

Abstract
Background
Viral-based immunotherapy can overcome resistance to immune checkpoint blockade (ICB) and fill the unmet needs of many patients with cancer. Oncolytic viruses (OVs) are defined as engineered or naturally occurring viruses that selectively replicate in and kill cancer cells. OVs also induce antitumor immunity. The purpose of this study was to compare the antitumor effects of live oncolytic vaccinia viruses versus the inactivated versions and elucidate their underlying immunological mechanisms.
Methods
We engineered a replication-competent, oncolytic vaccinia virus (OV-GM) by inserting a murine GM-CSF gene into the thymidine kinase locus of a mutant vaccinia E3L∆83N, which lacks the Z-DNA-binding domain of vaccinia virulence factor E3. We compared the antitumor effects of intratumoral (IT) delivery of live OV-GM versus heat-inactivated OV-GM (heat-iOV-GM) in a murine B16-F10 melanoma bilateral implantation model. We also generated vvDD, a well-studied oncolytic vaccinia virus, and compared the antitumor effects of live vvDD vs heat-inactivated vvDD (heat-ivvDD) in a murine A20 B-cell lymphoma bilateral tumor implantation model.
Results
Heat-iOV-GM infection of dendritic cells (DCs) and tumor cells in vitro induced type I interferon and proinflammatory cytokines and chemokines, whereas live OV-GM did not. IT live OV-GM was less effective in generating systemic antitumor immunity compared with heat-iOV-GM. Similar to heat-iOV-GM, the antitumor effects of live OV-GM also require Batf3-dependent CD103+ dendritic cells. When combined with systemic delivery of ICB, IT heat-iOV-GM was more effective in eradicating tumors, compared with live OV-GM. IT heat-ivvDD was also more effective in treating murine A20 B-cell lymphoma, compared with live vvDD.
Conclusions
Tumor lysis induced by the replication of oncolytic vaccinia virus has a limited effect on the generation of systemic antitumor immunity. The activation of Batf3-dependent CD103+ DCs is critical for antitumor effects induced by both live OV-GM and heat-iOV-GM, with the latter being more potent than live OV-GM in inducing innate and adaptive immunity in both locally injected and distant, non-injected tumors. We propose that evaluations of both innate and adaptive immunity, induced by IT oncolytic viral immunotherapy at injected and non-injected tumors, should be included as potential biomarkers for host responses to viral therapy.
Keywords: tumor microenvironment, oncolytic virotherapy, interferon inducers, immunotherapy, dendritic cells
Background
Oncolytic viral therapy has the potential to overcome resistance to immune checkpoint blockade (ICB), a form of immunotherapy increasingly being used in patients with advanced cancers.1–3 In 2015, the U.S. Food and Drug Administration approved the first oncolytic virus (OV) for the treatment of advanced melanoma: talimogene laherparepvec (T-VEC) is an engineered herpes simplex virus-1 that selectively replicates in tumors and expresses human granulocyte-macrophage colony-stimulating factor (GM-CSF).4–6 Clinical trials on the combination of intratumoral (IT) injection of T-VEC with systemic delivery of ICB agents—including ipilimumab and pembrolizumab, which blocks the cytotoxic T cell-associated antigen 4 (CTLA-4) and programmed death protein 1 (PD-1), respectively—showed enhanced therapeutic efficacy and increased cytotoxic T cell infiltration into tumors.7–9
Poxviruses are large cytoplasmic DNA viruses.10 Preclinical studies and clinical trials have demonstrated the efficacy of using oncolytic vaccinia viruses for the treatment of advanced cancers.11–15 For example, JX594, also known as pexastimogene devacirepvec (Pexa Vec), an oncolytic vaccinia virus featuring the deletion of the thymidine kinase (TK) gene to enhance tumor selectivity and the expression of human GM-CSF, has demonstrated therapeutic efficacy in a phase II clinical trial for patients with advanced hepatocellular carcinoma (HCC).15 Unfortunately, a phase III clinical trial (NCT02562755) comparing Pexa Vec followed by sorafenib (an FDA-approved tyrosine kinase inhibitor for HCC) versus sorafenib alone was discontinued after an interim futility analysis showed lack of efficacy.
While the tumor-killing effects of OVs have traditionally been attributed to their selective replication within tumor cells, the ability of oncolytic virotherapy to elicit host antitumor immunity plays a crucial role as well.16–20 However, the mechanisms by which oncolytic virotherapy induces antitumor immunity remain largely unknown. Modified vaccinia virus Ankara (MVA), a highly attenuated vaccinia strain, is an important vaccine vector against various infectious agents.21–26 We previously reported that MVA infection of conventional dendritic cells (cDCs) upregulates type I interferon (IFN), via the cGAS/STING-mediated cytosolic DNA-sensing pathway.27 The cGAS/STING and type I IFN pathways represent innate sensing mechanisms critical for antiviral and antitumor immunity.28–38 Infection of cDCs with heat-inactivated MVA (heat-iMVA), generated by heating the virus at 55°C for 1 hour, leads to higher levels of type I IFN than with live MVA. IT delivery of heat-iMVA leads to tumor eradication, as well as the development of systemic antitumor immunity, which requires CD8+ T cells, Batf3-dependent CD103+ DCs, as well as the STING-mediated cytosolic DNA-sensing pathway.39 Our results demonstrate that IT heat-iMVA alters the immunosuppressive tumor microenvironment (TME), by inducing the production of IFN, proinflammatory cytokines, and chemokines both in tumor cells and immune cells, via STING, and by activating tumor-infiltrating CD103+ DCs that contribute to the priming, expansion, and recruitment of activated antitumor CD4+ and CD8+ T cells and eventual tumor eradication.39
In this study, we engineered a replication-competent, oncolytic vaccinia virus (OV-GM) by inserting murine GM-CSF gene into the TK locus of a mutant vaccinia E3L∆83N (Western Reserve strain), which lacks the Z-DNA-binding domain of vaccinia virulence factor E3. E3L∆83N replicates efficiently in many cell lines but is highly attenuated, with reduced virulence of about 1000-fold, compared with wild type (WT) vaccinia using in vivo infection models.40 We compared the antitumor effects of IT delivery of live OV versus live OV-GM versus heat-iOV-GM, in bilateral and unilateral murine tumor models, in immune-competent syngeneic mice. Expression of murine GM-CSF by live OV-GM slightly improved the generation of effector CD8+ and CD4 T+ cells in both the injected and non-injected tumors. Although heat-iOV-GM does not express murine GM-CSF, we compared the antitumor effects of heat-iOV-GM with live OV-GM because many oncolytic viral platforms express GM-CSF as a transgene, including JX594/Pexa Vec, a clinical oncolytic vaccinia candidate. That comparison showed that IT heat-iOV-GM more effectively eradicated tumors and generated systemic antitumor immunity, than live OV-GM, in both unilateral and bilateral tumor implantation models. The antitumor effects of both live OV-GM and heat-iOV-GM required Batf3-dependent CD103+/CD8α+ DCs, which are efficient in cross-presenting tumor antigens.
To substantiate our findings, we also compared the antitumor effects of live vvDD, a well-studied oncolytic vaccinia virus (Western Reserve strain) that lacks genes encoding vaccinia growth factor (VGF) and TK,41–43 with its heat-inactivated version (heat-ivvDD), in a murine A20 B-cell lymphoma bilateral tumor implantation model. Our results showed that heat-ivvDD generated stronger antitumor effects and better survival than live vvDD.
Our results provide strong evidence that viral replication and viral-mediated oncolysis are not required for the generation of antitumor effects by vaccinia virus. Rather, the activation of the host’s immune system—including Batf3-dependent CD103+ DCs, likely via the cGAS/STING-mediated cytosolic DNA-sensing pathway—is crucial for the therapeutic efficacy of vaccinia-based immunotherapy. Thus, our findings have important clinical implications for the future design of optimal vaccinia-based cancer immunotherapeutics.
Methods
Study design
In this study, we used unilateral and bilateral tumor implantation models to compare the antitumor activities of live or heat-inactivated oncolytic vaccinia viruses. We also determined the relative contributions of the cytosolic DNA-sensing pathway, and CD103+ DCs, in inducing antitumor effects in STINGGt/Gt and Batf3-/- mice. In all experiments, animals were randomly assigned to various experimental groups. For survival studies, sample sizes of 8–10 mice were used. For experiments designed to evaluate the tumor immune cell infiltrates, 3–5 mice were used for each experiment and the experiments were performed two or three times. For the experiments designed to assess the induction of IT type I IFN and proinflammatory cytokines and chemokines levels, 3–5 mice were used for tumor collection and quantitative real-time PCR analyses were performed on each tumor sample.
Viruses and cell lines
E3L∆83N virus was kindly provided by Bertram Jacobs (Arizona State University). E3L∆83N (OV-TK+), OV, or OV-GM viruses were propagated in BSC-40 (Africa green monkey kidney cells, ATCC-CRL2761). Viruses were purified through a 36% sucrose cushion. Heat-iOV-GM virus was generated by incubating purified OV-GM virus at 55°C for 1 hour. vSC20 virus was kindly provided by Bernard Moss (National Institutes of Health).44 vvDD was generated by homologous recombination at the TK locus using mCherry as a fluorescent marker in this study. BSC-40 cells were maintained in DMEM medium containing 5% FBS, penicillin, and streptomycin. The murine melanoma B16-F10 cell line was originally obtained from I. Fidler (MD Anderson Cancer Center) and was maintained in RPMI 1640 medium supplemented with 10% FBS, penicillin, and streptomycin.
Mice
Female C57BL/6J mice were purchased from the Jackson Laboratory (Stock # 000664). Batf3-/- mice were from Dr Kenneth Murphy (Washington University). STINGGt/Gt mice were a kind gift from Dr Russell Vance (University of California, Berkeley). These mice were maintained in the animal facility at the Sloan Kettering Institute, and all procedures were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Our animal protocol was approved by the Institutional Animal Care and Use Committee at Sloan Kettering Institute.
