

Abstract
ENL is a histone acetylation reader essential for disease maintenance in acute leukemias, in particular the mixed-lineage leukemia (MLL)-rearranged leukemia. In this study, we carried out high-throughput screening of a small molecule library to identify inhibitors for the ENL YEATS domain. Structure-activity relationship studies of the hits and structure-based inhibitor design led to two compounds, 11 and 24, with IC50 values below 100 nM in inhibiting the ENL-acetyl-H3 interaction. Both compounds, and their precursor compound 7, displayed strong selectivity toward the ENL YEATS domain over all other human YEATS domains. Moreover, 7 exhibited on-target inhibition of ENL in cultured cells and a synergistic effect with the BET bromodomain inhibitor JQ1 in killing leukemia cells. Together, we have developed selective chemical probes for the ENL YEATS domain, providing the basis for further medicinal chemistry-based optimization to advance both basic and translational research of ENL.
Graphical Abstract
This document is the Accepted Manuscript version of a Published Work that appeared in final form in Journal of Medicinal Chemistry, copyright® American Chemical Society after peer review and technical editing by the publisher. To access the final edited and published work see Discovery of Selective Small-Molecule Inhibitors for the ENL YEATS Domain, DOI: 10.1021/acs.jmedchem.1c00367.
INTRODUCTION
Post-translational modifications (PTMs) of histones play an important role in the epigenetic regulation of gene expression. These modifications serve as binding sites to recruit reader proteins, which in turn transduce the epigenetic signals into downstream functional outcomes1–2. In addition to small compounds that modulate enzymatic activities of the histone-modifying enzymes, perturbations of reader-histone interactions also provide attractive therapeutic potentials. One such example is the BET bromodomain inhibitors3–4. Bromodomains are known as readers of histone acetylation5. Recent studies from our laboratories and others have identified the YEATS domains as a new family of epigenetic readers that bind to not only histone acetylation but also other types of acylations such as crotonylation6–16.
The YEATS domain, named after its five founding members (Yaf9, ENL, AF9, Taf14 and Sas5), is evolutionarily conserved from yeast to human17. The human genome encodes four YEATS domain-containing proteins: ENL, AF9, YEATS2 and GAS41 that all associate with chromatin-associated protein complexes18–19. ENL and AF9 are paralogues that share a similar protein structure including a highly conserved YEATS domain.
Both ENL and AF9 are subunits of the super elongation complex (SEC) and the complex of the histone H3K79 methyltransferase DOT1L, but mutually exclusive20–21. We and others previously showed that ENL, but not AF9, is required for disease maintenance in acute leukemias, in particular the MLL-rearranged leukemia14,22. Depletion of ENL or disrupting the interaction between its YEATS domain and acetylated histones suppresses leukemia progression. In addition, hotspot ENL YEATS domain mutations were found in Wilms’ tumor patients23–24. We showed that the reader function of the ENL YEATS domain is indispensable for the gain-of-function mutations in the oncogenesis of Wilms’ tumor25. Together, all these studies suggest that the YEATS domain of ENL is an attractive therapeutic target.
The acetyllysine binding pocket of the ENL YEATS domain is a long and narrow hydrophobic channel, making it a potentially good target for developing small-molecule inhibitors14. Indeed, recent publications of acetyllysine competitive small compounds and peptide-mimic chemical probes demonstrate that the ENL YEATS domain is pharmacologically tractable26–32. The peptide-mimic chemical probes showed slightly higher potency to the ENL YEATS domain than other YEATS domains, largely due to interactions outside of the acetyllysine binding pocket30. In contrast, the small molecule ENL inhibitors reported so far failed to distinguish ENL from its close paralogue AF9. In addition, none of these small molecule compounds showed significant impact on ENL-dependent leukemia cell growth, suggesting that development of potent, selective ENL YEATS domain inhibitors is in great need. Here we report the discovery of small-molecule compounds that exhibit preferential binding to ENL compared to AF9 and other YEATS domain proteins. Two compounds, 11 and 24, displayed IC50 values below 100 nM in inhibiting the ENL-acetyl-H3 interaction in vitro. In leukemia cells, compound 7 reduced ENL target gene expression and suppressed leukemia cell growth. In addition, 7 exhibited a synergistic effect with the BET bromodomain inhibitor JQ1 in killing leukemia cells. Our study provided valuable selective ENL chemical probes and potential leads for further medicinal chemistry-based optimization to advance both basic and translational research of ENL.
RESULTS
High-throughput library screen for ENL YEATS domain inhibitors.
In order to identify small molecule inhibitors for the YEATS domain of ENL, we first established an AlphaScreen assay system for high-throughput screening (HTS) of small molecule compounds. In this assay system, two analytes, a 6×His-tagged ENL YEATS domain (His-ENL) and a biotin-H3K9ac peptide (histone H3 residues 1–21 with an acetylation at Lys 9) were immobilized on Perkin Elmer Ni2+-chelating acceptor and streptavidin donor beads, respectively (Supporting Information, Figure S1A). Protein and peptide dose-response assays determined optimal concentrations of His-ENL and biotin-H3K9ac to be 100 nM and 30 nM, respectively (Supporting Information, Figure S1B and S1C). We also determined the optimized Alpha-beads concentration to be 10 μg/mL. This assay system was further evaluated in a high-throughput setting in 384-well plates. Inter-plate variations were measured between two separate plates and on two separate days, yielding robust and highly reproducible results with high signal/background (S/B) ratio (39.02), low coefficient of variation (3.5%), and an excellent Z’ factor (0.92) (Supporting Information, Figure S1D). DMSO tolerance of the assay (0.1–1%) indicated that the Alpha signals were maintained at 95% and 85% in the presence of 0.1% and 0.5% DMSO, respectively. We also set up a counter assay using a biotin-14xHis peptide to eliminate compounds that interfere with AlphaScreen assay components. Together, these data demonstrate that the AlphaScreen assay we developed is suitable for high-throughput screening of ENL inhibitors, with superior sensitivity and reproducibility.
After adapting the AlphaScreen-based HTS system to an automated format for ENL (100 nM His-ENL, 10 nM biotin-H3K9ac, 0.1% DMSO, and 2.5 μg/mL Alpha beads), we proceeded to screen a small molecule library of 66,625 compounds with diverse chemical scaffolds. Non-fragment compounds were screened at a concentration of 10 μM and fragment-based compounds were screened at a concentration of 50 μM. In the primary screen, we obtained 4648 hits with above 50% inhibition. Confirmation and counter assays yielded 524 compounds with above 60% inhibition of the His-ENL–H3K9ac interaction and below 20% inhibition of the counter screen. We then subjected the top 100 compounds to full dose-response curve validation and obtained 37 compounds with IC50 values below 5 μM, including 8 compounds with IC50 below 1 μM (Supporting Information, Table S1).
Structure-based inhibitor design and structure-activity relationship studies.
Among the top 8 hits that have an IC50 value below 1 μM (Supporting Information, Table S1), five, named as 1-5, are structurally similar and share a same pharmacophore [1,2,4]triazolo[4,3-a]pyridine-6-amide, suggesting a preferential binding of this pharmacophore to the ENL YEATS domain (Figures 1A and Supporting Information, Figure S2). All these five compounds also contain an aryl substituent at the amide nitrogen side, allowing them to be generally defined as N,C-diarylamides. To understand how these compounds interact with ENL, we performed docking analysis using an existing crystal structure of the ENL YEATS domain (the PDB entry: 5j9s). The results showed that all 5 compounds fit nicely to the acetyllysine binding channel of ENL (Figure 1B). The compounds are bound to the ENL YEATS domain with a similar orientation as an acetyllysine in a native histone ligand. Similar to acetyllysine side chain amide, the amide in 1-5 is poised to form two hydrogen bonds with S58 and Y78. Although the two aromatic rings can flip to bind either side of the channel, both potentially form pi stacking and van der Waals interactions with residues F28, H56, F59, Y78, and F81 in ENL for preferential binding (Figure 1C). The modelling analysis also indicated that 1-5 occupy almost fully the acetyllysine binding channel of ENL.
Figure 1. Structural modeling of the five initial HTS hits, 1–5, and an amine analog 6 with the ENL YEATS domain.

(A) Chemical structures of compounds 1-6 (left) and their IC50 values in inhibiting the His-ENL-H3K9ac interaction in AlphaScreen assay (right). (B) Structural model showing binding of 1-5 to the ENL YEATS domain. Modeling was based on the crystal structure of the ENL YEATS domain (PDB entry: 5j9s). 1-5 are shown in stick representation and the ENL YEATS domain is shown in contoured surface structure. Atoms in ENL are colored in gray, compound 1 in green, 2 in hotpink, 3 in yellow, 4 in cyan, and 5 in orange. (C) The modeled interaction of 2 with ENL. The Cα atoms of 2 are colored in orange and two hydrogen bonds (dashed lines) to S58 and Y78 of ENL are colored in yellow. Acetyllysine in the H3K27ac ligand in the original crystal structure is colored in hotpink and its two hydrogen bonds with E58 and Y78 are shown for comparison. (D) The modeled interaction of 6 with ENL. The Cα atoms of 6 are colored in orange. The amine in 6 shows a salt-bridge interaction with E26 in ENL.
Since ENL has relatively flat interfaces on the two sides of the acetyllysine binding channel, there is a little space for chemical maneuvers of 1-5 for improved binding. However, we noticed that E26, a residue at the edge of the acetyllysine binding channel can potentially flip its side chain toward the acetyllysine binding channel to interact with a ligand such as 2 (Figure 1C). We deemed that by adding a positively charged amine or amidine to 2 it is possible that a salt-bridge interaction with E26 can be introduced for strong binding to ENL. Therefore, we synthesized compound 6 (Scheme 1A) and tested its inhibition of the interaction between His-ENL and biotin-H3K9ac. The determined IC50 value for 6 was 0.63 μM, which is similar to that for 2 (Figure 1A and Supporting Information, Figure S2). Since the introduction of an amine makes the compound more favorable to dissolve in water, the salt-bridge interaction may compensate the energy loss due to desolvation when 6 binds ENL, resulting no improvement. Indeed, in the modelled structure, 6 interacts with ENL similar to 2 except that it engages E26 for a salt-bridge interaction (Figure 1D). We have also attempted to co-crystalize ENL with 6 for crystal structure determination, but unfortunately it has not been successful.
Scheme 1.

Synthesis of compounds 6–14.
Encouraged by the results from 6, we expanded the scope of substitution groups on both sides of the amide bond of hit compounds for a comprehensive structure–activity relationship (SAR) study. A major focus was to maintain a positive charged amine, amidine or guanidine as in 6 but tune the ENL binding as well as lower the energy loss due to desolvation by adding different alkyl substituents to the amine, amidine or guanidine. The inhibition potency of all compounds was first tested at 1 μM and 0.1 μM. Promising compounds were then subjected to a more accurate AlphaScreen assay for IC50 determination. We first started with replacing the primary amine of 6 with different kinds of tertiary amines through reductive amination of key aldehyde intermediate 46 (Scheme 1B), which resulted in 7-11 (Table 1 and Supporting Information, Figure S2). IC50 measurement showed that compounds tended to be more potent as the ring size of the substitutional groups on the tertiary amine decreased. Among them, 11 that has a four-membered ring azetidine moiety exhibited the most potency with an IC50 value as 51 nM in inhibiting the interaction between His-ENL and biotin-H3K9ac. These results suggest that the azetidine ring assists the binding to ENL. Further modifications were then introduced to 11 to afford 12-14 with different alkyl groups on the 2’ position of azetidine, which we wished to increase the electron density on the N atom and enhance the interaction between azetidine and Glu26. However, these compounds displayed lower potency than 11. Given that Glu26 is located at a loop area with much conformational flexibility, we moved the azetidine moiety from the para to meta position affording 15 but did not result in any increase of potency. We also attempted to increase the rigidity of the molecule by adding a methyl group to the benzylic carbon affording 16. However, it greatly reduced the inhibition potency. We also substituted the triazolopyridine moiety with similar heterocycles to afford 17-19, but none of these compounds outcompeted 11 (Table 1, Scheme 1C and Supporting Information, Figure S2).
Table 1.
Chemical structures and IC50 values of 7–28

|
In addition to amine derivatives, we also designed a series of amidine derivatives based on compound 6 to afford 20-23. These compounds were synthesized from corresponding nitrile intermediates followed by acid-catalyzed ethanolysis and then ammonolysis (Scheme 2A). However, none of these compounds showed improved potency. Although an amidine or guanidine tend to form a stronger salt bridge with a carboxylate than an amine, it may have a higher desolvation energy than an amine, contributing to weaken binding to ENL. For this reason, we focused the synthesis of additional amidine and guanidine derivates 24-28 that have higher hydrophobicity than 20-23. These compounds were synthesized by directly reacting 6 with corresponding N-heterocycle building blocks, except for compound 27, which was made through 5-fluoro-2-aminopyridine due to the inadequate reactivity of 5-fluoro-2-chloropyridine in the reaction with 6 (Scheme 2B). Among them, 24 exhibited an IC50 value as 85 nM. 11 and 24 are the two most potent compounds in our compound series. We further evaluated their binding to ENL using the surface plasmon resonance (SPR) analysis. His-ENL was immobilized on dextran-coated Au chips through EDC/NHS coupling, followed by flow-through of a buffer containing different concentrations of 11 and 24. The responses in sensorgrams were fitted to the Langmuir 1:1 binding kinetics model to obtain both association and dissociation rate constants, from which Kd values were then determined (Supporting Information, Figure S3A). Compared to the kinetics of typical small molecule-protein interactions, both association and dissociation of 11 and 24 toward ENL are relatively slow (association: 1800 and 1600 M−1·s−1; dissociation: 8.3 × 10−5 and 7.0 × 10−5 s−1 respectively). Their determined Kd values by SPR were 45 and 46 nM, respectively. As far as we know, 11 and 24 are the two most potent inhibitors for ENL that have so far been developed.
