

Abstract
Background
Modulation of adaptive immunity may underscore the efficacy of trans-arterial chemoembolization (TACE). We evaluated the influence of TACE on T-cell function by phenotypic lymphocyte characterization in samples of patients undergoing surgery with (T+) or without (T-) prior-TACE treatment.
Methods
We profiled intratumoral (IT), peritumoral (PT) and non-tumoral (NT) background tissue to evaluate regulatory CD4+/FOXP3+ (T-reg) and immune-exhausted CD8+/PD-1+ T-cells across T+ (n=58) and T− (n=61). We performed targeted transcriptomics and T-cell receptor sequencing in a restricted subset of samples (n=24) evaluated in relationship with the expression of actionable drivers of anti-cancer immunity including PD-L1, indoleamine 2,3 dehydrogenase (IDO-1), cytotoxic T-lymphocyte associated protein 4 (CTLA-4), Lag-3, Tim-3 and CD163.
Results
We analyzed 119 patients resected (n=25, 21%) or transplanted (n=94, 79%) for Child-Pugh A (n=65, 55%) and Barcelona Clinic Liver Cancer stage A (n=92, 77%) hepatocellular carcinoma. T+ samples displayed lower IT CD4+/FOXP3+ (p=0.006), CD8+ (p=0.002) and CD8+/PD-1+ and NT CD8+/PD-1+ (p<0.001) compared with T−. Lower IT (p=0.005) and NT CD4+/FOXP3+ (p=0.03) predicted for improved recurrence-free survival. In a subset of samples (n=24), transcriptomic analysis revealed upregulation of a pro-inflammatory response in T+. T+ samples were enriched for IRF2 expression (p=0.01), an interferon-regulated transcription factor implicated in cancer immune-evasion. T-cell clonality and expression of PD-L1, IDO-1, CTLA-4, Lag-3, Tim-3 and CD163 was similar in T+ versus T−.
Conclusions
TACE is associated with lower IT density of immune-exhausted effector cytotoxic and T-regs, with significant upregulation of pro-inflammatory pathways. This highlights the pleiotropic effects of TACE in modulating the tumor microenvironment and strengthens the rationale for developing immunotherapy alongside TACE.
Keywords: immunotherapy, liver neoplasms
Introduction
Treatment of unresectable, non-metastatic hepatocellular carcinoma (HCC) on the background of compensated hepatic reserve is dominated by the use of trans-arterial chemoembolization (TACE).1 By combining direct cytotoxic effect of intra-arterial chemotherapy delivery with the ischemic damage induced by blood inflow arrest, TACE can cause tumor regression in up to 50% of patients.2 Evolving experience in the delivery of TACE has made this loco-regional therapy the treatment of choice in patients with intermediate-stage Barcelona Clinic Liver Cancer (BCLC) stage B HCC according to clinical guidelines.3 The overall survival (OS) of patients treated with TACE in contemporary clinical studies extends beyond 2 years, underscoring the capacity of this therapy to significantly delay tumor progression, prevent systemic spread and prolong life expectancy of patients who are ineligible to radical therapies. The proven efficacy of TACE has extended its utility to disease downstaging prior to liver transplantation and avoid drop-out due to interval tumor progression.4
While efficacious, TACE is not universally successful in controlling HCC growth and is mostly delivered with palliative intent.5 Over the past few decades, numerous attempts have been made to improve the long-term outlook of HCC patients treated with TACE by combining loco-regional therapy with molecularly targeted anti-cancer agents such as sorafenib6 or bevacizumab,7 guided by the rationale that sequential or concomitant inhibition of pro-angiogenic pathways might further enhance the clinical efficacy of TACE. The TACTICS study has recently demonstrated that concurrent sorafenib therapy might delay tumor progression following TACE,8 however evidence of OS benefit is lacking and large phase II/III studies have failed primary efficacy endpoints.9 10
The development of immune checkpoint inhibitors (ICIs) targeting programmed-cell death 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) is progressing at rapid pace in liver cancer.11 However, the efficacy of CTLA-4 and PD-1 monotherapy is limited in HCC, where the forerunner anti-CTLA-4 inhibitor tremelimumab12 and the PD-1 inhibitors nivolumab13 14 and pembrolizumab15 16 have been demonstrated to induce radiologically measurable tumor rejection in approximately 20% of patients, although without demonstrable improvement in survival. Conversely, demonstration of a 42% risk reduction in mortality from combined PD-L1 and vascular endothelial growth factor (VEGF) inhibition has recently elevated atezolizumab and bevacizumab to the role of new standard of care in unresectable HCC.17 Results of the ImBrave150 study highlight the necessity of combination approaches to improve patient outcome.
The efficacy of TACE relates at least in part to treatment-induced modulation of innate and adaptive immunity.18 19 The release of cellular debris, pro-inflammatory cytokines and danger-associated molecular patterns after intra-arterial chemotherapy deliver and tumor embolization has a priming effect on adaptive immunity20 and the presence of spontaneous TACE-induced T-cell responses is predictive of improved outcome.21–24 A number of ongoing clinical trials are currently evaluating whether ICIs can synergize with TACE to promote more effective anti-tumor immune reconstitution and improve anti-tumor control.25 However, the precise functional characteristics of the tumor microenvironment in response to TACE are unknown. Taking advantage of a large clinically annotated series of archival HCC tissue samples, we have designed this study with the following aims: (1) phenotypically characterize the cellular subsets that enrich the tumor microenvironment after TACE; (2) evaluate the expression of PD-ligands and other co-inhibitory receptors and their respective ligands in response to TACE; (3) decipher clinically meaningful differences in the transcriptomic features of HCC following treatment by TACE with the aim to discover novel therapeutically actionable pathways involved in the anti-tumor immune response and prospectively rationalize translational studies of immunotherapy combinations in HCC.
