

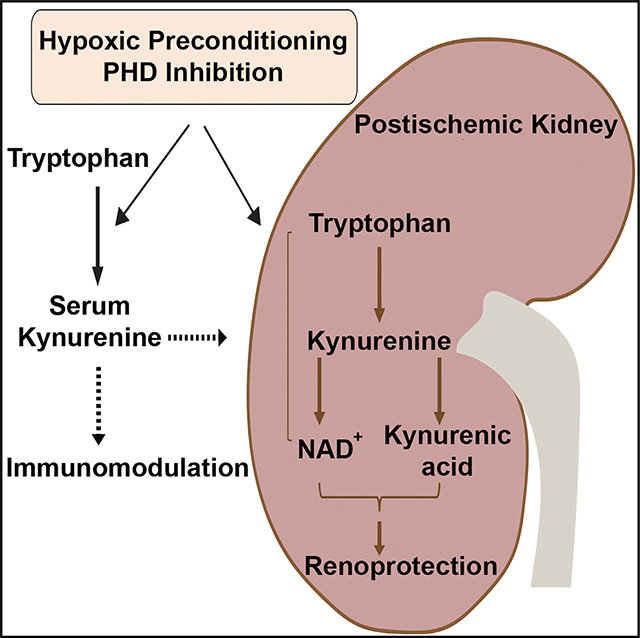
SUMMARY
Prolonged cellular hypoxia leads to energetic failure and death. However, sublethal hypoxia can trigger an adaptive response called hypoxic preconditioning. While prolyl-hydroxylase (PHD) enzymes and hypoxia-inducible factors (HIFs) have been identified as key elements of oxygen-sensing machinery, the mechanisms by which hypoxic preconditioning protects against insults remain unclear. Here, we perform serum metabolomic profiling to assess alterations induced by two potent cytoprotective approaches, hypoxic preconditioning and pharmacologic PHD inhibition. We discover that both approaches increase serum kynurenine levels and enhance kynurenine biotransformation, leading to preservation of NAD+ in the post-ischemic kidney. Furthermore, we show that indoleamine 2,3-dioxygenase 1 (Ido1) deficiency abolishes the systemic increase of kynurenine and the subsequent renoprotection generated by hypoxic preconditioning and PHD inhibition. Importantly, exogenous administration of kynurenine restores the hypoxic preconditioning in the context of Ido1 deficiency. Collectively, our findings demonstrate a critical role of the IDO1-kynurenine axis in mediating hypoxic preconditioning.
In brief
Torosyan et al. show that two potent cytoprotective strategies, hypoxic preconditioning and PHD inhibition, enhance kidney tissue resilience against ischemic injury by activating the kynurenine metabolic pathway. Specifically, they promote systemic kynurenine increase and enhance kynurenine metabolism, leading to preservation of NAD+ and generation of kynurenic acid in the post-ischemic kidney.
Graphical Abstract
INTRODUCTION
Oxygen deprivation results in energy failure in cells and is associated with significant mortality in the context of ischemic disorders. Sublethal hypoxia can induce hypoxic preconditioning (HP), an evolutionarily conserved adaptive response that confers resistance to diverse cellular insults (Kapitsinou and Haase, 2015; Murry et al., 1986).
The cellular response to hypoxia is predominantly controlled by hypoxia-inducible factors (HIFs), transcription factors consisting of an oxygen-labile alpha subunit (HIF-α) and a stable beta subunit (HIF-β). Under normoxia, HIF-α is hydroxylated at specific proline residues by prolyl-4-hydroxylase domain (PHD) proteins 1–3 (Epstein et al., 2001; Schofield and Ratcliffe, 2004) and targeted for rapid proteasomal degradation. Under hypoxia, HIF-α subunits translocate to the nucleus, dimerize with HIF-β, and induce the expression of genes regulating cellular metabolism, angiogenesis, erythropoiesis, apoptosis, and proliferation (Majmundar et al., 2010; Semenza, 2012). Remarkably, HIF activation by pharmacologic PHD inhibition maintains tissue homeostasis in different models of organ injury, including acute kidney injury (AKI) (Bernhardt et al., 2006; Eltzschig et al., 2014; Hill et al., 2008; Kapitsinou et al., 2012; Olenchock et al., 2016).
