

Abstract
Assemblies of racemic β-sheet-forming peptides have attracted attention for biomedical applications because racemic forms of peptides can self-associate more avidly than do single enantiomers. In 1953, Pauling and Corey proposed “rippled β-sheet” modes of H-bond-mediated interstrand assembly for alternating L- and D-peptide strands; this structural hypothesis was complementary to their proposal of “pleated β-sheet” assembly for L-peptides. Although no high-resolution structure has been reported for a rippled β-sheet, there is strong evidence for the occurrence of rippled β-sheets in racemic peptide assemblies. Here we compare propensities of peptide diastereomers in aqueous solution to form a minimum increment of β-sheet in which two antiparallel strands associate. β-Hairpin folding is observed for homochiral peptides with aligned nonpolar side chains, but no β-hairpin population can be detected for diastereomers in which one strand contains L residues and the other contains D residues. These observations suggest that rippled β-sheet assemblies are stabilized by interactions between β-sheet layers rather than interactions within these layers.
Keywords: Peptide conformational, β-hairpin, Pleated β-sheet, Rippled β-sheet, Diastereomers
Graphical Abstract

As predicted by Pauling and Corey, pairing of homochiral (all-L) peptide strands leads to pleated β-sheets, while pairing of L and D strands leads to rippled β-sheets. Our evidence indicates that heterochiral strand pairing in aqueous solution is less favorable than homochiral strand pairing.
β-Sheet is a common secondary structural motif within folded proteins, and intermolecular β-sheet interactions are crucial for pathogenic amyloid formation.[1,2] The β-sheets found in natural proteins and natural amyloids necessarily feature associations between homochiral strand segments because ribosomal biosynthesis incorporates exclusively L enantiomers of α-amino acids.[3] However, early modeling of H-bond-mediated associations between extended peptide strands led Pauling and Corey to predict not only homochiral modes of assembly (“pleated-sheets”) but also heterochiral modes (“rippled-sheets”), in which L- and D-peptide strands alternate.[4,5]
High-resolution structural characterization of folded proteins and more recently of amyloid assemblies has provided ample characterization of pleated β-sheets formed via intra- or intermolecular associations of homochiral peptide strands.[2,6] There have been far fewer studies of systems containing both L- and D-peptide segments, a requirement for rippled β-sheet formation. Schneider et al. and Nilsson et al. have provided strong evidence for rippled β-sheet occurrence in racemic peptide assemblies.[7–9] These groups employed isotopic labeling to show that L-strands formed H-bonding interactions with D-strands, as required for a rippled β-sheet. Racemic co-assembly for sheet-forming peptides can be used to inhibit pathological amyloid formation[10,11] or to enhance materials properties.[7] However, it is also possible for racemic peptides to form assemblies in which H-bonding interactions occur between homochiral strands. Rodriguez et al. have determined crystal structures for a racemic hexapeptide that feature pleated β-sheets, formed by exclusively L-peptides or exclusively D-peptides, with packing of L-β-sheets against D-β-sheets mediated by side chains.[12] Even when spectroscopic evidence supports H-bonding interactions in a rippled β-sheet mode (i.e., alternating L- and D-strands along the H-bonding direction), Schneider et al. have argued that a preference for heterochiral vs. homochiral peptide assembly is determined by side chain interactions involved in the packing of β-sheet layers against one another.[7]
The factors that influence formation of pleated β-sheet secondary structure have been widely explored with designed peptides that form two-stranded β-sheet conformations (“β-hairpins”).[13,14] This experimental strategy avoids the influence of a specific tertiary structure context, which is unavoidable when model proteins are employed.[15] Here, we use a β-hairpin-based approach to compare intramolecular association of homochiral vs. heterochiral peptide segments, i.e., antiparallel strand association in a pleated vs. a rippled β-sheet mode, in aqueous solution.
In an important precedent to our work, Chung and Nowick showed that in a nonpolar solvent, where interstrand associations should be driven by H-bonding, homochiral interactions are significantly more favorable than heterochiral interactions.[16] β-Hairpin folding in aqueous solution, however, is strongly influenced by favorable interactions among side chains on neighboring strands,[13,14,17] and it is therefore possible that the relative preference for homochiral vs. heterochiral interstrand association differs between aqueous and nonpolar solvents. Our work is motivated by this consideration.