Methods
Patient samples
We established a clinically annotated collection of archival paraffin-embedded HCC tissues from six tertiary academic centers for the care of HCC in UK, Italy and Spain, including a total of 119 patients (online supplemental table 1). Among all consecutive patients who received liver resection or transplantation for HCC, we identified 58 patients who had TACE prior to surgery and matched them with 61 who did not. Matching was performed on the basis of BCLC stage, Child-Pugh class and alfa-foetoprotein levels. All patients included had histological diagnosis of HCC, were staged preoperatively by dynamic CT and/or MRI and were followed up postoperatively with periodic imaging and clinical review as in accordance with current guidelines.3 Eligibility for TACE followed clinical guidelines and included: (1) diagnosis of HCC by means of histology or non-invasive criteria according to The European Association of the Study of the Liver and the American Association for the Study of Liver Diseases guidelines; (2) liver function within Child-Pugh B7 stage; (3) absence of main or branched portal vein thrombosis or extra-hepatic spread; (4) general contraindications to TACE (clinically significant artero-venous shunting, poor cardiac ejection fraction (<50%) or any other comorbid condition that in the opinion of the treating physician might have posed a significant risk to TACE). The decision to TACE was made by each respective multidisciplinary team in the context of bridging/downstaging strategy prior to surgery.26 Histopathologists at each site reviewed archival specimens to define key pathologic features on H&E slides. Number, location and size of neoplastic nodules were recorded and for each case, a number of sections representative of the full surface of the tumor mass were taken. Archival samples underwent central review (FAM, RDG) to guide tissue microdissection of target tumor areas27–29 and identify histopathological features of embolized tumors including necrosis and histologic alterations that have been previously described.30 To allow for maximal reproducibility in case of multifocal tumors, the dominant lesion was chosen for sampling.
jitc-2021-003311supp001.pdf (732.7KB, pdf)
Clinical variables including patients’ demographics, etiology of chronic liver disease, liver functional reserve and, wherever possible, recurrence and survival status were reconstructed retrospectively from review of medical records. OS was calculated from the date of surgery until last follow-up or patients’ death. Recurrence-free survival (RFS) was calculated from date of radical therapy to radiologically proven recurrence.
TACE procedure
At all participating institutions, indication and suitability for TACE was confirmed in every case following multidisciplinary review of clinical data and staging CT and confirmed on pre-treatment diagnostic visceral angiography of the celiac trunk and superior mesenteric artery as previously reported.31 32 Depending on tumor burden and vascular anatomy, TACE was administered super-selectively whenever possible (ie, with a segmental or subsegmental approach) using a 2.7–3.0 Fr micro-cathether, which served for intra-arterial infusion of doxorubicin (50 mg, fixed dose) emulsified in lipiodol (10 mL) followed by embolization with Gelfoam powder (Pfizer, New York, New York, USA) with particles ranging from 40 to 60 µm in size administered as embolic material.
Multi-colour immunohistochemistry (IHC)
Multiplex immunostaining for CD4, CD8, FOXP3 and PD-1 was performed on 2 µm tissue sections using a pre-optimized protocol.33 Individual count of CD8+ and/FOXP3, CD8+/PD-1+ co-immunopositive cells was performed in tissue photomicrographs assessed at 450× magnification across tumorous and non-tumoral areas and reported as cellular density per mm2 of tissue. Every sample was scored on the basis of the mean of three independent readings per region. A subset of samples with viable residual tumor post TACE paired with TACE-untreated controls underwent single marker immunostaining for biomarkers of T-cell exhaustion including PD-L1 (E1L3N, CST Cat. Nr. 13684), indoleamine 2,3 dehydrogenase (IDO-1, D5J4E, Cat. Nr. 82723), Tim-3 (Cat Nr. AF2365), Lag-3 (EPR20261, Cat Nr. Ab209236), CTLA-4 (F-9, Cat Nr. sc-376016) and CD163 (10D6, Cat Nr. NCL-L-CD163) according to a previously optimized protocol.34 Lag-3 and CTLA-4 were scored as number of immune-positive lymphocytes at 450× magnification. PD-L1 expression was scored categorically using a 1% cut-off value, as routinely employed in clinical trials of ICI. IDO-1, Tim-3 were evaluated using a semi-quantitative score (H-score, range 0–300) derived from multiplying the percentage of positive cells (1% cut-off) by chromogenic intensity (ranked from 0 to 3).
DNA/RNA purification
Tissue specimens were quality controlled by a consultant histopathologist (FAM) on H&E sections to identify target areas containing >20% of viable tumor prior to molecular profiling. DNA and RNA were purified from 10 µM-thick FFPE sections using the AllPrep DNA/RNA FFPE Kit (Qiagen, Venlo, NL, Cat. 80234). DNA/RNA quantification was performed on an ND2000 Nanodrop spectrophotometer (Thermo Fisher Scientific, Loughborough, UK).
