

Abstract
Mutations in the kinase LRRK2 and impaired endocytic trafficking are both implicated in the pathogenesis of Parkinson’s disease (PD). Expression of the PD-associated LRRK2 mutant in mouse dopaminergic neurons was shown to disrupt clathrin-mediated endocytic trafficking. Here, we explored the molecular mechanism linking LRRK2 to endocytosis and found that LRRK2 bound to and phosphorylated the µ1 subunit of the adaptor protein AP2 (AP2M1), a core component of the clathrin-mediated endocytic machinery. Analysis of human SH-SY5Y cells and mouse hippocampal neurons and tissues revealed that loss of LRRK2 abundance or kinase function resulted in decreased phosphorylation of AP2M1, which is required for the initial formation of clathrin-coated vesicles (CCVs). In contrast, overexpression of LRRK2 or expression of a Parkinson’s disease-associated gain-of-function mutant LRRK2 (G2019S) inhibited the uncoating of AP2M1 from CCVs at later stages and prevented new cycles of CCV formation. Thus, the abundance and activity of LRRK2 must be calibrated to ensure proper endocytosis. Dysregulated phosphorylation of AP2M1 from the brain but not thyroid tissues of LRRK2 knockout and G2019S-knockin mice suggests a tissue-specific regulatory mechanism of endocytosis. Furthermore, we found that LRRK2-dependent phosphorylation of AP2M1 mediated dopaminergic neurodegeneration in a Drosophila model of PD. Together, our findings provide a mechanistic link between LRRK2, AP2, and endocytosis in the pathogenesis of PD.
Introduction
Advances in genetics and multiple model systems have revealed an important role of intracellular trafficking defects in Parkinson’s disease (PD) (1–3). A systematic analysis of pathway-specific genetic risk factors revealed that endocytic membrane-trafficking pathway plays a major role in the risk of PD (2). Moreover, leucine rich repeat kinase 2 (LRRK2), the mutations in which are the most genetic causes of both familial and sporadic PD (4, 5), has been implicated in the vesicle trafficking including endocytosis (1, 3, 6, 7). Specific expression of the most prevalent LRRK2 mutation, G2019S, in dopaminergic neurons in the mouse brain induces a robust reduction in the number of synaptic vesicles and an accumulation of clathrin-coated vesicles (CCVs) at synapses, suggesting a clathrin-mediated endocytic (CME) trafficking defect in vivo (8). Despite these advances, it is currently unclear how LRRK2 regulates endocytosis or how the endocytic defects caused by LRRK2 mutations contribute to dopaminergic neurodegeneration.
Neuronal activity is highly reliant on efficient CME to retrieve and maintain synaptic vesicle proteins on the plasma membrane. Therefore, proper regulation of key proteins related to this process is critical for normal neuronal activity. The adaptor protein complex 2 (AP2) and clathrin constitute the major coat constituents in CME (9). AP2 is a heterotetrameric complex and is required for binding the clathrin coat to the membrane and recruiting cargoes into the pit. It consists of two large subunits (α and β2, also known as A1/2 and B1 respectively), a medium subunit (μ2, also known as M1), and a small subunit (σ2, also known as S1) (10–12). The large subunits bind to clathrin and recruit other accessory proteins in plasma membrane targeting. The M1 subunit interacts with cargo molecules, including transferrin (Tfn) receptor, and the S1 subunit appears to stabilize the AP2 complex (12, 13). Proper phosphorylation-dephosphorylation cycling of the AP2 complex has been implicated in synaptic vesicle recycling and is required for efficient endocytosis (13). Phosphorylation of AP2M1 is critical for its membrane association and, hence, initial clathrin-coated vesicle (CCV) formation. After CCV scission, AP2M1 dephosphorylation promotes its uncoating from CCVs, a critical process required for a new cycle of CCV formation. Notably, AP2M1 has been associated to PD risk (2). Here, using a yeast screen and analyses in neuronal cultures, mouse tissues, and animal PD models, we investigated how LRRK2 regulates endocytosis through interaction with the AP2 complex and, consequently, how LRRK2 mutation contributes to endocytic defects and the degeneration of dopaminergic neurons in PD.
Results
AP2M1 interacts with LRRK2.
To investigate how LRRK2 mutations mediate cell toxicity, we performed a genome-wide genetic screen using diploid-based synthetic lethality analysis (dSLA), performed as previously described (14–16), in which we identified deletion mutant strains of yeast that suppressed or enhanced LRRK2-induced toxicity. Deletion mutants of four AP2 subunits (apl3∆, apl1∆, apm4∆, aps2∆) rescued LRRK2-induced toxicity that was further confirmed with viability assays (Fig. 1A). Based on these results, we examined the physical interactions between LRRK2 and the four subunits of human AP2 (A2, B1, M1, and S1) by co-immunoprecipitation (co-IP) using overexpressed MYC-tagged LRRK2 and respective HA-tagged AP2 subunits in human embryonic kidney (HEK) 293T cells. LRRK2 preferentially pulled down AP2A2 and AP2M1 in cells (Fig. 1B). To further determine if LRRK2 directly interacts with AP2A2 and AP2M1 in an in vitro system, we purified GST-tagged recombinant AP2A2 and AP2M1 and incubated them with purified MYC-LRRK2. Antibody to MYC abundantly coimmunoprecipitated GST-AP2M1 but not GST-AP2A2, nor the negative control GST protein (Fig. 1C). Thus, we concluded that LRRK2 preferentially interacts with the AP2 subunit M1. We further examined whether the M1 and A2 subunits form a hemicomplex, wherein only M1 directly bound to LRRK2. Without M1, the A2 subunit did not bind to LRRK2 directly (Fig. 1C, and fig. S1), and adding M1 to an incubation of A2 and LRRK2 enabled both M1 and A2 to be immunoprecipitated with LRRK2 (fig. S1), suggesting that M1 and A2 indeed form a hemicomplex with only M1 directly binding to LRRK2. We then tested whether familial LRRK2 mutants differentially interacted with AP2M1. We found no significant difference in pulldown of wild-type (WT) LRRK2 with AP2M1 compared with that of the familial LRRK2 mutants R1441C (hereafter, RC), R1441G (RG), G2019S (GS), or I2020T (IT) (fig. S2). To examine whether LRRK2 interacts with AP2M1 in vivo in the mouse brain, we conducted co-IP of whole brain lysates from LRRK2 WT and knockout (KO) mice. Accordingly, endogenous LRRK2 pulled down endogenous AP2M1 in LRRK2 WT but not KO mice (Fig. 1D). To determine the domain of AP2M1 that interacts with LRRK2, we generated AP2M1 deletion mutants to generate each of its four structural domains according to SCOP (Structural Classification of Proteins): D1, D2, D3, and D4 (Fig. 1E, schematic). Only the D2 domain of AP2M1 interacted with LRRK2 (Fig. 1E). Reciprocally, the binding domain on LRRK2 that interacted with AP2M1 was determined using our available deletion mutation series spanning the entire LRRK2 proteins with different functional domains (17). We found that AP2M1 predominantly bound to the GTPase domain of LRRK2 (Fig. 1F). Together, the data suggest LRRK2 preferentially interacts with AP2M1 of the AP2 complex.
Figure 1. AP2M1 interacts with LRRK2.
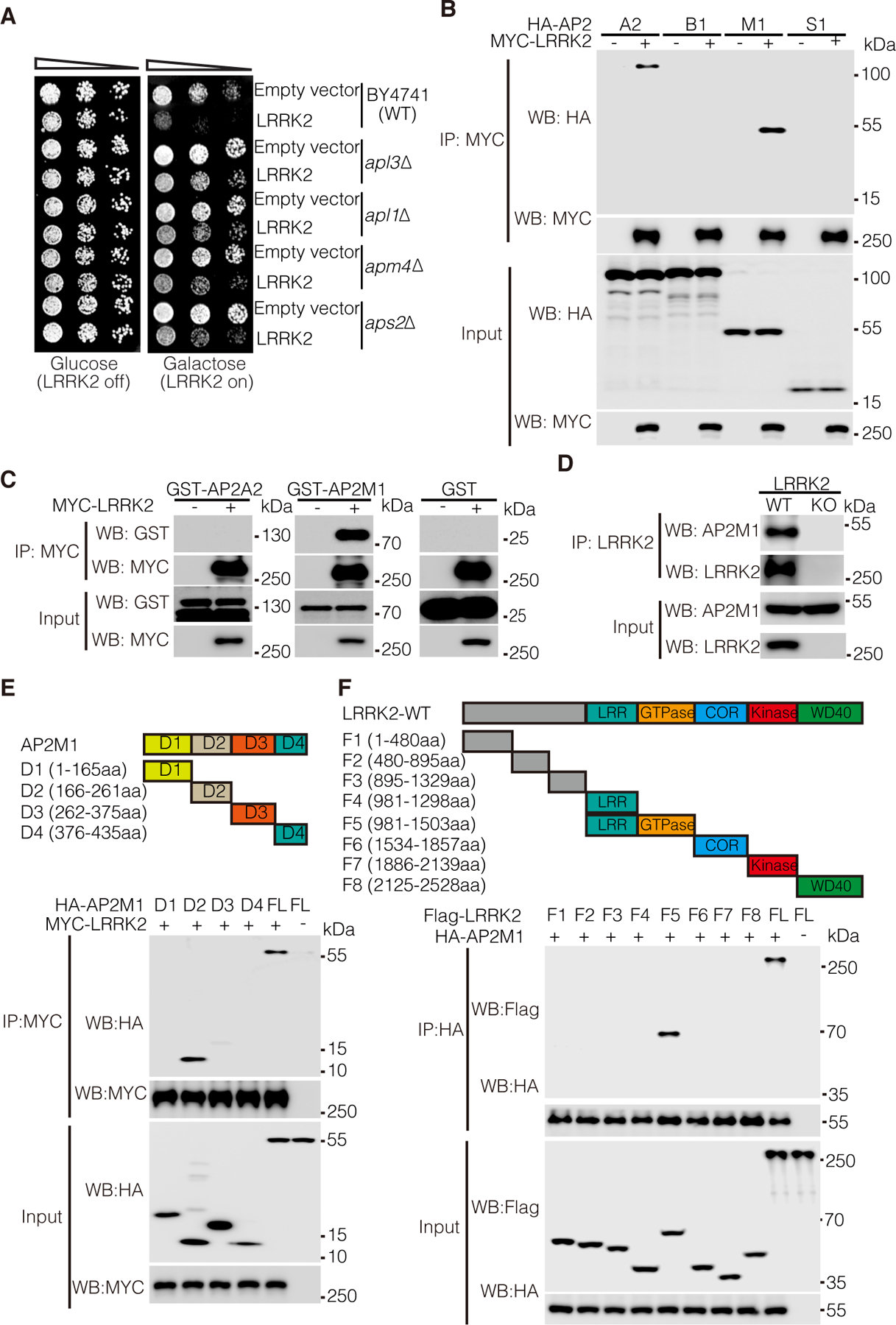
(A) Cell viability assay by cell number quantitation in response to expression of GTP-COR-Kin fragment of WT LRRK2 (pYES2-LRRK2-GCK) or empty vector in WT and AP2 subunit-deleted (apl3∆, apl1∆, apm4∆, aps2∆) BY4741 yeast strains. Cells were spotted onto media containing glucose (LRRK2 Off, repressed, left panel) or galactose (LRRK2 On, induced, right panel) and incubated at 30°C for 2 to 3 days. Shown are five-fold serial dilutions of yeast cells from left to right as indicated by graded open box. (B) Coimmunoprecipitation (co-IP) analysis for interaction between MYC-tagged LRRK2 and HA-tagged AP2 subunits (A2, B1, M1, or S1) in co-transfected HEK 293T cells. Co-IP with antibody to MYC was followed by immunoblotting for HA or MYC. (C) Co-IP analysis of the interactions between AP2 subunits (A2, M1) and LRRK2 using recombinant GST-tagged AP2M1 and A2 subunits and purified MYC-LRRK2. Co-IP with antibody to MYC was followed by immunoblotting for GST. GST protein was as a negative control. (D) Co-IP analysis of the interactions between AP2M1 and LRRK2 in mouse brain lysates. Whole brain lysates prepared form wild-type (WT) and LRRK2 knockout (KO) mice were subjected to co-IP with antibody to LRRK2 (Neuromab) followed by immunoblotting for LRRK2 and AP2M1. (E) Co-IP analysis of the interactions between MYC-LRRK2 and HA-tagged D1, D2, D3, D4 or full-length (FL) AP2M1 in co-transfected HEK 293T cells. Co-IP with antibody to MYC was followed by immunoblotting for HA or MYC. A schematic representation of AP2M1 D1, D2, D3, D4 domains is shown. (F) Co-IP analysis of the interactions between HA-tagged AP2M1 and Flag-tagged LRRK2 fragments in co-transfected HEK 293T cells. Co-IP with antibody to HA was followed by immunoblotting for Flag or HA. A schematic representation of the LRRK2 fragments is shown. Blots are representative of 3 experiments.
