

Abstract
Pharmacologic agonism of the β2-adrenergic receptor (β2AR) induces bronchodilation by activating the enzyme adenylyl cyclase to generate cyclic adenosine monophosphate (cAMP). β2AR agonists are generally the most effective strategy to relieve acute airway obstruction in asthmatic patients, but they are much less effective when airway obstruction in young patients is triggered by infection with respiratory syncytial virus (RSV). Here, we investigated the effects of RSV infection on the abundance and function of β2AR in primary human airway smooth muscle cells (HASMCs) derived from pediatric lung tissue. We showed that RSV infection of HASMCs resulted in proteolytic cleavage of β2AR mediated by the proteasome. RSV infection also resulted in β2AR ligand–independent activation of adenylyl cyclase, leading to reduced cAMP synthesis compared to that in uninfected control cells. Last, RSV infection caused stronger airway smooth muscle cell contraction in vitro due to increased cytosolic Ca2+ concentrations. Thus, our results suggest that RSV infection simultaneously induces loss of functional β2ARs and activation of multiple pathways favoring airway obstruction in young patients, with the net effect of counteracting β2AR agonist–induced bronchodilation. These findings not only provide a potential mechanism for the reported lack of clinical efficacy of β2AR agonists for treating virus-induced wheezing but also open the path to developing more precise therapeutic strategies.
INTRODUCTION
The β2-adrenergic receptor (β2AR) is a prototypical member of the heptahelical transmembrane family of G protein–coupled receptors (GPCRs) (1). This family includes three receptor subtypes, β1, β2, and β3, with β2AR having predominant expression in the respiratory tract (2, 3). Activation of βARs by binding to β-agonists results in the activation of a heterotrimeric stimulatory G protein (GS) and release of its α and βγ subunits (4–6). The α subunit then activates the enzyme adenylyl cyclase (AC), which generates the second messenger cyclic adenosine monophosphate (cAMP), which, in turn, mediates the activation of protein kinase A (PKA) (4, 5). Activated β2ARs are phosphorylated and desensitized by GPCR kinases (GRKs) and PKA (7, 8). Desensitized β2ARs are then internalized into endosomes, undergo dephosphorylation (that is, resensitization), and are recycled back to the plasma membrane as naïve receptors ready to engage again with agonist through a complex multistep signaling process (9). When activated by cAMP, PKA stimulates downstream signaling events that mediate the relaxation of airway smooth muscle cells in the respiratory tract (10) by inhibition of calcium-activated contraction (11). Short-acting β2AR agonists, such as albuterol, have for decades been the first-line pharmacological strategy for the treatment of acute bronchospasm in patients with asthma and other obstructive airway diseases.
Bronchiolitis, a disease that is characterized by obstruction of the distal airways and is commonly caused by a viral lower respiratory tract infection (LRTI), is currently the most common cause of hospitalization among infants and young children worldwide (12). Despite many similarities with the clinical manifestations and pathophysiology of asthma, several randomized controlled trials and systematic reviews have failed to demonstrate any consistent benefit from the use of β2AR agonists in the setting of viral bronchiolitis (13). Furthermore, a report indicates that virus-infected asthmatic children have a higher risk of treatment failure during acute exacerbations (14). The most common etiology of LRTI in children is the respiratory syncytial virus (RSV), an enveloped, nonsegmented, negative-sense RNA virus belonging to the Paramyxoviridae family (13, 15).
Human airway smooth muscle cells (HASMCs) are susceptible to RSV infection, which leads to alterations in their production of cAMP (16). Because little is known about the mechanisms limiting the response to β2AR agonists in children with viral LRTI, we postulated that β2AR engagement or signaling in HASMCs might be altered during RSV infection. To test this hypothesis, we examined β2AR signaling in HASMCs derived from pediatric donors and infected with red fluorescent protein (RFP)–expressing recombinant RSV (rrRSV). In particular, in addition to assessing the immediate downstream events that mediate smooth muscle cell relaxation in vitro, we analyzed the posttranslational modification and function of the β2AR in rrRSV-infected HASMCs and its response to pharmacological agonists, finding modifications that provide insights into the clinical manifestations of this common infection.
RESULTS
RSV infection results in posttranslational modifications of the β2AR
HASMCs derived from pediatric donors were incubated for 24 hours with sterile medium from Hep-2 cell cultures, ultraviolet (UV)–inactivated rrRSV, or rrRSV at a multiplicity of infection (MOI) of 1. After incubation, the cells were fixed and stained to detect total β2AR with a specific antibody and were visualized by confocal microscopy. Negative controls incubated without primary antibody showed minimal background fluorescence (Fig. 1A, first column). Compared to HASMCs incubated with sterile medium (Fig. 1A, second column), no substantial change in β2AR signal was visualized after exposure to UV-inactivated rrRSV (Fig. 1A, third column) or actively replicating rrRSV (Fig. 1A, fourth column). As expected, no RFP expression was detected in cells incubated with sterile medium or UV-inactivated rrRSV (Fig. 1A, third column), whereas RFP was observed in cells infected with replicating rrRSV (Fig. 1A, fourth column). For a more quantitative analysis, mean fluorescence intensity was measured in various visual fields of the same preparations and again showed no appreciable differences in β2AR abundance among the different groups (Fig. 1B).
Fig. 1. Analysis of β2AR expression in uninfected and rrRSV-infected HASMCs.

(A and B) Cultured HASMCs were incubated with sterile medium, UV-inactivated rrRSV (UVrrRSV), or replicating rrRSV at an MOI of 1 for 24 hours as indicated, and β2AR expression was visualized by immunocytochemistry. (A) Representative confocal microscopy images showing DAPI-stained nuclei (blue), β2AR (green), and rrRSV replication (red). Magnification, ×63. Scale bar, 20 μm. (B) Quantitation of β2AR mean fluorescence as measured with ImageJ software. Measurements from multiple fields are presented as box-and-whisker plots (N = 5 donors per group; all experiments were performed with HASMCs from three to eight donors). Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons.
To determine whether RSV infection altered β2AR phosphorylation, pediatric HASMCs incubated for 24 hours with sterile medium, UV-inactivated rrRSV, or rrRSV at an MOI of 1 were analyzed with a specific antibody against phosphorylated β2AR (pβ2AR) (17). Confocal microscopy showed that pediatric HASMCs incubated in sterile medium had minimal β2AR phosphorylation (Fig. 2A, second column) compared to the negative control (Fig. 2A, first column). There appeared to be a small increase in pβ2AR signal in cells exposed to UV-inactivated rrRSV (Fig. 2A, third column). In contrast, infection with actively replicating rrRSV (Fig. 2A, fourth column) resulted in a substantial increase in pβ2AR signal (Fig. 2A, fourth column). Consistently, a significant increase in the mean fluorescence of pβ2AR was detected in rrRSV-infected HASMCs compared to that in uninfected control cells (P < 0.001), confirming the increased extent of β2AR phosphorylation (Fig. 2B).
Fig. 2. Analysis of β2AR phosphorylation in response to RSV infection.

