

Abstract
Many metabolic phenotypes in cancer cells are also characteristic of proliferating non-transformed mammalian cells, and attempts to distinguish between phenotypes resulting from oncogenic perturbation from those associated with increased proliferation are limited. Here, we examined the extent to which metabolic changes corresponding to oncogenic KRAS expression differed from those corresponding to epidermal growth factor (EGF)-driven proliferation in human mammary epithelial cells (HMECs). Removal of EGF from culture medium reduced growth rates and glucose/glutamine consumption in control HMECs despite limited changes in respiration and fatty acid synthesis, while the relative contribution of branched-chain amino acids to the TCA cycle and lipogenesis increased in the near-quiescent conditions. Most metabolic phenotypes measured in HMECs expressing mutant KRAS were similar to those observed in EGF-stimulated control HMECs that were growing at comparable rates. However, glucose and glutamine consumption as well as lactate and glutamate production were lower in KRAS-expressing cells cultured in media without added EGF, and these changes correlated with reduced sensitivity to GLUT1 inhibitor and phenformin treatment. Our results demonstrate the strong dependence of metabolic behavior on growth rate, and provide a model to distinguish the metabolic influences of oncogenic mutations and non-oncogenic growth.
Keywords: Cancer metabolism, cell growth, cell proliferation, KRAS, branched-chain amino acids
Introduction
Numerous similarities exist in the metabolic behaviors of cancer cells and normal proliferating cells. Tumors, unlike most differentiated tissues, generally consume glucose at high rates and preferentially convert it to lactate regardless of oxygen availability [1]. This phenotype, known as aerobic glycolysis or the Warburg effect, is also observed in activated lymphocytes [2], dividing fibroblasts [3], and stem cells [4], and is similar to behavior found in many growing microbes [5,6], suggesting that fermentative metabolism is found in many proliferating cells. Growth-associated metabolic behaviors such as aerobic glycolysis, amino acid uptake, and nucleotide synthesis are activated by mitogenic signaling downstream of the MAPK and PI3K/Akt/mTOR pathways [7–9]. In healthy tissues, specific growth factors regulate proliferation through activating these signaling pathways to selectively effect anabolic metabolism [10]. However, the same signaling and metabolic pathways can also be activated in tumors through elevated expression or mutation of effector proteins involved in growth signaling.
Given the link between metabolism and mitogenic signaling, one hypothesis is that the metabolic rewiring in tumor cells upon transformation promotes proliferation and survival [11–13]. Many cancer cells are susceptible to antimetabolite drugs that impair nucleotide metabolism, and this strategy has been used clinically for more than half a century [14]. However, as with other chemotherapies that target cell division, these drugs can also affect normal proliferating cells [15]. Better understanding how proliferative metabolism differs between normal and cancer cells may help reduce the unwanted effects of antimetabolite drugs related to normal tissue proliferation [16].
Previous reports have examined how metabolic behavior differs between normal tissue and tumors using cultured cells [17], in vivo models [18], and human patients [19], but in these studies, differentiated and neoplastic cells can differ considerably in their proliferative rates. Other investigations have used growth factor removal, serum starvation, and confluence to induce quiescence in cell lines and compared their metabolic profiles to those of their exponentially growing counterparts [20–22]; however, fewer studies have studied the differences in metabolism in matched transformed and non-transformed cells proliferating at the same rate. This comparison may shed light on how metabolism is rewired to support proliferation in each context, and suggest potential druggable metabolic phenotypes that differentially affect cancerous and non-cancerous proliferating cells.
In this study, we investigated how metabolic changes induced by an oncogene compared to metabolic changes associated with non-oncogenic proliferation using human mammary epithelial cells (HMECs). To identify how metabolic behaviors change with both physiological and pathological proliferation rate, we stimulated growth through addition of increasing concentrations of epidermal growth factor (EGF) and by expression of oncogenic mutant KRAS. While KRAS mutations occur infrequently in breast cancer [23], mutant HRAS has been used among oncogene panels to transform primary HMECs [24,25] and confer HMECs with EGF-independent growth [26]; therefore, we believed that HMECs undergoing mutant RAS-driven growth could be considered as a proof-of-concept model for our approach. To distinguish metabolic differences of KRAS-expressing cells from EGF-stimulated control cells proliferating at equal rates, we developed a statistical model to analyze extracellular fluxes, intracellular metabolite levels, and labeling of metabolites from 13C-labeled nutrients. Consistent with other studies, we find changes in substrate use associated with increasing proliferation. Unexpectedly, however, we also observed an increase in biomass yield from glucose and glutamine as well as a reduced growth sensitivity to treatment with metabolic inhibitors in cells expressing mutant KRAS.
Results
Establishing an epithelial cell model to distinguish normal growth factor-driven metabolism and KRAS-driven metabolism
We used immortalized 184A1 human mammary epithelial cells (HMECs) to study the influence of growth rate and oncogenic KRAS expression on metabolism. We then generated empty-vector-expressing (EV) and KRAS(G12V)-expressing (KRAS) HMECs, cultured these cells in varying concentrations of epidermal growth factor (EGF), and measured the change in cell protein content over time to calculate the specific growth rate for each condition (Fig. 1a). (Protein content per cell and growth rate could be described by a linear relationship (Fig. 1b), and the use of protein content in place of cell counts predicted by this relationship did not impact the conclusions of our study.) Spent media was collected over the course of the experiment to assess changes in metabolite levels in the media. An additional set of HMECs was also simultaneously cultured under identical conditions for assessment of 13C labeled nutrient fate and other assays (Fig. 1a).
Figure 1. Cell culture system for distinguishing EGF- and oncogenic KRAS-stimulated metabolism.
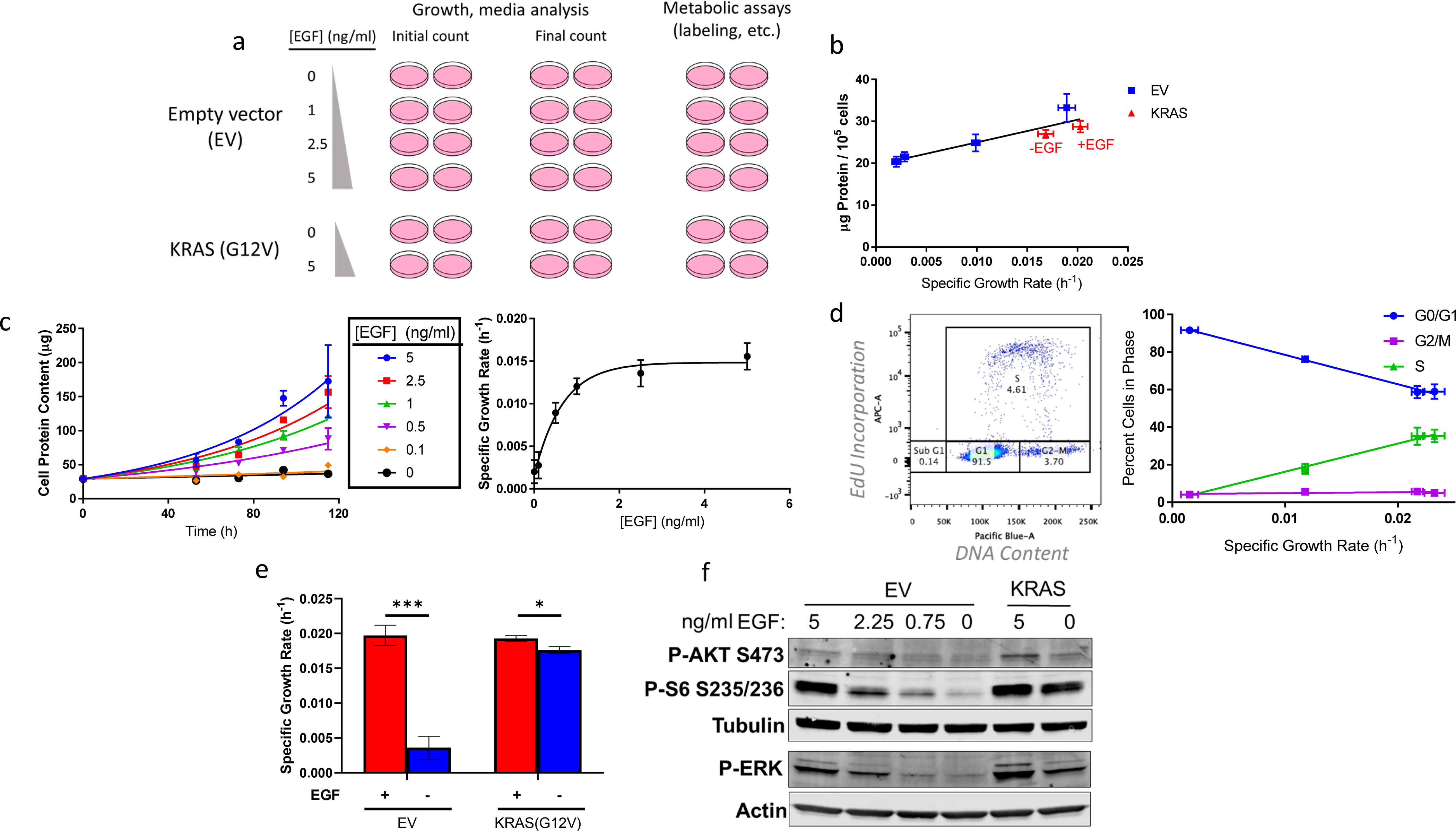
a, Schematic representing the workflow for this investigation. Experiments include plates corresponding to initial and final counts (assessed through quantifying total cellular protein content) to determine growth rate and extracellular fluxes and additional plates grown in parallel for other metabolic assays. EV-HMECs were incubated in four EGF concentrations to vary growth rate and KRAS-HMECs were incubated in 0 and 5 ng/ml EGF.
b, Protein content per cell vs. growth rate for EGF-titrated EV-HMECs and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
c, (left) Growth curves of EV-HMECs treated with varying concentrations with EGF and (right) estimated specific growth rate as a function of EGF concentration.
d, (left) Percent of cells in cell cycle phase, as assessed by flow cytometry, as a function of growth rate and (right) ratio of G0/G1 to G2/M/S phase EGF-titrated EV-HMECs as a function of growth rate.
e, Specific growth rates for EV-HMECs and KRAS-HMECs under 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
f. Western blot representing phospho-Akt (S473), phospho-S6 (S235/236), and phospho-ERK with tubulin and actin loading controls for EV- and KRAS-HMECs under a range of EGF concentrations.
For b, error bars represent mean ± SD for n = 3 wells; a single experiment was performed. For c, error bars indicate mean ± SD for cell counts, with n = 2 wells per time point (10 wells per each condition in total), and 95% confidence intervals for growth rates. Results are representative of three replicate experiments. For d, error bars indicate mean ± SD for n =3 wells; a single experiment was performed. For e, error bars represent mean ± SD for n = 3 wells, and results are representative of more than three replicate experiments. * p < 0.05, ** p < 0.01, *** p < 0.001 (Multiple t-test with adjusted p-value). For f, a single experiment was performed.
EGF addition to culture medium modulated growth rate in a dose-dependent manner from near-quiescence up to a “saturation” level EV-HMECs (Fig. 1c). Flow cytometry to assess EdU incorporation into DNA and DNA content of EGF-titrated EV-HMECs confirmed that increasing EGF levels shifted the cell cycle distribution from G0/G1 toward S phase (Fig. 1d), indicating that a larger fraction of cells was actively undergoing DNA synthesis with increased growth rate. Expression of the KRAS(G12V) mutant in HMECs conferred an ability to grow in EGF-free medium (Fig. 1e) [27], and elicited constitutive activation of the proliferation-linked MAPK pathway as indicated by phosphorylated ERK, and PI3K/Akt/mTOR signaling as indicated by phosphorylated Akt and ribosomal protein S6 (Fig. 1f). Although addition of 5 ng/ml EGF slightly increased growth rate in KRAS-HMECs, the difference was small compared to the proliferative shift elicited in EV-HMECs (Fig. 1e).
Glucose-to-lactate conversion decreases only slightly upon quiescence
We first examined how metabolism differs between EGF-stimulated and quiescent EV-HMECs.
Glucose is typically the most highly consumed carbon substrate for dividing mammalian cells under typical culture conditions [27]; as the primary glycolytic fuel, it can enter many biosynthetic pathways but is mostly excreted as lactate in cultured cells [28]. Consistent with previous reports [20,21], the glucose consumption rate in EGF-stimulated EV-HMECs correlated closely with growth rate, increasing three-to-four-fold as cells went from near-quiescent to maximum proliferation (Fig. 2a). Lactate production closely mirrored glucose consumption, but increased more sharply with faster growth (Fig. 2a). Contrary to the idea that aerobic glycolysis is initiated strictly by proliferation, the relative amount of lactate produced per glucose consumed increased only mildly (about 15%), over the range of measured growth rates (Fig. 2b), and this relationship held when considering only glucose-derived lactate as assessed through [U-13C6]glucose labeling (Fig. 2c). These results are similar to behavior observed previously in fibroblasts [20] and another HMEC line [21], but unlike fibroblasts, near-quiescent HMECs did not significantly increase protein secretion into the culture medium (Fig. 2d), indicating that increased protein production was not responsible for the continued aerobic glycolysis in these quiescent cells.