LRRK2 phosphorylates AP2M1 in vitro and in vivo.
LRRK2 mutations are the most common genetic causes of both familial and sporadic PD and LRRK2 kinase activity is highly relevant to PD pathogenesis. Notably, proper phosphorylation of AP2M1 is critical for its function in endocytosis. Thus, based on our observed strong interaction between LRRK2 and AP2M1, we asked whether LRRK2 phosphorylates AP2M1. To answer this question, we first performed an in vitro kinase assay with [32Pγ]-ATP using purified recombinant proteins of LRRK2 and AP2M1. We found that a kinase-active G2019S (GS) LRRK2 mutant, but not a kinase-deficient D1994A (DA) LRRK2 mutant, phosphorylated AP2M1 in vitro (Fig. 2A). A known LRRK2 kinase substrate Rab8a was included as a positive control. To determine the phosphorylation sites, we further subjected the phosphorylated AP2M1 by LRRK2 to mass spectrometry (MS) analysis and identified threonine at residue 156 (Thr156) of AP2M1 as the major residue phosphorylated by LRRK2 (fig. S3). Accordingly, we generated a phospho-deficient AP2M1-T156A (TA) mutant. Using a phospho-specific antibody against AP2M1-Thr156 in an in vitro kinase assay, we further confirmed that WT LRRK2 phosphorylates WT but not TA-mutant AP2M1 (Fig. 2, B and C). Consistent with the greater kinase activity of LRRK2 G2019S, the AP2M1 Thr156 phosphorylation was two-fold greater with LRRK2 G2019S as compared to LRRK2 WT control (Fig. 2, B and C). Moreover, the kinase-deficient LRRK2 mutant did not induce AP2M1 phosphorylation in our in vitro kinase assay (Fig. 2, B and C). These data confirmed that LRRK2 phosphorylates AP2M1 at Thr156 in vitro. To confirm our in vitro findings, we investigated the cellular phosphorylation of AP2M1 by overexpressing AP2M1 with hyperactive and kinase dead LRRK2, respectively, in SH-SY5Y cells. Consistent with our in vitro results, we found that LRRK2 hyperactive G2019S, but not LRRK2 kinase dead mutant D1994A, significantly increased AP2M1 cellular phosphorylation level (Fig. 2, D and E). To further extend our findings to more physiological conditions, we examined the effect of LRRK2 on the phosphorylation of endogenous AP2M1. We first treated SH-SY5Y cells with MLi-2, a known specific LRRK2 kinase inhibitor, at different concentrations. As expected, MLi-2 largely inhibited LRRK2 kinase activity, as evidenced by abolished LRRK2 S935 phosphorylation. Consistently, endogenous AP2M1 Thr156 phosphorylation level decreased to about 70% upon MLi-2 treatment at 0.2 μM compared to the control (Fig. 2, F and G), suggesting LRRK2 regulates AP2M1 phosphorylation at endogenous level. Next, we generated human neuroblastoma SH-SY5Y cell lines with depletion of LRRK2 using the CRISPR/Cas9 editing system. The resulting LRRK2 KO cells led to no detectable LRRK2 protein (fig. S4). While endogenous AP2M1 Thr156 phosphorylation was detected in both WT and LRRK2 KO cells, the phosphorylation level was much lower in LRRK2 KO cells than in WT cells (Fig. 2, H and I). To further confirm that the reduced AP2M1 Thr156 phosphorylation is specific due to LRRK2 knockout, we performed a rescue experiment by transfecting WT LRRK2 back into LRRK2 KO cells and examined the resulting AP2M1 phosphorylation. Indeed, we found that WT LRRK2 transfection completely rescued AP2M1 phosphorylation to a level of about twofold that of non-transfection LRRK2 KO cells (Fig. 2, J and K), suggesting the specificity of LRRK2 on AP2M1 phosphorylation. Prompted by the above results, we next examined endogenous AP2M1 phosphorylation levels in LRRK2 WT, KO and G2019S knock-in (GSKI) mouse brains. We found that the levels of endogenous AP2M1 phosphorylation were significantly lower in LRRK2 KO brain lysate compared to WT brain lysates while the levels of endogenous AP2M1 phosphorylation were significantly higher in LRRK2 GSKI brain lysate compared to WT brain lysates (Fig. 2, L and M). Taken together, we concluded that LRRK2 phosphorylates AP2M1 at Thr156 both in vitro in cell cultures and in vivo in mouse brains.
Figure 2. LRRK2 phosphorylates AP2M1 in vitro and in vivo.
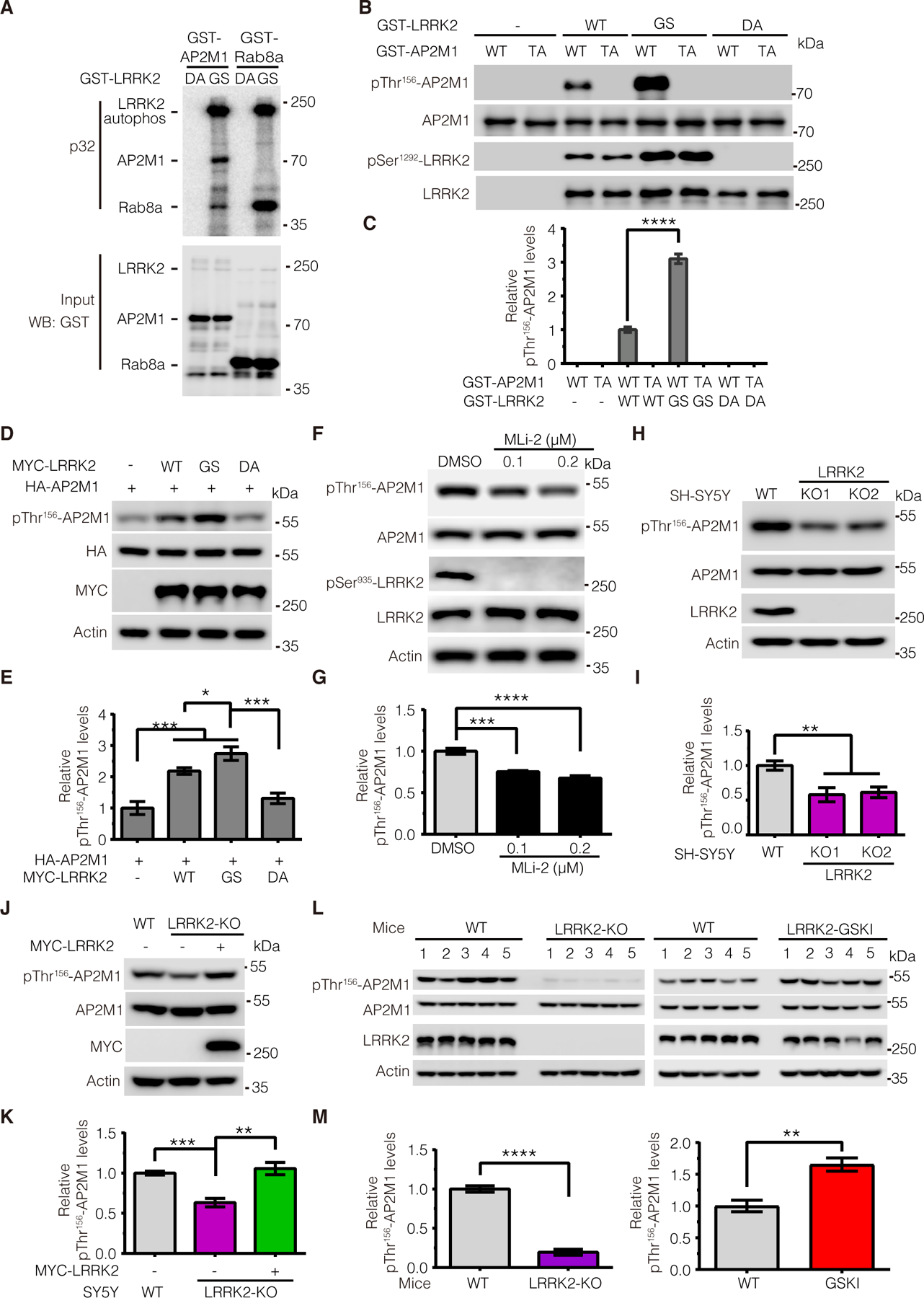
(A) In vitro kinase assays assessing the amount of 32P-ATP incorporated by GST-tagged AP2M1 upon incubation with recombinant GST-tagged LRRK2 protein—either the PD-associated G2019S (GS) mutant or the kinase-deficient D1994A (DA; 970–2527aa) mutant. Rab8a was as a positive control. (B and C) In vitro kinase assays assessing phosphorylation of recombinant GST-tagged WT or phospho-deficient T156A (TA) mutant AP2M1 by recombinant GST-tagged WT, GS mutant, or DA mutant LRRK2. Immunoblots were performed with antibody to pThr156-AP2M1. (D and E) Immunoblotting analysis of AP2M1 phosphorylation at Thr156 upon overexpression of WT, GS mutant, or DA-mutant LRRK2. (F and G) Immunoblotting analysis of endogenous AP2M1 phosphorylation level at Thr156 upon MLi-2 inhibitor treatment. Lysates from SH-SY5Y cells treated with MLi-2 (0.1 or 0.2 μM for 2 hours) were subjected to immunoblotting with antibodies to pThr156-AP2M1 and pSer935-LRRK2. (H and I) Immunoblotting analysis of endogenous AP2M1 phosphorylation at Thr156 in WT and LRRK2-KO SH-SY5Y cells. (J and K) Immunoblotting analysis of endogenous AP2M1 phosphorylation at Thr156 upon overexpression of MYC-LRRK2 in LRRK2 KO cells. (L and M) Immunoblotting analysis of endogenous AP2M1 phosphorylation at Thr156 in brain tissue from WT, LRRK2 knockout (KO), and LRRK2 G2019S-knock-in (GSKI) mice. Images were quantified by ImageJ software. All quantification data are mean ± SEM from n=5 independent experiments or mice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Student’s t tests for two comparisons or one-way ANOVA followed by a Tukey’s post hoc test for multiple comparisons.
Tissue-specific role of LRRK2 on phosphorylation of AP2M1.
