

Abstract
Screening for early‐stage disease is vital for reducing colorectal cancer (CRC)‐related mortality. Methylation of circulating tumor DNA has been previously used for various types of cancer screening. A novel cell‐free DNA (cfDNA) methylation‐based model which can improve the early detection of CRC is warranted. For our study, we collected 313 tissue and 577 plasma samples from patients with CRC, advanced adenoma (AA), non‐AA and healthy controls. After quality control, 187 tissue DNA samples (91 non‐malignant tissue from CRC patients, 26 AA and 70 CRC) and 489 plasma cfDNA samples were selected for targeted DNA methylation sequencing. We further developed a cfDNA methylation model based on 11 methylation biomarkers for CRC detection in the training cohort (area under curve [AUC] = 0.90 (0.85–0.94]) and verified the model in the validation cohort (AUC = 0.92 [0.88–0.96]). The cfDNA methylation model robustly detected patients pre‐diagnosed with early‐stage CRC (AUC = 0.90 [0.86–0.95]) or AA (AUC = 0.85 [0.78–0.91]). Here we established and validated a non‐invasive cfDNA methylation model based on 11 DNA methylation biomarkers for the detection of early‐stage CRC and AA. The utilization of the model in clinical practice may contribute to the early diagnosis of CRC.
Keywords: advanced adenoma, cell‐free DNA, colorectal cancer, early detection, methylation, sequencing
Cell‐free DNA (cfDNA) methylation is promising for colorectal cancer (CRC) screening. In this study, after DNA methylation sequencing of tissue and plasma samples, we established a cfDNA methylation model based on 11 DNA methylation biomarkers for the detection of early‐stage CRC and advanced adenoma. The utilization of the model may contribute to the non‐invasive early diagnosis of CRC.
Abbreviations
- AA
advanced adenoma
- AUC
area under curve
- AUROC
area under receiver operating characteristic curve
- BHP
Benjamini–Hochberg procedure
- CA19‐9
carbohydrate antigen 19‐9
- CEA
carcino‐embryonic antigen
- cfDNA
cell‐free DNA
- CpG
cytosine‐phosphoric‐guanine
- CRC
colorectal cancer
- ctDNA
circulating tumor DNA
- FFPE
formalin‐fixed paraffin‐embedded
- FIT
fecal immunological test
- gFOBT
guaiac‐based fecal occult blood test
- LASSO
Least Absolute Shrinkage and Selection Operator
- mt‐sDNA
multi‐target stool DNA
- NAA
non‐advanced adenoma
- NGS
next‐generation sequencing
- PCM
percentage of co‐methylated reads
- QC
quality control
- SD
standard deviation
1. Introduction
Colorectal cancer (CRC) is the third most common malignant neoplasm globally [1]. Despite improvements in treatment, the prognosis of CRC patients with advanced TNM stage remains poor. The 5‐year survival rate of stage IV CRC patients is 14% [2], in contrast to 91% for stage I CRC patients [3]. Therefore, screening for early‐stage CRC is one of the key strategies for reducing CRC‐associated mortality. Currently, the approaches for CRC screening can mainly be separated into two types, invasive colonoscopy and non‐invasive stool‐based CRC screening, such as guaiac‐based fecal occult blood test (gFOBT), fecal immunological test (FIT) and multi‐target stool DNA (mt‐sDNA) test [4]. The compliance rate of colonoscopy is fairly low due to its invasiveness, high cost and the requirement for extensive bowel preparation [5]. The mt‐sDNA test has been criticized for its relatively high false‐positive rate, which sometimes leads to unnecessary treatment [6]. Although gFOBT and FIT are much more cost‐effective and convenient than colonoscopy, they have a relatively low sensitivity for detecting advanced adenoma (AA) and early‐stage CRC [7]. Therefore, a non‐invasive CRC screening test with high sensitivity and specificity is urgently needed.
DNA methylation is one of the epigenetic mechanisms that cells use to regulate gene expression [8]. It is well recognized that aberrant hypermethylation of cytosine‐phosphoric‐guanine (CpG) islands in tumor suppressor genes can result in transcriptional silencing and carcinogenesis [9]. Hypermethylation of tumor suppressor genes has been found to be an early event in many cancers [10, 11, 12, 13]. Moreover, aberrant methylation is dynamic and potentially reversible, making it a potential target for treatment [14]. Therefore, DNA methylation pattern holds a profound potential as a biomarker in cancer screening and monitoring.
Circulating tumor DNA (ctDNA) consists of extracellular nucleic acid fragments that are released into the blood via necrosis, apoptosis or active DNA secretion by tumor cells [15]. The quantity of cell‐free DNA (cfDNA) has been reported to be higher in several tumors, especially in those patients with advanced cancer stage, than in healthy individuals [16, 17, 18]. The amount of cfDNA was shown to be related to tumor size and clinical stage [19, 20]. Moreover, it has been reported that methylation levels of cfDNA in plasma are consistent with those in the primary tumor [21, 22, 23]. These findings suggest that alterations to the cfDNA methylation signature might be able to serve as ideal biomarkers for non‐invasive cancer screening and diagnosis [24].
In this study, we aimed to evaluate the potential value of cfDNA methylation pattern as a biomarker for the screening and diagnosis of CRC. We developed a novel CRC‐specific cfDNA methylation model using high‐throughput targeted DNA methylation sequencing. The CRC‐specific cfDNA methylation model was generated and further refined using a cohort of plasma cfDNA samples. An independent validation cohort was used to validate the robustness and accuracy of this model.
2. Materials and methods
2.1. Patient enrollment and sample collection
The DNA samples used in this study were obtained from fresh‐frozen tissues, formalin‐fixed paraffin‐embedded (FFPE) tissues, and plasma. All of the specimens were collected at The Sixth Affiliated Hospital of Sun Yat‐sen University from August 2016 to May 2018. This study was approved by the ethics committee of The Sixth Affiliated Hospital of Sun Yat‐sen University (2016ZSLYEC‐056). The experiments were undertaken with the understanding and written consent of each subject and the study methodologies conformed to the standards set by the Declaration of Helsinki.