Scheme 2.

Synthesis of compounds 20–28.
The small molecule inhibitors occupy the same binding site of the ENL YEATS domain as the histone acyl-lysine.
To study the molecular basis of 7, 11, and 24 binding to ENL, we attempted to co-crystalize the ENL YEATS domain with these compounds, but it was not successful. We then modelled these compounds to the acetyllysine binding pocket of the ENL YEATS domain by docking analysis. 7 and 11 were docked in their protonated form while 24 was docked in the neutral form given it is less likely to be much protonated under physiological pH. In the modelled structures (Figures 2A–2D), all three compounds interact with ENL similar as 6 (Figure 1D). The amide forms two hydrogen bonds with S58 and Y78. The triazolopyridine ring was involved in pi stacking interactions with H56 in a parallel configuration and with Y78 in a T-shaped configuration. The phenyl group was also involved in pi stacking interactions with F28 and F59, both in a T-shaped configuration. Importantly, the amine of 7, the azetidine of 11, and the guanidine of 24 are all within 3 Å to E26, suggesting a common salt bridge or hydrogen bond interaction that stabilizes their interactions with ENL (Figure 2B–2D).
Figure 2. Compounds 7, 11 and 24 and their docking models bound to the ENL YEATS domain.

(A) Chemical structures of compounds 7, 11 and 24 and their IC50 values in inhibiting the His-ENL-H3K9ac interaction in AlphaScreen assay. (B-D) The molecular docking models of compounds 7 (B), 11 (C), and 24 (D) bound to the ENL YEATS domain. Modeling was based on the crystal structure of the ENL YEATS domain (PDB: 5j9s). Compounds are shown in stick representation and the ENL YEATS domain is shown as cartoon in gray. Compound-interacting residues of ENL are highlighted and shown in stick representation.
To experimentally validate that 7 binds to the acyl-lysine binding pocket of ENL, we compared the binding of 7 and a H3K27cr peptide (histone H3 residues 22–31 with a crotonylation at K27) to ENL by nuclear magnetic resonance (NMR) spectroscopy33. We expressed 15N-labelled His-ENL and recorded its 1H, 15N heteronuclear single quantum coherence (HSQC) spectra while 7 or the H3K27cr peptide was titrated into the sample (Figure 3 and Supporting Information, Figure S3B). As expected, the H3K27cr peptide induced large chemical shift perturbations (CSPs) in the ENL YEATS domain, which were in the intermediate to fast exchange regime on the NMR timescale. Addition of 7 caused CSPs in the intermediate to slow exchange regime, indicating that the ENL YEATS domain binds to 7 tighter than to the H3K27cr peptide. An overall similar pattern of CSPs observed in both experiments suggest that 7 and the H3K27cr peptide occupy the same binding site in the ENL YEATS domain.
Figure 3. Compound 7 and the H3K27cr peptide occupy the same binding site of the ENL YEATS domain.

Superimposed 1H,15N HSQC spectra of His-ENL collected as H3K27cr (H3 residues 22–31, left) or 7 (right) was added stepwise. Spectra are color-coded according to the protein:ligand molar ratios.
Compounds 7, 11 and 24 are highly selective toward the ENL YEATS domain over all other human YEATS domains.
To determine whether the small molecule inhibitors are selective toward ENL among the four human YEATS domains, we assessed 7, 11, and 24 in their inhibition of ENL, AF9, GAS41, and YEATS2 in peptide pulldown assays. We also included the original hit 1 in the assays for comparison. We used the H3K9ac peptide for AF9 and ENL, the H3K27ac peptide for GAS41, and the H3K27cr peptide for YEATS2, as these peptides are the preferred ligands of the corresponding YEATS domains7, 11, 14, 16. We found 1 μM of 11 and 24 and 5 μM of 1 and 7 strongly inhibited the binding of ENL to H3K9ac, whereas at even a 20 μM concentration, none of these compounds showed notable inhibition to AF9, GAS41, or YEATS2 binding to their corresponding acylated histone peptides (Figure 4A). We further measured the IC50 values of 1, 7, 11 and 24 in their inhibition of the binding of four human YEATS domains to their preferred histone peptide ligands using AlphaScreen assays. All four compounds displayed preferential inhibition of ENL over the other three YEATS domains. Compound 1 showed ~4-fold higher potency toward ENL than AF9, whereas no detectable inhibition was measured for the YEATS2 or GAS41 YEATS domain. Compounds 7, 11, and 24 exhibited even higher specificity to ENL. Particularly, the IC50 value of 11 to ENL was ~20-fold lower than that to AF9 (ENL IC50 51 nM and AF9 IC50 984 nM) (Figure 4B). As the previously reported small molecule ENL inhibitors are not able to differentiate between ENL and AF9, our compounds provide promising scaffolds for further development of ENL-specific inhibitors for the study of ENL biology and for disease intervention.
Figure 4. Compounds 7, 11, and 24 are highly specific to ENL over other YEATS domains.

(A) Peptide pulldowns of ENL, AF9, YEATS2, and GAS41 with the indicated acylated histone peptides with or without 1, 7, 11, and 24. Unmodified histone peptides were used as negative controls to the acylated peptides and DMSO as a negative control to compound treatment. (B) AlphaScreen measurement of IC50 of 1, 7, 11, and 24 in inhibition of YEATS domains binding to the corresponding acylated histone peptides as in (A). Data represent mean ± SEM, n = 4.
Compound 7 inhibits the endogenous ENL protein in MLL-rearranged cell lines.
To explore the small molecule ENL inhibitors we developed in biological applications, we first analyzed their cellular effects in MV4;11 and MOLM13 cells, two MLL-rearranged cell lines whose growth is dependent on ENL14, 22. We screened 15 compounds with in vitro IC50 values lower than 2 μM, and we found 7 as the most potent compound in cell growth inhibition (Supporting Information, Figure S4A). The discrepancy between in vitro IC50 and cellular efficacy is not due to cell permeability, as 7 was comparable to 11 in the standard Caco-2 permeability assay (Supporting Information, Figure S4B). Compound 7 exhibited ~40% inhibition of MOLM13 cell growth at 5 μM and 80% inhibition at 10 μM concentrations, while about double amounts of the compound were needed to achieve similar levels of inhibition in MV4;11 cells (Figure 5A). In contrast, U2OS cells, an ENL-independent cell line, showed little or no response to the treatment with 7, even at 50 μM.
Figure 5. Compound 7 exhibits on-target effect of ENL inhibition in MLL-rearranged leukemia cell lines.

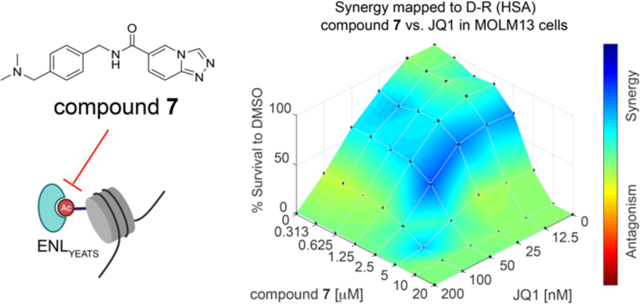
(A) 7 inhibits leukemia cell growth. Cell growth inhibition of 7 at various concentrations in MV4;11, MOLM13, and U2OS cells. Survived cells were calculated as % relative to DMSO treated cells. (B-C) Cellular thermal shift assays in MV4;11 (B) and MOLM13 (C) cells treated with 20 μM 7 at the indicated temperatures. β-actin was used as a loading control. (D) qRT-PCR analysis of HOXA9 and MYC gene expression in MOLM13 cells treated with 7 or the DMSO negative control. (E-F) 7 shows a synergistic effect with JQ1 in MOLM13 cells. (E) 3D synergy distribution of 7 and JQ1. MOLM13 cells were treated with indicated doses of 7 and JQ1 or DMSO for 6 days. Survived cells were calculated as % relative to DMSO treated cells. Synergistic interactions were analyzed using the Combenefit software. (F) GI50 of 7 when co-treated with indicated concentrations of JQ1. Data in (A) and (D) are shown as mean ± SEM, n = 3, two-tailed Student’s t test, ns, not significant, * P < 0.05, ** P < 0.005, *** P < 0.001, **** P < 0.0001. Data in (E) and (F) represent mean ± SEM, n = 3.
Next, we asked whether the growth inhibition effect was caused by on-target inhibition of the endogenous ENL protein. In this regard, we carried out cellular thermal shift assay (CETSA) to evaluate thermal stability of the ENL protein in MV4;11 and MOLM13 cells treated with 7. As the AF9 protein is undetectable in these cells with commercial antibodies, we evaluated thermal stability of the GAS41 protein for comparison. Compared to the DMSO-treated cells, we detected higher abundance of soluble ENL proteins in cells treated with 7, indicating that 7 bound and stabilized ENL proteins34. In contrast, the thermal stability of GAS41 proteins showed little or no difference between DMSO and 7 treatment (Figure 5B and 5C). Similarly, 7 stabilized ENL but not AF9 proteins in HeLa cells (Supporting Information, Figure S4C). The CETSA results suggest specific engagement of ENL with 7 in living cells.
We also evaluated the expression of two ENL target genes, HOXA9 and MYC, in MOLM13 cells. Compound 7 effectively suppressed HOXA9 gene expression at as low as 2.5 μM of drug concentration. At 10 μM, it suppressed ~ 80% of the expression of both HOXA9 and MYC genes, comparable to the levels of gene suppression in ENL knock-down cells (Figure 5D and Supporting Information, S4D), suggesting potent and on-target effect of the ENL inhibitor.
Compound 7 exhibits a synergistic effect with JQ1.
Previously we found that CRISPR/Cas9-mediated ENL knockout sensitized leukemia cells to JQ1, an effective inhibitor of BET bromodomain proteins including BRD44,14. An intriguing question was whether 7 has any synergy with JQ1 in killing leukemia cells. To answer this question, we carried out combinatory treatment of MV4;11 and MOLM13 cells with series of concentrations of 7 (0 to 20 μM) and JQ1 (0–200 nM). In both cell lines, we observed synergistic effect between ENL inhibition and JQ1 (Figure 5E and Supporting Information, Figure S4E)35. Particularly, at a concentration of 200 nM of JQ1, the GI50 of compound 7 in MOLM13 cells was reduced from 3.64 μM to 1.34 μM (Figure 5F). Together, these results demonstrate therapeutic potentials of the ENL inhibitors for future exploration in disease treatment.
DISCUSSION
The YEATS domain is a newly identified family of histone acylation readers. The four human YEATS domain-containing proteins, ENL, AF9, YEATS2, and GAS41, are subunits of protein complexes involved in chromatin and transcription regulation18–19. The evolutionally conserved histone-reading function of the YEATS domains is essential for the functionality of all the YEATS domain proteins in both yeast and human6–16. Dysregulation of the YEATS domain-containing proteins has been associated with various human diseases, including cancers. We and others showed that ENL and particularly its YEATS domain is essential for disease maintenance and progression of acute leukemias14, 22. Recently, we also found that the reader function of the ENL YEATS domain is indispensable for the aberrant gene activation and tumorigenesis caused by the gain-of function ENL YEATS domain mutations identified in Wilms’ tumor patients25. In addition, YEATS2 and GAS41 are frequently amplified in various types of human cancers36–38. All these studies suggest that the YEATS domains are promising drug targets and, therefore, targeting the YEATS domains may provide a novel therapeutic approach for a broad spectrum of human cancers.
Developing YEATS domain inhibitors has been a research focus of the epigenetic reader field in recent years. The initial efforts were focused on targeting the YEATS domain of ENL, because of great therapeutic potentials. Both small molecule chemical compounds and peptide-mimic probes have recently been developed as acetyllysine competitive inhibitors of the ENL YEATS domain26–32. However, target selectivity has been a big challenge because the YEATS domains share high structural similarity, especially between ENL and its close homologue AF9. The few small molecule ENL inhibitors reported so far have poor specificity that fail to distinguish ENL from AF9. The peptide-mimic chemical probes developed by the Li group showed slightly higher potency to the ENL YEATS domain than other YEATS domains, largely due to interactions outside of the acetyllysine binding pocket30. These results suggest that targeting both the acyllysine-binding pocket and additional proximal sites outside of the binding pocket might be a good approach to develop specific inhibitors. Indeed, based on this concept, the Li group has recently developed a conformationally preorganized cyclopeptide that showed a 38-fold higher binding affinity toward AF9 YEATS over ENL29.
