

SUMMARY
Natural killer (NK) cells are innate cytolytic effectors that target HIV-infected CD4+ T cells. In conjunction with antibodies recognizing the HIV envelope, NK cells also eliminate HIV-infected targets through antibody-dependent cellular cytotoxicity (ADCC). However, how these NK cell functions impact infected macrophages is less understood. We show that HIV-infected macrophages resist NK cell-mediated killing. Compared to HIV-infected CD4+ T cells, initial innate NK cell interactions with HIV-infected macrophages skew the response towards cytokine production, rather than release of cytolytic contents, causing inefficient elimination of infected macrophages. Studies with chimeric antigen receptor (CAR) T cells demonstrate that the viral envelope is equally accessible on CD4+ T cells and macrophages. Nonetheless, ADCC against macrophages is muted compared to ADCC against CD4+ T cells. Thus, HIV-infected macrophages employ mechanisms to evade immediate cytolytic NK cell function, while preserving inflammatory cytokine responses. These findings emphasize the importance of eliminating infected macrophages for HIV-cure efforts.
eTOC Blurb
While CD4+ T cells are major targets for HIV, macrophages are also infected. Clayton et al. find that NK cells have muted cytolytic and ADCC responses to infected macrophages, contributing to their inefficient elimination. Strategies to enhance macrophage susceptibility to killing will be essential for HIV cure efforts.
Graphical abstract
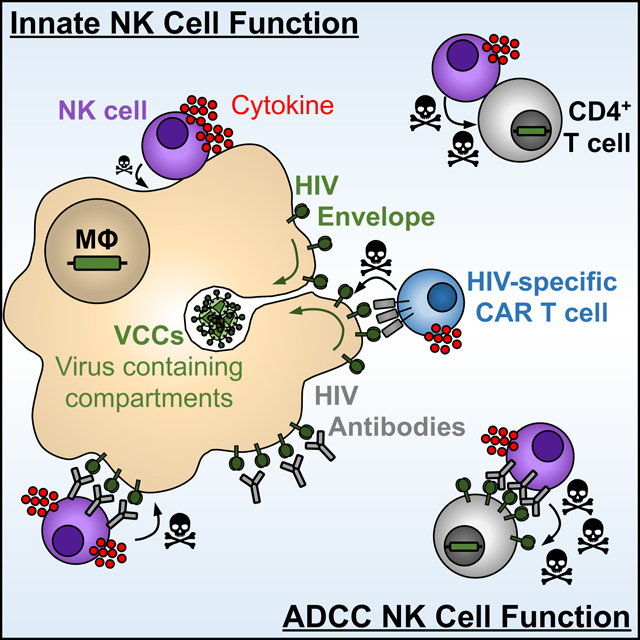
INTRODUCTION
The HIV reservoir is defined as any cell that harbors infectious virus, can reseed infection, and persist despite antiretroviral therapy (ART). While CD4+ T cells are the main targets of HIV infection, macrophages also become infected, persist in tissues during ART (Cribbs et al., 2015; Deleage et al., 2011; Ganor et al., 2019; Hansen et al., 2016; Ko et al., 2019; Tso et al., 2018; Zalar et al., 2010) and can act as a source of virus following treatment interruption (Andrade et al., 2020).
Macrophages resist the cytopathic effects of HIV infection (Campbell et al., 2019; Castellano et al., 2017; Reynoso et al., 2012; Swingler et al., 2007; Yuan et al., 2017) and exhibit poor penetrance by antiretrovirals (Gavegnano et al., 2013; Gavegnano and Schinazi, 2009; Perno et al., 1998). Following their assembly at the macrophage plasma membrane, virions bud internally into virus-containing compartments (VCC) (Deneka et al., 2007; Hammonds et al., 2017; Jouve et al., 2007; Jouvenet et al., 2006; Welsch et al., 2007), some of which can be transiently accessed from the extracellular surface through narrow microchannels, but are shielded from HIV-specific neutralizing antibodies (Chu et al., 2012; Deneka et al., 2007; Gaudin et al., 2013; Koppensteiner et al., 2012). Furthermore, macrophages can reseed new CD4+ T cell infections via cell-to-cell spread that is resistant to some neutralizing antibodies (Collins et al., 2015; Duncan et al., 2014). Finally, the persistence of infected macrophages during ART suggests a lack of immune-mediated clearance. These characteristics together contribute to the persistence of infected macrophages during ART.
Compared to infected CD4+ T cells, HIV/SIV-infected macrophages are intrinsically resistant to killing by cytotoxic CD8+ T cells (CTLs) (Clayton et al., 2018; Rainho et al., 2015; Vojnov et al., 2012). Furthermore, inefficient killing of macrophages during CTL recognition drives prolonged cell-cell contact, resulting in excessive TCR stimulation and hypersecretion of pro-inflammatory cytokines, including IFN-γ. These cytokines then further propagate inflammation through activation of macrophages (Clayton et al., 2018). Because macrophages are professional antigen presenting cells that help coordinate downstream immune responses in the acute setting, their resistance to killing makes intuitive sense. However, it also makes macrophages ideal environments for pathogens to evade killing, while promoting inflammation to drive pathology in the chronic setting. This emphasizes the need for new strategies that efficiently eliminate infected macrophages.
Natural killer (NK) cells are cytolytic effectors that, unlike CTL, can recognize HIV-infected cells through innate, antigen-independent mechanisms. With the help of antigen-specific antibodies that recognize the surface-exposed viral envelope, NK cells can also eliminate infected targets through a process termed antibody-dependent cellular cytotoxicity (ADCC) (Bernard et al., 2017). Studies of NK cell interactions with HIV-infected cells have focused almost exclusively on CD4+ T cells (Alsahafi et al., 2017; Bonaparte and Barker, 2003, 2004; Fogli et al., 2008; Norman et al., 2011; Richard and Cohen, 2010; Richard et al., 2010; Shah et al., 2010; Tomescu et al., 2015; Tremblay-McLean et al., 2017; Ward et al., 2004). However, one study by Quillay et al. showed that NK cells exert an antiviral effect on macrophages in an antigen-independent manner, but only when NK cell contact is initiated prior to, or within a few hours of infection; pre-established macrophage infections were poorly controlled (Quillay et al., 2016). While NK cells may be deficient in their ability to control infection in macrophages, it is not known how this compares to NK cell-mediated control of CD4+ T cell infection and whether the same antiviral mechanisms are at play. Furthermore, while the HIV envelope may be expressed transiently at the surface of infected macrophages, whether this can be exploited for ADCC to enhance killing of infected macrophages is unclear.
Here, we report that contact-mediated killing of HIV-infected macrophages is inefficient compared to killing of infected CD4+ T cells. Compared to the early NK cytolytic responses elicited by infected CD4+ T cells, interactions of NK cells with infected macrophages favors NK cell production of TNF-α over cytolytic degranulation. Since NK cells can also mediate ADCC, we first determined that the HIV envelope is transiently expressed at the surface of infected macrophages prior to internalization into the VCC. Using HIV-specific chimeric antigen receptor (CAR) T cells, we show that recognition of infected CD4+ T cells and macrophages is comparable. However, although ADCC enhances NK cell responses to infected macrophages, these responses are significantly muted compared to ADCC responses to CD4+ T cells. Macrophages thus employ mechanisms to evade efficient elimination. They do so by limiting immediate innate cytolytic NK cell effector function and ADCC.
RESULTS
Infected macrophages resist efficient innate NK cell killing
To study the interactions between NK cells and HIV-infected cells, peripheral blood mononuclear cells (PBMCs) from healthy donors were used to isolate CD14+ monocytes and CD4+ T cells. Monocytes were matured into macrophages. In parallel the CD4+ T cells were activated. After 7 days of maturation/activation, cells were infected with HIV89.6 for 2–3 days (Clayton et al., 2018). Autologous PBMCs were rested overnight and NK cells were then isolated and immediately co-cultured with infected targets. The overall experimental setup is depicted in Fig. 1A. Using flow cytometry to detect infected targets by intracellular Gag p24 staining (gating strategies for the elimination assays are shown in Fig. S1A and B), co-cultures with autologous NK cells over 4 hours led to significant elimination of infected CD4+ T cells but not infected macrophages (Fig. 1B and C). While elimination of infected macrophages was enhanced upon overnight and 48-hour co-culture, differences in target cell killing remained across multiple effector-to-target (E:T) ratios but achieved parity as more effectors were used (Fig. 1D and E). As expected, no killing was observed when autologous CD8+ T cells from healthy donors (which lack HIV memory responses) were used as effector cells (Fig. S1C and D). Thus, compared to HIV-infected CD4+ T cells, HIV-infected macrophages are more resistant to killing by NK cells.
Fig. 1. HIV-infected macrophages evade efficient NK cell-mediated elimination.

(A) Schematic of the experimental process to coordinate maturation, activation, and infection of target cells with co-culture of autologous ex vivo NK cells. Details are described in the STAR Methods. (B-E) NK cell innate-based elimination assay. HIV-infected CD4+ T cells and macrophages were co-cultured with NK cells for 4 hours, overnight, or 48-hours at different effector-to-target (E:T) ratios, followed by measurement of Gag p24+ target cell elimination by flow cytometry. (B) Representative elimination assay plots. (C) Summary data from 4-hour co-cultures at an E:T of 2 from 9 independent experiments (n=15 biological replicates). (D) Summary data from overnight co-cultures at different E:T ratios. Shown are data from 5 independent experiments (n=8 biological replicates). (E) Summary data from 48-hour co-cultures at different E:T ratios. Shown are data from 4 independent experiments (n=6 biological replicates). Statistical analysis for (C-E): paired t tests, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant. See also Fig. S1.
Infected macrophages skew towards early production of TNF-α over degranulation by NK cells
We next investigated whether resistance to NK cell killing might be due to differential recognition of infected targets, or attributable to inefficient killing due to their resistance to granzymes (Clayton et al., 2018). To characterize recognition of infected cells by NK cells, co-cultures were performed with a ratio of ten targets to one effector. For elimination assays, high E:T ratios are used to detect maximal killing. However, for recognition assays, an excess of targets is required to detect the maximal NK cell response. Furthermore, given that HIV infection frequencies averaged 14%, a T:E ratio of 10 equals ~1 infected target per 1 effector. After 6 hours of co-culture, NK cell degranulation (CD107a) and cytokine (TNF-α and IFN-γ) production were assessed by flow cytometry (gating strategies for the recognition assays are shown in Fig. S2A). Only minor amounts of IFN-γ were produced by NK cells in short-term co-cultures compared to the predominant cytokine, TNF-α (Fig. S2B); we observed significant amounts of IFN-γ only after overnight co-culture (Fig. S2C). NK cells exhibited both degranulation and TNF-α production in response to short-term co-culture with infected CD4+ T cells and infected macrophages (Fig. 2A and B), while autologous CD8+ T cells from healthy donors did not recognize infected cells (Fig. S2D and E). Baseline NK cell responses to uninfected CD4+ T cells were significantly higher compared to NK cell responses to uninfected macrophages (Fig. 2C). This may be due to the activation state of CD4+ T cells, resulting in higher expression of NK-activating ligands (Cerboni et al., 2007). When corrected for the baseline responses to uninfected cells and normalized based on the fraction of target cells infected, NK cell responses to infected CD4+ T cells and macrophages were comparable (Fig. 2D). We conclude that there are no deficiencies in the ability of NK cells to sense HIV-infected macrophages. However, effector functions of NK cells that respond to infected CD4+ T cells showed more degranulation than TNF-α production when compared to infected macrophages (Fig. 2E). Notably, a dominant population of NK cells responding to infected macrophages showed a lack of degranulation but a strong TNF-α response (“Single TNF-α” – Fig. 2F). Given that CD56brightCD16- NK cells are characterized by greater cytokine production and less cytotoxicity compared to their CD56dimCD16+ counterparts (reviewed in (Poli et al., 2009)), the CD56 phenotype of NK cells that responded to infected target cells was assessed (Fig. S2F). There were no significant differences in the CD56 phenotype of the NK cell populations that responded to infected CD4+ T cells and infected macrophages (Fig. S2G). Taken together, NK cell responses to infected macrophages versus CD4+ T cells are skewed towards production of TNF-α versus degranulation. This may contribute to the differential susceptibility of these target cells to efficient killing.
Fig. 2. Initial NK cell responses to HIV-infected macrophages are skewed towards TNF-α production over degranulation.

