

Abstract
Alcoholic fatty liver disease (AFLD) is one of the major causes of liver morbidity and mortality worldwide. We have previously shown that whole-body, but not hepatocyte-specific, deficiency of farnesoid X receptor (FXR) in mice worsens AFLD, suggesting that extra-hepatic FXR deficiency is critical for AFLD development. Intestinal FXR is critical in suppressing hepatic bile acid (BA) synthesis by inducing fibroblast growth factor 15 (FGF15) in mice and FGF19 in humans. We hypothesized that intestinal FXR is critical for reducing AFLD development in mice. To test this hypothesis, we compared the AFLD severity in wild type (WT) and intestine-specific Fxr knockout (FXRInt−/−) mice following treatment with control or ethanol-containing diet. We found that FXRInt−/− mice were more susceptible to ethanol-induced liver steatosis and inflammation, compared to WT mice. Ethanol treatment altered the expression of hepatic genes involved in lipid and bile acid homeostasis, and ethanol detoxification. Gut FXR deficiency increased intestinal permeability, likely due to reduced mucosal integrity, as revealed by decreased secretion of Mucin 2 protein and lower levels of E-cadherin protein. In summary, intestinal FXR may protect AFLD development by maintaining gut integrity.
Keywords: alcoholic fatty liver disease, bile acid, intestine, FXR, intestinal permeability
Introduction
Chronic alcohol drinking, especially in combination with binge drinking, leads to alcoholic fatty liver disease (AFLD), which is one of the major causes of chronic liver diseases worldwide1. The mechanisms underlying alcohol-induced liver damages are multifactorial: alcohol consumption alters metabolic pathways in the liver, impairs energy metabolism, and increases oxidative stress and inflammatory response, all of which could contribute to the development of AFLD1. Furthermore, alcohol consumption impairs gut integrity, permeability, and promotes bacterial translocation. Increased systemic absorption of endotoxin (lipopolysaccharide-LPS) from bacterial overgrowth is believed to be a major mechanism leading to liver injury2.
Despite significant advances in understanding the pathogenesis and clinical characteristics of AFLD, there are no FDA-approved treatments for this disease. As a transcription factor belonging to the nuclear receptor superfamily, farnesoid X receptor (FXR) appears to be a promising therapeutic target for treating fatty liver diseases, including AFLD. Several drugs targeting FXR are being investigated in clinical trials for liver diseases, including cholangitis and nonalcoholic steatohepatitis (NASH)3. In animal models, activation of FXR has been shown to be protective against the development of high-fat diet-induced non-alcoholic fatty liver disease (NAFLD), liver inflammation, acetaminophen-induced liver toxicity4–6, and recently, AFLD7, 8. Increased levels of bile acids (BAs), the endogenous ligands of FXR, may promote liver regeneration, liver repair, and improve insulin sensitivity9, 10. However, during cholestasis, elevated BAs can be cytotoxic, inducing apoptosis and necrosis9, 10.
FXR is highly expressed in the liver and intestines, and is a central regulator of BA homeostasis11. Activation of FXR in the small intestine induces fibroblast growth factor 15 (FGF15) in mice and FGF19 in humans12–14. The BA-FXR-FGF15/FGF19 axis has emerged to be an important pathway in mediating gut-liver crosstalk and maintaining normal liver and intestinal functions13–15. Studies have revealed that alcohol disrupts enterohepatic circulation of BAs in both mice and humans, and FXR deficiency in mice exacerbates AFLD, suggesting an important protective role of FXR and/or maintaining BA homeostasis in AFLD16–19. Our recent study showed that hepatic FXR deficiency didn’t affect the severity of AFLD, suggesting that deficiency in intestinal FXR and/or non-hepatic FXR-regulated pathways may be critical in the development of AFLD18, 20. Additionally, FXR signaling in the intestine has been shown to protect the integrity and barrier functions of the intestines, and FGF15 has been shown to regulate a variety of liver functions, including suppressing BA synthesis and promoting liver regeneration15, 21–25. Therefore, in the current study, we determined the effects of intestinal FXR deficiency on the development of AFLD in mice.
MATERIALS AND METHODS
Animals and treatments
The generation of intestine-specific Fxr knockout (KO) mice (FXRInt−/−) has been described previously14, 23. Briefly, the FXRInt−/− mice were generated via cross-breeding FXR floxed/floxed mice with villin Cre (+) mice. FXR floxed/floxed Cre (−) mice were used as control wild-type (WT) mice. All animal breeding, experiments and procedures were conducted at the Rutgers University with the approval of the Institutional Animal Care and Use Committee (IACUC).
Ethanol treatment was adopted from the widely used NIAAA model26. In detail, male, 10- to 12-week old WT and FXRInt−/− mice were fed a Lieber-DeCarli ethanol-containing diet (5% (v/v) ethanol) or isocaloric control liquid diet (vehicle, isocaloric maltose-dextrin) for 10 days before administered a single bolus dose of ethanol (5g/kg body weight (BW)) or maltose solution (9g maltose/kg of body weight) via oral gavage (n = 5–10/genotype/diet). After the treatment, mice were euthanized with liver, intestines and blood collected for analysis.
To evaluate the intestinal permeability in vivo, WT and FXRInt−/− mice (n = 5) were fed the aforementioned diets for 10 days. On day 11, these mice were fasted for 4 hours before oral gavage with 100 mg/ml FITC-dextran (FD4, dissolved in PBS, Sigma) at 0.44 mg/g body weight. Five hours later, serum was collected, and the levels of fluorescence in serum were measured using a microplate reader with excitation and emission filters set at 490 and 530 nm, respectively27.
Serum biochemical assays
Serum activities of alanine aminotransferase (ALT), aspartate transaminase (AST) and alkaline phosphatase (ALP), as well as serum levels of triglyceride (TG), total cholesterol (TC), and total BAs were measured using commercially available kits (Pointe Scientific, Canton, MI; BAs-Diazyme Laboratories, Poway, CA).
Hepatic lipid analysis
Frozen liver (100mg) was homogenized in 1 ml buffer containing 18 mM Tris⋅HCl (pH 7.5), 300 mM d-mannitol, 5 mM EGTA, and 0.1 mM PMSF. Afterwards, 400 μl homogenate was added to glass tube containing 2 ml 2:1 chloroform:methanol solution and shaked overnight at room temperature. One ml ddH2O was then added to the tube, vortexed, and centrifuged for 5 mins at 3,000 × g to separate different phases. Two ml organic phase was transferred to a new glass tube and dried within a Speed-Vac. The lipid pellets were then dissolved in a 60 μl tert-butanol and 40 μl of Triton X-114: methanol (2:1) mix. TG and TC were measured using commercially available assay kits (Pointe Scientific, Canton, MI) according to the manufacturer’s instructions. Hepatic lipid concentration was presented as mg/g liver weight.