It has been reported that AP2M1 is phosphorylated by AP2-associated protein kinase 1 (AAK1) at Thr156 (18). We therefore investigated the regulatory relationship between LRRK2 and AAK1 on AP2M1 phosphorylation. To this end, we first performed in vitro kinase assay to compare the amount of 32P-ATP incorporated by AP2M1 upon equimolar amounts of LRRK2 and AAK1 (Fig. 3, A and B). Our results showed that AAK1 was more potent than LRRK2 in phosphorylating AP2M1 in vitro. To test whether LRRK2 and AAK1 have synergistic effects on AP2M1 phosphorylation, we examined AP2M1 phosphorylation levels upon LRRK2 inhibition with MLi-2, AAK1 inhibition with LP-935509, or combined inhibition with MLi-2 and LP-935509. We found that while either MLi-2 or LP-935509 treatment reduced endogenous AP2M1 phosphorylation, treatment with both MLi-2 and LP-935509 had a greater effect on reducing AP2M1 phosphorylation (Fig. 3, C and D), suggesting an additive effect between LRRK2 and AAK1 on AP2M1 phosphorylation. Thus, we next explored whether the regulation of LRRK2 and AAK1 on AP2M1 is interdependent. To this end, we first overexpressed AAK1 in LRRK2 WT and KO SH-SY5Y cells and determined the endogenous AP2M1 phosphorylation level. We found that AAK1 increased the amount of endogenous AP2M1 phosphorylation in both LRRK2 WT and KO cells (Fig. 3, E and F), suggesting that AAK1 can phosphorylate AP2M1 independently of LRRK2. We noticed that LRRK2 KO cells exhibited a lower level of endogenous AP2M1 phosphorylation than did WT cells with or without AAK1 overexpression (Fig. 3, E and F), suggesting LRRK2 contributes a major part of endogenous AP2M1 phosphorylation. To investigate whether the effect of LRRK2 on AP2M1 phosphorylation relies on AAK1, we knocked down AAK1 expression in LRRK2 WT and KO cells. While the total phosphorylation of AP2M1 was decreased upon AAK1 knockdown, AP2M1 phosphorylation in LRRK2 WT cells was more than that in LRRK2 KO cells, suggesting endogenous LRRK2 still phosphorylates AP2M1 despite AAK1 knockdown (Fig. 3, G and H). Therefore, this suggests an independent regulation of AP2M1 phosphorylation by LRRK2. Notably, we failed to obtain AAK1 knockout cell lines even with multiple rounds of efforts using the CRISPR/Cas9 editing system, and shRNAs could only knock down about half of AAK1 abundance, suggesting an essential role of AAK1 in SH-SY5Y cells.
Figure 3. LRRK2 and AAK1 play an independent and tissue-specific role on phosphorylation of AP2M1.
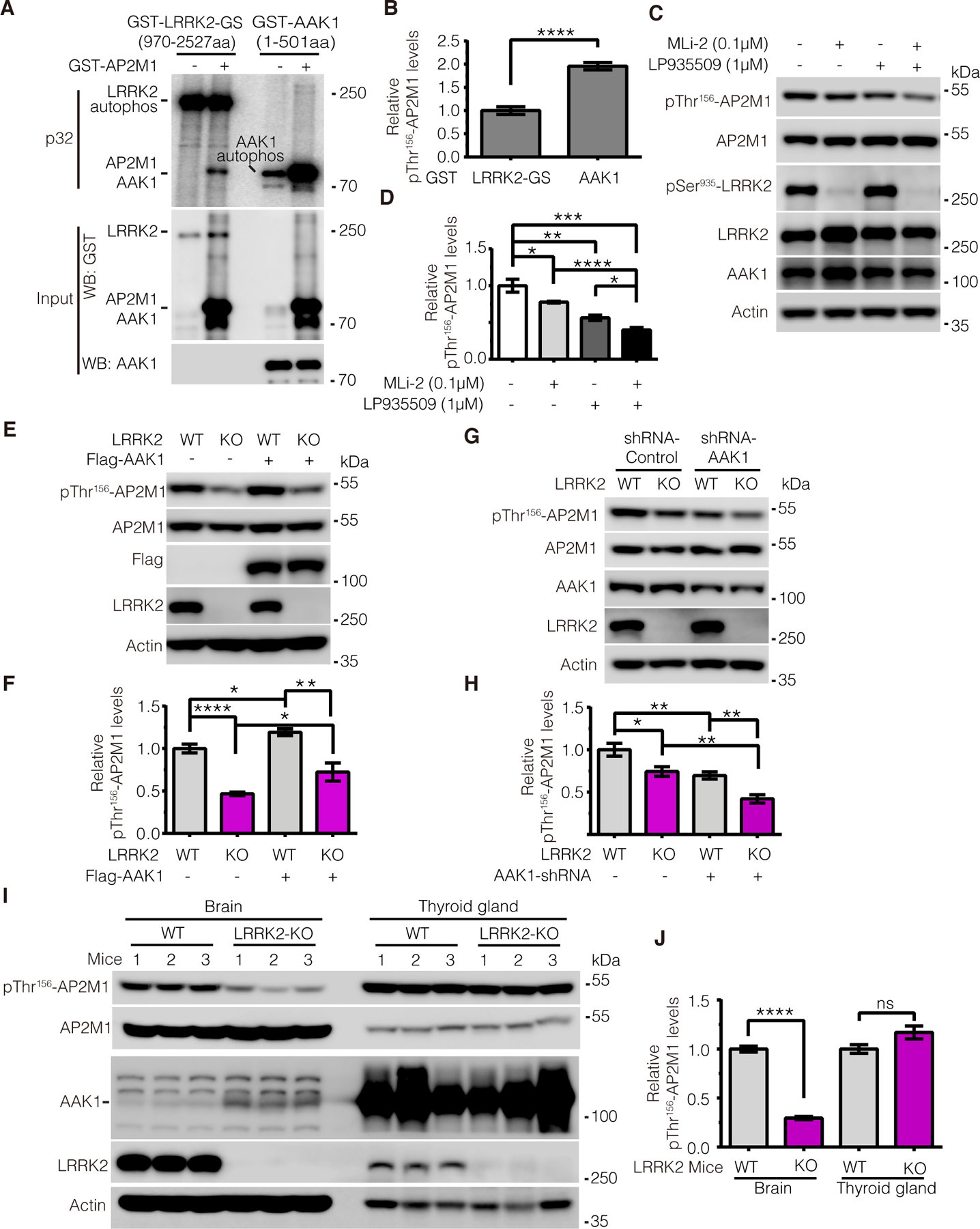
(A and B) In vitro kinase assays assessing the amount of 32P-ATP incorporated by AP2M1 upon equimolar amounts of GST-tagged LRRK2 G2019S (GS) (970–2527aa) and GST-tagged AAK1 (1–501aa). Note that because GST-AAK1 (1–501aa) fragment and GST-AP2M1 have similar molecular weights, the signal in the lane of GST-AAK1 with AP2M1 includes both AAK1 autophosphorylation and AP2M1 phosphorylation by AAK1; thus, to calculate AP2M1 phosphorylation by AAK1, the intensity was subtracted by the AAK1 autophosphorylation signal. (C and D) Immunoblotting analysis of endogenous AP2M1 phosphorylation at Thr156 upon LRRK2 inhibition with MLi-2 and AAK1 inhibition with LP-935509 (0.1 and 1 μM, respectively, for 2 hours). Immunoblotting was performed with antibodies to pThr156-AP2M1 and pSer935-LRRK2. (E and F) Immunoblotting analysis of endogenous AP2M1 Thr156 phosphorylation in LRRK2 WT and KO SH-SY5Y cells with overexpression of AAK1. (G and H) Immunoblotting analysis of endogenous AP2M1 Thr156 phosphorylation in LRRK2 WT or KO SH-SY5Y cells upon transfection of AAK1 shRNA. (I and J) Immunoblotting analysis of endogenous AP2M1 Thr156 phosphorylation in brain tissue and thyroid gland lysates from LRRK2 WT or KO mice. All quantification data in (A to J) are mean ± SEM from n=5 independent experiments; each experiment in (I and J) consisted of 3 WT and 3 KO mice. Blots were quantified by ImageJ software. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Student’s t tests for two comparisons or one-way ANOVA followed by a Tukey’s post hoc test for multiple comparisons.
We next asked the physiological relevance of AP2M1 phosphorylation by LRRK2 versus AAK1. LRRK2 is known for its relatively high expression in the brain, where the PD pathologies are observed. Notably, we found that unlike LRRK2, the expression of AAK1 was relatively low in mouse brain tissue (Fig. 3, I and J). In contrast, AAK1 was highly expressed in the mouse thyroid gland where LRRK2 expression was relatively low. The differential expression of LRRK2 and AAK1 observed in the brain versus the thyroid gland in mice prompted us to ask whether LRRK2 and AAK1 have tissue-specific roles in regulating AP2M1 phosphorylation. Indeed, knocking out the expression of LRRK2 in the mouse brain markedly decreased AP2M1 phosphorylation, suggesting LRRK2 plays a key role in regulating AP2M1 phosphorylation in brain. However, knocking out LRRK2 had no obvious effects on AP2M1 phosphorylation in the thyroid gland where AAK1 is dominantly expressed, suggesting a minor role of LRRK2 on AP2M1 phosphorylation in thyroid gland (Fig. 3, I and J). Notably, we noticed a mild increase of AAK1 protein level in LRRK2-knockout brains (Fig. 3I), suggesting a feedback regulation between LRRK2 and AAK1 protein levels may exist. Together, our results suggest an AAK1-independent and tissue-specific role of LRRK2 on AP2M1 phosphorylation.
LRRK2 phosphorylation of AP2M1 promotes AP2M1 membrane association.
Having established a phosphorylation regulation of AP2M1 by LRRK2, we next asked the physiological function of this regulation. It is known that phosphorylation of AP2M1 enhances AP2 recruitment to the plasma membrane during coated pit assembly (19). We therefore asked whether phosphorylation of AP2M1 by LRRK2 regulates AP2M1 membrane association. To minimize the effect of endogenous LRRK2 on AP2M1, we overexpressed MYC-LRRK2 and HA-AP2M1 in LRRK2 KO SY5Y cells, and subsequently fractionated these cells into cytosol and membrane fractions and determined the abundance of soluble or membrane-associated AP2M1 by Western blot (WB). AP2M1 phospho-deficient mutant AP2M1-T156A was introduced as a control. Our results showed that LRRK2-GS, the kinase active mutant, potentiated AP2M1-WT membrane localization but not AP2M1-T156A mutant (Fig. 4A and B). To further investigate whether the phosphorylation of endogenous AP2M1 by endogenous LRRK2 regulates AP2M1 membrane association, we treated the cells with LRRK2 inhibitor MLi-2 and fractionated these cells into cytosol and membrane fractions and determined the abundance of soluble or membrane-associated AP2M1 by Western blotting. Treatment of cells with MLi-2 decreased the membrane association of endogenous AP2M1 (Fig. 4, C and D). To further confirm this finding, we used two additional approaches involving microscopy (Fig. 4, E to H, and data file S1). First, we used an established liquid nitrogen coverslip freeze-thaw method (20, 21) to deplete cytosolic proteins and fix the membrane fraction on the coverslip, enabling us to further examine the localization of membrane-associated AP2M1. LRRK2 KO SY5Y cells were transfected with DsRed-LRRK2 WT, GS or kinase dead mutant DA and transduced with lentiviruses carrying eGFP-AP2M1-WT or T156A. Forty-eight hours after transfection, cells were permeabilized by liquid nitrogen freeze–thawing to deplete cytosol and then fixed and visualized by confocal microscopy. Membrane associated AP2M1-WT, but not AP2M1-T156A, was significantly increased with LRRK2 WT or GS co-expression (Fig. 4, E and G). The second approach we used was total internal reflection fluorescence (TIRF) microscopy. Consistently, WT and GS-mutant LRRK2 significantly increased the membrane association of WT but not T156A AP2M1(Fig. 4, F and H, and fig. S5). Together, these results suggest that LRRK2 phosphorylation of AP2M1 promotes AP2M1 membrane association.
Figure 4. LRRK2 phosphorylation of AP2M1 increases AP2M1 membrane association.