Despite the success in developing peptide-mimic chemical probes specific to AF9, ENL specific inhibitors were still lacking. Because ENL, but not AF9, is essential for MLL-rearranged acute leukemias and ENL mutant Wilms’ tumors, it is in urgent need to develop ENL specific inhibitors for further drug development. In our study, through HTS we identified compound 1, which showed a 4-fold preference towards ENL over AF9 YEATS. After several rounds of structure-based inhibitor design and structure-activity relationship studies, we were able to develop several compounds with much better selectivity. In particular, the IC50 value of compound 11 to ENL was ~20-fold lower over AF9, ~360-fold lower over YEATS2, and more than 1,000-fold lower over GAS41, providing a good lead for future drug development.
The selectivity of our compounds to ENL over AF9 is intriguing, given that the AF9 YEATS domain has a 10-fold higher affinity than ENL YEATS in acyllysine binding. The YEATS domains of AF9 and ENL share high degree of structural similarity11,14. It is not clear what interactions contribute to the ENL selectivity. By comparing the modelled structures of compounds 1, 7, 11 and 24 docked to the acetyllysine binding pocket of the ENL and AF9 YEATS domains, we observed that the triazolopyridine pharmacophore of these compounds adopts conformations to form stronger pi-pi interaction with H56 residue in ENL than in AF9 YEATS domain. When bound with the ENL YEATS domain, the distances between the triazole rings and the imidazole rings range from 3.4 to 3.6 Å and their dihedral angles range from 20° to 22°, whereas in the case of AF9 YEATS domain, the distances between them increase to 4.3–4.6 Å and their dihedral angles also increase to a range of 28°−37°, both leading to weaker pi-pi interactions compared to those in the ENL YEATS domain (Supporting Information, Figure S5). Additionally, the salt bridge interaction in the case of 7 and 11 and hydrogen bond for 24 with E26 in the ENL YEATS domain may also contribute to selectivity. Further structural study of YEATS domains in complex with these compounds will provide insights to guide future development of more potent and selective ENL YEATS domain inhibitors.
In leukemia cells, our synthesized compound 7 exhibited clear on-target cellular effects in reducing ENL target gene expression and suppressing leukemia cell growth. In addition, consistent with previous results of genetic ENL ablation, 7 exhibited a synergistic effect with the BET bromodomain inhibitor JQ1 in killing leukemia cells. The cellular effects of our compounds are superior to all reported ENL inhibitors. Overall, our study provides valuable selective ENL small molecule inhibitors that can serve as potential leads for further medicinal chemistry-based optimization to advance both basic and translational research of ENL. It also provides a molecular platform for the development of more complicated, multifunctional probes for applications such as visualization or targeted degradation in cells.
CONCLUSION
In this study, we carried out high-throughput screening of a small molecule library of > 66,000 compounds against the ENL YEATS domain and identified a series of hit molecules that share a [1,2,4]triazolo[4,3-a]pyridine-6-amide pharmacophore and a common N,C-diarylamide scaffold. By introducing a potential salt bridge interaction with E26 in ENL, we were able to generate compounds with IC50 and Kd values less than 100 nM. Importantly, our compounds outcompeted the previously reported ENL inhibitors by showing high selectivity toward ENL over AF9, the close paralogue of ENL. Furthermore, compound 7 exhibited on target effect in inhibiting ENL target gene expression and leukemia cell growth. Our ENL-specific YEATS domain inhibitors provide the basis for development of potent ENL-specific chemical probes in the future.
EXPERIMENTAL SECTION
Materials.
The biotinylated histone peptides used in the AlphaScreen assay: H3 (aa 1–22, ARTKQTARKSTGGKAPRKQLAT), H3K9ac (aa 1–22, ARTKQTARK(ac)STGGKAPR-KQLAT), H3 (aa 21–44, ATKAARKSAPATGGVKKPHRYRPG), H3K27ac (aa 21–44, ATKAARK(ac)SAPSTGGVKKPHRYRPG), H3K27cr (aa 21–44, ATKAARK(cro)SAPS-TGGVKKPHRYRPG) and the biotin-14xHis peptide used in counter assay were purchased from CPC Scientific. Anti-ENL (14893S) antibody was from Cell Signaling, anti-GAS41 (sc-393708) and anti-GST (sc-459) antibodies were from Santa Cruz, and anti-AF9 (HPA001824) and anti-β-actin (A1978) antibody was from Sigma. Human cell lines MV4;11, MOLM13, U2OS and HeLa cells were purchased from ATCC.
Protein expression and purification.
The cDNA encoding sequences of four human YEATS domains: ENL (aa 1–145), AF9 (aa 1–145), full-length GAS41 and YEATS2 (aa 201–332) were cloned in pGEX-6P-1 and pET19b expression vectors, respectively. The His-tagged YEATS proteins were expressed in E. coli Rosetta-2 (DE3) pLysS cells in the presence of 0.2 mM isopropyl-β-D-1-thioglactopyranoside (IPTG) for 18 h at 16 ºC. The His-tagged YEATS proteins were purified using Ni-NTA resins following the manufacture’s instruction. The eluted protein was dialyzed in a buffer containing 50 mM HEPES (pH7.4), 100 mM NaCl and 20% glycerol to remove imidazole. Proteins were adjusted to 0.5 mg/mL, aliquoted and stored at −80 ºC. Each batch of purified protein was tested in AlphaScreen assay conditions discussed as following. The GST-tagged proteins used in peptide pulldown assays were expressed in the same way and purified using Glutathion Sepharose resins (GE Healthcare).
AlphaScreen assay setup.
The AlphaScreen assay was carried out in 384-well plates. Manual assay setup was performed in 30 uL reaction in Alpha Reaction Buffer (50 mM HEPES pH7.4, 100 mM NaCl, 0.1% BSA, and 0.05% CHAPS) with final concentrations of 100 nM His-ENL YEATS, 30 nM Biotin-H3K9ac, and 10 μg/mL of Alpha donor and acceptor beads. During the automation step, we were able to reduce the assay volume to 20 μL per well while maintained the quality and robustness of the assay, with the optimal final concentrations of His-ENL YEATS (100 nM), biotin-H3K9Ac (10 nM), DMSO (0.1%), and Alpha-beads (2.5 μg/mL). Protein, peptide and compounds were mixed and incubated for 1 h at room temperature before adding the Alpha beads. Alpha signals were detected by an EnVision microplate reader equipped with an Alpha laser (PerkinElmer).
High-throughput screening using AlphaScreen.
High-throughput screen was performed at the Texas Screening Alliance for Cancer Therapeutics (TxSACT) facility. The 66,625 compounds screened were from Maybridge HitFinder Set (14,080), Chembridge Diversity Set (12,900), Chembridge Kinase Set (11,250), Chembridge Fragment Library (4,000), ChemDiv Fragment Collection (14,143), Legacy Collection (2,092), MicroSource Spectrum Collection (2,000), LOPAC Collection (1,275), Selleck Kinase and Bioactive Collection (2,260), NCI Diversity (1595), NCI Mechanistic collection (820), and NCI natural products (210). In the primary HTS, fragments were screened at 50 μM and non-fragment compounds were screened at 10 μM. After the single shot screen and hits triage, 990 hits were picked for confirmation assay, and counter assay with Biotin-14xHis peptide.
IC50 determination with AlphaScreen assay.
The AlphaScreen assay conditions are essential the same as the one used in high-throughput screen. The protein concentrations of AF9, Gas41 and YEATS2 are 30, 100, and 100 nM, respectively, and the peptide concentrations are 30 nM. All assays have been validated using protein and peptide competitors. For IC50 determination, compounds were subjected to eight 3-fold serial dilutions, for a total of nine concentrations ranging from 50 μM to 8 nM for dose response curve AlphaScreen assays. IC50 values were determined from the plot using nonlinear regression of variable slope (four parameters) and curve fitting performed using GraphPad Prism.
Modeling of inhibitors bound with ENL and AF9 YEATS domains.
Molecular docking of target compounds was carried out using AutoDock 439. The initial conformations of target compounds were first generated and MM2 minimized by PerkinElmer Chem3D software. Structures of the ENL and AF9 YEATS domains were obtained from PDB 5J9S and 4TMP respectively, with H3K27Ac and H3K9Ac deleted from the complexes. Structures of the YEATS domains were then pre-processed in MGLTools 1.5.6 to remove water molecules and add polar hydrogens. The grid box was set to be centred at coordinate (x = 27.352, y = −42.139, z = 3.0) for ENL, and at coordinate (x = 52.734, y = 10.522, z = −11.134) for AF9, with a size of 40 × 40 × 40 npts, which is big enough to contain the binding channel and surrounding amino acid residues. Glu26 residue of ENL YEATS domain was set to be flexible. Target compounds were then docked in the grid box. The conformations with lowest binding energies were converted to PDB files for visualization.
Compound synthesis.
All reagents and solvents for synthesis were purchased from commercial sources and used without purification. All glassware was flame-dried prior to use. Thin layer chromatography (TLC) was carried out on aluminium plates coated with 60 F254 silica gel. TLC plates were visualized under UV light (254 nm or 365 nm) or stained with 5% phosphomolybdic acid. Normal phase column chromatography was carried out using a Yamazen Smart Flash AKROS system. Analytical reverse-phase high pressure liquid chromatography (RP-HPLC) was carried out on Shimadzu LC20 HPLC system with an analytical C18 column. Semi-preparative HPLC was carried out on the same system with a semi-preparative C18 column. The mobile phases for were H2O with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B) if not mentioned otherwise. NMR spectra were recorded on a Bruker Avance Neo 400 MHz or Varian INOVA 300 MHz spectrometer in specified deuterated solvents. High resolution electrospray ionization mass spectrometry (HRMS-ESI) was carried out on a Thermo Scientific Q Exactive Focus system. The purities of compounds were confirmed by NMR and analytical HPLC-UV as ≥ 95%.
tert-Butyl (4-(([1,2,4]triazolo[4,3-a]pyridine-6-carboxamido)methyl)benzyl) carbamate (41).
To a solution of 39 (1 mmol, 163 mg) and 40 (1 mmol, 236 mg) in dry DMF (5 mL), was added DIPEA (2 mmol, 258 mg), and EDCI (1.2 mmol, 230 mg). The resulting solution was stirred under room temperature overnight. Then the solution was diluted with EtOAc (50 mL) and washed with saturated NaHCO3 solution (2 × 50 mL), 1 M HCl (2 × 50 mL) and saturated brine (50 mL). The organic layers were then dried with anhydrous Na2SO4 and then concentrated. The residue was purified by column chromatography (silica gel, 10% MeOH/DCM as eluent) to yield 41 as light yellow solid (250 mg, 66%).
(4-(([1,2,4]Triazolo[4,3-a]pyridine-6-carboxamido)methyl)phenyl)methanamine hydrochloride (6).
To a solution of 41 (0.5 mmol, 190 mg) in 5 mL of 1,4-dioxane was added 10 mL of 4 M HCl solution in 1,4-dioxane. The resulting solution was stirred under room temperature for 2 h. Then the reaction mixture was concentrated to dryness in vacuo to yield 6 as light yellow solid (150 mg, 95%). 1H NMR (400 MHz, Deuterium Oxide) δ 9.49 (s, 1H), 9.24 (s, 1H), 8.29 (dt, J = 9.7, 1.6 Hz, 1H), 8.10 (d, J = 9.6 Hz, 1H), 7.52 – 7.42 (m, 4H), 4.66 (s, 2H), 4.19 (s, 2H). 13C NMR (101 MHz, D2O) δ 165.4, 145.4, 138.5, 137.8, 134.1, 131.8, 129.2, 128.0, 127.4, 125.1, 112.1, 43.5, 42.7. ESI-HRMS (m/z): calculated for C15H16N5O (M+H)+: 282.1349, found: 282.1344.
4-(1,3-Dioxolan-2-yl)benzonitrile (43).
To a solution of 42 (38 mmol, 5.0 g) and ethylene glycol (76 mmol, 4.2 mL) in toluene (50 mL) was added pyridinium p-toluenesulfonate (4 mmol, 0.96 g). The resulting solution was heated to reflux with a Dean-Stark trap for 4 h. The resulting solution was then concentrated in vacuo and the residue was then purified by column chromatography (silica gel, 10% EtOAc/hexanes as eluent) to yield 43 as white solid (5.25 g, 79%). 1H NMR (300 MHz, Chloroform-d) δ 7.67 (d, J = 8.3 Hz, 2H), 7.58 (d, J = 8.3 Hz, 2H), 5.84 (s, 1H), 4.17 – 3.99 (m, 4H).
(4-(1,3-Dioxolan-2-yl)phenyl)methanamine (44).