(A and B) Cultured HASMCs were incubated with sterile medium, UVrrRSV, or rrRSV at an MOI of 1 for 24 hours before pβ2AR expression was visualized by immunocytochemistry. (A) Representative confocal microscopy images showing DAPI-stained nuclei (blue), pβ2AR (green), and rrRSV replication (red). Magnification, ×63. Scale bar, 20 μm. (B) Quantitation of pβ2AR mean fluorescence as measured with ImageJ software. Measurements from multiple fields are presented as box-and-whisker plots (N = 4 per group; all experiments were performed with HASMCs from three to eight donors). Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. ***P < 0.01 compared to untreated cells.
To further validate the increase in β2AR phosphorylation induced by RSV, HASMCs incubated with sterile medium, UV-inactivated rrRSV, or rrRSV were lysed and analyzed by Western blotting with an antibody against pβ2AR. Consistent with the confocal microscopy findings, this analysis revealed significantly increased pβ2AR abundance in infected cells compared to that in cells treated with medium or UV-inactivated virus (P < 0.01; Fig. 3A). Unexpectedly, we also observed the appearance of a 35-kDa band reactive with the antibody against pβ2AR and present only in the rrRSV-infected cells, possibly resulting from cleavage of pβ2ARs. To validate the identity of the 35-kDa fragment, UV-inactivated rrRSV controls and rrRSV-infected cells were lysed, subjected to immunoprecipitation (IP) with an anti-β2AR antibody, and analyzed by Western blotting with an anti-pβ2AR antibody lysates (Fig. 3B). This analysis showed a 35-kDa reactive band that was present in the samples from rrRSV-infected HASMCs but not in samples from the control cells.
Fig. 3. Analysis of the effects of RSV infection on pβ2AR protein.

(A) HASMCs were incubated for 24 hours with sterile medium, UVrrRSV, or rrRSV at an MOI of 1. Left: Cell lysates were analyzed by Western blotting with antibody against pβ2AR. Actin was used as a loading control. Western blot is representative of five experiments. Arrow indicates the position of the 35-kDa fragment reactive to anti-pβ2AR. The black horizontal bar indicates that the blots are not contiguous. Middle: Analysis of the fold change in the pβ2AR band intensity normalized to that of actin under the indicated conditions. Results from five individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. **P < 0.01. Right: Analysis of the fold change in the 35-kDa pβ2AR band intensity normalized to that of actin under the indicated conditions. Results from five individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. **P < 0.01. (B) HASMCs that were left uninfected or were infected with rrRSV for 24 hours, as indicated, were lysed, and 0.4 mg of cleared lysate was subjected to immunoprecipitation (IP) with an anti-β2AR antibody. Left: The immunoprecipitated samples were analyzed by Western blotting with an anti-pβ2AR antibody. Arrows indicate anti-pβ2AR–reactive proteins. Right: The SDS-PAGE gel was stained with Coomassie to ensure equal loading of lanes. Western blots are representative of three experiments. MW, molecular weight.
RSV infection increases the abundance of a 35-kDa protein product
To determine whether the 35-kDa fragment was a product of β2AR cleavage in response to infection with actively replicating RSV infection, pediatric HASMCs were infected with rrRSV at an MOI of 1. The cells were then lysed at different times and analyzed by Western blotting with anti-pβ2AR antibody. The 35-kDa protein product increased in abundance in a time-dependent fashion beginning at 4 hours after infection and continued to increase significantly for up to 24 hours (Fig. 4A). Because the abundance of the 35-kDa fragment was maximal at 24 hours after infection, we chose this time point to perform the remainder of the studies. To test whether β2AR cleavage is dependent on the amount of RSV used to infect the cells, we infected pediatric HASMCs for 24 hours with rrRSV at MOIs ranging from 0.01 to 1.0. Western blotting analysis showed that the abundance of the 35-kDa anti-pβ2AR–reactive protein increased with MOI (Fig. 4B), suggesting that increasing viral load caused a coincident increase in the breakdown of β2ARs.
Fig. 4. Time- and dose-dependent increase in the amount of the 35-kDa protein fragment in response to RSV infection.
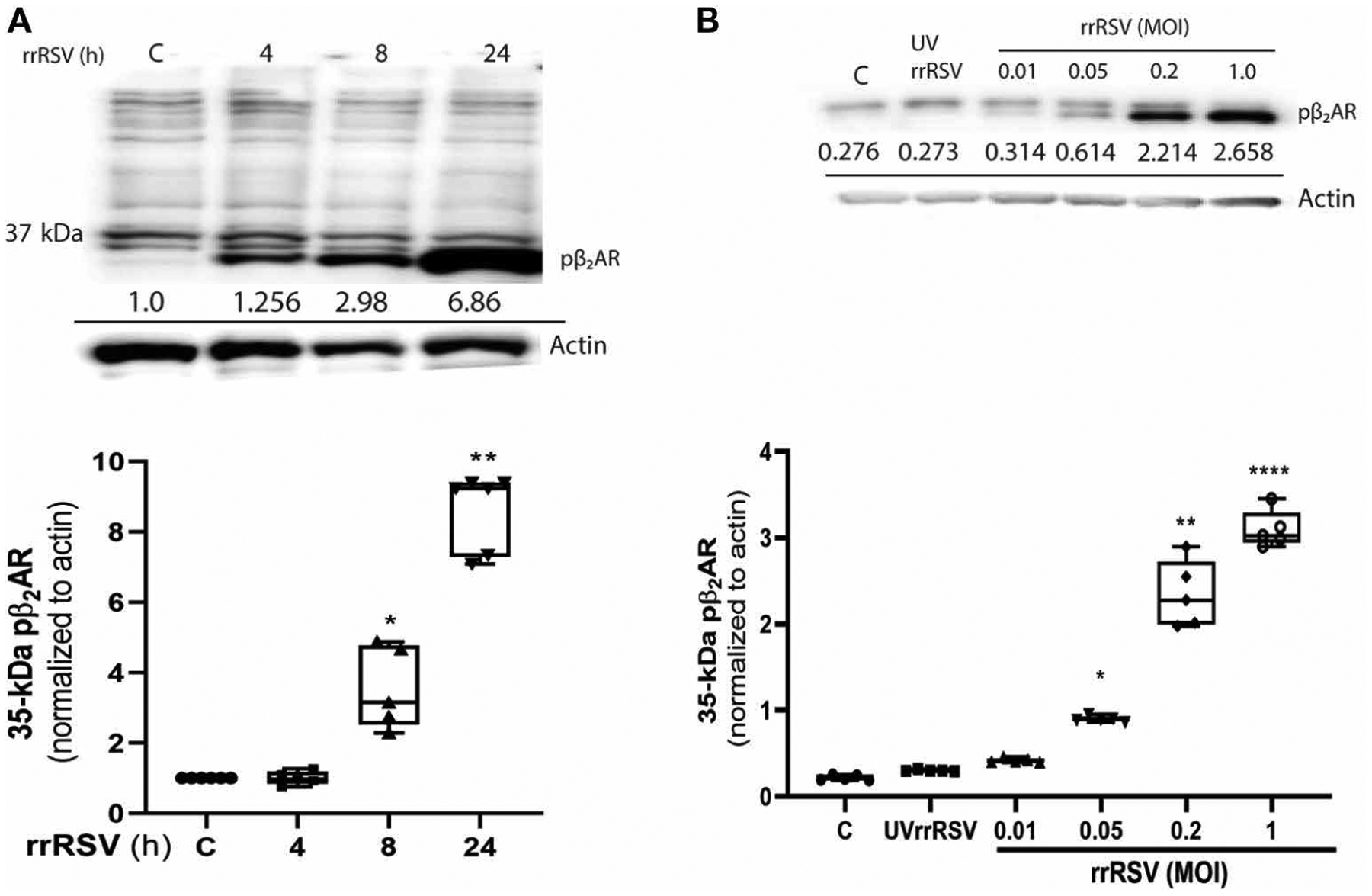
(A) Cultured HASMCs were incubated with sterile control medium (C) or were infected for the indicated times with rrRSV at an MOI of 1. Top: Cell samples were analyzed by Western blotting with an antibody against pβ2AR. Actin was used as a loading control. The black horizontal bar indicates that the blots are not contiguous. Bottom: The relative abundance of the 35-kDa protein fragment detected by anti-pβ2AR normalized to that of actin was determined with ImageJ software. Normalized amounts are shown below each band. (B) HASMCs were incubated with sterile control medium (C) or were infected with UVrrRSV or rrRSV at the indicated MOIs for 24 hours. Top: Cell samples were analyzed by Western blotting with antibody against pβ2AR. Actin was used as a loading control. The black horizontal bar indicates that the blots are not contiguous. Bottom: The relative abundance of the 35-kDa protein fragment detected by anti-pβ2AR normalized to that of actin was determined with ImageJ software. Normalized amounts are shown below each band. For the graphs in (A) and (B), results from three individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. P values were calculated by two-tailed test. *P < 0.05, **P < 0.01, and ****P < 0.0001 compared to control.