NanoString immune profiling and high-resolution T-cell receptor (TCR) sequencing
In a subset of samples (n=24), targeted transcriptomic profiling was performed on 200 ng of total RNA using the NanoString PanCancer-Immune panel on an nCounter Analysis System (NanoString Technologies, Seattle, Washington, USA) and analyzed using the nSolver Analysis Software (NanoString Technologies). Raw counts from gene expression data were normalized to the mean of the positive control probes for each assay and to the geometric mean of housekeeping genes in the codeset. We used unsupervised clustering to generate a heatmap of the normalized data to quantify the relative abundance of each transcript in cases versus controls (n=12 each). In total, 23/24 samples had sufficient DNA to undergo parallel sequencing of the CDR3 regions of human TCR-beta chain sequencing using the immunoSEQ Assay (Adaptive Biotechnologies, Seattle, Washington, USA).33 T-cell density was calculated by normalizing TCR template counts to the total amount of DNA that was usable for TCR sequencing. The amount of usable DNA was determined by PCR amplification and sequencing of housekeeping genes. Clonality was computed on productive rearrangements and defined as 1-Peilou’s evenness. Richness was calculated using the preseqR package.35
Statistical analysis
Descriptive statistics are presented as medians or means. Comparisons of proportions were performed using Pearson’s χ2 or Fisher’s exact tests and comparisons of medians were evaluated by Mann-Whitney U tests. Pearson’s or Spearman’s correlation coefficient tests were used to investigate correlations between clinicopathological variables. Analyses of survival were performed using Kaplan-Meier and Log-rank test followed by Cox regression analyses using a backwards elimination approach. Differential expression of specific genes in RNA expression experiments was determined using the false discovery rate method of Benjamini and Hochberg, with pre-defined q-value of 5%. All statistical analyses were performed using SPSS V.26.0 (IBM Inc) and GraphPad Prism (GraphPad software, La Jolla, California, USA). All estimates were reported with 95% CIs and a two-tailed level of significance of p≤0.05.
Results
Patient characteristics
In total, 119 archival tissue samples from patients who underwent liver resection (n=25, 21%) or transplantation (n=94, 79%) for HCC between 2005 and 2018 were included (100 men, 19 women). Median age at surgery was 54 (range 41–71). The majority of patients had cirrhosis (n=166, 97%) mostly secondary to hepatitis C virus infection (n=88, 74%) followed by alcohol excess (n=21, 18%). The majority of patients were in Child Pugh class A (n=65, 55%) had Tumour Node Metastasis (TNM) stage II disease (n=73, 61%) and had an overall BCLC stage A (n=92, 77%).
A total of 58 patients had TACE prior to surgery (T+, n=58, 49%), whereas 61 did not (T−, 51%). Median number of TACE procedures in T+ patients was 1 (range 1–5). Median interval from last TACE to surgery was 3.4 months (range 2–11 months). Twenty-one (17%) out of the 58 T+ patients had histological evidence of post-treatment necrosis in ≥50% of the examined tumor tissue. Compared with T−, T+ patients were balanced in terms of maximum tumor diameter (2.3 vs 2.5 cm, p=0.93), BCLC stage (proportion of BCLC A/B, 88% vs 95%, p=0.32), Alpha-foetoprotein levels (AFP)(median 8 vs 13.6 ng/mL, p=0.77) and Child-Pugh class (Child-Pugh A 67% vs 70%, p=0.41). However, T+ patients had higher T stage (T≥2, 84%) compared with T− (T≥2, 47%, p<0.001). In the 103 patients with recurrence data available, 26 (22%) had documented disease relapse after resection (n=12, 54%) and 14 after transplantation (17%). The median RFS time was not reached, the mean was 131 months (95% CI 109 to 153). Thirty-two patients died during follow-up (27%) and the median OS for the whole cohort was 130 months (95% CI 101 to 159 months).
Phenotypic characteristic of the T-cell infiltrate in HCC
For each sample, we evaluated phenotypic characteristics of the T-cell infiltrate on intratumoral (IT) and peritumoral (PT) areas of archival samples as well as in non-tumoral (NT) background liver. Representative sections are shown in figure 1. The overall distribution of CD8+, CD4+/FOXP3+ and CD8+/PD-1+ T-cells is reported in online supplemental figure 1. As shown in figure 2, IT areas of T+ samples had significantly lower median CD4+/FOXP3+ (8.2 vs 40.8 cells/mm2, p=0.006), total CD8+ (32.7 vs 118.4 cells/mm2, p=0.002), and CD8+/PD-1+co-immunopositive cells (16.3 vs 36.7 cells/mm2, p<0.001) compared with T− samples. The distribution of CD4+, CD8+, CD4+/FOXP3+ and CD8+/PD-1+ T-cell density in PT infiltrate was not associated with TACE pre-treatment, however, in NT areas, T− samples had a lower median density of CD8+/PD-1+ cells compared with T+ (9.4 vs 146.4 cells/mm2, p<0.001, online supplemental table 2), whereas all other NT cell subpopulations showed no difference in distribution.
Figure 1.

Representative sections illustrating architectural features of an HCC sample resected after TACE on standard H&E stain (Panel A). Red arrows highlight deposition of embolic material to the immediate vicinity of a viable HCC tumor deposit, marked by a blue dashed line (magnification 200×). Panel B highlights T-cell phenotypic characteristics by multiplex immunohistochemistry for CD4 (brown chromogen), FOXP3 (green chromogen), CD8+ (red chromogen), PD-1 (blue chromogen) and CD8+/PD-1 co-expression (purple chromogen). TACE pre-treatment (Panel B) is associated with less intense T-cell infiltrate compared with a TACE-untreated case (Panel C). Panels D–G illustrate representative sections of PD-L1 (Panel D), IDO-1 (Panel E), Tim-3/CD163 (Panel F) and Lag-3/CTLA-4 (Panel G) intratumoral staining corresponding to the H&E section shown in Panel B. CTLA-4, cytotoxic T-lymphocyte associated protein 4; HCC, hepatocellular carcinoma; IDO-1, indoleamine 2,3 dehydrogenase; PD-1, programmed-cell death 1; TACE, trans-arterial chemoembolization.
Figure 2.