Generating recombinant vaccinia virus expressing mGM-CSF
The murine GM-CSF (mGM-CSF) coding sequence was inserted into the pCB vector between Xba I and EcoR I sites. Vaccinia synthetic early and late promoter (PsE/L) was used to drive mGM-CSF (Csf2), and the vaccinia P7.5 promoter was used to express the drug selection gene, E. coli xanthine-guanine phosphoribosyl transferase gene (gpt). These two expression cassettes were flanked by a partial sequence of the TK gene on each side. To generate recombinant viruses OV (E3L∆83N-TK-) or OV-GM (E3L∆83N-TK--mGM-CSF), BSC40 cells were seeded into 6-well plates, and infected with E3L∆83N, at a multiplicity of infection (MOI) of 0.05. Two hours after virus infection, transfection mixtures containing plasmid DNA and Lipofectamine 3000 (Invitrogen) were added to wells, and the cells were then incubated at 37°C for 48 hours. The recombinant viruses were enriched in the gpt selection medium which contained mycophenolic acid, xanthine, and hypoxanthine and were plaque-purified in the gpt selection medium four times until the respective purified recombinant viruses were obtained. PCR reactions were used to verify the purity of these recombinant viruses. Primer sequences used for the PCR reactions were:
TK-F2: 5’-TGTGAAGACGATAAATTAATGATC-3’;
pCB-R3: 5’-ACCTGATGGATAAAAAGGCG-3’;
TK-F4: 5’-TTGTCATCATGAACGGCGGA-3’;
TK-R4: 5’-TCCTTCGTTTGCCATACGCT-3’;
GM-F: 5’-GGCATTGTGGTCTACAGCCT-3’;
GM-R: 5’-GTGTTTCACAGTCCGTTTCCG-3’;
TK-F5: 5’-GAACGGGACTATGGACGCAT-3’;
TK-R5: 5’-TCGGTTTCCTCACCCAATCG-3’.
Cytokine assays and western blot analysis
For the generation of bone marrow-derived dendritic cell (BMDCs), the bone marrow cells (5 million cells in each 15 cm cell culture dish) were isolated from WT or STINGGt/Gt mice and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), in the presence of 30 ng/ml GM-CSF (BioLegend), for 10-12 days.
For cytokine assays, BMDCs or B16-F10 melanoma cells were infected with various viruses at an MOI of 10 for 1 hour, or mock-infected. The inoculum was removed, and the cells were washed with PBS two times and incubated with a fresh medium. Supernatants were collected at various times postinfection. Cytokine levels were measured by using ELISA kits for murine IFN-α/β (PBL Biomedical Laboratories), IL-6, CCL5, CXCL10, or GM-CSF (R & D Systems).
For western blot analysis, BMDCs or B16-F10 cells were infected with the indicated viruses at an MOI of 10, and cell lysates were collected at different time points after virus infection. Polypeptides were separated by 15% SDS-PAGE, and western blot analysis was performed to determine the expression of mGM-CSF, using an anti-mGM-CSF antibody (Thermo Fisher), or to investigate the activation status of different components of the cGAS/STING pathway using antibodies against TBK-1, p-TBK-1, IRF-3, p-IRF-3, STING, p-STING, and cGAS (Cell Signaling Technology). GAPDH (Cell Signaling Technology) was used as a loading control.
mGM-CSF bioactivity assay
B16-F10 cells were infected with OV-GM at an MOI of 10 for 1 hour in a 6-well plate, and the inoculum was removed, and cells were washed with PBS. Fresh medium was added to the well, and the culture supernatants were collected at 24 hours after virus infection. The supernatant was UV irradiated and filtered through a 0.2 µm syringe filter (Nalgene). Different dilutions of the supernatants were added to bone marrow cells in RPMI medium. Generation of BMDCs was described previously.27 After 7 days, cultured DCs were fixed with Fix Buffer I (BD Biosciences) for 15 min at 37°C. Cells were washed, permeabilized with PermBuffer (BD Biosciences) for 30 min on ice and stained with antibodies against CD11c and CD11b for 30 min. Cells were analyzed using an LSRII Flow cytometer (BD Biosciences) for CD11c+ DCs. Data were analyzed using FlowJo software (Treestar).
Flow cytometry analysis of DC maturation
For DC maturation analysis, BMDCs were generated from C57BL/6J mice and infected with either live OV or live OV-GM at an MOI of 10, or with equivalent amounts of heat-iOV-GM. Cells were collected at 14 hours postinfection (hpi) and fixed with Fix Buffer I (BD Biosciences) for 15 min at 37°C. Cells were washed, permeabilized with PermBuffer (BD Biosciences) for 30 min on ice, and stained with antibodies against MHC Class I, CD40, CD86, and CD80 for 30 min. Cells were analyzed using an LSRII Flow cytometer (BD Biosciences) and data analyzed with FlowJo software (Treestar).
Tumor rechallenge to assess the development of systemic antitumor immunity
Surviving mice (8 weeks after tumor eradication) were rechallenged with intravenous delivery of a lethal dose of B16-F10 cells (1×105 cells in 50 µL PBS) and euthanized at 3 weeks postrechallenge to evaluate the presence of tumor foci on lung surfaces.
ELISPOT assay
Spleens were harvested from mice treated with different viruses and were mashed through a 70 µm strainer (Thermo Fisher Scientific). Red blood cells were lysed using ACK Lysis Buffer (Life Technology), and the cells were then resuspended in RPMI medium. CD8+ T cells were then purified using CD8a (Ly-2) MicroBeads (Miltenyi Biotechnology). Enzyme-linked ImmunoSpot (ELISPOT) assay was performed to measure IFN-γ+ CD8+ T cells, according to the manufacturer’s protocol (BD Bioscience). CD8+ T cells were mixed (1:1) with irradiated B16-F10 cells (250,000 cells each), in RPMI medium, and the ELISPOT plate was then incubated at 37°C for 16 hours before staining.
Preparation of single-cell suspensions from tumor samples
B16-F10 melanoma cells were implanted intradermally into the right and left flanks of C57BL/6J mice (5×105 cells into the right flank and 2.5×105 cells into the left flank). PBS, OV, live OV-GM, or heat-iOV-GM viruses (2×107 plaque forming unit (pfu)) were injected IT into the right-flank tumors 7 days after tumor implantation, and the injections were repeated once, 3 days later. Tumors were harvested 3 days after the second injection with forceps and surgical scissors and were weighed. They were then minced before incubation with Liberase (1.67 Wünsch U/mL) and DNase (0.2 mg/mL) in serum-free RPMI for 30 min at 37°C. Cell suspensions were generated by mashing through a 70 µm nylon filter and then washed with complete RPMI.
Flow cytometry analysis
For tumor-infiltrating lymphocytes analysis, single cell suspensions were generated and processed for surface labeling with anti-CD3, anti-CD45, anti-CD4, and anti-CD8 antibodies. Live cells were distinguished from dead cells by using fixable dye eFluor506 (eBioscience). They were further permeabilized using a permeabilization kit (eBioscience) and stained for granzyme B.
For PD-L1 expression analysis, B16-F10 cells were infected with different viruses at an MOI of 10. Uninfected cells were used as a control. Twenty-four hpi, cells were trypsinized and stained using an anti-PD-L1 antibody (eBioscience).
For the apoptosis/cell viability assays, B16-F10 cells were infected with different viruses at an MOI of 10. Cells were harvested at 6, 24, or 48 hpi, and stained for Annexin V and 7-AAD (BD Biosciences).
All flow data were acquired using either an LSRII flow cytometer (BD Biosciences) or an Aurora Five Laser flow cytometer (Cytek). Data were analyzed with FlowJo software (Treestar).
RNA isolation and quantitative real-time PCR
B16-F10 melanoma cells were implanted into the right and left flanks of C57BL/6J mice (5×105 cells into the right flank and 2.5×105 cells into the left flank). PBS, OV, live OV-GM, or heat-iOV-GM viruses were injected IT into the right-side tumors 7 days after tumor implantation. The injection was repeated once, 3 days after the first injection. Three days after the second injection, tumors were harvested from euthanized mice with forceps and surgical scissors and minced. RNAs were extracted from the tumor lysates with a RNeasy Mini kit (Qiagen) and were reverse transcribed with a First Strand cDNA synthesis kit (Fermentas). Quantitative real-time PCR was performed in triplicate with SYBR Green PCR Master Mix (Life Technologies) and an Applied Biosystems 7500 Real-time PCR Instrument (Life Technologies), using gene-specific primers. Relative expression was normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences for quantitative real-time PCR were:
Ifnb-F: 5’-TGGAGATGACGGAGAAGATG-3’;
Ifnb-R: 5’-TTGGATGGCAAAGGCAGT-3’;
Il6-F: 5’-AGGCATAACGCACTAGGTTT-3’;
II6-R: 5’-AGCTGGAGTCACAGAAGGAG-3’;
Ccl4-F: 5’-GCCCTCTCTCTCCTCTTGCT-3’;
Ccl4-R: 5’-CTGGTCTCATAGTAATCCATC-3’;
Ccl5-F: 5’-GCCCACGTCAAGGAGTATTTCTA-3’;
Ccl5-R: 5’-ACACACTTGGCGGTTCCTTC-3’;
Cxcl9-F: 5’-GGAACCCTAGTGATAAGGAATGCA-3’;
Cxcl9-R: 5’-TGAGGTCTTTGAGGGATTTGTAGTG-3’;
Cxcl10-F: 5’-GTCAGGTTGCCTCTGTCTCA-3’;
Cxcl10-R: 5’-TCAGGGAAGAGTCTGGAAAG-3’;
GAPDH-F: 5’-ATCAAGAAGGTGGTGAAGCA-3’;
GAPDH-R: 5’-AGACAACCTGGTCCTCAGTGT-3’
Unilateral intradermal tumor implantation and intratumoral injection of viruses
B16-F10 melanoma cells (1×105 cells in a volume of 50 µL PBS) were implanted intradermally into the shaved skin on the right flanks of WT C57BL/6J or Batf3-/- mice. Seven to eight days postimplantation, at 3 mm, tumors were injected with PBS, live OV-GM (2×107 pfu), or with equivalent amounts of heat-iOV-GM when the mice were under anesthesia. Viruses were injected two times a week, mouse survival monitored, and tumor measured two times a week and with tumor volumes calculated using the following formula: L (length) × W (width) × H (height)/2. Mice were euthanized for signs of distress or when tumor diameters reached 10 mm. Treatments were ended when mice died/euthanized, or tumors completely disappeared.