2.1.1. Tissue samples
The tumor tissues and the corresponding adjacent normal tissues were derived from patients receiving CRC resection. The adjacent normal tissues were collected from the normal intestine, which were more than 5 cm away from the primary tumor. The tissue samples of AA were obtained from FFPE specimens from The Sixth Affiliated Hospital of Sun Yat‐sen University.
2.1.2. Plasma samples
A 10‐mL aliquot of blood were drawn from healthy controls or treatment‐naive patients using BD Vacutainer® EDTA Tubes (Becton, Dickinson and Company, Plymouth, UK, Cat# 367525) and the plasma was immediately separated within 2 h after blood draw and was stored at −80 °C for a median of 9 days (range: 1–35 days) until DNA isolation and subsequent assays. The resultant plasma volume ranged from 2.0 to 3.2 mL. The healthy controls consisted of patients with benign anorectal diseases, such as hemorrhoids, anal fissures and perianal fistulae. To be included in this study, the healthy controls had to meet the following inclusion criteria: (1) age older than 18 years old, (2) no other significant medical history, such as cancer or chronic diseases, (c) willingness before the blood draw to have a colonoscopy which was normal.
2.2. Isolation of tissue genomic DNA and plasma cell‐free DNA
Tissue genomic DNA was isolated from fresh‐frozen and FFPE tissue samples using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany, Cat#: 69504) and the QIAamp DNA FFPE Tissue Kit (Qiagen, Cat# 56404), respectively.
Cell‐free DNA was isolated from plasma using the Bioo NextPrep‐Mag™ cfDNA Isolation Kit (Bioo Scientific, Austin, TX, USA, Cat# NOVA‐3825). Repeated freezing and thawing of plasma were avoided to prevent cfDNA degradation. The concentration and quality of cfDNA were determined using the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Eugene, OR, USA, Cat# Q32854) and the Agilent High Sensitivity DNA Kit (Agilent, Waldbrann, Germany, Cat# 5067‐4626) on a 2100 Bioanalyzer Instrument (Agilent), which assessed the size distribution of cfDNA. The cfDNA with yield > 3 ng and without overt genomic DNA contamination was used for sequencing library construction.
2.3. Bisulfite conversion
Bisulfite conversion was performed using the Zymo Lightning Conversion Reagent (Zymo Research, Irvine, CA, USA, Cat# D5031) according to the manufacturer’s protocols. For tissue samples, 2 μg of genomic DNA was fragmented into ~ 200‐bp fragments (peak size) by an M220 Focused‐ultrasonicator (Covaris, Inc., Boston, MA, USA) following the manufacturer’s instructions, and 800 ng of purified fragmented genomic DNA was then used for the following bisulfite conversion. After bisulfite conversion, the purified bisulfite‐converted DNA was quantified at A260 by NanoDrop (Thermo Fisher Scientific). Then, 100 and 150 ng of the bisulfite‐converted products were applied for library preparation for fresh‐frozen and FFPE tissue samples, respectively. For plasma samples, the recommended input of cfDNA for bisulfite conversion was 10 ng. If cfDNA yield was between 3 and 10 ng, all the purified cfDNA was used for bisulfite conversion. After bisulfite conversion, we used all the bisulfite‐converted cfDNA for library preparation without DNA quantification to avoid cfDNA loss. Following DNA bisulfite conversion, the bisulfite‐converted DNA was run through a Zymo‐Spin™ IC Column (Zymo Research, Cat# D5031), washed and desulfonated, and then eluted twice using the M‐Elution buffer to a final volume of 17 μL.
2.4. AnchorIRIS™ pre‐library construction
AnchorIRIS™ (Guangzhou, Guangdong) pre‐hyb library construction was performed using AnchorDx EpiVisioTM Methylation Library Prep Kit (AnchorDx, Guangzhou, China, Cat# A0UX00019) and AnchorDx EpiVisioTM Indexing PCR Kit (AnchorDx, Cat# A2DX00025). Following the procedure of end pair reparation, 3′ end adaptor ligation, and amplification of reverse complement DNA (Liang et al. [25]), the amplified DNA was purified using 1 : 6 Agencourt AMPure XP Magnetic Beads (Beckman Coulter, Brea, CA, USA, Cat# A63882). After 3′ end adaptor ligation of reverse complement DNA and indexing PCR (i5 and i7; Liang et al. [25]), the amplified pre‐libraries were subsequently purified using XP Magnetic Beads. Pre‐hyb libraries containing more than 800 ng DNA were used for target enrichment assay.
2.5. AnchorIRIS™ target enrichment
Target Enrichment was performed using AnchorDx EpiVisioTM Target Enrichment Kit (AnchorDx, Cat# A0UX00031) and methylation panels, AnchorDx PanMet V1 or V2. A total of 1000 ng of DNA containing up to four pre‐hyb libraries were pooled for target enrichment using AnchorDx PanMet V1 or V2 methylation panel. AnchorDx PanMet V2 included 12 624 pre‐selected regions which were enriched for cancer‐specific methylation and contained all the 9921 regions of AnchorDx PanMet V1. The total size of the genomic regions targeted by the AnchorDx PanMet V1 and V2 panel was 563 272 and 733 057 bp, which covered 45 566 and 55 369 CpG sites, respectively. Probe hybridization, purification and final PCR amplification were carried out according to the protocols from Liang [25].
2.6. DNA methylation level calculation
Enriched libraries were sequenced by Illumina HiSeq X Ten Sequencing System. Percentage of co‐methylated reads (PCM) was calculated by the analysis pipeline developed by Liang [25].
Reads having at least three methylated CpGs within a sliding window of five CpGs were designated as co‐methylated reads and used for subsequent analysis of methylation pattern and predictive modeling of malignant/normal states of patient samples. Log2 PCM was used for the model construction to optimize the model’s performance and stability.