Histology and Immunohistochemistry (IHC) analysis
Liver sections were stained with hematoxylin and eosin (H&E) using method previously described18, 20 and Masson’s Trichrome using a kit from PolyScientific (K037) 18, 20. The small intestines were Swiss-rolled and fixed overnight in 10% PBS-buffered formalin and embedded in paraffin. Five μm-thick sections were stained with H&E. For IHC, sections were deparaffinized and rehydrated prior to antigen retrieval, removal of endogenous peroxidase, and blocking with normal goat serum. Incubated overnight at 4°C with Ym1 (1:400, Stemcell Technologies,MA), Mucin 2 (1:500, Santa Cruz, Dallas, TX), E-cadherin (1:300, Cell signaling, Danvers, MA) or rabbit IgG (ProSci Inc., Poway, CA). Tissue sections were incubated for 30 min with a biotinylated goat anti-rabbit secondary antibody (Vector Labs, Burlingame, CA). Antibody was visualized using a DAB Peroxidase Substrate Kit (Vector Labs). Photomicrographs were taken using the VS120-S5 System (Olympus, Center Valley, PA). Five random areas (200X) from each IHC specimen were selected and the images were acquired. Quantification of the staining and hepatocyte nuclei were calculated. Data were presented as the ratio of stained cells to the hepatocyte nucleus count.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of liver sections was performed using In situ Cell Death Detection Kit, POD (11684817910, Roche) according to the manufacturer’s instructions. Briefly, 5 μm liver sections were deparafinized, rehydrated, and treated for 30 minutes at room temperature with 20μg/ml Proteinase K solution in 10 mM Tris-HCl buffer (pH 7.5). Slides were then incubated with 100 μl TUNEL reaction mixture for 60 mins and 100 μl Converter-POD solution for 30 mins at 37 °C. Slides were stained with DAB substrate until developed (Vector Laboratories Inc.) and counterstained with Mayer’s hematoxylin for 30 seconds.
For Periodic acid-Schiff (PAS) staining, deparaffinized and rehydrated intestine sections were stained with a commercially available kit (Thermo Scientific, Waltham, MA). Sections were then stained with Schiff’s reagent for 15 minutes at room temperature, followed by a 5-minute wash in running tap water. The sections were counterstained with hematoxylin, washed in running tap water for 2 minutes, blued with phosphate buffered saline, dehydrated and cover-slipped.
Total RNA isolation and quantitative RT-PCR assay
Total RNA was isolated from frozen tissues by TRIzol reagent (Invitrogen, Carlsbad, CA), and reverse-transcribed according to the manufacturer’s protocols. The mRNA expression levels of genes were quantified by quantitative real-time RT-PCR (qPCR) using the SYBR green chemistry and normalized to β-actin mRNA levels using the ΔΔCt method. Primers for qPCR assay are provided in supplementary Table 1.
Western blot analysis
Liver lysates (20μg/well) were separated on the 10% SDS-polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride membranes, and the blots were then probed with antibodies against CYP2E1 (1:2500, Abcam, Cambridge, MA). Bands were visualized using Enhanced Chemiluminescence reagents (Thermo Scientific, Rockford, IL), and then normalized against β-actin (1:1000, Cell signaling, Danvers, MA). The density of the bands was quantified using ImageJ.
Statistical analysis
The data are expressed as mean ± SD. Difference among groups was assessed by the two-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test. Differences were considered significant at p < 0.05 and indicated by an asterisk (*) for difference between treatments and a pound (#) for difference among different genotypes.
Results
Intestinal FXR deficiency worsened alcohol-induced liver steatosis
Ethanol treatment markedly increased the serum activities of ALT, AST and ALP in FXRInt−/− but not in WT mice (Fig. 1A). For both strains of mice, ethanol treatment didn’t alter serum levels of TGs and TC but increased total BA concentrations, with statistical significance reached only in WT mice (Fig. 1A). In agreement with previous findings, the histology of the livers of WT and FXRInt−/− mice with the vehicle control appeared normal (Fig. 1B). Alcohol feeding resulted in only mild hepatic microsteatosis in WT mice, but marked microsteatosis and macrosteatosis, as well as hepatocyte ballooning and Mallory-Denk bodies (MDBs) in FXRInt−/−mice (Fig. 1B). Ethanol treatment increased hepatic TG but not TC levels. However, no significant difference was found between WT and FXRInt−/− mice (Fig. 1C). Ethanol feeding increases number of TUNEL-positive cells28. There were increased numbers of cells in WT mouse livers with TUNEL positive staining (Fig. 1D), and the positively stained cells were both hepatocytes and non-hepatocytes. Interestingly, there were more TUNEL positive cells in FXRInt−/− mice on control diet, which was further increased by ethanol treatment.
Figure 1:
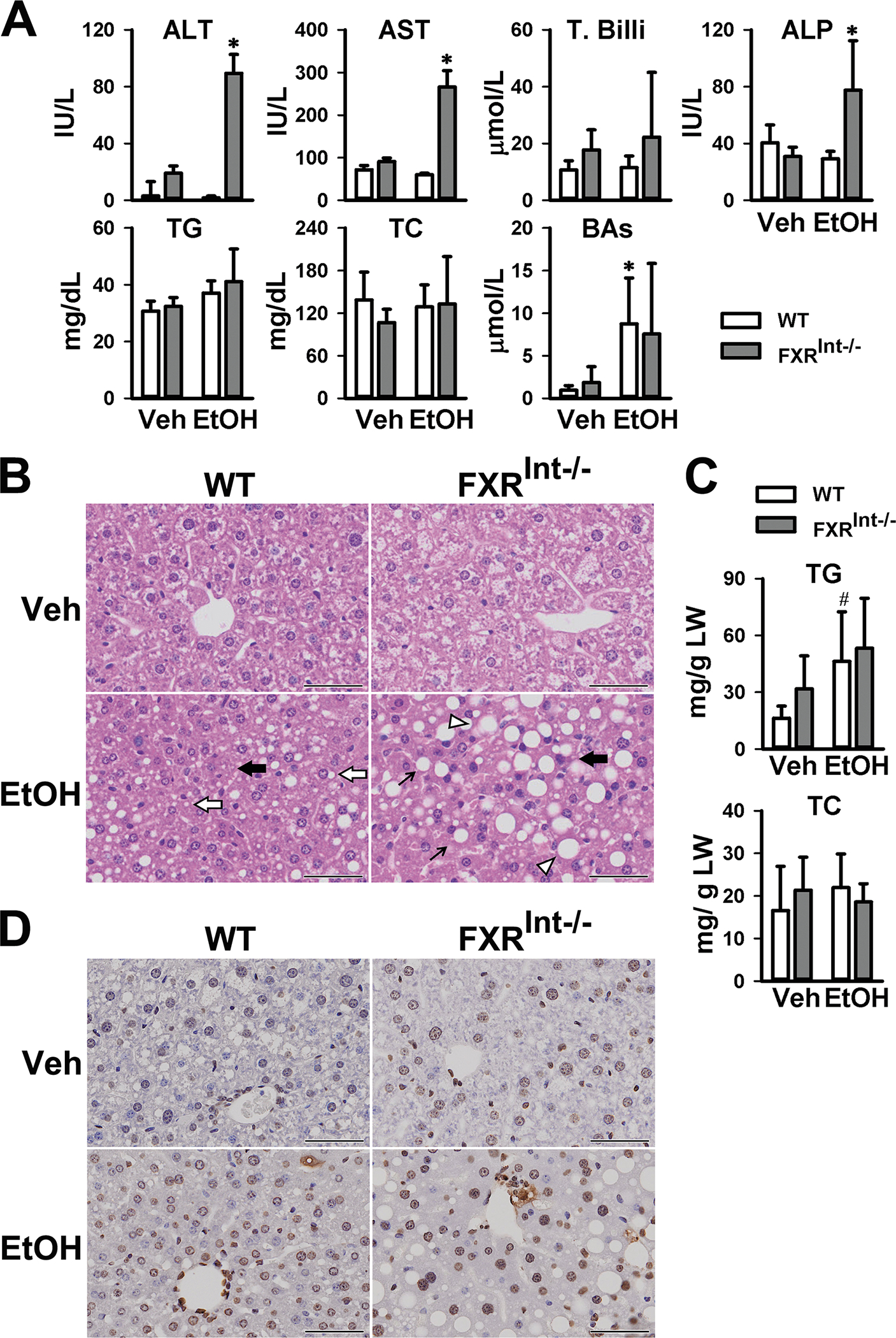
Assessment of liver injury in WT and FXRInt−/− mice fed control or chronic (the Lieber-Decali diet containing 5% ethanol)-binge ethanol. (A) Activities of serum ALT, AST, total bilirubin (T. Billi), ALP and serum levels of TG, TC, and total BAs; (B) H&E staining of WT and FXR Int−/− mice. Top figures no EtOH and bottom micrographs show ethanol induced changes including microvesicular steatosis (open arrow) in WT mice, macrovesicular steatosis (arrow) in FXRInt−/− mice, hepatocyte ballooning (open triangle), and MDBs (black arrow); (C) Hepatic levels of TG and TC; (D) Hepatic staining of TUNNEL. Values are presented as mean ± SD (n=11, 15, 10 and 15 mice/group for WT/Veh, WT/EtOH, FXRInt−/−/Veh, and FXRInt−/−/EtOH, respectively). * indicates statistical significance (p <0.05) of ethanol treatment (EtOH) compared to vehicle (Veh) control group in the same strain. # indicates statistical significance between WT and FXRInt−/− mice (p <0.05) with the same treatment. The scale bars show 50 μm for 200x magnification.