RSV infection induces the cleavage and loss of β2ARs
Because RSV infection increased the relative abundance of the 35-kDa β2AR cleavage product that was detectable with anti-pβ2AR antibodies, we tested whether RSV infection caused a loss of cell surface β2ARs on HASMCs in experiments with [125I]cyanopindolol radioligand binding assays. Plasma membrane fractions were isolated from paired pediatric HASMCs that were either uninfected or infected with rrRSV, and saturation receptor binding assays were performed. Infection resulted in a significant loss of β2ARs from the plasma membrane compared to paired uninfected pediatric HASMCs (Fig. 5A). This finding suggests that RSV infection leads to the loss of cell surface receptors, which was accompanied by the time- and dose-dependent increase in the 35-kDa anti-pβ2AR–reactive protein.
Fig. 5. Loss of β2AR in response to RSV infection.

(A) Cultured HASMCs were treated with sterile control medium or were infected with rrRSV at an MOI of 1. Twenty-four hours later, isolated plasma membranes were subjected to [125]I-CYP (cyanopindolol, a β2AR agonist) β2AR binding at a saturation concentration of 250 pM. A Wilcoxon matched-pair signed-rank test was performed, with P values calculated by two-tailed test. *P < 0.05; N = 3 experiments. CPM, counts per minute. (B) pβ2AR-reactive protein expression was analyzed by Western blotting in HASMCs pretreated with vehicle, 100 nM MG-132, or 2.5 μM batimastat (Bat) before being treated with sterile medium or being infected with rrRSV at an MOI of 1 for 24 hours. Top: The cells were then analyzed by Western blotting with antibody against pβ2AR. Actin was used as a loading control. Bottom: The bar graph shows the intensity of the band corresponding to the 35-kDa anti-pβ2AR–reactive band normalized to that of the actin band. Results from five individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. P values were calculated by two-tailed test. **P < 0.01 and ****P < 0.0001 compared to the RSV only treatment. (C and D) Cultured HASMCs were pretreated with the indicated concentrations of propranolol with or without albuterol (Alb), as indicated, for 1 hour before being incubated with sterile control medium (C), UVrrRSV, or rrRSV (MOI of 1) for 24 hours. Top: The cells were lysed and analyzed by Western blotting with antibodies against pβ2AR (C) and RSV-G protein (D). Actin was used as a loading control. Bottom: The graphs show the relative band intensities, normalized to that of actin, for pβ2AR (C) and RSV-G protein (D). Results from three individual blots are presented as box-and-whisker plots (N = 3). Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. P values were calculated by two-tailed test. *P < 0.05 and **P < 0.01 compared to the RSV only treatment. (E) HASMCs embedded in a 1.5% collagen matrix were incubated with sterile medium or rrRSV for 48 hours before being subjected to a 30-min treatment with vehicle or 200 μM albuterol followed by 200 μM methacholine. Images of the collagen gels were taken at 10-min intervals for 30 min. Data are from three independent experiments and are expressed as the percentage change in gel contraction from time zero. Data are means ± SEM and were analyzed by one-way analysis of variance (ANOVA) using Bonferroni’s multiple comparison test. *P < 0.05 and ***P < 0.001 compared to the methacholine treatment only. The black horizontal bars in the Western blots indicate that the blots are not contiguous.
Because we observed the loss of β2ARs from the plasma membranes of rrRSV-infected cells, we isolated endosomes from the same paired set of rrRSV-infected and uninfected pediatric HASMCs and measured β2AR abundance by radioligand binding assays. Consistent with the recycling of β2ARs, we observed low receptor abundance in the endosomes of uninfected pediatric HASMCs (fig. S1). However, despite the loss of β2ARs from the plasma membranes, we observed no significant accumulation of receptors in the endosomes of rrRSV-infected pediatric HASMCs. This finding suggests that RSV infection leads to increased β2AR phosphorylation but, rather than targeting the receptors for endosomal recycling, the virus promotes receptor cleavage. Therefore, we hypothesize that the 35-kDa band that is recognized by the anti-pβ2AR antibody derives from the cleavage of the pβ2ARs, thereby accounting for the loss of receptors from the plasma membranes of rrRSV-infected pediatric HASMCs.
To test whether the 35-kDa anti-pβ2AR–reactive band was a cleavage product derived from the proteasomal degradation of pβ2AR or through cleavage by matrix metalloproteinases (MMPs), we pretreated HASMCs with the proteasome inhibitor MG-132 (18) or the pan-MMP inhibitor batimastat (19) before infecting them with rrRSV. Western blotting analysis of the cell lysates with anti-pβ2AR antibody showed that batimastat had no effect on the rrRSV-mediated generation of the 35-kDa anti-pβ2AR–reactive band (Fig. 5B). In contrast, pretreatment with MG-132 prevented the production of the 35-kDa fragment, suggesting that pβ2AR undergoes proteasomal degradation. Because proteasomal inhibition with MG-132 inhibits RSV replication and budding in human nasal airway epithelial cells (20), we sought to determine its effect on RSV infection in HASMCs using the RSV-G protein as a surrogate biomarker of viral replication. Cells were pretreated with MG-132 or batimastat and then infected with rrRSV at an MOI of 1. Twenty-four hours later, the cells were analyzed by Western blotting to detect RSV-G protein. Consistent with previous findings, RSV-G protein abundance was reduced in cells pretreated with MG-132 but was increased in cells treated with batimastat (fig. S2).