Figure 2. Glycolytic and protein production fluxes in EGF-titrated EV-HMECs.
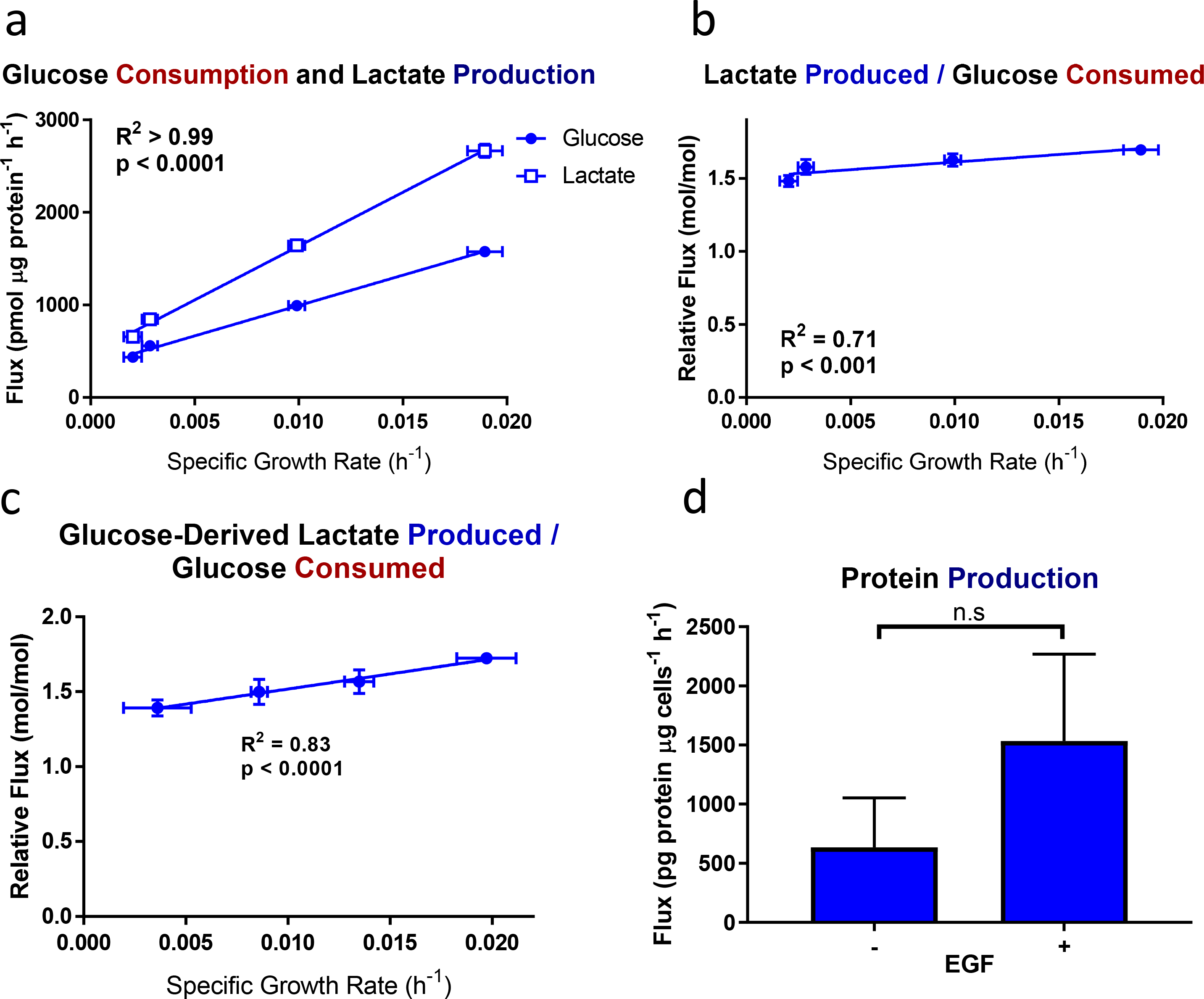
a, Glucose consumption and lactate production, b, lactate produced per glucose consumed, c, glucose-derived lactate produced per glucose consumed, and d, net protein production as functions of specific growth rate in EV-HMECs under variable concentrations of EGF.
For a-b, error bars represent mean ± SD for n = 3 wells, and results are representative of three replicate experiments. For c, error bars represent ± SD for n = 3 wells; results are representative of two replicate experiments. P-values for a-c reflect the likelihood that the regression line slopes differ from zero and were determined using the extra sum-of-squares F-test. For d, error bars represent ± SD for n = 2 or 3 wells; a single experiment was performed. n.s., not significant (Student’s t-test).
Glutamine consumption decreases with quiescence despite continued TCA cycle activity
After glucose, glutamine is typically the most highly consumed carbon substrate in cultured mammalian cells [27]; it is a major nitrogen donor and TCA cycle substrate, and much of it is deaminated and secreted as glutamate [29]. In contrast to glucose and lactate fluxes, glutamine consumption and glutamate production both approached zero as proliferation ceased in EV-HMECs (Fig. 3a). The relative glutamate production per glutamine consumed ranged from half of glutamine consumption at maximal growth rates to apparent net glutamate consumption in EGF-starved cells (Fig. 3b). Actual glutamate consumption was difficult to establish given that this substrate, along with pyruvate and aspartate, is not included in base DMEM, although trace amounts may come from undefined media components. However, glutamate consumption was apparent at low growth rates when the cells were cultured in pyruvate/aspartate/glutamate-supplemented medium (Fig. 3c). This growth-associated shift in substrate use was also evident when considering the ratio of glutamine uptake to glucose uptake, which shifted from 13% to 1% in cells growing at the highest to lowest rates (Fig. 3d).
Figure 3. Glutamine- and TCA cycle-associated fluxes in EGF-titrated EV-HMECs.
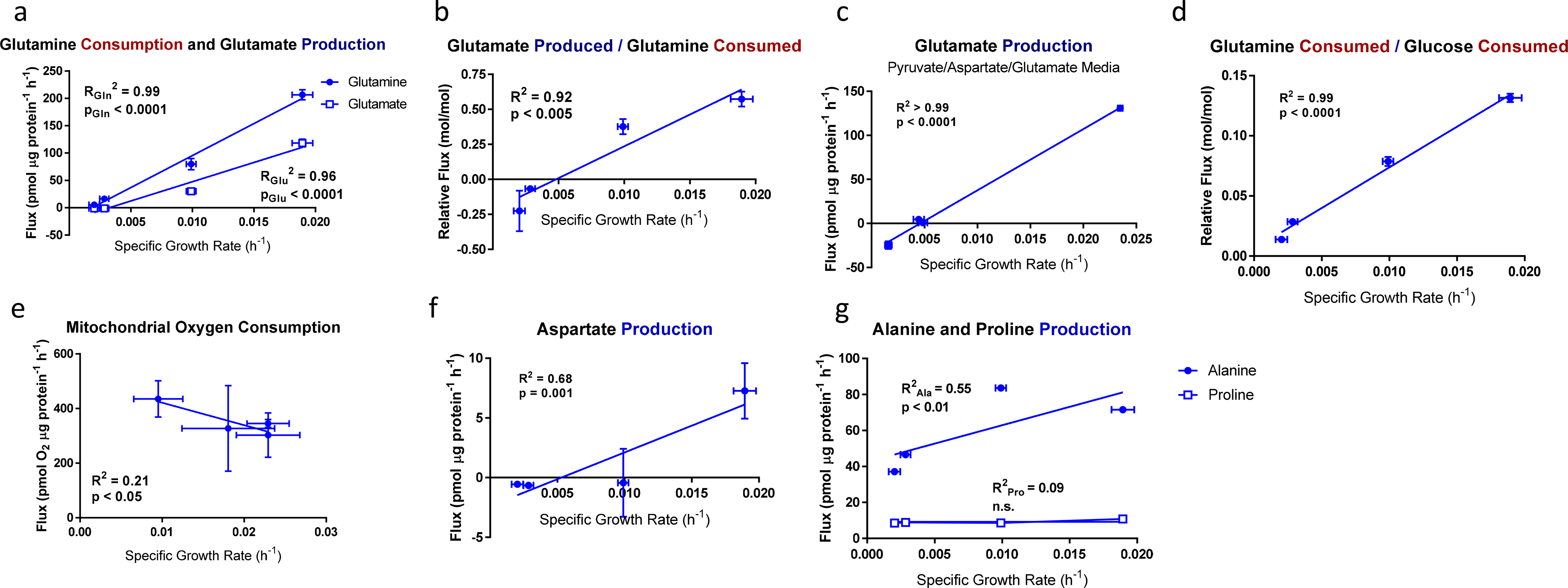
a, Glutamine consumption and glutamate production; b, glutamate produced per glutamine consumed; c, Glutamate production in medium supplemented with glutamate, aspartate, and pyruvate; d, glutamine consumed per glucose consumed; e, mitochondrial oxygen consumption f, aspartate production; and g, alanine and proline production as functions of specific growth rate for EV-HMECs under variable concentrations of EGF.
For a-b, d, and f-g, error bars represent mean ± SD for n = 3 wells, and results are representative of three replicate experiments. For c, error bars represent ± SD for n = 3 wells; a single experiment was performed. For e, error bars represent mean ± SD for n = 5 wells; a single experiment was performed. P-values reflect the likelihood that the regression line slopes differ from zero and were determined using the extra sum-of-squares F-test.
For most cancer cells in culture, glutamine is the major anaplerotic substrate for the TCA cycle and hence is important for maintaining respiration [30]. However, despite a stark decrease in glutamine consumption, mitochondrial oxygen consumption increased as growth rate decreased (Fig. 3e). This shift, along with the mild association of glucose to lactate conversion with growth, typifies the oxidative-to-glycolytic transition associated with increased proliferation [11]. One explanation for how respiration could be maintained despite a lack of glutamine consumption in near-quiescence is that biosynthetic and secretory fluxes (e.g. glutamate and aspartate production) decreased markedly with reduced growth (Figs. 3a, 3f) thereby requiring less anaplerosis to maintain respiration, although alanine and proline were still secreted at relatively high rates (Fig. 3g). Regardless, sustained respiratory activity requires a continuous influx of acetyl-CoA (AcCoA) into the TCA cycle [12], which could be from glucose-derived pyruvate or other sources such as β-oxidation [31]. Given the decrease in glucose consumption observed at low growth (Fig. 2a), alternative substrates may be necessary for continued respiration in these near-quiescent cells.
Lipogenesis is maintained under quiescence with reduced contribution from glucose
AcCoA is also polymerized to form fatty acids and cholesterol, and lipogenesis can be substantial in non-proliferating cells despite reduced biomass production [32]. We used isotopomer spectral analysis (ISA) [34] to quantify the fractional substrate contributions to palmitate synthesis (also called DS, where S refers to the substrate measured) as well as the proportion of palmitate synthesized de novo following 13C label addition (also called g(t)). In brief, DS is a normalized measure of the contribution of a substrate S of a particular biosynthetic product; and g(t) is a measure of overall production rate, indicating the fraction of that has been newly synthesized from the time t of tracer addition to metabolite extraction [33]. ISA using [U-13C6]glucose and [5-13C]glutamine showed that, while de novo synthesis of palmitate increased with growth rate, it remained considerable even in near-quiescent conditions, as an eightfold reduction in growth rate gave only an estimated 34 to 38% decrease in g(t) (Figs. 4a–b). Nominal contribution of [5-13C]glutamine via reductive carboxylation to lipogenic AcCoA (DGln,RC) changed little, ranging nonlinearly over a span of 10% across different growth rates (Fig. 4c); and total glutamine contribution (DGln,tot), as assessed by [U-13C5]glutamine labeling, was similar in magnitude and trend (Fig. 4d). Given the drastic decrease in glutamine consumption with slower growth, the apparent lipogenic glutamine contribution likely resulted in part from rapid exchange of intracellular and extracellular glutamine pools [34]. However, the contribution of glucose to the lipogenic AcCoA pool did decrease with lower growth rates (Fig. 4e), which suggested that, in addition to respiration, continued lipid biosynthesis also required additional carbon sources in quiescent cells.
Figure 4. ISA estimates of lipogenic behavior in EV-HMECs under varying rates of EGF-stimulated proliferation.