Intestinal FXR deficiency worsened the alcohol-induced liver inflammation
The hepatic expression of genes involved in inflammation were determined (Fig. 2A), including interleukin 6 (Il6), Il-1β, tumor necrosis factor α (Tnfα), monocyte chemoattractant protein 1 (Mcp1), toll like receptor 4 (Tlr4), cluster of differentiation 14 (Cd14) and lipocalin 2 (Lcn2). Surprisingly, in WT mice, ethanol treatment reduced the mRNA levels of Il-6 and Il-1β, had no effects on Tnfα, Tlr4 and Cd14, and tended to increase the mRNA levels of Mcp1 and Lcn2. In contrast, ethanol significantly increased the mRNA levels of Mcp1, Cd14, and Lcn2 in FXRInt−/− mice, while having little effects on the expression of other genes.
Figure 2:
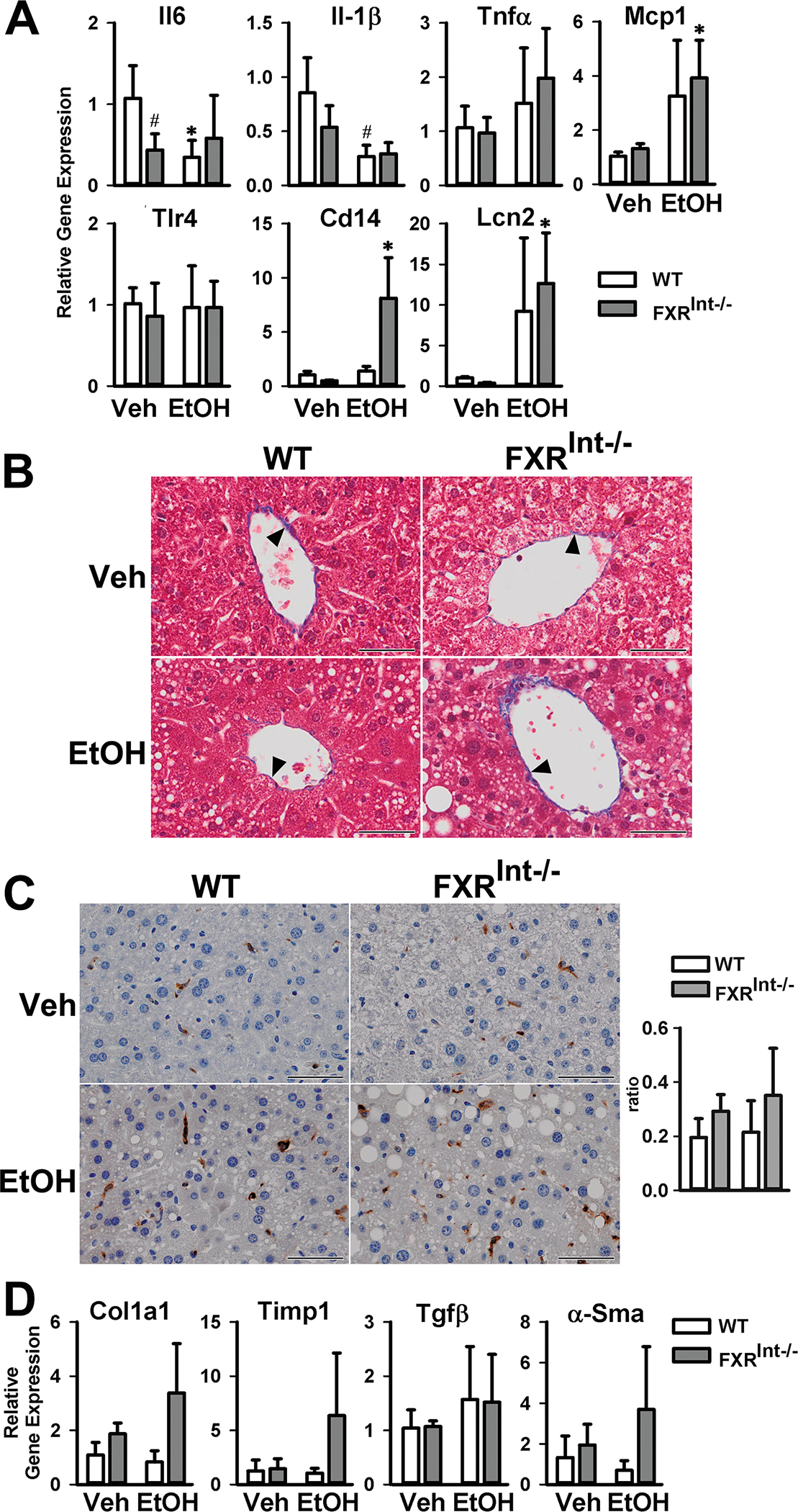
Effects on hepatic inflammation and fibrosis by ethanol in WT and FXRInt−/− mice. (A) Relative mRNA levels of genes involved in inflammation; (B) Representative Masson’s trichrome staining of the livers (200X); solid triangle indicating the collagen stained blue (C) IHC and semi-quantification of YM1 positive cells in the livers (200x); (D) Relative mRNA levels of fibrogenic genes. Values are presented as mean ± SD (n=6, 10, 5 and 10 mice/group for WT/Veh, WT/EtOH, FXRInt−/−/Veh, and FXRInt−/−/EtOH, respectively). * indicates statistical significance (p <0.05) of ethanol treatment compared to vehicle (Veh) control group in the same strain. # indicates statistical significance between WT and FXRInt−/− mice (p <0.05) with the same treatment. The scale bars show 50 μm for 200x magnification.
The effects of ethanol treatment on liver fibrosis were determined by Masson’s trichrome staining in both WT and FXRInt−/− mice (Fig. 2B). As expected, no obvious evidence of liver fibrosis was found in mice fed the control diet, nor in WT mice under ethanol. However, ethanol treatment seems to slightly increase the collagen deposition in the livers of FXRInt−/− mice. Because this change was no clear, we determined other markers associated with liver fibrosis, including YM1 positive M2 macrophage (Fig. 2C) and mRNA levels of fibrotic genes (Fig. 2D) such as collagen 1a1 (Col1α1), tissue inhibitor of metalloproteinase 1 (Timp1), and alpha smooth muscle actin (α-Sma), and transforming growth factor beta (Tgfβ). However, similar to results of IHC, there was a trend of increased expression of fibrosis markers in ethanol-treated FXRInt−/− mice, but did not reach statistical significance.