2.7. Statistical analysis
2.7.1. CRC‐specific cfDNA methylation biomarkers for diagnostic analysis
With the AnchorDx PanMet methylation panels, we first performed a differential methylation analysis in normal, AA and CRC tissue samples using Wilcoxon signed‐rank test, where the P‐value for each methylation biomarker was corrected by multiple testing through the Benjamini–Hochberg procedure (BHP) to control the false‐discovery rate at a significance level of 0.05. We also calculated the distinguishing power by the area under receiver operating characteristic curve (AUROC) and absolute methylation change for each biomarker. CRC‐specific DNA methylation biomarkers were identified using the following criteria:
significant difference between CRC and normal tissue samples (adjusted P < 0.05) with relatively large absolute change (> 0.2);
significant difference between AA and normal tissue samples (adjusted P < 0.05) with relatively large absolute change (> 0.2);
same trend for CRC and AA compared with normal controls.
We also added DNA methylation biomarkers that were significantly different between AA and CRC to improve the potential differentiation power. Consequently, a total of 667 CRC‐specific DNA methylation biomarkers were obtained in this tissue cohort analysis.
2.7.2. Development and validation of the CRC cfDNA methylation screening model
After obtaining these 667 CRC‐specific DNA methylation biomarkers based on the tissue samples, we then analyzed the plasma samples, where we identified 545 biomarkers with differential DNA methylation levels in the plasma between CRC patients and healthy controls (P < 0.05). Then, the plasma cfDNA methylation dataset consisting of CRC patients and healthy controls was randomly split into a training and a validation cohort with matched gender, age and TNM stage. In the training cohort, 545 differentially expressed DNA methylation biomarkers were used for subsequent biomarker shrinkage and model construction. Least Absolute Shrinkage and Selection Operator (LASSO) was applied for variable selection. Lambda with minimum error estimated by 10‐fold cross‐validation was used. Eleven DNA methylation biomarkers were obtained for model building based on a binary prediction. Eventually, we constructed a logistic regression model using these 11 biomarkers as the covariate in the training cohort. A CRC diagnosis risk score was calculated by multiplying the unbiased coefficient estimate and the biomarker methylation value matrix in both the training and validation cohort. The predictability of the model was evaluated by AUROC, which calculated the proportion of concordant pairs among all pairs of observations. The model’s diagnosis performance for non‐AA (NAA), AA, and CRC patients was then evaluated. All data were shown as mean ± standard deviation (SD).
3. Results
3.1. Patient and sample characteristics
To characterize DNA methylation biomarkers which were specific to CRC, a total of 313 tissue samples were collected (139 normal, 30 AA, 144 CRC). After DNA extraction and AnchorIRIS™ library construction, 212 DNA samples passed quality control (QC) and were subsequently used for DNA methylation next‐generation sequencing (NGS), and 101 samples were excluded due to failure of DNA extraction QC (DNA yield and quality; n = 2) or low library yield (n = 99). A further 25 samples did not pass the sequencing QC metrics. Ultimately, 187 samples (91 normal, 26 AA, and 70 CRC) were analyzed for the discovery of DNA methylation biomarkers specific to CRC (Fig. 1).
Fig. 1.
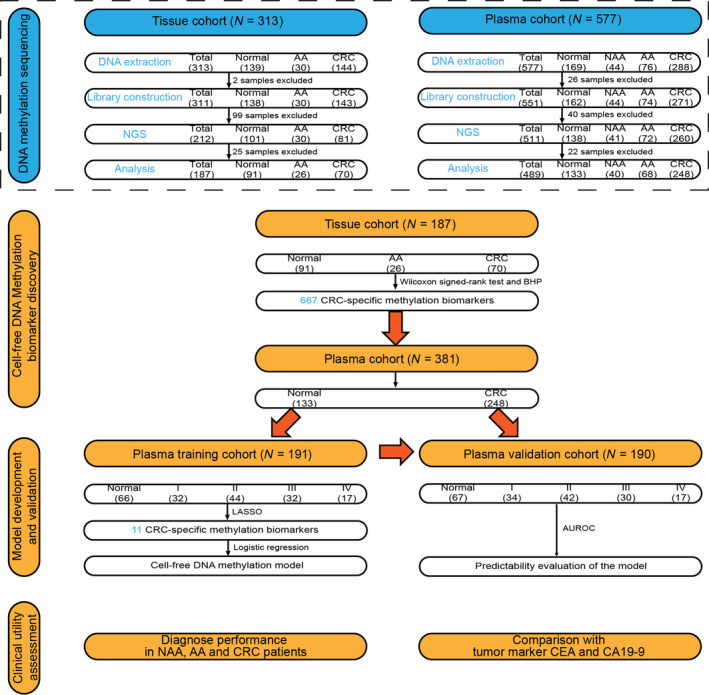
The study workflow chart. In the DNA methylation sequencing phase, 313 tissue samples (139 Normal, 30 AA and 144 CRC) were collected for NGS. Additionally, 577 plasma samples (169 Healthy controls, 44 NAA, 76 AA and 288 CRC) were collected for NGS. After DNA extraction, library construction and DNA methylation sequencing, 187 tissue samples and 489 plasma samples were eventually analyzed. Wilcoxon signed‐rank test and BHP were applied to screen the CRC‐specific methylation biomarkers in the tissue cohort, which led to the discovery of 667 DNA methylation biomarkers. In all, 133 normal plasma samples and 248 CRC plasma samples were randomly assigned to the training and validation cohort, respectively, and were then analyzed to further identify CRC‐specific methylation biomarkers from these 667 biomarkers. After LASSO selection, 11 CRC‐specific methylation biomarkers were obtained using the training cohort, which were then further confirmed using the validation cohort. Ultimately, the clinical value of the model was assessed by performing diagnostic tests in NAA, AA and CRC patients. The robustness of the model in the management of CRC was evaluated by comparison with CEA and CA19‐9. I, CRC stage I; II, CRC stage II; III, CRC stage III; IV, CRC stage IV.