(A and B) Cellular fractionation assays of AP2M1 intensity on membrane and cytosol fractions in LRRK2 KO SH-SY5Y cells transfected with HA-AP2M1-WT or T156A (TA) with or without MYC-LRRK2-GS. After 48 hours, cells were harvested and fractionated into cytosol and membrane fractions. Data are mean ± SEM from n=5 independent experiments. *P < 0.05 by Student’s t tests. (C and D) Cellular fractionation assays of AP2M1 intensity on membrane and cytosol fractions upon LRRK2 inhibitor MLi-2 treatment. Data are mean ± SEM, n=3 independent experiments. *P < 0.05 by Student’s t tests. (E) Liquid nitrogen coverslip freeze–thaw methods assessing AP2M1 membrane association. LRRK2 KO SH-SY5Y cells were transfected with DsRed-LRRK2 WT, GS, or DA and transduced with lentiviruses carrying eGFP-AP2M1 WT or TA. After 48 hours, cells were permeabilized by liquid nitrogen freeze–thaw to deplete cytosol and then fixed and visualized by confocal microscopy. Scale bars, 20 μm. (F) TIRF microscopy assessing AP2M1 membrane association. LRRK2 KO SH-SY5Y cells were cotransfected with DsRed-LRRK2 WT, GS, or DA and eGFP-AP2M1 WT or TA at a plasmid ratio of 5:1. After 48 hours, cells were fixed and imaged by TIRF microscopy. Scale bars, 20 μm. (G and H) Quantification of AP2M1 intensity on the membrane by the methods described and results represented in panels (E) and (F), respectively. Images were quantified by ImageJ software. Data are mean ± SEM from three independent experiments, with the means from the n ≥ 10 cells quantified in each experiment (data file S1) shown in the triangles. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 by one-way ANOVA followed by a Tukey’s post hoc test.
LRRK2 phosphorylation of AP2M1 mediates LRRK2-induced endocytosis defects in neurons.
Next, we explored the functional consequence of increased AP2M1 membrane association by LRRK2 phosphorylation. AP2M1 phosphorylation specifically enhances its association with cargos such as transferrin (Tfn) receptor, which in turn, mediates endocytosis (22, 23). Therefore, we monitored endocytic trafficking using Tfn-594 dye internalization. To study in a more physiological condition, we examined the endocytosis in wildtype, LRRK2 KO and GSKI neurons by monitoring Tfn internalization. Surface Tfn was removed by acid stripping. The internalized Tfn in neuronal cell bodies was monitored (Fig. 5). Overexpression of AP2M1-WT or T156A in wildtype neurons induced decreased Tfn internalization, indicating a defect in endocytosis. Both LRRK2 GSKI and KO neurons also induced defects in Tfn internalization (Fig. 5, and data file S1). Notably, Tfn internalization defects in LRRK2 GSKI neurons were enhanced by overexpression of AP2M1-WT but not AP2M1-TA mutant (Fig. 5, A and C). However, no significant difference was observed when AP2M1-WT or T156A was overexpressed in LRRK2 KO neurons (Fig. 5, A and C). This suggests that increased AP2M1 phosphorylation by LRRK2 induces more severe defects in endocytosis. To further examine if LRRK2 phosphorylation impairs AP2M1 mediated endocytosis, we inhibited endogenous LRRK2 kinase activity in primary neurons by MLi-2 treatment and examined the subsequent endocytosis of neurons overexpressing AP2M1-WT or T156A (Fig. 5, B and D). Indeed, inhibition of LRRK2 kinase rescued AP2M1-WT mediated endocytic defects (Fig. 5, B and D). Together, these data suggest LRRK2 phosphorylation of AP2M1 mediates LRRK2-induced endocytosis defects in neurons.
Figure 5. LRRK2 phosphorylation of AP2M1 mediates LRRK2 induced endocytosis defects in neurons.
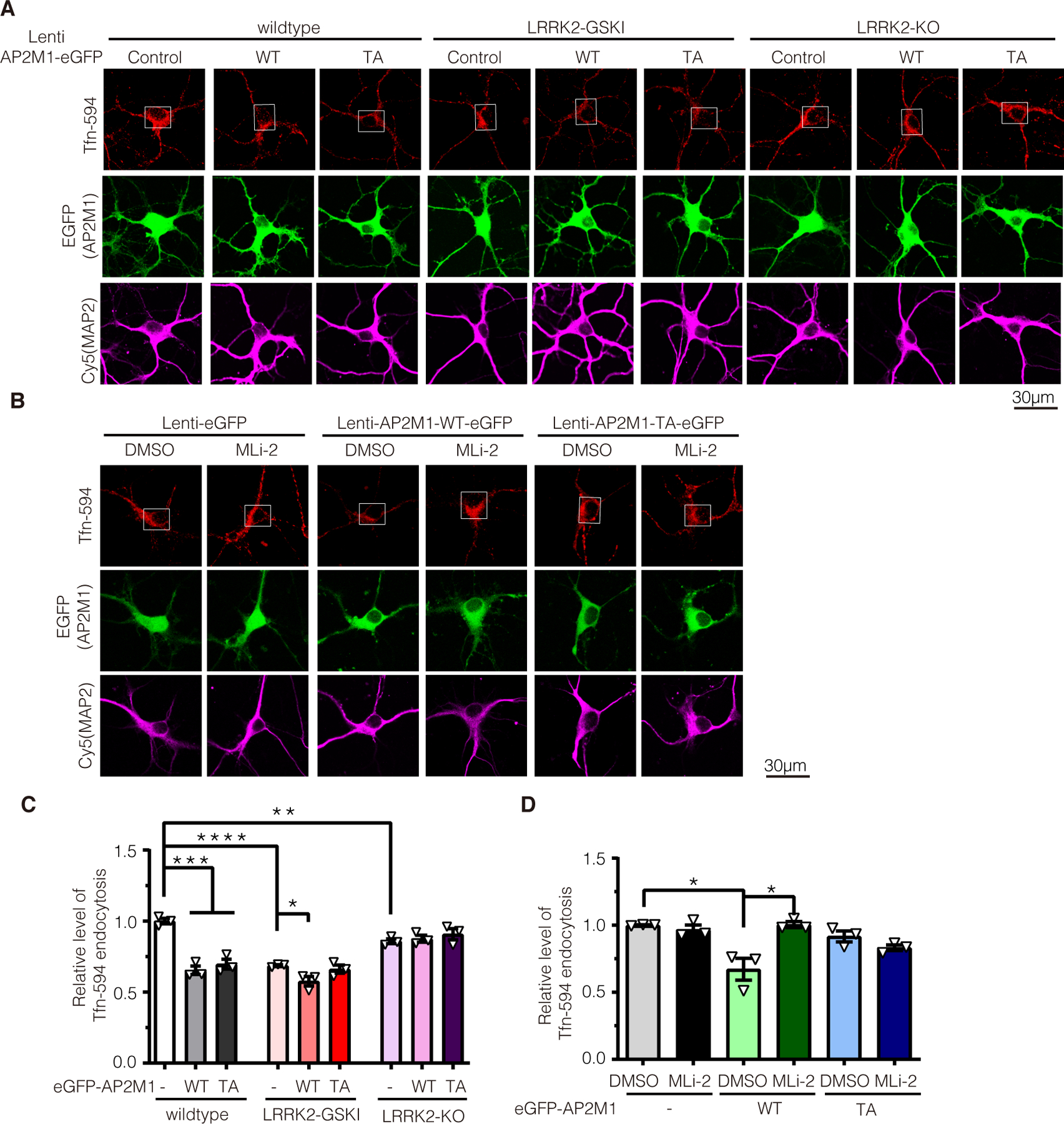
(A) Lentiviruses carrying eGFP-AP2M1 WT or T156A (TA) were transduced into LRRK2 WT, KO or GSKI hippocampal neurons, and Tfn internalization was monitored. (B) Lentiviruses carrying eGFP-AP2M1 WT or T156A (TA) were transduced into LRRK2 GSKI hippocampal neurons pretreated with 0.1 μM MLi-2 for 2 hours, and Tfn internalization was monitored. Scale bars, 30 μm. (C and D) Quantification of Tfn internalization at the cell bodies (indicated in white boxes) represented and described in panels (A) and (B), respectively. Data are means ± SEM from three independent experiments, with the means from the n ≥ 15 cells (C) and n ≥ 40 cells (D) quantified in each experiment (data file S1) indicated by the triangles. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Student’s t tests for two comparisons or one-way ANOVA followed by a Tukey’s post hoc test.
Excessive LRRK2 kinase activity inhibits AP2M1 uncoating.
How do both LRRK2 KO and GSKI induce endocytic defects? One explanation could be that LRRK2 regulates AP2M1 phosphorylation and dephosphorylation cycle. Phosphorylation of AP2M1 enhances AP2 recruitment to the plasma membrane during initial coated pit assembly (19). After coated pits are assembled, coated pits will be scissored and endocytosed into cytosol. Following this process, AP2M1 needs to be dephosphorylated and uncoated from the pits, and then recycled back to the plasma membrane for a new cycle of coated pit endocytosis (13). Thus, we hypothesized that LRRK2 KO could impair basal level of AP2M1 phosphorylation for initial coated pit assembly, while excessive LRRK2 kinase activity could inhibit AP2M1 dephosphorylation, which in turn may inhibit AP2M1 uncoating from the pits. To test this possibility, we examined the effects of GS-mutant and DA-mutant LRRK2 on AP2M1 uncoating using an in vitro uncoating assay (24). Purified CCVs from mouse brains (Fig. 6, A to C) were incubated with purified GST-tagged GS or DA LRRK2 in the presence of an ATP-regenerating system including ATP and Hsc70, GST protein was used as a control, and AP2M1 presence on CCV pellets was analyzed by Western blotting. The amount of AP2M1 on CCV indicates the amount of AP2M1 that was not successfully uncoated and released. LRRK2 GS retained more AP2M1 on CCV pellets (Fig. 6, D and E), indicating an inhibition of AP2M1 uncoating. These data are consistent with our previous study by transmission electron microscopy (TEM), which showed that specific expression of LRRK2 GS in dopaminergic neurons in the mouse brain induces a robust reduction in the number of synaptic vesicles and an accumulation of CCVs at synapses in vivo (8).
Figure 6. Excessive LRRK2 kinase activity inhibits AP2M1 uncoating.