Effects of ethanol on expression levels of genes involved in BA and lipid homeostasis
Because intestinal FXR is critical in regulating BA synthesis, mRNA levels of the genes involved in BA homeostasis were determined in both the liver (Fig. 3A) and ileum (Fig. 3B). In WT mice, ethanol tended to reduce the mRNA levels of all FXR target genes in the liver, regardless of mode of regulation, including small heterodimer partner (Shp or Nr0b2), bile salt export pump (Bsep or Abcb11), lipocalin 13 (Lcn13), cholesterol 7α-Hydroxylase (Cyp7a1), and cholesterol 12α-hydroxylase (Cyp8b1). However, only reduction of mRNA levels of Lcn13 reached statistical significance. Surprisingly, FXRInt−/− mice did not show upregulation of expression of Cyp7a1 and Cyp8b1 compared to WT mice, presumably due to elevated basal expression of these genes by the high-carbohydrate-containing control diet Furthermore, ethanol treatment did not alter the hepatic expression of the BA related genes in FXRInt−/− mice. In the ileums of WT mice, ethanol markedly reduced Fgf15 mRNA levels but had no effects on the expression of other FXR target genes, including Ostβ and intestine bile acid binding protein (Ibabp) (Fig. 3C). The intestinal expression of FXR and its target genes were undetectable or very low in FXRInt−/− mice, which was not further reduced by ethanol treatment (Fig. 3C).
Figure 3:
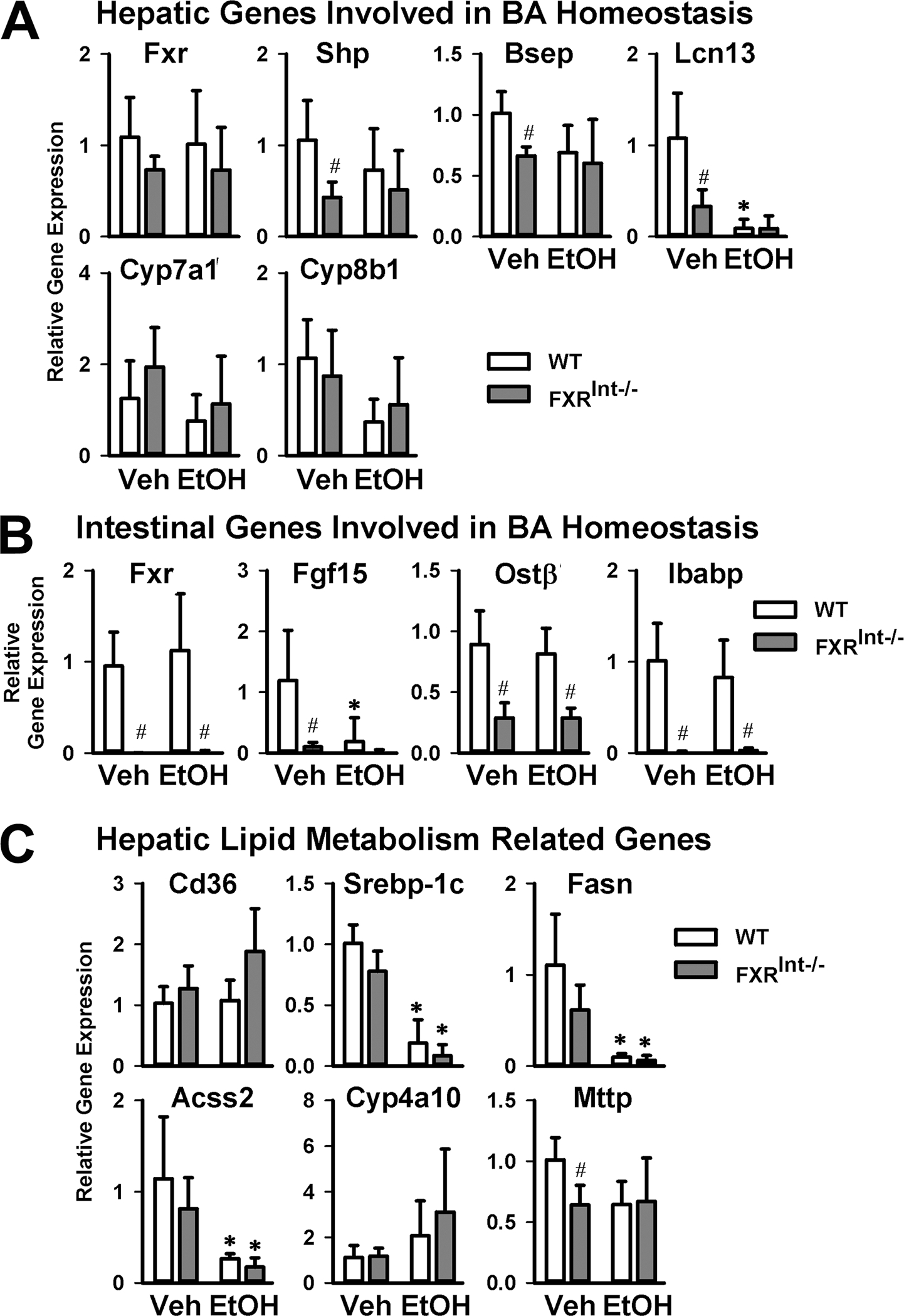
Expression of genes involved in BA homeostasis and regulating lipid homeostasis. (A) Relative liver mRNA levels of genes involved in BA homeostasis; (B) Relative intestinal mRNA levels of genes involved in BA homeostasis; (C) Relative hepatic mRNA levels of genes involved in lipid metabolism, including fatty acid uptake, fatty acid synthesis, fatty acid β-oxidation and VLDL secretion. Values are presented as mean ± SD (n=6, 10, 5 and 10 mice/group for WT/Veh, WT/EtOH, FXRInt−/−/Veh, and FXRInt−/−/EtOH, respectively). * indicates statistical significance (p <0.05) of ethanol treatment compared to vehicle (Veh) control group in the same strain. # indicates statistical significance between WT and FXRInt−/− mice (p <0.05) with the same treatment.
Alcohol drinking is known to affect lipid metabolism in the liver. The mRNA levels of genes critically involved in hepatic lipid homeostasis were therefore determined (Fig. 3C), including uptake of long-chain free fatty acids (cluster of differentiation 36, Cd36), de novo lipogenesis (sterol regulatory element-binding protein 1c, Srebp-1c; fatty acid synthase, Fasn; and acyl-CoA synthetase short-chain family member 2, Acss2), β-oxidation of fatty acids (a PPARα target gene, Cyp4a10), and very low density lipoprotein (VLDL) secretion (microsomal triglyceride transfer protein, Mttp). The results showed that alcohol significantly decreased the mRNA levels of Srebp-1c, Fasn, and Acss2, and increased those of Cyp4a10 in both WT and FXRInt−/− mice. The mRNA levels of Mttp were slightly lower in FXRInt−/− mice than in WT mice, but no alcohol-dependent changes were seen for Cd36 or Mttp.
Expression of genes involved in alcohol metabolism
Alcohol dehydrogenases (ADHs), catalase and CYP2E1 convert ethanol to acetaldehyde that is metabolized to acetate by aldehyde dehydrogenases (ALDHs)1. During these metabolic processes, a large amount of reactive oxygen species are generated, which contribute to liver injury1, 29. No changes were found between WT and FXRInt−/− mice on control nor ethanol diet, although catalase expression was reduced by ethanol in WT mice (Fig. 4A). Ethanol induced CYP2E1 protein levels in both WT and FXRInt−/− mice, as expected. However, the degree of induction seemed less in FXRInt−/− mice (Fig. 4B). Components of the nuclear factor E2-related factor 2 (NRF2) signaling pathway, including NAD(P)H:quinone oxidoreductase 1 (Nqo1), glutathione transferase A3 (Gsta3), and glutamate-Cysteine Ligase Catalytic Subunit (Gclc), protect cells against oxidative stress and glutathione depletion29, 30. The basal expression levels of these genes were similar in both strains of mice. Ethanol significantly induced the mRNA levels of Nqo1, but not Gsta3 and Gclc, in both WT and FXRInt−/− mice (Fig. 4C).