To explore the clinical application of cfDNA methylation biomarkers for the detection of early‐stage CRC, 577 plasma samples (169 Normal , 44 NAA, 76 AA and 288 CRC) were collected for DNA methylation sequencing. All of the patients and healthy controls were treatment‐naive before the blood draw. After excluding 66 samples that did not pass DNA extraction QC (n = 26) or that had limited library yield (n = 40), a total of 511 samples were used for NGS. Finally, 489 plasma samples (133 Normal, 40 NAA, 68 AA and 248 CRC) that passed sequencing QC were subsequently analyzed for model development and validation (Fig. 1). Detailed information about sample exclusion is shown in Table S1. Clinical characteristics of the 489 patients or healthy controls with plasma analyzed are listed in Table 1.
Table 1.
The demographic and clinical characteristics of the healthy controls and patients in the plasma cohort. IA, inapplicable.
| Characteristics | Normal | NAA | AA | CRC |
|---|---|---|---|---|
| Total (n) | 133 | 40 | 68 | 248 |
| Gender | ||||
| Male, n (%) | 76 (57.14) | 25 (62.50) | 43 (63.24) | 143 (57.66) |
| Female, n (%) | 57 (42.86) | 15 (37.50) | 25 (36.76) | 105 (42.34) |
| Age (years) | ||||
| Mean | 44 | 56 | 59 | 60 |
| Range | 18–78 | 38–86 | 23–86 | 24–89 |
| Stage | ||||
| I, n (%) | IA | IA | IA | 66 (26.61) |
| II, n (%) | IA | IA | IA | 86 (34.68) |
| III, n (%) | IA | IA | IA | 62 (25.00) |
| IV, n (%) | IA | IA | IA | 34 (13.71) |
3.2. Measurement of cell‐free DNA concentration
As shown in Fig. 1, the concentration of 551 cfDNA samples (162 Normal, 44 NAA, 74 AA and 271 CRC) that passed DNA extraction QC was measured using a Qubit® fluorescent dye method. The concentration of cfDNA from CRC and AA samples was significantly higher than that from healthy control samples (Fig. 2). CRC and AA patients yielded a mean cfDNA concentration of 6.43 ng ± 0.45 and 6.08 ng ± 0.65 per 1 mL plasma, respectively, whereas healthy controls had a mean concentration of 3.94 ng ± 0.24 per 1 mL plasma (Table S2). Overall, the cfDNA concentration in CRC and AA patients was higher than that of healthy controls.
Fig. 2.
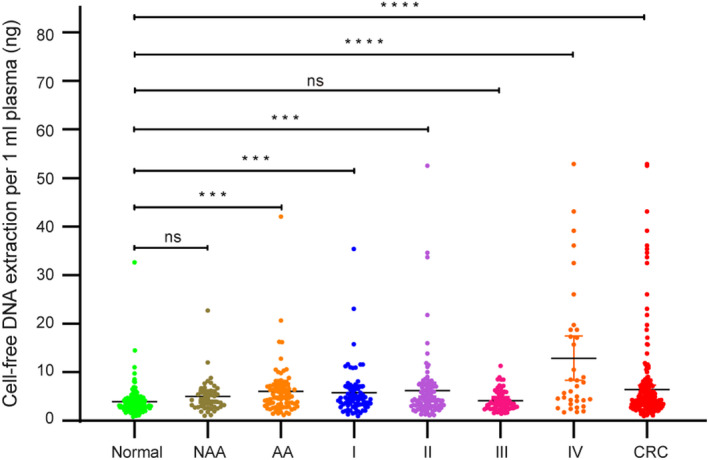
The cfDNA extraction analysis in healthy controls, NAA, AA and CRC patients. A total of 551 (162 Healthy controls [Normal], 44 NAA, 74 AA, 69 CRC stage I [I], 97 CRC stage II [II], 70 CRC stage III [III], 35 CRC stage IV [IV]) cfDNA extraction QC‐qualified samples were measured and compared for the cfDNA concentration (paired Student’s t‐test). Data are shown as mean ± SD; ns, not significant; ***P < 0.001; ****P < 0.0001.
3.3. Characterization of methylation biomarkers specific to CRC
After analyzing the tissue cohort’s sequencing data using Wilcoxon signed‐rank test and BHP‐adjust method, 667 CRC‐specific DNA methylation biomarkers that distinguished CRC and AA from normal tissue samples were identified (Fig. 3A). The correlation analysis result of these 667 biomarkers is shown in Fig. S1. These biomarkers were found to be distributed frequently in the intronic (28.34%) and promoter (25.19%) region of the genome (Table S3). The methylation levels in the normal cohort differed significantly from those in the AA and CRC cohort (Fig. 3B). Moreover, the levels of DNA methylation biomarkers in the AA group, which were calculated by dividing the normal group’s PCM and Log2 transformed values, were similar to those in the malignant group, which suggests that DNA methylation might be an early epigenetic event in CRC (Fig. 3C).
Fig. 3.

Characterization of the tissue DNA methylation landscape. (A) Unsupervised hierarchical clustering of the 667 CRC‐specific DNA methylation biomarkers in 187 tissue samples. (B) Principal component analysis of CRC, AA and Normal cohort. (C) Correlation of the methylation pattern between CRC and AA group. The mean methylation level was calculated based on 9921 sequenced biomarkers. The values plotted were generated by dividing PCM of the Normal cohort followed by log2 transformation.
3.4. Development and validation of the cfDNA methylation model for CRC detection
After the discovery of 667 CRC‐specific DNA methylation biomarkers in the tissue cohort, we further analyzed the methylation of these 667 biomarkers in the plasma cohort (Fig. 1). In all, 133 normal and 248 CRC plasma samples were randomly assigned to the training and validation cohort, respectively, after matching for gender, age and TNM stage. The clinical characteristics of the training and validation cohort are shown in Table S4. The LASSO method was applied to shrink the number of CRC‐specific DNA methylation biomarkers in the training cohort. Eleven age‐independent methylation biomarkers were eventually obtained (Table S5, Fig. S2). The cfDNA methylation model with these 11 biomarkers was then constructed using logistic regression. With a risk score threshold of 0.58 defined by Youden’s indexing in the training cohort, the cfDNA methylation model yielded a sensitivity of 82.4% and a specificity of 84.8% for CRC detection in the training cohort, and a sensitivity of 84.6% and a specificity of 86.6% in the validation cohort, respectively (Table S6). This model could distinguish CRC patients from healthy controls in both the training (AUC = 0.90) and the validation cohort (AUC = 0.92; Fig. 4A,B). We then evaluated the performance of this model in separating NAA, AA and stage I CRC patients from healthy controls, and found that the AUC was 0.77, 0.85 and 0.90, respectively (Fig. 4D–F). Furthermore, the risk score that was calculated based on the 11 DNA methylation biomarkers was demonstrated to be significantly higher in NAA, AA and CRC patients than in healthy controls (Fig. 4I). These data indicated that the cfDNA methylation model could serve as a robust and non‐invasive method for CRC detection.