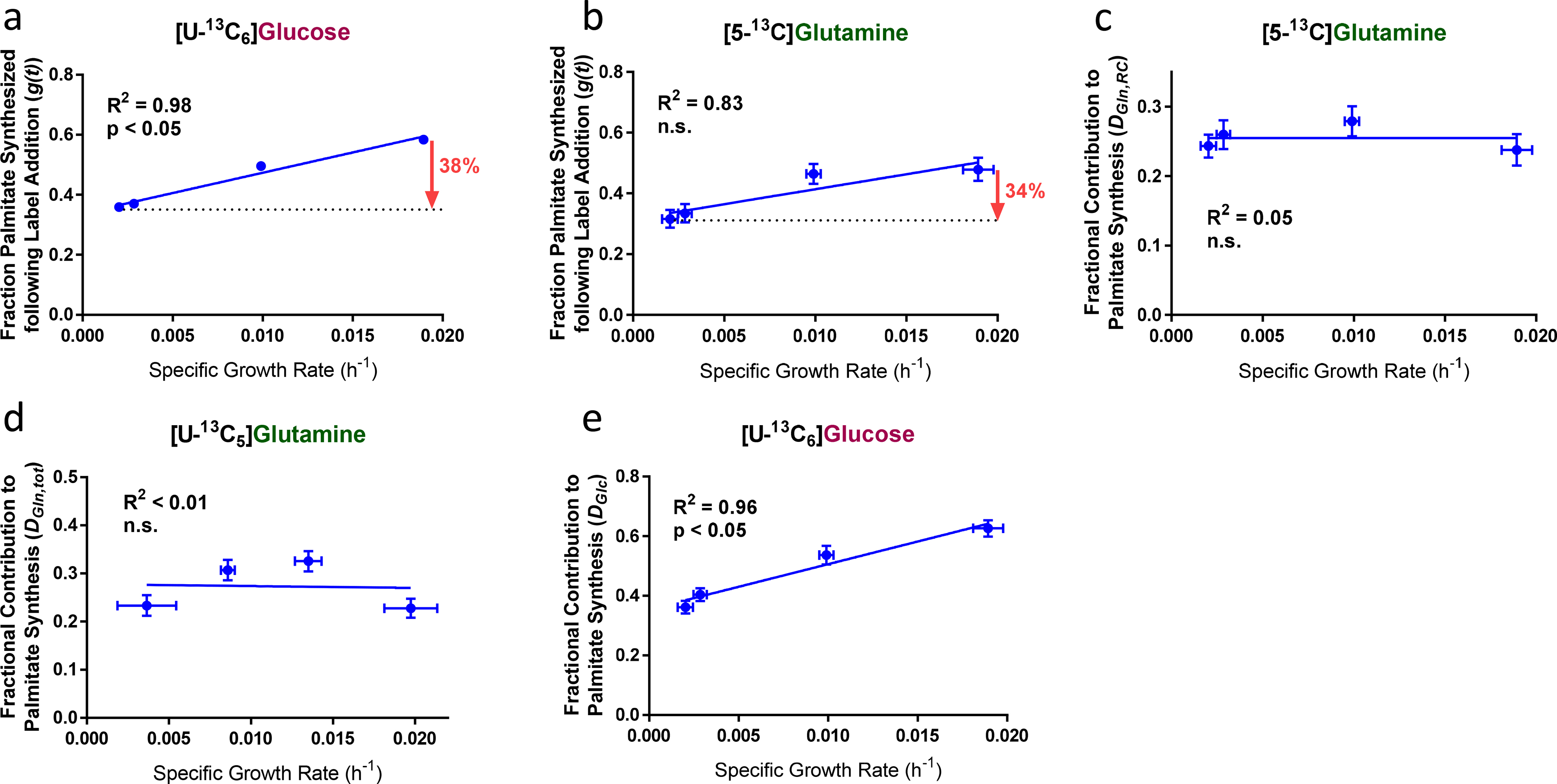
ISA estimates for a, fraction of palmitate synthesized following [U-13C6]glucose addition, b, fraction of palmitate synthesized following [5-13C]glutamine addition, c, fraction of palmitate contributed by [5-13C]glutamine, d, fraction of palmitate contributed by [U-13C5]glutamine, and e, fraction of palmitate contributed by [U-13C6]glucose as functions of growth rate in EV-HMECs under variable concentrations of EGF.
Analyzed MIDs were averages of n = 3 wells, error bars represent 95% confidence intervals, and results are representative of two replicate experiments. P-values reflect the likelihood that the regression line slopes differ from zero and were determined using the extra sum-of-squares F-test.
Branched chain amino acids undergo a catabolic-to-proteinogenic shift with growth rate
We next investigated what other substrates may sustain continued respiration and fatty acid synthesis in quiescent cells. Although fatty acids in culture medium or serum can be oxidized to AcCoA, palmitate consumption did not increase with decreased growth (Fig. 5a). However, the consumption rates of branched-chain amino acids (BCAAs) leucine, isoleucine, and valine, which can also contribute AcCoA and succinyl-CoA to the TCA cycle, remained substantial as growth rates decreased (Fig. 5b). Compared to other measured substrates, BCAAs had the smallest reduction in consumption upon EGF withdrawal, and isoleucine consumption appeared to increase under near-quiescence (Figs. 5b–c).
Figure 5. BCAA and fatty acid metabolism in near-quiescent and actively proliferating EV-HMECs.
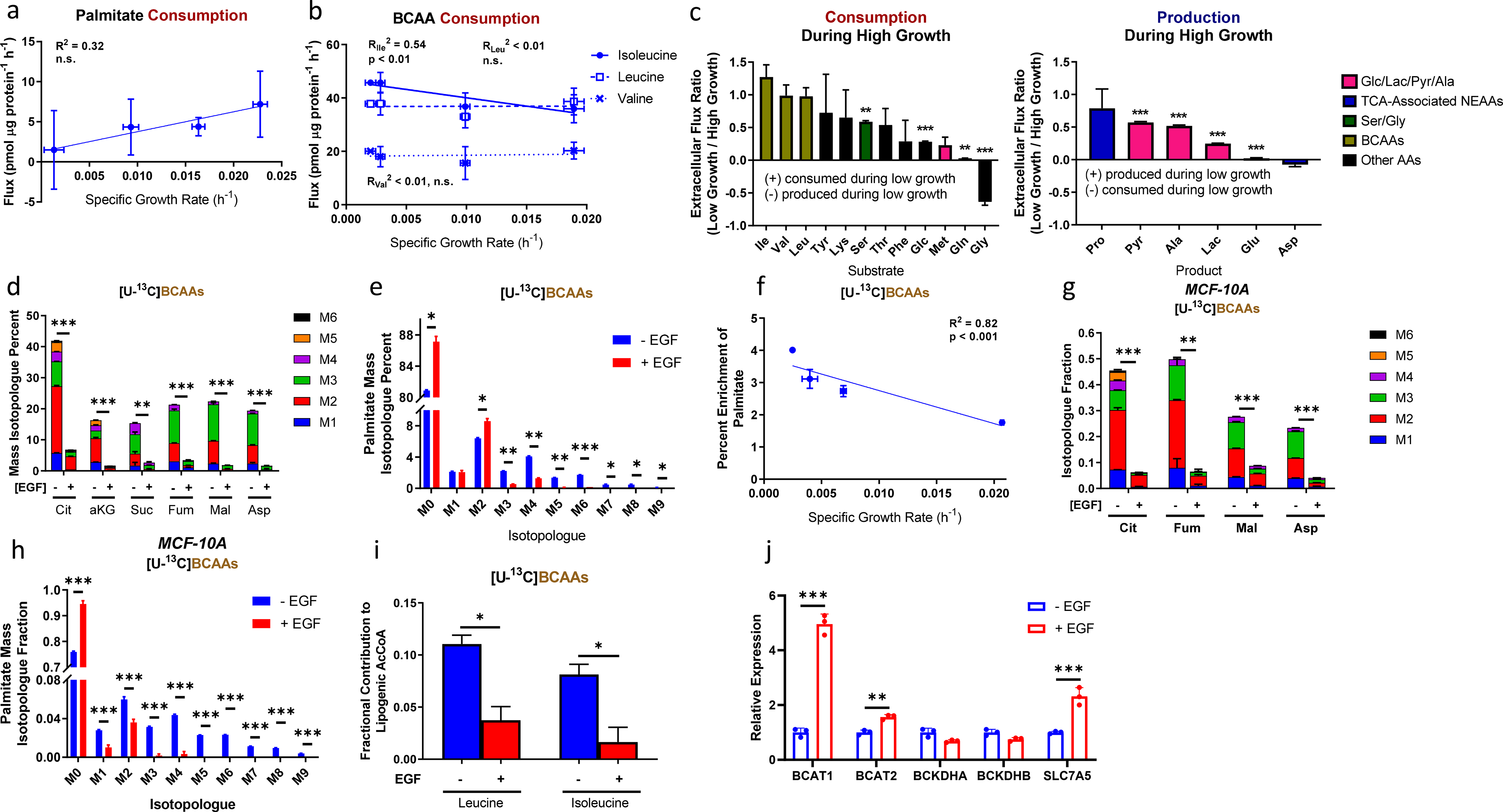
a, Palmitate consumption as a function of specific growth rate for EGF-titrated EV-HMECs.
b, BCAA consumption fluxes in EV-HMECs under variable levels of EGF stimulation as functions of growth rate.
c, Fluxes of metabolite (left) consumption and (right) production for low-growth (EGF-starved) relative to high-growth (5 ng/ml EGF) HMECs. Negative values represent flux directionality reversing upon EGF withdrawal.
d, Mass isotopologue distributions of labeled TCA cycle metabolites for EV-HMECs in growth-saturating EGF (+EGF) and no EGF (−EGF) cultured in [U-13C]BCAAs. Legend denotes different states of isotopologue enrichments (M1 = one 13C atom, M2 = two 13C atoms, etc.).
e, Mass isotopologue distribution of palmitate for EV-HMECs in 5 ng/ml EGF (+EGF) and no EGF (−EGF) cultured in [U-13C]BCAAs.
f, Percent enrichment of palmitate by [U-13C]BCAAs as a function of specific growth rate in EV-HMECs under variable concentrations of EGF.
g-h. Mass isotopologue distributions of g, TCA cycle metabolites and h, palmitate for MCF-10A cells in 5 ng/ml EGF (+EGF) and no EGF (−EGF) cultured in [U-13C]BCAAs.
i, ISA estimates of fractional contributions to lipogenic AcCoA by [U-13C6]leucine and [U-13C6]isoleucine for HMECs cultured in 5 ng/ml EGF (+EGF) or media without EGF (−EGF).
j. Relative mRNA levels of the BCAA catabolic enzymes BCAT1, BCAT2, BCKDHA, BCKDHB, and transporter SLC7A5 from EV-HMECs cultured in 5 ng/ml EGF (+EGF) or media without EGF (−EGF).
For a, error bars represent mean ± SD for n = 2 or 3 wells; a single experiment was performed. For b-c, error bars represent mean ± SD with n = 3 wells; results are representative of three replicate experiments. For d-h, error bars represent mean ± SD for n = 2 (for d-f) or 3 (for g-h) wells; results are representative of two replicate experiments. For i, error bars represent 95% confidence intervals and analyzed MIDs were averages of n = 3 wells; a single experiment was performed. For j, error bars represent mean ± SD for n = 3 wells for a single experiment. P-values for a, b, and f reflect the likelihood that the regression line slopes differ from zero and were determined using the extra sum-of-squares F-test. * p < 0.05, ** p < 0.01, *** p < 0.001 (Multiple t-test with adjusted p-value, except Fig. 5i, for which statistical significance is based on 95% confidence interval non-overlap).
We cultured HMECs under varying EGF concentrations in [U-13C]BCAAs to observe their incorporation into central carbon metabolism. Compared to conditions with 5 ng/ml EGF, the fractions of TCA cycle metabolites labeled from BCAAs were increased upon EGF withdrawal (Fig. 5d), indicating that contribution of BCAA-derived AcCoA to the TCA cycle is one way to maintain respiration when growth decreases. Additionally, citrate serves as a source of lipogenic AcCoA via conversion through ATP-citrate lyase, and labeling of citrate from BCAAs in media lacking added EGF suggested an increase in BCAA contribution to fatty acid synthesis under near-quiescence. This hypothesis was supported by palmitate labeling from BCAAs, which showed a net shift from lighter to heavier isotopologues upon EGF withdrawal (Fig. 5e) as well as an inverse correlation between total 13C enrichment and growth rate (Fig. 5f). We observed similar increases in enrichment from [U-13C]BCAAs in TCA cycle metabolites and palmitate in MCF-10A cells upon EGF starvation (Figs. 5g–h), demonstrating that this behavior is not specific to the 184A1 HMEC line. Of the three BCAAs, leucine and isoleucine can contribute most directly to lipogenesis through catabolism to AcCoA. We used ISA to quantify the contribution of these two substrates to palmitate production and found that, following growth factor withdrawal, the use of the amino acids increased roughly four-fold (Fig. 5i), which accounted for approximately half of the corresponding decrease in glucose contribution to lipogenesis (Fig. 4e).