NK cell innate-based recognition of HIV-infected targets. NK cells were incubated with targets at a target-to-effector ratio of 10 for 6 hours, followed by flow cytometry analysis of degranulation/CD107a and TNF-α production. (A) Representative NK cell recognition assay plots. (B) Data summaries for NK cell responses (%CD107a + %TNF-α) to uninfected and infected targets from twelve independent experiments (n=19 biological replicates). (C) Comparison of NK cell responses to uninfected CD4+ T cells and macrophages from twelve independent experiments (n=19 biological replicates). (D) Comparison of NK cell responses to infected CD4+ T cells and macrophages. Values are corrected for background recognition of uninfected target cells and normalized to target cell infection frequencies. NK cell responses that were less than 2% above background (n=5) were excluded from the analysis. (E and F) Data summaries for the quality of the NK cell response to infected CD4+ T cells and macrophages. Values are corrected for background recognition of uninfected target cells. (E) Ratios of CD107a versus TNF-α production were calculated as described in the STAR Methods. Shown are results from ten independent experiments (n=14 biological replicates). (F) The “% Total Response” was calculated as described in the STAR Methods. Values are corrected for background recognition of uninfected target cells. Shown are results from ten independent experiments (n=14 biological replicates). Statistical analysis for (B-F): paired t tests, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant. See also Fig. S2.
The HIV envelope is present transiently on the surface of infected macrophages in addition to the virus-containing compartment (VCC)
One mechanism to enhance NK cell recognition of infected target cells is by engagement of the Fc receptor CD16 (FcγRIII) with HIV envelope-specific antibodies. This physically connects the NK cell to its target to initiate ADCC. However, accessibility of the HIV envelope on the surface of infected macrophages is a controversial issue. While HIV virions bud and are released from the surface of CD4+ T cells (Jolly and Sattentau, 2007), virions that bud from macrophages are instead stored in intracellular VCCs, which show only transient connections with the plasma membrane (Deneka et al., 2007; Gaudin et al., 2013). Virions in VCCs are largely inaccessible to HIV neutralizing antibodies (Koppensteiner et al., 2012), however, the HIV envelope may be transiently expressed at the cell surface, which may provide a window of opportunity for HIV antibody binding prior to internalization of the envelope.
To determine whether HIV-specific antibodies could detect infected macrophages, and thus contribute to NK cell ADCC, live infected CD4+ T cells and macrophages were stained with a panel of broadly neutralizing antibodies. These experiments were performed at 4°C to prevent antibody-induced envelope internalization (Anand et al., 2019), and analyzed by flow cytometry (gating strategies shown in Fig. S3A). An anti-CD19 antibody was used as a negative control. Three CD4 binding site-specific antibodies (VRC01, 3BNC117 and N6) and two V3 glycan supersite-specific antibodies (10–1074 and PGT121), in addition to a single domain CD4-binding site-specific antibody fragment that lacks an Fc domain (J3 VHH - (McCoy et al., 2012)), were tested. HIV-specific antibodies detected both infected CD4+ T cells and macrophages, but to varying degrees (Fig. 3A). PGT121 exhibited the largest fold change in HIV-specific antibody MFI for CD4+ T cells and macrophages, while VRC01 exhibited the weakest (Fig. 3B). Some antibodies (VRC01, N6 and J3 VHH) stained macrophages more strongly than CD4+ T cells, which may affect the outcome of ADCC responses. Thus, only antibodies with comparable levels of specific staining on infected CD4+ T cells and macrophages (3BNC117 and PGT121) were used for subsequent ADCC assays.
Fig. 3. HIV assembles into plasma membrane contiguous VCCs in macrophages, but the HIV envelope is also transiently accessible at the cell surface.

(A and B) HIV antibody-based detection of the HIV envelope on the surface of infected CD4+ T cells and macrophages. (A) Representative flow cytometry plots of uninfected and infected CD4+ T cells (black) and macrophages (red) stained with HIV envelope-specific antibodies. Histograms of HIV-specific antibody staining show the shifts in MFI between the uninfected and infected populations. (B) Summary of the “Fold change in HIV-specific Antibody MFI” for each antibody for HIV89.6-infected CD4+ T cells and macrophages. This fold change was calculated as described in the STAR Methods. Data were collected across a total of 23 independent experiments. Statistical analysis, unpaired t test, **p<0.01, ns=not significant. (C and D) Kinetics of the HIV envelope internalization on infected macrophages. (C) Representative images of the Gag+Envelope+ population over the 60-minute time course showing the distribution of CD33, the HIV Gag and the envelope. Additional representative images of 10–1074 staining are shown in Fig. S3C. (D) Summary of the internalization scores over the 60-minute time course for the Gag and Envelope. Shown are the mean +/− SEM data from 7 independent experiments (J3 VHH staining; n=8 biological replicates) and 3 independent experiments (10–1074 staining; n=3 biological replicates). Statistical analysis: paired t test between the 0-minute time point and the 10, 30 and 60-minute time points within either the J3 VHH or 10–1074 groups, *p<0.05, **p<0.01, ns=not significant. (E and F) Characterization of the internalized HIV envelope on infected macrophages using confocal microscopy. (E) Representative images of infected macrophages stained for the HIV Gag and Envelope (J3 VHH) and compartment markers EEA1 (top row), LAMP1 (middle row), or Siglec-1 (bottom row). Blue staining is Hoescht/Nuclei. The bar denotes 10μm. (F) Summary of the HIV Envelope co-colocalization with the HIV Gag, EEA1, LAMP1, and Siglec-1. Shown are the calculated Pearson Correlation Coefficients for the Envelope vs Gag (n=106 images over 14 independent experiments), Envelope vs EEA1 (n=42 images over 7 independent experiments), Envelope vs LAMP1 (n=23 images over 3 independent experiments), and Envelope vs Siglec-1 (n=29 images over 7 independent experiments). Statistical analysis: unpaired t test, ****p<0.0001, ns=not significant. See also Fig. S3.
To determine the persistence of the HIV envelope surface expression on macrophages, infected cells were stained at 4°C with AF647-conjugated J3 VHH or 10-1074, followed by washing and a time course of subsequent incubation at 37°C. Cells were then stained at 4°C for surface CD33, fixed, stained for intracellular Gag and then analyzed via imaging flow cytometry. Surface localization of the HIV envelope, detected by J3 VHH staining, was assessed on Gag+ macrophages, and the internalization of the J3 VHH probe (or the Gag probe as a control) relative to surface CD33 staining was calculated using the IDEAS software Internalization Wizard (Fig. S3B). HIV Gag- macrophages either in the uninfected samples or the infected samples showed only minor staining for J3 VHH, indicating that there was minimal non-specific staining and endocytosis of the J3 VHH (Fig. S3B). The HIV envelope was detected at the surface of infected macrophages when staining was performed at 4°C (Fig. 3C). Subsequent incubation at 37°C showed delivery of the HIV envelope into an intracellular compartment. While Gag staining showed no significant change in internalization score over 60 minutes, the HIV envelope was internalized in as little as 10 minutes at 37°C (Fig. 3D). Similar kinetics of internalization were observed when 10–1074 was used to detect the HIV envelope on infected macrophages (Fig. 3D and Fig. S3C). We conclude that in infected macrophages, the HIV envelope can be expressed at the plasma membrane, from which it is then rapidly trafficked to cell-internal compartments.
To confirm the identity of the intracellular compartment to which the HIV envelope traffics, live infected macrophages on coverslips were incubated with J3 VHH-AF647 at 37°C for one hour, followed by washing, fixing, and intracellular staining for Gag and resident proteins for different intracellular compartments, including EEA1 (early endosome), LAMP1 (lysosome), and Siglec-1 (VCC - (Hammonds et al., 2017)). The HIV envelope was found in intracellular compartments, which co-localized with HIV Gag (0.731 ± 0.013 mean ± SEM Pearson Correlation Coefficient, Fig. 3E and F). Significantly less co-localization was observed with EEA1 and with LAMP1. Although lower than co-localization with Gag, HIV envelope co-localization with Siglec-1 was significantly greater than with either EEA1 or LAMP1. This surface expression and subsequent internalization into the VCC was also confirmed with macrophages infected with CCR5-tropic HIVADA (Fig. S3D–F). Thus, following initial surface exposure on infected macrophages, the HIV envelope is delivered to VCCs, which are distinct from early endosomes and lysosomes (Deneka et al., 2007).
HIV-specific CAR T cells recognize infected CD4+ T cells and macrophages equally well
To determine whether transient surface expression of the HIV envelope is sufficient to induce receptor clustering and intracellular signaling of a chimeric antigen receptor (CAR), we constructed three HIV-specific CAR T cells and a CD19-specific CAR T cell as a negative control (Fig. 4A). Following confirmation of the specificity and functionality of each CAR T cell culture using plate-bound antigens (Fig. S4A–C), two separate co-culture assays were performed to characterize the HIV envelope’s accessibility on infected macrophages (Fig. 4A). The first consisted of a co-culture assay with CAR T cells and suspended CD4+ T cells or macrophages to compare CAR T cell responses to both targets. The second consisted of a co-culture of CAR T cells with adherent macrophages to confirm that detection of HIV envelope on suspended macrophages is not an artifact of disrupted VCCs due to the physical displacement of macrophages from the culture plate (a process required for antibody staining and co-culture assays). For the suspended target co-cultures, CAR T cells and uninfected or infected CD4+ T cells and macrophages were incubated for 6 hours, followed by an assessment of CD107a and TNF-α production by flow cytometry (gating strategy shown in Fig. S4D). While CD19-specific CAR T cells did not respond, all three HIV-specific CAR T cells responded to infected targets (Fig. 4B and C). Furthermore, there were no significant differences in the CAR T cell responses to infected CD4+ T cells and macrophages (Fig. 4C). We conclude that the HIV envelope is equally accessible on the surface of both types of target cells. While all three HIV-specific CAR T cells overall exhibited more efficient killing of infected CD4+ T cells than of macrophages, certain cultures of CAR T cells efficiently eliminated infected macrophages, even after just 4 hours of co-culture (Fig. S4E and F). Overnight co-cultures enhanced elimination of infected macrophage by PGT121 CAR T cells to levels comparable with killing of CD4+ T cells, even at an E:T as low as 0.5 (Fig. S4G). This is an improvement over NK cell-mediated elimination, which requires and E:T of 2 to reach comparable levels of CD4+ T cell and macrophage killing after overnight co-culture (Fig. 1D). Still, at lower E:T ratios, macrophages are more difficult to kill than infected CD4+ T cells, in agreement with previous reports of CTL-mediated killing (Clayton et al., 2018; Rainho et al., 2015; Vojnov et al., 2012).
Fig. 4. CAR T cells recognize and kill HIV-infected macrophages.

(A) Schematic of the CAR constructs and different co-culture assays used to characterize CAR T cell interactions with infected cells. “CD8 TM”: CD8 transmembrane region. (B and C) CAR T cell recognition assays with suspended target co-cultures. Infected CD4+ T cells and macrophages were co-cultured with CAR T cells at a E:T of 0.5 or 1 for 6 hours, followed by flow cytometry-based analysis of CAR T cell function. (B) Representative flow cytometry plots of CAR T cell CD107a and TNF-α production. (C) Summary of the CD107a and TNF-α responses to infected CD4+ T cells and macrophages. Responses to infected targets are corrected for the responses to uninfected targets. Shown are data from 3 independent experiments (n=6 biological replicates). (D and E) Adherent macrophage co-culture CAR T cell recognition and killing assays. CAR T cells were added to the plate-bound macrophages at an E:T of 2 and incubated overnight, followed by ELISA measurement of IFN-γ in the culture supernatants and assessment of infected macrophage elimination by flow cytometry. Shown are data from three independent experiments (n=5 biological replicates). (D) Summaries of IFN-γ production in the macrophage only cultures (gray dashed lines) and co-cultures. (E) Summaries of infected macrophage elimination. Statistical analysis for (C-D), paired t tests: *p<0.05, **p<0.01, ns=not significant. See also Fig. S4.
As a final test to confirm that the HIV envelope is accessible on plate-adherent macrophages, a more physiological state than macrophages maintained in suspension, CAR T cells were added to adherent uninfected and infected macrophage cultures and incubated overnight. CAR T cells co-cultured with uninfected macrophages (white boxes) produced more IFN-γ than macrophage-only cultures (gray lines) (Fig. 4D). This represents the background CAR T cell response. Co-culture of the control CD19 CAR T cells with HIV-infected macrophages showed no increase in IFN-γ production compared to co-cultures with uninfected macrophages. However, co-cultures of J3 and PGT121 CAR T cells with infected macrophages showed a significant increase in IFN-γ production (Fig. 4D), suggesting that HIV-specific CAR T cells can recognize adherent macrophages. Furthermore, all three HIV-specific CAR T cells killed adherent, HIV-infected macrophages at a level that significantly exceeded killing observed with the CD19 control CAR T cell (Fig. 4E). We conclude that expression of the HIV envelope on macrophages is sufficient to enable CAR-mediated T cell recognition. Analogous antibody-based recognition of the HIV envelope is therefore unlikely to be a limiting factor for NK cell-mediated ADCC.