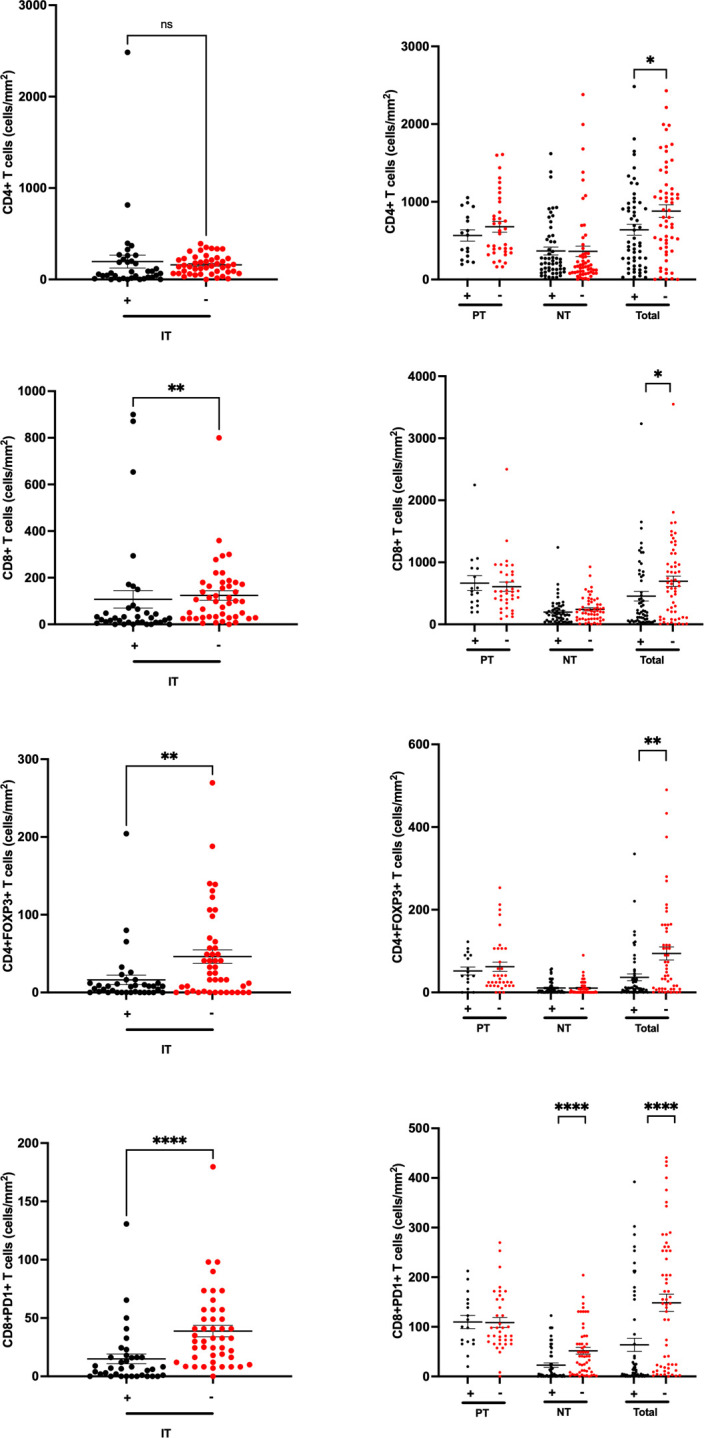
Histograms illustrating the relationship between CD4+, CD8+, CD4+/FOXP3+ and CD8+/PD-1+tumor infiltrating lymphocytes and prior-treatment with TACE (T+/T−) in patients with HCC, assayed across intratumoral (IT), peritumoral (PT) and non-tumoral (NT) background liver areas. TACE pre-treatment (T+) was associated with lower T-cell density values for IT CD8+, CD4+/FOXP3+, CD8+/PD-1+ cells and NT CD4+/FOXP3+ cells. **p<0.01, ****p<0.001. HCC, hepatocellular carcinoma; PD-1, programmed-cell death 1; TACE, trans-arterial chemoembolization.
In patients that were radiologically evaluable for relapse (n=103), samples with higher (ie, ≥median) CD4+/FOXP3+ density in IT were characterized by a significantly shorter RFS (HR 4.2, 95% CI 1.4 to 12.5, median RFS not reached vs 57 months, Log rank p=0.005, figure 3). We found no relationship between CD4+/FOXP3+ density in PT areas, nor CD8+/PD-1+ density in either IT, PT nor NT areas with either RFS or OS (p>0.05). Multivariate Cox regression analyses of RFS confirmed IT CD4+/FOXP3+ density to predict for shorter RFS (HR 3.5, 95% CI 1.1 to 10.8, p=0.028) following adjustment for TNM stage, Child-Pugh class and median AFP levels (online supplemental table 3). Neither characteristics of the IT nor NT T-cell infiltrate were associated with the presence of necrosis post-TACE (online supplemental figure 2).
Figure 3.
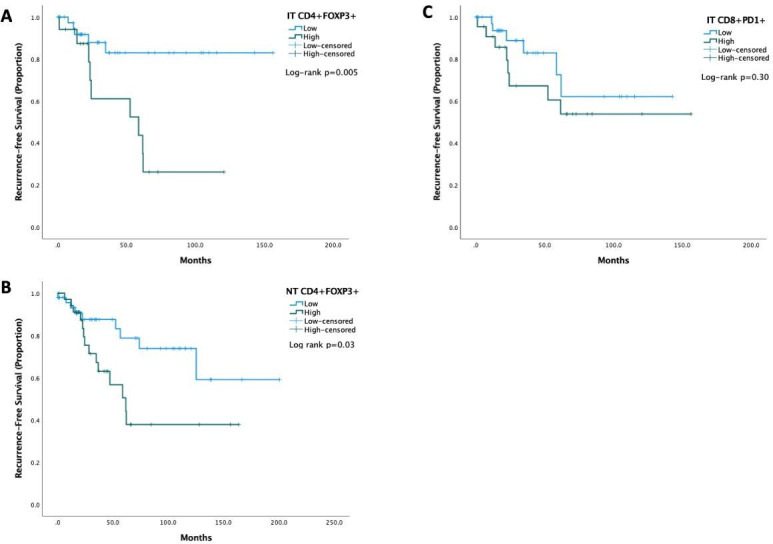
Kaplan-Meier curves illustrating the relationship between characteristics of the T-cell infiltrate found differentially regulated in association with TACE and recurrence-free survival of patients with HCC. IT and CD4+/FOXP3+ and NT CD4+/FOXP3+ but not IT CD8+/PD-1+ T-cell density were significantly associated associated with RFS. HCC, hepatocellular carcinoma; IT, intratumoral; NT, non-tumoral; PD-1, programmed-cell death 1; RFS, recurrence-free survival; TACE, trans-arterial chemoembolization.