As a first step to explore the relationship between residue chirality and strand pairing in water, in the absence of higher-order assembly, we examined the diastereomeric peptide pair in Figure 1. Peptide LL1 features the “tryptophan zipper” (Trpzip) motif,[18] which leads to formation of β-hairpins that are stabilized by cross-strand interactions among Trp side chains. In addition to two segments intended to form strands (residues 1–5 and 8–12), this peptide contains a central Aib-Gly unit, which has a high propensity to form a reverse turn. The Aib-Gly turn has been shown to support β-hairpin folding when embedded in peptides otherwise comprised of L residues.[19,20] This locally achiral turn segment was chosen to support comparisons that involve strand segments of differing configurations, e.g., between homochiral LL1 and heterochiral isomer LD1.
Figure 1.

Trpzip peptides designed to compare homochiral and heterochiral strand association. L-Trp residues are highlighted in purple, and D-Trp residues are highlighted in blue. LL1 features a stable homochiral β-hairpin motif.
If the Aib-Gly segment forms a two-residue loop, as implied in the drawing of LL1, then the Trp residues occupy “non-H-bonded” positions in the strand segments, i.e., the backbone N-H and C=O groups of these residues are oriented away from the neighboring strand. For homochiral peptides, side chains of residues in non-H-bonded positions within β-hairpins project from the same side of the β-hairpin and are oriented toward the other strand.[13,14,21] This positioning promotes cross-strand interactions of side chains from residues that are aligned in non-hydrogen bonded positions, as is the case for the Trp2/Trp11 pair and the Trp4/Trp9 pair in LL1.
Peptide conformational behavior was examined via NMR spectroscopy with 1 mM samples dissolved in 9:1 H2O:D2O. Resonances were assigned based on two-dimensional NMR data, as explained in the Supporting Information. The chemical shifts of amide NH resonances showed little or no change between 0.3 and 5 mM (Fig. S60 and S61), which suggests that data acquired with 1 mM samples were not influenced by peptide self-association.