HIV antibody binding enhances NK cell responses to infected macrophages, but less well than ADCC responses to infected CD4+ T cells
To determine whether HIV antibody binding can enhance NK cell recognition and killing of infected macrophages, we incubated uninfected or infected target cells either with polyclonal IgG from a healthy donor, or with the HIV-specific antibodies 3BNC117 and PGT121. Autologous NK cells were then co-cultured with antibody-bound targets for 6 hours, followed by analysis of CD107a and TNF-α production by flow cytometry. Both 3BNC117 and PGT121 induced a dramatic NK cell response to infected CD4+ T cells compared to control IgG (Fig. 5A and B). 3BNC117 and PGT121 also enhanced NK cell responses to infected macrophages. However, ADCC responses to infected CD4+ T cells were significantly greater than the responses to infected macrophages, after we applied a correction for the background responses to uninfected cells, normalized for the levels of infection, and corrected for the innate NK cell responses observed with IgG treatment (Fig. 5C). Furthermore, when compared to NK responses to infected CD4+ T cells, ADCC responses to infected macrophages were significantly skewed towards production of TNF-α over degranulation (Fig. 5D). To determine whether these differences in ADCC responses applied to other strains of HIV, we measured NK cell responses to HIVADA-infected CD4+ T cells and macrophages. PGT121 significantly enhanced NK cell responses to both HIVADA-infected CD4+ T cells and macrophages (Fig. S5A), however, similar to HIV89.6-infected targets, the responses to infected macrophages were significantly less compared to the responses to infected CD4+ T cells (Fig. S5B). Furthermore, the NK cell responses to HIVADA-infected macrophages were significantly skewed towards production of TNF-α (Fig. S5C). Thus, while HIV-specific antibodies can enhance NK cell responses to infected macrophages, these responses are significantly weaker compared to ADCC responses to infected CD4+ T cells.
Fig. 5. Antibody-mediated NK cell responses to HIV-infected macrophages versus CD4+ T cells are muted.

NK cell ADCC recognition assays using normal human IgG (control), 3BNC117, or PGT121 at a T:E of 10 for 6 hours followed by flow cytometry analysis of NK cell function. (A) Representative recognition assay plots. (B) HIV-specific ADCC enhances responses to both infected CD4+ T cells and macrophages. Data summaries for NK cell responses (%CD107a + %TNF-α) from six independent experiments (n=9 biological replicates). These raw values were not corrected for background recognition of uninfected cells. (C) Total NK cell ADCC responses to infected macrophages are significantly lower compared to ADCC responses to CD4+ T cells. “%CD107a + %TNF-α” values are initially corrected for background recognition of uninfected targets and normalized to target cell infection frequencies, followed by corrections of recognition in IgG conditions. Shown are data from six independent experiments (n=9 biological replicates). (D) Data summaries for the quality of the NK cell response from six independent experiments (n=9 biological replicates). Values are corrected for background recognition of uninfected target cells. Ratios of CD107a versus TNF-α production were calculated as described in the STAR Methods. Statistical analysis for (B-D): paired t tests, **p<0.01, ***p<0.001, ****p<0.0001. See also Fig. S5.
In line with the recognition assay data, 3BNC117 and PGT121 significantly enhanced killing of infected CD4+ T cells after a 4-hour co-culture (Fig. 6A and B). Both antibodies also modestly but significantly enhanced killing of infected macrophages. However, ADCC-mediated elimination of infected CD4+ T cells was substantially greater than elimination of infected macrophages during this short-term co-culture (Fig. 6C). While ADCC enhanced killing of CD4+ T cells after overnight co-culture, ADCC did not significantly enhance the killing of infected macrophages after overnight co-culture (Fig. 6D). However, at an E:T ratio of 2, there was a trend towards ADCC-enhanced killing. This translated to significantly greater ADCC-mediated killing of infected CD4+ T cells versus infected macrophages across all E:T ratios after overnight co-culture (Fig. 6E). PGT121 significantly enhanced NK-mediated elimination of HIVADA-infected CD4+ T cells, and modestly but significantly enhanced NK-mediated elimination of HIVADA-infected macrophages (Fig. S6A and B). However, elimination of HIVADA-infected macrophages was significantly less than that of infected CD4+ T cells across multiple E:T ratios (Figure S6C). This suggests that differences in ADCC responses to HIV-infected targets is not envelope-dependent, at least in the case of these two strains of HIV. Thus, while expression of the HIV envelope can be exploited to enhance responses of NK cells to infected CD4+ T cells, ADCC responses to infected macrophages are significantly muted, rendering macrophages less susceptible to NK cell ADCC-enhanced killing.
Fig. 6. ADCC-mediated killing of HIV-infected macrophages is less efficient than killing of HIV-infected CD4+ T cells.

NK cell ADCC target elimination assays using normal human IgG (control), 3BNC117, or PGT121 at different E:T ratios, followed by flow cytometry-based analysis of residual live target cell infection by intracellular Gag p24 staining. (A) Representative flow cytometry plots. (B and C) Summary elimination assay data for a 4-hour co-culture from 4 independent experiments (n=7 biological replicates) showing (B) ADCC-induced elimination of infected targets and (C) the differences in ADCC elimination of infected CD4+ T cells and macrophages. (D and E) Summary of PGT121 ADCC elimination assays for overnight co-cultures from 4 independent experiments (n=5 biological replicates) showing (D) PGT121-specific ADCC elimination of infected targets and (E) the differences in ADCC elimination of infected CD4+ T cells and macrophages. Statistical analysis for (B-E): paired t tests, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant. See also Fig. S6.
DISCUSSION
Macrophages are targets of HIV infection, store virus in long-lived VCCs, persist in tissues during ART, survive the cytopathic effects of HIV infection, and can seed new infections by cell-to-cell spread. While CTLs can recognize these infected targets, their elimination is inefficient (Clayton et al., 2018; Rainho et al., 2015; Vojnov et al., 2012). This contributes to poor viral suppression and amplification of inflammation through prolonged CTL-macrophage interactions (Clayton et al., 2018), which may add to chronic inflammation during HIV infection. Here we show that NK cells, the innate cytolytic counterpart to CTL, are inefficient at killing of HIV-infected macrophages compared to HIV-infected CD4+ T cells as targets. We show that the magnitude of NK cell sensing of HIV infection, likely through innate receptors, is similar for infected macrophages and CD4+ T cells. However, the output of this sensing differs. Responses to infected CD4+ T cells skew towards cytolytic degranulation compared to those against infected macrophages that skew more towards production of TNF-α, a mechanism that may accommodate the antigen-presenting function of macrophages upon initial encounter with NK cells.
NK cells use multiple mechanisms to kill their targets. Perforin-dependent killing is rapid and is the first line of defense; however, for scenarios in which NK cells cannot kill their targets immediately or in which NK cells engage in serial killing, the mode of killing may switch instead to death receptor engagement through Fas ligand (Prager et al., 2019; Zhu et al., 2016). NK cell-mediated killing of infected macrophages requires overnight co-culture, whereas killing of infected CD4+ T cells is detected already after 4 hours. Poor degranulation and limited release of perforin/granzymes upon interaction with infected macrophages may force NK cells to rely on death receptor engagement to mediate killing. Similar mechanisms have been described for HIV-infected immature dendritic cells, which require overnight co-cultures to detect NK cell-mediated killing that depends on the TRAIL death receptor pathway and not on perforin (Melki et al., 2010). Cell size must also be considered when interpreting elimination assay data for two different types of target cell. The macrophage is significantly larger than the CD4+ T cell, but whether this results in a requirement for the release of more perforin/granzymes to be killed is not known. This may be the case for CAR T cells, which show comparable recognition of both cell types, and can efficiently kill macrophages, but still less well than killing of CD4+ T cells at lower E:T ratios. In contrast, the NK cell response to infected macrophages versus CD4+ T cells is significantly skewed towards production of TNF-α over degranulation. This suggests that initial interactions with macrophages yield less release of NK cell-derived cytolytic contents than interactions with CD4+ T cells. Thus, macrophage killing is likely mediated by engagement of death receptors instead of by the immediate release of perforin/granzymes.
The receptor-ligand interactions on NK-macrophage pairs that promote TNF-α while suppressing degranulation is of great interest for the development of cure strategies as a means of eliminating reservoirs of infected macrophage. In the case of CTL effectors, TCR stimulation is sufficient to induce degranulation. Strength and duration of TCR signaling can dictate the extent of cytokine production from cells that have degranulated (Clayton et al., 2018; Jenkins et al., 2015). In contrast, NK cells require combinations of signals from both activating and inhibitory receptors to induce effector function (Bryceson et al., 2006a, b). The ability of target cells to skew NK effector functions may therefore be a result of differential engagement of NK cell receptors. It is unclear whether modulation of NK cell ligands on CD4+ T cells by HIV accessory proteins (Alsahafi et al., 2017; Apps et al., 2016; Jost and Altfeld, 2012; Richard and Cohen, 2010; Richard et al., 2010; Schwartz et al., 1996; Shah et al., 2010; van Stigt Thans et al., 2019; Ward et al., 2007) is similar on infected macrophages and infected CD4 T cells. However, here we have shown that NK cell degranulation and TNF-α production are more potently stimulated by CD4+ T cells than by macrophages in the absence of infection. Thus, differential killing of infected targets may stem from underlying differences in contact-mediated degranulation. This may be due to higher levels of activating ligands or lower levels of inhibitory ligands. Indeed, activation of CD4+ T cells, required for productive HIV infection, upregulates the display of activating NKG2D ligands (Cerboni et al., 2007). For macrophages, TLR stimulation can upregulate NKG2D ligands and sensitize macrophages to NK cell killing (Eissmann et al., 2010; Hamerman et al., 2004; Nedvetzki et al., 2007). However, in our work we allow maturation of macrophages in the absence of any TLR stimulus to prevent their polarization, which can limit HIV infection (Cassol et al., 2010). Together, the different CD4+ T cell and macrophage activation states, while ideal for HIV infection, may yield target cells that differ in their ability to activate NK cells. When comparing infected CD4+ T cells and macrophages, many more ligand-receptor pairs beyond NKG2D may be differentially engaged on NK cells. This is a topic obvious for future study.
One method of enhancing NK cell responses is through ADCC, which has been characterized extensively with CD4+ T cell targets (Lee and Kent, 2018), but ADCC responses to HIV-infected macrophages have not been characterized. Although surface expression of the HIV envelope on infected macrophages has been controversial, our CAR T cell assays establish that it is equally accessible at the surface of both CD4+ T cells and macrophages. Exposure of the envelope should therefore not be a limiting factor for ADCC. However, antibody-enhanced NK cell responses to infected macrophages were significantly muted compared to CD4+ T cells. When the HIV envelope is not associated with Gag protein at the membrane, it can be internalized from the CD4+ T cell surface (Egan et al., 1996) into the early endosomes (Anand et al., 2019), a mechanism that helps protect the cells from ADCC (Anand et al., 2019; von Bredow et al., 2015). Furthermore, broadly neutralizing antibodies, such as PGT121, can induce internalization of the envelope, but their monomeric Fabs cannot (Anand et al., 2019). Using J3 VHH, a monomeric single domain antibody, we showed that the envelope on both HIV89.6 and HIVADA-infected macrophages traffics to the VCCs. However, trafficking may shift to early endosomes if full-sized antibodies are used, which may limit ADCC responses to infected macrophages. Still, ADCC responses to infected CD4+ T cells were potent, suggesting that even in the presence of antibody-induced internalization, surface levels of envelope-antibody complexes are sufficient to induce NK cell effector function. Expression of activating ligands can co-activate NK cells to enhance ADCC (Parsons et al., 2016), and expression of HLA-C and HLA-E can inhibit ADCC (Ward et al., 2004). Thus, higher expression of activating ligands on activated CD4+ T cells than on macrophages or higher expression of MHC-I on macrophages than CD4+ T cells may explain the differences in ADCC responses to infected targets. We conclude that infected macrophages inhibit early NK cell cytolytic effector function by skewing responses towards TNF-α production and limiting ADCC-based recognition of infected targets, emphasizing their ability to use multiple mechanisms to persist and contribute to the HIV reservoir.