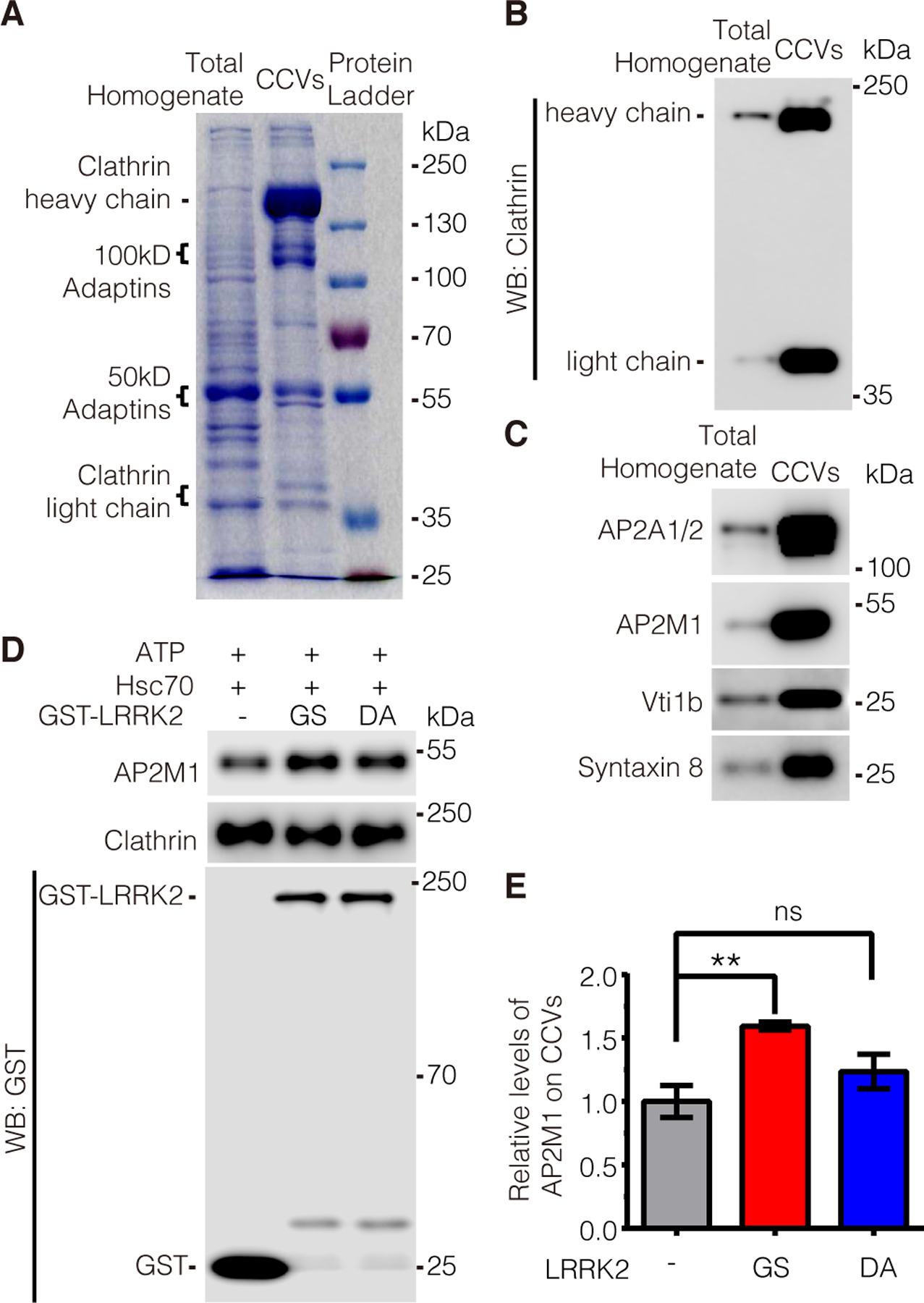
(A) Protein profile of the total homogenates and CCVs purified from mouse brains after SDS-PAGE and Coomassie blue staining. (B and C) Characterization of purified CCVs by immunoblotting with antibodies to clathrin heavy and light chains (B) and with antibodies to CCV-enriched proteins as indicated (C). (D and E) Immunoblotting analysis of uncoated AP2M1 from CCVs. Purified CCVs from mouse brains were incubated with purified GST-LRRK2 GS or DA in the presence of an ATP-regenerating system including ATP and Hsc70. Images was quantified by ImageJ software. Data are the means ± SEM, n=5 independent experiments, with CCVs purified from brain tissue homogenates from 10 mice in each experiment. **P < 0.01 by one-way ANOVA followed by a Tukey’s post hoc test.
LRRK2 regulated AP2M1 phosphorylation modulates LRRK2-induced dopaminergic neurodegeneration in vivo.
To determine whether AP2M1 phosphorylation by LRRK2 modulates LRRK2 dopaminergic neurodegeneration in vivo, we used a LRRK2 Drosophila model in which flies express human WT or GS-mutant LRRK2. We also generated transgenic flies carrying UAS-AP2M1-WT and T156A. The transgenic flies were crossed with dopa decarboxylase (Ddc)-Gal4>UAS-GFP driver flies to achieve specific expression of transgene in dopaminergic and serotonin neurons. Dopaminergic neuron number as revealed by GFP fluorescence was monitored in four major dopaminergic neuronal clusters in Drosophila adult brain (PPM1/2, PPM3, and PPL1) (Fig. 7, A and B) (29). Consistent with previous LRRK2 Drosophila models (30), our LRRK2 WT and GS flies exhibited substantial dopaminergic neurodegeneration in the major dopaminergic clusters (Fig. 7, C to E) and total dopaminergic neurons (Fig. 7F). Overexpression of AP2M1-WT or T156A alone also induced dopaminergic neurodegeneration in the major dopaminergic clusters (Fig. 7, C to E) and total dopaminergic neurons (Fig. 7F). Notably, WT AP2M1 enhanced both LRRK2 WT- and GS–induced dopaminergic neurodegeneration, as indicated by the decreased total number of dopaminergic neurons, whereas AP2M1-T156A had little effect (Fig. 7F).
Figure 7. LRRK2 phosphorylation of AP2M1 modulates LRRK2-induced dopaminergic neurodegeneration in vivo.
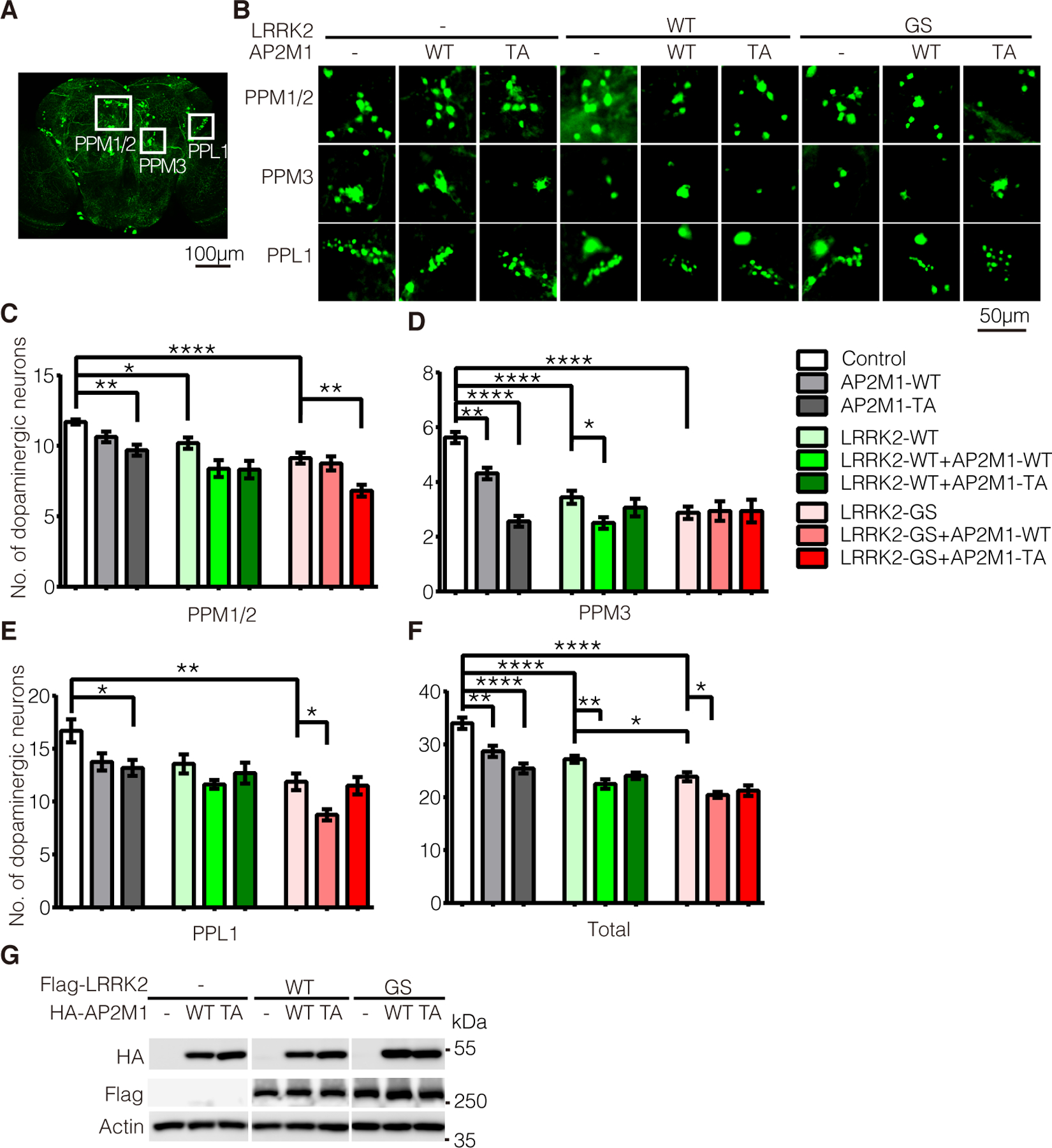
(A) Diagram of dopaminergic neuronal clusters (PPM1/2, PPM3, PPL1) in the posterior areas of the adult fly brain. Scale bars, 100 μm. (B) Representative confocal images (GFP) of dopamine neurons in each dopaminergic cluster from 7-week-old flies of the indicated genotypes. Scale bars, 50 μm. (C to E) Quantification of dopamine neurons per dopaminergic cluster in 7-weeks old flies of the indicated genotypes. (F) Total numbers of dopamine neurons in four major dopaminergic clusters of the flies at 7-weeks old. Data are the means ± SEM, n=8 flies per genotype, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by one-way ANOVA followed by a Tukey’s post hoc test. (G) Levels of overexpressed LRRK2 and AP2M1 in flies driven by GMR-Gal4. Lysates prepared form whole heads of one-week old flies of the indicated genotypes were subjected to immunoblotting with antibodies to HA-HRP, Flag-HRP and fly actin. Blots are representative of n=3 independent experiments.
Discussion
The major finding of this study is that LRRK2 interacts with and directly regulates the phosphorylation cycle of AP2M1, one of the major coat constituents in CME and recently implicated in PD risk (2), leading to the dysregulation of CME and dopaminergic neurodegeneration. We demonstrated that either knockout or excessive LRRK2 kinase activity induces endocytic defects, which is caused by LRRK2-induced abnormal AP2M1 phosphorylation cycle. These findings are consistent with a model in which knockout of LRRK2 decreases the basal level of AP2M1 phosphorylation (which is required for the initial CCV formation), while excessive LRRK2 kinase activity inhibits AP2M1 dephosphorylation after CCV scission and results in inhibition of AP2M1 uncoating for the new cycle of CCV formation (Fig. 8). Too much AP2M1 also impairs endocytosis. This might be mainly due to a dominant negative effect given that phosphorylation and dephosphorylation cycle of AP2M1 is tightly regulated during the endocytosis. Further, AP2M1-WT but not the T156A mutant enhanced LRRK2 WT- and GS–induced dopaminergic neurodegeneration in vivo. This suggests that LRRK2 phosphorylation of AP2M1 mediates LRRK2-induced dopaminergic neurodegeneration in vivo and indicates that AP2M1 phosphorylation by LRRK2 plays an important role in modulation of mutant LRRK2 pathogenesis.
Figure 8. Model of LRRK2 regulation of AP2M1 phosphorylation cycles to mediate endocytosis.