Two-dimensional NMR analysis of LL1 revealed several cross-strand NOEs that are consistent with formation of the expected β-hairpin conformation (Figure 2A).[22,23] Specifically, the backbone NH of Thr10 displayed NOEs with the side chains of Trp2 and Trp4 as well as with the backbone NH of Thr3 and Glu5. Such cross-strand NOEs are generally regarded as the strongest evidence for β-hairpin folding in solution. The presence of a few NOEs between adjacent backbone NH groups (Trp4-Glu5, Trp9-Thr10 and Thr10-Trp11) suggests that LL1 equilibrates rapidly between the β-hairpin and alternative (“unfolded”) conformations on the NMR timescale, as is common for peptides of this size in water, if they fold at all.[13,14,17,18]
Figure 2.
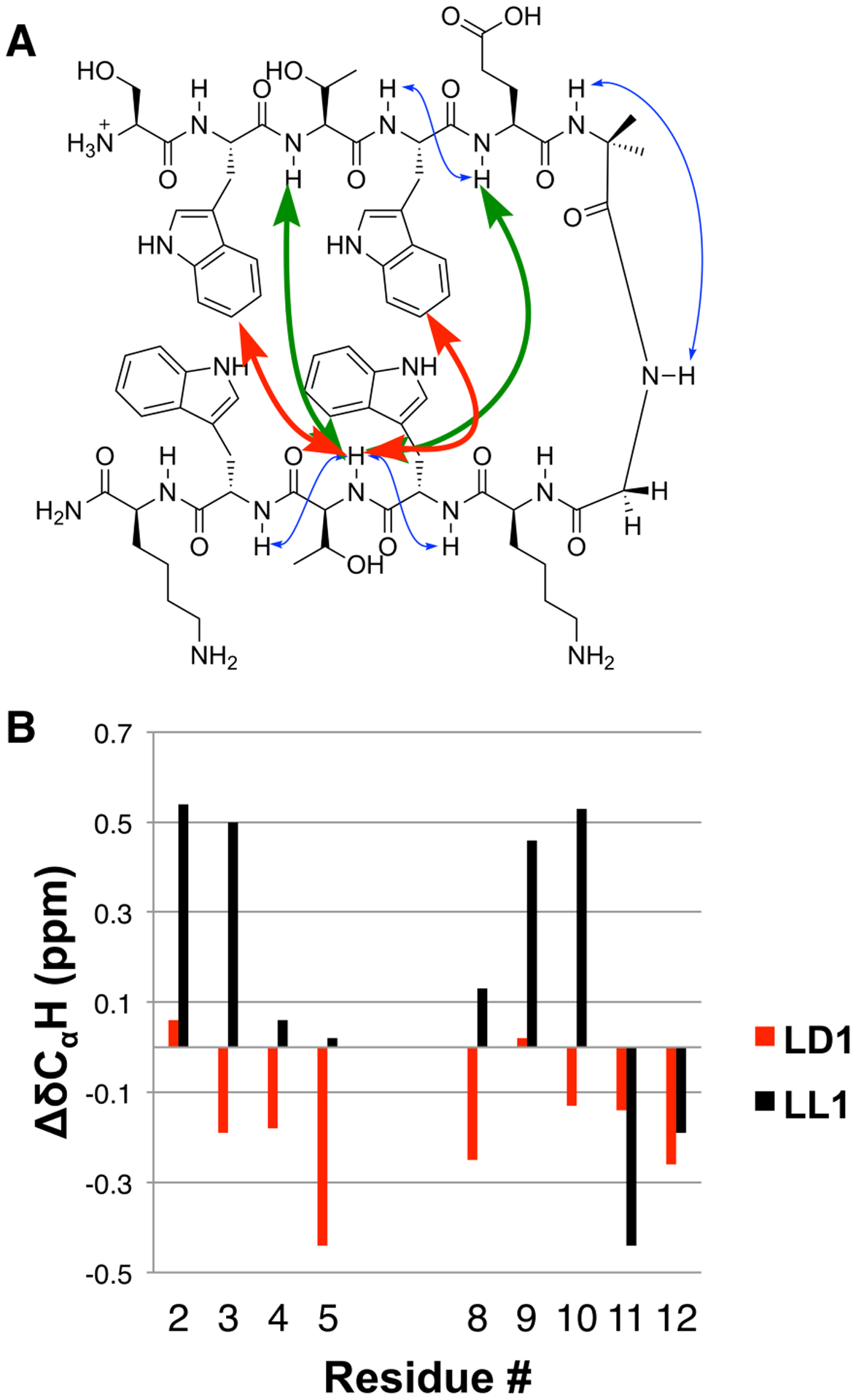
(A) Selected NOEs for LL1 (1 mM in 9:1 H2O:D2O). Red arrows indicate non-sequential NOEs involving side chains. Green arrows indicate non-sequential NOEs involving only backbone protons. Blue arrows indicate sequential backbone NOEs. (B) CαH chemical shift patterns for peptides LL1 and LD1. ΔδCαH=δCαH(obs)− δCαH(RC), with δCαH (RC) values obtained from ref. [25]. No value is shown for residue 1 because the N-terminus is not acylated. No values are shown for Aib-Gly to highlight the strand region.
Further evidence of significant β-hairpin folding was provided by CαH chemical shift patterns.[24,25] Relative to the “random coil” value for a given residue, δCαH is usually upfield for residues in an α-helix and downfield for residues in a β-sheet.[24] For LL1, ΔδCαH [= δCαH(obs) − δCαH(RC)] values of > 0.1 ppm (Figure 2B) suggest that segment Lys8-Trp9-Thr10 participates in β-sheet secondary structure, as expected for the predicted β-hairpin conformation. In the N-terminal segment, Trp2 and Thr3 show strong positive ΔδCαH values. Shielding effects from aromatic side chains can cause deviations from the intrinsic effects of local secondary structure on ΔδCαH values, which may explain the small ΔδCαH values of residues near the Aib-Gly turn segment.