Figure 4:
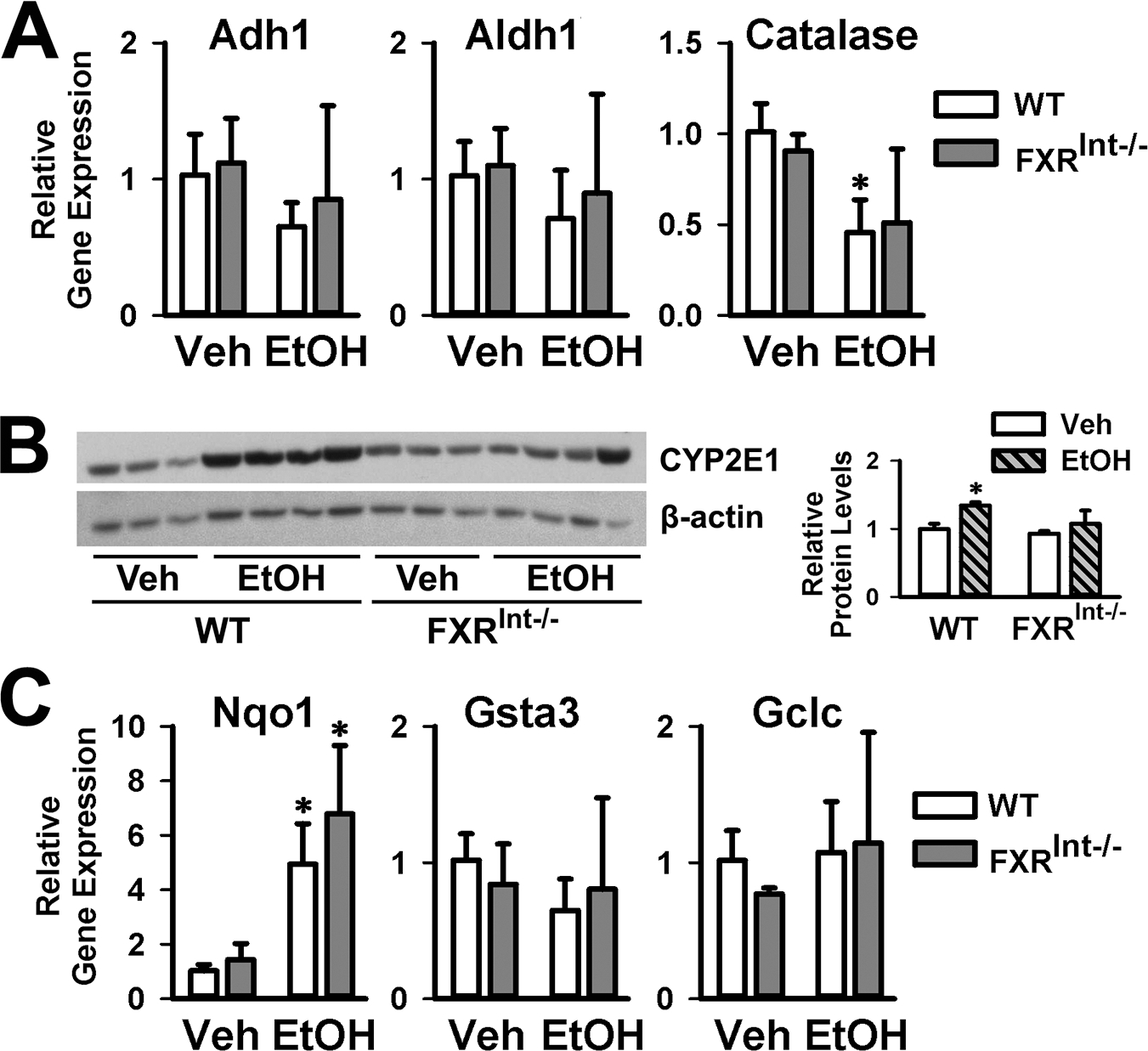
Hepatic expression of genes in ethanol metabolism and oxidative stress. (A) Relative hepatic mRNA levels of Adh1, Aldh1 and Catalase; (B) Representative immunoblots of liver CYP2E1 protein levels. Densitometric values were calculated and normalized with internal loading control, β-actin, and represented within the bar graphs; (C) Relative hepatic mRNA levels of Nqo1, Gsta3 and Gclc. Values are presented as mean ± SD (n=6, 10, 5 and 10 mice/group for WT/Veh, WT/EtOH, FXRInt−/−/Veh, and FXRInt−/−/EtOH, respectively). * indicates statistical significance (p <0.05) of ethanol treatment compared to vehicle (Veh) control group in the same strain. # indicates statistical significance between WT and FXRInt−/− mice (p <0.05) with the same treatment.
FXR deficiency modified intestinal morphology and exacerbated ethanol induced mucosal damage
Since FXR is highly expressed in the ileum, the histology of the distal ileum was examined (Fig. 5A). The ileum of FXRInt−/− mice on control diet revealed a distorted mucosal lining with increased sub-epithelial space and signs of degeneration with breaches along the mucosal lining, including openings of the epithelium to the lamina propria. Following alcohol feeding, both WT and FXRInt−/− mice demonstrated changes of villi morphology, including widening and shorting of villi, and shrinkage of the submucosa and muscular layer. These histopathological findings were more severe in FXRInt−/− mice. In addition, FXR deletion and ethanol administration caused the shortening and loss of crypts, as well as the opening of crypts to the lumen, suggesting a possible direct exposure of the intestinal endothelium to intestinal microbiota.