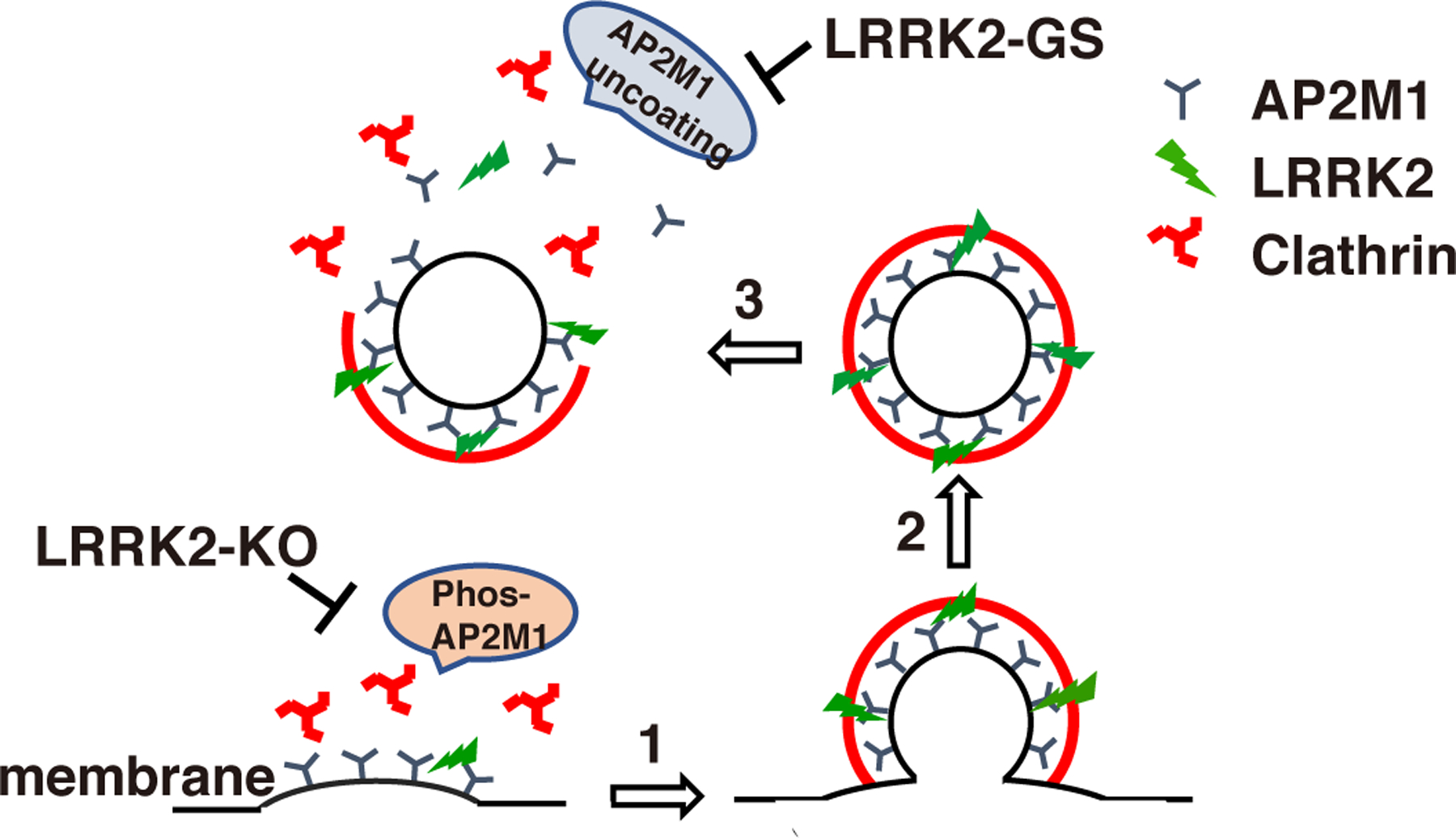
Endogenous LRRK2 phosphorylates AP2M1 at Thr156 at plasma membrane, increases AP2M1 membrane association and, in turn, promotes the initiation of CCV formation (“1”). Knockout of LRRK2 inhibits this initial step. After CCV formation and subsequent scission (“2”), AP2M1 dephosphorylation promotes its uncoating from CCVs, a critical process required for the new cycle of CCV formation (“3”). However, overexpression of LRRK2 or excessive LRRK2 kinase activity such as LRRK2 GS mutant inhibits AP2M1 dephosphorylation after CCV scission and results in inhibition of AP2M1 uncoating for the new cycle of CCV formation.
A recent systematic analysis of pathway-specific genetic risk factors revealed that the endocytic membrane trafficking pathway plays a major role in the risk of PD (2). Of note, AP2M1 is one of the risk genes among the most contributors of endosomal membrane-trafficking pathway to the risk of PD (2). Sequential screenings have also nominated LRRK2 as a regulator for clathrin-mediated endocytosis and have reported an initial functional interaction between LRRK2 and different AP2 subunits (31). However, the biological process and the molecular mechanism underlying how AP2M1 involves in neurodegeneration and PD pathogenesis are unknown. Our study provides an attractive mechanistic link that AP2M1-mediating endocytosis is regulated through phosphorylation at Thr156 by LRRK2, the most commonly mutated protein of both sporadic and familial PD, leading to dopaminergic neurodegeneration. We and others previously reported that ArfGAP1 acts as a GTPase-activating protein (GAP) and also a kinase substrate for LRRK2 to regulate neuronal toxicity in vitro and in vivo (17, 32). ArfGAP1 was reported to promote AP2-dependent endocytosis (33). It would be interesting to test whether LRRK2, ArfGAP1 and AP2 form a protein complex to regulate vesicular endocytosis and mediate neurodegeneration.
Mutations in LRRK2 have been demonstrated to cause defects in endocytosis (4, 5). Specific expression of the most prevalent LRRK2 mutation, G2019S, in dopaminergic neurons in mouse brain induces a robust reduction in the number of synaptic vesicle and an accumulation of CCVs at synapses, indicating a CME trafficking defect in vivo (8). But how LRRK2 mediates endocytosis is largely unclear. LRRK2 has been reported to interact with and phosphorylate several endocytic proteins, including endophilin A1 (EndoA) (34–36), synaptojanin (SYNJ1) (37, 38), auxilin (DNAJC6) (39), and Rabs (20, 40–44). These proteins and AP2M1 perform distinct functions at different steps in the endocytic pathway, raising the possibility that LRRK2 acts as a regulatory kinase for multiple proteins at endocytic trafficking and LRRK2-induced neuronal toxicity could be a result of a combination of several aspects of endocytic trafficking defects. Interestingly, SYNJ1, auxilin, and AP2M1 are also risk factors for PD and are linked to CME (2, 45–51).
LRRK2 phosphorylates AP2M1 at Thr156, the site that has been reported to be phosphorylated by a known kinase, adaptor-associated kinase 1 (AAK1) (18, 52, 53). Interestingly, our study suggests that LRRK2 and AAK1 have independent roles on AP2M1 Thr156 phosphorylation. Importantly, our study also revealed a tissue-specific role of LRRK2 on AP2M1 phosphorylation, suggesting potential pathophysiologic relevance of AP2M1 phosphorylation by LRRK2. Notably, a single nucleotide polymorphism in an intron of the AAK1 gene has been associated with the age of onset of PD (54, 55), further suggesting impaired AP2M1 phosphorylation at Thr156 may be associated with PD and that maintaining proper phosphorylation cycle of AP2M1 may play an important role in PD pathogenesis.
Collectively, we show that LRRK2 acts as a kinase for AP2M1 and regulates the phosphorylation cycle of AP2M1, a major player in CME and a newly implicated factor in PD risk, leading to defects in CME and in turn dopaminergic neurodegeneration. This regulation is tissue specific. Our study provides a detailed molecular mechanism and further supports the importance of endocytic pathway in PD pathology.
Materials and methods
Animals.
LRRK2 GSKI mice were purchased from Taconic (13940) (56) and LRRK2 KO mice were purchased from JAX Laboratory (16121) (57). Mice were housed and treated in accordance with the National Institutes of Health (NIH) ‘Guide for the Care and Use of Laboratory Animals’ and Institutional Animal Care and Use Committees of Kansas State University and University of Connecticut School of Medicine. Animals were housed in a 12-hour dark and light cycle with free access to water and food.
Plasmids.
Entry clones carrying AP2A2, AP2B1, AP2M1, AP2S1, AAK1 full-length cDNA were obtained from DNASU (clones HsCD00042465, HsCD00042299, HsCD00041654, HsCD00399704, HsCD00718566). Full-length human AP2A2, AP2B1, AP2M1, and AP2S1 were cloned by Gateway technology into the mammalian expression vector pcDNA3.1–3HA or 3Flag-DEST, which was generating by replacing a V5 tag with a 3HA tag or a 3Flag tag from pcDNA3.1-nV5-DEST vector (Invitrogen), and E. coli expression vector pDEST15 with GST tag Gateway Vector (Invitrogen). MYC-LRRK2-WT was a gift from Dr. Ted Dawson (Addgene plasmid # 17609, http://n2t.net/addgene:17609; RRID: Addgene_17609) (58). pENTR221 LRRK2 WT was a gift from Michael J Fox Foundation MJFF (Addgene plasmid # 39529). Full-length human LRRK2 and AP2M1, were cloned by Gateway technology into the mammalian expression vector pcDNA3.1-eGFP-DEST or pcDNA3.1-DsRed-DEST, which was generating by replacing a V5 tag with an eGFP or DsRed (Clontech) tag from pcDNA3.1-nV5-DEST vector (Invitrogen). To generate AP2M1 or LRRK2 single or multiple mutants, site-directed mutagenesis were carried out using the In-Fusion HD Cloning Plus (Clontech). AP2M1 domain fragments (D1, D2, D3, D4) were cloned into pcDNA3.1 vector with a N-terminal HA tag. Truncated mutants for human LRRK2 were cloned into the mammalian expression vector pcDNA3.1 with three flag tags at N-terminus as described previously (17). Full-length AP2M1 WT and T156A were cloned into Drosophila Gateway expression vector pUAST vector with an N-terminal HA tag (Drosophila Genomics Resource Center, Bloomington, IN). Full-length AP2M1 WT and T156A were cloned into pFUGW lentiviral vector with a C-terminal eGFP tag. All cDNAs or mutation sites were confirmed by DNA sequencing analysis.
Antibodies and reagents.
Rabbit monoclonal antibody to AP2M1 (Ab759976), rabbit monoclonal antibody to p-Thr156 AP2M1 (ab109397), rabbit monoclonal antibody to p-Ser1292-LRRK2 (ab203181), rabbit polyclonal antibody to GFP, rabbit polyclonal antibody to GST-HRP (ab3416) were obtained from Abcam. Mouse monoclonal antibody to LRRK2 (clone N138/6) was from UC Davis/NIH NeuroMab facility. Rabbit monoclonal antibody to clathrin heavy chain (CHC, 4796) and mouse monoclonal antibody to E-cadherin (4A2, 14472) were obtained from Cell Signalling Technology. Mouse monoclonal antibody to AAK1 was obtained from Santa Cruz Biotechnology (sc-134242). Rabbit polyclonal antibodies to syntaxin 8 (110083) and Vti1b (164002) were obtained from Synaptic Systems. Mouse monoclonal antibody to LAMP1 was obtained from Developmental Studies Hybridoma Bank (clone H4A3, DSHB). Mouse monoclonal antibody to V5 antibody (R96025) was purchased from Thermo Fisher. Mouse monoclonal antibodies to MYC, V5-HRP, Flag, Flag-HRP, HA, HA-HRP, and actin were obtained from Sigma-Aldrich. HRP-linked antibodies to rabbit or mouse IgG were obtained from Jackson Immuno Research Labs. AlexaFluor-488–conjugated antibodies to mouse or rabbit IgG, cyanine5 (CY5)-conjugated antibody to mouse IgG, and AlexaFluor-594–conjugated Tfn were obtained from Molecular Probes (Thermo Fisher).
Yeast strains and genetic procedures.
Yeast haploid WT strain BY4741 (YSC1048), deletion mutants of yeast AP2 four subunits (apl3∆, YSC6273–201930327; apl1∆, YSC6273–201937758; apm4∆, YSC6273–201922921; aps2∆, YSC6273–201937795) at genetic background (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) were obtained from Dharmacon (Horizon Discovery). Transformations of yeast were performed using a standard high efficiency lithium acetate procedure (59). Yeast cells carrying galactose-inducible expression constructs containing the central GTP-COR-Kin fragment of WT LRRK2 (pYES2-LRRK2-GCK, (60)) or empty vector were grown in synthetic complete media lacking uracil (SC-URA) containing glucose (2% dextrose) to repress the GAL1 promoter, or in medium containing 2% galactose (to induce the GAL1 promoter) to enable the induction of expression.
Yeast cell viability assays (spotting assays).
WT BY4741 yeast cells and deletion mutants of yeast AP2 four subunits carrying galactose-inducible LRRK2 expression constructs were grown overnight at 30°C in liquid media (SC-URA) containing raffinose to log phase, followed by growth in media containing galactose for a further 6 hrs. Cultures were then normalized for OD 600nm, 5-fold serially diluted and spotted onto plates containing solid media (SC-URA) with either glucose or galactose as the sole carbon source. Cells were grown at 30°C for at least 2 days before imaging.
Yeast genome-wide genetic screen.