TACE pre-treatment is associated with distinctive phenotypic features of T-cell infiltrate but not clonality
To complement the multiplex-immunohistochemistry data showing depletion of CD4+/FOXP3+ cells in HCC samples pre-treated with TACE, we performed an exploratory targeted transcriptomic analysis of a smaller subset of 24 patient samples with viable tumor tissue. We used the nCounter PanCancer Immune Profiling panel in 12 T+ and 12 T− samples to provide mechanistic insight into the molecular drivers characterizing the tumor immune microenvironment in association with TACE response (online supplemental figure 3). As shown in figure 4, directed gene set analyses demonstrated increased expression of transcripts belonging to a number of functional domains including secretion of chemokines, regulation of immune cell function, complement cascade activation and production of interleukins and cytokines. We subsequently assessed differential expression of individual genes across T+/T− groups and demonstrated significant upregulation of IRF2, an interferon-regulated transcription factor acting downstream of alpha and beta interferons in T+ (figure 4B). We next evaluated whether TACE pre-treatment was associated with differential immunohistochemical expression of a panel of co-inhibitory signals in the same sample group. As shown in online supplemental figure 4, there was no difference in the expression of co-inhibitory proteins including PD-L1, IDO-1, Lag-3, Tim-3 or CTLA-4, nor differential distribution of CD163 cells intratumorally or peritumorally. Lastly, we evaluated whether TACE pre-treatment might influence measures of T-cell clonality by high-resolution TCR-beta chain sequencing using the ImmunoSEQ assay. As shown in figure 4, we found no evidence for an association between prior treatment with TACE and T-cell clonality within the intratumoral infiltrate as measured by a number of reproducible readouts including productive clonality, productive entropy and evaluation of the most frequently identified clonotypes (top, top 10 and top 100) across sample groups.
Figure 4.

Targeted transcriptomic analysis of tumorous HCC tissue (n=24) using Nanostring PanCancer Immune profiling illustrates the differential regulation of 11 gene expression signatures on the basis of TACE pre-treatment using directed gene set analysis (GSA) and confirms significant upregulation of IRS2 in TACE-pre-treated samples (Panel B). Panel C: histograms illustrating the distribution of productive clonality, productive entropy and number of most frequently identified clonotypes as assayed by deep sequencing of the TCR-beta chain. HCC, hepatocellular carcinoma; TACE, trans-arterial chemoembolization; TCR, T-cell receptor.
Discussion
Immunogenic cell death (ICD) is a cell death modality characterized by elicitation of an immune response against dead cell antigens. The majority of anti-cancer therapeutic strategies such as chemotherapy36 or targeted therapy are inducers of apoptosis,37 a process that by definition is poorly immunogenic and may in some instances promote tolerogenic responses.
There is long-standing evidence to suggest that local ablative therapy induces T-cell responses against tumor as well as recall antigens in HCC,21 however these are insufficient to induce protective immunity against disease relapse. Limited but compelling evidence suggests the presence of immune-exhausted, antigen-specific CD8+ T-cell responses in intratumoral and peri-tumoral infiltrating lymphocytes in HCC,38 and that relative abundance of tumour-associated antigen responses is higher in patients pre-treated with TACE. However, the relative contribution of regulatory T-cells in shaping the phenotypic characteristics of such heavily immune-exhausted effector response has not been fully appreciated.
Our study on retrospective archival samples of HCC shows that patients treated with prior TACE are characterized by lower proportion of cytotoxic T-cell infiltrate, as evidenced by reduced CD8+ cell density in TACE-pretreated patients. Phenotype studies of the cytotoxic T-cell branch showed a reduction in immune-exhausted CD8+/PD-1+ populations within the tumor and in the surrounding cirrhotic parenchyma compared with TACE-untreated controls, a population linked with resistance to immunotherapy in HCC.39 In addition, TACE pre-treatment is associated with lower regulatory T-cell density within the tumor. Interestingly, patients with lower intratumoral CD4+/FOXP3+ cells had a significantly lower risk of relapse following radical therapy for HCC, providing an important link in the interplay between TACE-associated reduction in the regulatory infiltrate and long-term prognosis of these patients. These results trace an interesting parallel with studies of ICD in other malignancies, where density of T-regs and exhausted cytotoxic lymphocytes measured in situ have been demonstrated as strong prognostic factors for response to anti-cancer therapy.40
In attempting to provide better functional insight to the differences in T-cell infiltration observed in our in situ profiling experiments, we used high throughput sequencing technologies to verify whether prior treatment with TACE was associated with broad differences in gene expression within the tumour microenvironment (TME) or diversity in T-cell clonality.