We examined whether this apparent increase in BCAA-derived carbon contribution to the TCA cycle and lipogenesis was mediated by changes in expression of the BCAA catabolic enzymes BCAT1/2, BCKDHA/B, or the major BCAA transporter SLC7A5. Unexpectedly, we observed decreases in BCAT1, BCAT2, and SLC7A5 mRNA expression upon EGF removal (80, 36, and 57%, respectively; Fig. 5j). These data suggest that mitogen withdrawal transcriptionally downregulates elements of BCAA metabolism, similar to glucose and glutamine metabolism [13,36,37]. However, unlike glucose and glutamine metabolism, reduced transcriptional activation of BCAA metabolism does not decrease pathway flux within the time period assessed in this study (Fig. 5b).
Some metabolic phenotypes in oncogenic KRAS HMECs are not explained by growth rate
We next performed a similar metabolic analysis in KRAS-HMECs and compared the findings to those observed in EV-HMECs, allowing an assessment of how mutant KRAS expression changes metabolism and to what extent this was accounted for by growth rate. Linear regression was first applied to metabolic indicators observed in EV-HMECs cultured under variable EGF concentrations as functions of specific growth rate. These regression models served to “predict” metabolic behavior in HMECs based on growth rate. The outlier shift model, which describes the likelihood that a given result may be an outlier of a particular data set, was then used to evaluate whether metabolic changes resulting from constitutively active KRAS in HMECs in either 5 or 0 ng/ml EGF (designated +EGF and –EGF respectively) matched those predicted by growth rate alone. We established two thresholds to designate KRAS-associated differences from growth-based trends: (a) magnitude difference from the EV-HMEC regression line, which varied depending on the measured parameter (± 10% difference in the predicted value for extracellular fluxes and pool sizes, and ± 5% difference from the predicted value for metabolite percent enrichment from 13C tracers), and (b) statistical significance, which was fixed at p = 0.025 (p = 0.05 ÷ 2 to account for both + and –EGF tests). To be considered noteworthy, measurements had to exceed both thresholds. (The R code used to generate the linear regression models and calculated magnitude difference and statistical significance parameters is given in Supp. File 1, and the inputs and outputs to this code are given in Supp. File 2.) Compared to EV-HMECs at maximal growth, KRAS+EGF cells grew at roughly the same rate, whereas KRAS-EGF cells tended to grow about 15% more slowly.
Most measured differences in KRAS(G12V)-expressing HMECs from growth-dependent trends in EV-HMECs were small and/or statistically insignificant. No intracellular metabolite concentrations exceeded both the statistical significance and 10% difference thresholds with the exception of lactate, which decreased 27% in KRAS-EGF cells (Figs. 6a–b). We detected no differences in percent carbon enrichment in metabolites labeled by [U-13C6]glucose, [U-13C5]glutamine, or [U-13C]BCAAs (Figs. 6c–d) that exceeded either the statistical significance or 5% enrichment difference thresholds. Additionally, ISA parameters for palmitate in KRAS-HMECs during culture in [U-13C6]glucose and [5-13C]glutamine fell or were projected to fall within 95% confidence bands from growth-dependent trends (Figs. 6e–h). These results suggested that KRAS-HMECs behaved largely similar to what EV-HMECs undergoing EGF stimulation would predict with respect to metabolite levels, relative contributions of major substrates, and lipogenesis. Most extracellular fluxes followed a similar pattern. No KRAS+EGF cell fluxes consistently registered as statistically significant from predicted rates (Fig. 7a). Collectively, our data suggested that KRAS- and EV-HMECs exhibited similar metabolic behaviors when cultured under maximal EGF stimulation.
Fig. 6. Comparison of metabolic indicators in EV- and KRAS-HMECs.
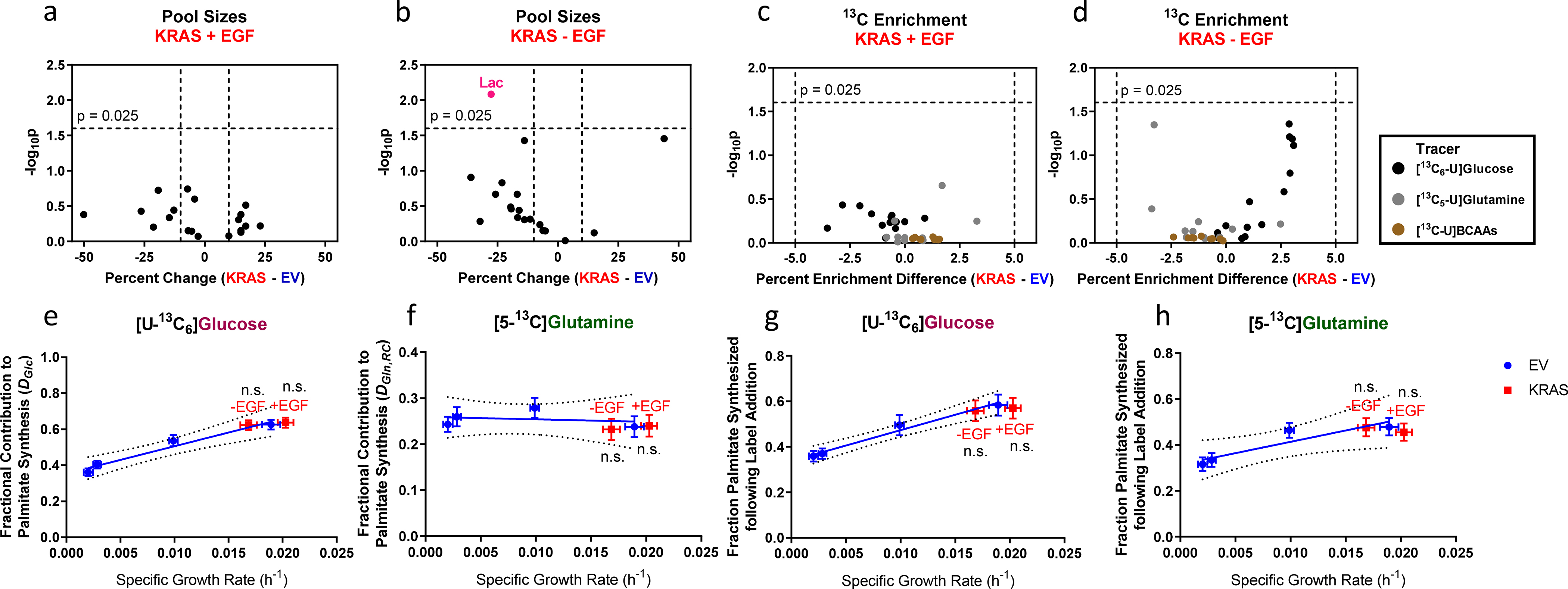
a-d. a-b, Volcano plots of pool sizes in KRAS-HMECs cultured in a, 5 ng/ml EGF and b, 0 ng/ml EGF; and c-d, 13C enrichment from [U-13C6]glucose, [U-13C5]glutamine, and [U-13C]BCAAs in KRAS-HMECs cultured in c, 5 ng/ml EGF and d, 0 ng/ml EGF. Plots show percent change and statistical significance of differences from growth-based trends in EV-HMECs.
e-h. ISA estimates for e, fraction of palmitate contributed by [U-13C6]glucose, f, fraction of palmitate contributed by [5-13C]glutamine, g, fraction of palmitate synthesized following [U-13C6]glucose addition, and h, fraction of palmitate synthesized following [5-13C]glutamine addition in EGF-titrated EV-HMECs and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
In a-d, values in pink indicate where statistical significance and magnitude difference thresholds are exceeded. In e-h, analyzed MIDs were averages of n = 3 wells; error bars represent 95% confidence intervals. P-values were determined using outlier shift model. Results are representative of two replicate experiments.
Figure 7. Comparison of metabolic fluxes in EV- and KRAS-HMECs.
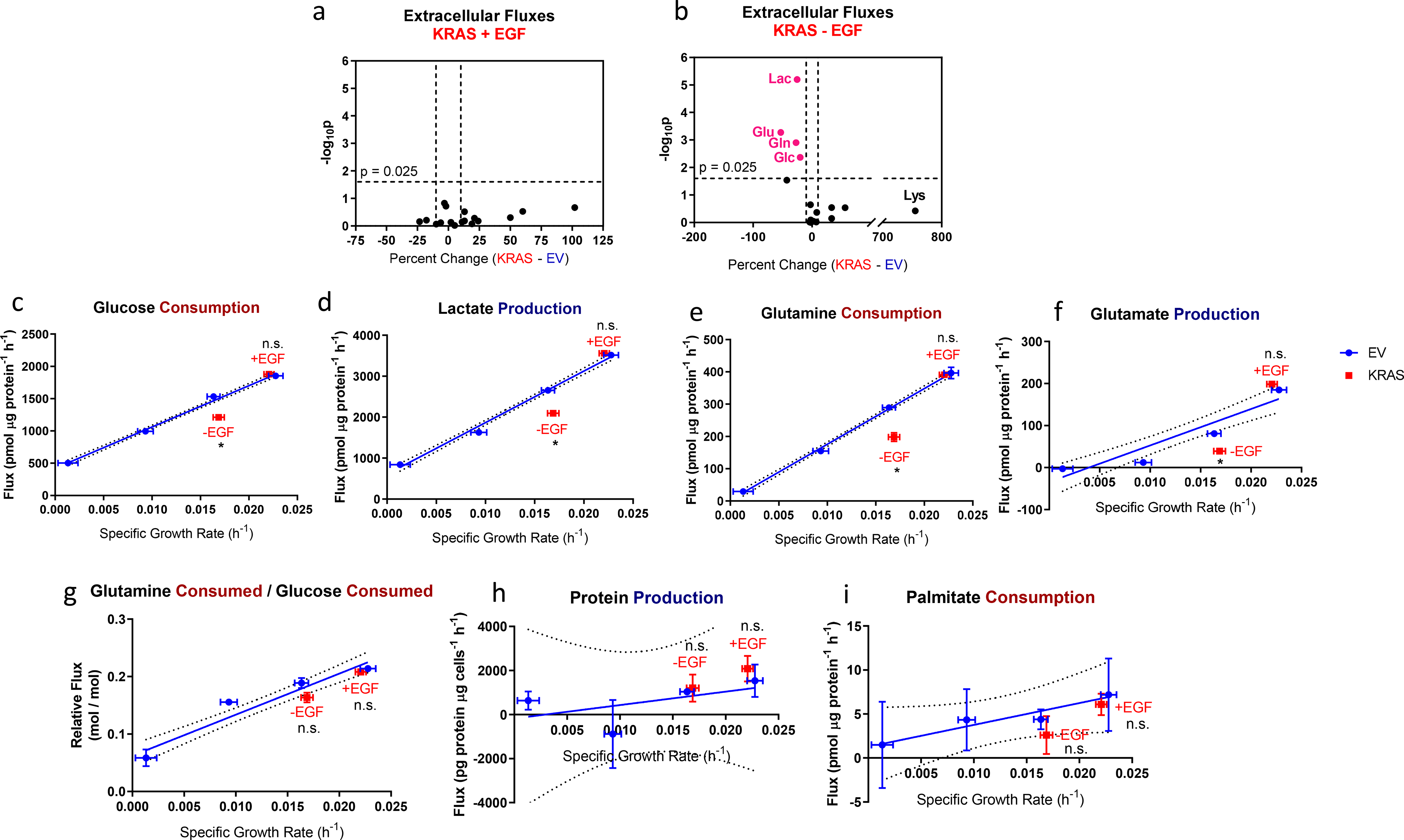
a-b. Volcano plots of extracellular fluxes from KRAS-HMECs cultured in a, 5 ng/ml EGF and b, 0 ng/ml EGF showing percent change and statistical significance of differences from growth-based trends in EV-HMECs. Values in pink indicate where statistical significance and magnitude difference thresholds are exceeded.
c-f. c, Glucose consumption, d, lactate production, e, glutamine consumption, and f, glutamate production vs. growth rate for EGF-titrated EV-HMECs and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
g. Glutamine consumed per glucose consumed vs. growth rate for EGF-titrated EV-HMECs and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
h-i. h, Net protein production and i, palmitate consumption vs. growth rate for EGF-titrated EV-HMECs and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF).
Error bars in c-f represent mean ± SD for n = 2 or 3 wells. Results are representative of three replicate experiments. In g, error bars represent ± SD for n = 2 or 3 wells; results are representative of three replicate experiments. In h-i, error bars represent ± SD for n = 2 wells. A single experiment was performed for each. n.s., not significant, p < 0.025 (Outlier shift model).