Fig. 4.
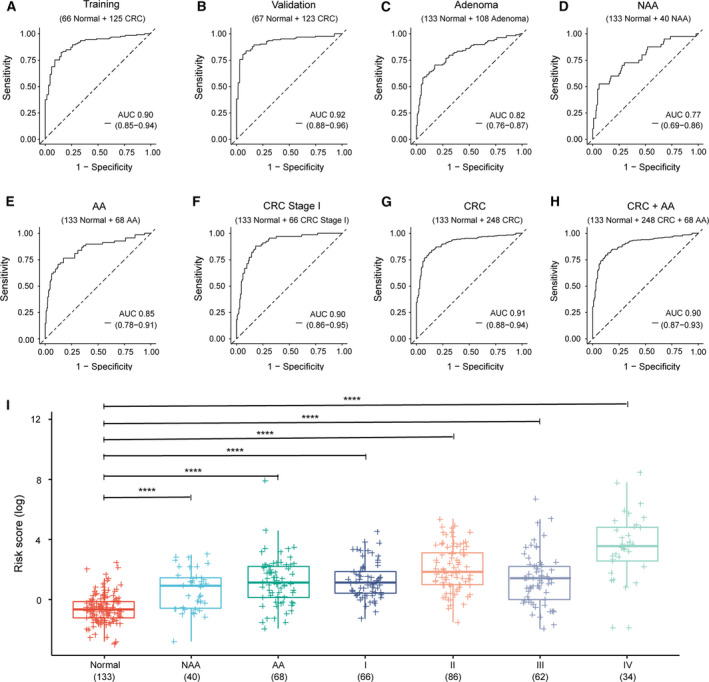
The performance and risk score of the cfDNA methylation model in detecting adenoma and CRC patients. (A,B) AUC of the model was 0.90 (0.85–0.94) and 0.92 (0.88–0.96) in the training and validation cohort, respectively. (C–E) When applied to the diagnosis of adenoma patients, the model achieved an AUC of 0.82 (0.76–0.87), 0.77 (0.69–0.86) and 0.85 (0.78–0.91) in adenoma, NAA and AA patients, respectively. (F) AUC of the model in the detection of CRC stage I was 0.90 (0.86–0.95; n = 199). (G) The model performed robustly in diagnosing CRC patients, which achieved an AUC of 0.91 (0.88–0.94; n = 381). (H) The overall AUC of the model was 0.90 (0.87–0.93) in the detection of CRC and AA cohort (n = 449). (I) The risk score of the model in healthy controls (Normal) and in patients with NAA, AA and CRC stage I–IV (n = 489, paired Student’s t‐test). The error bars indicate confidence interval; ****P < 0.0001.
3.5. Comparison of clinical utility in CRC detection between cfDNA methylation model and conventional tumor biomarker
Currently, carcino‐embryonic antigen (CEA) and carbohydrate antigen 19‐9 (CA19‐9) are among the most commonly used blood tumor biomarkers for therapy monitoring in CRC. In addition, the abnormal increase of CEA and CA19‐9 leads to the suspicion of CRC in clinical practice. A comparison in the performance in CRC detection was carried out between the cfDNA methylation model and CEA and CA19‐9 monitoring. The cfDNA methylation model was shown to be superior to CEA and CA19‐9 in CRC detection in terms of both sensitivity and specificity (AUC 0.91 versus 0.77 and 0.59, respectively, Fig. 5, Table 2). In addition, cfDNA methylation model as opposed to CEA and CA19‐9 monitoring, correlated well with AA and tumor stage (Table 2), showcasing the advantage of the cfDNA methylation model over CEA and CA19‐9 in the early detection of CRC.
Fig. 5.
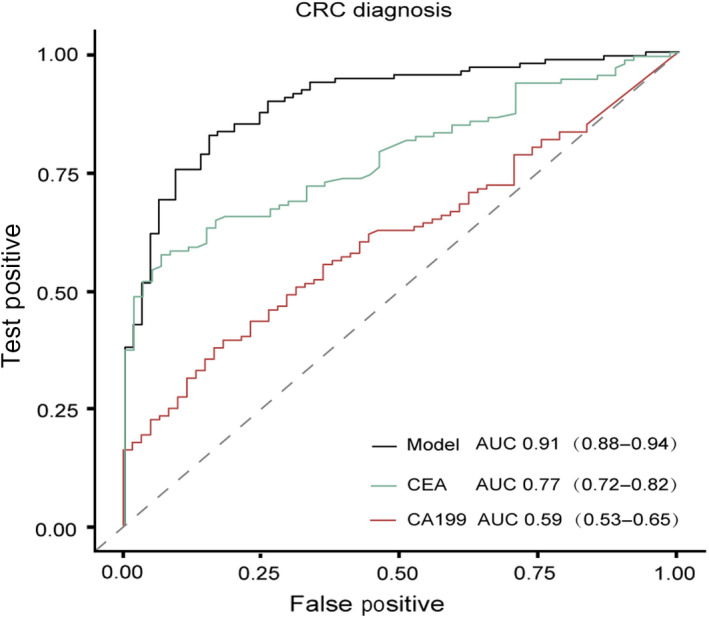
Comparison of CRC diagnostic performance between the cfDNA methylation model and the tumor biomarker CEA and CA19‐9. AUC in detecting CRC of the cfDNA methylation model, CEA and CA19‐9 was 0.91 (0.88–0.94), 0.77 (0.72–0.82) and 0.59 (0.53–0.65), respectively.