To further investigate whether the 35-kDa fragment was a product of pβ2AR cleavage, we tested whether a β2AR antagonist could reduce the generation of the cleaved product. Pediatric HASMCs were pretreated with the β2AR antagonist propranolol for 1 hour before undergoing rrRSV infection. Twenty-four hours later, Western blotting analysis was performed on cell lysates with the anti-pβ2AR antibody. Consistent with our previous observation, rrRSV infection resulted in a significant increase in the amount of the 35-kDa cleavage product generated, which was not altered by the lower concentration of propranolol but was significantly reduced (~60%) by the higher concentration (Fig. 5C). These findings suggest that propranolol, by specifically binding to β2AR, reduced the availability of substrate for degradation upon rrRSV infection. Note that pretreatment with propranolol before infection with rrRSV also led to a reduction in RSV-G protein abundance (Fig. 5D).
Because the infection of HASMCs with rrRSV led to the loss of β2AR from the plasma membrane, we used a collagen gel contraction assay to assess changes in contractility in the cells and whether the selective β2AR agonist albuterol could inhibit methacholine-induced contraction. Pediatric HASMCs embedded in 1.5% collagen matrix were incubated with sterile medium or rrRSV at an MOI of 1 for 48 hours and then were pretreated with or without albuterol for 30 min before being stimulated with methacholine. Infection was confirmed by the visualization of RFP by fluorescence microscopy (fig. S3). Methacholine induced a 20% contraction in rrRSV-infected cells, as compared to 15% contraction in uninfected cells. Pretreatment with albuterol led to a 50% reduction in the methacholine-induced contraction of uninfected cells (8% contraction), whereas methacholine-induced contraction was reduced by 25% (15% contraction) in RSV-infected cells pretreated with albuterol.
RSV infection increases cAMP concentrations
Given the loss of β2ARs from the plasma membranes of RSV-infected cells, we investigated whether the amount of intracellular cAMP was less in RSV-infected cells than in uninfected cells. Thus, lysates from uninfected and rrRSV-infected pediatric HASMCs were tested for intracellular cAMP. Unexpectedly, a significant increase in cAMP concentration (P < 0.05) was observed in rrRSV-infected cells compared to that in uninfected cells (Fig. 6A). In addition, isoproterenol failed to increase the cAMP concentration in RSV-infected cells as compared to in uninfected cells. Last, propranolol blocked the isoproterenol-induced increase in cAMP production in uninfected cells but failed to affect cAMP production in RSV-infected cells (Fig. 6A).
Fig. 6. Activation of AC in response to RSV infection.

(A) Cultured HASMCs were left uninfected or were infected with rrRSV (MOI of 1) for the indicated times before being left untreated or treated with the indicated concentrations of propranolol (Prop). The amounts of cAMP generated in the cells were determined as indicated in Materials and Methods. Data were analyzed using the mixed-effects model with Geisser-Greenhouse correction. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared to the corresponding zero time point for each treatment. #P < 0.05 and ##P < 0.01 compared to the control zero time point. (B) Cultured HASMCs were left uninfected or were infected with rrRSV for the indicated times. Top: The cells were then analyzed by Western blotting with antibodies against total and phosphorylated CREB (pCREB) protein. Forskolin was used as a positive control for CREB phosphorylation. Bottom: Relative abundance of pCREB to total CREB normalized to that of GAPDH. Results from three individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. P values were calculated by two-tailed test. ***P < 0.001 and ****P < 0.0001 between uninfected and infected cells. (C) HASMCs were left uninfected or were infected with rrRSV for the indicated times. Top: The cells were analyzed by Western blotting with antibody against AC5/6. Actin was used as a loading control. Bottom: The relative abundance of AC5/6, normalized to that of actin, was determined. Results from eight individual blots are presented as box-and-whisker plots. Data are means ± SEM and were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons. (D) HASMCs were left uninfected or were infected with rrRSV. Cell-free membranes were prepared and treated with vehicle control (C) or 4 mM NaF, and AC activity was assessed using radioactive [γ-32P]ATP to measure cAMP generation. Data are means ± SEM of three independent experiments and were analyzed by one-way ANOVA with Dunnett T3 correction for multiple comparisons. **P < 0.01 and ****P < 0.0001, compared to the uninfected control. The black horizontal bars in the Western blots indicate that the blots are not contiguous.
Because we observed an increased cAMP production in the RSV-infected cells, we sought to determine whether this increase had any classical downstream effects, such as PKA activation and the consequent phosphorylation of cAMP response element–binding protein (CREB) (21). Pediatric HASMCs were incubated with sterile medium or rrRSV, and cell lysates were then analyzed by Western blotting for phosphorylated CREB (pCREB). Consistent with the increased amounts of cAMP after rrRSV infection, there was a significant increase in the amount of pCREB (Fig. 6B). We found no significant change in the amount of total CREB protein after rrRSV infection.
Given the increased amount of cAMP and the phosphorylation of its downstream mediator CREB upon RSV infection, we studied whether viral infection led to increases in the abundance of AC, the key molecule involved in cAMP generation. After 6 or 24 hours of infection with rrRSV, cell lysates were analyzed by Western blotting for AC5 and AC6 (AC5/6), the most commonly expressed AC isoforms in HASMCs (22). Compared to uninfected controls, infected cells showed no significant differences in AC5/6 abundance either 6 or 24 hours after rrRSV infection (Fig. 6C). Because RSV infection led to increased cAMP production and the activation of cAMP-dependent signaling without altering AC5/6 abundance, we wondered whether rrRSV infection might instead lead to AC activation. To test this hypothesis, we measured AC activity in plasma membrane fractions isolated from paired uninfected and rrRSV-infected pediatric HASMCs by directly stimulating the Gs protein with NaF. AC activity was significantly increased in rrRSV-infected plasma membranes compared to that in uninfected controls (Fig. 6D), suggesting that RSV activates AC in an agonist-independent manner in host cells, leading to increased cAMP generation despite the loss of β2ARs from the plasma membrane.
RSV infection predisposes cells to increased Ca2+ influx
Because a transient increase in intracellular Ca2+ is required for muscle contraction, we tested whether rrRSV infection also altered intracellular Ca2+ concentrations in HASMCs. First, the response to Gαq-stimulating ligands was assessed by measuring the effect of increasing doses of carbachol or methacholine on intracellular free Ca2+ using the Calcium-6 Assay Kit with a FlexStation 3 fluorescent plate reader (fig. S4). Because carbachol generated a greater response than did methacholine, we then infected pediatric HASMCs with rrRSV and monitored the cells for changes in intracellular Ca2+ concentration in response to carbachol (23). Carbachol-induced increases in intracellular Ca2+ concentration were enhanced as a result of RSV infection, but only 6 hours after infection and not at 24 hours after infection (Fig. 7). This observation suggests that RSV infection potentiates intracellular Ca2+ mobilization in pediatric HASMCs. Future studies will be needed to determine whether the resulting increased intracellular Ca2+ concentration contributes to the airway hyperreactivity observed in pediatric patients during RSV infection.
Fig. 7. RSV infection increases the intracellular Ca2+ concentration.

Cultured HASMCs were incubated with sterile medium or rrRSV at an MOI of 1 for 6 or 24 hours before being assessed for changes in cytoplasmic free Ca2+ concentration. HASMCs were treated with Calcium-6 reagent for 3 hours before they were incubated with vehicle or the indicated concentrations of carbachol (Cch) (100 nM to 100 μM). Readings were taken every 7 s for 2 min and are expressed as the area under the curve (AUC; N = 4 donors each with three experimental replicates; error bars indicate the SEM). Dose-response curve fitting was performed with three-parameter, nonlinear regression.
