gersbach CA; Duvall CL An anionic, endosome-escaping polymer to potentiate intracellular delivery of cationic peptides, biomacromolecules, and nanoparticles. Nat Commun 2019, 10, 5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gwak SJ; Macks C; Bae S; Cecil N; Lee JS Physicochemical stability and transfection efficiency of cationic amphiphilic copolymer/pDNA polyplexes for spinal cord injury repair. Sci Rep 2017, 7, 11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bäckström A; Kugel L; Gnann C; Xu H; Aslan JE; Lundberg E; Stadler C A sample preparation protocol for high throughput immunofluorescence of suspension cells. bioRxiv 2020, 2020.01.05.895201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tsang M; Gantchev J; Ghazawi FM; Litvinov IV Protocol for adhesion and immunostaining of lymphocytes and other non-adherent cells in culture. Biotechniques 2017, 63, 230–233. [DOI] [PubMed] [Google Scholar]
- (45).Pearson RM; Sen S; Hsu H.-j.; Pasko M; Gaske M; Král P; Hong S Tuning the Selectivity of Dendron Micelles Through Variations of the Poly(ethylene glycol) Corona. ACS Nano 2016, 10, 6905–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Johnson DW New Applications for Poly(ethylene-alt-maleic anhydride). Durham University, 2010. [Google Scholar]
- (47).Weber EW; Maus MV; Mackall CL The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhao Z; Ukidve A; Kim J; Mitragotri S Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.