Table 2.
AUC, sensitivity, specificity and accuracy of the cfDNA methylation model, CEA and CA19‐9 in disease diagnosis.
| Characteristics | AA | CRC Stage I | CRC Stage II | CRC |
|---|---|---|---|---|
| AUC | ||||
| Model | 0.85 | 0.90 | 0.94 | 0.91 |
| CEA | 0.64 | 0.68 | 0.78 | 0.77 |
| CA19‐9 | 0.54 | 0.48 | 0.60 | 0.59 |
| Sensitivity | ||||
| Model | 76.5% | 87.9% | 83.7% | 83.9% |
| CEA | 40.0% | 55.6% | 58.8% | 63.6% |
| CA19‐9 | 57.9% | 31.6% | 39.5% | 29.8% |
| Specificity | ||||
| Model | 82.7% | 82.0% | 91.7% | 85.7% |
| CEA | 82.6% | 81.0% | 89.3% | 81.0% |
| CA19‐9 | 55.4% | 81.8% | 81.0% | 90.1% |
| Accuracy | ||||
| Model | 80.6% | 83.9% | 88.6% | 84.5% |
| CEA | 67.7% | 66.9% | 76.7% | 69.3% |
| CA19‐9 | 56.2% | 53.8% | 63.8% | 49.6% |
4. Discussion
Detection at the early stage is pivotal for the successful treatment of various cancer types, including CRC [26]. Colonoscopy accompanied by tissue biopsy remains the gold standard in the diagnosis of CRC [27]. However, colonoscopy is less than perfect for the purpose of CRC screening due to its invasiveness, high cost, time consumption and unpleasant examination experience [28]. Furthermore, the accuracy of detecting early‐stage CRC patients by endoscopy can vary significantly between different endoscopists [29]. Therefore, an accurate, robust and non‐invasive test is highly desirable for CRC screening.
DNA methylation aberration usually occurs early in the progression of many tumors, suggesting that detection of altered DNA methylation patterns could be a promising strategy in cancer screening [30]. Moreover, the genomic profile of ctDNA was shown to share features with that of concurrent tumor in the same cancer patient, which has important implications for non‐invasive cancer screening [31]. Progress in high‐throughput sequencing technology and the availability of multi‐omics have contributed to developing diagnostic tools for the early detection of cancers [32, 33]. However, to our knowledge, the methylation profile of different stages of CRC progression, which comprises normal tissue, AA and various CRC stages, is rarely available, even though the risk of progression from AA to CRC has been commonly acknowledged [34]. Intending to construct a cfDNA methylation model for the early detection of CRC, we first analyzed DNA methylation status of tissue samples from CRC, AA and normal mucosa, which helped identify DNA methylation biomarkers that could distinguish CRC tumor from normal tissue. Moreover, we evaluated the capability of a blood‐based cfDNA methylation test for early detection of CRC. A diagnostic model based on 11 cfDNA methylation biomarkers showed high performance in distinguishing CRC from normal individuals. The overall sensitivity of CRC detection was 83.9% in the validation cohort at a specificity of 85.7%. The model, especially, achieved high sensitivities on AA (76.5%) and stage I CRC (87.9%), which is critical for detecting CRC in the stages with curative treatments.
At present, several blood‐based DNA methylation biomarkers have been assessed for early detection of CRC. The Epi proColon assay (SEPT9) yielded an overall sensitivity of 48.2% at a specificity of 91.5% for CRC detection in a prospective clinical trial of 7941 asymptomatic individuals [35]. In a cohort of 2105 individuals, a two‐biomarker blood test (BCAT1/IKZF1) identified 66% of CRC at a specificity of 95% [36]. A single methylation biomarker, cg10673833, was demonstrated to be superior to other previous reported methylation biomarkers in CRC detection, with a sensitivity and specificity of 89.7% and 86.8%, respectively [37].
The benefit of all screening tests for reducing CRC morbidity and mortality relies on the test performance in detecting stage I CRC and AA [38]. The sensitivity of SEPT9 (Epi proColon assay) was 35.0% and 11.2% for detecting stage I CRC and AA, respectively [35], although a recent study reported improved performance by analyzing multiple regions of this gene [39]. The BCAT1/IKZF1 methylation test identified 38% of stage I CRC but only 6% of AA [36]. The single methylation biomarker, cg10673833, had a sensitivity of 33.3% against CRC advanced precancerous leision which included AA [37]. A pan‐cancer detection test based on cfDNA methylation profiling yielded a sensitivity of ~ 25% for stage I CRC [40]. Although a head‐to‐head comparison of the performance between our 11‐biomarker CRC diagnostic model and other screening tests has not been made, this approach showed superior sensitivities of 87.9% in stage I CRC and 76.5% in AA. Nevertheless, further verification using colonoscopy remains recommended in cases of positive results as suggested by the 11 methylation biomarkers, due to the false‐positive results. Overall, these results suggested that this model could serve as a promising screening test for non‐invasive detection of CRC at an early and curable stage, which needs further validation in real clinical settings.
There are certain limitations associated with this study. First, the plasma samples were retrospectively collected and less than optimal, as most of the CRC patients enrolled in the plasma cohort were symptomatic and were older than healthy controls, which might influence the model’s clinical accuracy.
Secondly, the specimens used in this study were collected from a single institution despite its large sample size. Therefore, the robustness of the cfDNA methylation model should be further validated in a prospective, multi‐center trial.
Thirdly, the cfDNA methylation model was constructed based on hypermethylated regions in CRC tissue. However, some CRC‐specific methylation regions may not be hypermethylated [41] and might have been excluded from our biomarker discovery step. Therefore, it is plausible that a plasma‐based discovery step is needed to identify other cfDNA signatures that have not been included in the current panel.
Fourthly, the clinical utility of the cfDNA methylation model in CRC detection was compared with CEA and CA19‐9, which are not used as CRC screening biomarkers in clinical practice due to their low sensitivity. Hence, it is necessary to conduct a prospective study to compare our model with more appropriate non‐invasive CRC screening tests, such as FIT.