High-resolution T-cell receptor beta chain sequencing revealed no evidence of a difference in T-cell clonality in T+ versus T− samples using multiple readouts reflecting the diversity of T-cell receptor V–D–J sequence rearrangements within the intratumoral infiltrate. This is an important translational finding, in view of the importance of T-cell clonality as a mechanism underscoring the efficacy of cancer immunotherapy across indications.41 On the other hand, transcriptomic analysis showed evidence of a more intensely inflamed TME in TACE-pre-treated samples, as demonstrated by the significant upregulation of a number of pro-inflammatory gene signatures in T+ compared with to T−. Candidate gene analysis revealed IRF2, a transcriptional repressor of PD-L1 and active regulator of a number of components of the Major Histocompatibility Complex-I pathway,42 to be enriched in T+ samples. Because IRF2 loss leads to the positive regulation of co-inhibitory signals, we performed targeted IHC experiments on the same subset of T+/T− samples to verify the differential expression of a panel of actionable drivers of T-cell exhaustion alongside CD163 a marker identifying M2-polarized macrophage, an abundant myeloid component within the HCC TME.43 Interestingly, we found no difference in the expression of any of the assayed co-inhibitory signals in relationship with TACE pre-treatment, a finding that leads us to postulate the independence of these pathways from IRF2-mediated transcriptional regulation: a point that should be explored in future mechanistic studies. Recent evidence in KRAS-mutant colorectal cancer models, a notoriously immunotherapy-resistant tumor, suggests IRF2 to downregulate chemoattraction of myeloid suppressive cells, underscoring the multifaceted role of this transcription factor in regulating anti-cancer immunity beyond T-cell biology.44
Taken together, our study provides important evidence to suggest that TACE pre-treatment may be characterized by profound and prognostically important changes in the functional characteristics of the immune cell infiltrate.
These findings are of key translational importance, as they provide a rationale for the evaluation of ICIs alongside TACE. This is an area of active investigation, where combinations of PD-1/PD-L1 inhibitors with either CTLA-4 or anti-vascular agents are being pursued in absence of a strong immunologic rationale and simply based on evidence of efficacy in advanced disease.
While early-phase studies of locally ablative therapies in combination with CTLA-4 antagonist have shown evidence of CD8+ T cell enrichment in paired pre/post-treatment biopsies,45 the modest effect of PD-1 and CTLA-4 inhibitors as a monotherapy in improving outcome of patients with advanced HCC casts legitimate doubts as to their potential to improve survival in intermediate-stage disease.
Our study provides renovated emphasis on strategies to deplete T-reg cells and control of T-reg cell functions to increase antitumor immune responses in the context of TACE.46
Interestingly, VEGF inhibition can reduce the differentiation of T-regs47 and unsurprisingly, combination of PD-L1 inhibitors with bevacizumab has shown evidence of enhanced activity in advanced HCC compared with PD-1 monotherapy.17 A number of approaches are being tested to improve selectivity of immune-based interventions towards T-reg depletion or functional modulation: our result provide a rational basis for their development in combination with TACE.48
We acknowledge the retrospective, case–control study design as an important limitation to our observation. Unfortunately, none of the patients on this study had pre-TACE tumor biopsies available for analyses. In clinical practice, tumor biopsies are in fact very rarely performed in patients amenable to radical therapies if they satisfy non-invasive diagnostic criteria.3 In our participating centers, pre-treatment biopsies are generally not performed in patients who are candidates for resection or liver transplantation due to the potential risk of seeding.49
Another important limitation is the lack of standardization in terms of interval from TACE to surgery as well as in the number of TACE procedures performed, factors that we could not control for given the retrospective and observational nature of our study. Patients in the T+ group had also higher TNM stage. This is perhaps unsurprising given the predominant indication for TACE in this population was downstaging and small tumors would have been more likely to undergo upfront resection or transplantation.
Standardization of times between TACE to surgery, although scientifically desirable, would be impossible to achieve even in the context of a prospective study given that time from downstaging/bridging TACE to liver transplantation is dependent on donor organ availability and cannot be controlled for. Longitudinal monitoring of T-cell characteristics pre-TACE and post-TACE at pre-defined intervals was only prospectively feasible in a study of subtotal embolization or ablation: procedures that are therapeutically suboptimal and may not reflect the full extent of immune activation within the tumor microenvironment.45 In our study, we concentrated immune phenotyping analyses on restricted T-cell populations that are functionally related to the mechanism of action of ICIs. Other adaptive and innate mechanisms of immune response may be involved and underscore the efficacy of TACE. Prospective studies on fresh frozen samples may shed more light on the role of tumor and host determinants of immune-exclusion using multi-omics technologies.50 Lastly, it should be acknowledged that patients evaluated in this study were surgical candidates mostly within BCLC A-stage criteria and that TACE was performed as bridging or downstaging therapy prior to radical approaches. Therefore, caution should be made in extrapolating our results to BCLC-B HCC patients who routinely receive TACE as a palliative rather than a downstaging oncological therapy and that are at the focus of clinical trials. The immune regulation of the microenvironment after TACE may in fact be different as a result of advancing stage of the disease and ongoing clinical trials of TACE/immunotherapy combinations will define whether immunotherapy exerts truly synergistic effects in combination with TACE.
In spite of the acknowledged limitations, this is the first study to comprehensively evaluate the influence of TACE in the TME from resected specimens. Demonstration of the key role of regulatory T-cell responses should prioritize targeting of negative regulators of anti-cancer immunity alongside TACE, with a particular emphasis of T-reg targeting, in an attempt to improve response and survival following loco-regional therapies for HCC.
Acknowledgments
The authors would like to acknowledge the infrastructure support provided by Imperial Experimental Cancer Medicine Centre, the NIHR Imperial College BRC, the Cancer Research UK Imperial Centre and the Imperial College Healthcare NHS Trust Tissue Bank.
Footnotes
Contributors: DJP contributed to study concept and design. SMM, AF, TK, PF, PT, BM, VC, CA, DA, RDG, AUA, TM, FAM, EG, FG, SB, VM contributed to acquisition of data. SMM, TK, RJB, DMM, DJP contributed to analysis and interpretation of data. DJP, SMM contributed to drafting of the manuscript. All the authors contributed to critical revision of the manuscript for important intellectual content. DJP, SMM contributed to statistical analysis. DJP obtained funding, contributed to administrative, technical or material support, study supervision.