For combination therapy of large tumors, the first injection started when tumor diameters reached 5 mm. Anti-PD-L1 (200 µg per mouse), or isotype control, were then given intraperitoneally (i.p.), to the mice concurrent with virus treatment, throughout the course of study.
Bilateral tumor implantation model and assessment of the therapeutic efficacy of combination therapy, with IT injection of viruses plus ICB
B16-F10 melanoma cells were implanted intradermally into the left and right flanks of C57BL/6J mice (5×105 into the right flanks and 1×105 into the left flanks). Seven to eight days after tumor implantation, when tumor diameters reached 3 mm in the right flanks, live OV-GM (2×107 pfu), or equivalent amounts of heat-iOV-GM, were then injected IT into right flank tumors. The tumors were injected two times a week, concurrently with i.p. delivery of anti-CTLA-4 (100 µg per mouse) or isotype control antibodies. Tumor sizes were measured, and mouse survival was monitored. Mice were euthanized at signs of distress or when tumor diameters reached 10 mm. Treatments were ended when mice died/euthanized, or tumors completely disappeared.
Bilateral tumor implantation model for A20 murine B-cell lymphoma and evaluation of treatment efficacy of live vvDD versus. heat-ivvDD
A20 B-cell lymphoma cells were implanted intradermally into the left and right flanks of BALB/c mice (5×105 into the right flanks and 1×105 into the left flanks). Eleven days later, when tumor diameters reached 5 mm in the right flanks, live vvDD (4×107 pfu), or equivalent amounts of heat-ivvDD, were injected IT into the larger tumors on the right flanks. The tumors were injected two times a week for a total of four injections. Tumor sizes were then measured, and mouse survival was monitored.
Statistics
Two-tailed unpaired Student’s t-tests were used for comparisons of two groups in the studies. Survival data were analyzed by log-rank (Mantel-Cox) test. The p values deemed significant are indicated in the figures as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. The numbers of animals included in the study are discussed in each figure legend and were strictly limited by the necessary statistical power.
Reagents
The commercial sources for reagents were as follows: Antibodies used for flow cytometry were purchased from eBioscience (Live/Dead eFluor 506, CD45.2 Alexa Fluor 700, CD3 PE-Cy7, CD4 Pacific blue-eFluor 450, CD8 PerCP-efluor710, CD11b APC-eFluor 780, MHC Class I APC, CD40 APC, CD80 APC, CD86 APC), Invitrogen (granzyme B PE-Texas Red), and BD Pharmingen (CD11c-PE-Cy7). Murine anti-GM-CSF antibody was purchased from Thermo Fisher. DNAse I and Liberase TL were purchased from Roche. Recombinant murine GM-CSF protein was purchased from GenScript. Therapeutic anti-CTLA4 (clone 9H10 and 9D9) and anti-PD-L1 (clone 10F.9G2) were purchased from BioXcell.
Results
Generation of oncolytic vaccinia virus expressing murine granulocyte-macrophage colony-stimulating factor (mGM-CSF)
Oncolytic vaccinia viruses deleted of TK- are more attenuated and more tumor-selective than TK+ viruses.44 45 Here, we generated a recombinant TK- oncolytic vaccinia virus expressing mGM-CSF under the control of a vaccinia synthetic early/late promoter (PsE/L) (figure 1A). VACV E3L∆83N virus was used as the parental virus (OV-TK+). E3L∆83N replicates efficiently in B16-F10 murine melanoma cells and human melanoma cell lines SK-MEL39, SK-MEL188, and SK-MEL90 (online supplemental figure 1A–D). Two recombinant viruses with the loss of part of the TK gene and with and without mGM-CSF (OV and OV-GM) were generated and verified by PCR analyses and sequencing (online supplemental figure 2A). The replication capacities of OV-TK+, OV, and OV-GM in murine B16-F10 cells were determined by infecting them at an MOI of 0.01. OV-TK+ replicated efficiently in B16-F10 cells, with viral titers increasing by 20,000-fold, at 72 hpi, compared with the viral titers at 1 hour postinfection (figure 1B). Deletion of the TK gene resulted in threefold decreased viral replication efficiency in B16-F10 melanoma cells, compared with the parental virus. In addition, OV-GM replicated efficiently in murine B16-F10 cells, with a 2800-fold increase of viral titers at 72 hpi (figure 1B).
Figure 1.

Live oncolytic vaccinia virus fails to induce IFN-β, and CCL5 from infected bone marrow-derived dendritic cells (BMDCs), or B16-F10, or MC38 cells. (A) Schematic diagram of homologous recombination between a pCB-mGM-CSF plasmid and E3L∆83N vaccinia viral DNA at the thymidine kinase (TK) locus to generate the recombinant virus E3L∆83N-TK- (OV) and E3L∆83N-TK--mGM-CSF (OV-GM). mGM-CSF was expressed under the control of the vaccinia synthetic early and late promoter (PsE/L). (B) Fold changes of viral titers of recombinant viruses in murine B16-F10 melanoma cells at 72 h post-infection compared with those at 1 h post-infection. B16-F10 melanoma cells were infected with OV-TK+, OV, or OV-GM at an MOI of 0.01. Cells were collected at 1, 24, 48, and 72 h post-infection, and viral yields (log pfu) were determined by titration in BSC40 cells. (C) mGM-CSF expression of live OV-GM or heat-iOV-GM in B16-F10 or SK-MEL31 cells as verified by ELISA. Supernatants were collected at 24 hours post-infection. (D) BMDCs were infected with either live OV or live OV-GM at an MOI of 10, or with an equivalent amount of heat-iOV-GM, and supernatants were collected 22 h post-infection. The levels of secreted IFN-β, CCL5 and CXCL10 in the supernatants were determined by ELISA. (E) Western blot analyses of BMDCs from WT or STINGGt/Gt mice infected with either live OV-GM at an MOI of 10 or with an equivalent amount of heat-iOV-GM. The levels of p-TBK1, TBK1, p-IRF3, IRF3, and STING are shown. GAPDH was used as a loading control. hpi: hours post-infection. (F) Expression levels of DC surface markers, MHCI, CD40, CD86, and CD80, on BMDCs infected with either live OV, live OV-GM, or heat-iOV-GM as determined by FACS. NT: no treatment control. (G) The concentrations of secreted IFN-β and CCL5 in the supernatants of murine B16-F10 melanoma cells infected with either live OV or live OV-GM at an MOI of 10, or with an equivalent amount of heat-iOV-GM for 24 hours. (H) The concentrations of secreted IFN-β and CCL5 in the supernatants of murine MC38 colon adenocarcinoma cells infected with either live OV, live OV-GM, or heat-iOV-GM.
jitc-2021-002569supp001.pdf (3.7MB, pdf)
To test the expression of mGM-CSF from the OV-GM recombinant viruses, we infected B16-F10 murine melanoma cells with OV-GM at an MOI of 10. Western blot analyses showed expression levels of mGM-CSF, in both the cell lysates and the supernatants (online supplemental figure 2B) at 24 hpi. The bioactivity of the secreted mGM-CSF was tested by culturing murine bone marrow cells (2.5×105) with serial dilutions of supernatants obtained from B16-F10 cells infected with OV-GM (collected at 24 hpi) or with recombinant mGM-CSF protein (20 ng/mL), for 7 days. Total numbers of CD11c+ cells, cultured under different conditions, are shown (online supplemental figure 2C). We found that 1:400 dilution of the supernatants collected from OV-GM-infected B16-F10 cells had similar bioactivity to recombinant mGM-CSF (20 ng/mL) (online supplemental figure 2C). ELISA was used to determine the concentrations of mGM-CSF in the supernatants collected from B16-F10 murine melanoma cells and SK-MEL31 human melanoma cells infected with either live OV, or live OV-GM, at an MOI of 10, or with equivalent amounts of heat-iOV-GM, at 22 hpi. The concentrations of mGM-CSF in the supernatants of B16-F10 and SK-MEL31 cells infected with live OV-GM were determined to be 1400 ng/mL and 1200 ng/mL, respectively (figure 1C). As expected, heat-iOV-GM infection failed to induce mGM-CSF secretion (figure 1C).
Heat-iOV-GM induces innate immunity in BMDCs and tumor cells in contrast to live OV or live OV-GM
We compared the abilities of live OV, live OV-GM, or heat-iOV-GM to induce innate immunity in BMDCs, B16-F10 murine melanoma cells, and MC38 murine colon cancer cells. BMDCs from WT and STINGGt/Gt mice were infected with either live OV or live OV-GM at an MOI of 10, or with equivalent amounts of heat-iOV-GM. Supernatants were collected at 22 hpi, and the concentrations of IFN-β, CCL5, and CXCL10 were determined by ELISA. Whereas live OV or live OV-GM infection failed to induce IFN-βor CXCL10, and only slightly induced CCL5 above background levels, heat-iOV-GM strongly induced IFN-β, CCL5, and CXCL10 in a STING-dependent manner (figure 1D). Western blot analyses showed that infection of BMDCs with live OV-GM triggered only nominal phosphorylation of TBK1 and IRF3, at 4 and 6 hpi, which is dependent on STING. By contrast, infection of BMDCs with heat-iOV-GM strongly induced phosphorylation of TBK1 and IRF3, at 4 and 6 hpi, which is largely dependent on STING (figure 1E). Fluorescence-activated cell sorting (FACS) analyses of BMDCs infected with either live OV, or live OV-GM, or heat-iOV-GM for 14 hours revealed that heat-iOV-GM infection-induced cell surface expression markers of DC maturation, such as MHC class I, CD40, CD86, and CD80, on BMDCs. By contrast, live OV or live OV-GM infection only modestly induced CD86 and CD80, downregulated MHC class I, compared with mock treatment control (NT) (figure 1F). These results indicate that while heat-iOV-GM infection of BMDCs induces innate immune responses, via the STING-mediated cytosolic DNA-sensing pathway, and activates DC maturation, live OV or live OV-GM infection fails to do so.
We also observed that similar findings in murine B16-F10 melanoma and MC38 colon cancer cells infected with either live OV, live OV-GM, or heat-iOV-GM. Heat-iOV-GM infection potently induced IFN-β and CCL5 secretion from B16-F10 (figure 1G) and MC38 (figure 1H) cells, while live OV or live OV-GM infection did not.