LiAlH4 (0.99 g, 26 mmol) was suspended in dry THF (50 mL) and was cooled under 0 °C. A solution of 43 (4.5 g, 25.7 mmol) in dry THF (50 mL) was added dropwise to the LiAlH4 suspension under the same temperature. After the addition, the reaction mixture was warmed up to room temperature and stirred for 4 h. Water (3 mL) was added dropwise followed by 2 M aqueous NaOH solution (3 mL) and then water (3 mL). The precipitate was filtered and washed with THF. The combined filtrate was then dried with anhydrous Na2SO4 and evaporated in vacuo. The residue was then purified by column chromatography (silica gel, 100% EtOAc as eluent) to yield 44 as colorless oil to white solid (3.0 g, 67%). 1H NMR (300 MHz, Chloroform-d) δ 7.44 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 5.78 (s, 1H), 4.15 – 3.95 (m, 4H), 3.85 (s, 2H).
N-(4-(1,3-dioxolan-2-yl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (45).
To a solution of 44 (2 mmol, 358 mg) and 39 (2 mmol, 326 mg) in DMF (10 mL) was added DIPEA (4 mmol, 516 mg) and EDCI (2.4 mmol, 460 mg). The resulting solution was then stirred under room temperature overnight. The reaction mixture then diluted with EtOAc (50 mL), then washed with saturated NaHCO3 solution (2 × 50 mL), 1 M HCl (2 × 50 mL), and brine (50 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to yield 45 as white solid (520 mg, 80%). 1H NMR (400 MHz, DMSO-d6) δ 9.36 (d, J = 0.8 Hz, 1H), 9.25 (t, J = 5.9 Hz, 1H), 9.14 (t, J = 1.4 Hz, 1H), 7.90 – 7.76 (m, 2H), 7.44 – 7.33 (m, 4H), 5.70 (s, 1H), 4.52 (d, J = 5.8 Hz, 2H), 4.09 – 3.88 (m, 4H).
N-(4-formylbenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (46).
To a solution of 45 (520 mg, 1.6 mmol) in1,4-dioxane (5 mL) was added 5 mL of 4 M HCl solution in 1,4-dioxane. The resulting solution was stirred for 2 h and concentrated in vacuo. The residue was suspended in H2O and basified with saturated NaHCO3 solution. The precipitation was filtered and dried to yield 46 as yellowish solid (390 mg, 87%). 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 9.42 – 9.31 (m, 2H), 9.16 (t, J = 1.4 Hz, 1H), 7.89 (d, J = 8.1 Hz, 2H), 7.87 – 7.78 (m, 2H), 7.57 (d, J = 7.9 Hz, 2H), 4.61 (d, J = 5.9 Hz, 2H).
N-(4-((dimethylamino)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (7).
To a solution of 46 (35 mg, 0.12 mmol) in THF (3 mL) was added a solution of dimethylamine in THF (1 M, 0.25 mL) and Ti(OiPr)4 (0.36 mmol, 102 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.36 mmol, 76 mg) was added and the mixture was refluxed under N2. The reaction was monitored by TLC. Small additional portions of NaBH(OAc)3 were added to drive the reaction to completion. Upon complete consumption of 46, the reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 7 as white solid (11 mg, 30%). 1H NMR (400 MHz, Methanol-d4) δ 9.29 (d, J = 0.8 Hz, 1H), 9.09 (t, J = 1.4 Hz, 1H), 7.91 – 7.76 (m, 2H), 7.57 – 7.44 (m, 4H), 4.65 (s, 2H), 4.29 (s, 2H), 2.83 (s, 6H). 13C NMR (101 MHz, DMSO) δ 164.1, 148.8, 141.0, 138.1, 131.6, 129.5, 128.1, 127.4, 127.0, 121.5, 114.9, 59.5, 42.9, 41.8. ESI-HRMS (m/z): calculated for C17H20N5O (M+H)+: 310.1662, found: 310.1654.
N-(4-(piperidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (8).
To a solution of 46 (35 mg, 0.12 mmol) in THF (3 mL) was added piperidine (20 mg, 0.24 mmol) and Ti(OiPr)4 (0.36 mmol, 102 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.36 mmol, 76 mg) was added and the mixture was refluxed under N2. The reaction was monitored by TLC. Small portions of NaBH(OAc)3 were added to drive the reaction to completion. Upon complete consumption of 46, the reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 8 as white solid (12 mg, 28%). 1H NMR (400 MHz, Methanol-d4) δ 9.26 (d, J = 0.8 Hz, 1H), 9.04 (t, J = 1.4 Hz, 1H), 7.87 – 7.76 (m, 2H), 7.38 – 7.29 (m, 4H), 4.60 (s, 2H), 3.50 (s, 2H), 2.37 (t, J = 5.1 Hz, 4H), 1.58 (p, J = 5.6 Hz, 4H), 1.45 (p, J = 5.6 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 164.8, 149.0, 137.5, 137.5, 135.9, 129.9, 127.4, 127.3, 126.4, 122.5, 114.2, 62.9, 53.8, 43.1, 25.0, 23.7. ESI-HRMS (m/z): calculated for C20H24N5O (M+H)+: 350.1975, found: 350.1970.
N-(4-(morpholinomethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (9).
To a solution of 46 (35 mg, 0.12 mmol) in THF (3 mL) was added morpholine (21 mg, 0.24 mmol) and Ti(OiPr)4 (0.36 mmol, 102 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.36 mmol, 76 mg) was added and the mixture was refluxed under N2. The reaction was monitored by TLC. Small portions of additional NaBH(OAc)3 were added to drive the reaction to completion. Upon complete consumption of 46, the reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 9 as white solid (15 mg, 43%). 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 9.22 (t, J = 5.9 Hz, 1H), 9.14 (t, J = 1.4 Hz, 1H), 7.87 – 7.75 (m, 2H), 7.32 – 7.24 (m, 4H), 4.49 (d, J = 5.8 Hz, 2H), 3.55 (t, J = 4.7 Hz, 4H), 3.42 (s, 2H), 2.37 – 2.27 (m, 4H). 13C NMR (101 MHz, DMSO) δ 163.5, 148.4, 137.8, 137.6, 136.5, 129.0, 127.3, 126.8, 126.4, 121.1, 114.4, 66.2, 62.1, 53.1, 42.5. ESI-HRMS (m/z): calculated for C19H22N5O2 (M+H)+: 352.1768, found: 352.1764.
N-(4-(pyrrolidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (10).
To a solution of 46 (35 mg, 0.12 mmol) in THF (3 mL) was added pyrrolidine (17 mg, 0.24 mmol) and Ti(OiPr)4 (0.36 mmol, 102 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.36 mmol, 76 mg) was added and the mixture was refluxed under N2. The reaction was monitored by TLC. Small portions of NaBH(OAc)3 were added to drive the reaction to completion. Upon complete consumption of 46, the reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 10 as white solid (15 mg, 38%). 1H NMR (300 MHz, Methanol-d4) δ 9.28 (s, 1H), 9.07 (d, J = 1.6 Hz, 1H), 7.86 (dd, J = 9.7, 1.7 Hz, 1H), 7.79 (d, J = 9.7 Hz, 1H), 7.41 (s, 4H), 4.61 (s, 2H), 3.94 (s, 2H), 3.02 – 2.76 (m, 4H), 1.97 – 1.88 (m, 4H). 13C NMR (75 MHz, CD3OD) δ 164.8, 149.0, 138.8, 137.5, 134.0, 129.7, 127.7, 127.4, 126.4, 122.4, 114.2, 58.8, 53.4, 43.0, 22.6. ESI-HRMS (m/z): calculated for C19H22N5O (M+H)+: 336.1819 (M+H); found: 336.1810.
N-(4-(azetidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (11).
To a solution of 46 (35 mg, 0.12 mmol) in THF (3 mL) was added azetidine hydrochloride (22 mg, 0.24 mmol) and Ti(OiPr)4 (0.36 mmol, 102 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.36 mmol, 76 mg) was added and the mixture was refluxed under N2. The reaction was monitored by TLC. Small portions of additional NaBH(OAc)3 were added to drive the reaction to completion. Upon complete consumption of 46, the reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 11 as white solid (15 mg, 40%). 1H NMR (400 MHz, Methanol-d4) δ 9.29 (d, J = 0.8 Hz, 1H), 9.09 (t, J = 1.4 Hz, 1H), 7.91 – 7.76 (m, 2H), 7.52 – 7.42 (m, 4H), 4.63 (s, 2H), 4.30 (s, 2H), 4.06 (t, J = 8.1 Hz, 4H), 2.47 (p, J = 8.1 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 166.3, 150.4, 141.7, 138.9, 131.2, 130.9, 129.5, 128.8, 127.8, 123.7, 115.6, 59.2, 55.2, 44.3, 17.2. ESI-HRMS (m/z): calculated for C18H20N5O (M+H)+: 322.1662; found: 322.1654.
N-(4-((2-methylazetidin-1-yl)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (12).
To a solution of 46 (58 mg, 0.2 mmol) in DMF (1 mL) was added 2-methylazetidine hydrochloride (43 mg, 0.4 mmol) and Ti(OiPr)4 (0.6 mmol, 170 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.6 mmol, 127 mg) was added and the mixture was heated to 70~75 °C for 24 h under N2. The reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 12 as white solid (58 mg, 88%). 1H NMR (400 MHz, Methanol-d4) δ 9.22 (d, J = 0.8 Hz, 1H), 9.02 (t, J = 1.4 Hz, 1H), 7.80 (dd, J = 9.6, 1.6 Hz, 1H), 7.71 (dt, J = 9.6, 1.0 Hz, 1H), 7.31 – 7.20 (m, 4H), 4.54 (s, 2H), 3.58 (d, J = 12.4 Hz, 1H), 3.46 (d, J = 12.4 Hz, 1H), 3.35 – 3.27 (m, 1H), 3.24 – 3.15 (m, 1H), 2.89 (dt, J = 9.8, 7.7 Hz, 1H), 2.04 (dtd, J = 10.2, 7.8, 2.3 Hz, 1H), 1.72 (tt, J = 10.0, 8.5 Hz, 1H), 0.97 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 167.0, 151.2, 139.8, 139.7, 138.3, 131.5, 129.7, 129.6, 128.6, 124.7, 116.4, 64.8, 63.7, 53.1, 45.3, 27.4, 22.1. ESI-HRMS (m/z): calculated for C19H22N5O (M+H)+: 336.1819; found: 336.1814.
N-(4-((2-isopropylazetidin-1-yl)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (13).
To a solution of 46 (58 mg, 0.2 mmol) in DMF (1 mL) was added 2-isopropylazetidine hydrochloride (54 mg, 0.4 mmol) and Ti(OiPr)4 (0.6 mmol, 170 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.6 mmol, 127 mg) was added and the mixture was heated to 70~75 °C for 24 h under N2. The reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 13 as white solid (61 mg, 84%). 1H NMR (400 MHz, Methanol-d4) δ 9.29 (dt, J = 1.9, 0.9 Hz, 1H), 9.08 (ddt, J = 4.2, 2.8, 1.4 Hz, 1H), 7.91 – 7.77 (m, 2H), 7.46 – 7.33 (m, 4H), 4.62 (s, 2H), 4.18 – 4.03 (m, 1H), 3.90 – 3.65 (m, 1H), 3.43 (s, 1H), 3.39 – 3.34 (m, 1H), 3.22 (d, J = 8.8 Hz, 1H), 2.25 (d, J = 10.6 Hz, 1H), 2.07 – 1.95 (m, 1H), 1.85 (dd, J = 14.8, 8.4 Hz, 1H), 0.95 (dd, J = 6.7, 0.8 Hz, 3H), 0.85 (dd, J = 6.7, 2.8 Hz, 3H). ESI-HRMS (m/z): calculated for C21H25N5O (M+H)+: 364.2132; found: 364.2128.
N-(4-((2-(tert-butyl)azetidin-1-yl)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (14).
To a solution of 46 (48 mg, 0.165 mmol) in DMF (1 mL) was added 2-tert-butyl azetidine hydrochloride (50 mg, 0.33 mmol) and Ti(OiPr)4 (0.495 mmol, 141 mg). The mixture was stirred under room temperature for 10 min. Then NaBH(OAc)3 (0.495 mmol, 105 mg) was added and the mixture was heated to 70~75 °C for 24 h under N2. The reaction mixture was diluted with saturated NaHCO3 solution (20 mL) and the precipitate was filtered. The filtrate was then extracted by EtOAc (2 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to give 14 as white solid (50 mg, 80%). 1H NMR (400 MHz, Methanol-d4) δ 9.30 (d, J = 0.8 Hz, 1H), 9.07 (t, J = 1.4 Hz, 1H), 7.92 – 7.79 (m, 2H), 7.40 – 7.29 (m, 4H), 4.61 (s, 2H), 3.95 (d, J = 12.9 Hz, 1H), 3.47 – 3.37 (m, 1H), 3.19 – 2.98 (m, 2H), 2.76 (d, J = 9.0 Hz, 1H), 2.01 – 1.85 (m, 2H), 0.93 (s, 9H). ESI-HRMS (m/z): calculated for C22H27N5O (M+H)+: 378.2288; found: 378.2286.
N-(3-(azetidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (15).