While killing of infected macrophages by NK cells may be inefficient, HIV-specific CAR T cells showed robust degranulation in response to infected macrophages, on a par with recognition of infected CD4+ T cells. This suggests a potential therapeutic avenue for an HIV cure that would target macrophages in addition to CD4+ T cells. Interestingly, while all three HIV-specific CAR T cells exhibited more efficient killing of infected CD4+ T cells versus macrophages, certain cultures of CAR T cells were able to efficiently eliminate infected macrophages even after just 4 hours of co-culture. Such variability may stem from the expression of effector cell granzyme B, high levels of which are required for macrophage killing (Clayton et al., 2018). Together, our findings support the potential use of CAR T cell therapies to overcome barriers induced by macrophages that promote resistance to endogenous TCR and NK elimination. This work also emphasizes the need to refine and optimize CAR T cell approaches for HIV cure strategies that include modifications to limit IFN-γ production and increase expression of granzyme B as a means to mitigate unwanted inflammation and enhance viral clearance.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Bruce Walker (BWALKER@mgh.harvard.edu)
Materials Availability
All custom materials, including the soluble HIVJRCSF envelope gp140, are available upon request. There are restrictions to the availability of the CAR T cell lentiviral vector constructs due to proprietary sequences in the plasmid backbone and as such, material transfer agreements are required for any sharing of these resources. Please contact Dr. Marcela Maus (MGH) regarding access to these lentiviral vectors.
Data and Code Availability
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author on request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Buffy coat blood products from anonymous HIV-uninfected healthy donors were acquired from the Massachusetts General Hospital Blood Bank. No identifying information, including the age and gender of the donors, was available. All human subjects gave written consent for use of their blood products for research purposes. The institutional review board of Massachusetts General Hospital approved the use of Buffy Coats for research purposes. Sample sizes for each set of experiments is found in the figure legends.
Cell Lines, Primary Human Cells, and Microbes
Both cell lines (HEK293T/17 cells) and primary human immune cells were used to make stocks of infectious HIV virus and create target/effectors cells for co-culture assays. For all primary human samples, peripheral blood mononuclear cells (PBMCs) were collected by Ficoll gradient separations from buffy coat samples, cryopreserved and stored at −150°C for future use. All HIV and CAR T cell plasmids were grown up in Stbl3 E.coli (ThermoFisher, Cat#C737303).
Preparation of HIV Stocks
HIV89.6 proviral plasmid DNA (NIH AIDS Reagent Program, Cat#3552; contributed by Dr. Ron Collman) was transformed into Stbl3 cells and grown in LB-carbenicillin maxi cultures (250mL) for 48 hours at room temperature with shaking at 180 rpm (these conditions were used to prevent recombination). CAR T cell lentiviral construct plasmid DNA was transformed into Stbl3 cells and grown in LB-carbenicillin maxi cultures (250mL) for 24 hours at 37°C. DNA was maxiprepped (ThermoFisher, Cat#K210017) according to the manufacturer’s instructions. HEK293T/17 cells (ATCC, Manassas, VA, Cat#CRL-11268) were used to make stocks of HIV89.6, an R5X4 dual-tropic strain of HIV. Briefly, HEK293T/17 cells (<10 passages) were plated overnight at 25 million cells per T225 flask in 50 mL of D10 media: DMEM with high glucose and pyruvate (ThermoFisher, Cat#1995065) containing 10% FBS (Sigma-Aldrich), 1 U/ml penicillin and 100 mg/ml streptomycin (Corning Inc), and 2 mM L-glutamine (Corning Inc). The next day, the culture media was exchanged for 44mL of fresh D10 media. 50 μg of HIV89.6 proviral plasmid DNA was prepared in 6.5mL of serum-free DMEM and then mixed with 200 μg of 25kDa polyethylenimine (PEI - stock at 1mg/mL; Polysciences Inc, Warrington, PA, Cat#23966–1). Following a 10-minute incubation, the DNA:PEI mixture was added dropwise to the cells. 72 hours post-transfection, culture supernatants were collected, centrifuged at 3000 xg for 5 minutes to remove cell debris, filtered through a 0.45 μm membrane (Millipore, Burlington, MA, Cat#SLHV033RS) to remove smaller aggregates, concentrated with PEG-it™ Virus Precipitation Solution (System Biosciences, Palo Alto, CA, Cat#LV825A-1) as per the manufacturer’s instructions, resuspended in R10 media at a 100x lower volume than the original volume of virus (ie. 0.4mL of R10 for 40mL of virus), and frozen at −80°C.
CCR5, M-tropic HIVADA stocks were propagated on activated PBMCs. Briefly, ~40 million PBMCs were thawed, plated in a T225 flask in R10 media, and stimulated with 1 μg/mL of PHA-M (Sigma-Aldrich, Cat#L8902) for three days. 10 million activated PBMCs were harvested and resuspended in 1 mL of HIVADA cell-free virus stock (NIH AIDS Reagent Program, Cat#416; contributed by Dr. Howard Gendelman). The cells were incubated for two hours at 37°C with occasional shaking/strumming, followed by removal of the virus, two washes in R10 media, and resuspension in 10 mL of R10 + 10 ng/mL IL-2 in a T25 flask. After 4 days, the cells were pelleted and the virus-containing supernatant was filtered through a 0.45 μm membrane, concentrated using PEG-it™ Virus Precipitation Solution as described for HIV89.6 virus stocks, and frozen at −80°C. The cells were resuspended in fresh R10 + 10 ng/mL IL-2 and incubated at 37°C for a full 24 hours. This was repeated for an additional seven days until a total of eight concentrated virus stocks were acquired. Each stock of HIV89.6 and HIVADA virus was titered on activated CD4+ T cells and macrophages to determine the volume of virus that yielded saturated levels of infection for each cell type 2–3 days post infection.
Preparation of HIV-infected targets
Monocyte-derived macrophages (termed “macrophages” in this manuscript) and CD4+ T cells, were prepared as previously described (Clayton et al., 2018). Briefly, monocytes were isolated from frozen PBMCs using the EasySep Human CD14 Positive Selection Kit II (STEMCELL Technologies, Vancouver, Canada, Cat#17858) as per the manufacturer’s instructions. CD4+ T cells were enriched from the remaining CD14-depleted PBMCs using the EasySep Human CD4+ T cell Isolation Kit (STEMCELL Technologies, Cat#17952) as per the manufacturer’s instructions. Monocytes were matured into macrophages over seven days in 24-well or 6-well low attachment plates (0.5 million/well or 2 million/well respectively; Corning Inc, Corning, NY, Cat#CLS3471 and CLS3473) in the presence of 50ng/mL recombinant GM-CSF (Biolegend, San Diego, CA, Cat#572904) and 50ng/mL recombinant M-CSF (Biolegend, Cat#574806) in R10 Media: RPMI-1640 (Sigma-Aldrich, St. Louis, MO) containing 10% Certified FBS (ThermoFisher, Waltham, MA, Cat#16000044), 1 U/ml penicillin and 100 mg/ml streptomycin (Corning Inc), 2 mM L-glutamine (Corning Inc), and 10mM HEPES (Corning Inc). Certified FBS lots were chosen based on low endotoxin and their ability to mature macrophages that yielded efficient levels of HIV infection. Half of the R10 media containing fresh GM-CSF/M-CSF was exchanged on the fourth day of maturation. Successful maturation was assessed via spreading of the cells onto the surface of the plate. In parallel, CD4+ T cell targets were activated by anti-CD3 and anti-CD28 antibodies. Briefly, the day before the isolation, one 24-well non-treated TC plate (Corning, Inc, Cat#CLS3738) was coated overnight with 2 μg/mL of anti-CD3 antibody (Biolegend, Cat#317326, RRID_AB_11150592) in sterile bicarbonate coating buffer (8.4g NaHCO3, 3.56g Na2CO3, 1L of ddH2O, pH to 9.5 – 500ul per well). The next day, the wells were each washed twice with 1mL of sterile PBS and given 400ul of R10 media containing 4 μg/mL of anti-CD28 antibody (Biolegend, Cat#302934, RRID:AB_11148949) and 10 ng/mL IL-2 (R&D Systems, Minneapolis, MN, Cat#202-IL-500). Note that IL-2 media was made fresh every 7–10 days (older IL-2 resulted in inefficient activation and expansion of the cells). The isolated CD4+ T cells were resuspended to 2 million/mL in R10 + 10ng/mL IL-2, and then 400ul of cells was added to each well of the 24 well plate (0.8 million/well in a final concentration of 2ug/mL soluble anti-CD28 antibody). After 4–5 days of activation, the cells were removed from the plate, washed in R10 media, and rested for an additional 3 days in R10 + 10ng/mL IL-2 at a seeded concentration of approximately 0.5 million/mL (this allowed for cell expansion). Note that the cell activation on the plate was allowed to proceed until the cells had blasted, exhibited some adherence to the bottom of the plate, and the media was yellow. Cells lifted too early from the plate resulted in lower levels of downstream infection. In general, half a 24-well plate of harvested, activated CD4+ T cells was seeded into a T75 flask (incubated in a horizontal position) in 40mL of R10/IL-2 media. Together, the macrophages were matured and the CD4+ T cells were activated/expanded for a total of 7 days.
HIV89.6 and HIVADA were used to infect macrophages and CD4+ T cells for co-culture assays. Briefly, for macrophages in 6-well and 24-well plates, the media was removed, and 1.2 mL or 0.3 mL of fresh macrophage media (R10 containing GM-CSF/M-CSF) was added back to each well, respectively. The volume of virus added to each well of macrophages was determined based on the viral tittering of each stock that yielded saturated levels of infection, which typically ranged from ~10% to 50%, depending on the donor. 1.5–2 plates of macrophages were infected per experiment. The cells were incubated for six hours at 37°C, followed by removal of the virus and addition of the original conditioned media diluted 1-in-2 with fresh macrophage media. In parallel, activated CD4+ T cells were washed once in R10 media and then infected in a 96-well flat bottom plate at 1 million cells/well in 50ul of R10 + 10ng/mL IL-2 with the appropriate volume of virus that yielded saturated levels of infection (~10–50%, depending on the donor). Typically, 3 million cells were infected per experiment. The cells were spinoculated at 800 xg for one hour, incubated at 37°C for three hours, followed by resuspension of the cells to 2 million/mL in R10 + 10 ng/mL IL-2. After 2–3 days, levels of infection were assessed for each target cell type via flow cytometry in the morning before setting up the co-culture assays. Macrophages were washed in PBS and lifted from the low attachment plates using Cell Dissociation Buffer (ThermoFisher, Cat#13151014) for 10 minutes at 37°C and transferred over to 96 well V-bottom plates. Before addition of antibodies, macrophage samples were Fc blocked with Human TruStain FcX (Biolegend, Cat#422302, RRID:AB_2818986) at 5ul of FcX per sample in 50ul of FACS Buffer (2% FBS, 1mM EDTA in PBS). Once the Fc blocking was completed, macrophages and CD4+ T cells were surface stained with anti-CD14-APC/Cy7 (Biolegend, Cat#301820, RRID:AB_493695) or anti-CD3-APC/Cy7 (Biolegend, Cat#300318, RRID:AB_314054), respectively, anti-CD4-APC (Biolegend, Cat#317416, RRID:AB_571945), and LIVE/DEAD Fixable Blue (ThermoFisher, Cat#L34962). Note, for the macrophages, the staining mix was made up in 50ul of FACS Buffer and added directly to the macrophages in the 50ul of FcX-FACS Buffer. Following a 20 minute incubation at 4°C, the samples were washed, fixed and permeabilized using BD CytoFix/CytoPerm Fixation/Permeabilization Kit (BD Biosciences, Woburn, MA, Cat#554714), and stained for the HIV Gag protein using anti-Gag p24-RD1 (Beckman Coulter, Brea, CA, Cat#6604667, RRID:AB_1575989). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software (BD Biosciences). All data were analyzed using FlowJo 10.6.0 software (FlowJo, LLC, Ashland, OR). Gating strategies to assess for infection are shown in Fig. S1A and B. Uninfected cell samples were used to draw the infected cell gate. Infected cells were considered LIVE/DEADNeg, CD3+CD4-p24+ for CD4+ T cells and CD14Int/PosCD4-p24+ for macrophages. Once the levels of infection in each sample were determined, when appropriate, uninfected cells were added to the infected cell sample to help normalize infection levels between the two target cell types before co-cultures. Target cell infections ranged from 3% to 63% across all experiments.
Preparation of effector NK and CD8+ T cells
Autologous PBMCs (100–150 million) were thawed and cultured in 25mL R10 media with 50 ng/mL recombinant IL-15 (Biolegend, Cat#570304) overnight in a T75 flask (horizontal) before the co-culture assays. IL-15 was added to enhance the survival of NK cells in unfractionated PBMCs during the overnight resting period (Huntington et al., 2007). Following confirmation of target cell infection, NK cells were isolated from rested PBMCs using the EasySep Human NK Cell Isolation Kit (Stemcell Technologies, Cat#17955) as per the manufacturer’s instructions. Yields ranged from 1.0% to 3.6%. For experiments involving autologous CD8+ T cells and NK cells, 12.5 million rested PBMCs were used to isolate CD8+ T cells using the EasySep Human CD8+ T Cell Isolation Kit (Stemcell Technologies, Cat#17953) as per the manufacturer’s instructions, which provided yields of ~8–16%, while the remainder PBMCs were used to isolate NK cells. For all co-culture assays, effector cells were loaded with CellTrace Violet (ThermoFisher, Cat#C34557) to distinguish them from target cells. Briefly, cells were resuspended to ~2 million/mL in R10 and incubated with CellTrace Violet (1:200 dilution of 5mM reconstituted reagent) for 5 minutes at 37°C. To quench the reaction, a volume of cold FBS equal to the volume of media was added to cells. The cells were pelleted, washed once in R10, and then resuspended to the appropriate concentration for the assays.