To rule out the possibility that cell death induced by virus infection might affect cytokine production, we performed cell apoptosis and viability assays using B16-F10 cells infected by live OV-GM or heat-iOV-GM, at an MOI of 10. Cells were harvested at 6, 24, and 48 hpi and stained for cell apoptotic markers. Even though around 16% of live OV-GM-infected tumor cells were late-apoptotic or dead (ie, Annexin V+7-AAD+) at 48 hpi, at the time we collected samples for cytokine analysis (24 hpi), most of the live-OV-GM-infected tumor cells were still alive or in early-apoptosis (Annexin V+7-AAD-) (online supplemental figure 1D, E). Infection with heat-iOV-GM did not result in increased cell death, compared with no virus control (online supplemental figure 1D, E).
Antitumor effects induced by IT live OV-GM are dependent on Batf3-dependent CD103+ dendritic cells (DCs)
IT heat-iMVA-induced antitumor effects require Batf3-dependent DCs.39 Here, we used a B16-F10 melanoma unilateral implantation model to test whether live OV-GM also requires Batf3-dependent DCs for antitumor effects. Briefly, B16-F10 melanoma cells (5×105 cells) were implanted intradermally into the right flanks of Batf3-/- or WT C57BL/6J mice. Seven days later, we injected live OV-GM (2×107 pfu), or equivalent amounts of heat-iOV-GM, into right flank tumors, two times a week (figure 2A). We found that IT live OV-GM effectively delayed tumor growth, or eradicated tumors in WT mice, resulting in a 64% survival rate (figure 2B, C). By contrast, IT live OV-GM was ineffective in Batf3-/- mice, with a 0% survival rate. These results are almost indistinguishable from the PBS-treated group, with a median survival of 17 days, in both groups (figure 2B, C). IT heat-iOV-GM was highly effective in WT mice, resulting in a 92% survival rate, but its efficacy was abrogated in Batf3-/- mice, to 0% survival. However, the median survival extended from 17 days in the PBS-treated WT mice to 25 days in the heat-iOV-GM-treated Batf3-/- mice (figure 2B, C). These results are similar to what we reported previously for heat-iMVA.39 These findings indicate that the antitumor effects of oncolytic DNA virus in a unilateral tumor implantation model require Batf3-dependent CD103+ DCs, but not viral replication and oncolysis itself. Thus, IT heat-iOV-GM was more effective than live OV-GM in eradicating injected tumors, likely due to its enhanced ability to induce DC activation and the induction of type I IFN, proinflammatory cytokines, and chemokines, in both DCs and tumor cells.39 Both heat-iOV-GM and heat-iMVA failed to express viral inhibitory proteins that antagonize innate immune sensing mechanisms. We expect that these two inactivated viruses behave similarly, in that: (i) they enter the tumor, stromal, and immune cells in the injected tumors, and (ii) viral DNAs gain access to the cytoplasm of infected cells to trigger potent innate immune responses, partly through the activation of the cytosolic DNA-sensing pathway. Therefore, IT heat-iOV-GM alters the tumor immunosuppressive microenvironment and enhances tumor antigen presentation by the CD103+ DCs.
Figure 2.
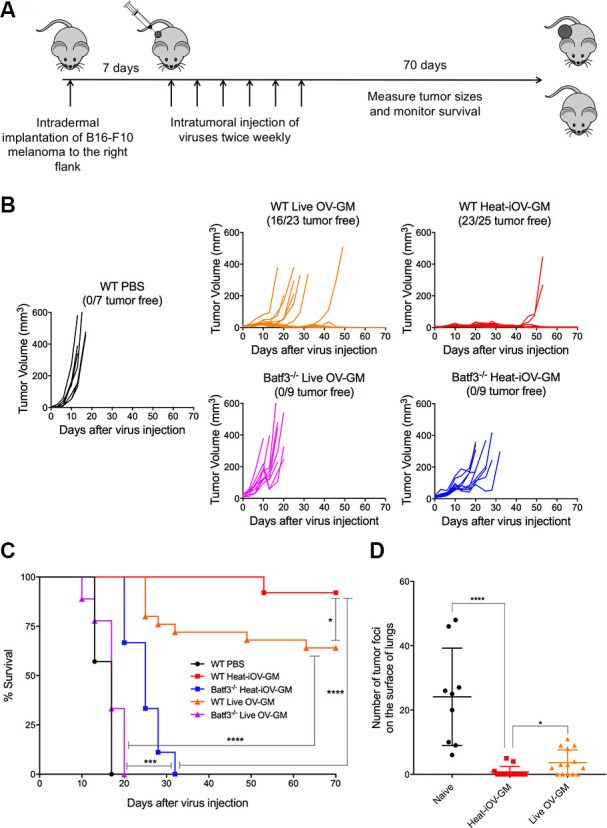
Batf3-dependent CD103+ dendritic cells play an important role in the anti-tumor effects of IT live OV-GM and heat-iOV-GM. (A) Tumor implantation and treatment plan in a unilateral B16-F10 intradermal implantation tumor model. (B) Tumor volumes of injected tumors in WT mice treated with either live OV-GM (n=23), heat-iOV-GM (n=25), or PBS control (n=7), or Batf3-/- mice treated with live OV-GM (n=9) or heat-iOV-GM (n=9) over days post-treatment. (C) Kaplan-Meier survival curve of WT and Batf3-/- mice treated with PBS, live OV-GM, or heat-iOV-GM. (*P < 0.05; ***P < 0.001; ****P < 0.0001). (D) The number of tumor foci on the surface of lungs collected at 3 weeks from either naïve (n=9), or heat-iOV-GM-treated (n=13), or live OV-GM-treated mice (n=14), after intravenous delivery of 1 x 105 B16-F10 cells (*P < 0.05; ****P < 0.0001, t test).
IT heat-iOV-GM is more effective in generating long-lasting memory responses against tumor rechallenge in a different organ system compared with IT live OV-GM
IT heat-iMVA-treatment of murine B16-F10 tumors generates potent systemic antitumor immunity, resulting in the rejection of tumor rechallenge through the intravenous route.39 Here, we compared the efficacy of IT heat-iOV-GM vs IT live OV-GM in generating systemic antitumor memory responses. Intravenous injection of B16-F10 melanoma cells (1×105 cells per mouse) into the surviving mice that were treated previously either with IT heat-iOV-GM or live OV-GM was performed at 8 weeks after the original tumors were eradicated. Mice were euthanized 3 weeks after rechallenge and the lung surfaces were then evaluated under a dissecting microscope for tumor foci. Whereas naïve mice developed an average of 24 tumor foci per lung surface, 5 of 14 live OV-GM-treated mice failed to develop tumors (with an average of five tumor foci on each of the nine mice), and 10 out of 13 heat-iOV-GM mice rejected tumor challenges (with an average of three tumor foci on each of the three mice) (figure 2D). These results indicate that IT heat-iOV-GM generated stronger systemic antitumor long-lasting memory immune responses than IT live OV-GM.
IT heat-iOV-GM induces higher levels of activated CD8+ and CD4+ T cells in the non-injected distant tumors compared with IT live OV-GM
To understand why IT heat-iOV-GM is more effective than live OV-GM in generating antitumor effects, especially in the non-injected distant tumors, we investigated the immune cell infiltrates in both the injected and non-injected tumors in IT heat-iOV-GM or live OV-GM-treated mice. We intradermally implanted 2.5×105 B16-F10 melanoma cells into the left flanks and 5×105 B16-F10 melanoma cells into the right flanks of the mice. Seven days post-tumor implantation, we injected 2×107 pfu of OV, OV-GM, heat-iOV-GM, or PBS into the larger tumors in the right flanks. The injection was repeated 3 days later. Both the injected and non-injected tumors were harvested, and cell suspensions were generated (figure 3A). We analyzed the live immune cell infiltrates in the tumors by FACS. IT live OV-GM generated higher percentages of Granzyme B+ CD8+ T cells compared with IT live OV in the distant non-injected tumors (78% in the OV-GM group compared with 56% in the OV group and 54% in the PBS mock-treatment group), although both viruses were highly efficient in generating Granzyme B+ CD8+ T cells in the injected tumors (figure 3B, C). In addition, IT live OV-GM generated higher percentages of Granzyme B+ CD4+ T cells compared with IT live OV in the distant non-injected tumors (31% in the OV-GM group compared with 16% in the OV group and 13% in the PBS mock-treatment group) (figure 3D, E). In the injected tumors, IT live OV-GM also generated higher percentages of Granzyme B+ CD4+ T cells compared with IT live OV (96% in the OV-GM group vs 79% in the OV group and 11% in the PBS mock-treatment group) (figure 3D, E). These results indicate that the expression and secretion of GM-CSF from OV-GM-infected tumor cells have an immune adjuvant effect. However, IT heat-iOV-GM induced higher percentages of Granzyme B+ CD8+ T cells and Granzyme B+ CD4+ T cells compared with live OV-GM or live OV in the distant non-injected tumors (94% Granzyme B+ CD8+ T cells and 62% Granzyme B+ CD4+ T cells in the heat-iOV-GM group compared with 78% Granzyme B+ CD8+ T cells and 31% Granzyme B+ CD4+ T cells in the live OV-GM group) (figure 3B–E).
Figure 3.