However, for KRAS-HMECs cultured in the absence of EGF, the four largest fluxes measured – glucose consumption, lactate production, glutamine consumption, and glutamate production – met both magnitude change and statistical significance thresholds in every experiment we performed (Figs. 7b–f). Each of these fluxes was lower than the predicted values, with glucose, lactate, and glutamine fluxes decreasing by about 20 to 30% and glutamate production decreasing by about 35 to 50%. Despite these differences, the ratio of glutamine to glucose consumption failed to register as significantly different from the growth-based trend (Fig. 7g). This suggested robust coordination between the two nutrients in both the EV and KRAS cells and potentially explained the lack of apparent differences in labeling data, which reflect relative, rather than absolute, differences in substrate use. We also found lysine consumption to be consistently higher than predicted, but these differences were only statistically significant in one experiment. Aside from one experiment in which isoleucine consumption in KRAS-EGF cells was 11% lower than predicted (p = 0.021), differences in BCAA fluxes failed to exceed fold change and statistical significance thresholds (Table 1).
Table 1.
BCAA extracellular flux behavior of KRAS-HMECs relative to EV-HMEC trends expected for corresponding growth rates. Percent changes are reported, with p-values given in parentheses.
| KRAS + EGF | KRAS - EGF | |||||
|---|---|---|---|---|---|---|
| Experiment | Isoleucine | Leucine | Valine | Isoleucine | Leucine | Valine |
| 1 | +13% (0.66) | +18% (0.72) | +21% (0.52) | +19% (0.24) | +18% (0.26) | +15% (0.70) |
| 2 | −7.5% (0.038) | −2.3% (0.39) | +7.0% (0.45) | −2.9% (0.22) | −2.4% (0.81) | +8.0% (0.43) |
| 3 | −8.6% (0.12) | −8.7% (0.032) | −5.1% (0.70) | −11% (0.021) | −7.6% (0.56) | −5.1% (0.73) |
Although oncogenic KRAS expression has been previously associated with scavenging of nontraditional substrates such as bulk protein [35] and lysophospholipids [36], net protein efflux into the media and palmitate uptake did not deviate notably from growth-dependent trends (Figs. 7h–i). Therefore, the decreases in major carbon substrate fluxes represent increases in the yield of cell biomass substrate consumed, suggesting that HMECs undergoing oncogenic-KRAS-driven growth may be more metabolically efficient compared to those undergoing EGF-stimulated proliferation under the culture conditions tested.
Oncogenic KRAS-driven HMECs are more resistant to metabolic inhibitor treatment than EGF-driven control cells
Numerous reports have described selective tumor cell death or cytostasis caused by blocking metabolic pathways that are hyperactivated due to mutation or gene overexpression [37,38]. We probed how KRAS-HMECs and EV-HMECs would respond to glucose transport inhibition with STF-31, which blocks GLUT1 [42], and respiratory inhibition with phenformin, which blocks mitochondrial complex I. GLUT1 is the most highly expressed glucose transporter in 181A1 cells [43]. The ratio of growth rates of inhibitor-treated to control DMSO-treated EV-HMECs was approximately linear over moderate-to-high control growth rate values (Fig. 8), so regression models were constructed across this range and compared to KRAS-HMEC behavior. As expected, inhibitor treatment resulted in slower growth for all HMEC/EGF treatment combinations, although the fractional reduction in EV-HMEC growth rate tended to decrease with lower EGF concentrations (Fig. 8). KRAS+EGF cells responded to these inhibitors in a manner predicted by growth-based trends in EV-HMECs (Fig. 8). However, KRAS-EGF cells were more resistant to inhibitor treatment (Fig. 8). These results, along with the lower-than-predicted glucose and glutamine uptake rates of KRAS-EGF cells (Figs. 7b–c, e), suggest that metabolic differences in oncogenic KRAS-driven cells can also improve their resistance to metabolic inhibition in the absence of EGF. However, the decreased sensitivity to inhibitor treatment might also occur for reasons other than differences in metabolic inhibition between EGF- and oncogenic KRAS-stimulated cells. For instance, these cells may exhibit differences in apoptosis resistance, drug efflux activity, or metabolism of the small molecule inhibitors.
Figure 8. Comparison of metabolic inhibitor sensitivities in EV- and KRAS-HMECs.
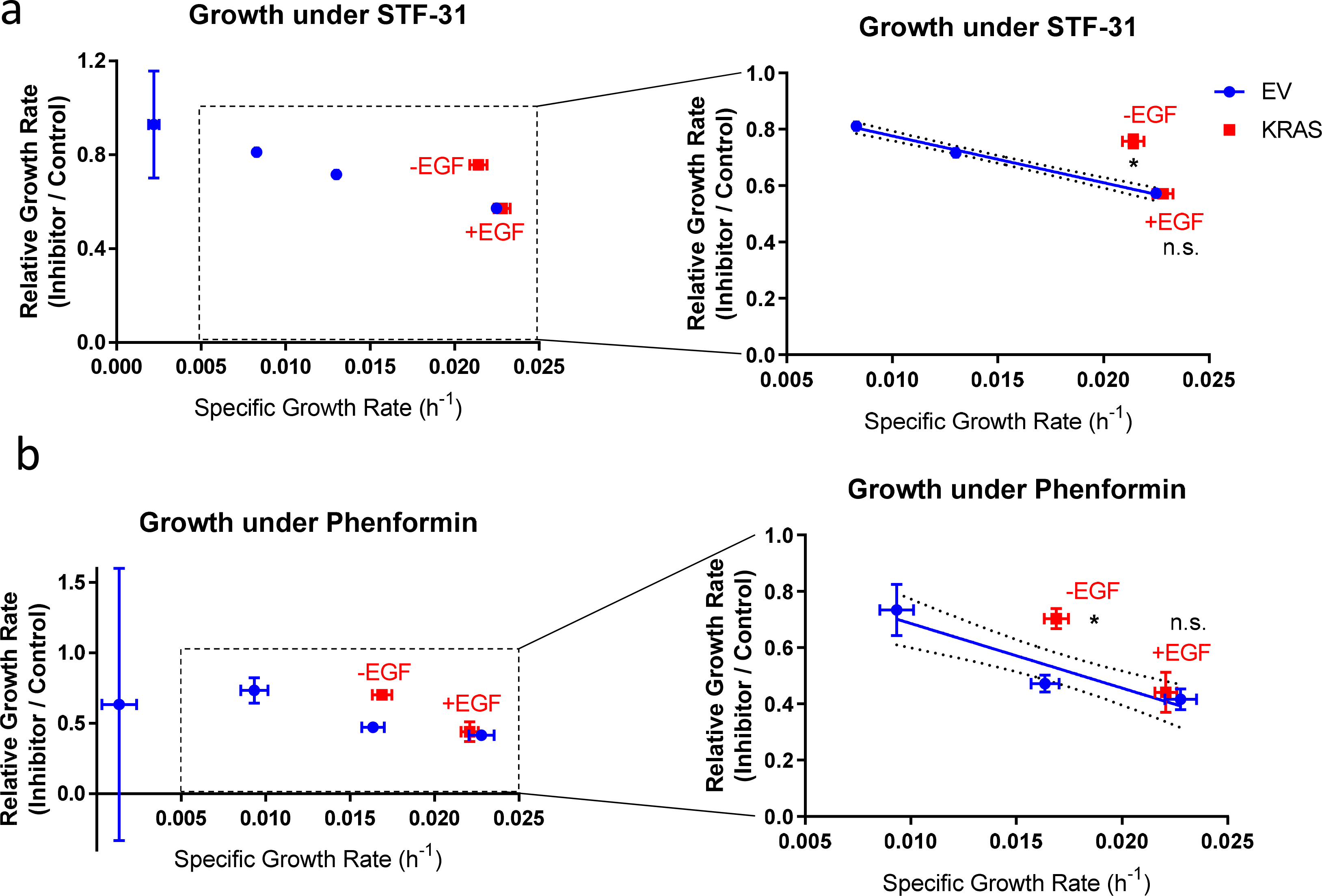
Ratio of inhibitor-treated to DMSO control growth rates for EGF-titrated EV-HMECs (excluding EGF-free condition) and KRAS-HMECs cultured in 5 ng/ml EGF (+EGF) and 0 ng/ml EGF (−EGF). Inhibitors: a, 10 μM STF-31 (GLUT1 inhibitor) and b, 10 μM phenformin (respiratory complex I inhibitor). The complete set of data are shown on the left, and zoomed perspectives on the KRAS-HMEC points along with regression line through the EV-HMEC points at the three highest growth rates are shown on the right.
Error bars represent mean ± SD for n = 2 or 3 wells. A single experiment was performed for each inhibitor. n.s., not significant, p < 0.025 (Outlier shift model).
Discussion
Growth is a biochemically intensive process, and dramatic metabolic remodeling is required to provide the cofactors and building blocks necessary to create new cells [40]. Our observation that growth in EGF-stimulated HMECs was generally associated with larger nutrient consumption rates (Fig. 5c) supports this principle and mirrors the results of prior studies [20,21]. Yet, as with quiescent fibroblasts [20] and contact-inhibited HMECs [21], glucose-to-lactate conversion persisted with only slight abatement in cells upon EGF starvation (Fig. 2b), suggesting that aerobic glycolysis in mammalian cells cannot be solely explained by growth. Furthermore, net protein secretion did not increase with reduced growth (Fig. 2d), which indicated that quiescence-associated extracellular matrix protein production could not explain the highly fermentative metabolism in this system [20]. The observed decline in glutamine consumption and glutamate production upon growth inhibition (Fig. 3a) matches behavior seen in contact-inhibited MCF-10A HMECs [21], but differs from results in quiescent fibroblasts, where no such reduction occurred [20]. The discrepancy in the extent of this quiescence-associated decrease in substrate uptake may result from the differential roles of glucose and glutamine in fueling cell proliferation. In cultured mammalian cells, glucose oxidation is proposed to be the predominant source of ATP, while glutamine is the single greatest source of nitrogen, and glucose and glutamine can both be significant sources of carbon [29,45,46]. If proliferation stops, the requirements of producing new biomass might be expected to result in reductions in both glucose and glutamine uptake. However, maintenance ATP costs not associated with macromolecular biosynthesis can still be substantial under quiescence, potentially even exceeding growth-associated energetic needs [44,47]. Quiescent cells may therefore maintain glucose consumption and oxidation for survival, but reduce glutamine uptake to levels approaching zero, as observed in EGF-starved HMECs.
BCAAs were one class of substrates whose uptake did not increase with faster proliferation (Figs. 5b–c), and their proteinogenic-to-catabolic shift in metabolic fate closely paralleled similar behavior in growing preadipocytes upon their differentiation into quiescent adipocytes [41]. However, additional studies are necessary to understand the extent to which, for instance, quiescence-associated BCAA catabolism and contribution to fatty acid synthesis is general to other mammalian cells, as well as the underlying mechanism behind this growth-associated shift in substrate use. Despite maintaining constant rates of BCAA consumption, HMECs exhibited decreased expression of BCAT1, BCAT2, and the BCAA transporter SLC7A5 transcripts (Fig. 5j). It is unclear why a discrepancy between transcription and metabolic flux is observed, but possible reasons include a disconnect between protein expression and mRNA levels or persistence of high enzyme activity despite reduced expression. Many metabolic enzymes are expressed in excess and are not rate limiting for pathway flux, and several factors may maintain activity despite lower expression, including post-translational-mediated changes in enzyme activity (e.g. BCKDH (de)phosphorylation [49]). BCKDH has been described as the rate-limiting step of BCAA catabolism [50], and though the differences were not statistically significant, a trend toward elevated transcript levels of the BCKDHA/B subunits following EGF removal was observed (Fig. 5j). Although we anticipate the growth rate reduction upon EGF withdrawal may also lead to the redirection of BCAA flux from protein synthesis toward oxidation and lipogenesis, the average 8-fold increase in TCA cycle metabolite labeling by [U-13C]BCAAs (Fig. 5d) likely primarily reflects the approximate 4-fold and greater than 10-fold reductions in glucose and glutamine uptake, respectively (Fig. 5c). Regardless, these data suggest a distinct shift in the relative contribution of BCAAs to metabolism in proliferating versus quiescent HMECs. Although our experiments did not identify additional BCAA metabolic alterations characteristic to KRAS-HMECs, other works suggest that alterations in BCAA metabolism contribute to cancer growth and survival in a manner that is influenced by the cancer tissue of origin [42–45].