Funding: DJP is supported by grant funding from the Wellcome Trust Strategic Fund (PS3416) and by the Cancer Research UK Postdoctoral bursary (C57701/A26137). AF is supported by grant from Instituto de Salud Carlos III (PI18/00542).
Competing interests: DJP received lecture fees from ViiV Healthcare, Roche, Falk and Bayer Healthcare and travel expenses from BMS, MSD and Bayer Healthcare; consulting fees for Mina Therapeutics, EISAI, H3B, DaVolterra, Roche and Astra Zeneca; received research funding (to institution) from MSD and BMS. AF received lecture fees from Bayer HealthCare, Gilead and MDS; Consulting fees from Bayer HealthCare, Roche, Guerbert and Astra-Zeneca. EG received lecture fees from Bayer HealthCare, Gilead, AbbVie, MSD, Eisai.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
Ethical approval to conduct this study was granted by the Imperial College Tissue Bank (Reference Number R16008) and confirmed at every participating center before study initiation. All study procedures were conducted in accordance with the Declaration of Helsinki and in accordance with Good Clinical Practice. Institutional Review Board approval was obtained for the use of clinical data at each participating institution.
References
- 1.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003;37:429–42. 10.1053/jhep.2003.50047 [DOI] [PubMed] [Google Scholar]
- 2.Forner A, Gilabert M, Bruix J, et al. Treatment of intermediate-stage hepatocellular carcinoma. Nat Rev Clin Oncol 2014;11:525–35. 10.1038/nrclinonc.2014.122 [DOI] [PubMed] [Google Scholar]
- 3.European Association for the Study of the Liver . Electronic address eee, European association for the study of the L. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182–236. [DOI] [PubMed] [Google Scholar]
- 4.Bryce K, Tsochatzis EA. Downstaging for hepatocellular cancer: harm or benefit? Transl Gastroenterol Hepatol 2017;2:106. 10.21037/tgh.2017.11.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinato DJ, Howell J, Ramaswami R, et al. Review article: delivering precision oncology in intermediate-stage liver cancer. Aliment Pharmacol Ther 2017;45:1514–23. 10.1111/apt.14066 [DOI] [PubMed] [Google Scholar]
- 6.Erhardt A, Kolligs F, Dollinger M, et al. TACE plus sorafenib for the treatment of hepatocellular carcinoma: results of the multicenter, phase II SOCRATES trial. Cancer Chemother Pharmacol 2014;74:947–54. 10.1007/s00280-014-2568-8 [DOI] [PubMed] [Google Scholar]
- 7.Pinter M, Ulbrich G, Sieghart W, et al. Hepatocellular carcinoma: a phase II randomized controlled double-blind trial of transarterial chemoembolization in combination with biweekly intravenous administration of bevacizumab or a placebo. Radiology 2015;277:903–12. 10.1148/radiol.2015142140 [DOI] [PubMed] [Google Scholar]
- 8.Kudo M, Ueshima K, Ikeda M, et al. Randomised, multicentre prospective trial of transarterial chemoembolisation (TACE) plus sorafenib as compared with TACE alone in patients with hepatocellular carcinoma: TACTICS trial. Gut 2020;69:1492–501. 10.1136/gutjnl-2019-318934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lencioni R, Llovet JM, Han G, et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: the space trial. J Hepatol 2016;64:1090–8. 10.1016/j.jhep.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 10.Meyer T, Fox R, Ma YT, et al. Sorafenib in combination with transarterial chemoembolisation in patients with unresectable hepatocellular carcinoma (TACE 2): a randomised placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol 2017;2:565–75. 10.1016/S2468-1253(17)30156-5 [DOI] [PubMed] [Google Scholar]
- 11.Pinato DJ, Guerra N, Fessas P, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020;39:3620–37. 10.1038/s41388-020-1249-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81–8. 10.1016/j.jhep.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 13.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. 10.1016/S0140-6736(17)31046-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yau T, Park JW, Finn RS, et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (PTS) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology 2019;30:v874–5. 10.1093/annonc/mdz394.029 [DOI] [Google Scholar]
- 15.Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 2018;19:940–52. 10.1016/S1470-2045(18)30351-6 [DOI] [PubMed] [Google Scholar]
- 16.Finn RS, Ryoo B-Y, Merle P, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol 2020;38:193–202. 10.1200/JCO.19.01307 [DOI] [PubMed] [Google Scholar]
- 17.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. 10.1056/NEJMoa1915745 [DOI] [PubMed] [Google Scholar]
- 18.Singh P, Toom S, Avula A, et al. The immune modulation effect of locoregional therapies and its potential synergy with immunotherapy in hepatocellular carcinoma. J Hepatocell Carcinoma 2020;7:11–17. 10.2147/JHC.S187121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tampaki M, Ionas E, Hadziyannis E, et al. Association of Tim-3 with BCLC stage, serum PD-L1 detection, and response to transarterial chemoembolization in patients with hepatocellular carcinoma. Cancers 2020;12 10.3390/cancers12010212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pol J, Vacchelli E, Aranda F, et al. Trial Watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015;4:e1008866. 10.1080/2162402X.2015.1008866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zerbini A, Pilli M, Penna A, et al. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res 2006;66:1139–46. 10.1158/0008-5472.CAN-05-2244 [DOI] [PubMed] [Google Scholar]
- 22.Hiroishi K, Eguchi J, Baba T, et al. Strong CD8(+) T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J Gastroenterol 2010;45:451–8. 10.1007/s00535-009-0155-2 [DOI] [PubMed] [Google Scholar]