The yeast LRRK2 toxicity modifier screen was performed using diploid-based synthetic lethality analysis (dSLA) as previously described (14). Briefly, we transformed pYES2-LRRK2-GCK plasmid into a heterozygous deletion diploid Magic Marker collection including about 6000 genes (15, 16). The transformants carrying LRRK2 plasmid were sporulated and then underwent selection for haploid mutants that also harbored the LRRK2 plasmid on solid media containing G418 and canavanine but lacking uracil [Magic Medium, (15)]. Haploid deletion mutants carrying LRRK2 plasmid were grown on selectable media containing glucose to suppress LRRK2 expression or on selectable media containing galactose to induce LRRK2 expression. After comparing colony sizes on galactose plates to those on glucose plates, the clones that suppressed or enhanced LRRK2 toxicity were identified. Initial hits from the screen were identified by PCR and sequencing. Each hit was then individually verified by fresh transformations and spotting assays.
Cell culture, transfections, and coimmunoprecipitation.
SH-SY5Y cells were cultured in DMEM medium supplemented with 10% fetal bovine serum. Transient transfection with MYC-LRRK2 and HA-AP2A2/B1/M1/S1 or HA-AP2M1 truncated fragments, or HA-AP2M1 with Flag-LRRK2 truncated fragments was carried out using LipoD293 (Signagen) as per the manufacturer’s introductions. After 48 hours, cells were washed by PBS once, lysed in immunoprecipitation (IP) buffer [1% Triton X-100, 0.5% NP40, 150 mM NaCl, 20 mM HEPES, pH 7.4, 1 mM EGTA, 1 × Complete mini protease inhibitor cocktail (Pierce)] by rotation at 4 °C for 1 hour. Cell lysates were centrifuged at 15,000 rpm for 15 mins. Supernatants were incubated with protein-G Dyna beads (Bio-Rad) pre-coated with antibodies to MYC or HA followed by overnight rotation at 4 °C. The Dyna beads were pelleted and stringently washed five times with IP buffer supplemented with 500 mM NaCl. The immunoprecipitated proteins were resolved on SDS-PAGE and subjected to immunoblotting.
In vitro kinase assay.
An in vitro kinase assay was performed as previously described (17, 28). GST-tagged AP2M1 WT and T156A were expressed in E. coli BL21 and purified by Glutathione beads as per the manufacturer’s instructions (GE healthcare). The purity of the GST-tagged proteins was assessed with denaturing SDS-PAGE followed by Coomassie blue staining. Equimolar amounts of GST-LRRK2-WT, -GS, or -DA recombinant proteins (970–2527aa) (Thermofisher PV4873, PV4881, and PV6051 respectively, concentration provided), GST-AAK1 (1–501aa) (Thermofisher A30967, concentration provided) with or without GST-AP2M1-WT or T156A protein were subjected to a kinase reaction in kinase buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EGTA pH 8.0, 20 mM β-glycerol phosphate, 10 μM ATP, 0.5 μCi γ- 32P-ATP, and 20 mM MgCl2). The reactions were incubated at 30 °C for 20 mins, put on ice, resolved on SDS-PAGE. LRRK2 autophosphorylation and AP2M1 phosphorylation were imaged using a Typhoon Phosphoimager. Input levels of protein were determined by immunoblotting of a GST-HRP antibody. The reactions without radioisotope were subjected to mass spectrometric analysis to identify AP2M1 phosphorylation sites by LRRK2.
Mass spectrometric analysis.
Mass spectrometry analysis of the sites of AP2M1 phosphorylation was performed by the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, MA, USA).
Generation of LRRK2 knockout (KO) SH-SY5Y cell line by the CRISPR/Cas9 system.
The gRNA (GAGTCCAAGACGATCAACAG) that target exon 2 of human LRRK2 genomic sequence was subcloned into plasmid pSpCas9(BB)-2A-Puro(PX459) V2.0 (a gift from Feng Zhang, Addgene, Plasmid #62988, http://n2t.net/addgene:62988; RRID:Addgene_62988) according to previously described protocol (61). SH-SY5Y cells were transfected with LRRK2 gRNA plasmids. After 48 hours, single cells were plated and maintained in a 96-well plate. Deletion of LRRK2 was verified by Western blot with LRRK2-specific antibody and sequencing confirmation (fig. S4). Multiple LRRK2 KO SH-SY5Y cell lines were selected.
Subcellular fractionation.
Cells were collected in fractionation buffer (20 mM HEPES pH=7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 × Complete mini protease inhibitor cocktail (Pierce)), incubated on ice for 15 mins following by passing through a 27-gauge needle 10 times and leaving on ice for 20 mins. The lysates were centrifuged at 8000g for 5 min. The resulting supernatant, which are the cytoplasm and membrane fractions, was further centrifuged in an ultracentrifuge at 100,000 g for 2 hours to produce the pellet (membrane fraction) and the supernatant (cytosol fraction).
Liquid nitrogen coverslip freeze-thaw protocol.
Liquid nitrogen coverslip freeze-thaw protocol to deplete cytosol was performed as previously described (21). Briefly, cells were plated on poly-L-ornithine coated coverslips, transfected with indicated DsRed-LRRK2 plasmids and transduced with lentiviruses carrying eGFP-AP2M1 WT or T156A. After 48 hours, cells were chilled on ice, washed twice with ice-cold PBS, and incubated in ice-cold glutamate buffer (25 mM KCl, 25 mM HEPES, pH 7.4, 2.5 mM magnesium acetate, 5 mM EGTA, and 150 mM potassium glutamate). The coverslip was then dipped in liquid nitrogen for 5 s and allowed to thaw for few seconds, followed by gentle washes with ice-cold glutamate buffer and rehydration for 5 mins in ice-cold PBS. Cells were then fixed in cold 4% paraformaldehyde (PFA) for 20 mins, washed, and mounted onto slides using Vectashield mounting medium (Vector Laboratories). Imaging was conducted on a double-blind basis using a Zeiss confocal 880 microscope (Carl Zeiss). The images were selected at random. The intensity of membrane-associated fluorescence was quantified by ImageJ software (NIH) (62). A minimum of 10 cells was quantified.
Primary neuronal cultures.
Primary hippocampal neurons from postnatal (1-day old) pups (28, 60). Briefly, hippocampus tissue was dissected and dissociated by trypsin (Invitrogen). The cells were seeded into 24-well plates pre-coated with poly-L-ornithine and were maintained in Neurobasal-A medium (Invitrogen) supplemented with B27 supplement and L-glutamine. The glial cells were inhibited by adding 5-fluoro-20-deoxyuridine (5F2DU, 30 µM, Sigma) at 4 days in vitro (DIV 4).
Transferrin (Tfn) uptake Assay.
Transferrin uptake assay was performed as previously described (63). Briefly, mouse primary hippocampal neurons at DIV 5 were transduced with the indicated lentiviruses for 48 or 72 hours. The cells were then washed once with Neurobasal-A medium and incubated for 1 hour at 37 °C in Neurobasal-A medium without serum before incubated with 20 ug/mL Alexa Fluor 594–conjugated transferrin (Molecular Probes) for 16 mins at 37 °C. The cells were chilled on ice, washed once with ice-cold PBS, incubated with ice-cold acid wash buffer (0.2 M acetic acid, 0.5 M NaCl) for 5 mins on ice, then washed once with PBS and fixed with 4% PFA. Imaging was conducted on a double-blind base on a Zeiss confocal 880 microscope (Carl Zeiss). The images were selected at random. The intensity of internalized Tfn-594 fluorescence in neuronal cell body was quantified by ImageJ software. A minimum of 10 cells was quantified. For those groups with a bigger variation, a larger sample size (more cells in each group) were quantified to increase confidence in the results.
Isolation of clathrin-coated vesicles.
CCVs were isolated as previously described (64, 65). Briefly, the homogenates from 10 whole mouse brains were centrifuged at 19,000g for 40mins. The resulting supernatant was centrifuged at 43,000g for 60 mins. The pellet containing the CCVs was resuspended in an equal volume of 12.5% Ficoll and 12.5% sucrose and centrifuged at 43,000g for 40min. The supernatant was then pelleted by centrifugation at 88,000 g for 1 hour at 4 °C. The pellet was resuspended in 4 ml homogenization buffer (100 mM MES, pH 6.5, 0.5 mM MgCl2, 1 mM EGTA, 1 mM DTT, and 0.1 mM PMSF) and layered over 6 ml of 8 % sucrose-D2O solution in the homogenization buffer and centrifuged at 100,000 g for 2 hours at 4 °C. The resulting pellet containing CCVs was collected, washed, and resuspended in the homogenization buffer. The suspension was further centrifuged at 20,000 g for 10 mins at 4 °C and the resulting supernatant containing CCVs was collected, snap frozen in aliquots and stored at −80 °C for uncoating assays.
Uncoating assays
Uncoating assays were performed as previously described (24, 64). In brief, 5 to 6 μg of the above-described purified CCVs were incubated with recombinant proteins GST-LRRK2 G2019S or D1994A, or GST protein as a control, in an ATP-regenerating system (1.3 μg Hsc70, 0.8 mM ATP, 5 mM of creatine phosphate, and 0.2 IU of creatine phosphokinase, 10 mM ammonium sulfate, 20 mM HEPES, pH 7.0, 2 mM magnesium acetate, and 25 mM KCl) in a final volume of 50 μl. The mixture was incubated for 10 mins at 25 °C, put on ice, centrifuged at 100,000 g for 20 mins at 4 °C. Pellets were resolved by SDS-PAGE and coated AP2M1 on CCVs was analyzed by Western blot.
Preparations of lentiviral eGFP-tagged AP2M1-WT or AP2M1-TA.
The second-generation lentiviral packaging system was employed to produce high-titer lentiviruses. Briefly, pFUGW-AP2M1-WT-eGFP and pFUGW-AP2M1-T156A-eGFP lentiviral plasmids were transfected into HEK 293FT cells along with viral packaging plasmids (psPAX2 and pMD2.G). After 48 hours, the culture media were collected, and viral particles were precipitated by centrifugation at 35,000 g for 2 hours. Viral particles were resuspended into serum free medium and stored at −80 °C.
Immunocytochemistry.
Neurons transfected with indicated plasmids were fixed in 4% PFA for 20 mins, washed with PBS three times, permeabilized and blocked for 1 hour with PBS containing 5% goat serum and 0.3% Triton X-100. The neurons were then incubated with mouse anti-MAP2 antibody at 1: 1000 dilution at 4 °C overnight. After washing with PBS three times, the neurons were incubated with goat anti-mouse IgG conjugated to CY5 at room temperature for 2 hours. The coverslips were mounted onto slides using Vectashield mounting medium (Vector Laboratories). The imaging was conducted on a Zeiss confocal 880 microscope with Zen black software or a Zeiss Automatic stage microscope with Zen blue software.
TIRF microscopy.
TIRF imaging experiments were performed on an inverted wide-field microscope operated in the TIRF mode as described previously in more detail (66). Briefly, cells were plated on poly-L-ornithine coated coverslips, transfected with indicated DsRed-LRRK2 and eGFP-AP2M1 plasmids. After 48 hours, cells were fixed in 4% PFA for 20 mins, washed and mounted onto slides using Vectashield mounting medium (Vector Laboratories). Imaging was conducted on a TIRF microscope on a double-blind base. The images were selected at random. The intensity of membrane-associated fluorescence was quantified by ImageJ software (NIH). A minimum of 10 cells was quantified.
Drosophila genetics.
Ddc-GAL4 (Stock No. 7010) and UAS-GFP (Stock No. 5430) fly lines were obtained from Bloomington Stock Center. pUAST-HA-AP2M1, T156A, and pUAST-attB-LRRK2 WT and G2019S were microinjected into Drosophila embryos (BestGene Inc.). The insertion site is at 99F8 for pUAST-attB plasmid injection. The transgenic AP2M1 were crossed with flies carrying LRRK2 different forms. The resulting bigenic flies were crossed with Ddc-GAL4>UAS-GFP, which induces the co-expression of GFP, AP2M1 and LRRK2 in dopamine and serotonin neurons.