The clustering of side chain indole groups that is characteristic of Trpzip β-hairpin conformations gives rise to a characteristic maximum at 228 nm in the circular dichroism (CD) spectrum.[26] LL1 at 50 μM manifests a strong signal of this type (Fig. S62). The intensity of the 228 nm diminishes as the sample is heated (Fig. S63), which suggests thermal denaturation of the β-hairpin conformation. The denaturation process was reversible. The melting curve was fitted to a two-state transition model assuming there is a difference in heat capacity between the folded and unfolded forms.[27] An equimolar mixture of peptides SWTWE-Aib and GKWTWK was used to provide a CD standard for the unfolded state. This analysis suggested that LL1 is ≥ 90% folded at 25 °C. Overall, the NMR and CD data are consistent with conclusion that a two-stranded antiparallel pleated β-sheet conformation is substantially populated for peptide LL1 in aqueous solution, as expected based on precedent.[18]
The diastereomeric peptide in which the five residues nearest the C-terminus are derived from D-amino acids, LD1, displays no evidence of β-hairpin formation in aqueous solution. No cross-strand NOEs were detected (Fig. S32), and ΔδCαH < 0 for all residues (Figure 2B). Thus, the evidence suggests that LD1 does not form a two-stranded antiparallel rippled β-sheet in aqueous solution.
Because of the differences in configuration for residues 8–12 between LL1 and LD1, formation of a β-hairpin centered on the Aib-Gly turn, as implied by the drawings in Figure 1, would lead to different spatial arrangements of the Trp side chains. All four side chains of LL1 project from the same side of the hairpin, but for LD1, the side chains from L-Trp2 and L-Trp4 would project from the opposite side of the hairpin relative to those from D-Trp9 and D-Trp11. Thus, this hypothetical hairpin conformation of LD1 would not benefit from favorable cross-strand Trp side chain interactions that are presumably a major driving force for folding of LL1.[18] This consideration prompted us to consider two addition diastereomeric peptide pairs, LL2/LD2 and LL3/LD3.
In heterochiral peptides LD2 and LD3, there are sequential shifts of the D-Trp residues along the C-terminal segment, relative to LD1, that would allow all four Trp side chains to project from the same side of β-hairpins anchored by an Aib-Gly reverse turn, as implied by the drawings in Figure 3A. The N-terminal Trp side chain pair would be offset relative to the C-terminal Trp side chain pair in terms of the interstrand H-bonding pattern, but this offset would not necessarily preclude favorable cross-strand interactions. In all-L β-hairpins, side chains that are offset in terms of the H-bonding pattern can nevertheless engage in favorable “diagonal” interstrand interactions.[13,17]