Intratumoral injection of heat-iOV-GM induces higher levels of activated CD8+ and CD4+ T cells in the non-injected distant tumors. (A) B16-F10 melanoma cells were intradermally implanted into the left and right flanks of mice (2.5 x 105 and 5 x 105 cells, respectively). PBS, OV, live OV-GM, or heat-iOV-GM (2 x 107 pfu) were injected IT into the right-side tumors on day 8 and day 11 after tumor implantation. Tumors were harvested 3 days post last virus injection and were analyzed for immune cell infiltration by FACS. (B) Representative flow cytometry plot of CD8+ T cells expressing Granzyme B in the non-injected or injected tumors from mice treated with PBS, OV, live OV-GM, or heat-iOV-GM. (C) Percentages of CD8+ T cells expressing Granzyme B within non-injected and injected tumors. (D) Representative flow cytometry plot of CD4+ T cells expressing Granzyme B in the non-injected and injected tumors from mice treated with PBS, OV, live OV-GM, or heat-iOV-GM. (E) Percentages of CD4+ T cells expressing Granzyme B within non-injected and injected tumors (n=5, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001) (F-I) CD8+ T cells from splenocytes of mice treated with different viruses were analyzed for anti-tumor interferon-γ (IFN-γ+) T cells using ELISPOT assay. (F) IFN-γ+ spots per 250,000 purified CD8+ T cells from the spleens of the mice treated with IT PBS, OV, live OV-GM, or heat-iOV-GM (n=5, *P < 0.05; **P < 0.01). (G) Representative images from an ELISPOT assay from (F). (H) IFN-γ+ spots per 250,000 purified CD8+ T cells from WT, Batf3-/-, or STINGGt/Gt mice treated with IT heat-iOV-GM. (I) ELISPOT images from pooled CD8+ T cells of WT, Batf3-/-, or STINGGt/Gt mice treated with IT heat-iOV-GM from (H). (n=3, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
IT heat-iOV-GM induces higher numbers of antitumor CD8+ T cells in the spleens of treated tumor-bearing mice compared with IT live OV-GM
To test whether IT heat-iOV-GM is more effective in generating systemic antitumor immunity compared with IT live OV-GM, we analyzed tumor-specific CD8+ T cells in the spleens of tumor-bearing mice treated with either OV, live OV-GM, heat-iOV-GM, or PBS control, as described above in a murine B16-F10 bilateral tumor implantation model using an ELISPOT assay. Briefly, CD8+ T cells were isolated from splenocytes and 2.5×105 cells were cultured overnight at 37°C in an anti-IFN-γ-coated BD ELISPOT microwells plate. CD8+ T cells were stimulated by γ-irradiated B16-F10 cells, and cytokine secretion was detected by an anti-IFN-γ antibody. Whereas CD8+ T cells from PBS or OV-treated tumor-bearing mice barely reacted to B16-F10 cells, CD8+ T cells from live OV-GM-treated mice had some reactivity to B16-F10 cells (figure 3F, G). By contrast, heat-iOV-GM-treated mice showed much higher numbers of IFN-γ+ spots compared with OV, live OV-GM, or PBS-treated mice, with an average of 126 IFN-γ+ spots in the heat-iOV-GM group, vs 16 IFN-γ+ spots in the live OV-GM group, vs 4 IFN-γ+ spots in the OV or PBS group (figure 3F, G). Similar experiments were performed in an MC38 murine colon cancer model and we found that IT heat-iOV-GM generated higher numbers of IFN-γ+ spots compared with live OV-GM-treated mice (online supplemental figure 3A, B). Taken together, these results indicate that IT heat-iOV-GM is more potent than live OV-GM in generating tumor-specific activated CD8+ and CD4+ T cells, which are then recruited to inflamed, non-injected distant tumors.
Batf3-dependent CD103+ DCs and the STING-mediated cytosolic DNA-sensing pathway are required for the induction of tumor-specific CD8+ T cells in the spleens of IT heat-iOV-GM-treated mice.
We have previously shown that Batf3-dependent CD103+ DCs are critical for the generation of antitumor CD8+ T cells in the tumor-draining lymph nodes (TDLNs) and for the recruitment of CD8+ and CD4+ T cells into injected and non-injected distant tumors in response to IT heat-iMVA.39 The STING pathway plays an important role in this process.39 Here, we tested whether Batf3-dependent CD103+ DCs and STING are involved in the generation of tumor-specific CD8+ T cells in the spleens. We found that IT heat-iOV-GM resulted in higher numbers of IFN-γ+ spots in WT mice compared with STINGGt/Gt mice, with an average of 80 IFN-γ+ spots in the heat-iOV-GM-treated WT mice vs 47 IFN-γ+ spots in the heat-iOV-GM-treated STINGGt/Gt mice (figure 3H, I). As expected, IT heat-iOV-GM failed to generate IFN-γ+ spots in the Batf3-/- mice (figure 3H, I). These results further support that IT heat-iOV-GM activates the STING-mediated cytosolic DNA-sensing pathway in Batf3-dependent CD103+ DCs to generate systemic antitumor immunity.
IT heat-iOV-GM induces stronger innate immune responses in the injected tumors than live OV-GM
We hypothesized that IT heat-iOV-GM leads to stronger induction of innate immunity in the infected tumor cells and tumor-infiltrating immune cells, compared with IT live OV-GM. To test this hypothesis, we intradermally implanted B16-F10 melanoma cells into the right flanks of C57BL/6J mice; once the tumors were 3–4 mm in diameter, they were injected with either 2×107 pfu of live OV-GM, or equivalent amounts of heat-iOV-GM. PBS was used as a control. Tumors were harvested 1 day postinjection and mRNAs were extracted. Quantitative real-time PCR analyses showed that whereas IT live OV-GM modestly induced innate immune responses in the injected tumors compared with IT PBS control, IT heat-iOV-GM even more strongly induced the expression of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 genes compared with IT live OV-GM (online supplemental figure 4A–F).
IT heat-iOV-GM induces higher levels of IFN and proinflammatory cytokines and chemokines in distant non-injected tumors compared with live OV or live OV-GM
Here, we compared the innate immunity generated in non-injected distant tumors in mice treated with either IT heat-iOV-GM, live OV-GM, or live OV. Briefly, B16-F10 melanoma cells were implanted intradermally into the left and right flanks of C57BL/6J mice (2.5×105 and 5×105 cells, respectively). Seven days later, IT injection of 2×107 pfu of OV, OV-GM, heat-iOV-GM, or PBS was carried out into the larger tumors in the right flanks. The injections were repeated 3 days later. The non-injected tumors on the left flank were harvested 2 days after the last injection, followed by tumor mRNAs extraction (figure 4A). Quantitative real-time PCR analyses showed that IT heat-iOV-GM resulted in the induction of higher levels of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 gene expression in the non-injected distant tumors compared with those mice treated with either live OV-GM, live OV, or PBS control (figure 4B). These results indicate that IT heat-iOV-GM induces stronger innate immune activation at the non-injected distant tumors compared with IT live OV-GM. Whereas IT live OV is not effective in inducing innate immunity at the non-injected distant tumors compared with PBS mock-treatment control, IT live OV-GM induces slightly higher innate immune responses in the distant non-injected tumors compared with IT live OV (figure 4B).
Figure 4.

IT heat-iOV-GM induces higher levels of IFN and proinflammatory cytokines and chemokines in distant non-injected tumors than live OV-GM. (A) Tumor implantation and treatment schedule in a bilateral intradermal tumor implantation model. (B) B16-F10 melanoma cells were implanted intradermally into the left and right flanks of C57BL/6J mice. After the tumors were established, the larger tumors on the right flank were injected with either PBS, live OV, live OV-GM, or heat-iOV-GM twice weekly. The non-injected tumors were harvested 2 days after the second injection and RNAs were extracted. Quantitative real-time PCR analyses of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 gene expression in non-injected B16-F10 tumors isolated from mice treated with either PBS, OV, live OV-GM, or heat-iOV-GM (n=4-5, *P < 0.05, **P < 0.01, t-test). (C) Expression of IFN, proinflammatory cytokines, and chemokines in non-injected B16-F10 tumors from WT, Batf3-/-, or STINGGt/Gt mice treated with heat-iOV-GM were analyzed. Relative expression of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 genes was measured by quantitative real-time RT-PCR and was normalized to the expression of GAPDH. Each panel shows the fold changes of the mRNA levels in non-injected tumors from WT, Batf3-/-, or STINGGt/GT mice treated with heat-iOV-GM, compared with those from WT mice treated with PBS (n=4, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). (D) Schematic diagram of a bilateral intradermal tumor implantation model with CD4 and/or CD8 depletion. (E) Relative expression levels of Ifnb, Il6, Ccl5, Cxcl10 in non-injected tumors from each treatment groups were measured by quantitative real-time RT-PCR and were normalized to the expression of GAPDH (n=4, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
To make sure our observation was not limited to B16-F10 melanoma, we performed similar experiments in an MC38 murine colon cancer model. We confirmed that IT heat-iOV-GM induced higher levels of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 gene expression in both injected tumors (harvested at 1 day post-first injection) and non-injected tumors (harvested 2 days post-second injection) compared with IT live OV-GM (online supplemental figure 5A–F).
STING and Batf3-dependent CD103+ DCs contribute to the induction of IFN and proinflammatory cytokines and chemokines by IT heat-iOV-GM in distant non-injected tumors
We hypothesized that the cytosolic DNA-sensing pathway in the Batf3-dependent CD103+ DCs might be important for inducing type I IFN and proinflammatory cytokines and chemokines in response to tumor DNA released from the dying tumor cells. To test that, we intradermally implanted B16-F10 melanoma cells into the left and right flanks of Batf3-/-, STINGGt/Gt, and WT C57BL/6J mice (2.5×105 and 5×105 cells, respectively). Seven days after tumor implantation, heat-iOV-GM (an equivalent amount of 2×107 pfu of the live virus) or PBS was injected into the larger tumors in the mouse right flanks, with a total of two injections, 3 days apart. The non-injected tumors from the left flanks of Batf3-/-, STINGGt/Gt, and WT mice were harvested at day 3 post-last injection (figure 4A). Quantitative real-time PCR analyses showed that the induction of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 gene expression in the non-injected distant tumors of WT mice treated with IT heat-iOV-GM was reduced in STINGGt/Gt mice and abolished in Batf3-/- mice (figure 4C). These results indicate that STING and Batf3-dependent CD103+ DCs play important roles in the induction of IFN and proinflammatory cytokines and chemokines by IT heat-iOV-GM in distant non-injected tumors.
CD8+ T cells are required for the induction of innate immune responses in the distant non-injected tumors
We previously showed that CD8+ T cells are required for heat-iMVA-induced antitumor effects,39 whereas CD4+ T cells are important for the generation of antitumor memory responses. To determine the relative contribution of CD8+ and CD4+ T cells in mediating innate-immune activation in the non-injected tumors, we depleted either CD8+ or CD4+ T cells individually, or together, by i.p. administering anti-CD8 and/or anti-CD4 antibodies 1 day prior to IT injection of heat-iOV-GM. Two days after the second injection, we isolated the non-injected tumor mRNAs for RT-PCR analyses (figure 4D). Flow cytometry analyses confirmed that IT CD4+ and CD8+ T cells were depleted (online supplemental figure 6). We found that depleting CD8+ T cells alone abolished Ifnb, Il6, Ccl5, and Cxcl10 gene expression in non-injected tumors, whereas depleting CD4+ T cells had moderate reduction (figure 4E). These results indicate that cytotoxic CD8+ T cells induced after heat-iOV-GM injection elicit tumor killing in the non-injected tumors and resulting in the induction of innate immunity.