Preparation of CAR T cell effectors
HIV-specific Chimeric Antigen Receptor (CAR) constructs were generated using the sequences for the human CD4 D1D2 domains (aa 26–208; GenBank: NP_000607.1), J3 VHH (McCoy et al., 2012), and the single-chain variable fragment (scFv) derived from PGT121 (VH domain GenBank: JN201894.1; VL domain GenBank: JN201911.1). Each construct was designed with an N-terminal fusion to the human CD8 leader/signal sequence (aa 1–21; GenBank: AAH25715.1), and a C-terminal fusion to the CD8 hinge and transmembrane region (aa 138–206; GenBank: AAH25715.1), followed by a human 4–1BB intracellular signaling domain (aa 214–255; GenBank: NP_001552), human CD3 zeta chain intracellular signaling domain (aa 52–163; GenBank: NP_000725.1), T2A sequence (EGRGSLLTCGDVEENPGPR), and mCherry fluorescent reporter sequence (GenBank: ANF29837.1). These fusion constructs were cloned into a 3rd generation self-inactivating lentiviral vector backbone plasmid (Marcela Maus, MGH) using In-fusion protocols (Takara Bio, Cat#638947) as per the manufacturer’s instructions. Successful cloning was determined through whole plasmid sequencing. The transfer plasmids were co-transfected with second-generation helper plasmids into HEK293T/17 cells using Lipofectamine® P3000 (ThermoFisher, Cat#L3000015) as per the manufacturer’s instructions. Lentiviral supernatants were harvested at 72 hours post-transfection, filtered through a 0.45 μm membrane and concentrated PEG-it™ Virus Precipitation Solution as per the manufacturer’s instructions. Lentivirus containing transfer plasmids encoding a CD19-specific scFv was provided by Marcela Maus.
To create CAR-transduced T cells, frozen PBMCs from healthy donors were used to isolate total T cells using the EasySep Human T Cell Isolation Kit (Stemcell Technologies, Cat#17951) as per the manufacturer’s instructions, and activated at a 1:3 cell:bead ratio using CD2/CD3/CD28 MACSiBead™ Particles from the T Cell Activation/Expansion Kit (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat#130–091-441) in CTS™ OpTmizer™ T Cell Expansion SFM (ThermoFisher, Cat#A1048501), supplemented with 1 U/ml penicillin and 100 mg/ml streptomycin (Corning Inc), 2 mM L-glutamine (Corning Inc), 10mM HEPES (Corning Inc), and 10 ng/mL IL-2. One day after activation, T cells were transduced with each of the 4 lentiviral stocks encoding the CD19, CD4 D1D2, J3, and PGT121 CAR constructs in the media containing the MACSiBead™ Particles. Three days after transduction, the cells were passed through a magnet to remove the activating particles and left to expand for 14 days in R10 media + 10 ng/mL IL-2.
Confirmation of transduction and functionality of the CAR T cells was performed using plate-bound antigen recognition assays with recombinant CD19 and soluble HIV envelope. To construct the soluble HIV envelope (gp140) protein, the codon optimized leader sequence and full ectodomain of HIV envelope (strain JR-CSF aa1–677, GenBank: AAT67498.1) with mutations to render the gp140 cleavage deficient (R495E and K502E for JR-CSF) were given a C-terminal a FactorXa site (IEGR), a Fibritin Foldon sequence (GSGGYIPEAPRDGQAYVRKDGEWVLLSTFL) to aid trimerization, and a 6XHIS tag for purification (Kovacs et al., 2014). gBlocks of these constructs were obtained from Integrated DNA Technologies (Coralville, IA) and cloned into a proprietary mammalian expression vector using In-fusion protocols (Takara Bio, Cat#638947) as per the manufacturer’s instructions. Successful cloning was confirmed via sanger sequencing. The protein was produced via mammalian expression suspension culture and purified by Ni-NTA column chromatography (Qiagen, Hilden, Germany, Cat#30230) and size exclusion chromatography using a Superdex 200 Increase 10/300 GL (Cytiva Life Sciences, Marlborough, MA, Cat#28990944) the Lingwood Lab (Ragon Institute of MGH, MIT and Harvard).
Recombinant CD19-Fc (R&D Systems, Cat#9269-CD-050) and the purified soluble envelope protein were coated overnight on a Not Treated, sterile 96 well plate (Corning, Cat#351172) at 10 μg/mL in sterile bicarbonate coating buffer (8.4g NaHCO3, 3.56g Na2CO3, 1L of ddH2O, pH to 9.5 – 100ul per well). After washing the plate in PBS, the CAR T cells were plated onto the appropriate wells in the presence of anti-CD107a-BV421 (Biolegend, Cat#328626, RRID:AB_11203537), and a 1:1000 dilution of GolgiPlug and GolgiStop (BD Biosciences, Cat#555029 and 554724, respectively) and incubated for 6 hours at 37°C. The cells were then surface stained with anti-CD3-PerCp/Cy5.5 (Biolegend, Cat#300328, RRID:AB_15775008), anti-CD8-APC (Biolegend, Cat#344722, RRID:AB_2075388), and LIVE/DEAD Fixable Blue (ThermoFisher, Cat#L34962). The cells were fixed and permeabilized using the BD CytoFix/CytoPerm Kit and intracellular stained using anti-IFN-γ-AF700 (Biolegend, Cat#502520, RRID:AB_528921). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software. All data were analyzed using FlowJo 10.6.0 software. Gating strategies for this assay can be found in Fig. S4A and B. CD19 and HIV-specific CAR T cells (LIVE/DEADNeg, mCherry+CD3+CD8+) that showed reactivity (CD107a and IFN-γ responses above background) to the CD19-Fc or the HIV envelope, respectively, were used for downstream co-culture assays. Bulk CAR T cell cultures were stained with CellTrace Violet as described for the ex vivo NK cells and CD8+ T cells above to distinguish them from the target cells in co-culture assays.
METHODS DETAILS
Elimination assays
Uninfected and infected CD4+ T cell and macrophage target cells, and CellTrace Violet-stained effector cells (autologous ex vivo NK cells, CD8+ T cells, or CAR T cells) were prepared as described above. In 96 well round bottom low-attachment plates (Corning, Cat#CLS7007), effectors were co-cultured with uninfected and infected target cells at different effector-to-target ratios (E:T) as indicated in the figure legends for 4 hours, overnight, or for 48 hours at 37°C. For ADCC assays, targets were first plated and incubated with normal human IgG (R&D Systems, Cat#1–001-A, RRID:AB_907192), 3BNC117 or PGT121 (produced and purified in-house with the help of the Balazs and Lingwood laboratories) at a final concentration of 30 μg/mL at room temperature for 15–20 minutes, followed by addition of NK cells and incubation at 37°C for 4 hours or overnight. For CAR T cell co-culture assays using adherent macrophages, uninfected and infected macrophages were lifted and replated at 250,000 cells per well of a 24 well low-attachment plate (Corning Inc, Cat#CLS3471) in 500ul of macrophage media and allowed to adhere overnight. The next day, the cells were washed in R10 media to remove any non-adherent cells and one representative well was harvested and counted to determine the remaining number of macrophages per well (~50,000 cells on average). CellTrace Violet CAR T cells were then added at an E:T of 2 to the adherent macrophages in 500ul of R10 + 10ng/mL of IL-2 and incubated overnight. An E:T of 2 was selected to make sure enough IFN-γ was produced by the CAR T cells in the culture supernatants for detection by ELISA. The next day, the culture supernatants were saved for IFN-γ analysis by ELISA and the cells used for flow cytometry analysis. To remove all cells from the low attachment plate, cells were first mixed to disrupt the pellets and then transferred over to 96 well V-bottom plates. 100ul of Cell Dissociation Buffer (ThermoFisher, Cat#13151014) was added to each of the sample wells in the low attachment plate and incubated for 10 minutes at 37°C. During the last half of the incubation, the 96 well V-bottom plate was centrifuged to pellet the cells and the culture supernatant was aspirated. The Cell Dissociation Buffer was pushed around the low attachment plate wells to disrupt any remaining cells, and then transferred over and mixed with the corresponding cell pellets in the 96 well V-bottom plates. The cells were centrifuged in the 96 well V bottom and transferred over to 96 well V-bottom plates. The cells were centrifuged and then Fc blocked using Human TruStain FcX (Biolegend, Cat#422302, RRID:AB_2818986) at 5ul of FcX per sample in 50ul of FACS Buffer (2% FBS, 1mM EDTA in PBS). Once the Fc blocking was completed, macrophages and CD4+ T cells were surface stained with anti-CD14APC/Cy7 (Biolegend, Cat#301820, RRID:AB_493695) or anti-CD3-APC/Cy7 (Biolegend, Cat#300318, RRID:AB_314054), respectively, anti-CD4-APC (Biolegend, Cat#317416, RRID:AB_571945), and LIVE/DEAD Fixable Blue (ThermoFisher, Cat#L34962). Note, the staining mix was made up in 50ul of FACS Buffer and added directly to the cells in the 50ul of FcX-FACS Buffer. Following a 20 minute incubation at 4°C, the samples were washed, fixed and permeabilized using BD CytoFix/CytoPerm Fixation/Permeabilization Kit (BD Biosciences, Woburn, MA, Cat#554714), and stained for the HIV Gag protein using anti-Gag p24-RD1 (Beckman Coulter, Brea, CA, Cat#6604667, RRID:AB_1575989). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software (BD Biosciences). All data were analyzed using FlowJo 10.6.0 software (FlowJo, LLC).
Gating strategies for the elimination assays are found in Fig. S1A and B. Standard forward and side scatter settings for lymphocyte gating were used to capture T/NK cell events. Following gating of cells based on forward and side scatter area, doublets were excluded via a singlet gate using forward scatter area and width. Live CD4+ T cell targets were defined as LIVE/DEAD BlueNeg on forward scatter area versus LIVE/DEAD Blue plots, and CD3+CellTrace VioletNeg based on CD3 versus CellTrace Violet plots (Fig. S1A). For all macrophage elimination assay cultures, forward and side scatter PMT settings were decreased to capture the larger macrophage events. A gate was created to select this macrophage population, which excluded cells within the condensed lymphocyte population (NK cells, CD8+ T cells or CAR T cells – in Fig. S1B). Log transformation of the initial SSC-A and FSC-A plots show the effector NK cells are excluded from the macrophage gate. Downstream gating of the effector cells shows these are a distinct CD14-Violet+ population or cells and not debris. Thus, effector cell populations were not present in any subsequent macrophage populations. This initial gating based on cell size allows for cleaner separations of the macrophage targets and effector cell populations. Following singlet gating, live macrophage targets were defined as LIVE/DEAD BlueNeg on forward scatter area versus LIVE/DEAD Blue plots, and CD14Int/Pos VioletNeg based on CD14 versus CellTrace Violet plots. Note that some macrophage cultures had CellTrace Violet/CD14 dual positive cells, which represent macrophage targets which have phagocytosed a small amount of effector cells. As shown in Fig. S1B, this only represented ~4.7% of total live effector cells assuming macrophages were phagocytosing NK cells at a 1:1 ratio. The majority of the NK cells were initially excluded from the macrophages in the SSC-A versus FSC-A plots. These Violet+ macrophages were included in the macrophage target gate. Within this target cell gate, infected cells were defined as CD4-p24+ target cells. “%Residual Gag+ Targets” was calculated by dividing the %CD4-p24+ cells with effectors (E:T 0.1, 0.25, 0.5, 1, 2, or 4) by the %CD4-p24+ cells without effectors (E:T 0) and multiplying by 100.