Despite the organ-wide protection generated by activation of hypoxic signaling, gaps in understanding the underlying cellular and systemic mechanisms have hindered clinical translation. In the present study, we hypothesized that activation of hypoxia response leads to systemic metabolic alterations, which play a crucial role in mediating protection against ischemic kidney injury and inflammation. To test this hypothesis, we used metabolomic profiling to identify common alterations in serum metabolome induced by two cytoprotective approaches, HP and pharmacologic PHD inhibition. By integrating functional genomics studies, we identified a critical role for tryptophan metabolism in promoting resistance against post-ischemic kidney injury.
RESULTS
HP attenuates post-ischemic kidney injury and inflammation
While hypoxia is the major inhibitory stimulus of the PHD catalytic activity, the effect of hypoxia exposure as preconditioning against renal ischemia-reperfusion injury (IRI) has only recently been explored (Johnsen et al., 2020). To investigate the impact of HP on renal IRI outcomes, we used a model of normobaric hypoxia in which mice were subjected to 8% O2 for 2 days. We have previously reported that this model activates HIF-signaling in the lungs (Kapitsinou et al., 2016). Furthermore, consistent with the activation of hypoxic response, renal erythropoietin (Epo) mRNA and serum Epo protein levels were increased 22- and 25-fold, respectively (Figure S1A). Following hypoxia exposure, mice were subjected to unilateral renal IRI and sacrificed on day 3 after IRI (Figure S1B). Remarkably, post-ischemic kidneys from HP mice showed attenuated kidney injury as indicated by significantly reduced injury scores and 2.3-fold suppression in kidney injury molecule 1 (Kim1) mRNA compared to normoxic controls. Furthermore, HP significantly reduced the infiltration of myeloid cells in post-ischemic kidneys by 85% and suppressed the expression of the proinflammatory genes vascular cell adhesion molecule 1 (Vcam1) and Tnfa. Taken together, these results suggest that HP protects against IRI-induced kidney injury and inflammation.
Hypoxia and pharmacologic inhibition of PHDs increase circulating levels of the tryptophan metabolite kynurenine
We have previously reported that pre-ischemic administration of PHD inhibitor (PHI) activates HIFs and attenuates post-ischemic renal injury and inflammation (Kapitsinou et al., 2012, 2014). To identify candidate metabolites involved in cytoprotection provided by HP and PHI, we compared alterations in serum metabolome induced by these approaches. Unbiased metabolomic profiling was performed by liquid chromatography-mass spectrometry (LC/MS) and gas chromatography-mass spectrometry (GC/MS) platforms (Figure 1A). Among 369 named biochemicals detected, 82 metabolites (54 upregulated and 28 downregulated) were determined to be significantly altered in the serum of PHI-treated mice compared to controls (Table S1). For the HP, we detected 362 named biochemicals, of which 140 (55 upregulated and 85 downregulated) were found to be significantly dysregulated in the serum of hypoxia exposed mice compared to controls (Figure 1A; Table S1). Notably, when we compared the significantly changed metabolites between the two datasets, we identified a high number of unique metabolomic alterations (118/135), of which 15 metabolites showed discordant changes (Figure 1A; data not shown). For instance, while PHI increased serum levels of pyruvate and ribose and reduced levels of ribulose, the opposite was the case in HP. These findings suggest that HP and PHI induced distinct metabolic changes in glucose metabolism. On the other hand, the presence of 17 overlapping metabolites revealed a shared metabolomic response between the two approaches, which mainly involved alterations in amino acid metabolism (Figure 1B). Among the overlapping metabolites, kynurenine, a breakdown product of tryptophan, showed a 2.7-fold and 1.8-fold increase, respectively, in HP and PHI compared to corresponding controls (Figure 1B). These results were validated by a targeted LC-MS/MS assay, which quantified serum kynurenine concentrations on independent experiments using a calibration curve made with known concentrations of kynurenine (Figure 1C). Consistently, the serum kynurenine to tryptophan ratio was significantly increased by both HP and PHD inhibition (Figure 1C), whereas no significant change in serum tryptophan levels was noted for both conditions (data not shown).