IT heat-iOV-GM generated stronger therapeutic efficacy than IT live OV-GM in a B16-F10 bilateral tumor implantation model in the presence or absence of anti-CTLA-4 antibody
We showed in an earlier publication that treatment with either anti-CTLA-4 or anti-PD-L1, in B16-F10 melanoma, has limited therapeutic effects,39 as observed by many other groups as well.16 46 47 Here, we investigated the therapeutic efficacy induced by heat-iOV-GM in comparison with live OV-GM in a bilateral tumor implantation model, and test whether combining it with systemic delivery of ICB could further improve the treatment outcome. We implanted B16-F10 cells intradermally into the flanks of C57BL/6J mice, with 5×105 into the right flanks and 1×105 into the left flanks, and started virus treatment 7–8 days later, when tumor diameters reached 3 mm in the right flanks. IT injection of either PBS, live OV-GM, or heat-iOV-GM were given to the tumors on the right flanks two times a week, combined with i.p. delivery of either anti-CTLA-4 antibody or isotype control. Tumors in the left flanks were not injected with the virus. We then monitored tumor growth and mouse survival (figure 5A). Without anti-CTLA-4 antibody, heat-iOV-GM-treated mice showed improved tumor growth control and survival compared with those treated with live OV-GM, extending median survival from 16.5 days in live OV-GM-treated group to 28 days in heat-iOV-GM treated group (figure 5B–D). Combination with ICB further enhanced the abscopal antitumor effect induced by heat-iOV-GM. The heat-iOV-GM and anti-CTLA4 combination treatment resulted in delayed tumor growth and a higher rate of tumor regression in the distant tumor compared with the live OV-GM and anti-CTLA-4 combination therapy (figure 5D). The cure rate in the heat-iOV-GM plus anti-CTLA4 group was 80%, which is higher than the 40% cure rate in the live OV-GM plus anti-CTLA-4 treatment group (figure 5B).
Figure 5.

Combination with immune checkpoint blockade further enhances the anti-tumor effects induced by heat-iOV-GM on non-injected tumors and large established tumors. (A) WT C57BL/6J mice were intradermally implanted with B16-F10 tumors into their left and right flanks. Starting from day 7 post-implantation, tumor-bearing mice were treated twice weekly with IT injection of live OV-GM or heat-iOV-GM in the combination of i.p. delivery of isotype control or anti-CTLA-4 antibody (n=10 for all groups). PBS was used as a control. Tumor volumes and mouse survival was monitored throughout the course of study. (B) Kaplan-Meier survival curves of WT mice treated with PBS, live OV-GM, or heat-iOV-GM with or without anti-CTLA-4 antibody (*P < 0.05; ***P < 0.001; ****P < 0.0001). (C) Tumor volumes of injected tumors over days of treatment. (D) Tumor volumes of non-injected tumors over days of treatment. (E) WT C57BL/6J mice were intradermally implanted with B16-F10 melanoma cells in their right flanks. When tumor diameters reached 5 mm, intratumoral injection of live OV-GM, or heat-iOV-GM combined with i.p. delivery of anti-PD-L1 or isotype control was initiated and continued twice a week. PBS was used as a control. Tumor growth and mouse survival were monitored throughout the course of the study. (F) Tumor volumes of injected tumors over days of treatment. (G) Kaplan-Meier survival curve of WT mice treated with PBS, live OV-GM or heat-iOV-GM with or without anti-PD-L1 (n=9 or 10; *P < 0.05; ***P < 0.001; ****P < 0.0001). (H) PD-L1 expression on B16-F10 cells infected by live OV-GM or heat-iOV-GM was measured by flow cytometry. Cells were infected at an MOI of 10 for 24 hours before staining.
Heat-iOV-GM and immune checkpoint blockade combination therapy improves therapeutic efficacy in a large established murine B16-F10 tumor model
We investigated the therapeutic effect of the combination therapy in an aggressive large tumor model. We implanted the B16-F10 cells into the right flanks of WT C57BL/6J mice and started virus treatment at a later time point when the tumor diameters reached 5 mm (figure 5E). While neither the two viruses alone nor live OV-GM in combination with anti-PD-L1 eradicated the injected tumors, the heat-iOV-GM combined with anti-PD-L1 generated strong antitumor effects leading to tumor regression and elimination in 50% of treated mice (figure 5F, G). There was an extension of median survival from 14 days in the live OV-GM plus anti-PD-L1-treated mice to 32 days in the heat-iOV-GM plus anti-PD-L1-treated mice (figure 5G). Consistent with these in vivo findings, heat-iOV-GM infection of B16-F10 melanoma cells in vitro induces PD-L1 expression, whereas live OV-GM infection slightly reduces PD-L1 expression (figure 5H). These results collectively support that heat-iOV-GM is more immunogenic and generates stronger antitumor effects when combined with ICB compared with live OV-GM plus ICB in both bilateral tumor implantation and large established aggressive tumor models.
IT heat-ivvDD generates stronger antitumor effects compared with live oncolytic vaccinia vvDD in a murine A20 B-cell lymphoma bilateral tumor implantation model
vvDD is a well-studied oncolytic vaccinia virus with deletion of the TK and VGF genes.42–44 It is highly attenuated but generates strong antitumor effects in nude mice.44 We first generated vvDD expressing mCherry from the parental vSC20 virus (with deletion of both copies of the VGF gene) through homologous recombination at the TK locus. We then compared the induction of innate immune responses in BMDCs infected with either live vvDD or heat-inactivated vvDD (heat-ivvDD). Heat-ivvDD induced much higher levels of Ifnb and Cxcl10 gene expression, and IFN-β and CXCL10 protein secretion in WT BMDCs compared with live vvDD (figure 6A, B). The induction effects of heat-ivvDD were largely diminished in STING-deficient BMDCs (figure 6A, B). Western blot analyses demonstrated that heat-ivvDD induced higher levels of phosphorylation of TBK1, STING, and IRF3 compared with live vvDD (figure 6C). As expected, heat-ivvDD-induced IRF3 phosphorylation is dependent on STING (figure 6C). Here, we used an A20 murine B-cell lymphoma bilateral tumor implantation model to assess the antitumor effects of live vvDD versus heat-ivvDD in both injected and non-injected tumors. The initial tumor volumes at the injected sites for the PBS, live vvDD, and heat-ivvDD were 51, 31, and 45 mm3, respectively, and the tumor volumes at the non-injected sites were 4, 6, and 6.2 mm3, respectively (figure 6D). IT heat-ivvDD eradicated nine out of nine injected tumors, whereas IT live vvDD cured five out of nine injected tumors (figure 6F). IT heat-ivvDD also generated stronger abscopal effects on the non-injected tumors compared with live vvDD, with growth in only one of nine non-injected tumors in the heat-ivvDD group and five of nine non-injected tumors in the live vvDD group (figure 6G). IT live vvDD extended median survival from 11 days in the PBS mock treatment group to 41 days post-IT viral treatment (p<0.001). IT Heat-ivvDD conferred 89% survival while IT live vvDD rendered 44% survival (p<0.05) (figure 6E). Taken together, these findings show that heat-ivvDD is more immunogenic and more effective in eradicating tumors than live vvDD.
Figure 6.

Heat-ivvDD activates the cGAS/STING pathway in BMDCs and induces stronger antitumor immune responses in murine murine A20 B-cell lymphoma bilateral implantation model compared with live vvDD. (A, B) WT or STINGGt/Gt BMDCs were infected with either live vvDD or heat-ivvDD at an MOI of 10. Cells were harvested at 16 hpi and prepared for RT-PCR analysis for Ifnb and Cxcl10 gene expressions (A). Supernatants were collected at the same time for ELISA assays of IFNβ and CXCL10 protein production (B). (C) Western blot assay of WT or STINGGt/Gt BMDCs infected by the indicated viruses at an MOI of 10. Cells were harvested at different time points after infection. The levels of p-TBK1, TBK1, p-IRF3, IRF3, p-STING, STING, and cGAS are shown. GAPDH was used as a loading control. (D-G) A20 tumor cells were intradermally implanted into both flanks of WT C57BL/6J mice. When tumor diameters reached 5 mm, intratumoral injection of live vvDD or heat-ivvDD was initiated and continued twice a week for a total of 4 injections. PBS was used as a control. Tumor growth and mouse survival were monitored throughout the course of the study. (D) Initial tumor volumes before the first injection. (E) Kaplan-Meier survival curves of WT mice treated with PBS, live vvDD or heat-ivvDD (n=9 or 10; *P < 0.05; ***P < 0.001; ****P < 0.0001). (F) Tumor volumes of injected tumors over days of treatment. (G) Tumor volumes of non-injected tumors over days of treatment.
Discussion
Although IT delivery of the OV talimogene laherparepvec (T-VEC) has been approved for the treatment of advanced melanoma as a single agent and IT delivery of T-VEC in combining with systemic administration of ICB are being investigated in clinical trials for melanoma and other cancers, our understanding of the contribution of viral replication and oncolysis to the generation of antitumor immunity by oncolytic DNA viruses remains limited.
In this study, we designed an oncolytic vaccinia virus, E3L∆83N-TK--mGM-CSF (OV-GM), similar to JX594, in which the TK locus was deleted, and an mGM-CSF expression cassette was inserted. JX594 is a leading oncolytic vaccinia virus that has been tested in many clinical trials for various cancers.11–15 We compared the antitumor immunity of IT live OV-GM versus IT heat-iOV-GM in both unilateral and bilateral B16-F10 melanoma models and found that IT heat-iOV-GM was more effective than IT live OV-GM in eradicating or delaying the growth of both injected and non-injected distant tumors in both models. In the bilateral tumor implantation model, IT heat-iOV-GM induced higher expression of Ifnb, Il6, Ccl4, Ccl5, Cxcl9, and Cxcl10 genes and higher numbers of activated CD4+ and CD8+ T cells in the non-injected distant tumors, compared with IT live OV-GM. These results were confirmed in two additional murine cancer models, demonstrating that our findings are not limited to one tumor type or microenvironment.