It is likely that BCAA metabolism is strongly influenced by nutrient levels [55] and, more generally, by the cell microenvironment [56]. Optimal HMEC culture requires serum-free media, which is thus lipid-depleted compared to media supplemented with 10% serum; nonetheless, our HMEC culture media contains approximately 0.5% by volume bovine pituitary extract as an alternative source of lipids. The lipid levels found in serum-containing media may enable higher rates of lipid scavenging, and hence potentially lower levels of BCAA contribution to lipid synthesis than those observed in our study. Likewise, although uptake of lipid-derived palmitate was used as a surrogate for lipid scavenging (Fig. 7i), the complexity of lipid species makes this an imperfect measure. More extensive lipidomic analyses have identified preferential uptake of select lipid species, such as lysophospholipids containing unsaturated fatty acids, following oncogenic KRAS expression [39]. Thus, a more extensive characterization of lipid metabolism in lipid-enriched media will be important for resolving growth- and oncogene-dependent features of lipid metabolism in future studies.
In comparing metabolic indicators between HMECs driven by oncogenic KRAS to EGF-stimulated control cells, we found that few required factors beyond growth rate for their prediction under the designated statistical thresholds. This result, shown across a single parental cell line, mirrored the strong influence of proliferation on metabolism that other studies have demonstrated across multiple cell lines and tissue types [21,27]. However, we also unexpectedly observed that that HMECs driven by oncogenic KRAS reduce substrate uptake rates relative to EGF-stimulated control cells growing at comparable rates. The means through which KRAS-expressing HMECs are able to do this are unclear, given the energy demands for proliferation, but may include involve a greater coupling between respiration and ATP production or a decrease in ATP synthesis/expenditure futile cycles. Though we were unable to confidently identify a selective metabolic vulnerability in KRAS-HMECs, given the ability of pyruvate kinase M2 to shunt glycolytic flux toward NADPH production via the pentose phosphate pathway (PPP) following oxidative stress, the relatively reduced glucose uptake rate of KRAS-driven HMECs may sensitize them to acute PPP inhibition or pro-oxidant treatment compared to similarly growing control HMECs [57]. Previous reports have described increases in glucose and glutamine uptake upon induction of mutant KRAS [17,58]. KRAS is associated with mitogenic downstream signaling that effects enhanced glucose and glutamine metabolism [7,11,13], so increased activity of these pathways could reasonably follow from elevated proliferation alone. Correspondingly, compared to nearly quiescent EV-HMECs under EGF withdrawal, major metabolic fluxes are considerably elevated in EGF-starved KRAS-HMECs, which continue to grow relatively fast (Figs. 7c–f). However, contrary to our results, several of these reports observed faster glucose consumption and lactate production even when oncogenic KRAS expression does not appear to lead to a substantial increase in proliferation [17,47]. The reasons for these discrepancies are unclear, but may include differences in cell line tissue of origin, culture conditions, or perhaps even degree of KRAS overexpression or activation of other signaling pathways.
A major motivation for understanding the metabolic rewiring of cancer cells has been the hope that this effort will lead to new therapies. The established record of antimetabolites in the clinic bolsters the hypothesis that essential metabolic pathways can be successfully targeted to slow tumor formation or induce apoptosis. Many metabolic pathways are used by both healthy and neoplastic tissues, and the abundance of branchpoints and alternative nutrients contribute to the difficulty of maintaining cytotoxic pressure within an acceptable therapeutic window. We have described how increased growth rate via EGF stimulation in an HMEC model extensively remodels substrate use and shown that metabolic behavior in oncogenic KRAS-driven largely follows growth-dependent trends. We also observed that KRAS-HMECs cultured in media lacking added EGF possess lower than expected extracellular fluxes of major substrates based on growth rate alone. However, the absence of statistically significant changes in other indicators of metabolic behavior in oncogenic KRAS-expressing cells suggests a degree of fundamental robustness in metabolism with respect to growth. Our observation that the impact of growth rate on metabolism is much stronger than that of an oncogenic mutation indicates that it may be difficult to distinguish pathways involved in each, consistent with the considerable overlap in signaling networks active in both proliferation and cancer.
To our knowledge, our study provides the first comprehensive metabolic comparison between models of physiological and oncogenic growth that controls for proliferation rate. This framework can be applied to any cell culture system in which growth rate can be tuned and used to control for any cancer-associated genetic or environmental perturbation. In the context of therapeutics that target metabolism, our approach can give insight into the specificity of such targets for cancer cells versus non-transformed proliferating cells. Our results here demonstrate the commanding role that growth plays in influencing metabolism while underscoring the complexity of how oncogenic signaling interacts with each.
Methods & Materials
All supplies were purchased from Sigma unless otherwise noted.
Cell culture
The immortalized HMEC line 184A1 (ATCC) was cultured in a humidified incubator controlled at 37°C and 5% CO2. In between experiments, cells were grown in MCDB 170 medium, which comprised a mammary epithelial basal medium (MEBM) base (Lonza) supplemented with 5 ng/ml EGF (Peprotech), 5 μg/ml insulin, 0.5 μg/ml hydrocortisone, 5 μg/ml transferrin, 70 μg/mL bovine pituitary extract (Hammond), and 10 μM isoproterenol. Cells were cultured on tissue-culture treated polystyrene 10 cm dishes (Corning Falcon), media was replaced every 2–3 days, and cells were subcultured via trypsinization at roughly 80% confluence for up to 4 passages. All experiments were performed in a supplemented Dulbecco’s Modified Eagle’s Medium (DMEM) (Corning Mediatech) containing 10 mM glucose, 4 mM glutamine, and no pyruvate. This supplemented DMEM possessed both the supplements added to MEBM to constitute MCDB 170 (excluding EGF, which varied in each experiment), 10 mM HEPES, 0.1 mM ethanolamine, and 0.1 mM phosphoethanolamine. In some experiments, 0.25 μg/ml anti-EGFR antibody (Sigma, MABF120) was added in EGF-free media to neutralize potential autocrine EGFR signaling and further reduce growth [48]; cells under this condition experienced the same media-change regimen as other variable EGF treatments. Cryostocks were created by resuspending roughly 2–3 × 106 cells into a 1 ml 15:3:2 MEBM:fetal bovine serum:glycerol mixture in cryotubes; cryotubes were then cooled to −80°C in an isopropanol bath and stored in the vapor phase of liquid nitrogen.
For all extracellular flux (excluding oxygen consumption) and labeling experiments, cells were plated in MCDB 170 on tissue-culture treated polystyrene 6-well plates (Corning Falcon). Roughly 24 h later, cells were washed once or twice with phosphate-buffered saline (PBS) (Corning Falcon), and 2.4 ml supplemented DMEM with EGF treatment was added to each well. (To ensure that the phenotypes observed were not principally the result of differences in confluency levels, cell seeding density was varied to give roughly equal confluencies at the conclusion of each experiment. For instance, fast-growing cells in 5 ng/ml EGF were seeded at 62.5 × 103 per well, while slow growing cells in EGF-free medium were seeded at 300–400 × 103 per well.) Roughly 48 h later, cells were again washed once or twice with PBS and 2.4 ml fresh supplemented DMEM with EGF treatment was again added to each well; at this time, cells were lysed for initial protein quantification and for extracellular flux experiments, fresh medium was collected and frozen. After an additional approximate 48 h, cells were again lysed for final protein quantification, spent medium was collected and frozen for extracellular flux experiments, and intracellular metabolites were extracted for labeling experiments.
When pyruvate, aspartate, and glutamate were added to supplemented DMEM, they were incorporated to concentrations of 1 mM, 0.15 mM, and 0.14 mM respectively. For inhibitor experiments, the compounds STF-31 and phenformin were added to 10 μM after determining that these concentrations corresponded to an approximately 50% reduction in growth rate for empty vector-expressing HMECs (EV-HMECs) growing in 5 ng/ml EGF. For cells growing in parallel control conditions, the vehicle DMSO was added to 0.1% to match its concentration in treatment conditions.
Transfection and transduction
Unless otherwise noted, all data shown are from 184A1 cells that have been infected with γ-retrovirus containing either an empty vector or a vector with the oncogenic G12V KRAS mutant under the CMV promoter.
First, HEK293T cells at 50–70% confluency on a 10 cm plate in serum-free DMEM were treated with 26 μl X-tremeGENE Transfection Reagent (Roche) mixed with 3 μg pBABE-puro EV or pBABE-puro-G12V KRAS, 2.7 μg gag/pol vector, and 0.3 μg VSV-G vector (all plasmids provided by Addgene) in DMEM in a total aliquot volume of 186 μl. Cells were incubated with the transfection complex for 36–48 h to produce EV- or KRAS-containing γ-retrovirus. Virus-containing medium was processed through 0.45 μm-pore filters (Pall), mixed with polybrene (Millipore) to a final concentration of 8 μg/ml, and added to HMECs at 35–50% confluency in a 10 cm dish. HMECs were incubated with virus for roughly 24 h before being washed 3 times with PBS, and they were then cultured for at least two passages in 0.5–2 μg/ml puromycin in MEBM to select for transduced cells before being frozen as cryostocks.
Protein extraction for growth rate measurements
Growth rates were calculated for cells in every experiment using initial and final protein contents. Specifically, at each time point, media was aspirated from 6-well plates, cells were washed with PBS, and 500 μl of 0−4°C RIPA buffer was added to each well. The 6-well plates were then stored at 4°C overnight. Lysates were subsequently transferred to microcentrifuge tubes, which were vortexed briefly and centrifuged at 21,000 x g at 4°C for 10 min, and supernatants were transferred to new microcentrifuge tubes. Protein concentrations in the cleared lysates were quantified within 1 week from harvest using a BCA Protein Assay Kit (Thermo Pierce) following manufacturer’s instructions.
Preliminary experiments suggested cell growth to follow exponential kinetics. Therefore, average specific growth rate μ was calculated using following formula:
| ( 1 ) |
where Xf represents final protein content, Xi represents initial protein content, and t represents the time between harvesting cells for initial and final protein counts [49].
Extracellular fluxes
Fresh media (prior to addition to cells) and spent media were collected for the measurement of nutrient consumption and byproduct secretion rates. Samples were centrifuged for 350–500 x g for 10 min and supernatant was transferred to new microcentrifuge tubes and stored at −20°C. Glucose, lactate, glutamine, and glutamate concentrations were measured using a YSI 7100 Biochemistry Analyzer (Xylem).
Pyruvate, citrate, and most amino acid concentrations were measured by spiking equal quantities of 13C-labeled forms (Cambridge Isotope Laboratories) of each into 100 μl of either media or external standards containing known concentrations of each metabolite. To each media sample and external standard, 300 μl of −20°C acetone was added to precipitate proteins; the mixtures were quickly vortexed and centrifuged at 21,000 x g for 10 min, and supernatants were transferred to new tubes. These supernatants were then dried under air and stored at −20°C until gas chromatography-mass spectrometry (GC-MS) analysis. Following GC-MS analysis and correction for natural abundance, the ratios of unlabeled to labeled metabolites for external standards were used to construct standard curves, which had R2 > 0.99 and almost entirely spanned the range of metabolite concentrations in media.
Preliminary experiments suggested that extracellular fluxes were largely constant over the course of the experiment. For glucose and most organic and amino acids, the average specific consumption rate qS for substrate S was therefore calculated as:
| ( 2 ) |
where [S]i is initial substrate concentration, [S]f is final substrate concentration, and V is media volume per well ( = 2.4 ml) [61]. For secreted products such as lactate and glutamate, the negative of this equation is taken to give average specific production rate.
In the case of glutamine, which undergoes spontaneous thermal degradation to pyroglutamate and ammonia, average specific consumption qGln was calculated using a modified equation that accounts for this process:
| ( 3 ) |
where k is the first-order degradation constant, [Gln]f is the final glutamine concentration, and [Gln]i is the initial glutamine concentration [49]. This constant was determined for each experiment by measuring the final glutamine concentration of media in wells without cells [Gln]f,0 relative to the initial concentration:
| ( 4 ) |
To measure the concentration of palmitate in culture medium, 100 μl medium was added to 200 μl methanol and 225 μl chloroform in a microcentrifuge tube. At room temperature, this mixture was vortexed for 10 minutes and then centrifuged at 21,000 x g for 10 min. The bottom nonpolar layer was carefully transferred to a new microcentrifuge tube, and 225 μl additional chloroform was added to the methanol/culture medium mixture for a second extraction. Following an additional round of vortexing and centrifugation, 1 μl of a 200 mM [U-13C16]palmitic acid (Cambridge Isotope Laboratories) in ethanol solution was added to each tube containing nonpolar extracts. These extracts were dried under air, frozen at −80°C, and derivatized to give fatty acid methyl esters (FAMEs), which were subsequently analyzed on GC-MS. After correcting for natural abundance, the ratio of unlabeled-to-labeled palmitate methyl ester was used to estimate palmitate media concentrations, and the values were used in equation (2) to calculate consumption rates.
The BCA Protein Assay Kit (Thermo Pierce) was used to measure total protein concentration in culture medium. The values were used in equation (2) to calculate net efflux rates.