Recognition assays
Uninfected and infected CD4+ T cell and macrophage target cells and CellTrace Violet-stained effector cells (autologous ex vivo NK cells, CD8+ T cells, or CAR T cells) were prepared as described above. In 96 well round bottom low-attachment plates(Corning, Cat#CLS7007), effectors were co-cultured with uninfected and infected target cells at the E:Ts indicated in the figure legends for 6 hours at 37°C in the presence of anti-CD107a-AF488 (Biolegend, Cat#328610, RRID:AB_1227504) and 1:1000 dilution of GolgiPlug and GolgiStop. An effector-only sample was included as a control. For all NK cell recognition assays, a target-to-effector (T:E) of 10 (or E:T of 0.1) was used to increase the chances of detecting responses to infected cells above background, as the frequencies of HIV-infected target cells were on average 14%. This resulted in ~1 infected target to ~1 effector cell. Responses to infected CD4+ T cells were barely detected above background at an E:T of 1. Higher E:T ratios (0.5 and 1) were used for the CAR T cell recognition assays as the frequency of mCherry+CD8+ in the effector population were on average 16%. This resulted in ~1 infected target to ~1 CAR T cell. For ADCC assays, the targets were first plated and incubated with normal human IgG, 3BNC117 or PGT121 at a final concentration of 30 μg/mL at room temperature for 15–20 minutes, followed by addition of NK cells and incubation at 37°C for 6 hours. All cells were harvested from the low attachment plate as described above for the elimination assay and Fc blocked using Human TruStain FcX (Biolegend, Cat#422302, RRID:AB_2818986) at 5ul of FcX per sample in 50ul of FACS Buffer (2% FBS, 1mM EDTA in PBS). Once the Fc block was completed, the cells were surface stained with LIVE/DEAD Fixable Blue (ThermoFisher, Cat#L34962), anti-CD56-BV650 (Biolegend, Cat#362532, RRID:AB_2562602) and anti-CD16-BUV395 (BD Bioscience, Cat#563785, RRID:AB_2744293) for NK cell cultures, anti-CD3-BV650 (Biolegend, Cat#317324, RRID:AB_2563352) and anti-CD8-BUV395 (BD Biosciences, Cat#563795, RRID:AB_2722501) for CD8+ T cell and CAR T cell cultures. Note, the staining mix was made up in 50ul of FACS Buffer and added directly to the cells in the 50ul of FcX-FACS Buffer. Following a 30 minute incubation at 4°C, the samples were washed, fixed and permeabilized using BD CytoFix/CytoPerm Fixation/Permeabilization Kit (BD Biosciences, Woburn, MA, Cat#554714). The cells were then intracellular stained using anti-TNF-α-BV711 (Biolegend, Cat#502940, RRID:AB_2563885) and anti-IFN-γ-BV510 (Biolegend, Cat#502544, RRID:AB_2563883). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software. All data were analyzed using FlowJo 10.6.0 software.
The gating strategies for the NK cell and CD8+ T cell recognition assays are shown in Fig. S2A. The gating strategies for the CAR T cell recognition assay are shown in Fig. S4D. Live effector cells were defined as LIVE/DEAD BlueNeg, CellTrace VioletPos, CD56Int/Pos (NK cells), or CD3+CD8+ (CD8+ T cells), or mCherry+CD8+ (CAR T cells). Conditions with effector cells only were used to draw the gating for CD107a, TNF-α, and IFN-γ responses. CD107a, TNF-α, IFN-γ frequencies were corrected for the background frequencies observed in uninfected conditions when indicated. In cases where the levels of productive CD4+ T cell and macrophage infection were different, the combined %CD107a+%TNF-α responses to the target cells were normalized based on differences in infection frequencies. Ratios of degranulation versus cytokine production were calculated by dividing the total CD107a response (%CD107a+TNF-α- + %CD107+TNF-a+) by the total cytokine response (%CD107a-TNF-α+ + %CD107+TNF-a+). The “% Total Response” was calculated as either single function %CD107a+TNF-α- (“CD107a”) or %CD107a-TNF-α+ (“TNF-α”) or “Dual” function %CD107+TNF-a+ divided by the total sum of these responses and multiplied by 100.
For overnight recognition assays, 50,000 HIV-infected CD4+ T cells or macrophages were co-cultured with autologous NK cells at an E:T of 1 or 2 for 18 hours, followed by measurement of IFN-γ in the culture supernatants using IFN-γ ELISA kits (Biolegend). An E:T of 1 or 2 was selected to make sure enough IFN-γ was produced by the NK cells in the culture supernatants for detection by ELISA. For CAR T cell overnight recognition assays, the number of remaining adherent macrophages (~50,000 on average) in each well of a 24 well low-attachment plate (Corning Inc, Cat#CLS3471) were determined and the CAR T cells were added at an E:T of 2 based on the number of macrophages. After an overnight co-culture, the IFN-γ in the culture supernatants was measured via ELISA (Biolegend, Cat#430101). IFN-γ values were interpolated from the standard curve and normalized based on differences in infection frequencies.
HIV Envelope Antibody Staining Assays
HIV-specific antibodies were obtained from the NIH AIDS Reagent Program: VRC01 (Cat#12033 from Dr. John Mascola), 3BNC117 (Cat#12474 from Dr. Michel Nussenzweig), N6 (Cat#12968 from Dr. Mark Conners), 10–1074 (Cat#12477 from Dr. Michel Nussenzweig), and PGT121 (Cat#12343 from IAVI). The HIV envelope CD4-bindings site-specific VHH, J3 (McCoy et al., 2012) was cloned, produced, purified and directly conjugated to AlexaFluor647 (AF647) using sortase chemistry as described below:
The tetrapeptide Gly3-Cys was synthesized by standard solid-phase peptide synthesis. Maleimide-AF647 (ThermoFisher, Cat#A20347) was dissolved in 20 mM NaHCO3 buffer (pH 8.3). The tetrapeptide Gly3Cys was added and left to stir at room temperature for 3 hours until LC-MS analysis indicated near-complete conversion to the product. The solution was then filtered and purified by reverse phase-HPLC with a semi-preparative column (C18 column, Gemini, 5 μm, 10×250 mm - Phenomenex, Torrance, CA, Cat#00G-4435-N0) at a flow rate of 4.0 mL/min; solvent A: 0.1% trifluoroacetic acid in H2O, solvent B: 0.1% trifluoroacetic acid in CH3CN. The J3 VHH was cloned into a pHEN6 vector and was engineered to have an LPETGG sortase tag followed by a 6XHIS tag for purification. The J3 VHH was expressed periplasmically in E. Coli and was purified via Ni-NTA column chromatography (Qiagen, Cat#30230). For the enzymatic reaction, we used the penta-mutant sortase A (Chen et al., 2011), which has an improved kcat. The reaction mixture (200 μL) contained Tris·HCl (50 mM, pH 7.5), CaCl2 (10 mM), NaCl (150 mM), triglycine-AF647 probe (750 μM), J3-VHH (200 μM), and sortase (5 μM). The reaction was left to proceed at 4°C for 2 hours, and the products were analyzed by LC-MS. Yields were generally >80%. The excess of the Gly3-AF647 substrate was purified with a PD-10 size-exclusion column (Cytiva, Cat#17085101) followed by Ni-NTA beads to remove sortase and any remaining unreacted 6XHIS-tagged J3 VHH. The AF647-labeled J3 VHH was stored at −80°C with 5% glycerol.
HIV antibodies were used to detect HIV envelope on the surface of CD4+ T cells and macrophages infected with HIV89.6 or HIVADA. For CD4+ T cells, 2–3 days post-infection, the cells were washed in R10 media, counted, ~100,000–150,000/sample transferred to a 96 well V-bottom plate followed by incubation with HIV antibodies at 200 nM for 30 minutes at 4°C (on ice, to prevent internalization). The cells were then washed and stained with anti-CD3-AF700 (Biolegend, Cat#300324, RRID:AB_493739), anti-human IgG Fc-AF647 (Biolegend, Cat#409320, RRID:AB_2563330), anti-CD4-BV510 (Biolegend, Cat#317444, RRID:AB_2561866), and LIVE/DEAD Fixable Blue (ThermoFisher, Cat#L34962) for 30 minutes at 4°C. The cells were then washed, fixed and permeabilized using CytoFix/CytoPerm and intracellular stained with anti-Gag p24-RD1. For macrophages, 2–3 days post-infection, the cells were washed in PBS and lifted from the low attachment plates using Cell Dissociation Buffer (ThermoFisher, Cat#13151014) for 10 minutes at 37°C, counted, and then ~100,000–150,00/sample transferred over to 96 well V-bottom plates. Before addition of antibodies, macrophage samples were Fc blocked with Human TruStain FcX (Biolegend, Cat#422302, RRID:AB_2818986) at 5ul of FcX per sample in 50ul of FACS Buffer (2% FBS, 1mM EDTA in PBS). Once the Fc blocking was completed, the cells were incubated with HIV antibodies, which were directly conjugated to AF647 using the Zenon Alexa Fluor 647 Human IgG Labeling Kit (ThermoFisher, Cat# Z25408, RRID:AB_2736598), at 200nM for 30 minutes at 4°C. Note, the staining mix was made up in 50ul of FACS Buffer and added directly to the macrophages in the 50ul of FcX-FACS Buffer. Directly conjugated HIV-specific antibodies were required as secondary detection using anti-human IgG Fc-AF647 would have reacted to the human Fc block. The cells were then washed and stained with anti-CD14-AF700 (Biolegend, Cat#301822, RRID:AB_493747), anti-CD4-BV510, and LIVE/DEAD Fixable Blue for 30 minutes at 4°C. The cells were then washed, fixed and permeabilized using CytoFix/CytoPerm and intracellular stained with anti-Gag p24-RD1 (Beckman Coulter, Brea, CA, Cat#6604667, RRID:AB_1575989). Flow cytometric data were acquired using a FACSCanto instrument with FACSDiva software. All data were analyzed using FlowJo 10.6.0 software. The “Fold Change in HIV-specific Antibody MFI” was calculated as the HIV-specific antibody mean fluorescence intensity (MFI) of infected cells (CD4-Gagp24+) divided by the HIV-specific MFI of uninfected cells (CD4+Gagp24-).
Imaging Flow Cytometry Analysis
For the analysis of HIV envelope internalization kinetics, uninfected or HIV89.6-infected macrophages (2–3 days post-infection), were harvested with Cell Dissociation Buffer (ThermoFisher, Cat#13151014), incubated for 10 minutes at 37°C, counted, and then ~100,000–150,00/sample transferred over to 4× 96 well V-bottom plates (one plate for each time point – 0, 10, 30 and 60 minutes). Before addition of antibodies, macrophage samples were Fc blocked with Human TruStain FcX (Biolegend, Cat#422302, RRID:AB_2818986) at 5ul of FcX per sample in 50ul of FACS Buffer (2% FBS, 1mM EDTA in PBS). Once the Fc blocking was completed, the cells were then incubated with either J3-AF647 or 10–1074 (directly conjugated to AF647 using the Zenon Alexa Fluor 647 Human IgG Labeling Kit), at 1000nM or 200nM, respectively, which were made up in 50ul of FACS Buffer and added directly to the cells in the FcX/FACS Buffer. After 30 minutes on ice, the cells were washed, resuspended in 50ul FACS buffer with FcX, and then either put on ice immediately (0-minute timepoint) or incubated at 37°C for 10, 30 or 60 minutes, followed by transfer to ice. For each of the timepoints, after 10 minutes on ice, anti-CD33-BV421 (Biolegend, Cat#366622, RRID:AB_2716148) and LIVE/DEAD Fixable Near-IR (ThermoFisher, Cat#L34976) were added to the cells followed by an incubation on ice for 20 minutes. The cells were washed once, fixed, and permeabilized using CytoFix/CytoPerm. The cells were intracellular stained using anti-Gag p24-FITC (Beckman Coulter, Cat#6604665, RRID:AB_1575987). 5,000 events were collected from each sample on the Image-Stream X Mark II imaging flow cytometer (Amnis, EMB Millipore) and analyzed using IDEAS 6.2 software (Amnis, EMD Millipore). Single-stained macrophages were used to calculate the compensation matrix. The Internalization Wizard was used to calculate the internalization scores of Gag p24 and HIV envelope antibody staining using CD33 staining as the surface probe.