DISCUSSION
The most typical clinical manifestations of RSV bronchiolitis in infants include wheezing and coughing and the need for an increased effort in breathing through the use of accessory respiratory muscles. RSV is increasingly recognized as a frequent cause of similar symptoms in adults, particularly in the elderly and immunocompromised individuals (24). In most clinical settings, nebulized albuterol (a selective β2AR agonist) is administered as first-line therapy to alleviate these symptoms by enabling the constricted airways to relax, despite the fact that most controlled trials have shown this approach to be ineffective (25). The lack of clinical response to albuterol during RSV infection remains an unresolved conundrum, because the wheezing of most atopic asthmatic patients does improve within minutes of a nebulized albuterol treatment (26). Furthermore, studies showed that patients with acute asthma exacerbations triggered by RSV are more likely to fail the standard therapy protocol based on albuterol treatment (14). Our study sought to test the hypothesis that RSV infection directly alters β2AR structure and function, thereby impairing the clinical response to albuterol.
We found that RSV infection led to increased β2AR phosphorylation, mimicking the effect of β-agonists, such as albuterol. However, this increased phosphorylation did not result from an increase in total β2AR abundance, because our fluorescence microscopy study showed no appreciable differences in endogenous β2ARs between uninfected and infected cells. In this context, we also found that a 35-kDa protein fragment that was detected by an anti-pβ2AR antibody was generated in airway smooth muscle cells infected with replicating RSV but not after incubation with UV-inactivated virus. Furthermore, our data showed that generation of this protein fragment was time- and dose-dependent on RSV replication. Because RSV infection enhances proteasomal degradation (27, 28), we hypothesized that the 35-kDa fragment was generated by β2AR cleavage. Previous work showed that inhibition of proteasomal degradation by MG-132 results in impaired RSV replication, which is reflected in reduced synthesis of RSV-G protein (20). Thus, reduced viral load after MG-132 treatment might underlie the reduced accumulation of the 35-kDa fragment that we observed. RSV infection also induces loss of signal transducer and activator of transcription 2 (STAT2) and NF-E2–related factor 2 (NRF2) in human alveolar cells by proteasomal degradation involving an E3 ubiquitin ligase (27, 28). Similarly, RSV inhibition of T helper cell differentiation can be reversed with the proteasome inhibitor PS-341 (29). On the other hand, the β1AR is cleaved at the extracellular N terminus, both constitutively and by agonist-induced mechanisms mediated by metalloproteinases upon O-glycosylation (30).
Canonical β2AR stimulation by its agonist isoproterenol leads to the receptor being shuttled to lysosomes for degradation or being cleaved by metalloproteases (31, 32). In this context, the isoproterenol-induced degradation of β2ARs is not inhibited by proteasomal inhibition with MG-132 (32). In contrast, we found that production of the 35-kDa protein fragment was completely blocked by MG-132 but not by the metalloprotease inhibitor batimastat. The reduction in the abundance of the 35-kDa fragment upon propranolol treatment, despite RSV infection, supports the hypothesis that this protein fragment is a cleaved product of mature β2ARs and not a protein generated after RSV infection.
Parallel with impairing the generation of the 35-kDa protein fragment, propranolol also significantly reduced the accumulation of RSV-G protein, indicating that it reduced the extent of RSV infection. This suggests that there may be cross-talk between the β2AR and RSV infection such that the unbound receptor may promote the infection, whereas the effect of propranolol in locking β2AR into an antagonist-bound conformation reduces viral replication and RSV-G protein synthesis. Our radioligand binding assays showed a significant reduction in plasma membrane β2AR abundance after RSV infection, further supporting this hypothesis. Moreover, fluorescence microscopy studies of endogenous β2AR showed no appreciable difference in receptor abundance after RSV infection, which is probably due to the antibody recognizing the C-terminal region of the β2AR, a hypothesis also supported by the IP studies that showed unique enrichment of the 35-kDa protein fragment. Nevertheless, we cannot exclude completely the possibility that the 35-kDa fragment is part of an immature, newly synthesized receptor localized within the endoplasmic reticulum.
In contrast to the loss of β2ARs in pediatric HASMCs infected with RSV, this infection resulted in a marked increase in cAMP abundance even in the absence of stimulus. However, the increase in cAMP amounts did not result from increased expression of AC but rather from an increased enzymatic activity. This finding suggests that RSV infection leads to AC activation that is independent of β2AR stimulation. However, the β2AR pathway may be required for sustenance of RSV infection, because propranolol treatment resulted in a decrease in RSV infection. Consistent with this finding, RSV-infected HASMCs did not generate cAMP in response to stimulation of β2AR with agonist. Previous studies showed that ACs can be activated by phosphorylation mediated by the kinase cRaf (33) and that RSV can activate Raf (34), which is suggestive of one possible mechanism whereby RSV infection might activate AC and lead to increased cAMP generation. Because we observed increased cAMP generation in RSV-infected cells, it is also possible that, in addition to leading to AC activation, RSV may also lead to the inhibition of a phosphodiesterase, an enzyme required for the degradation of cAMP.
Counterintuitive to the observation of the reduced number of β2ARs, yet consistent with AC activation, cAMP abundance was increased in RSV-infected cells, suggesting that cAMP-mediated mechanisms may support active viral replication. Consistent with the increase in cAMP abundance, we also found increased activation of CREB, as measured by its phosphorylation. CREB is phosphorylated by PKA, which is activated by AC-generated cAMP (35). Thus, RSV-mediated regulation of AC underlies the activation of CREB. In addition, increased cAMP abundance could also be due to simultaneous inhibition of a phosphodiesterase or of protein phosphatase 1 (PP1), which could account for increased CREB phosphorylation (36). It is also possible that RSV infection alters cyclooxygenase-2 (COX-2) expression and activity and, in turn, may lead to the synthesis of prostanoids [prostaglandin E2 (PGE2)] that bind to EP2/4 receptors and drive cAMP generation through AC activation (37). However, on the basis of the observation of AC activation, our findings support the hypothesis that RSV infection drives CREB activation through the cAMP-PKA axis. Our findings raise a potential paradox in the mechanism of cAMP-mediated airway relaxation (10). Whereas RSV infection activates the cAMP-PKA axis, it does not mediate airway relaxation (38), suggesting that this effect of the virus is not the same as β2AR–G protein–mediated activation of AC.
Ca2+ is a critical second messenger involved in the contraction of smooth muscle in the airways. Notwithstanding the increased cAMP-PKA functional axis, RSV-infected airways show a pro-contractile phenotype (38). Consistently, we found increased cytosolic free Ca2+ concentrations in pediatric HASMCs infected with RSV. Furthermore, RSV-infected HASMCs were still capable of signaling through Gαq-mediated pathways, which resulted in a significant increase in intracellular free Ca2+. These findings suggest that RSV infection promotes the release of intracellular Ca2+, which might explain the pro-contractile phenotype. Although an increased intracellular Ca2+ concentration inhibits AC activity (39), in our experiments, RSV infection led to simultaneous increases in Ca2+ concentration and AC activity, suggesting that this infection delinks their regulatory cross-talk.