Figure 1. Hypoxia and pharmacologic PHD inhibition promote tryptophan metabolism to kynurenine in an IDO1-dependent manner.
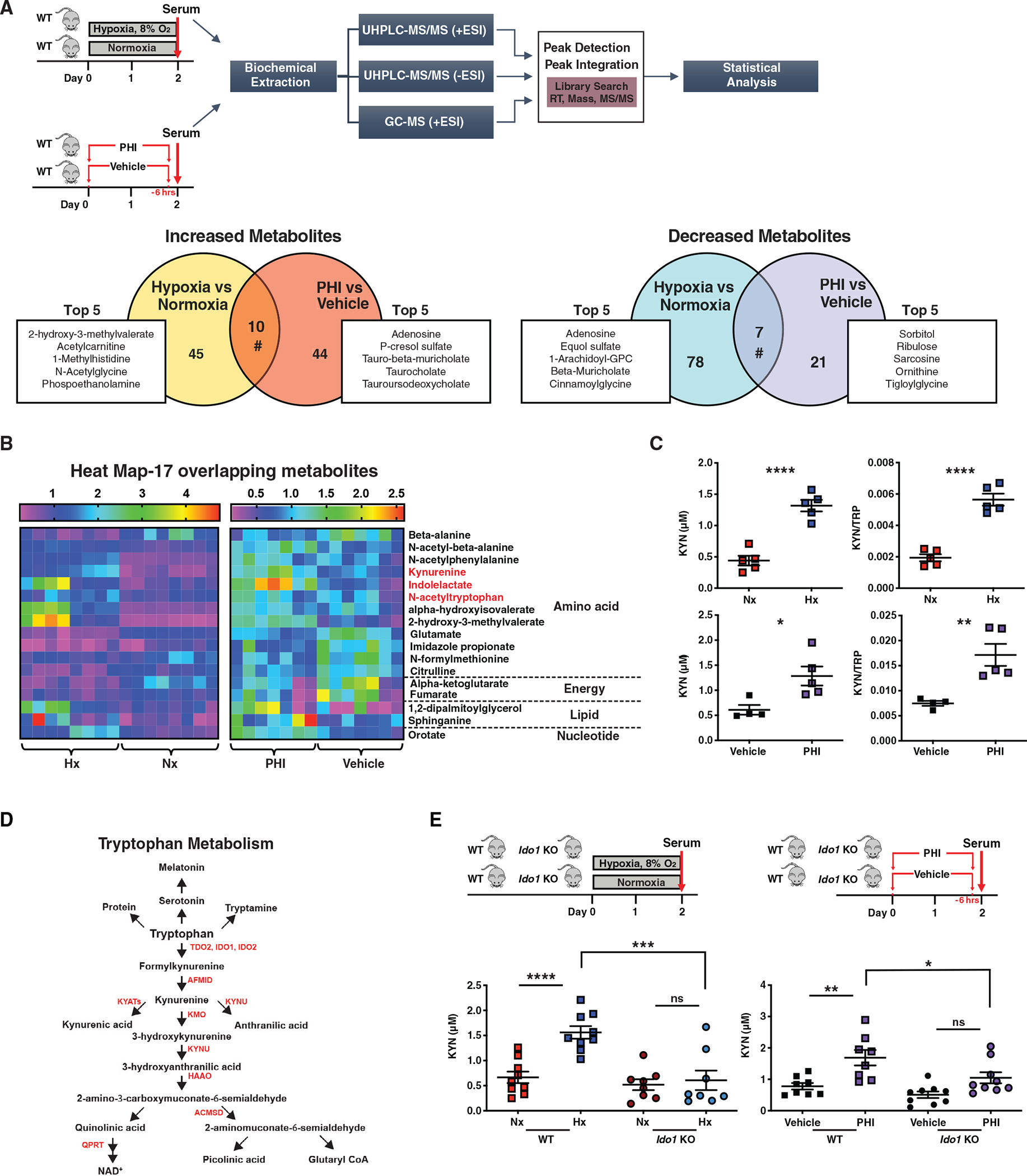
(A) Top graph summarizes the experimental workflow for the comparative metabolomic analysis of HP and pharmacologic PHD inhibition. Sera from mice exposed to 2 days of hypoxia 8% or two doses of PHI were analyzed using liquid chromatography-mass spectrometry (LC/MS) and gas chromatography-mass spectrometry (GC/MS) platforms. On the bottom, Venn diagrams show the differentially accumulated metabolite features, revealing uniquely or commonly increased (left graph) or decreased (right graph) metabolites under exposure to hypoxia versus normoxia and PHI versus vehicle. The top five uniquely regulated metabolites are listed for each condition in the attached boxes. n = 8 for HP and n = 7 for PHI.
(B) Heatmap visualization of the 17 overlapping metabolites between the two experimental sets (hypoxia versus normoxia and PHI versus vehicle).
(C) Serum kynurenine (KYN) concentration and kynurenine/tryptophan (KYN/TRP) ratio in the indicated conditions. n = 5 for HP and n = 4 (vehicle) and 5 (PHI).
(D) Diagram of the tryptophan metabolic pathway.
(E) Upper schemes illustrate the experimental protocols employed. Lower graphs show the serum kynurenine concentrations in wild-type (WT) and Ido1 KO mice exposed to hypoxia (left) or PHI (right) compared to corresponding controls. n = 9 (WT, Nx and WT, Hx) and n= 8 (Ido1 KO, Hx and Ido1 KO, Nx).
Error bars represent SEM. For (A) and (B), statistics were determined using Welch’s two-sample t tests and random effects ANOVA. For (C), two-tailed t test was used. For (E), statistics were determined using one-way ANOVA with Sidak correction. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not statistically significant. Nx, normoxia; Hx, hypoxia; PHI, prolyl-hydroxylase inhibitor; WT, wild-type; KYN, kynurenine; KYN/TRP: kynurenine/tryptophan.