Oxygen consumption rate measurement
Mitochondrial oxygen consumption rate was measured using a Seahorse Bioscience Extracellular Flux Analyzer X24. Cells were seeded in 100 μl supplemented MEBM on Seahorse Bioscience 24-well plates at densities that gave a roughly 80% confluence at the time of measurement (2–20 × 103 per well, depending on EGF concentration) and allowed to attach for 1 hour in the sterile hood before moving to the incubator. As with 6-well plate experiments, this process was performed for two sets of plates per conditions: one for initial cell protein measurements and one for final cell protein measurements and oxygen consumption rates. Approximately 12 hours later, cells were washed once with 500 μl sterile PBS, and 500 μl supplemented DMEM with EGF treatment was added to each well. 48 hours later, this process was repeated for one plate per condition, and the other was used to measure initial cell protein levels (with 100 μl cold RIPA buffer per well used to lyse cells). Following an additional 48 h, basal oxygen consumption was measured in each well of the remaining plate. Saturating concentrations of antimycin and rotenone (2 μM each) were then added to each well to inhibit mitochondrial respiration and oxygen consumption was measured once again. These inhibitor-treated rates were subtracted from the basal rates to give mitochondrial-specific oxygen consumption values. The plate was then used to measure final cell protein levels. Mitochondrial oxygen consumption rates were normalized to final protein levels, and initial and final protein contents were used to calculate specific growth rates for each EGF treatment.
Intracellular metabolite extraction
Labeling experiments using 13C-labeled substrates were performed as previously described, with the following modification: At the final media change, the supplemented DMEM with EGF treatment media contained either [U-13C6]glucose, [1,2-13C2]glucose, [U-13C5]glutamine, [5-13C]glutamine, or branched-chain amino acids ([U-13C6]isoleucine, [U-13C6]leucine, and/or [U-13C5]valine) (Cambridge Isotope Laboratories for all except [U-13C5]glutamine, which came from Sigma) in place of their unlabeled analogs.
For metabolite extraction, media was first aspirated from the 6-well plate and cells were washed with ice-cold saline. Saline was then aspirated, and 500 μl of −20 to −80°C methanol was added to each well to quench metabolism. Next, 300 μl of ice cold MilliQ water (containing a total of 2 μg norvaline as an internal standard) was added to a well, and a pipette tip was used to scrape the cells, wash the well, and transfer this methanol-water extract to a microcentrifuge tube. This process was repeated for each well of the plate. Afterward, 600 μl −20°C chloroform was added to each tube, and all tubes were vortexed at 4°C for 10 min. The metabolite extracts were then centrifuged at 21,000 x g for 10 min, and the resulting upper (polar) and lower (nonpolar) phases of the extract were transferred to separate tubes. These separate polar and nonpolar metabolite extracts were then dried under air and stored at −80°C until derivatization for GC-MS analysis.
Metabolite derivatization
Polar metabolites (both intracellular and medium extracts) were derivatized by a two-step process: 15 μl of methoxyamine in pyridine (MOX Reagent, Thermo Pierce) was added before incubation at 40°C for 1.5 h. Next, 20 μl N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamide, with 1% tert-Butyldimethylchlorosilane (TBDMS) (Sigma) was added, and samples were incubated at 60°C for 1 h. The reaction mixtures were quickly vortexed, centrifuged at 21,000 x g for 1 min, and supernatant was transferred to GC-MS vials for analysis.
Nonpolar metabolites were converted to FAMEs by addition of 500 μl 2% (v/v) sulfuric acid in methanol and incubation at 60°C for 2–3 h. Next, 600 μl hexane and 175 μl saturated aqueous sodium chloride solution were added to each tube, and all tubes were vortexed for 10 min and centrifuged at 21,000 x g for 1 min. The upper phases of the resulting biphasic mixtures were transferred to new microcentrifuge tubes and then dried under air. Finally, the dried FAME samples were resuspended in 30–50 μl hexane and transferred to GC-MS vials for analysis.
GC-MS analysis
Both polar metabolite and FAME samples were analyzed on a 6890N GC with a DB-35ms Ultra Inert capillary column coupled to a 5975B Inert XL MS (Agilent). The flow rate of the helium carrier gas (Airgas) was maintained at 1 ml/min. The inlet temperature was held at 270°C. Injection volumes and split ratios ranged from 2 μl splitless to 1 μl with a 1:10 split, depending on sample concentration and detected ion abundances. Both scan and selected ion monitoring (SIM) modes were used to detect measured ions (with SIM parameters identical to previously published values [62]). The instrument was operated in electron ionization mode with an energy of 70 eV.
For polar metabolite samples, the GC oven was first held at 100°C for 3 min, then ramped at 2.5°C/min to 300°C; masses were profiled from 150 to 625 amu when in scan mode. For FAME samples, the oven was first held at 80°C for 1 min and then ramped at 5°C/min to 300°C. Raw abundance data was converted to mass isotopologue distributions (MIDs) and corrected for natural abundance [63] using an in-house software operating in Matlab (MathWorks). To calculate pool sizes, total metabolite abundances were normalized by total norvaline abundance and total cellular protein content.
Isotopomer spectral analysis
Isotopomer spectral analysis (ISA) [34] was used to determine the fractional contributions of glucose, glutamine, leucine, and isoleucine to lipogenic acetyl-CoA (AcCoA), as well the fraction of palmitate synthesized de novo during culture. Cells were grown in 13C tracer-containing medium as described for 2–3 days. Nonpolar metabolites were extracted, derivatized, and analyzed on the GC-MS. The resulting MIDs for palmitate were averaged within each growth condition, and they were used as inputs for the ISA algorithm.
ISA was implemented as a truncated metabolic flux analysis (MFA) model in the elementary metabolite unit-based software Metran [52], which operates in Matlab (MathWorks). The model is identical as described in previous literature from our lab [53]. In brief, the MID for palmitate was modeled as a function of two free fractional flux parameters: DS, the fractional contribution of a substrate S to lipogenic AcCoA, and g(t), the fraction of palmitate synthesized de novo following addition of the 13C tracer (here, t is 48–72 h). Weighted nonlinear least-squares regression was used to predict best-fit values and 95% confidence intervals for these parameters.
Fractional enrichment calculation
Percent enrichment (PE) of metabolites by 13C-labeled tracers (i.e. the percent of carbon atoms enriched by the 13C tracer) was calculated from MIDs corrected for natural 13C abundance [51] using the following formula
| ( 5 ) |
where n is the total number of carbon atoms in the metabolite, i represents the number of enriched atoms in a particular isotopologue, and mi represents the fractional abundance of a metabolite isotopologue with i enriched atoms.
Western blots
Cells were lysed in ice-cold RIPA buffer (Alfa Aesar) with protease and phosphatase inhibitor cocktails (BioVision), 1 mM vanadate, and 10 μM of the proteasome inhibitor MG132. Cells were then quickly scraped and lysates were added to microcentrifuge tubes, which were gently rocked at 4°C for 15 min and then centrifuged at 21,000 x g at 4°C for 10 min. The cleared lysate was then transferred to separate tubes and stored at −80°C until analysis.
The BCA assay was used to quantify protein concentration for each sample tube. Equal protein amounts were boiled in sample buffer (Boston Bioworks), loaded onto and run on 4–20% SDS-PAGE gels (Life Technologies), and transferred to polyvinylidene fluoride membranes (EMD Millipore). Primary antibodies included anti-phospho-Akt S473 (Cell Signaling, 4060), anti-phospho-S6 S240/244 (Cell Signaling, 5364), anti-phospho-ERK1/2 Y202/204 (Cell Signaling, 9101), anti-tubulin (EMD Millipore, CBL270), and anti-β-actin (Sigma, A1978). Following incubation with secondary antibody, membranes were imaged on the Odyssey CLx infrared imaging system (LI-COR).
RT-qPCR
RNA was extracted from empty vector-HMECs cultured in 5 ng/ml (+EGF) or 0 ng/ml (−EGF) EGF in 6-well plates using the Quick-RNA Miniprep Kit (Zymo R1054) according to manufacturer’s recommendations. RNA concentration was determined via Qubit RNA BR Assay Kit (ThermoFisher Q10211), and 1 μg of cDNA was synthesized using iScript™ cDNA Synthesis Kit (BioRad 1708891). Quantitative PCR was performed with 10 ng of cDNA and LuminoCt SYBR Green qPCR ReadyMix (Sigma-Aldrich L6544) on a Roche LightCycler 480 Instrument II. Primers were used at a final concentration of 250 nM. Expression of each gene was normalized to the endogenous control gene RPLP0.
Primer sequences are as follows: RPLP0 Fwd TGGTCATCCAGCAGGTGTTCGA Rev ACAGACACTGGCAACATTGCGG, BCAT1 Fwd CTACGACCCTTGGGATCTGC Rev GCATCCGTTACTGCAATCCTTC, BCAT2 Fwd CGCTGAATGGTGTTATCCTGCC Rev CAGCAACTGCTTCATGGTGATCG, BCKDHA Fwd CAGAGAACCAGCCCTTCCTCAT Rev AGCCTTGGCTCAGCAGATAGTG, BCKDHB Fwd TGCACTGTTGGCTTGCGAGACA Rev TTCCGCAATGGCAGTAGCTCCA, SLC7A5 Fwd GCCACAGAAAGCCTGAGCTTGA Rev ATGGTGAAGCCGATGCCACACT.
Testing KRAS deviation from growth-based trends
To identify metabolic behaviors in KRAS-HMECs distinct from EV-HMEC growth-dependent trends, we fit linear regression models of the EV-HMEC behavior to assess the magnitude and statistical significance of the difference for the KRAS-HMEC measurements. Specifically, we tested the ability of the EV-HMEC data to predict the extracellular fluxes, pool sizes, and metabolite PE from [U-13C6]glucose, [U-13C5]glutamine, and [U-13C]BCAA tracers of KRAS-HMECs cultured in both 5 and 0 ng/ml EGF. Specifically, we used the statistical software R [66] to fit the outlier-shift model [55], testing whether the KRAS-HMEC measurements are outliers from the EV-HMEC trend. The threshold for statistical significance was set at p = 0.025 (corresponding to an overall significance level of 0.05, with the factor of 2 used to correct for multiple testing of two KRAS observations per metabolite/flux). The threshold for magnitude difference was ± 10% difference of the predicted value for extracellular fluxes and pool sizes, and ± 5% difference from the predicted value for metabolite PEs from 13C tracers.
For extracellular fluxes, which could be paired with individual growth rate measurements by measuring spent medium from well replicates used to determine final cell protein content, ordinary least-squares regression was used to fit individual replicates of flux vs. growth rate (rather than calculating average and standard deviations of replicates). However, due to the destructive nature of metabolite extraction, individual pool size and PE measurements could not be paired with growth rate values from individual well replicates. Instead, tissue culture plates were grown in parallel to measure pool size/PE and growth rates separately. Average and standard deviation values of these measurements were used to fit iteratively reweighted general Deming regression models [56] in order to account for variability in both the growth rate (x-value) and pool size/PE (y-value) when estimating the corresponding trendlines.
Percent change of extracellular fluxes and pool sizes of KRAS-HMEC values relative to those predicted by a linear regression fit to the EV-HMEC measurements at the measured KRAS-HMEC growth rate μKRAS was calculated using the following expression:
| ( 6 ) |
For tracer PEs, which are already normalized for the entire metabolite pool, differences were simply taken between the measured KRAS-HMEC value and the predicted value from the EV-HMEC growth-based trend:
| ( 7 ) |
The outputs of these analyses were graphed on volcano plots, where statistical significance is shown on the y-axis as –log10(p-value) and magnitude difference is shown on the x-axis as either percent change or PE difference.
The R code used to generate linear regression models and calculate statistical significance and magnitude difference values for KRAS(G12V) cells relative to EGF-stimulated EV cells is provided as Supp. File 1. The inputs (extracellular flux, labeling, and pool size data) and outputs (magnitude difference and statistical significance parameters) to this code are given in Supp. File 2.
Cell cycle analysis
DNA synthesis was determined by incorporation of the thymidine analog 5-ethynyl-2’-deoxyuridine (EdU), using the Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (ThermoFisher). EV-HMECs cultured for a total of 72 h in variable EGF concentrations (with a medium replacement at 48 h) were pulsed with 10 μM EdU for 2 h. Cells were then fixed and labeled following manufacturer’s instructions. DNA content was assessed by subsequently staining cells with FxCycle Violet (ThermoFisher). Cells were analyzed on a BD FACSCanto II using FACS Diva Software to quantify signal from FxCycle Violet and Alexa Fluor 488-labeled EdU, and gates corresponding to G0/G1, G2/M, and S phases were constructed based on fluorescence intensity.