Statistical analysis.
Statistical analysis was performed with Prism 6.0 software (GraphPad). One-way ANOVA followed by a Tukey’s post hoc test was used for data analysis for multiple comparisons and Student’s t tests (unpaired, two-tailed) was used for two comparisons as described in the figure legends. Data represent mean ± SEM, and P ≤ 0.05 was considered statistically significant: *P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgements:
The authors acknowledge Confocal Core and Molecular Biological Core supported by CVM-KSU. The authors thank Dr. Ping Li of the Department of Chemistry (KSU) for assistance on MS data analysis. The authors thank Dr. Xiuqin Bai at Eastern Washington University for consulting on using the appropriate statistical tests. J.B.-G. is currently affiliated with the Virginia Commonwealth University.
Funding:
This work was supported, in part, by grants from NIH/NINDS R01 NS112506, NIH/NIA K01 AG046366 award, Parkinson’s Foundation Stanley Fahn Junior Faculty award PF-JFA-1934, American Parkinson Disease Association (APDA) Research Grant, NIH/NIGMS P20 GM113109 pilot grant, SUCCESS-FYI Intramural Grant from Kansas State University College of Veterinary Medicine, UConn Startup fund.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Some of the materials require material transfer agreements with a third party: Entry clones carrying AP2A2, AP2B1, AP2M1 and AP2S1 full-length cDNA are available from DNASU under a master MTA with Arizona State University; MYC-LRRK2-WT is available from Addgene under a MTA with Johns Hopkins University; pENTR221-LRRK2-WT is available from Addgene under a MTA with Michael J. Fox foundation; pSpCas9(BB)-2A-Puro(PX459) V2.0 is available from Addgene under a MTA with Broad Institute; and the Drosophila Gateway expression vector pUAST vector with an N-terminal HA tag is available from Drosophila Genomics Resource Center (Bloomington, IN), which is under a MTA with Carnegie Institution of Washington.
References and Notes
- 1.Abeliovich A, Gitler AD, Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, Billingsley K, Vitale D, Blauwendraat C, Gibbs JR, Pihlstrom L, Gan-Or Z., Consortium International Parkinson’s Disease Genomics, M. R. Cookson, M. A. Nalls, A. B. Singleton, The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov Disord 34, 460–468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connor-Robson N, Booth H, Martin JG, Gao B, Li K, Doig N, Vowles J, Browne C, Klinger L, Juhasz P, Klein C, Cowley SA, Bolam P, Hirst W, Wade-Martins R., An integrated transcriptomics and proteomics analysis reveals functional endocytic dysregulation caused by mutations in LRRK2. Neurobiol Dis 127, 512–526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lees AJ, Hardy J, Revesz T., Parkinson’s disease. Lancet 373, 2055–2066 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Cookson MR, The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci 11, 791–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiong Y, Dawson TM, Dawson VL, Models of LRRK2-Associated Parkinson’s Disease. Adv Neurobiol 14, 163–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong Y, Yu J., Modeling Parkinson’s Disease in Drosophila: What Have We Learned for Dominant Traits? Front Neurol 9, 228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong Y, Neifert S, Karuppagounder SS, Liu Q, Stankowski JN, Lee BD, Ko HS, Lee Y, Grima JC, Mao X, Jiang H, Kang SU, Swing DA, Iacovitti L, Tessarollo L, Dawson TM, Dawson VL, Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc Natl Acad Sci U S A 115, 1635–1640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ, Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Conner SD, Schmid SL, Differential requirements for AP-2 in clathrin-mediated endocytosis. J Cell Biol 162, 773–779 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Traub LM, Downs MA, Westrich JL, Fremont DH, Crystal structure of the alpha appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc Natl Acad Sci U S A 96, 8907–8912 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE, Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol 17, 517–568 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Smythe E., Regulating the clathrin-coated vesicle cycle by AP2 subunit phosphorylation. Trends Cell Biol 12, 352–354 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Meluh PB, Pan X, Yuan DS, Tiffany C, Chen O, Sookhai-Mahadeo S, Wang X, Peyser BD, Irizarry R, Spencer FA, Boeke JD, Analysis of genetic interactions on a genome-wide scale in budding yeast: diploid-based synthetic lethality analysis by microarray. Methods Mol Biol 416, 221–247 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Pan X, Yuan DS, Ooi SL, Wang X, Sookhai-Mahadeo S, Meluh P, Boeke JD, dSLAM analysis of genome-wide genetic interactions in Saccharomyces cerevisiae. Methods 41, 206–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan X, Yuan DS, Xiang D, Wang X, Sookhai-Mahadeo S, Bader JS, Hieter P, Spencer F, Boeke JD, A robust toolkit for functional profiling of the yeast genome. Mol Cell 16, 487–496 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Xiong Y, Yuan C, Chen R, Dawson TM, Dawson VL, ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J Neurosci 32, 3877–3886 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricotta D, Conner SD, Schmid SL, von Figura K, Honing S., Phosphorylation of the AP2 mu subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol 156, 791–795 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honing S, Ricotta D, Krauss M, Spate K, Spolaore B, Motley A, Robinson M, Robinson C, Haucke V, Owen DJ, Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell 18, 519–531 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, Alessi DR, Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J 37, 1–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seaman MN, Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol 165, 111–122 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fingerhut A, von Figura K, Honing S., Binding of AP2 to sorting signals is modulated by AP2 phosphorylation. J Biol Chem 276, 5476–5482 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Rohde G, Wenzel D, Haucke V., A phosphatidylinositol (4,5)-bisphosphate binding site within mu2-adaptin regulates clathrin-mediated endocytosis. J Cell Biol 158, 209–214 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh P, Kornfeld S., AP-1 binding to sorting signals and release from clathrin-coated vesicles is regulated by phosphorylation. J Cell Biol 160, 699–708 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR, Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23, 329–341 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, Andrabi SA, Xiong Y, Dickson DW, Wszolek ZK, Pandey A, Dawson TM, Dawson VL, Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 157, 472–485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA, Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9, 1231–1233 (2006). [DOI] [PubMed] [Google Scholar]
- 28.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM, Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16, 223–232 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S., Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol 54, 618–627 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW, A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A 105, 2693–2698 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heaton GR, Landeck N, Mamais A, Nalls MA, Nixon-Abell J, Kumaran R, Beilina A, Pellegrini L, Li Y, Consortium International Parkinson Disease Genomics, Harvey K, Cookson MR, Sequential screening nominates the Parkinson’s disease associated kinase LRRK2 as a regulator of Clathrin-mediated endocytosis. Neurobiol Dis 141, 104948 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stafa K, Trancikova A, Webber PJ, Glauser L, West AB, Moore DJ, GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet 8, e1002526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai M, Gad H, Turacchio G, Cocucci E, Yang JS, Li J, Beznoussenko GV, Nie Z, Luo R, Fu L, Collawn JF, Kirchhausen T, Luini A, Hsu VW, ARFGAP1 promotes AP-2-dependent endocytosis. Nat Cell Biol 13, 559–567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chavez-Gutierrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P., LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, Geens A, De Strooper B, Verstreken P., A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 92, 829–844 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Soukup SF, Verstreken P., EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy 13, 971–972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan PY, Li X, Wang J, Powell J, Wang Q, Zhang Y, Chen Z, Wicinski B, Hof P, Ryan TA, Yue Z., Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J Neurosci 37, 11366–11376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islam MS, Nolte H, Jacob W, Ziegler AB, Putz S, Grosjean Y, Szczepanowska K, Trifunovic A, Braun T, Heumann H, Heumann R, Hovemann B, Moore DJ, Kruger M., Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet 25, 5365–5382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen M, Krainc D., LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A 115, 5576–5581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A., RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M., Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, West AB, LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum Mol Genet 27, 385–395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonet-Ponce L, Cookson MR, The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr Opin Cell Biol 59, 73–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeong GR, Jang EH, Bae JR, Jun S, Kang HC, Park CH, Shin JH, Yamamoto Y, Tanaka-Yamamoto K, Dawson VL, Dawson TM, Hur EM, Lee BD, Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol Neurodegener 13, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olgiati S, Quadri M, Fang M, Rood JP, Saute JA, Chien HF, Bouwkamp CG, Graafland J, Minneboo M, Breedveld GJ, Zhang J, Network International Parkinsonism Genetics, Verheijen FW, Boon AJ, Kievit AJ, Jardim LB, Mandemakers W, Barbosa ER, Rieder CR, Leenders KL, Wang J, Bonifati V., DNAJC6 Mutations Associated With Early-Onset Parkinson’s Disease. Ann Neurol 79, 244–256 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, Pappata S, Quarantelli M, Annesi G, Quattrone A, Chien HF, Barbosa ER, Network International Parkinsonism Genetics, B. A. Oostra, P. Barone, J. Wang, V. Bonifati, Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34, 1208–1215 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Ng J, Cortes-Saladelafont E, Abela L, Termsarasab P, Mankad K, Sudhakar S, Gorman KM, Heales SJR, Pope S, Biassoni L, Csanyi B, Cain J, Rakshi K, Coutts H, Jayawant S, Jefferson R, Hughes D, Garcia-Cazorla A, Grozeva D, Raymond FL, Perez-Duenas B, De Goede C, Pearson TS, Meyer E, Kurian MA, DNAJC6 Mutations Disrupt Dopamine Homeostasis in Juvenile Parkinsonism-Dystonia. Mov Disord, (2020). [DOI] [PMC free article] [PubMed]
- 48.Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O., A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7, e36458 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisan-Ruiz C., The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34, 1200–1207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olgiati S, De Rosa A, Quadri M, Criscuolo C, Breedveld GJ, Picillo M, Pappata S, Quarantelli M, Barone P, De Michele G, Bonifati V., PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15, 183–188 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Kirola L, Behari M, Shishir C, Thelma BK, Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Parkinsonism Relat Disord 31, 124–128 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Conner SD, Schmid SL, Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol 156, 921–929 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wrobel AG, Kadlecova Z, Kamenicky J, Yang JC, Herrmann T, Kelly BT, McCoy AJ, Evans PR, Martin S, Muller S, Salomon S, Sroubek F, Neuhaus D, Honing S, Owen DJ, Temporal Ordering in Endocytic Clathrin-Coated Vesicle Formation via AP2 Phosphorylation. Dev Cell 50, 494–508e411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdel-Magid AF, Inhibitors of Adaptor-Associated Kinase 1 (AAK1) May Treat Neuropathic Pain, Schizophrenia, Parkinson’s Disease, and Other Disorders. ACS Med Chem Lett 8, 595–597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Latourelle JC, Pankratz N, Dumitriu A, Wilk JB, Goldwurm S, Pezzoli G, Mariani CB, DeStefano AL, Halter C, Gusella JF, Nichols WC, Myers RH, Foroud T., Coordinators Progeni Investigators, Laboratories Molecular Genetic, Coordinators GenePd Investigators, Laboratories Molecular Genetic, Genomewide association study for onset age in Parkinson disease. BMC Med Genet 10, 98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, Benson DL, Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J Neurosci 36, 7128–7141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, Shen J., Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A 107, 9879–9884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM, Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102, 16842–16847 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gietz RD, Woods RA, Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350, 87–96 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ, GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet 6, e1000902 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Motley A, Bright NA, Seaman MN, Robinson MS, Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol 162, 909–918 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Semerdjieva S, Shortt B, Maxwell E, Singh S, Fonarev P, Hansen J, Schiavo G, Grant BD, Smythe E., Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J Cell Biol 183, 499–511 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Campbell C, Squicciarini J, Shia M, Pilch PF, Fine RE, Identification of a protein kinase as an intrinsic component of rat liver coated vesicles. Biochemistry 23, 4420–4426 (1984). [DOI] [PubMed] [Google Scholar]
- 66.Li Z, Kumarasinghe R, Collinson MM, Higgins DA, Probing the Local Dielectric Constant of Plasmid DNA in Solution and Adsorbed on Chemically Graded Aminosilane Surfaces. J Phys Chem B 122, 2307–2313 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.