To a solution of 47 (22 mg, 0.125 mmol) and 39 (27 mg, 0.14 mmol) in DMF (0.4 mL) was added HBTU (52 mg, 0.14 mmol) and DIPEA (33 mg, 0.25 mmol). The resulting solution was then stirred under room temperature overnight. The reaction mixture then diluted with EtOAc (30 mL), then washed with saturated NaHCO3 solution (2 × 5 mL), and brine (5 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to yield 15 as white solid (25 mg, 62%). 1H NMR (400 MHz, Methanol-d4) δ 9.30 (d, J = 0.9 Hz, 1H), 9.09 (t, J = 1.4 Hz, 1H), 7.94 – 7.79 (m, 2H), 7.38 – 7.30 (m, 3H), 7.24 (d, J = 1.0 Hz, 1H), 4.63 (s, 2H), 3.63 (s, 2H), 3.31 (t, J = 7.2 Hz, 4H), 2.21 – 2.07 (m, 2H). ESI-HRMS (m/z): calculated for C18H20N5O (M+H)+: 322.1662; found: 322.1656.
Methyl 4-(1-aminoethyl)benzoate (49).
Methyl 4-acetylbenzoate (500 mg, 2.8 mmol), ammonium acetate (1.29 g, 16.8 mmol) and sodium cyanoborohydrate (263 mg, 4.2 mmol) were dissolved in 10 ml methanol and solution was stirred at room temperature for 16 h. The reaction mixture was concentrated and acidified with 2 M HCl (5 mL), then extracted with DCM. The aqueous layer was basified with solid NaHCO3 and extracted with DCM (2 × 30 mL). the combined DCM layers were dried over Na2SO4 and concentrated. The residue was used without further purification.
Methyl 4-(1-((tert-butoxycarbonyl)amino)ethyl) benzoate (50).
Methyl 4-(1-aminoethyl)benzoate 49 (250 mg, 1.39 mmol) was dissolved in DCM (5 mL) and Boc anhydride (348 mg, 1.6 mmol), DIPEA (0.5 ml, 2.7 mmol) and DMAP (17 mg, 0.139 mmol) were added and stirred for overnight. The reaction was washed with water and extracted with ethyl acetate (2 × 20 mL). the combined organic layers were dried over Na2SO4, concentrated and purified by silica gel column chromatography (20% EtOAc/Hexane) to yield 50 as white solid (300 mg, 77%).
tert-Butyl (1-(4-(hydroxymethyl)phenyl)ethyl) carbamate (51).
Methyl 4-(1-((tert-butoxycarbonyl)amino)ethyl)benzoate 50 (0.3 g, 1.0 mmol) was dissolved in THF (3 mL) and the solution cooled to below −5 °C in an ice/salt bath. LiAlH4 (2 M in THF, 1 mL) was added dropwise over 10 min. Upon completion of addition, the reaction was stirred at 0 °C for 75 min. Water (0.16 mL) was added dropwise followed by 2 M aqueous NaOH solution (0.16 mL) and then water (0.16 mL). The suspension was stirred for 15 min and then diluted with EtOAc (15 mL). The mixture was dried over Na2SO4 and filtered and the resulting filtrate was concentrated in vacuo to afford the title compound, which was used without further purification (215 mg, 80%).
tert-Butyl (1-(4-(chloromethyl)phenyl)ethyl) carbamate (52).
To a stirred solution of 51 (200 mg, 0.8 mmol) in DCM (5 mL) was added methanesulfonyl chloride (108 mg, 0.95 mmol) and triethylamine (0.23 mL, 1.6 mmol). The solution was stirred for 16 h at room temperature then washed with water and brine. After separation, the organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography (EtOAc/hexane 0 to 80%) to yield 52 white solid (100 mg, 46% yield).
tert-Butyl (1-(4-(azetidin-1-ylmethyl)phenyl)ethyl) carbamate (53).
To a stirred solution of 52 (100 mg, 0.37 mmol) in acetonitrile (5 mL) was added azetidine hydrochloride (41 mg, 0.44 mmol) and DIPEA (0.2 ml, 1.1 mmol). The solution was stirred at 80 °C for 16 h. The reaction mixture was diluted with water and extracted with DCM. The DCM layers were dried over Na2SO4 and concentrated to yield the crude product, which was used without further purification (100 mg).
1-(4-(Azetidin-1-ylmethyl)phenyl)ethan-1-amine (54).
To a stirred solution of 53 (100 mg. 0.3 mmol), 4 M HCl in 1,4-dioxane (0.5 ml, 1.8 mmol) was added and stirred at room temperature for 1 h. The reaction mixture was concentrated to yield 54 as off-white solid. ESI-HRMS (m/z): calculated for C12H19N2 (M+H)+: 191.1543; found: 191.1539.
N-(1-(4-(azetidin-1-ylmethyl)phenyl)ethyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (16).
To a stirred solution of 39 (50 mg, 0.3 mmol) and 54 (58 mg, 0.3 mmol) in DMF (1 ml) was added HBTU (136 mg, 0.36 mmol) and DIPEA (0.1 ml, 0.6 mmol). The solution was stirred at room temperature for 16 h. The reaction mixture was diluted with water and extracted with DCM. The DCM layers were dried over Na2SO4 and concentrated to give the crude product, which was purified by flash chromatography to yield compound 16 as off-white solid (30 mg, 34%). 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 9.13 (s, 1H), 8.99 (d, J = 7.9 Hz, 1H), 7.90–7.72 (m, 2H), 7.32 (d, J = 7.5 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 5.14 (dd, J = 13.9, 7.1 Hz, 1H), 3.47 (s, 2H), 3.08 (t, J = 6.8 Hz, 4H), 1.94 (dd, J = 13.4, 6.9 Hz, 2H), 1.47 (d, J = 7.0 Hz, 3H). ESI-HRMS (m/z): calculated for C19H22N5O (M+H)+: 336.1819; found: 336.1808.
N-(4-(azetidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-7-carboxamide (17).
To a solution of 55 (44 mg, 0.25 mmol) and 56 (45 mg, 0.275 mmol) in DMF (1 mL) was added HBTU (104 mg, 0.275 mmol) and DIPEA (65 mg, 0.5 mmol). The resulting solution was then stirred under room temperature overnight. The reaction mixture then diluted with EtOAc (30 mL), then washed with saturated NaHCO3 solution (2 × 5 mL), and brine (5 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to yield 17 as white solid (52 mg, 65%). 1H NMR (400 MHz, Methanol-d4) δ 9.27 (s, 1H), 8.58 (d, J = 7.2 Hz, 1H), 8.26 (s, 1H), 7.44 (d, J = 7.3 Hz, 1H), 7.37 (d, J = 7.8 Hz, 2H), 7.29 (d, J = 7.1 Hz, 2H), 4.61 (s, 2H), 3.61 (s, 2H), 3.32 – 3.26 (m, 4H), 2.12 (p, J = 7.2 Hz, 2H). ESI-HRMS (m/z): calculated for C18H20N5O (M+H)+: 322.1662; found: 322.1653.
N-(4-(azetidin-1-ylmethyl)benzyl)-[1,2,4]triazolo[1,5-a]pyridine-6-carboxamide (18).
To a solution of 55 (44 mg, 0.25 mmol) and 57 (45 mg, 0.275 mmol) in DMF (1 mL) was added HBTU (104 mg, 0.275 mmol) and DIPEA (65 mg, 0.5 mmol). The resulting solution was then stirred under room temperature overnight. The reaction mixture then diluted with EtOAc (30 mL), then washed with saturated NaHCO3 solution (2 × 5 mL), and brine (5 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to yield 18 as white solid (49 mg, 61%). 1H NMR (400 MHz, Methanol-d4) δ 9.34 (s, 1H), 8.52 (d, J = 1.4 Hz, 1H), 8.14 (dd, J = 9.3, 1.9 Hz, 1H), 7.86 (d, J = 9.3 Hz, 1H), 7.38 (d, J = 7.7 Hz, 2H), 7.30 (d, J = 7.3 Hz, 2H), 4.62 (s, 2H), 3.65 (s, 2H), 3.36 (d, J = 4.2 Hz, 4H), 2.14 (p, J = 7.2 Hz, 2H). ESI-HRMS (m/z): calculated for C18H20N5O (M+H)+: 322.1662; found: 322.1655.
N-(4-(azetidin-1-ylmethyl)benzyl)-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide (19).
To a solution of 55 (44 mg, 0.25 mmol) and 58 (49 mg, 0.275 mmol) in DMF (1 mL) was added HBTU (104 mg, 0.275 mmol) and DIPEA (65 mg, 0.5 mmol). The resulting solution was then stirred under room temperature overnight. The reaction mixture then diluted with EtOAc (30 mL), then washed with saturated NaHCO3 solution (2 × 5 mL), and brine (5 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 20% methanol/EtOAc as eluent) to yield 19 as white solid (49 mg, 59%). 1H NMR (400 MHz, Methanol-d4) δ 9.29 (d, J = 2.2 Hz, 1H), 8.88 (d, J = 2.2 Hz, 1H), 7.38 (d, J = 7.9 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.59 (s, 1H), 4.61 (s, 2H), 3.62 (s, 2H), 3.29 (d, J = 7.2 Hz, 4H), 2.53 (s, 3H), 2.13 (p, J = 7.2 Hz, 2H). ESI-HRMS (m/z): calculated for C19H22N5O (M+H)+: 336.1819; found: 336.1812.
tert-Butyl (4-(bromomethyl)benzyl)carbamate (60).
To a solution of 59 (238 mg, 1 mmol) in DCM (2 mL) was added triphenylphosphine (316 mg, 1.2 mmol). Then carbon tetrabromide (400 mg, 1.2 mmol) was added in portions under ice water bath. The reaction was left under ice water bath for another 3 h. Then the reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography (10:1 hexanes/EtOAc) to yield 60 as white solid (252 mg, 83%).
tert-Butyl (4-(cyanomethyl)benzyl)carbamate (61).
To a solution of 60 (150 mg, 0.5 mmol) in 2 mL of DMF was added NaCN (50 mg, 1 mmol). The reaction mixture was then stirred under 60 °C for 4 h. The reaction mixture was cooled and diluted with water and extracted with DCM. Combined DCM layers were dried over Na2SO4 and then concentrated to yield 61 as white solid, which was used without further purification (96 mg, 78%).
N-(4-(cyanomethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (63).
To a solution of 61 (96 mg, 0.39 mmol) in 1 mL of 1,4-dioxane was added 4 M HCl solution in dioxane (4 mL). The reaction mixture was stirred at room temperature for 1 h and then concentrated to dryness in vacuo. The residue was dissolved in 2 mL of DMF, to which was added 39 (64 mg, 0.39 mmol), DIPEA (155 mg, 1.2 mmol) and HBTU (175 mg, 0.46 mmol). The reaction mixture was stirred at room temperature overnight and then diluted with DCM (20 mL), washed with saturated NaHCO3 solution (2 × 20 mL), 1 M HCl (2 × 20 mL) and brine. The combined DCM layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (0–10% MeOH/DCM) to yield 63 as pale yellow solid (58 mg, 52%). 1H NMR (300 MHz, DMSO-d6) δ 9.37 (s, 1H), 9.29 (t, J = 5.7 Hz, 1H), 9.15 (t, J = 1.2 Hz, 1H), 7.90–7.66 (m, 2H), 7.43–7.23 (m, 4H), 4.50 (d, J = 5.8 Hz, 2H), 4.01 (s, 2H).
N-(4-(2-amino-2-iminoethyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (20).
To 1.5 mL of absolute EtOH was dropwise added 1 mL of acetylchloride under N2 at 0 °C. The solution was stirred at 0 °C for 5 min before a solution of 63 (20 mg, 0.069 mmol) in absolute EtOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 36 h. The reaction mixture was then evaporated to dryness under high vacuum. To the residue was added a 7 M NH3 solution in methanol (1 mL). The reaction mixture was then stirred overnight and concentrated in vacuo. The residue was dissolved in 1 M HCl solution, and washed with EtOAc to remove residual 63, then evaporated to dryness to yield 20 as its hydrochloride salt (17 mg, 71%). 1H NMR (300 MHz, D2O) δ 9.47 (s, 1H), 9.22 (s, 1H), 8.24 (dt, J = 9.6, 1.8 Hz, 1H), 8.08 (d, J = 9.6 Hz, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 1H), 4.65 (s, 2H), 3.89 (s, 2H). ESI-HRMS (m/z): calculated for C16H17N6O (M+H)+ : 309.1458; found: 309.1451.
tert-Butyl (3-(hydroxymethyl)-4-methylbenzyl) carbamate (65).