Ido1 deficiency abolishes the systemic accumulation of kynurenine and the subsequent renoprotection provided by HP and PHD inhibition
Most of the cellular free tryptophan (>95%) is degraded through the kynurenine pathway (KP) in which the rate-limiting step is the conversion of tryptophan to N-formylkynurenine. This reaction is catalyzed by either tryptophan-2,3-dioxygenase (TDO2) mainly in liver or indoleamine-2,3-dioxygenase (IDO1 and the more recently discovered IDO2) extrahepatically (Cervenka et al., 2017) (Figure 1D). To assess how HP and PHI increased serum kynurenine levels, we quantified the transcripts of several genes encoding KP enzymes in liver, kidney, and lung, tissues with robust transcriptional responses to activation of hypoxia signaling. In the liver, where most of the tryptophan degradation occurs through Tdo2 under normal conditions, neither HP nor PHI induced Tdo2 mRNA (Figure S2A). In fact, we detected a minor suppression in Tdo2 by PHI and a 1.8-fold reduction in Ido2 transcript by HP, whereas hepatic expression levels for Ido1 were too low to analyze. Notably, an upward trend was detected for renal Ido1 by both HP and PHI but did not reach statistical significance. Among the differentially expressed KP genes in the lung, Ido1 mRNA was significantly upregulated by PHI (Figure S2A). Overall, while there was not a uniform pattern of transcriptional responses in KP genes, the lack of induction in hepatic Tdo2 suggested an extrahepatic mechanism for the serum kynurenine increase by HP and PHI.