Figure 5:
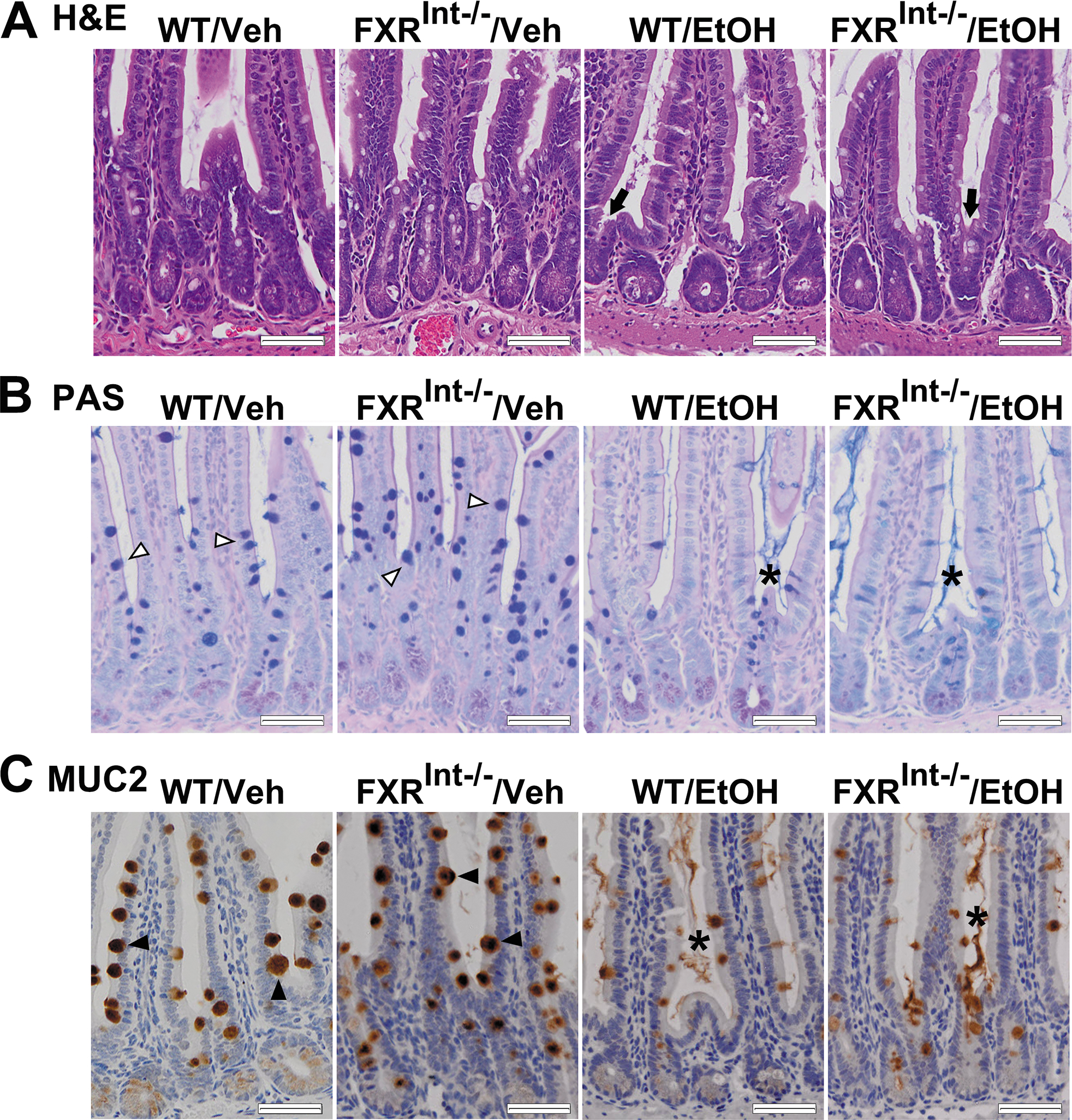
Effects of ethanol feeding on the intestinal mucus layer and villi morphology. (A) Representative images of H&E staining of the distal ileum. The ileum of vehicle-fed FXRInt−/− mice showed distorted mucosal lining with breaches along the mucosal lining. The shortening and opening of crypts to the lumen were observed in ethanol-fed mice (black arrow); (B) PAS staining of the intestine. Mucus were produced in villi (open triangle), and were detected in the intestinal lumen between villi (star) after ethanol fed. For neutral glycoproteins (magenta), mucins that are both acidic and neutrally charged (purple) and acidic glycoproteins (blue); (C) MUC2 IHC of the small intestine (black triangle) demonstrated release of MUC2 from goblet cells in the villi following alcohol consumption (star). The scale bars show 50 μm for 200x magnification.
The mucus layer acts as a physical barrier to protect against intestinal bacterial translocation1 and mucus production in the ileum was assessed by PAS-staining method (Fig. 5B). In WT mice, PAS-positive goblet cells were observed in the intestinal lumen between villi, at the apical sites of villi epithelial cells. Secretion of mucus from goblet cells was elicited primarily by irritating stimuli. Control diet-fed FXRInt−/− mice revealed dark purple staining in the villus with less staining in the intestinal lumen. After ethanol feeding, mucus was released in the lumen between the villi in both WT and FXRInt−/− mice. In addition, PAS-staining within goblet cells and the intestinal lumen was blue in FXRInt−/− mice, indicating higher acidic glycoproteins composition in the secreted mucins and mucus.
Mucin 2 protein (MUC2) is the major gel-forming mucin in the intestine mucus, and was observed in goblet cells of the ileum (Fig. 5C). Compared to WT mice, FXRInt−/− mice showed upregulated MUC2 expression, as well as more numbers and larger size of goblet cells (Fig. 5C). Interestingly, ethanol feeding appeared to reduce the MUC2 expression in WT and FXRInt−/−, while it promoted MUC2 secretion from the goblet cells into the intestinal lumen.
FXR deficiency reduced E-cadherin levels and increased intestinal epithelial barrier permeability
E-cadherin is a cell-cell adhesion molecule which is necessary to form adherens junctions in epithelial tissues. E-cadherin expression was downregulated in the livers of whole-body FXR knockout mice, which might contribute to liver tumorigenesis in these mice31. In this study, the small intestines of WT and FXRInt−/− mice were stained for E-cadherin protein (Fig. 6A). The results showed that the basal E-cadherin protein levels in the ileum crypts were lower in FXRInt−/− mice compared to WT mice (Fig. 6A). Alcohol feeding reduced E-cadherin expression in both strains of mice (Fig. 6A). The expression of other genes involved in tight junctions, including zonula occludens-1 (Zo1), occludin, claudin 1, and junction adhesion molecule (Jam), were not affected by FXR deficiency or ethanol feeding (data not shown).
Figure 6:
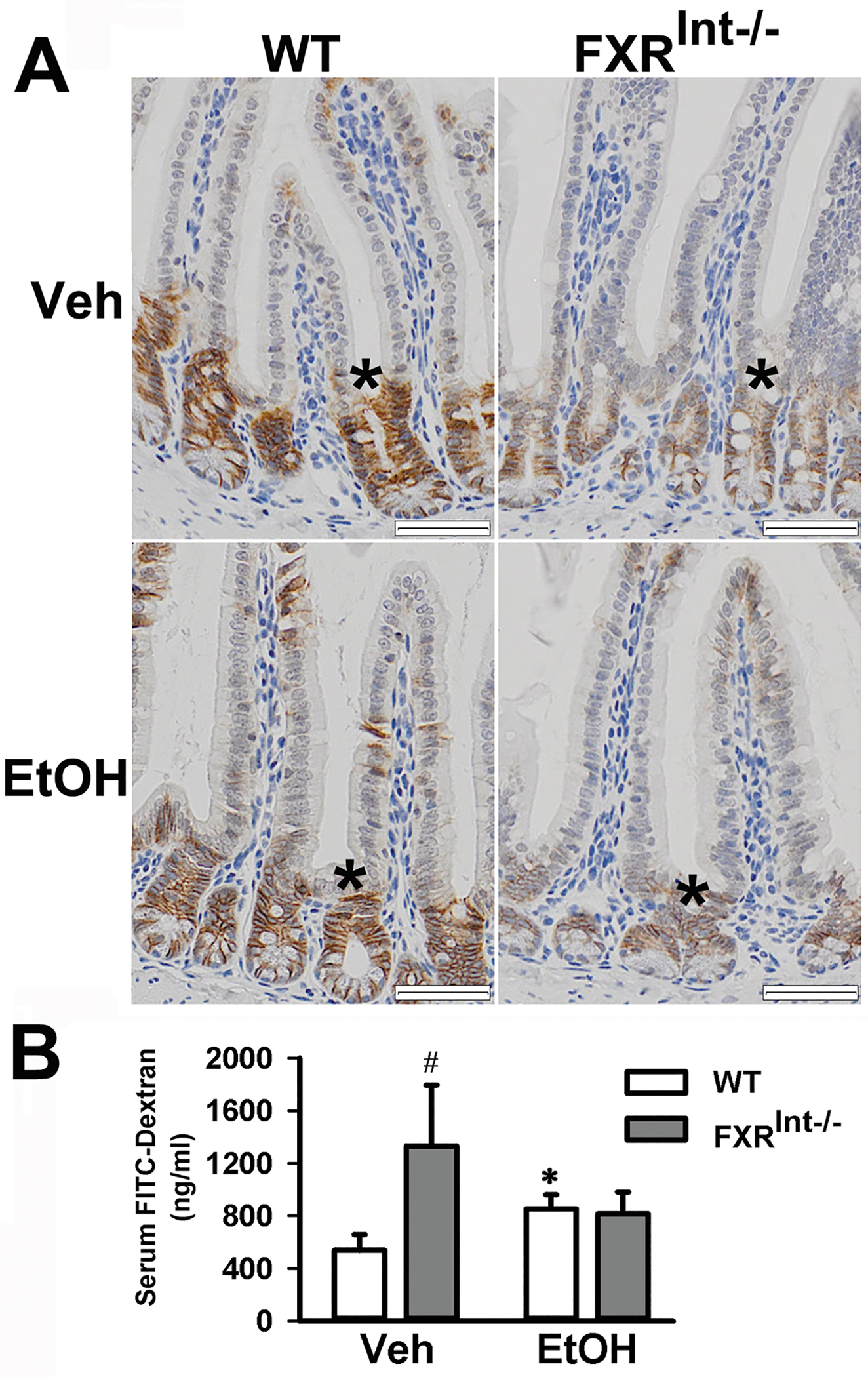
Effects of ethanol feeding on the intestinal permeability. (A) Representative stained images of E-cadherin along the ileal crypt-villus axis (star) from all strains fed either vehicle or ethanol. (B) Plasma levels of FITC-dextran in vehicle and ethanol-fed mice. Mice (n = 5) were administered FITC-dextran via oral gavage to determine intestinal permeability. Values are presented as mean ± SD. * indicates statistical significance (p <0.05) of ethanol treatment compared to vehicle (Veh) control group in the same strain. # indicates statistical significance between WT and FXRInt−/− mice (p <0.05) with the same treatment. The scale bars show 50 μm for 200x magnification.