2-Methyl-5-cyanobenzoic acid 64 (5 mmol, 0.81 g) was dissolved in anhydrous THF (15 mL). A solution of LiAlH4 in THF (1.0 M, 20 mL) was added dropwise to the solution under N2 at 0 °C. After completion of addition, the reaction mixture was heated to reflux overnight. The solution was then cooled to room temperature and then to 0 °C. Water (5 mL) was added dropwise, followed by 2 M NaOH solution (5 mL). After stirring for another 10 min, the mixture was filtered over celite. To the filtrate was added Boc2O (5 mmol, 1.09 g) and the solution was stirred at room temperature for 4 h. The solution was then concentrated in vacuo to yield crude 65 as yellow oil (0.75 g, 60%). 1H NMR (300 MHz, Chloroform-d) δ 7.28 (s, 1H), 7.16–7.09 (m, 2H), 4.84 (brs, 1H), 4.68 (d, J = 5.2 Hz, 2H), 4.27 (d, J = 5.9 Hz, 2H), 2.32 (s, 3H), 1.66 (s, 1H), 1.45 (s, 9H).
tert-Butyl (3-(bromomethyl)-4-methylbenzyl) carbamate (66).
To a solution of 65 (1.2 mmol, 300 mg) in DCM (10 mL) was added triphenylphosphine (1.44 mmol, 380 mg). Then carbon tetrabromide (1.44 mmol, 480 mg) was added in portions at 0 °C. The reaction was stirred at 0 °C for 3 h. Then the reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography (hexanes/EtOAc=10:1) to yield 66 as white solid (284 mg, 76%). 1H NMR (300 MHz, Chloroform-d) δ 7.23 (s, 1H), 7.19–7.13 (m, 2H), 4.81 (brs, 1H), 4.59 (s, 2H), 4.27 (d, J = 5.9 Hz, 2H), 2.40 (s, 3H), 1.46 (s, 9H).
tert-Butyl (3-(cyanomethyl)-4-methylbenzyl) carbamate (67).
To a solution of 66 (156 mg, 0.5 mmol) in 5 mL of DMF was added NaCN (50 mg, 1 mmol). The reaction mixture was then stirred under 60 °C for 4 h. The reaction mixture was cooled and diluted with water and extracted with DCM. Combined DCM layers were dried over Na2SO4 and then concentrated to yield 67 as white solid, which was used without further purification (78 mg, 60%).
N-(3-(cyanomethyl)-4-methylbenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (69).
To a solution of 67 (78 mg, 0.3 mmol) in 1 mL of 1,4-dioxane was added 4 M HCl solution in dioxane (4 mL). The reaction mixture was stirred at room temperature for 1 h and then concentrated to dryness in vacuo. The residue was dissolved in 2 mL of DMF, to which was added 39 (49 mg, 0.3 mmol), DIPEA (116 mg, 0.9 mmol) and HBTU (137 mg, 0.36 mmol). The reaction mixture was stirred at room temperature overnight and then diluted with DCM (20 mL), washed with saturated NaHCO3 solution (2 × 20 mL), 1 M HCl (2 × 20 mL) and brine. The combined DCM layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (0–10% MeOH/DCM) to yield 69 as white solid (46 mg, 50%). 1H NMR (300 MHz, DMSO-d6) δ 9.37 (t, J = 0.7 Hz, 1H), 9.24 (t, J = 5.9 Hz, 1H), 9.14 (s, 1H), 7.88–7.76 (m, 2H), 7.32 (s, 1H), 7.25–7.18 (m, 2H), 4.47 (d, J = 5.9 Hz, 2H), 3.98 (s, 2H), 2.27 (s, 3H).
N-(3-(2-amino-2-iminoethyl)-4-methylbenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (21).
To 1.5 mL of absolute EtOH was dropwise added 1 mL acetylchloride under N2 at 0 °C. The solution was stirred at 0 °C for 5 min before a solution of 69 (20 mg, 0.066 mmol) in absolute EtOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 36 h. The reaction mixture was then evaporated to dryness under high vacuum. To the residue was added a 7 M NH3 solution in methanol (1 mL). The reaction mixture was then stirred overnight and concentrated in vacuo. The residue was dissolved in 1 M HCl solution, and washed with EtOAc to remove residual 69, then evaporated to dryness to yield 21 as its hydrochloride salt (14 mg, 60%). 1H NMR (300 MHz, D2O) δ 9.46 (s, 1H), 9.22 (s, 1H), 8.33 (d, J = 9.3 Hz, 1H), 8.08 (d, J = 9.2 Hz, 1H), 7.31 – 7.13 (m, 3H), 4.50 (s, 2H), 3.83 (s, 2H), 2.14 (s, 3H). ESI-HRMS (m/z): calculated for C17H19N6O (M+H)+: 323.1615; found: 323.1611.
tert-Butyl (3-fluoro-4-(hydroxymethyl)benzyl) carbamate (71).
2-Fluoro-4-cyanobenzoic acid 70 (5 mmol, 0.83 g) was dissolved in anhydrous THF (15 mL). A solution of LiAlH4 in THF (1.0 M, 20 mL) was added dropwise to the above solution under N2 at 0 °C. After completion of addition, the reaction mixture was heated to reflux overnight. The solution was then cooled to room temperature and then to 0 °C. Water (5 mL) was added dropwise, followed by 2 M NaOH solution (5 mL). After stirring for another 10 min, the mixture was filtered over celite. To the filtrate was added Boc2O (5 mmol, 1.09 g) and the solution was stirred at room temperature for 4 h. The solution was then concentrated in vacuo to yield crude 71 as yellow oil (0.71 g, 55%).
tert-Butyl (4-(bromomethyl)-3-fluorobenzyl) carbamate (72).
To a solution of 71 (1.2 mmol, 306 mg) in DCM (10 mL) was added triphenylphosphine (1.44 mmol, 380 mg). Then carbon tetrabromide (1.44 mmol, 480 mg) was added in portions at 0 °C. The reaction was stirred at 0 °C for 3 h. Then the reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography (hexanes/EtOAc=10:1) to yield 72 as white solid (250 mg, 66%). 1H NMR (300 MHz, Chloroform-d) δ 7.34 (t, J = 7.8 Hz, 1H), 7.13 – 6.95 (m, 2H), 4.88 (brs, 1H), 4.50 (s, 2H), 4.30 (d, J = 6.2 Hz, 2H), 1.46 (s, 9H).
tert-Butyl (4-(cyanomethyl)-3-fluorobenzyl) carbamate (73).
To a solution of 72 (159 mg, 0.5 mmol) in 5 mL of DMF was added NaCN (50 mg, 1 mmol). The reaction mixture was then stirred under 60 °C for 4 h. The reaction mixture was cooled and diluted with water and extracted with DCM. Combined DCM layers were dried over Na2SO4 and then concentrated to yield 73 as white solid, which was used without further purification (85 mg, 64%).
N-(4-(cyanomethyl)-3-fluorobenzyl)-[1,2,4]triazolo [4,3-a]pyridine-6-carboxamide (75).
To a solution of 73 (79 mg, 0.3 mmol) in 1 mL of 1,4-dioxane was added 4 M HCl solution in dioxane (4 mL). The reaction mixture was stirred at room temperature for 1 h and then concentrated to dryness in vacuo. The residue was dissolved in 2 mL of DMF, to which was added 39 (49 mg, 0.3 mmol), DIPEA (116 mg, 0.9 mmol) and HBTU (137 mg, 0.36 mmol). The reaction mixture was stirred at room temperature overnight and then diluted with DCM (20 mL), washed with saturated NaHCO3 solution (2 × 20 mL), 1 M HCl (2 × 20 mL) and brine. The combined DCM layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (0–10% MeOH/DCM) to yield 75 as white solid (51 mg, 55%). 1H NMR (300 MHz, Methanol-d4) δ 9.29 (s, 1H), 9.09 (s, 1H), 7.83 (t, J = 8.7 Hz, 2H), 7.46 (t, J = 7.8 Hz, 1H), 7.34 – 7.16 (m, 2H), 4.63 (s, 2H), 3.92 (s, 2H).
N-(4-(2-amino-2-iminoethyl)-3-fluorobenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (22).
To 1.5 mL of absolute EtOH was added dropwise 1 mL of acetylchloride under N2 at 0 °C. The solution was stirred at 0 °C for 5 min before a solution of 75 (20 mg, 0.065 mmol) in absolute EtOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 36 h. The reaction mixture was then evaporated to dryness under high vacuum. To the residue was added a 7 M NH3 solution in methanol (1 mL). The reaction mixture was then stirred overnight and concentrated in vacuo. The residue was dissolved in 1 M HCl solution, and washed with EtOAc to remove residual 75, then evaporated to dryness to yield 22 as its hydrochloride salt (10 mg, 47%). 1H NMR (300 MHz, D2O) δ 9.46 (s, 1H), 9.22 (s, 1H), 8.22 (dd, J = 9.6, 1.5 Hz, 1H), 8.06 (d, J = 9.6 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 7.33 – 7.14 (m, 2H), 4.63 (s, 2H), 3.93 (s, 2H). ESI-HRMS (m/z): calculated for C16H16FN6O (M+H)+: 327.1364; found: 327.1352.
tert-Butyl (4-fluoro-3-(hydroxymethyl)benzyl) carbamate (77).
2-Fluoro-5-cyanobenzoic acid 76 (5 mmol, 0.83 g) was dissolved in anhydrous THF (15 mL). A solution of LiAlH4 in THF (1.0 M, 20 mL) was added dropwise to the solution under N2 at 0 °C. After completion of addition, the reaction mixture was heated to reflux overnight. The solution was then cooled to room temperature and then to 0 °C. Water (5 mL) was added dropwise, followed by 2 M NaOH solution (5 mL). After stirring for another 10 min, the mixture was filtered over celite. To the filtrate was added Boc2O (5 mmol, 1.09 g) and the solution was stirred at room temperature for 4 h. The solution was then concentrated in vacuo to yield crude 77 as yellow oil (0.81 g, 59%).
tert-Butyl (3-(bromomethyl)-4-fluorobenzyl) carbamate (78).
To a solution of 77 (1.2 mmol, 306 mg) in DCM (10 mL) was added triphenylphosphine (1.44 mmol, 380 mg). Then carbon tetrabromide (1.44 mmol, 480 mg) was added in portions at 0 °C. The reaction was stirred at 0 °C for 3 h. Then the reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography (hexanes/EtOAc=10:1) to yield 78 as white solid (234 mg, 62%). 1H NMR (300 MHz, Chloroform-d) δ 7.34 – 7.27 (m, 1H), 7.25–7.18 (m, 1H), 7.05–6.96 (m, 1H), 4.87 (brs, 1H), 4.49 (s, 2H), 4.29 (d, J = 6.2 Hz, 2H), 1.46 (s, 9H).
tert-Butyl (3-(cyanomethyl)-4-fluorobenzyl) carbamate (79).
To a solution of 78 (159 mg, 0.5 mmol) in 5 mL of DMF was added NaCN (50 mg, 1 mmol). The reaction mixture was then stirred under 60 °C for 4 h. The reaction mixture was cooled and diluted with water and extracted with DCM. Combined DCM layers were dried over Na2SO4 and then concentrated to yield 79 as white solid, which was used without further purification (79 mg, 60%).
N-(3-(cyanomethyl)-4-fluorobenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (81).
To a solution of 79 (79 mg, 0.3 mmol) in 1 mL of 1,4-dioxane was added 4 M HCl solution in dioxane (4 mL). The reaction mixture was stirred at room temperature for 1 h and then concentrated to dryness in vacuo. The residue was dissolved in 2 mL of DMF, to which was added 39 (49 mg, 0.3 mmol), DIPEA (116 mg, 0.9 mmol) and HBTU (137 mg, 0.36 mmol). The reaction mixture was stirred at room temperature overnight and then diluted with DCM (20 mL), washed with saturated NaHCO3 solution (2 × 20 mL), 1 M HCl (2 × 20 mL) and brine. The combined DCM layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (0–10% MeOH/DCM) to yield 81 as white solid (57 mg, 62%). 1H NMR (300 MHz, DMSO-d6) δ 9.37 (s, 1H), 9.29 (t, J = 5.6 Hz, 1H), 9.14 (s, 1H), 7.85 (d, J = 9.6 Hz, 1H), 7.78 (dd, J = 9.7, 1.5 Hz, 1H), 7.48 – 7.20 (m, 3H), 4.50 (d, J = 6.1 Hz, 2H), 4.05 (s, 2H).
N-(4-(2-amino-2-iminoethyl)-3-fluorobenzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (23).
To 1.5 mL of absolute EtOH was dropwise added 1 mL of acetylchloride under N2 at 0 °C. The solution was stirred at 0 °C for 5 min before a solution of 81 (20 mg, 0.065 mmol) in absolution EtOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 36 h. The reaction mixture was then evaporated to dryness under high vacuum. To the residue was added a 7 M NH3 solution in methanol (1 mL). The reaction mixture was then stirred overnight and concentrated in vacuo. The residue was dissolved in 1 M HCl solution, and washed with EtOAc to remove residual 81, then evaporated to dryness to yield 23 as its hydrochloride salt (13 mg, 60%). 1H NMR (300 MHz, D2O) δ 9.51 (s, 1H), 9.27 (s, 1H), 8.35 (dd, J = 9.6, 1.6 Hz, 1H), 8.13 (d, J = 9.6 Hz, 1H), 7.50 – 7.34 (m, 2H), 7.27 – 7.10 (m, 1H), 4.62 (s 2H), 3.91 (s, 2H). ESI-HRMS (m/z): calculated for C16H16FN6O (M+H)+ : 327.1364; found: 327.1359.