To examine whether reduced degradation drives the increase in serum kynurenine by HP and PHI, we used a semiquantitative high-performance liquid chromatography (HPLC) tandem mass spectrometry (MS/MS)-based assay to measure the levels of downstream KP metabolites. Compared to normoxia, the serum increase in kynurenine by hypoxia was associated with 34% increase in the serum levels of xanthurenic acid (XA), a metabolite formed by transamination of 3-hydroxykynurenine (3-HK) (Figure S2B). Furthermore, modest reductions were noted for the metabolites downstream of 3-HK, 3-hydroxyanthranilic acid (3-HAA), quinolinic acid (QUIN), and picolinic acid (PIC). In the setting of PHI, besides the expected increase in serum kynurenine, the other metabolites showed no significant changes (Figure S2B). For the kidney, hypoxia led to 54% reduction in 3-HAA, while QUIN and PIC showed significant increases (by 22% and 61%, respectively) compared to normoxia. Treatment with PHI led to significant increases of kynurenine (by 119%) and QUIN (by 14%) in kidney tissue compared to vehicle. Neither hypoxia nor PHI changed significantly the kidney NAD+, which was validated by an independent biochemical assay (Figure S2C). Overall, these results rule out significant inhibition of kynurenine degradation by HP and PHI. Finally, serum blood urea nitrogen (BUN) levels remained normal following HP and PHI (Figure S2D), excluding reduced kidney function leading to kynurenine accumulation.
Because IDO1 is a rate-limiting enzyme in the conversion of tryptophan to kynurenine in extrahepatic tissues, we next examined the role of IDO1 in regulating serum kynurenine levels in the setting of hypoxic and pharmacologic PHD inhibition (Figure 1E). To this end, we used mice with germline inactivation of Ido1 (Baban et al., 2004). Under normoxia, Ido1 knockout (KO) mice had serum kynurenine levels comparable to wild-type mice, but upon exposure to hypoxia, they did not show a significant increase in serum kynurenine levels compared to normoxic littermates (Figure 1E). Likewise, Ido1 KO mice treated with PHI failed to reach serum kynurenine levels observed in PHI-treated wild-type mice (Figure 1E). Therefore, these data indicate that Ido1 drives the increase in serum kynurenine induced by both HP and PHI. Nevertheless, we cannot exclude that additional enzymatic activities in tryptophan metabolism may contribute to serum kynurenine levels following HP and PHD inhibition.
We next investigated whether the IDO1-dependent increase in serum kynurenine following HP or PHI was critical for the renoprotection generated by these approaches. Remarkably, post-IRI kidneys from Ido1 KO mice subjected to HP showed comparable injury with non-preconditioned controls as indicated by Kim1 mRNA and tubular injury scores (Figure 2A). Furthermore, infiltration by Ly6-B.2+ve cells and Tnfa mRNA were not reduced by HP in Ido1 KO mice, whereas Vcam1 transcript was upregulated by 1.8-fold. Accordingly, PHI failed to attenuate post-ischemic kidney injury and inflammation in Ido1 KO mice based on Kim-1, Vcam1, and Tnfa transcripts; tubular injury scores; and quantitative analysis of Ly6.B2+ve cells in post-ischemic kidneys compared to corresponding controls (Figure 2B). The lack of renoprotection in Ido1 KO mice was in sharp contrast to our prior studies on wild-type mice. In summary, the IDO1-kynurenine axis is essential in renoprotection provided by HP and pharmacologic PHD inhibition.
Figure 2. IDO1 deficiency abolishes the renoprotective and anti-inflammatory effects mediated by HP and pharmacologic PHD inhibition against renal IRI.