We recognize that our studies were performed in vitro and may not fully recapitulate what happens in vivo. In addition, the HASMCs used in these studies were from deidentified donors, for whom little medical history was available. Although it would be interesting to reproduce these results in a relevant animal model, murine models of RSV infection have shown many shortcomings, such as the lack of tropism for the bronchiolar epithelium, the failure to propagate infection from the upper to lower airway, the extremely high inoculum required to establish the infection, and the persistence of virus in the respiratory tract well beyond the typical timeline of human infection (40). Thus, we believe that the present study provides mechanistic insights into the underpinnings of the limited therapeutic efficacy of traditional β-agonists in the setting of viral bronchiolitis and virus-induced asthma exacerbations (14).
In conclusion, our data suggest that RSV infection of pediatric airway smooth muscle cells generates a pro-contractile phenotype by increasing the intracellular Ca2+ concentration while inducing the proteasomal degradation of β2ARs. This loss of functional β2ARs could account for the lack of clinical response of RSV-infected patients to β-agonists. Our findings provide insights for the potential development of more effective and precise therapeutic strategies for viral-induced wheezing, especially in the pediatric population.
MATERIALS AND METHODS
HASMC isolation
Human trachea or whole lungs were obtained from deceased donors within 24 hours of death. A tracheobronchial airway segment (from 1 cm proximal to the carina to the second bifurcation distal to the carina) was dissected from the lungs and rinsed with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with antibiotics and antimycotics before the removal of the epithelial layer by overnight digestion followed by manual scraping. The remainder of the tissue specimen was minced and then transferred to digestion buffer containing collagenase (0.1 mg/ml), deoxyribonuclease (DNase) I, and 2.5 mM Hepes in Hank’s balanced salt solution supplemented with antibiotics and antimycotics. Digestion was performed overnight at 37°C with shaking. Digested tissue was sterile-filtered with a 100-μm cell strainer, and then the cells were pelleted, washed, and transferred to culture dishes for 1 hour to remove the fibroblasts. Nonattached cells were transferred to fresh culture dishes containing DMEM-F12 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics/antimycotics. Culture purity was assessed by immunofluorescent staining for α-isoactin, which is specific for smooth muscle cells.
HASMC culture
Primary HASMCs isolated from the lungs of deidentified deceased pediatric donors were used between the third and sixth passages. Cells were maintained at 37°C in 5% CO2 with DMEM-F12 growth medium supplemented with 10% FBS and the antibiotics penicillin, streptomycin, and amphotericin. Serum was withdrawn for a minimum of 12 hours before all experiments. Four to six pediatric donors were used for each experiment with ages ranging from 34 weeks to 4 years old. The purity of the cell cultures was assessed by immunocytochemistry for smooth muscle actin and visual morphology through passaging. Experiments were repeated with cells from different donors throughout the study to control for host genetics and environment.
Ethics statement
Our experiments were performed with primary HASMCs derived from anonymous patient donors. Human tissue was provided by the International Institute for the Advancement of Medicine and processed according to protocols approved by the Cleveland Clinic Foundation.
Reagents
MG-132, batimastat, propranolol, isoproterenol, NaF, albuterol (salbutamol), forskolin, methacholine, carbachol, and isoproterenol were all purchased from Sigma-Aldrich.
Western blotting
HASMCs at 70 to 80% confluency were infected with rrRSV (at an MOI of 1) at the times indicated in the figure legends. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 25 mM tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS (Thermo Fisher Scientific), protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific), and DNase (Qiagen). Protein concentration was determined with the bicinchoninic acid (BCA) protein assay reagent (Thermo Fisher Scientific). Proteins were resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Scientific). The membranes were blocked in tris-buffered saline/Tween 20–5% bovine serum albumin for 1 hour at room temperature and incubated with the following primary antibodies: rabbit anti-β2AR (Abcam), rabbit anti-pβ2AR (355,356; Santa Cruz Biotechnology), CREB (Cell Signaling Technology), pCREB (Ser133; Cell Signaling Technology), AC (GeneTex), and RSV-G protein (GeneTex). β-Actin (Sigma-Aldrich) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam) was used to ensure equal loading. Luminol-generated signals (Thermo Fisher Scientific) or infrared-labeled secondary antibodies (LICOR) were used to detect the bands.
IP of β2AR
Cells were lysed in IP lysis buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific) and placed on ice for 5 min before undergoing sonication. Protein debris was removed by centrifugation, and 400 μg of total lysate was used for IP. The cell lysate was precleared with protein G magnetic beads (Bio-Rad), which were then removed before incubation with an anti-β2AR antibody (Bio-Rad) overnight at 4°C with rotation. Protein G magnetic beads were added to the lysate for 20 min with rotation at room temperature. Beads were washed with IP lysis buffer and then boiled in Laemmli buffer for 10 min. Beads were then pelleted, and the supernatant was resolved by SDS-PAGE and transferred to PVDF membranes before undergoing Western blotting analysis with anti-pβ2AR (Santa Cruz Biotechnology).
Purification of early endosomes and plasma membranes
Purification of plasma membranes and endosomes was performed as described previously (9, 41). Briefly, cells were homogenized in ice-cold lysis buffer containing 5 mM tris-HCl (pH 7.5), 5 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride together with leupeptin and aprotinin (each at 2 μg/ml). Cell debris and nuclei were removed by centrifugation at 1000g for 5 min at 4°C. The supernatant was transferred to a new tube and subjected to centrifugation at 37,000g for 30 min at 4°C. The pellet containing plasma membrane was resuspended in 75 mM tris-HCl (pH 7.5), 2 mM EDTA, and 12.5 mM MgCl2, whereas the supernatant underwent another round of centrifugation at 200,000g for 1 hour at 4°C. The pellet containing the endosomes was recovered in 75 mM tris-HCl (pH 7.5), 2 mM EDTA, and 12.5 mM MgCl2 for 1 hour at 4°C and was resuspended in 75 mM tris-HCl (pH 7.5), 2 mM EDTA, and 12.5 mM MgCl2.
Measurement of β2AR density and AC activity
β2AR density was determined by incubating 20 μg of membranes (plasma membranes or endosomes) with saturating concentrations of [125I]cyanopindolol (250 pM), either alone or in addition to 40 μM alprenolol to assess nonspecific binding as described previously (42, 43). AC activity assays were performed by incubating 20 μg of membranes (isolated plasma membranes or endosomes) at 37°C for 15 min with labeled [α-32P]adenosine triphosphate (ATP) as described previously (44, 45). Isoproterenol was used instead of albuterol in the plasma membrane/endosomal resensitization experiments because it is a full receptor agonist, which enables greater G protein activation and results in measurable amounts of cAMP generated in vitro, especially in the endosomal fractions. The cAMP content was determined in the cytosol with a catch point cAMP kit (Molecular Devices) according to the manufacturer’s instructions (43).