Figure 3.
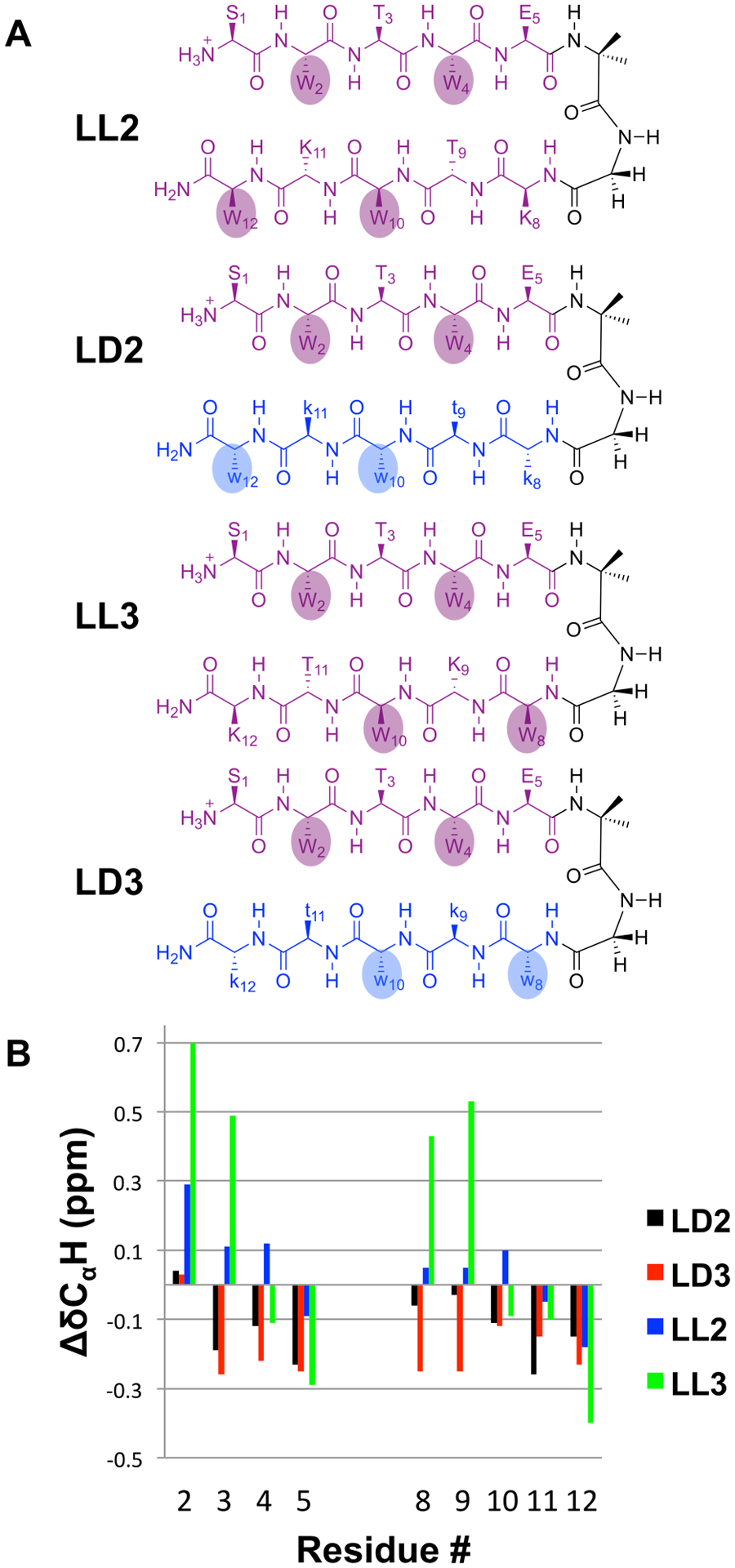
(A) Sequence isomers of LL1 and LD1, with alternative locations of Trp residues in the C-terminal segment. The positioning of Trp residues in LD2 and LD3 is intended to promote Trp side chain interaction. (B) CαH chemical shift patterns for peptides LD2, LD3, LL2, and LD3.
In homochiral peptides LL2 and LL3, the sequential shifts of the two Trp residues that follow the Aib-Gly segment would cause the N-terminal pair of Trp side chains to project from the opposite side of the β-hairpin relative to the C-terminal pair of Trp side chains, if the hairpins formed as implied by the drawings. These side chain orientations could discourage formation of a β-hairpin centered on the Aib-Gly reverse turn for LL2 or LL3. However, β-hairpin conformations centered on alternative reverse turns would allow clustering of the four Trp side chains from LL2 or LL3 in a Trpzip motif.
Collectively, NMR and CD data for LL2 and LL3 indicate that neither has a β-hairpin propensity as strong as that of LL1. Thus, LL2 and LL3 display a CD maximum at 228 nm, but in both cases the normalized intensity is lower than the maximum for LL1 (Fig. S62). Neither LL2 nor LL3 displays a cluster of cross-strand NOEs consistent with a single β-hairpin conformation (Fig. S41 and S47), in contrast to LL1. For LL3, multiple residues display large, positive ΔδCαH values (Figure 3B, S58), which may indicate a modest extent of folding in this case.
NMR data for LD2 and LD3 do not provide any evidence for formation of rippled β-sheet secondary structure. For most residues in each heterochiral peptide, ΔδCαH < 0 (Figure 3B).[24] No NOEs between protons from non-sequentially adjacent residues were detected for LD3, and only a single NOE of this type was observed for LD2 (Fig. S38). Overall, the NMR data suggest that none of the LD peptides has a significant propensity to adopt a specific secondary structure.
Collectively, the data for homochiral peptides LL1-LL3 and heterochiral isomers LD1-LD3 show that well-established design principles for generating a two-stranded pleated β-sheet in aqueous solution, based on the Trpzip motif,[18] cannot be extended to generate a two-stranded rippled β-sheet. These principles deliver the expected β-hairpin folding for LL1, but none among the three heterochiral isomers displays evidence of β-sheet secondary structure.