N-(4-(((5-fluoropyrimidin-2-yl)amino)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (24).
To a stirred solution of 2-chloro-5-fluoropyrimidine (24 mg, 0.18 mmol) and amine 6 (48 mg. 0.15 mmol) in ethanol (1 mL) was added DIPEA (0.1 mL, 0.57 mmol) and the reaction mixture was heated to 75 °C for 36 h. The reaction was concentrated and purified by flash chromatography (silica gel, 5% methanol/EtOAc as eluent) to yield 24 as light-yellow solid (22 mg, 39%). 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 9.21 (t, J = 5.9 Hz, 1H), 9.12 (t, J = 1.4 Hz, 1H), 8.32 (d, J = 1.0 Hz, 2H), 7.86 – 7.73 (m, 3H), 7.26 (s, 4H), 4.46 (d, J = 5.9 Hz, 2H), 4.43 (d, J = 6.4 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 163.9, 159.9, 153.3, 150.9, 148.8, 146.1, 145.8, 139.4, 138.0, 137.8, 127.8, 127.5, 127.2, 126.9, 121.6, 114.9, 44.7, 43.0. ESI-HRMS (m/z): calculated for C19H17FN7O (M+H)+: 378.1473; found: 378.1465.
N-(4-(((5-isopropylpyrimidin-2-yl)amino)methyl) benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (25).
To a stirred solution of amine 6 (50 mg, 0.17 mmol) and 2-chloro-5-isopropylpyrimidine (15 mg, 0.17 mmol) in ethanol (2 mL) was added DIPEA (68 mg, 0.53 mmol) and heated to 80 °C for 48 h. The reaction mixture was concentrated and purified by flash chromatography (0–10% methanol/DCM) to yield 25 as off-white solid (10 mg, 18%). 1H NMR (400 MHz, DMSO-d6) δ 9.35 (d, J = 0.8 Hz, 1H), 9.21 (t, J = 5.8 Hz, 1H), 9.13 (t, J = 1.4 Hz, 1H), 8.16 (s, 2H), 7.86 – 7.73 (m, 2H), 7.49 (t, J = 6.4 Hz, 1H), 7.26 (s, 4H), 4.49 – 4.40 (m, 4H), 2.71 (hept, J = 6.9 Hz, 1H), 1.15 (d, J = 6.9 Hz, 7H). 13C NMR (101 MHz, DMSO-d6) δ 163.9, 161.7, 156.5, 148.8, 139.9, 138.0, 137.7, 129.2, 127.7, 127.5, 127.2, 127.0, 121.6, 114.9, 44.3, 43.0, 28.7, 24.0. ESI-HRMS (m/z): calculated for C22H23N7ONa (M+Na)+: 424.1856, found: 424.1849.
N-(4-(((4,5-dihydro-1H-imidazol-2-yl)amino) methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (26).
To a stirred solution of amine 6 (95 mg, 0.3 mmol) and 82 (70 mg, 0.6 mmol) in DMF (2 mL) was added triethylamine (91 mg, 0.9 mmol) and heated to 100 °C for 16 h. The reaction mixture was concentrated under reduce pressure. Water was added to the crude product and the solid was filtered and concentrated under reduced pressure to provide the crude compound 26 (20 mg, 19%). The crude compound was purified by RP-HPLC (HPLC gradient: 0–70 min: 95% A to 50% A). 1H NMR (300 MHz, DMSO-d6) δ 9.38 (s, 1H), 9.28 (s, 1H), 9.14 (s, 1H), 8.67 (s, 1H), 7.82 (q, J = 9.7 Hz, 2H), 7.46 – 7.18 (m, 4H), 4.49 (d, J = 5.9 Hz, 2H), 4.35 (d, J = 5.9 Hz, 2H), 3.60 (s, 4H). ESI-HRMS (m/z): calculated for C18H20N7O (M+H)+: 350.1724; found: 350.1724.
tert-Butyl (4-(((5-fluoropyridin-2-yl)amino)methyl) benzyl)carbamate (84).
To a stirred solution of 83 (1 mmol, 300 mg) in 5 mL DMF was added 60 (1 mmol, 112 mg) and K2CO3 (1 mmol, 138 mg). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (25 mL) and then extracted with DCM (2 × 20 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated. The residue was then purified by flash chromatography to yield 84 as white solid (100 mg, 30%). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 3.0 Hz, 1H), 7.31 (d, J = 7.9 Hz, 2H), 7.25 (d, J = 8.2 Hz, 2H), 7.19 (ddd, J = 9.0, 7.9, 3.0 Hz, 1H), 6.32 (dd, J = 9.1, 3.4 Hz, 1H), 4.84 (s, 2H), 4.46 (d, J = 5.6 Hz, 2H), 4.30 (d, J = 6.0 Hz, 2H), 1.46 (s, 9H).
N-(4-(aminomethyl)benzyl)-5-fluoropyridin-2-amine dihydrochloride (85).
To a stirred solution of 84 (0.3 mmol, 100 mg) in 1,4-dioxane (1 mL) was add a 4 M HCl solution in dioxane (4 mL). The solution was stirred at room temperature for 1 h. The reaction mixture was then concentrated in vacuo to yield 85 as white solid (85 mg, 95%), which was used without further purification.
N-(4-(((5-fluoropyridin-2-yl)amino)methyl)benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (27).
To a stirred solution of 85 (54 mg, 0.18mmol) and 39 (32 mg, 0.20 mmol) in DMF (2 mL) was added DIPEA (0.15 mL, 0.8 mmol) and HBTU (80 mg, 0.22 mmol). The solution was then stirred at room temperature overnight. The reaction mixture was then diluted with water (20 mL) and extracted with EtOAc (2 × 20 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated. The residue was then purified by flash chromatography to provide 27 as pale yellow solid (38 mg, 56%). 1H NMR (400 MHz, DMSO-d6) δ 9.36 (d, J = 0.8 Hz, 1H), 9.21 (t, J = 5.9 Hz, 1H), 9.13 (t, J = 1.4 Hz, 1H), 7.90 (d, J = 3.1 Hz, 1H), 7.87 – 7.73 (m, 2H), 7.33 (td, J = 8.8, 3.1 Hz, 1H), 7.28 (s, 4H), 7.04 (t, J = 6.0 Hz, 1H), 6.51 (dd, J = 9.1, 3.6 Hz, 1H), 4.47 (d, J = 5.9 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 163.5, 155.7, 153.7, 151.3, 148.4, 139.2, 137.6, 137.4, 133.5, 133.3, 127.3, 127.2, 126.7, 126.5, 125.1, 124.9, 121.1, 114.4, 108.8, 108.7, 44.4, 42.6. ESI-HRMS (m/z): calculated for C20H17FN6O (M+H)+: 377.1521; found: 377.1520.
N-(4-(((3,6-difluoropyridin-2-yl)amino)methyl) benzyl)-[1,2,4]triazolo[4,3-a]pyridine-6-carboxamide (28).
To a stirred solution of 2,3,6-trifluoropyridine (32 mg, 0.24 mmol) and amine 6 (63 mg. 0.2 mmol) in N-methyl-2-pyrrolidone (NMP, 0.5 mL) was added DIPEA (0.1 mL, 0.57 mmol), and the reaction mixture was heated to 90 °C overnight. The reaction mixture was cooled and then diluted with EtOAc (25 mL), then washed with brine (2 × 5 mL). The organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by column chromatography (silica gel, 5% methanol/EtOAc as eluent) to yield 28 as light-yellow solid (30 mg, 38%). 1H NMR (400 MHz, Methanol-d4) δ 9.27 (d, J = 1.0 Hz, 1H), 9.04 (q, J = 1.4 Hz, 1H), 7.90 – 7.76 (m, 2H), 7.41 – 7.25 (m, 5H), 6.02 (dtd, J = 8.2, 3.1, 2.6, 1.3 Hz, 1H), 4.59 (d, J = 1.1 Hz, 2H), 4.57 (s, 2H). ESI-HRMS (m/z): calculated for C20H17F2N6O (M+H)+: 395.1426; found: 395.1426.
Surface plasmon resonance (SPR) assay.
All SPR experiments were performed on a Biosensing BI-4500 instrument with 1 × PBS with 0.1% DMSO as running buffer and a flow rate at 60 μL/min. His-ENL YEATS was immobilized through EDC/NHS coupling on CM dextran coated sensor chips (Biosensing). Sensor chip was activated by flowing 200 μL 0.05 M NHS plus 0.2 M EDC solution through the surface. Then 200 μL of 6 μM His-ENL YEATS in 10 mM NaOAc/HOAc, 150 mM NaCl, pH 5.5 was injected and flowed through the activated surface. 100 μL of 1 M ethanolamine pH 7.8 solution was injected to block the remaining activated ester on the surface. 11 and 24 were dissolved in running buffer and subjected to a 2-fold serial dilution for a total of 4 concentrations ranging from 5.6 μM to 0.7 μM (for 24, 5.4 μM to 0.675 μM). For each cycle, 350 μL of compound solution was injected and flow through the surface followed by a 600 s delay for dissociation. Prior to the first cycle, 350 μL of running buffer was injected for baseline calibration. A control flow channel was set up in parallel without His-ENL YEATS immobilization to subtract non-specific binding signals. The data analysis was performed on the kinetic analysis software provided by Biosensing Instrument Inc. and fitted into the Langmuir 1:1 binding model.
Nuclear magnetic resonance (NMR) experiments for ENL YEATS domain.
For NMR experiments, the YEATS domain of ENL (aa 1–148) was expressed as a C-terminal, uncleavable 6xHis fusion protein (plasmid was a generous gift from Oleg Fedorov). The 15N-labeled protein was expressed in E. coli Rosetta-2 (DE3) pLysS cells grown in NH4Cl (Sigma-Aldrich) minimal media. After induction with IPTG (final concentration 0.5 mM) (Gold biotechnology) for 18 hrs at 16 °C, cells were harvested via centrifugation and lysed by sonication. The uniformly 15N-labeled YEATS domain was incubated with Ni-NTA resins (Thermo Fisher Sci), washed and eluted with imidazole. The protein was further purified by size exclusion chromatography, concentrated (Millipore) and stored at −80 °C. NMR experiments were performed at 298 K on a Varian INOVA 600 MHz spectrometer. The 1H,15N HSQC spectra of 0.2 mM uniformly 15N-labeled YEATS domain (25–50 mM Tris-HCl pH 7.5 buffer, supplemented with 150 mM NaCl, 0.2 mM TCEP and 10% D2O) were collected in the presence of increasing amount of H3K27cr (aa 22–31) peptide (synthesized by Synpeptide) or compound 7. NMR data were processed and analyzed with NMRPipe and NMRDraw as previously described33.
Competitive peptide pulldown assay.
Compounds at indicated concentrations were mixed with 2 μg of GST-fused YEATS proteins in 300 μL binding buffer (50 mM Tris-HCl pH7.5, 250 mM NaCl, 0.1% NP-40, 1 mM PMSF) and rotated at 4ºC for 1h. Then, 0.5 μg of biotinylated histone peptides with different modifications were added and incubated for 4 h. Streptavidin magnetic beads (Amersham) were added to the mixture, and the mixture was incubated for 1 h with rotation. The beads were then washed three times and analyzed using SDS-PAGE and western blotting.
IC50 determination in the inhibition of YEATS domains.
To assess the specificity of 7, 11 and 24, their IC50 values in inhibition of the four human YEATS domain proteins binding to targeted histone peptides were determined in AlphaScreen assays. Compounds were subjected to twelve 3-fold serial dilutions, for a total of thirteen concentrations ranging from 54 μM to 0.1 nM for dose response curve. IC50 values were determined from the plot using nonlinear regression of variable slope (four parameters) and curve fitting performed by the GraphPad Prism software.
Cell growth inhibition assay.
Human Leukemia MV4;11 and MOLM13 cells were maintained in RPMI-1640 (Cellgro) supplemented with 10% fetal bovine serum (Sigma). Human U2OS cells were maintained in DMEM (Cellgro) supplemented with 10% fetal bovine serum. Five thousand cells were seeded in 96-well plate in 100 μL medium, treated with DMSO or compounds at indicated concentrations for 3 days. Cell viability was measured using the CellTiter-Glo luminescent cell viability assay kit (Promega) according to the manufacturer’s instruction. Survived cells were calculated as % relative to DMSO treated cells.
RNA extraction and qRT-PCR.
Total RNA was extracted using the RNeasy plus kit (Qiagen) and reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR (qPCR) analyses were performed as described previously using PowerUp SYBR Green PCR Master Mix and the Bio-Rad CFX96 real-time PCR detection system. Gene expressions were calculated following normalization to GAPDH levels using the comparative Ct (cycle threshold) method.
Cellular thermal shift assay (CETSA)34.