Immunofluorescence and Microscopy Analysis
For microscopy analysis of HIV envelope-containing compartments, macrophages were matured on sterile glass coverslips in 6-well plates. Following 2–3 days of infection with HIV89.6 or HIVADA, coverslips with macrophages were transferred to a new 6-well plate, rinsed and washed three times (5 minutes each) in R10 media, and then incubated in 1mL of R10 with 200nM J3-AF647 for one hour at 37°C. Macrophages were rinsed and washed three times in R10 media, rinsed three times in PBS, and then fixed in 4% methanol-free formaldehyde in PBS (Thermo Scientific, Cat#28906) for 15 minutes at room temperature. Macrophages were then rinsed and washed three times in PBS, followed by permeabilization in 0.2% Triton X-100 in PBS (Sigma-Aldrich, Cat#234729) for five minutes at room temperature. Macrophages were then rinsed and washed three times with PBS, and then incubated for 30 minutes at room temperature with 5 drops of Image - IT FX Signal Enhancer (ThermoFisher, Cat# R37107). Macrophages were rinsed and washed three times in PBS, followed by blocking in 10% bovine serum albumin (BSA; Sigma-Aldrich, Cat#A9418) for one hour at room temperature. Cells were rinsed and washed three times for one minute in PBS. Macrophages were incubated with either anti-EEA1 (Cell Signaling Technologies, Cat#3288S, RRID:AB_2096811), anti-LAMP1 (Cell Signaling Technologies, Cat#9091S, RRID:AB_2687579), or anti-Siglec-1 (Sigma-Aldrich, Cat#MABT328) at 1:100, 1:200 and 1:100 dilutions, respectively, in 3% BSA for one hour at room temperature. Following a rinse and three washes in PBS, macrophages were incubated for one hour with anti-rabbit IgG-AF488 (Jackson ImmunoResearch, West Grove, PA, Cat#711-545-152, RRID:AB_2313584) and anti-Gag p24-RD-1 (Beckman Coulter) at 1:250 and 1:50 dilutions, respectively, in 3% BSA at room temperature for one hour. For experiments using anti-Siglec-1, the antibody was directly conjugated to AF488 using the Zenon Alexa Fluor 488 Mouse IgG1 Labeling Kit (ThermoFisher, Cat# Z25002, RRID:AB_2736914), and the cells were incubated with both the anti-Siglec-1 and anti-Gag p24-RD-1 antibody together for one hour. Macrophages were rinsed and washed three times in PBS, followed by staining with Hoescht (ThermoFisher, Cat#H3570) at a 1:200 dilution in PBS for five minutes. Finally, macrophages were rinsed once and then mounted onto microscope slides using ProLong Antifade Gold (ThermoFisher, Cat#P36934). The edges were sealed with clear nail polish and the allowed to dry overnight. Images were acquired on a Zeiss LSM 510 Confocal Microscope using Zen 2009 software (Carl Zeiss AG, Oberkochen, Germany). Image preparation for figures and co-localization analysis was performed using Zen 2009 software, ImageJ software and the EzColocalization plugin (Stauffer et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism version 8.0 was used to determine statistical significance between experimental conditions using the paired, or unpaired student’s t-test as indicated in each figure legend. Significance intervals for each experiment are also described in each figure legend. The figure legends also indicate the number of independent experiments and “n”, the number of biological replicates (individual donor samples).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF Purified Anti-human CD3 Antibody | Biolegend | Cat#317326, RRID:AB_11150592 |
| Ultra-LEAF Purified Anti-human CD28 Antibody | Biolegend | Cat#302934, RRID:AB_11148949 |
| APC/Cyanine 7 anti-human CD14 antibody | Biolegend | Cat#301820, RRID:AB_493695 |
| APC/Cyanine 7 anti-human CD3 antibody | Biolegend | Cat#300318, RRID:AB_314054 |
| APC anti-human CD4 antibody | Biolegend | Cat#317416, RRID:AB_571945 |
| HIV-1 core antigen-RD-1 antibody | Beckman Coulter | Cat#6604667, RRID:AB_1575989 |
| Brilliant Violet™ 421 anti-human CD107a (LAMP-1) antibody | Biolegend | Cat#328626, RRID:AB_11203537 |
| PerCP/Cyanine5.5 anti-human CD3 antibody | Biolegend | Cat#300328, RRID:AB_1575008 |
| APC anti-human CD8 antibody | Biolegend | Cat#344722, RRID:AB_2075388 |
| Alexa Fluor® 700 anti-human IFN-gamma antibody | Biolegend | Cat#502520, RRID:AB_528921 |
| Alexa Fluor® 488 anti-human CD107a (LAMP-1) antibody | Biolegend | Cat#328610, RRID:AB_1227504 |
| Brilliant Violet™ 650 anti-human CD56 (NCAM) antibody | Biolegend | Cat#362532, RRID:AB_2562602 |
| BD Horizon BUV395 Mouse Anti-Human CD16 | BD Biosciences | Cat#563785, RRID:AB_2744293 |
| Brilliant Violet™ 650 anti-human CD3 antibody | Biolegend | Cat#317324, RRID:AB_2563352 |
| BUV395 Mouse Anti-Human CD8 antibody | BD Biosciences | Cat#563795, RRID:AB_2722501 |
| Brilliant Violet 711™ anti-human TNF-alpha antibody | Biolegend | Cat#502940, RRID:AB_2563885 |
| Brilliant Violet 510™ anti-human IFN-gamma antibody | Biolegend | Cat#502544, RRID:AB_2563883 |
| Normal Human IgG Control Antibody | R&D Systems | Cat#1-001-A, RRID:AB_907192 |
| Anti-HIV- gp120 Monoclonal (VRC01) | NIH AIDS Reagent Program | Cat#12033 |
| Anti-HIV- gp120 Monoclonal (3BNC117) | NIH AIDS Reagent Program | Cat#12474 |
| Anti-HIV- gp120 Monoclonal (N6) | NIH AIDS Reagent Program | Cat#12968 |
| Anti-HIV- gp120 Monoclonal (10-1074) | NIH AIDS Reagent Program | Cat#12477 |
| Anti-HIV- gp120 Monoclonal (PGT121) | NIH AIDS Reagent Program | Cat#12343 |
| Anti-HIV Envelope Monoclonal 3BNC117 (for ADCC assays) | This paper | NA |
| Anti-HIV Envelope Monoclonal PGT121 (for ADCC assays) | This paper | NA |
| Alexa Fluor® 700 anti-human CD3 antibody | Biolegend | Cat#300324, RRID:AB_493739 |
| Alexa Fluor® 700 anti-human CD14 antibody | Biolegend | Cat#301822, RRID:AB_493747 |
| Brilliant Violet 510™ anti-human CD4 antibody | Biolegend | Cat#317444, RRID:AB_2561866 |
| Alexa Fluor® 647 anti-human IgG Fc antibody | Biolegend | Cat#409320, RRID:AB_2563330 |
| Brilliant Violet 421™ anti-human CD33 antibody | Biolegend | Cat#366622, RRID:AB_2716148 |
| HIV-1 core antigen-FITC antibody | Beckman Coulter | Cat#6604665, RRID:AB_1575987 |
| EEA1 (C45B10) Rabbit mAb antibody | Cell Signaling Technologies | Cat#3288S, RRID:AB_2096811 |
| LAMP1 (D2D11) XP Rabbit Antibody | Cell Signaling Technologies | Cat#9091S, RRID:AB_2687579 |
| Anti-Siglec-1 (CD169) Antibody, clone 5F1.1 | Sigma-Aldrich | Cat#MABT328 |
| Alexa Fluor 488-AffiniPure Donkey Anti-Rabbit IgG 9H+L) antibody | Jackson ImmunoResearch | Cat#711-545-152, RRID:AB_2313584 |
| Bacterial and Virus Strains | ||
| HIV 89.6 Proviral DNA | NIH AIDS Reagent Program | Cat#3552 |
| HIV ADA Virus | NIH AIDS Reagent Program | Cat#416 |
| Biological Samples | ||
| Human Blood Products (Buffy Coats) | Massachusetts General Hospital Blood Bank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human GM-CSF | Biolegend | Cat#572904 |
| Recombinant Human M-CSF | Biolegend | Cat#574806 |
| Recombinant IL-2 | R&D Systems | Cat#202-IL-500 |
| PHA-M | Sigma-Aldrich | Cat#L8902-5MG |
| 25kDa polyethylenimine | Polysciences | Cat#23966-1 |
| Recombinant IL-15 | Biolegend | Cat#570304 |
| Recombinant Human CD19-Fc Chimera Protein, CF | R&D Systems | Cat#9269-CD-050 |
| Recombinant HIV-JRCSF gp140 Trimer-Foldon | This paper | NA |
| ELISA Coating Buffer | Biolegend | Cat#421701 |
| Pierce 16% Formaldehyde, Methanol-Free | ThermoFisher | Cat#28906 |
| Triton X-100 | Sigma-Aldrich | Cat#234729 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A9418 |
| Hoescht 3342 trihydrochloride | ThermoFisher | Cat#H3570 |
| Critical Commercial Assays | ||
| EasySep Human CD14 Positive Selection Kit II | Stemcell | Cat#17858 |
| EasySep Human CD4+ T Cell Isolation Kit | Stemcell | Cat#17952 |
| PEG-It Virus Precipitation Solution | Systems Biosciences | Cat#LV825A-1 |
| Human TruStain FcX | Biolegend | Cat#422302, RRID:AB_2818986 |
| LIVE/DEAD Fixable Blue Dead Cell Stain Kit | ThermoFisher | Cat#L34962 |
| BD CytoFix/CytoPerm | BD Biosciences | Cat#554714 |
| EasySep Human NK Cell Isolation Kit | Stemcell | Cat#17955 |
| EasySep Human CD8+ T cell Isolation Kit | Stemcell | Cat#17953 |
| CellTrace Violet Proliferation Kit | ThermoFisher | Cat#C34557 |
| Lipofectamine 3000 | ThermoFisher | Cat#L3000015 |
| EasySep Human T Cell Isolation Kit | Stemcell | Cat#17951 |
| T Cell Activation/Expansion Kit | Miltenyi Biotech | Cat#130-091-441 |
| CTS™ OpTmizer™ T Cell Expansion SFM | ThermoFisher | Cat#A1048501 |
| GolgiPlug | BD Biosciences | Cat#555029 |
| GolgiStop | BD Biosciences | Cat#554724 |
| ELISA Max Standard Set Human IFN-g | Biolegend | Cat#430101 |
| Zenon Alexa Fluor 647 Human IgG Labeling Kit | ThermoFisher | Cat#Z25408, RRID:AB_2736598 |
| LIVE/DEAD Fixable Near-IR | ThermoFisher | Cat#L34976 |
| Image-iT FX Signal Enhancer ReadyProbes Reagent | ThermoFisher | Cat#R37107 |
| Zenon Alexa Fluor 488 Mouse IgG1 Labeling Kit | ThermoFisher | Cat#Z25002, RRID:AB_2736914 |
| ProLong Gold Antifade Mountant | ThermoFisher | Cat#P36934 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HEK293T/17 Cells | ATCC | Cat#CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| CD19 and HIV CAR T Cell Constructs | This paper | NA |
| Software and Algorithms | ||
| FlowJo 10.6.0 | FlowJo LLC | https://www.flowjo.com |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Zen 2009 | ZEISS | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| IDEAS 6.2 | Amnis, EMD Millipore, Luminex Corp | https://www.luminexcorp.com/imaging-flow-cytometry/ |
| Other | ||
| Certified FBS | ThermoFisher | Cat#16000044 |
| 24-well low-attachment plates | Sigma-Aldrich | Cat#CLS3473 |
| 6-well low-attachment plates | Sigma-Aldrich | Cat#CLS3471 |
| 24-well non-treated TC plates | Sigma-Aldrich | Cat#CLS3738 |
| 0.45um syringe filters | Millipore | Cat#SLHV033RS |
| Cell Dissociation Buffer | ThermoFisher | Cat#13151014 |
| Nunc MaxiSorp flat-bottom | Sigma-Aldrich | Cat#M9410-1CS |
| 96 well round bottom low attachment plates | Sigma-Aldrich | Cat#CLS7007 |
| DMEM with high glucose and pyruvate | ThermoFisher | Cat#1995065 |
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and Virus Strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; and Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
Highlights.
HIV infected macrophages resist efficient innate-based NK cell-mediated killing
The HIV envelope is equally accessible on infected CD4+ T cells and macrophages.