In order to determine whether the conclusions drawn above might be specific to tryptophan-rich sequences, we compared diastereomeric peptides based on a different design (Figure 4). Peptide LL4 contains a central Aib-Gly segment to promote the necessary reverse turn. Each strand has the same sequence, KVFFK, with each residue derived from an L-amino acid. The three central hydrophobic residues were inspired by the Val-Phe-Phe motif found in the amyloid-forming peptide Aβ(1–42);[9,10] interstrand interactions involving these hydrophobic side chains would be expected to stabilize a β-hairpin conformation in aqueous solution. The four Lys residues were incorporated to promote solubility and discourage aggregation. In the diastereomer LD4, the five residues nearest the C-terminus are derived from D-amino acids.
Figure 4.

Peptides containing the Val-Phe-Phe motif found in Aβ(1–42).
Several interstrand NOEs were detected for LL4 in aqueous solution (Figure 5A), and all were consistent with the predicted β-hairpin conformation.[22] In addition, we observed multiple NOEs between backbone NH groups on adjacent residues, which suggest that LL4 equilibrates rapidly between β-hairpin and unfolded conformations on the NMR timescale. ΔδCαH data for LL4 (Figure 5B) reveal several values > 0; it is possible that upfield shifts caused by aromatic side chain clustering in the folded state interferes with the downfield shifts that would be caused by β-sheet secondary structure at some positions.
Figure 5.

(A) Selected NOEs for LL4, 1 mM in 9:1 H2O:D2O. The red arrow indicates a non-sequential NOE involving a side chain. Green arrows indicate non-sequential NOEs involving only backbone protons. Blue arrows indicate sequential backbone NOEs in aqueous solution. (B) CαH chemical shift patterns for peptides LD4 and LL4. (C) Selected NOEs for LL4 in 1 mM in 9:1 H2O:D2O with 30% TFE-d3. Colors as in part (A), with purple arrows indicating additional non-sequential NOEs observed only in 30% TFE-d3.
The NMR data for heterochiral isomer LD4 suggest that the heterochiral peptide is completely unfolded in aqueous solution. No NOE was detected between sequentially non-adjacent residues (Fig. S49). The δCαH values were universally near or slightly below the random coil values.
Use of 2,2,2-trifluoroethanol (TFE) or another alcohol as a peptide co-solvent is known to enhance intrinsic secondary structure preferences, for both α-helix-prone[28] and β-sheet-prone[29] sequences. In 30% v/v TFE, LL4 displays a larger set of cross-strand NOEs relative to pure aqueous solution, and all NOEs are consistent with the expected β-hairpin conformation (Figure 5C). However, heterochiral peptide LD4 displays no cross-strand NOEs in 30% v/v TFE (Fig. S55).
The possibility that H-bond-mediated association of homochiral strands, to form an antiparallel pleated β-sheet, is more favorable than the comparable association of heterochiral strands, to form an antiparallel rippled β-sheet, does not conflict with recent observations that racemic peptides can undergo large-scale self-assembly involving rippled β-sheets. As noted by Schneider et al.,[7] the interactions of side chains from adjacent β-sheet layers in a higher-order assembly (i.e., side chain interactions along an axis that is orthogonal to the H-bonding direction within each sheet layer) can be more favorable for rippled β-sheet relative to pleated β-sheet. This insight highlights the balance of forces that controls higher-order peptide assembly, which does not always lead to rippled β-sheet formation for racemic peptides.[12]
Supplementary Material
Acknowledgements
This research was supported in part by NIH grant R01 GM061238. Instrumentation support was provided by NIH grant S10 OD012245 2013. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grant P41GM136463, old number P41GM103399 (NIGMS) and P41RR002301. Equipment was purchased with funds from the University of Wisconsin-Madison, the NIH P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, S10RR029220), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA. We are grateful to C. Clewett, P. F. Cobra, C. G. Fry, H. Hofstetter, and J. K. Vasquez for their help with NMR instrumentation.