MOLM13, MV4;11, and HeLa cells were incubated with 20 μM of compound 7 for 5 h. Cells were then collected and washed with PBS for three times. The cell pellets were resuspended in PBS containing protease inhibitors and aliquoted into PCR microtubes (approximately 3 million cells in 54 μL). Cells were heated at indicated temperatures for 3 min in a thermal cycler (Bio-Rad) and then incubated at room temperature for 2 min. 6 μL of 10x Cell Lysis Buffer (8% NP40, 50% glycerol, and 10 mM DTT) was added to each sample before subjected to three freeze-thaw cycles by liquid nitrogen and 37 ºC water bath incubations to lyse the cells. Cell lysates were centrifuged at 13,000 rpm at 4 ºC for 10 min, and the supernatants were analyzed using SDS-PAGE and Western blotting.
Caco-2 cell permeability assay.
The cell permeability assay was performed at Charles River Labs to measure the rate of flux of a compound across polarized Caco-2 cell monolayers. Ranitidine, Talinolol, and Warfarin were used as controls. All compounds were tested at 10 μM with 2 hours incubation in triplicates. Papp (apparent permeability) in apical to basolateral (A-B) direction was calculated and used to predict the in vivo absorption of a compound. Papp > 1×10−6 cm/s is considered as higher permeability.
Combinatorial treatment of compound 7 and JQ1.
MOLM13 or MV4;11 cells were treated with DMSO, compound 7, JQ1 or combination of compounds 7 and JQ1 at indicated concentrations for 6 days. Cell viability was measured using the CellTiter-Glo luminescent cell viability assay kit (Promega). Survived cells were calculated as % relative to DMSO treated cells. Synergistic interactions were analyzed and visualized using the Combenefit software35.
Quantification and statistical analysis.
All AlphaScreen assays were performed in 4–6 replicates. Raw signal readings were normalized to no compound controls. GraphPad Prism 9 was used for regression analysis for IC50 determination.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by funds from NIH grants (GM135671 and CA252707) to T.G.K., the Cancer Prevention & Research Institute of Texas (RP160237) to X.S., Welch Foundation (A-1715), the Texas A&M X Grant Program, and the Texas A&M College of Science Strategic Transformative Research Program to W.R.L., and NIH grant (CA255506) and the Thomas and Garretta Newhof/Prein & Newhorf Research Fund to H. Wen.
ABBREVIATIONS USED
- AF9
ALL1-fused gene from chromosome 9 protein
- BET
the Bromodomain and extra-terminal domain
- CETSA
cellular thermal shift assay
- CSP
chemical shift perturbations
- DOT1L
disruptor of telomeric silencing 1 like
- ENL
eleven-nineteen-leukemia protein
- GAS41
glioma-amplified sequence 41
- GI50
half growth inhibition concentration
- HTS
high-throughput screening
- MLL
mixed-lineage leukemia
- PTM
Post-translational modification
- SEC
super elongation complex
- SEM
standard errors of the mean
- SPR
surface plasmon resonance
- YEATS domain
Yaf9, ENL, AF9, Taf14, and Sas5 domain
- YEATS2
YEATS domain-containing protein 2
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Overview of screen assay development, IC50 of top compounds, SPR and NMR data, cell growth inhibition, cell permeability assay, CETSA result of 7 in HeLa, qRT-PCR data, synergistic effect of 7 and JQ1 in MV4;11, molecular docking models of compounds bound to AF9 and ENL YEATS domains, HPLC traces for lead compounds, and table of structure and IC50 of compounds from HTS with IC50 below 5 μM (PDF), Molecular formula strings (CSV) and 3D PDB files for docking models.
REFERENCES
- 1.Jenuwein T; Allis CD, Translating the histone code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD; Allis CD, The language of covalent histone modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 3.Dawson MA; Prinjha RK; Dittmann A; Giotopoulos G; Bantscheff M; Chan WI; Robson SC; Chung CW; Hopf C; Savitski MM; Huthmacher C; Gudgin E; Lugo D; Beinke S; Chapman TD; Roberts EJ; Soden PE; Auger KR; Mirguet O; Doehner K; Delwel R; Burnett AK; Jeffrey P; Drewes G; Lee K; Huntly BJ; Kouzarides T, Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE, Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhalluin C; Carlson JE; Zeng L; He C; Aggarwal AK; Zhou MM, Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [DOI] [PubMed] [Google Scholar]
- 6.Andrews FH; Shinsky SA; Shanle EK; Bridgers JB; Gest A; Tsun IK; Krajewski K; Shi X; Strahl BD; Kutateladze TG, The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol 2016, 12, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu CC; Shi J; Yuan C; Zhao D; Jiang S; Lyu J; Wang X; Li H; Wen H; Li W; Shi X, Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Genes Dev 2018, 32, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu CC; Zhao D; Shi J; Peng D; Guan H; Li Y; Huang Y; Wen H; Li W; Li H; Shi X, Gas41 links histone acetylation to H2A.Z deposition and maintenance of embryonic stem cell identity. Cell Discov 2018, 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein BJ; Ahmad S; Vann KR; Andrews FH; Mayo ZA; Bourriquen G; Bridgers JB; Zhang J; Strahl BD; Cote J; Kutateladze TG, Yaf9 subunit of the NuA4 and SWR1 complexes targets histone H3K27ac through its YEATS domain. Nucleic Acids Res 2018, 46, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y; Sabari BR; Panchenko T; Wen H; Zhao D; Guan H; Wan L; Huang H; Tang Z; Zhao Y; Roeder RG; Shi X; Allis CD; Li H, Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell 2016, 62, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y; Wen H; Xi Y; Tanaka K; Wang H; Peng D; Ren Y; Jin Q; Dent SY; Li W; Li H; Shi X, AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi W; Guan H; Lyu J; Zhao D; Xi Y; Jiang S; Andrews FH; Wang X; Gagea M; Wen H; Tora L; Dent SYR; Kutateladze TG; Li W; Li H; Shi X, YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat Commun 2017, 8, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shanle EK; Andrews FH; Meriesh H; McDaniel SL; Dronamraju R; DiFiore JV; Jha D; Wozniak GG; Bridgers JB; Kerschner JL; Krajewski K; Martin GM; Morrison AJ; Kutateladze TG; Strahl BD, Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes Dev 2015, 29, 1795–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wan L; Wen H; Li Y; Lyu J; Xi Y; Hoshii T; Joseph JK; Wang X; Loh YE; Erb MA; Souza AL; Bradner JE; Shen L; Li W; Li H; Allis CD; Armstrong SA; Shi X, ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q; Zeng L; Zhao C; Ju Y; Konuma T; Zhou MM, Structural insights into histone crotonyl-lysine recognition by the AF9 YEATS domain. Structure 2016, 24, 1606–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao D; Guan H; Zhao S; Mi W; Wen H; Li Y; Zhao Y; Allis CD; Shi X; Li H, YEATS2 is a selective histone crotonylation reader. Cell Res 2016, 26, 629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Masson I; Yu DY; Jensen K; Chevalier A; Courbeyrette R; Boulard Y; Smith MM; Mann C, Yaf9, a novel NuA4 histone acetyltransferase subunit, is required for the cellular response to spindle stress in yeast. Mol Cell Biol 2003, 23, 6086–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulze JM; Wang AY; Kobor MS, YEATS domain proteins: a diverse family with many links to chromatin modification and transcription. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire 2009, 87, 65–75. [DOI] [PubMed] [Google Scholar]
- 19.Schulze JM; Wang AY; Kobor MS, Reading chromatin Insights from yeast into YEATS domain structure and function. Epigenetics 2010, 5, 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas D; Milne TA; Basrur V; Kim J; ElenitobaJohnson KS; Allis CD; Roeder RG, Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A 2011, 108, 15751–15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He N; Chan CK; Sobhian B; Chou S; Xue Y; Liu M; Alber T; Benkirane M; Zhou Q, Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A 2011, 108, E636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erb MA; Scott TG; Li BE; Xie H; Paulk J; Seo HS; Souza A; Roberts JM; Dastjerdi S; Buckley DL; Sanjana NE; Shalem O; Nabet B; Zeid R; Offei-Addo NK; Dhe-Paganon S; Zhang F; Orkin SH; Winter GE; Bradner JE, Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadd S; Huff V; Walz AL; Ooms A; Armstrong AE; Gerhard DS; Smith MA; Auvil JMG; Meerzaman D; Chen QR; Hsu CH; Yan C; Nguyen C; Hu Y; Hermida LC; Davidsen T; Gesuwan P; Ma Y; Zong Z; Mungall AJ; Moore RA; Marra MA; Dome JS; Mullighan CG; Ma J; Wheeler DA; Hampton OA; Ross N; Gastier-Foster JM; Arold ST; Perlman EJ, A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 2017, 49, 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perlman EJ; Gadd S; Arold ST; Radhakrishnan A; Gerhard DS; Jennings L; Huff V; Guidry Auvil JM; Davidsen TM; Dome JS; Meerzaman D; Hsu CH; Nguyen C; Anderson J; Ma Y; Mungall AJ; Moore RA; Marra MA; Mullighan CG; Ma J; Wheeler DA; Hampton OA; Gastier-Foster JM; Ross N; Smith MA, MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 2015, 6, 10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan L; Chong S; Xuan F; Liang A; Cui X; Gates L; Carroll TS; Li Y; Feng L; Chen G; Wang SP; Ortiz MV; Daley SK; Wang X; Xuan H; Kentsis A; Muir TW; Roeder RG; Li H; Li W; Tjian R; Wen H; Allis CD, Impaired cell fate through gain-of-function mutations in a chromatin reader. Nature 2020, 577, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asiaban JN; Milosevich N; Chen E; Bishop TR; Wang J; Zhang Y; Ackerman CJ; Hampton EN; Young TS; Hull MV; Cravatt BF; Erb MA, Cell-Based ligand discovery for the ENL YEATS domain. ACS Chem Biol 2020, 15, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christott T; Bennett J; Coxon C; Monteiro O; Giroud C; Beke V; Felce SL; Gamble V; Gileadi C; Poda G; Al-Awar R; Farnie G; Fedorov O, Discovery of a selective inhibitor for the YEATS domains of ENL/AF9. SLAS Discov 2019, 24, 133–141. [DOI] [PubMed] [Google Scholar]
- 28.Heidenreich D; Moustakim M; Schmidt J; Merk D; Brennan PE; Fedorov O; Chaikuad A; Knapp S, Structure-based approach toward identification of inhibitory fragments for Eleven-Nineteen-Leukemia Protein (ENL). J Med Chem 2018, 61, 10929–10934. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y; Chen G; Li XM; Liu S; Tian G; Li Y; Li X; Li H; Li XD, Selective targeting of AF9 YEATS domain by cyclopeptide inhibitors with preorganized conformation. J Am Chem Soc 2020, 142, 21450–21459. [DOI] [PubMed] [Google Scholar]
- 30.Li X; Li XM; Jiang Y; Liu Z; Cui Y; Fung KY; van der Beelen SHE; Tian G; Wan L; Shi X; Allis CD; Li H; Li Y; Li XD, Structure-guided development of YEATS domain inhibitors by targeting pi-pi-pi stacking. Nat Chem Biol 2018, 14, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moustakim M; Christott T; Monteiro OP; Bennett J; Giroud C; Ward J; Rogers CM; Smith P; Panagakou I; Diaz-Saez L; Felce SL; Gamble V; Gileadi C; Halidi N; Heidenreich D; Chaikuad A; Knapp S; Huber KVM; Farnie G; Heer J; Manevski N; Poda G; Al-Awar R; Dixon DJ; Brennan PE; Fedorov O, Discovery of an MLLT1/3 YEATS domain chemical probe. Angew Chem Int Ed Engl 2018, 57, 16302–16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni X; Heidenreich D; Christott T; Bennett J; Moustakim M; Brennan PE; Fedorov O; Knapp S; Chaikuad A, Structural insights into interaction mechanisms of alternative piperazine-urea YEATS domain binders in MLLT1. ACS Med Chem Lett 2019, 10, 1661–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein BJ; Piao L; Xi Y; Rincon-Arano H; Rothbart SB; Peng D; Wen H; Larson C; Zhang X; Zheng X; Cortazar MA; Pena PV; Mangan A; Bentley DL; Strahl BD; Groudine M; Li W; Shi X; Kutateladze TG, The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep 2014, 6, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundback T; Nordlund P; Martinez Molina D, The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014, 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- 35.Di Veroli GY; Fornari C; Wang D; Mollard S; Bramhall JL; Richards FM; Jodrell DI, Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer U; Meltzer P; Meese E, Twelve amplified and expressed genes localized in a single domain in glioma. Hum Genet 1996, 98, 625–628. [DOI] [PubMed] [Google Scholar]
- 37.Pikor LA; Lockwood WW; Thu KL; Vucic EA; Chari R; Gazdar AF; Lam S; Lam WL, YEATS4 is a novel oncogene amplified in non-small cell lung cancer that regulates the p53 pathway. Cancer Res 2013, 73, 7301–7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmermann K; Ahrens K; Matthes S; Buerstedde JM; Stratling WH; Phi-van L, Targeted disruption of the GAS41 gene encoding a putative transcription factor indicates that GAS41 is essential for cell viability. J Biol Chem 2002, 277, 18626–18631. [DOI] [PubMed] [Google Scholar]
- 39.Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.