Since FXRInt−/− mice revealed a downregulation of E-cadherin protein levels, intestinal permeability was assessed in vivo by measuring FITC-dextran leakage in serum after an oral administration27 (Fig. 6B). On control diet, serum levels of FITC-dextran were significantly higher in FXRInt−/− mice than in WT mice. Serum FITC-dextran levels were increased in WT mice with alcohol feeding while no increased FITC-dextran was noted in FXRInt−/− mice (Fig. 6B).
Discussion
FXR is the major sensor for BA homeostasis by suppressing synthesis, promoting enterohepatic circulation, and regulating metabolism of BAs. FXR is highly expressed in the liver and ileum. Activation of FXR in the ileum strongly induces FGF15 in mice and FGF19 in humans12–14. We and other scientists have determined liver and gut-specific functions of FXR revealed by tissue specific regulation of target gene expression11, 24, 32.
Activation FXR has been shown to protect the liver from ethanol-induced steatosis and oxidative stress17, 33. Consistent to these studies, we and others have shown that whole-body deficiency of FXR worsens the severity of AFLD18, 34. First, we speculated that hepatic FXR is important in protecting the liver from ethanol-induced injury, because FXR deficiency is associated with reduced expression of several essential autophagy genes and other FoxO3 target genes35. However, to our surprise, the follow-up study showed that hepatic deficiency of FXR in mice did not affect the severity of AFLD development20, suggesting extrahepatic FXR is protective in the development of AFLD.
Intestinal FXR is critical in suppressing BA synthesis and in promoting liver regeneration by strongly inducing the expression of Fgf15 in mice and FGF19 in humans. FGF15/19 travels to the liver to activate hepatic fibroblast growth factor receptor 4 (FGFR4) to suppress BA synthesis and to promote cell cycle progression12, 14, 15. Meanwhile, gut FXR directly regulates many target genes in the intestine involved in BA transport (OSTβ), innate immunity (defensins), and lipid metabolism (ceramide) 11, 36. Moreover, BAs regulate microbiome population and diversity in the gut, which could further affect intestine integrity37, 38. Moreover, conversion from primary to secondary BAs by microbiome in the gut provides a ligand (lithocholic acid) of pregnane X receptor and Takeda G protein-coupled receptor 5 to generate another layer of protection and regulation of energy metabolism39, 40.
In addition to regulation of BA homeostasis, intestinal FXR helps maintain the integrity of intestinal barrier against bacterial overgrowth and translocation25. In this study, although we found minimal alterations in lipid metabolism and oxidative stress in FXRInt−/− mice following ethanol feeding using the NIAAA model, the intestine FXR deficient mice suffered more severe liver inflammation and injury. The underlying mechanism has been explored, with the data suggesting that intestine FXR deficient mice exhibited an increase in gut leakage compared to WT mice. Our previous study has revealed a marked decrease in E-cadherin expression in the livers of whole-body FXR KO mice31. Interesting, multiple FXR binding sites upstream of the Cdh1 gene that encodes E-cadherin have been identified by ChIP-seq, suggesting that Cdh1 is a directly induced by FXR activation32, 41. E-cadherin is the major component of the adherens junctions, a complex with β-catenin to maintaining cell-cell contacts in epithelial and endothelial tissues. In this study, we confirmed that intestinal E-cadherin was lower in FXRInt−/− mice compared to WT mice. Moreover, alcohol consumption decreased E-cadherin in the intestinal crypts, thereby disrupting the mucosal barrier in FXRInt−/− mice, which was confirmed by an in vivo functional assessment for leaky gut, and was associated with increased liver injury in FXRInt−/− mice. In agreement, a recent study by Hartmann et al. showed that activation of FXR in the gut protects against AFLD development in mice7.
In summary, loss of FXR function contributes to AFLD development, and the current study provides evidences showing that intestinal FXR is critical in maintaining gut integrity and epithelial barrier, which may prevent ethanol-induced bacterial translocation and increase in gut permeability. As results, intestinal FXR may protect the liver from gut endotoxin-induced inflammation (Fig. 7).