We have previously reported that, unlike live vaccinia, heat-inactivated vaccinia (55°C for 1 hour) induces type I IFN in murine and human plasmacytoid DCs (pDCs) and viral entry into host cells is required for the induction effects.48 In murine pDCs, the TLR7/MyD88 pathway is required for the induction of type I IFN by heat-inactivated vaccinia.48 We have also shown that heat-iMVA infection of GM-CSF-cultured BMDCs induces type I IFN via the cGAS/STING-mediated DNA-sensing pathway.39 Viral proteins, including inhibitors of the cGAS/STING pathway, are not produced in heat-iMVA-infected cells.39 As expected, heat-iOV-GM infection also activates STING-dependent IFN induction in BMDCs in this study.
The host type I IFN pathway plays an important role in antitumor immunity.49–51 Type I IFN signatures correlate with T cell markers in human melanoma metastases.50 Preclinical studies have shown that IFNAR signaling on dendritic cells, specifically CD103+/CD8α+ DCs, can affect antigen cross-presentation and the generation of antitumor immunity.50 51 Tumor-infiltrating CD103+ DCs are critical for the generation of antitumor immunity, including stimulating naïve and activated CD8+ T cells through antigen cross-presentation and the recruitment of antigen-specific T cells into TME.52 53 Our in vitro and in vivo results support our hypothesis that the inferiority of live OV or OV-GM stems from its expression of inhibitory viral genes, which leads to the dampening of type I IFN and proinflammatory cytokine and chemokine production in infected BMDCS and tumor cells. By contrast, heat-iOV-GM failed to express those inhibitory genes.39 48 Similar to what we observed with heat-iMVA, infection of heat-iOV-GM in BMDCs and tumor cells induced type I IFN and proinflammatory cytokine and chemokine production, while live OV or OV-GM infection of BMDCs, or B16-F10, or MC38 tumor cells, failed to induce those innate immune mediators. Heat-iOV-GM infection of BMDCs induced DC maturation, whereas live oncolytic vaccinia infection did not.
To strengthen our conclusion that the in vivo antitumor effects of vaccinia virus correlate with its potency of activation of the cGAS/STING pathway in immune cells, we constructed vvDD expressing mCherry and compared its innate immune activation with heat-ivvDD in BMDCs, as well as its antitumor effects in a syngeneic immune-competent murine tumor model. vvDD is a well-accepted oncolytic vaccinia with an excellent safety profile.41–43 vvDD only weakly induced the cGAS/STING-mediated type I IFN pathway, whereas heat-ivvDD robustly activated innate immune responses from BMDCs. IT heat-ivvDD also resulted in better control of the growth of both the injected and non-injected tumors compared with live vvDD.
Here, we propose the following model for induction of innate immunity by heat-inactivated vaccinia in the non-injected distant tumors and the immunological mechanisms underlying the superiority of IT heat-inactivated vaccinia over live oncolytic vaccinia (figure 7). First, compared with live oncolytic vaccinia, heat-inactivated vaccinia infection leads to stronger induction of type I IFN and proinflammatory cytokines and chemokines in the injected tumors via the cGAS/STING-dependent mechanism, which results in local activation of tumor-infiltrating CD8+ and CD4+ T cells, stronger activation of CD103+ DCs, and enhanced tumor-antigen presentation in the TDLNs and spleens. Second, more activated tumor-specific Granzyme B+ CD8+ and CD4+ T cells are then recruited to the distant non-injected tumors to engage in tumor cell killing in heat-inactivated vaccinia-treated mice compared with live oncolytic vaccinia-treated mice. Third, the cGAS/STING-dependent cytosolic-sensing of tumor DNA from dying tumor cells leads to the induction of innate immunity in the non-injected tumors. Finally, heat-inactivated vaccinia treatment generated stronger CD8+ T cells-mediated tumor cell killing and higher levels of Ifnb, Il6, Ccl4, Ccl5, Cxcl9 and Cxcl10 gene expression in the non-injected tumors compared with live oncolytic vaccinia. Based on our findings, we propose that evaluations of both innate and adaptive immunity induced by IT oncolytic viral immunotherapy at non-injected tumors should be included as potential biomarkers for comparing potency and efficacy of various oncolytic constructs in preclinical and clinical studies.
Figure 7.

Working model of heat-inactivated vaccinia as a stronger inducer of anti-tumor innate immunity especially in distant tumors compared with live OV-GM. (A) Schematic of induction of innate immunity by IT delivery of heat-inactivated vaccinia vs. live oncolytic vaccinia in both injected and non-injected distant tumors (created with biorender.com). (B) Comparison of immune activation by heat-inactivated vaccinia vs. live oncolytic vaccinia. IT delivery of heat-inactivated vaccinia induces (i) higher levels of type I IFN than live oncolytic vaccinia due to activation of the cGAS/STING-mediated cytosolic DNA-sensing pathway; (ii) stronger T cell priming in TDLNs and spleen; and (iii) more activated T cells, which then migrate to the distant tumors resulting in enhanced abscopal tumor cell killing and eventually tumor regression through the induction of a stronger innate immunity.
Our results support that the cGAS/STING-dependent cytosolic-sensing tumor DNA from dying tumor cells in the non-injected tumors leads to the induction of innate immunity. In the absence of STING, IT heat-iOV-GM-induced innate immunity in the non-injected tumors was reduced compared with WT controls, supporting a role for STING in this process. Furthermore, in the absence of Batf3-dependent CD103+ DCs, IT heat-iOV-GM-induced innate immunity in the non-injected tumors was abolished. This is consistent with our previous report that in the absence of CD103+ DCs, both injected and non-injected tumors failed to recruit antitumor CD4+ and CD8+ T cells in response to IT heat-iMVA treatment.39 Using ELISPOT assay, we showed that Batf3-dependent DCs are crucial for the generation of antitumor CD8+ T cells in the spleens of mice after IT heat-iOV-GM treatment. By contrast, IT live OV-GM has limited potency to induce innate immunity at the non-injected distant tumors, which correlates with the lower levels of activated CD8+ T cells in the non-injected tumors and spleens compared with those treated with IT heat-iOV-GM. Furthermore, depletion of CD8+ T cells from the circulation and tumors abolished IT heat-iOV-GM-induced innate immunity in the non-injected tumors.
Batf3 is a transcription factor that is critical for the development of CD103+/CD8α+ lineage DCs, which play an important role in cross-presentation of viral and tumor antigens.54 55 We were surprised by our finding that IT live OV-GM had no antitumor activity in the Batf3-/- mice, whereas IT heat-iOV-GM extended the median survival to 25 days in the Batf3-/- mice compared with 17 days in PBS-treated WT mice. These results suggest that: (i) viral-mediated oncolysis plays a little role (if any) in the Batf3-/- mice, which lack CD103+/CD8α+ DCs; (ii) IT heat-iOV-GM induces limited antitumor activity independent of CD103+ DCs. This could be related to its ability to induce the production of type I IFN and proinflammatory cytokines and chemokines in other myeloid cells such as CD11b+ DCs, pDCs, or tumor-associated macrophages, or inflammatory monocytes, as well as in infected tumors and stromal cells. Further studies to define the contributions of other myeloid cells to heat-iOV-GM-induced antitumor immunity are warranted.
We observed that both IT live OV-GM and IT heat-iOV-GM can generate long-lasting antitumor memory responses through an “in situ vaccination” effect, in which tumor antigens are presented by CD103+ DCs to generate tumor-specific CD4+ and CD8+ T cells in the TDLNs; these cells then return to the circulation, are recruited to non-injected distant tumors, or establish residence in secondary lymphoid organs such as the spleen or lymph nodes or in other tissues such as the skin or the lungs. IT heat-iOV-GM is more potent in inducing long-lasting memory responses compared with IT live OV-GM, as 77% of tumor-bearing mice successfully treated with IT heat-iOV-GM rejected tumor rechallenge through intravenous route, whereas only 36% of tumor-bearing mice successfully treated with IT live OV-GM rejected tumor rechallenge. This has important clinical implications because potential viral-based immunotherapy that generates stronger immunological memory will be more effective in preventing cancer recurrence and prolonging patient survival.
In this study, we found that heat-iOV-GM performs better than live OV-GM when combined with anti-CTLA-4 antibody in a murine B16-F10 bilateral tumor implantation model. The survival advantage of the heat-iOV-GM and anti-CTLA-4 antibody combination is largely due to better control of tumor growth of the non-injected tumors compared with Live-OV-GM plus anti-CTLA-4. This is consistent with the notion that IT heat-iOV-GM generates stronger innate immunity in the non-injected distant tumors compared with live OV-GM, which synergizes with systemic delivery of anti-CTLA-4 antibody. In addition, IT heat-iOV-GM plus anti-PD-L1 antibody is more effective in restraining tumor growth compared with IT live OV-GM plus anti-PD-L1 or IT heat-iOV-GM alone in a large established B16-F10 melanoma model. This is likely due to upregulation of PD-L1 expression in heat-iOV-GM-infected tumor or immune cells, which can be counteracted by anti-PD-L1 antibody. Together with other published studies, our results support the use of combination therapy of IT immunogenic viruses with systemic delivery of ICB to potentiate antitumor effects in both injected and non-injected tumors.16 39 56
Acknowledgments
We thank Dr Stewart Shuman for critical review of the manuscript. We thank Dr Katharina Shaw and Dr Curt Balch for editing. E3L∆83N virus was kindly provided by Bertram Jacobs (Arizona State University). vSC20 virus was kindly provided by Bernard Moss (National Institutes of Health).
Footnotes
Contributors: WW and LD designed and performed the experiments, analyzed the data, and prepared the manuscript. SL and PD performed the experiments, analyzed the data, and assisted in manuscript preparation. NY, YW, and RAG assisted in experiments and data interpretation. TM and JW assisted in experimental design, data interpretation, and manuscript preparation.