- 23.Liao Y, Wang B, Huang Z-L, et al. Increased circulating Th17 cells after transarterial chemoembolization correlate with improved survival in stage III hepatocellular carcinoma: a prospective study. PLoS One 2013;8:e60444. 10.1371/journal.pone.0060444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinato DJ, Karamanakos G, Arizumi T, et al. Dynamic changes of the inflammation-based index predict mortality following chemoembolisation for hepatocellular carcinoma: a prospective study. Aliment Pharmacol Ther 2014;40:1270–81. 10.1111/apt.12992 [DOI] [PubMed] [Google Scholar]
- 25.Viveiros P, Riaz A, Lewandowski RJ, et al. Current state of liver-directed therapies and combinatory approaches with systemic therapy in hepatocellular carcinoma (HCC). Cancers 2019;11 10.3390/cancers11081085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pompili M, Francica G, Ponziani FR, et al. Bridging and downstaging treatments for hepatocellular carcinoma in patients on the waiting list for liver transplantation. World J Gastroenterol 2013;19:7515–30. 10.3748/wjg.v19.i43.7515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinato DJ, Ramachandran R, Toussi STK, et al. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br J Cancer 2013;108:429–37. 10.1038/bjc.2012.538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulkarni S, Pfeiffer J. Clinical genomics. 1st edn. New York: Elsevier, 2014. [Google Scholar]
- 29.Pinato DJ, Tan TM, Toussi STK, et al. An expression signature of the angiogenic response in gastrointestinal neuroendocrine tumours: correlation with tumour phenotype and survival outcomes. Br J Cancer 2014;110:115–22. 10.1038/bjc.2013.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morisco F, Stigliano R, Godfrey A, et al. Efficacy of loco-regional ablation therapy of HCC in a population of liver transplanted patients. Dig Dis Sci 2008;53:1131–7. 10.1007/s10620-007-9986-y [DOI] [PubMed] [Google Scholar]
- 31.Pinato DJ, Arizumi T, Allara E, et al. Validation of the hepatoma arterial embolization prognostic score in European and Asian populations and proposed modification. Clin Gastroenterol Hepatol 2015;13:1204–8. 10.1016/j.cgh.2014.11.037 [DOI] [PubMed] [Google Scholar]
- 32.Facciorusso A, Mariani L, Sposito C, et al. Drug-eluting beads versus conventional chemoembolization for the treatment of unresectable hepatocellular carcinoma. J Gastroenterol Hepatol 2016;31:645–53. 10.1111/jgh.13147 [DOI] [PubMed] [Google Scholar]
- 33.Pinato DJ, Vallipuram A, Evans JS, et al. Programmed cell death ligand expression drives immune Tolerogenesis across the diverse subtypes of neuroendocrine tumours. Neuroendocrinology 2021;111:465–74. 10.1159/000506745 [DOI] [PubMed] [Google Scholar]
- 34.Marafioti T, Jones M, Facchetti F, et al. Phenotype and genotype of interfollicular large B cells, a subpopulation of lymphocytes often with dendritic morphology. Blood 2003;102:2868–76. 10.1182/blood-2003-03-0692 [DOI] [PubMed] [Google Scholar]
- 35.Reuben A, Gittelman R, Gao J, et al. Tcr repertoire Intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov 2017;7:1088–97. 10.1158/2159-8290.CD-17-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green DR, Ferguson T, Zitvogel L, et al. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009;9:353–63. 10.1038/nri2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellamy CO, Malcomson RD, Harrison DJ, et al. Cell death in health and disease: the biology and regulation of apoptosis. Semin Cancer Biol 1995;6:3–16. 10.1006/scbi.1995.0002 [DOI] [PubMed] [Google Scholar]
- 38.Flecken T, Schmidt N, Hild S, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014;59:1415–26. 10.1002/hep.26731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021;592:450–6. 10.1038/s41586-021-03362-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halama N, Michel S, Kloor M, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 2011;71:5670–7. 10.1158/0008-5472.CAN-11-0268 [DOI] [PubMed] [Google Scholar]
- 41.Aversa I, Malanga D, Fiume G, et al. Molecular T-cell repertoire analysis as source of prognostic and predictive biomarkers for checkpoint blockade immunotherapy. Int J Mol Sci 2020;21 10.3390/ijms21072378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kriegsman BA, Vangala P, Chen BJ, et al. Frequent loss of IRF2 in cancers leads to immune evasion through decreased MHC class I antigen presentation and increased PD-L1 expression. J Immunol 2019;203:1999–2010. 10.4049/jimmunol.1900475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yarchoan M, Xing D, Luan L, et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res 2017;23:7333–9. 10.1158/1078-0432.CCR-17-0950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019;35:559–72. 10.1016/j.ccell.2019.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545–51. 10.1016/j.jhep.2016.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci 2019;110:2080–9. 10.1111/cas.14069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukumura D, Kloepper J, Amoozgar Z, et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15:325–40. 10.1038/nrclinonc.2018.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dees S, Ganesan R, Singh S, et al. Regulatory T cell targeting in cancer: emerging strategies in immunotherapy. Eur J Immunol 2021;51:280–91. 10.1002/eji.202048992 [DOI] [PubMed] [Google Scholar]
- 49.Stigliano R, Marelli L, Yu D, et al. Seeding following percutaneous diagnostic and therapeutic approaches for hepatocellular carcinoma. What is the risk and the outcome? Seeding risk for percutaneous approach of HCC. Cancer Treat Rev 2007;33:437–47. 10.1016/j.ctrv.2007.04.001 [DOI] [PubMed] [Google Scholar]
- 50.Llovet JM, Montal R, Sia D, et al. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599–616. 10.1038/s41571-018-0073-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-003311supp001.pdf (732.7KB, pdf)
Data Availability Statement
Data are available upon reasonable request.