References
- [1].Tycko R, Neuron 2015, 86, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Meier BH, Riek R, Böckmann A, Trends Biochem. Sci 2017, 42, 777–787. [DOI] [PubMed] [Google Scholar]
- [3].Englander MT, Avins JL, Fleisher RC, Liu B, Effraim PR, Wang J, Schulten K, Leyh TS, Gonzalez RL, Cornish VW, Proc. Natl. Acad. Sci 2015, 112, 6038–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pauling L, Corey RB, Proc. Natl. Acad. Sci 1953, 39, 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Raskatov JA, Schneider JP, Nilsson BL, Acc. Chem. Res 2021, 54, 2488–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paravastu AK, Leapman RD, Yau WM, Tycko R, Proc. Natl. Acad. Sci 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nagy-smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP, ACS Cent. Sci 2017, 3, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Swanekamp RJ, Dimaio JTM, Bowerman CJ, Nilsson BL, J. Am. Chem. Soc 2012, 134, 5556–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Urban JM, Ho J, Piester G, Fu R, Nilsson BL, Molecules 2019, 24, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dutta S, Foley AR, Warner CJA, Zhang X, Rolandi M, Abrams B, Raskatov JA, Angew. Chem. Int. Ed 2017, 56, 11506–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Torbeev V, Grogg M, Ruiz J, Boehringer R, Schirer A, Hellwig P, Jeschke G, Hilvert D, J. Pept. Sci 2016, 22, 290–304. [DOI] [PubMed] [Google Scholar]
- [12].Zee C, Glynn C, Gallagher-Jones M, Miao J, Santiago CG, Cascio D, Gonen T, Sawaya MR, Rodriguez JA, IUCrJ 2019, 6, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hughes RM, Waters ML, Curr. Opin. Chem. Biol 2006, 16, 514–524. [DOI] [PubMed] [Google Scholar]
- [14].Gellman SH, Curr. Opin. Chem. Biol 1998, 2, 717–725. [DOI] [PubMed] [Google Scholar]
- [15].Minor DLJ, Kim PS, Nature 1994, 371, 264–267. [DOI] [PubMed] [Google Scholar]
- [16].Chung DM, Nowick JS, J. Am. Chem. Soc 2004, 126, 3062–3063. [DOI] [PubMed] [Google Scholar]
- [17].Syud FA, Stanger HE, Gellman SH, J. Am. Chem. Soc 2001, 123, 8667–8677. [DOI] [PubMed] [Google Scholar]
- [18].Cochran AG, Skelton NJ, Starovasnik MA, Proc. Natl. Acad. Sci 2001, 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Masterson LR, Etienne MA, Porcelli F, Barany G, Hammer RP, Veglia G, Pept. Sci 2007, 88, 746–753. [DOI] [PubMed] [Google Scholar]
- [20].Huang R, Setnička V, Etienne MA, Kim J, Kubelka J, Hammer RP, Keiderling TA, J. Am. Chem. Soc 2007, 129, 13592–13603. [DOI] [PubMed] [Google Scholar]
- [21].Berg JM, Tymoczko JL, Stryer L, Biochemistry, Freeman WH, 2002.
- [22].Wüthrich K, NMR of Proteins and Nucleic Acids, Wiley, 1986. [Google Scholar]
- [23].Dyson HJ, Wright PE, Annu. Rev. Biophys. Biophys. Chem 1991, 20, 519. [DOI] [PubMed] [Google Scholar]
- [24].Fesinmeyer RM, Hudson FM, Andersen NH, J. Am. Chem. Soc 2004, 126, 7238–7243. [DOI] [PubMed] [Google Scholar]
- [25].Wishart SD, Bigam CG, Holm A, Hodges RS, Skyes BD, Bio J. NMR 1995, 5, 67–81. [DOI] [PubMed] [Google Scholar]
- [26].Grishina IB, Woody RW, Faraday Discuss. 1994, 99, 245–262. [DOI] [PubMed] [Google Scholar]
- [27].Greenfield NJ, Nat. Protoc 2007, 1, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jasanoff A, Fersht AR, Biochemistry 1994, 33, 2129–2135. [DOI] [PubMed] [Google Scholar]
- [29].Rajan R, Balaram P, Int. J. Pept. Protein Res 1996, 48, 328–336. [DOI] [PubMed] [Google Scholar]
- [30].Raskatov JA, Biopolymers 2021, 112, e23391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.