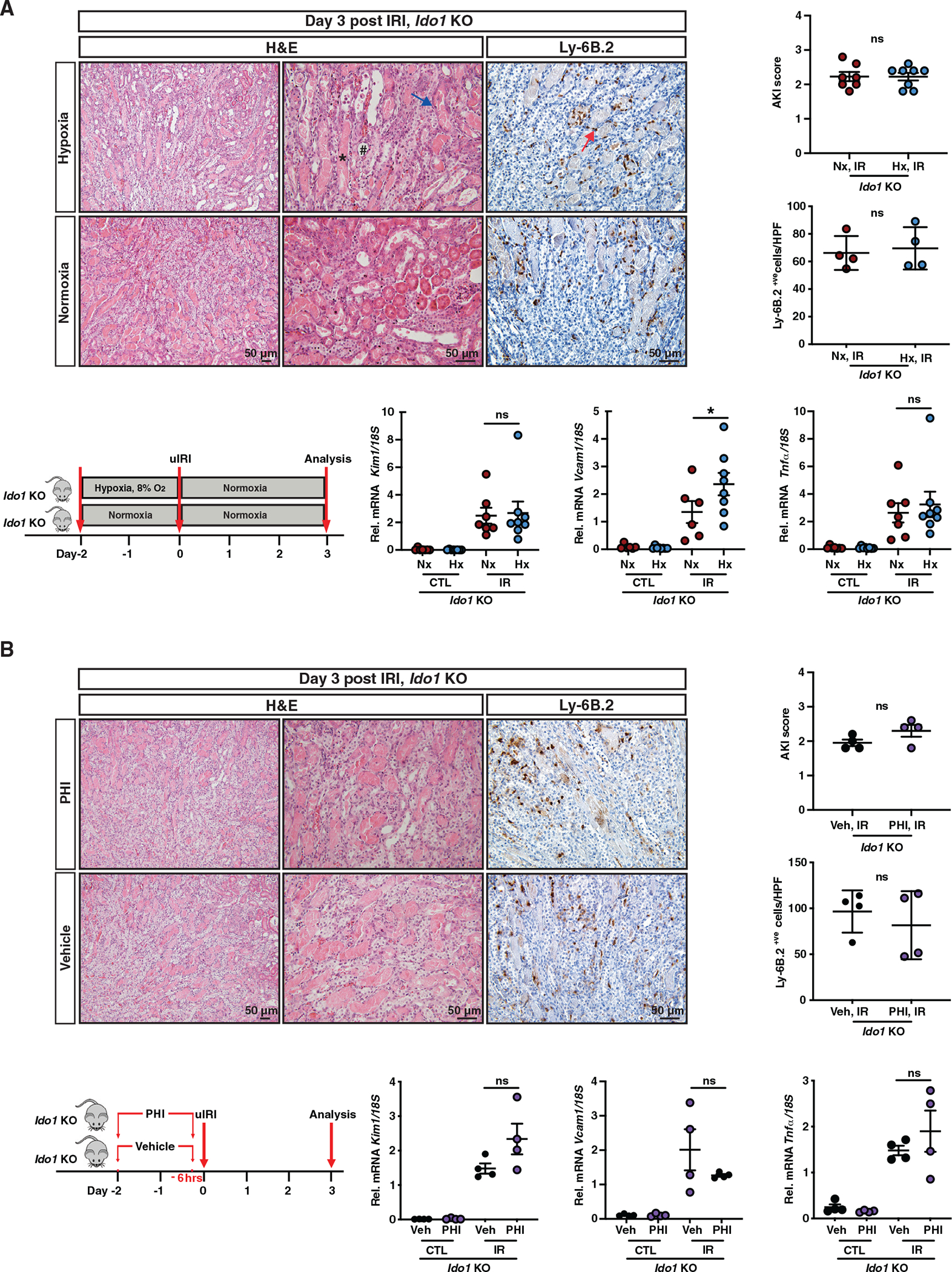
(A and B) Overview of the experimental protocol and representative images of H&E- and Ly-6B.2-stained sections of day 3 post-IRI kidneys from Ido1 KO mice subjected to HP compared to normoxia (A) or PHD inhibition compared to vehicle (B). Asterisk points to a tubule with cast formation, # indicates a dilated tubule, the blue arrow points to necrosis, and the red arrow points to a Ly-6B.2+ve cell. Right panels demonstrate scoring of tubular injury and quantification of Ly-6B.2+ve cells/HPF. Bottom graphs show Kim1, Vcam1, and Tnfa mRNA in IR and contralateral kidneys. (A) For AKI score, n = 7 (Nx, IR) and 8 (Hx, IR); for Ly-6B.2+ve cells/HPF, n = 4; for Kim1 and Tnfa mRNA, n = 7 (Nx) and 8 (Hx); and for Vcam1 mRNA, n = 6 (Nx) and 8 (Hx). (B) n = 4. Error bars represent SEM. For AKI scores and Ly-6B.2+ve cell counts in (A) and (B), statistics were determined by two-tailed t test. All other comparisons were performed by one-way ANOVA with Sidak correction. *p < 0.05; ns, not statistically significant. uIRI, unilateral IRI; IR, kidney subjected to uIRI; CTL, contralateral kidney. Scale bar indicates 50 μm.