Statistical analysis
GraphPad Prism (Version 9.0) was used for data visualization and statistical analysis. Curve fitting was performed by simple linear regression (unweighted). P-values for flux vs. growth rate trends were generated using the extra sum-of-squares F-test and reflect the likelihood that the associated slopes are completely uncorrelated (i.e. equal to zero). Student’s unpaired t-test was used to calculate a p-value for a single comparison. Multiple unpaired t-tests were used when performing multiple comparisons, which the Holm-Sidak method used to adjust p-values for multiple comparisons. Equal standard deviations were assumed for each population for 13C labeling experiments, where variance appeared generally consistent across measurement values, but not for extracellular fluxes measurements, where variance tended to increase with larger values. The threshold for statistical significance is given as p ≤ 0.05 aside from the outlier shift model test, which is p ≤ 0.025.
Supplementary Material
Supporting File 1. R code used to calculate statistical significance and magnitude difference of metabolic behavior in KRAS-HMECs from linear trends fit to data from EGF-stimulated EV-HMECs.
Supporting File 2. Extracellular flux, labeling, and pool size data; and calculated statistical parameters for KRAS- vs. growth-based trend comparisons.
Acknowledgements
This work was funded by NIH grants 1R01DK075850-01 and 1R01CA160458-01A1 to OI and GS. MAK was supported by David H. Koch Graduate Fellowship Fund and the Ludwig Fund for Cancer Research. WD was supported by the David H. Koch Graduate Fellowship Fund. MGVH acknowledges support from the MIT Center for Precision Cancer Medicine, SU2C, the Ludwig Center at MIT, and a faculty scholars award from HHMI. These funding bodies had no role in the design of the study; collection, analysis, and interpretation of the data; or in writing the manuscript. We thank Junyoung O. Park, Jordan A. Krall, Raven J. Reddy, Raphaël Ferreira, Woo Suk Ahn, Zhe Zhang, Thomas M. Wasylenko, Daniel Schmidt, and all other members of the GS and MVH labs for helpful discussions and feedback.
Abbreviations:
- 13C
carbon-13
- AcCoA
acetyl coenzyme A
- ATP
adenosine triphosphate
- BCAA
branched-chain amino acid
- BCAT1/2
branched-chain amino acid transaminase 1/2
- BCKDHA/B
branched-chain keto acid dehydrogenase E1 subunit alpha/beta
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
dimethyl sulfoxide
- EdU
5-ethynyl-2’-deoxyuridine
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- EV
empty vector
- FAME
fatty acid methyl ester
- GLUT1
glucose transporter 1
- HMEC
human mammary epithelial cell
- ISA
isotopomer spectral analysis
- MAPK
mitogen-activated protein kinase
- MEBM
mammary epithelial cell basal medium
- MID
mass isotopologue distribution
- mTOR
mechanistic target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced
- PBS
phosphate-buffered saline
- PE
percent enrichment
- PI3K
phosphatidylinositol 3‑kinase
- PPP
pentose phosphate pathway
- RT-qPCR
reverse transcription quantitative real-time PCR
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SIM
selected ion monitoring
- SLC7A5
solute carrier family 5 member 5
- TCA
tricarboxylic acid
- U-13C
uniformly labeled with carbon-13
Footnotes
Conflicts of Interest
The authors declare no conflicts of interests.
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Warburg O, Posener K & Negelein E (1924) On the metabolism of carcinoma cells. Biochem Z 152, 309–344. [Google Scholar]
- 2.Wang T, Marquardt C & Foker J (1976) Aerobic glycolysis during lymphocyte proliferation. Nature 261, 702–705. [DOI] [PubMed] [Google Scholar]
- 3.Munyon WH & Merchant DJ (1959) The relation between glucose utilization, lactic acid production and utilization and the growth cycle of L strain fibroblasts. Exp Cell Res 17, 490–498. [DOI] [PubMed] [Google Scholar]
- 4.Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C & Terzic A (2011) Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson A & Nielsen J (2016) Metabolic trade-offs in yeast are caused by F1F0-ATP synthase. Sci Rep 6, 22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basan M, Hui S, Zhang Z, Shen Y, Williamson JR & Hwa T (2015) Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 528, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cairns RA, Harris IS & Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM & Manning BD (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (80- ) 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González A & Hall MN (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg RA (2014) Growth Factors, Receptors, and Cancer. In The Biology of Cancer 2nd ed., pp. 131–174. Garland Science, New York, NY. [Google Scholar]
- 11.Vander Heiden MG, Cantley LC & Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBerardinis RJ, Lum JJ, Hatzivassiliou G & Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7, 11–20. [DOI] [PubMed] [Google Scholar]
- 13.Boroughs LK & DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farber S, Diamond LK, Mercer RD, Sylvester RF & Wolff JA (1948) Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N Engl J Med 238, 787–793. [DOI] [PubMed] [Google Scholar]
- 15.DeVita VT, Hellman S & Rosenberg SA (2005) Cancer, Principles and Practice of Oncology.
- 16.Vander Heiden MG (2011) Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10, 671–84. [DOI] [PubMed] [Google Scholar]
- 17.Gaglio D, Metallo CM, Gameiro P a, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G & Chiaradonna F (2011) Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, Condon KJ, Lammers A, Kellersberger KA, Stall BK, Stephanopoulos G, Bar-Sagi D, Han J, Rabinowitz JD, Cima MJ, Langer R & Vander Heiden MG (2017) Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE & DeBerardinis RJ (2016) Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemons JMS, Feng X-J, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina E a, Rabitz H a, Rabinowitz JD & Coller H a (2010) Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol 8, e1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, Gygi SP, Selfors LM & Brugge JS (2016) Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab 23, 867–880. [DOI] [PubMed] [Google Scholar]
- 22.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH & Thompson CB (2001) Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol 21, 5899–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karnoub AE & Weinberg RA (2008) Ras oncogenes: Split personalities. Nat Rev Mol Cell Biol 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC & Weinberg RA (2001) Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao JJ, Gjoerup O V., Subramanian RR, Cheng Y, Chen W, Roberts TM & Hahn WC (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3, 483–495. [DOI] [PubMed] [Google Scholar]
- 26.Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, Pauley R, Momiki S, Caamano J, Klein‐Szanto AJP, Koszalka M, Russo J & Klein-Szanto a J (1991) Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog 4, 25–35. [DOI] [PubMed] [Google Scholar]
- 27.Salomon DS, Perroteau I, Kidwell WR, Tam J & Derynck R (1987) Loss of growth responsiveness to epidermal growth factor and enhanced production of alpha-transforming growth factors in ras-transformed mouse mammary epithelial cells. J Cell Physiol 130, 397–409. [DOI] [PubMed] [Google Scholar]
- 28.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB & Mootha VK (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosios AM, Hecht VC, Danai L V., Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR & Vander Heiden MG (2016) Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell 36, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBerardinis RJ & Cheng T (2010) Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reitzer L, Wice B & Kennell D (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 253, 2669–2676. [PubMed] [Google Scholar]
- 32.Nelson DL, Lehninger AL & Cox MM (2008) Lehninger Principles of Biochemistry W. H. Freeman. [Google Scholar]
- 33.Ahn WS & Antoniewicz MR (2013) Parallel labeling experiments with [1,2-(13)C]glucose and [U-(13)C]glutamine provide new insights into CHO cell metabolism. Metab Eng 15, 34–47. [DOI] [PubMed] [Google Scholar]
- 34.Kelleher JK & Masterson TM (1992) Model equations for condensation biosynthesis using stable isotopes and radioisotopes. Am J Physiol 262, E118–25. [DOI] [PubMed] [Google Scholar]
- 35.Fan J, Kamphorst JJ, Rabinowitz JD & Shlomi T (2013) Fatty Acid Labeling from Glutamine in Hypoxia Can Be Explained by Isotope Exchange without Net Reductive Isocitrate Dehydrogenase (IDH) Flux. J Biol Chem 288, 31363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang M-R, Lu Y, You H, Kwiatkowski D & Zhang H (2011) Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB & Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105, 18782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin J a, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG & Bar-Sagi D (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB & Rabinowitz JD (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller S a, Su SM, de Botton S & Yen KE (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–6. [DOI] [PubMed] [Google Scholar]
- 41.Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, Blagih J, Vincent DF, Campbell KJ, Ceteci F, Sansom OJ, Blyth K & Vousden KH (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- 42.Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi J-T, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP & Giaccia AJ (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 3, 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, Pepin F, Durinck S, Korkola JE, Griffith M, Hur JS, Huh N, Chung J, Cope L, Fackler MJ, Umbricht C, Sukumar S, Seth P, Sukhatme VP, Jakkula LR, Lu Y, Mills GB, Cho RJ, Collisson EA, van’t Veer LJ, Spellman PT & Gray JW (2015) Modeling precision treatment of breast cancer. Genome Biol 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG & Stephanopoulos G (2016) Metabolic requirements for cancer cell proliferation. Cancer Metab 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lunt SY & Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–64. [DOI] [PubMed] [Google Scholar]
- 46.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S & Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104, 19345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheikh K, Förster J & Nielsen LK (2005) Modeling hybridoma cell metabolism using a generic genome-scale metabolic model of Mus musculus. Biotechnol Prog 21, 112–21. [DOI] [PubMed] [Google Scholar]
- 48.Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP & Metallo CM (2015) Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, Grenier-Larouche T, An J, Lapworth AL, Astapova I, Hannou SA, George T, Arlotto M, Olson LB, Lai M, Zhang GF, Ilkayeva O, Herman MA, Wynn RM, Chuang DT & Newgard CB (2018) The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab 27, 1281–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brosnan JT & Brosnan, Margaret E (2006) Branched-Chain Amino Acids: Enzyme and Substrate Regulation. J Nutr 136, 269–273. [DOI] [PubMed] [Google Scholar]
- 51.Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier S V, Bai AHC, Karra D, Piro RM, Felsberg J, Addington A, Lemke D, Weibrecht I, Hovestadt V, Rolli CG, Campos B, Turcan S, Sturm D, Witt H, Chan T a, Herold-Mende C, Kemkemer R, König R, Schmidt K, Hull W-E, Pfister SM, Jugold M, Hutson SM, Plass C, Okun JG, Reifenberger G, Lichter P & Radlwimmer B (2013) BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 19, 901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayers JR, Torrence ME, Danai L V., Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, Muir A, Chin CR, Freinkman E, Jacks T, Wolpin BM, Vitkup D & Vander Heiden MG (2016) Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science (80- ) 353, 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, Davidson SM, Papagiannakopoulos T, Yang A, Dayton TL, Ogino S, Stampfer MJ, Giovannucci EL, Qian ZR, Rubinson D a, Ma J, Sesso HD, Gaziano JM, Cochrane BB, Liu S, Wactawski-Wende J, Manson JE, Pollak MN, Kimmelman AC, Souza A, Pierce K, Wang TJ, Gerszten RE, Fuchs CS, Vander Heiden MG & Wolpin BM (2014) Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, McSkimming D, Kannan N, Tojo A, Edison AS & Ito T (2017) Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA & Sabatini DM (2017) Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB & Vander Heiden MG (2016) Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anastasiou D & Poulogiannis G (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Sci … 1278, 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Son J, Lyssiotis C a, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho R a, Cantley LC & Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T & Rabinowitz JD (2013) Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stampfer MR, Pan CH, Hosoda J, Bartholomew J, Mendelsohn J & Yaswen P (1993) Blockage of EGF receptor signal transduction causes reversible arrest of normal and immortal human mammary epithelial cells with synchronous reentry into the cell cycle. Exp Cell Res 208, 175–188. [DOI] [PubMed] [Google Scholar]
- 61.Murphy TA & Young JD (2013) ETA: robust software for determination of cell specific rates from extracellular time courses. Biotechnol Bioeng 110, 1748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahn WS & Antoniewicz MR (2011) Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab Eng 13, 598–609. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez CA, Des Rosiers C, Previs SF, David F & Brunengraber H (1996) Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom 31, 255–62. [DOI] [PubMed] [Google Scholar]
- 64.Antoniewicz MR, Kelleher JK & Stephanopoulos G (2007) Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab Eng 9, 68–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O & Stephanopoulos G (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Team RC (2017) R: A language and environment for statistical computing. . [Google Scholar]
- 67.Seber GAF & Lee AJ (2012) Departures from Underlying Assumptions. In Linear Regression Analysis pp. 227–263. John Wiley & Sons, Inc., Hoboken, NJ, USA. [Google Scholar]
- 68.Martin RF (2000) General deming regression for estimating systematic bias and its confidence interval in method-comparison studies. Clin Chem 46, 100–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting File 1. R code used to calculate statistical significance and magnitude difference of metabolic behavior in KRAS-HMECs from linear trends fit to data from EGF-stimulated EV-HMECs.
Supporting File 2. Extracellular flux, labeling, and pool size data; and calculated statistical parameters for KRAS- vs. growth-based trend comparisons.