Immunocytochemistry
HASMCs were grown on glass chamber slides and exposed to rrRSV at an MOI of 1.0 or sterile medium for 2.5 hours and then were incubated as indicated in the figure legends. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 for 5 min, blocked with 3% bovine serum albumin in phosphate-buffered saline, and then incubated with primary antibodies against β2AR or pβ2AR (355,356) for 1 hour, followed by Alexa Fluor–conjugated secondary antibody (Invitrogen). Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs) was applied, and coverslips were mounted and sealed before imaging. Images were collected with an upright fluorescent or confocal microscope (Leica Microsystems) with a 405-diode laser to excite DAPI and a HeNe laser to excite the Alexa Fluor 488–labeled secondary antibody. Cells were visualized with a 40× or 63×/1.4 oil objective. Densitometry was performed with National Institutes of Health (NIH) ImageJ software.
cAMP measurements
For intracellular cAMP measurements, cells lysates were analyzed at designated times after infection and in response to isoproterenol (1 μM) or propranolol (10 μM). Assays were performed according to the manufacturers’ instructions (Molecular Devices).
Collagen gel contraction assay
This assay was performed to determine changes in smooth muscle cell contractility in response to RSV infection. Cells were trypsinized, pelleted, suspended, and counted. Cells suspended in DMEM-F12 (5 × 105 cells/ml) were mixed with an equal volume of type I rat tail collagen solution to give a final collagen concentration of 1.5%, with 0.5 ml per well in 24-well cell culture plates. On the next day, serum was withdrawn for 24 hours before the cells were infected with rrRSV at an MOI of 1 for an additional 48 hours in serum-free medium. The collagen gels were then loosened from the plate, and the medium was removed and replaced with Krebs-Henseleit buffer (Sigma-Aldrich) with calcium chloride (0.373 g/liter) and sodium bicarbonate (2.1 g/liter). Cells were pretreated or not with 200 μM albuterol for 30 min before being treated with 200 μM methacholine to initiate contraction. The concentration of albuterol was increased two-log fold to enable its timely diffusion through the collagen gel matrix to the cells. The concentration of methacholine used was chosen on the basis of the previously published data to ensure that changes in contractility in either direction could be detected (46). Images were acquired with an Olympus SZ61 microscope with a Leica EC3 camera. NIH ImageJ software was used to analyze gel area. Data were expressed as percentage gel contraction and calculated using the following equation: [(T0 gel area − T30 gel area)/T0 gel area × 100%].
Measurement of intracellular Ca2+
The intracellular Ca2+ response to carbachol or methacholine was measured with the Fluorogenic Imaging Plate Reader Assay (Molecular Devices) in 80 to 90% confluent HASMCs grown in black-walled, clear-bottom Costar 96-well plates. Cells were loaded with Calcium-6 dye for 3 hours and incubated at 37°C in 5% CO2 according to the manufacturer’s instructions. Plates were read in a FlexStation 3 reader, and fluorescence was measured over 2 min at 6-s intervals upon the addition of carbachol or methacholine (100 nM to 100 μM in dimethyl sulfoxide; Sigma-Aldrich) or vehicle. The area under the curve (AUC) of the measured fluorescence was used to calculate changes in intracellular Ca2+. All samples were measured in quadruplicate, and the experiments were repeated with cells from three donors.
Virus propagation
To easily verify active viral replication in our cells, we used a recombinant RSV-A2 strain expressing the RFP gene (rrRSV), which was provided by M. Peeples (Nationwide Children’s Hospital, Columbus, OH) and P. Collins (NIH, Bethesda, MD). The virus stock was propagated with HEp-2 cells grown at 37°C and 5% CO2 in Eagle’s minimum essential medium supplemented with 10% FBS and 1% each of GlutaMAX, penicillin/streptomycin, and Hepes. Cells at approximately 50% confluence were inoculated with 3 ml per dish of virus stock diluted 1:10 with heat-inactivated FBS. After incubation for 2 hours at 37°C, the inoculum was removed and replaced with 25 ml of fresh medium. The medium was replaced again 2 days later, and the virus was harvested after 1 to 2 additional days of incubation, at which point all cells appeared bright red when viewed under a fluorescent microscope. To harvest the virus, infected cells were scraped from the plate, separated with a pipette, mixed at medium speed, and pelleted by centrifugation at 1200g for 5 min. The supernatant was collected, and cell debris was removed by centrifugation at 9500g for 20 min in a centrifuge at 4°C. Aliquots (1 ml each) of the supernatant were snap-frozen in liquid nitrogen and stored at −70°C until needed. The final titer was determined with a modified plaque-forming unit assay as previously described (47).
Statistical analysis
All data are expressed as means ± SEM. Multiple comparisons were performed with the nonparametric Kruskal-Wallis test and corrected for multiple comparisons with the Dunn’s test. Additional statistical analysis was performed with the mixed-effects model with Geisser-Greenhouse correction or one-way analysis of variance (ANOVA) with the Bonferroni’s multiple comparison test. All calculations were performed with GraphPad Prism software version 5.0. P < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments:
This article is dedicated to the memory of M. E. Lauer, whose diligence and passion for science is left imprinted on all those lucky enough to have met and worked with him. We are indebted to M. Peeples (Nationwide Children’s Hospital Research Institute, Columbus, OH) and P. Collins (NIH, Bethesda, MD) for providing the original batch of the RFP-expressing RSV.