Administration of kynurenine restores the HP in the context of Ido1 deficiency
We next wished to determine whether exogenous administration of kynurenine in hypoxia exposed Ido1 KO mice restores the HP-induced renoprotection. Therefore, we injected Ido1 KO mice subjected to HP with intraperitoneal (i.p.) kynurenine (50 mg/kg) 2 h prior to renal IRI, which led to serum kynurenine levels comparable to hypoxia and PHI (Figure S3A). Remarkably, administration of kynurenine ameliorated tissue injury in Ido1 KO mice subjected to HP resulting in significantly reduced tubular injury scores and Kim1 transcript levels compared to vehicle/HP-treated Ido1 KO mice (Figure 3A). Furthermore, in addition to an 87% reduction in infiltration by Ly6.B2+ve cells, Ido1 KO mice subjected to HP and kynurenine administration had 3.8-fold and 3.0-fold suppression in Vcam1 and Tnfa mRNA, respectively, compared to post-IRI kidneys of vehicle/HP-treated Ido1 KO mice (Figure 3A). Similar results were achieved with administration of 200 mg/kg i.p. kynurenine and using normoxic vehicle-treated Ido1 KO mice as controls (Figure S3B).
Figure 3. Exogenous administration of kynurenine is not sufficient to provide renoprotection under normoxia but restores the HP in the context of IDO1 deficiency.