Lastly, samples of sessile serrated poly (SSP), a type of premalignant lesion of CRC [42], should be included in future studies to test the suitability of our CRC risk model for detecting such lesion.
5. Conclusions
We established a promising and non‐invasive cfDNA methylation model for the detection of CRC, especially early‐stage CRC and AA. The diagnostic value of this model was validated in an independent cohort, highlighting its promising application for early detection of CRC.
Conflict of interest
J‐BF, DZZ, XYC and ZC are/were employees of AnchorDx Medical Co., Ltd. or AnchorDx, Inc. All other authors declare no competing financial interest.
Author contributions
PL, FG and J‐BF designed and supervised the study and revised the manuscript. XRW, YFZ, XYC and ZC developed the methodology including analyzing and organizing data. TH, XWH, YFZ, QLD, JK, LL, XSH, DZZ and XJW were responsible for acquiring and managing patients. YFZ, XRW, FG, ZC, JBF and PL wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.12942.
Supporting information
Fig. S1. The correlation analysis of the 667 CRC‐specific DNA methylation biomarkers.
Fig. S2. The correlation analysis between age and 11 CRC‐specific DNA methylation biomarkers.
Table S1. The reasons for tissue and plasma sample exclusion.
Table S2. The cfDNA extraction quantity in CRC, AA, NAA patients and healthy controls.
Table S3. The distribution of the 667 CRC‐specific DNA methylation biomarkers in the genome.
Table S4. The demographic and clinical characteristics of the training and validation cohort.
Table S5. The genomic characteristics of the 11 DNA methylation biomarkers.
Table S6. The AUC, sensitivity, specificity and accuracy of the cfDNA methylation model in diagnosis of CRC.
Acknowledgements
We are grateful for the sincere cooperation of the patients enrolled in the study for voluntarily providing samples and clinical information. This work was supported by National Key R&D Program of China (Nos 2017YFC1308800, 2017YFC1309002), National Natural Science Foundation of China (No. 81870383), Clinical Innovation Research Program of Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory; No. 2018GZR0201005), Guangdong Natural Science Foundation (Nos 2017A030313785, 2016A030310187), Science and Technology Planning Project of Guangzhou City (Nos 201804010014, 201803040019), Science and Technology Planning Project of Guangdong Province (Nos 2015B020229001, 20160916, 2017B020226005), Key‐Area Research and Development Program of Guangdong Province (2019B020229002), Scheme of Guangzhou Economic and Technological Development District for Leading Talents in Innovation and Entrepreneurship (No. 2017‐L152), Scheme of Guangzhou for Leading Talents in Innovation and Entrepreneurship (No. 2016007), Scheme of Guangzhou for Leading Team in Innovation (No. 201909010010), and Sun Yat‐sen University 5010 Project (No. 2010012).
Xianrui Wu and Yunfeng Zhang contributed equally to this article.
Data accessibility
The datasets which were used and analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA & Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA & Jemal A (2020) Colorectal cancer statistics, 2020. CA Cancer J Clin 70, 145–164. [DOI] [PubMed] [Google Scholar]
- 3.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL & Siegel RL (2019) Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 69, 363–385. [DOI] [PubMed] [Google Scholar]
- 4.Wolf AMD, Fontham ETH, Church TR, Flowers CR, Guerra CE, LaMonte SJ, Etzioni R, McKenna MT, Oeffinger KC, Shih Y‐CTet al. (2018) Colorectal cancer screening for average‐risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J Clin 68, 250–281. [DOI] [PubMed] [Google Scholar]
- 5.Han YD, Oh TJ, Chung TH, Jang HW, Kim YN, An S & Kim NK (2019) Early detection of colorectal cancer based on presence of methylated syndecan‐2 (SDC2) in stool DNA. Clin Epigenetics 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, Ahlquist DA & Berger BM (2014) Multitarget stool DNA testing for colorectal‐cancer screening. N Engl J Med 370, 1287–1297. [DOI] [PubMed] [Google Scholar]
- 7.Young GP, Symonds EL, Allison JE, Cole SR, Fraser CG, Halloran SP, Kuipers EJ & Seaman HE (2015) Advances in fecal occult blood tests: the FIT revolution. Dig Dis Sci 60, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13, 484–492. [DOI] [PubMed] [Google Scholar]
- 9.Flavahan WA, Gaskell E & Bernstein BE (2017) Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hata T, Dal Molin M, Hong SM, Tamura K, Suenaga M, Yu J, Sedogawa H, Weiss MJ, Wolfgang CL, Lennon AMet al. (2017) Predicting the grade of dysplasia of pancreatic cystic neoplasms using Cyst fluid DNA methylation markers. Clin Cancer Res 23, 3935–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee P‐Het al. (2012) Genome‐wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 55, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrawal A, Murphy RF & Agrawal DK (2007) DNA methylation in breast and colorectal cancers. Mod Pathol 20, 711–721. [DOI] [PubMed] [Google Scholar]
- 13.Hulbert A, Jusue‐Torres I, Stark A, Chen C, Rodgers K, Lee B, Griffin C, Yang A, Huang P, Wrangle J (2017) Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin Cancer Res 23, 1998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia‐Manero G, Jabbour E, Borthakur G, Faderl S, Estrov Z, Yang H, Maddipoti S, Godley LA, Gabrail N, Berdeja JGet al. (2013) Randomized open‐label phase II study of decitabine in patients with low‐ or intermediate‐risk myelodysplastic syndromes. J Clin Oncol 31, 2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aucamp J, Bronkhorst AJ, Badenhorst CPS & Pretorius PJ (2018) The diverse origins of circulating cell‐free DNA in the human body: a critical re‐evaluation of the literature. Biol Rev Camb Philos Soc 93, 1649–1683. [DOI] [PubMed] [Google Scholar]
- 16.Boddy JL, Gal S, Malone PR, Harris AL & Wainscoat JS (2005) Prospective study of quantitation of plasma DNA levels in the diagnosis of malignant versus benign prostate disease. Clin Cancer Res 11, 1394–1399. [DOI] [PubMed] [Google Scholar]