Funding: This work was supported by the Society of Memorial Sloan Kettering (MSK) research grant (LD), MSK Technology Development Fund (LD), and Parker Institute for Cancer Immunotherapy Career Development Award (LD). This work was supported in part by the Swim Across America (JW, TM), Ludwig Institute for Cancer Research (JW, TM), and National Cancer Institute grants R01 CA56821 (JW). This research was also funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Competing interests: Memorial Sloan Kettering Cancer Center has filed patent applications for the use of inactivated vaccinia as monotherapy or in combination with immune checkpoint blockade for solid tumors. LD, PD, WW, TM, and JW are authors on the patent, which has been licensed to IMVAQ Therapeutics. LD, NY, TM, and JW are cofounders of IMVAQ Therapeutics and hold equities in IMVAQ Therapeutics. TM is a consultant of Immunos Therapeutics and Pfizer. He has research support from Bristol Myers Squibb; Surface Oncology; Kyn Therapeutics; Infinity Pharmaceuticals, Inc.; Peregrine Pharmaceuticals, Inc.; Adaptive Biotechnologies; Leap Therapeutics, Inc.; and Aprea. He has patents on applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neoantigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4. JW is a consultant for Adaptive Biotech, Advaxis, Am-gen, Apricity, Array BioPharma, Ascentage Pharma, Astellas, Bayer, Beigene, Bristol Myers Squibb, Celgene, Chugai, Elucida, Eli Lilly, F Star, Genentech, Imvaq, Janssen, Kleo Pharma, Linnaeus, MedImmune, Merck, Neon Therapeutics, Ono, Polaris Pharma, Polynoma, Psioxus, Puretech, Recepta, Trieza, Sellas Life Sciences, Serametrix, Surface Oncology, and Syndax. Research support: Bristol Myers Squibb, Medimmune, Merck Pharmaceuticals, and Genentech. Equity: Potenza Therapeutics, Tizona Pharmaceuticals, Adaptive Biotechnologies, Elucida, Imvaq, Beigene, Trieza, and Linnaeus. Honorarium: Esanex. Patents: xenogeneic DNA vaccines, alphavirus replicon particles expressing TRP2, MDSC assay, Newcastle disease viruses for cancer therapy, genomic signature to identify responders to ipilimumab in melanoma, engineered vaccinia viruses for cancer immunotherapy, anti-CD40 agonist monoclonal antibody (mAb) fused to monophosphoryl lipid A (MPL) for cancer therapy, CAR+ T cells targeting differentiation antigens as means to treat cancer, anti-PD-1 antibody, anti-CTLA-4 antibodies, and anti-GITR antibodies and methods of use thereof.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not required.
References
- 1.Russell SJ, Barber GN. Oncolytic viruses as Antigen-Agnostic cancer vaccines. Cancer Cell 2018;33:599–605. 10.1016/j.ccell.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol 2018;18:498–513. 10.1038/s41577-018-0014-6 [DOI] [PubMed] [Google Scholar]
- 3.Zamarin D, Wolchok JD. Potentiation of immunomodulatory antibody therapy with oncolytic viruses for treatment of cancer. Mol Ther Oncolytics 2014;1:14004. 10.1038/mto.2014.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu BL, Robinson M, Han Z-Q, et al. Icp34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 2003;10:292–303. 10.1038/sj.gt.3301885 [DOI] [PubMed] [Google Scholar]
- 5.Andtbacka RHI, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015;33:2780–8. 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 6.Harrington KJ, Andtbacka RH, Collichio F, et al. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: subanalysis of the phase III OPTiM trial. Onco Targets Ther 2016;9:7081–93. 10.2147/OTT.S115245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puzanov I, Milhem MM, Minor D, et al. Talimogene Laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol 2016;34:2619–26. 10.1200/JCO.2016.67.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase II study evaluating the efficacy and safety of Talimogene Laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol 2018;36:JCO2017737379. 10.1200/JCO.2017.73.7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017;170:e10:1109–19. 10.1016/j.cell.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moss B. Poxviridae: The viruses and their replication. : Lippincott Williams & Wilkins, 2007. [Google Scholar]
- 11.Park B-H, Hwang T, Liu T-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 2008;9:533–42. 10.1016/S1470-2045(08)70107-4 [DOI] [PubMed] [Google Scholar]
- 12.Liu T-C, Hwang T, Park B-H, et al. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 2008;16:1637–42. 10.1038/mt.2008.143 [DOI] [PubMed] [Google Scholar]
- 13.Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011;477:99–102. 10.1038/nature10358 [DOI] [PubMed] [Google Scholar]
- 14.Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 2012;20:749–58. 10.1038/mt.2011.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013;19:329–36. 10.1038/nm.3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 2014;6:226ra32. 10.1126/scitranslmed.3008095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe 2014;15:260–5. 10.1016/j.chom.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015;14:642–62. 10.1038/nrd4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemay CG, Keller BA, Edge RE, et al. Oncolytic viruses: the best is yet to come. Curr Cancer Drug Targets 2018;18:109–23. 10.2174/1568009617666170206111609 [DOI] [PubMed] [Google Scholar]
- 20.Davola ME, Mossman KL. Oncolytic viruses: how "lytic" must they be for therapeutic efficacy? Oncoimmunology 2019;8:e1581528. 10.1080/2162402X.2019.1596006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutter G, Staib C. Vaccinia vectors as candidate vaccines: the development of modified vaccinia virus Ankara for antigen delivery. Curr Drug Targets Infect Disord 2003;3:263–71. 10.2174/1568005033481123 [DOI] [PubMed] [Google Scholar]
- 22.McCurdy LH, Larkin BD, Martin JE, et al. Modified vaccinia Ankara: potential as an alternative smallpox vaccine. Clin Infect Dis 2004;38:1749–53. 10.1086/421266 [DOI] [PubMed] [Google Scholar]
- 23.Vollmar J, Arndtz N, Eckl KM, et al. Safety and immunogenicity of IMVAMUNE, a promising candidate as a third generation smallpox vaccine. Vaccine 2006;24:2065–70. 10.1016/j.vaccine.2005.11.022 [DOI] [PubMed] [Google Scholar]
- 24.Gómez CE, Nájera JL, Krupa M, et al. The poxvirus vectors MVA and NYVAC as gene delivery systems for vaccination against infectious diseases and cancer. Curr Gene Ther 2008;8:97–120. 10.2174/156652308784049363 [DOI] [PubMed] [Google Scholar]
- 25.Goepfert PA, Elizaga ML, Sato A, et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis 2011;203:610–9. 10.1093/infdis/jiq105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gómez CE, Nájera JL, Krupa M, et al. MVA and NYVAC as vaccines against emergent infectious diseases and cancer. Curr Gene Ther 2011;11:189–217. 10.2174/156652311795684731 [DOI] [PubMed] [Google Scholar]
- 27.Dai P, Wang W, Cao H, et al. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog 2014;10:e1003989. 10.1371/journal.ppat.1003989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa H, Ma Z, Barber GN. Sting regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009;461:788–92. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–91. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013;339:826–30. 10.1126/science.1229963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Shu C, Yi G, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013;39:1019–31. 10.1016/j.immuni.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao P, Ascano M, Wu Y, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013;153:1094–107. 10.1016/j.cell.2013.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao P, Ascano M, Zillinger T, et al. Structure-function analysis of STING activation by c[G(2',5')pA(3',5')p] and targeting by antiviral DMXAA. Cell 2013;154:748–62. 10.1016/j.cell.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013;498:332–7. 10.1038/nature12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates sting. Nature 2013;498:380–4. 10.1038/nature12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diner EJ, Burdette DL, Wilson SC, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human sting. Cell Rep 2013;3:1355–61. 10.1016/j.celrep.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo S-R, Fuertes MB, Corrales L, et al. Sting-Dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830–42. 10.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng L, Liang H, Xu M, et al. Sting-Dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014;41:843–52. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai P, Wang W, Yang N, et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via sting and Batf3-dependent dendritic cells. Sci Immunol 2017;2. 10.1126/sciimmunol.aal1713. [Epub ahead of print: 19 May 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brandt TA, Jacobs BL. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol 2001;75:850–6. 10.1128/JVI.75.2.850-856.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 2001;61:8751–7. [PubMed] [Google Scholar]
- 42.Zeh HJ, Downs-Canner S, McCart JA, et al. First-In-Man study of Western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther 2015;23:202–14. 10.1038/mt.2014.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Downs-Canner S, Guo ZS, Ravindranathan R, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther 2016;24:1492–501. 10.1038/mt.2016.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buller RM, Chakrabarti S, Cooper JA, et al. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J Virol 1988;62:866–74. 10.1128/JVI.62.3.866-874.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puhlmann M, Brown CK, Gnant M, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther 2000;7:66–73. 10.1038/sj.cgt.7700075 [DOI] [PubMed] [Google Scholar]
- 46.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 1999;190:355–66. 10.1084/jem.190.3.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lasso P, Gomez-Cadena A, Urueña C, et al. An Immunomodulatory Gallotanin-Rich Fraction From Caesalpinia spinosa Enhances the Therapeutic Effect of Anti-PD-L1 in Melanoma. Front Immunol 2020;11:584959. 10.3389/fimmu.2020.584959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao H, Dai P, Wang W, et al. Innate immune response of human plasmacytoid dendritic cells to poxvirus infection is subverted by vaccinia E3 via its Z-DNA/RNA binding domain. PLoS One 2012;7:e36823. 10.1371/journal.pone.0036823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunn GP, Bruce AT, Sheehan KCF, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005;6:722–9. 10.1038/ni1213 [DOI] [PubMed] [Google Scholar]
- 50.Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011;208:2005–16. 10.1084/jem.20101159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011;208:1989–2003. 10.1084/jem.20101158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broz ML, Binnewies M, Boldajipour B, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014;26:638–52. 10.1016/j.ccell.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spranger S, Dai D, Horton B, et al. Tumor-Residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017;31:711–23. 10.1016/j.ccell.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322:1097–100. 10.1126/science.1164206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edelson BT, Kc W, Juang R, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 2010;207:823–36. 10.1084/jem.20091627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamarin D, Ricca JM, Sadekova S, et al. Pd-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest 2018;128:1413–28. 10.1172/JCI98047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-002569supp001.pdf (3.7MB, pdf)
Data Availability Statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.