Figure 7:
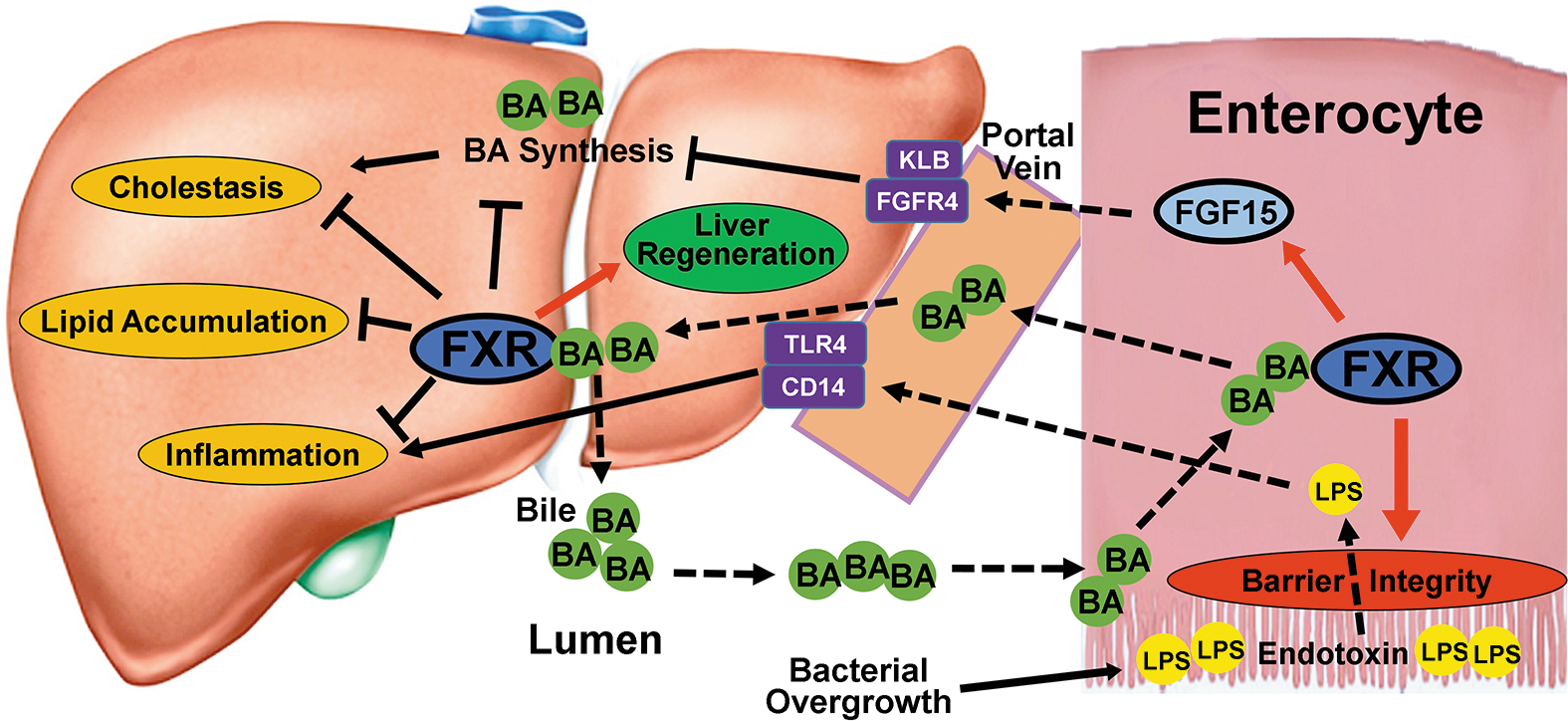
Schematic figure of the effects of FXR on BA homeostasis and ethanol induced injury. Intestinal FGF15 is induced by activated FXR, released into portal circulation and transported to the liver where it binds to its receptor complex of FGFR4 and β-Klotho on the hepatocyte membrane to suppress BA synthesis. Activation of hepatic FXR by BAs leads to minor suppression of BA synthesis, promotion of liver regeneration and protection of the liver against cholestasis, steatosis, and inflammation. Intestinal FXR is crucial for maintaining intestinal barrier integrity and protecting the liver against influx of toxic gut endotoxins.
Supplementary Material
Financial support:
NIH-R01GM104037; NIH-R21ES029258; NIH-T32ES007148; NIH-F32DK116495; NIH-AR005073; VA-BX002741; Rutgers Busch grant;
List of abbreviations:
- AFLD
Alcoholic fatty liver disease
- ALP
Alkaline phosphatase
- AST
Aspartate transaminase
- ALT
Serum alanine aminotransferase
- BA
Bile acid
- ECL
Enhanced Chemiluminescence
- FGF15/19
Fibroblast growth factor 15/19
- FXR
Farnesoid X receptor
- H&E
Hematoxylin and eosin
- IHC
Immunohistochemistry
- MDBs
Mallory-Denk bodies
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- PAS
Periodic acid-Schiff staining
- TC
Total cholesterol
- TGs
Triglycerides
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- VLDL
Very low density lipoprotein
- WT
Wild type
References
- 1.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 1987;4:8–14. [DOI] [PubMed] [Google Scholar]
- 3.Malhi H, Camilleri M. Modulating bile acid pathways and TGR5 receptors for treating liver and GI diseases. Curr Opin Pharmacol 2017;37:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther 2009;328:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee FY, de Aguiar Vallim TQ, Chong HK, Zhang Y, Liu Y, Jones SA, et al. Activation of the farnesoid X receptor provides protection against acetaminophen-induced hepatic toxicity. Mol Endocrinol 2010;24:1626–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iracheta-Vellve A, Calenda CD, Petrasek J, Ambade A, Kodys K, Adorini L, et al. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatol Commun 2018;2:1379–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006;312:233–236. [DOI] [PubMed] [Google Scholar]
- 10.Naugler WE. Bile acid flux is necessary for normal liver regeneration. PLoS One 2014;9:e97426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y, Li F, Guo GL. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol Res 2011;63:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217–225. [DOI] [PubMed] [Google Scholar]
- 13.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 2007;48:2664–2672. [DOI] [PubMed] [Google Scholar]
- 14.Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012;56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong B, Sun R, Huang M, Chow MD, Zhong XB, Xie W, et al. Fibroblast Growth Factor 15-Dependent and Bile Acid-Independent Promotion of Liver Regeneration in Mice. Hepatology 2018;68:1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J 2013;27:3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu W, Zhu B, Peng X, Zhou M, Jia D, Gu J. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem Biophys Res Commun 2014;443:68–73. [DOI] [PubMed] [Google Scholar]
- 18.Kong B, Zhang M, Huang M, Rizzolo D, Armstrong LE, Schumacher JD, et al. FXR deficiency alters bile acid pool composition and exacerbates chronic alcohol induced liver injury. Dig Liver Dis 2019;51:570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajaj JS, Kakiyama G, Zhao D, Takei H, Fagan A, Hylemon P, et al. Continued Alcohol Misuse in Human Cirrhosis is Associated with an Impaired Gut-Liver Axis. Alcohol Clin Exp Res 2017;41:1857–1865. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M, Kong B, Huang M, Wan R, Armstrong LE, Schumacher JD, et al. FXR deletion in hepatocytes does not affect the severity of alcoholic liver disease in mice. Dig Liver Dis 2018;50:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009;49:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013;62:899–910. [DOI] [PubMed] [Google Scholar]
- 23.Kong B, Huang J, Zhu Y, Li G, Williams J, Shen S, et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am J Physiol Gastrointest Liver Physiol 2014;306:G893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Liu H, Zhang M, Guo GL. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B 2016;6:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 2006;103:3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008;134:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Z, Sun X, Kang YJ. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol 2001;159:329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dianzani MU. Lipid peroxidation in ethanol poisoning: a critical reconsideration. Alcohol Alcohol 1985;20:161–173. [PubMed] [Google Scholar]
- 30.Nanji AA, Zhao S, Sadrzadeh SM, Dannenberg AJ, Tahan SR, Waxman DJ. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin Exp Res 1994;18:1280–1285. [DOI] [PubMed] [Google Scholar]
- 31.Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther 2011;338:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhan L, Liu HX, Fang Y, Kong B, He Y, Zhong XB, et al. Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS One 2014;9:e105930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livero FA, Stolf AM, Dreifuss AA, Bastos-Pereira AL, Chicorski R, de Oliveira LG, et al. The FXR agonist 6ECDCA reduces hepatic steatosis and oxidative stress induced by ethanol and low-protein diet in mice. Chem Biol Interact 2014;217:19–27. [DOI] [PubMed] [Google Scholar]
- 34.Wu WB, Chen YY, Zhu B, Peng XM, Zhang SW, Zhou ML. Excessive bile acid activated NF-kappa B and promoted the development of alcoholic steatohepatitis in farnesoid X receptor deficient mice. Biochimie 2015;115:86–92. [DOI] [PubMed] [Google Scholar]
- 35.Manley S, Ni HM, Williams JA, Kong B, DiTacchio L, Guo G, et al. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol 2014;2:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez FJ, Jiang C, Xie C, Patterson AD. Intestinal Farnesoid X Receptor Signaling Modulates Metabolic Disease. Dig Dis 2017;35:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018;68:1574–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 2018;24:1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keitel V, Stindt J, Haussinger D. Bile Acid-Activated Receptors: GPBAR1 (TGR5) and Other G Protein-Coupled Receptors. Handb Exp Pharmacol 2019;256:19–49. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, Xie W. A brief history of the discovery of PXR and CAR as xenobiotic receptors. Acta Pharm Sin B 2016;6:450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2010;51:1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.