- 17.Kamat AA, Baldwin M, Urbauer D, Dang D, Han LY, Godwin A, Karlan BY, Simpson JL, Gershenson DM, Coleman RLet al. (2010) Plasma cell‐free DNA in ovarian cancer: an independent prognostic biomarker. Cancer 116, 1918–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madhavan D, Wallwiener M, Bents K, Zucknick M, Nees J, Schott S, Cuk K, Riethdorf S, Trumpp A, Pantel Ket al. (2014) Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat 146, 163–174. [DOI] [PubMed] [Google Scholar]
- 19.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl Het al. (2005) Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA 102, 16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, Knudsen M, Nordentoft I, Wu H‐T, Tin ASet al. (2019) Analysis of plasma cell‐free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol 5, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu RH, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, Yi S, Shi W, Quan Q, Li Ket al. (2017) Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 16, 1155–1161. [DOI] [PubMed] [Google Scholar]
- 22.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RMet al. (2014) Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 6, 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danese E, Minicozzi AM, Benati M, Montagnana M, Paviati E, Salvagno GL, Lima‐Oliveira G, Gusella M, Pasini F, Lippi Get al. (2015) Comparison of genetic and epigenetic alterations of primary tumors and matched plasma samples in patients with colorectal cancer. PLoS One 10, e0126417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan JCM, Massie C, Garcia‐Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R & Rosenfeld N (2017) Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17, 223–238. [DOI] [PubMed] [Google Scholar]
- 25.Liang W, Zhao Y, Huang W, Gao Y, Xu W, Tao J, Yang M, Li L, Ping W, Shen Het al. (2019) Non‐invasive diagnosis of early‐stage lung cancer using high‐throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 9, 2056–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel RL, Miller KD & Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70, 7–30. [DOI] [PubMed] [Google Scholar]
- 27.Ladabaum U, Dominitz JA, Kahi C & Schoen RE (2020) Strategies for colorectal cancer screening. Gastroenterology 158, 418–432. [DOI] [PubMed] [Google Scholar]
- 28.Yang C, Sriranjan V, Abou‐Setta AM, Poluha W, Walker JR & Singh H (2018) Anxiety associated with colonoscopy and flexible sigmoidoscopy: a systematic review. Am J Gastroenterol 113, 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rees CJ, Bevan R, Zimmermann‐Fraedrich K, Rutter MD, Rex D, Dekker E, Ponchon T, Bretthauer M, Regula J, Saunders Bet al. (2016) Expert opinions and scientific evidence for colonoscopy key performance indicators. Gut 65, 2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esteller M (2008) Epigenetics in cancer. N Engl J Med 358, 1148–1159. [DOI] [PubMed] [Google Scholar]
- 31.Siravegna G, Marsoni S, Siena S & Bardelli A (2017) Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14, 531–548. [DOI] [PubMed] [Google Scholar]
- 32.Aravanis AM, Lee M & Klausner RD (2017) Next‐generation sequencing of circulating tumor DNA for early cancer detection. Cell 168, 571–574. [DOI] [PubMed] [Google Scholar]
- 33.Lennon AM, Buchanan AH, Kinde I, Warren A, Honushefsky A, Cohain AT, Ledbetter DH, Sanfilippo F, Sheridan K, Rosica Det al. (2020) Feasibility of blood testing combined with PET‐CT to screen for cancer and guide intervention. Science 369, eabb9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Click B, Pinsky PF, Hickey T, Doroudi M & Schoen RE (2018) Association of colonoscopy adenoma findings with long‐term colorectal cancer incidence. JAMA 319, 2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Church TR, Wandell M, Lofton‐Day C, Mongin SJ, Burger M, Payne SR, Castaños‐Vélez E, Blumenstein BA, Rösch T, Osborn Net al. (2014) Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 63, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedersen SK, Symonds EL, Baker RT, Murray DH, McEvoy A, Van Doorn SC, Mundt MW, Cole SR, Gopalsamy G, Mangira Det al. (2015) Evaluation of an assay for methylated BCAT1 and IKZF1 in plasma for detection of colorectal neoplasia. BMC Cancer 15, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, Wang W, Sheng H, Pu H, Mo Het al. (2020) Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med 12, eaax7533. [DOI] [PubMed] [Google Scholar]
- 38.Lauby‐Secretan B, Vilahur N, Bianchini F, Guha N & Straif K (2018) The IARC perspective on colorectal cancer screening. N Engl J Med 378, 1734–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin S, Zhu D, Shao F, Chen S, Guo Y, Li K, Wang Y, Ding R, Gao L, Ma Wet al. (2021) Efficient detection and post‐surgical monitoring of colon cancer with a multi‐marker DNA methylation liquid biopsy. Proc Natl Acad Sci USA 118, e2017421118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, Oxnard GR, Klein EA, Smith Det al. (2020) Sensitive and specific multi‐cancer detection and localization using methylation signatures in cell‐free DNA. Ann Oncol 31, 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irizarry RA, Ladd‐Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster Met al. (2009) The human colon cancer methylome shows similar hypo‐ and hypermethylation at conserved tissue‐specific CpG island shores. Nat Genet 41, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keum N & Giovannucci E (2019) Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 16, 713–732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The correlation analysis of the 667 CRC‐specific DNA methylation biomarkers.
Fig. S2. The correlation analysis between age and 11 CRC‐specific DNA methylation biomarkers.
Table S1. The reasons for tissue and plasma sample exclusion.
Table S2. The cfDNA extraction quantity in CRC, AA, NAA patients and healthy controls.
Table S3. The distribution of the 667 CRC‐specific DNA methylation biomarkers in the genome.
Table S4. The demographic and clinical characteristics of the training and validation cohort.
Table S5. The genomic characteristics of the 11 DNA methylation biomarkers.
Table S6. The AUC, sensitivity, specificity and accuracy of the cfDNA methylation model in diagnosis of CRC.
Data Availability Statement
The datasets which were used and analyzed during the current study are available from the corresponding author on reasonable request.