Funding:
These experiments were funded, in part, by grants from the U.S. NIH: NHLBI RO1 HL-61007 (to G.P.), NHLBI RO1 HL-127602 (to S.V.N.P.), and NIH-NHLBI RO1-HL-148057 (to F.R.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ, cDNA for the human β2-adrenergic receptor: A protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc. Natl. Acad. Sci. U.S.A 84, 46–50 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes PJ, Basbaum CB, Nadel JA, Roberts JM, Localization of β-adrenoreceptors in mammalian lung by light microscopic autoradiography. Nature 299, 444–447 (1982). [DOI] [PubMed] [Google Scholar]
- 3.Hamid QA, Mak JCW, Sheppard MN, Corrin B, Craig Venter J, Barnes PJ, Localization of β2-adrenoceptor messenger RNA in human and rat lung using in situ hybridization: Correlation with receptor autoradiography. Eur. J. Pharmacol 206, 133–138 (1991). [DOI] [PubMed] [Google Scholar]
- 4.Shore SA, Drazen JM, β-agonists and asthma: Too much of a good thing? J. Clin. Invest 112, 495–497 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shore SA, Moore PE, Regulation of β-adrenergic responses in airway smooth muscle. Respir. Physiol. Neurobiol 137, 179–195 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Müller S, Hekman M, Lohse MJ, Specific enhancement of beta-adrenergic receptor kinase activity by defined G-protein beta and gamma subunits. Proc. Natl. Acad. Sci. U.S.A 90, 10439–10443 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penn RB, Benovic JL, Regulation of heterotrimeric G protein signaling in airway smooth muscle. Proc. Am. Thorac. Soc 5, 47–57 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seibold A, Williams B, Huang Z-F, Friedman J, Moore RH, Knoll BJ, Clark RB, Localization of the sites mediating desensitization of the β2-adrenergic receptor by the GRK pathway. Mol. Pharmacol 58, 1162–1173 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Vasudevan NT, Mohan ML, Goswami SK, Naga Prasad SV, Regulation of β-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 10, 3684–3691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan SJ, Deshpande DA, Tiegs BC, Misior AM, Yan H, Hershfeld AV, Rich TC, Panettieri RA, An SS, Penn RB, β-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J. Biol. Chem 289, 23065–23074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oguma T, Kume H, Ito S, Takeda N, Honjo H, Kodama I, Shimokata K, Kamiya K, Involvement of reduced sensitivity to Ca2+ in β-adrenergic action on airway smooth muscle. Clin. Exp. Allergy 36, 183–191 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Piedimonte G, Perez MK, Respiratory syncytial virus infection and bronchiolitis. Pediatr. Rev 35, 519–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright M, Piedimonte G, Respiratory syncytial virus prevention and therapy: Past, present, and future. Pediatr. Pulmonol 46, 324–347 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Merckx J, Ducharme FM, Martineau C, Zemek R, Gravel J, Chalut D, Poonai N, Quach C; Pediatric Emergency Research Canada (PERC) DOORWAY team, Respiratory viruses and treatment failure in children with asthma exacerbation. Pediatrics 142, e20174105 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Shay DK, Holman RC, Newman RD, Liu LL, Stout JW, Anderson LJ, Bronchiolitis-associated hospitalizations among US children, 1980–1996. JAMA 282, 1440–1446 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Moore PE, Cunningham G, Calder MM, DeMatteo AD Jr., Peeples ME, Summar ML, Peebles RS Jr., Respiratory syncytial virus infection reduces β2-adrenergic responses in human airway smooth muscle. Am. J. Respir. Cell Mol. Biol 35, 559–564 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA, Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates β-1 adrenergic receptor endocytosis through different pathways. J. Biol. Chem 278, 35403–35411 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Borissenko L, Groll M, 20S proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem. Rev 107, 687–717 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Hu JL, Van den Steen PE, Sang Q-XA, Opdenakker G, Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov 6, 480–498 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Obata K, Kojima T, Masaki T, Okabayashi T, Yokota S, Hirakawa S, Nomura K, Takasawa A, Murata M, Tanaka S, Fuchimoto J, Fujii N, Tsutsumi H, Himi T, Sawada N, Curcumin prevents replication of respiratory syncytial virus and the epithelial responses to it in human nasal epithelial cells. PLOS ONE 8, e70225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naqvi S, Martin KJ, Arthur JS, CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem. J 458, 469–479 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Bogard AS, Xu C, Ostrom RS, Human bronchial smooth muscle cells express adenylyl cyclase isoforms 2, 4, and 6 in distinct membrane microdomains. J. Pharmacol. Exp. Ther 337, 209–217 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belvisi MG, Birrell MA, The emerging role of transient receptor potential channels in chronic lung disease. Eur. Respir. J 50, 1601357 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE, Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med 352, 1749–1759 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Ralston SL, Lieberthal AS, Meissner HC, Alverson BK, Baley JE, Gadomski AM, Johnson DW, Light MJ, Maraqa NF, Mendonca EA, Phelan KJ, Zorc JJ, Stanko-Lopp D, Brown MA, Nathanson I, Rosenblum E, Sayles S, Hernandez-Cancio S, Clinical practice guideline: The diagnosis, management, and prevention of bronchiolitis. Pediatrics 134, e1474–e1502 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Gadomski AM, Scribani MB, Bronchodilators for bronchiolitis. Cochrane Database Syst. Rev 2014, CD001266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Zheng J, Zheng K, Hou Y, Zhao F, Zhao D, Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 57, 65–73 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Komaravelli N, Ansar M, Garofalo RP, Casola A, Respiratory syncytial virus induces NRF2 degradation through a promyelocytic leukemia protein-ring finger protein 4 dependent pathway. Free Radic. Biol. Med 113, 494–504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin L, Peng D, Hu C, Xiang Y, Zhou Y, Tan Y, Qin X, Differentiation of Th subsets inhibited by nonstructural proteins of respiratory syncytial virus is mediated by ubiquitination. PLOS ONE 9, e101469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakalahti AE, Khan H, Vierimaa MM, Pekkala EH, Lackman JJ, Ulvila J, Kerkelä R, Petäjä-Repo UE, β-Adrenergic agonists mediate enhancement of β1-adrenergic receptor N-terminal cleavage and stabilization in vivo and in vitro. Mol. Pharmacol 83, 129–141 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schönbein GW, Matrix metalloproteinases cleave the β2-adrenergic receptor in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol 299, H25–H35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao K, Shenoy SK, β2-adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J. Biol. Chem 286, 12785–12795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Q, Gros R, Gray ID, Taussig R, Ferguson SSG, Feldman RD, Raf kinase activation of adenylyl cyclases: Isoform-selective regulation. Mol. Pharmacol 66, 921–928 (2004). [PubMed] [Google Scholar]
- 34.Monick MM, Staber JM, Thomas KW, Hunninghake GW, Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J. Immunol 166, 2681–2687 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Ihnatovych I, Novotny J, Haugvicova R, Bourova L, Mareš P, Svoboda P, Ontogenetic development of the G protein-mediated adenylyl cyclase signalling in rat brain. Brain Res. Dev. Brain Res 133, 69–75 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Taylor CT, Furuta GT, Synnestvedt K, Colgan SP, Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc. Natl. Acad. Sci. U.S.A 97, 12091–12096 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu T, Zaman W, Kaphalia BS, Ansari GAS, Garofalo RP, Casola A, RSV-induced prostaglandin E2 production occurs via cPLA2 activation: Role in viral replication. Virology 343, 12–24 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Morris DR, Ansar M, Ivanciuc T, Qu Y, Casola A, Garofalo RP, Selective blockade of TNFR1 improves clinical disease and bronchoconstriction in experimental RSV infection. Viruses 12, 1176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillou JL, Nakata H, Cooper DM, Inhibition by calcium of mammalian adenylyl cyclases. J. Biol. Chem 274, 35539–35545 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Peebles RS Jr., Graham BS, Pathogenesis of respiratory syncytial virus infection in the murine model. Proc. Am. Thorac. Soc 2, 110–115 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrino C, Naga Prasad SV, Schroder JN, Hata JA, Milano C, Rockman HA, Restoration of β-adrenergic receptor signaling and contractile function in heart failure by disruption of the betaARK1/phosphoinositide 3-kinase complex. Circulation 111, 2579–2587 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA, Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1: A role in receptor sequestration. J. Biol. Chem 276, 18953–18959 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Naga Prasad SV, Inhibition of protein phosphatase 2A activity by PI3Kγ regulates β-adrenergic receptor function. Mol. Cell 41, 636–648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi DJ, Koch WJ, Hunter JJ, Rockman HA, Mechanism of β-adrenergic receptor desensitization in cardiac hypertrophy is increased β-adrenergic receptor kinase. J. Biol. Chem 272, 17223–17229 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA, Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J. Clin. Invest 112, 1067–1079 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakota Y, Ozawa Y, Yamashita H, Tanaka H, Inagaki N, Collagen gel contraction assay using human bronchial smooth muscle cells and its application for evaluation of inhibitory effect of formoterol. Biol. Pharm. Bull 37, 1014–1020 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Othumpangat S, Gibson LF, Samsell L, Piedimonte G, NGF is an essential survival factor for bronchial epithelial cells during respiratory syncytial virus infection. PLOS ONE 4, e6444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.