HIV antibody-enhanced NK cell detection/killing of infected macrophages is muted
ACKNOWLEDGEMENTS
We thank Drs. Judy Lieberman, Alejandro Balazs, David Collins, Brad Jones, Julie Boucau, Wilfredo Garcia Beltran, Mary Carrington, Keith Reeves, and Gaurav Gaiha for their helpful discussions and comments. We thank Alicja Piechocka-Trocha for experimental help. We thank the Flow Cytometry and Sample Processing Cores at the Ragon Institute for their help with instrumentation and processing of the samples. We thank Dr. Thomas Diefenbach for help with microscopy and imaging flow cytometry analysis, and Dr. Musie Ghebremichael for helpful comments with statistical analysis. We thank the Lingwood Lab and Julia Bals for their assistance in producing and purifying the recombinant HIV envelope protein. We thank the Balazs Lab and Dr. Jackie Brady for the creation of HIV antibody-expressing cell lines. We thank Alexander B. Austin of the MIT Koch peptide facility for assistance with peptide synthesis. We thank Katherine A. Whang for technical assistance in the synthesis of AF647-VHH-J3. Funding support was provided by the Howard Hughes Medical Institute (K.L.C., G.M., and B.D.W.), the National Institutes of Health (AI149704 and AI118544 to B.D.W.; F32AI143480 to K.L.C.), and the Ragon Institute of MGH, MIT and Harvard (K.L.C. and B.D.W).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests
SUPPLEMENTAL INFORMATION
Please see the separate document for Supplemental Figures and Supplemental Figure Legends
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alsahafi N, Richard J, Prevost J, Coutu M, Brassard N, Parsons MS, Kaufmann DE, Brockman M, and Finzi A. (2017). Impaired Downregulation of NKG2D Ligands by Nef Proteins from Elite Controllers Sensitizes HIV-1-Infected Cells to Antibody-Dependent Cellular Cytotoxicity. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand SP, Grover JR, Tolbert WD, Prevost J, Richard J, Ding S, Baril S, Medjahed H, Evans DT, Pazgier M, et al. (2019). Antibody-Induced Internalization of HIV-1 Env Proteins Limits Surface Expression of the Closed Conformation of Env. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade VM, Mavian C, Babic D, Cordeiro T, Sharkey M, Barrios L, Brander C, Martinez-Picado J, Dalmau J, Llano A, et al. (2020). A minor population of macrophage-tropic HIV-1 variants is identified in recrudescing viremia following analytic treatment interruption. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, et al. (2016). HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe 19, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard NF, Kiani Z, Tremblay-McLean A, Kant SA, Leeks CE, and Dupuy FP (2017). Natural Killer (NK) Cell Education Differentially Influences HIV Antibody-Dependent NK Cell Activation and Antibody-Dependent Cellular Cytotoxicity. Front Immunol 8, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaparte MI, and Barker E. (2003). Inability of natural killer cells to destroy autologous HIV-infected T lymphocytes. AIDS 17, 487–494. [DOI] [PubMed] [Google Scholar]
- Bonaparte MI, and Barker E. (2004). Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood 104, 2087–2094. [DOI] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, and Long EO (2006a). Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev 214, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, and Long EO (2006b). Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, To RK, and Spector SA. (2019). TREM-1 Protects HIV-1-Infected Macrophages from Apoptosis through Maintenance of Mitochondrial Function. mBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassol E, Cassetta L, Alfano M, and Poli G. (2010). Macrophage polarization and HIV-1 infection. J Leukoc Biol 87, 599–608. [DOI] [PubMed] [Google Scholar]
- Castellano P, Prevedel L, and Eugenin EA (2017). HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep 7, 12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, and Santoni A. (2007). Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood 110, 606–615. [DOI] [PubMed] [Google Scholar]
- Chen I, Dorr BM, and Liu DR (2011). A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci U S A 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H, Wang JJ, Qi M, Yoon JJ, Wen X, Chen X, Ding L, and Spearman P. (2012). The intracellular virus-containing compartments in primary human macrophages are largely inaccessible to antibodies and small molecules. PLoS One 7, e35297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton KL, Collins DR, Lengieza J, Ghebremichael M, Dotiwala F, Lieberman J, and Walker BD (2018). Resistance of HIV-infected macrophages to CD8(+) T lymphocyte-mediated killing drives activation of the immune system. Nat Immunol 19, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DR, Lubow J, Lukic Z, Mashiba M, and Collins KL (2015). Vpr Promotes Macrophage-Dependent HIV-1 Infection of CD4+ T Lymphocytes. PLoS Pathog 11, e1005054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs SK, Lennox J, Caliendo AM, Brown LA, and Guidot DM (2015). Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses 31, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleage C, Moreau M, Rioux-Leclercq N, Ruffault A, Jegou B, and Dejucq-Rainsford N. (2011). Human immunodeficiency virus infects human seminal vesicles in vitro and in vivo. Am J Pathol 179, 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, and Marsh M. (2007). In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol 177, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CJ, Williams JP, Schiffner T, Gartner K, Ochsenbauer C, Kappes J, Russell RA, Frater J, and Sattentau QJ (2014). High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol 88, 2025–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MA, Carruth LM, Rowell JF, Yu X, and Siliciano RF (1996). Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J Virol 70, 6547–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissmann P, Evans JH, Mehrabi M, Rose EL, Nedvetzki S, and Davis DM (2010). Multiple mechanisms downstream of TLR-4 stimulation allow expression of NKG2D ligands to facilitate macrophage/NK cell crosstalk. J Immunol 184, 6901–6909. [DOI] [PubMed] [Google Scholar]
- Fogli M, Mavilio D, Brunetta E, Varchetta S, Ata K, Roby G, Kovacs C, Follmann D, Pende D, Ward J, et al. (2008). Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals. PLoS Pathog 4, e1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganor Y, Real F, Sennepin A, Dutertre CA, Prevedel L, Xu L, Tudor D, Charmeteau B, Couedel-Courteille A, Marion S, et al. (2019). HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol 4, 633–644. [DOI] [PubMed] [Google Scholar]
- Gaudin R, Berre S, Cunha de Alencar B, Decalf J, Schindler M, Gobert FX, Jouve M, and Benaroch P. (2013). Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PLoS One 8, e69450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavegnano C, Detorio MA, Bassit L, Hurwitz SJ, North TW, and Schinazi RF (2013). Cellular pharmacology and potency of HIV-1 nucleoside analogs in primary human macrophages. Antimicrob Agents Chemother 57, 1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavegnano C, and Schinazi RF (2009). Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir Chem Chemother 20, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, Ogasawara K, and Lanier LL (2004). Cutting edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol 172, 2001–2005. [DOI] [PubMed] [Google Scholar]
- Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, Melikyan GB, and Spearman P. (2017). Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog 13, e1006181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen EC, Ransom M, Hesselberth JR, Hosmane NN, Capoferri AA, Bruner KM, Pollack RA, Zhang H, Drummond MB, Siliciano JM, et al. (2016). Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, Tabarias H, Degli-Esposti MA, Dewson G, Willis SN, et al. (2007). Interleukin 15-smediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol 8, 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, Voskoboinik I, and Trapani JA (2015). Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med 212, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, and Sattentau QJ (2007). Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J Virol 81, 7873–7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost S, and Altfeld M. (2012). Evasion from NK cell-mediated immune responses by HIV-1. Microbes Infect 14, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouve M, Sol-Foulon N, Watson S, Schwartz O, and Benaroch P. (2007). HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2, 85–95. [DOI] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, and Bieniasz PD (2006). Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol 4, e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, and Kim WK (2019). Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J Neuroimmune Pharmacol 14, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppensteiner H, Banning C, Schneider C, Hohenberg H, and Schindler M. (2012). Macrophage internal HIV-1 is protected from neutralizing antibodies. J Virol 86, 2826–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs JM, Noeldeke E, Ha HJ, Peng H, Rits-Volloch S, Harrison SC, and Chen B. (2014). Stable, uncleaved HIV-1 envelope glycoprotein gp140 forms a tightly folded trimer with a native-like structure. Proc Natl Acad Sci U S A 111, 18542–18547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, and Kent SJ (2018). Anti-HIV-1 antibody-dependent cellular cytotoxicity: is there more to antibodies than neutralization? Curr Opin HIV AIDS 13, 160–166. [DOI] [PubMed] [Google Scholar]
- McCoy LE, Quigley AF, Strokappe NM, Bulmer-Thomas B, Seaman MS, Mortier D, Rutten L, Chander N, Edwards CJ, Ketteler R, et al. (2012). Potent and broad neutralization of HIV-1 by a llama antibody elicited by immunization. J Exp Med 209, 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki MT, Saidi H, Dufour A, Olivo-Marin JC, and Gougeon ML (2010). Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk--a pivotal role of HMGB1. PLoS Pathog 6, e1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedvetzki S, Sowinski S, Eagle RA, Harris J, Vely F, Pende D, Trowsdale J, Vivier E, Gordon S, and Davis DM (2007). Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 109, 3776–3785. [DOI] [PubMed] [Google Scholar]
- Norman JM, Mashiba M, McNamara LA, Onafuwa-Nuga A, Chiari-Fort E, Shen W, and Collins KL (2011). The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nat Immunol 12, 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MS, Richard J, Lee WS, Vanderven H, Grant MD, Finzi A, and Kent SJ. (2016). NKG2D Acts as a Co-Receptor for Natural Killer Cell-Mediated Anti-HIV-1 Antibody-Dependent Cellular Cytotoxicity. AIDS Res Hum Retroviruses 32, 1089–1096. [DOI] [PubMed] [Google Scholar]
- Perno CF, Newcomb FM, Davis DA, Aquaro S, Humphrey RW, Calio R, and Yarchoan R. (1998). Relative potency of protease inhibitors in monocytes/macrophages acutely and chronically infected with human immunodeficiency virus. J Infect Dis 178, 413–422. [DOI] [PubMed] [Google Scholar]
- Poli A, Michel T, Theresine M, Andres E, Hentges F, and Zimmer J. (2009). CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandstrom N, Fasbender F, Claus M, Eils R, Beaudouin J, et al. (2019). NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med 216, 2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillay H, El Costa H, Duriez M, Marlin R, Cannou C, Madec Y, de Truchis C, hmati M, Barre-Sinoussi F, Nugeyre MT, and Menu E. (2016). NK cells control HIV-1 infection of macrophages through soluble factors and cellular contacts in the human decidua. Retrovirology 13, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainho JN, Martins MA, Cunyat F, Watkins IT, Watkins DI, and Stevenson M. (2015). Nef Is Dispensable for Resistance of Simian Immunodeficiency Virus-Infected Macrophages to CD8+ T Cell Killing. J Virol 89, 10625–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso R, Wieser M, Ojeda D, Bonisch M, Kuhnel H, Bolcic F, Quendler H, Grillari J, Grillari-Voglauer R, and Quarleri J. (2012). HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J Virol 86, 10327–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, and Cohen EA (2010). HIV-1 Vpu disarms natural killer cells. Cell Host Microbe 8, 389–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Sindhu S, Pham TN, Belzile JP, and Cohen EA (2010). HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115, 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O, Marechal V, Le Gall S, Lemonnier F, and Heard JM (1996). Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 2, 338–342. [DOI] [PubMed] [Google Scholar]
- Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, and Barker E. (2010). Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer W, Sheng H, and Lim HN (2018). EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep 8, 15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingler S, Mann AM, Zhou J, Swingler C, and Stevenson M. (2007). Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog 3, 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomescu C, Mavilio D, and Montaner LJ (2015). Lysis of HIV-1-infected autologous CD4+ primary T cells by interferon-alpha-activated NK cells requires NKp46 and NKG2D. AIDS 29, 1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay-McLean A, Bruneau J, Lebouche B, Lisovsky I, Song R, and Bernard NF. (2017). Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4(+) T Cells. Viruses 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso FY, Kang G, Kwon EH, Julius P, Li Q, West JT, and Wood C. (2018). Brain is a potential sanctuary for subtype C HIV-1 irrespective of ART treatment outcome. PLoS One 13, e0201325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stigt Thans T, Akko JI, Niehrs A, Garcia-Beltran WF, Richert L, Sturzel CM, Ford CT, Li H, Ochsenbauer C, Kappes JC, et al. (2019). Primary HIV-1 Strains Use Nef To Downmodulate HLA-E Surface Expression. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojnov L, Martins MA, Bean AT, Veloso de Santana MG, Sacha JB, Wilson NA, Bonaldo MC, Galler R, Stevenson M, and Watkins DI (2012). The majority of freshly sorted simian immunodeficiency virus (SIV)-specific CD8(+) T cells cannot suppress viral replication in SIV-infected macrophages. J Virol 86, 4682–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, and Evans DT (2015). Envelope Glycoprotein Internalization Protects Human and Simian Immunodeficiency Virus-Infected Cells from Antibody-Dependent Cell-Mediated Cytotoxicity. J Virol 89, 10648–10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J, Bonaparte M, Sacks J, Guterman J, Fogli M, Mavilio D, and Barker E. (2007). HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 110, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JP, Bonaparte MI, and Barker E. (2004). HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. AIDS 18, 1769–1779. [DOI] [PubMed] [Google Scholar]
- Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, and Krausslich HG. (2007). HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog 3, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Fan X, Staitieh B, Bedi C, Spearman P, Guidot DM, and Sadikot RT (2017). HIV-related proteins prolong macrophage survival through induction of Triggering receptor expressed on myeloid cells-1. Sci Rep 7, 42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalar A, Figueroa MI, Ruibal-Ares B, Bare P, Cahn P, de Bracco MM, and Belmonte L. (2010). Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antiviral Res 87, 269–271. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Huang B, and Shi J. (2016). Fas ligand and lytic granule differentially control cytotoxic dynamics of natural killer cell against cancer target. Oncotarget 7, 47163–47172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author on request