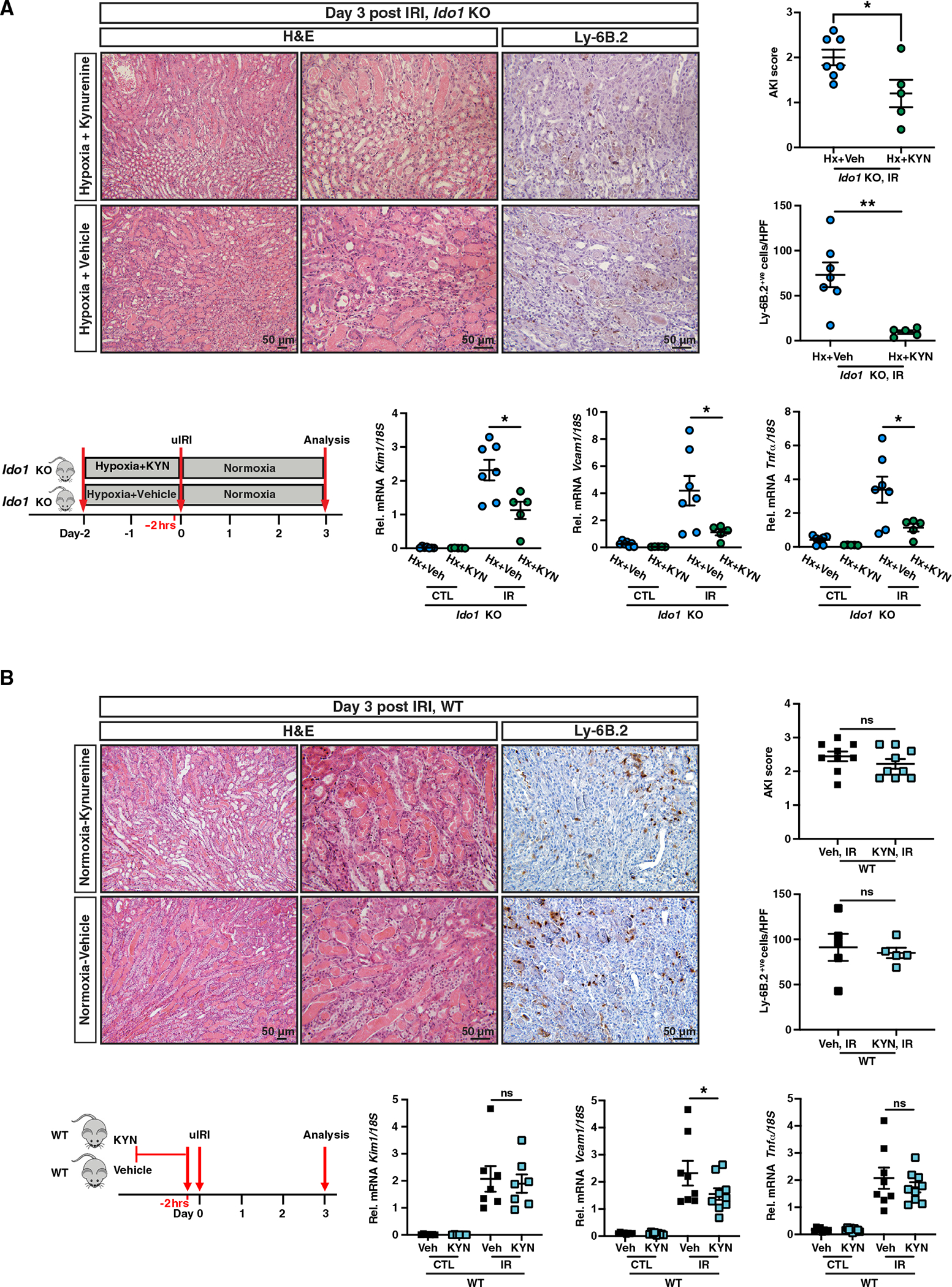
(A and B) Schematic of the experimental protocol, representative images of H&E- and Ly-6B.2-stained sections of day 3 post-IRI kidneys from Ido1 KO mice subjected to HP and kynurenine administration (50 mg/kg) compared to HP/vehicle-treated Ido1 KO mice (A) or WT mice treated with kynurenine (200 mg/kg) versus vehicle under normoxia (B). Right panels demonstrate scoring of tubular injury (A, n = 7 [Hx+Veh] and 5 [Hx+KYN]; B, n = 9) and quantification of Ly6B.2+ve cells/HPF (A, n = 7 [Hx+Veh] and 5 [Hx+KYN]; B, n = 5). Bottom graphs show Kim1, Vcam1, and Tnfa mRNA levels in IR and CTL kidneys (A, n = 7 [Hx+Veh] and 5 [Hx+KYN]; B, n = 7 for Kim1, n = 8 [Veh] and 9 [KYN] for Vcam1 and Tnfa). Error bars represent SEM. For AKI scores and Ly-6B.2+ve cell counts in (A) and (B), statistics were determined by two-tailed t test. All other comparisons were performed by one-way ANOVA with Sidak correction. *p < 0.05; **p < 0.01; ns, not statistically significant. Scale bar indicates 50 μm.
We next investigated whether an increase in serum kynurenine alone was sufficient to afford renoprotection under normoxia. Treatment of wild-type mice with an i.p. injection of kynurenine (200 mg/kg) 2 h prior to renal IRI led to a 1.5-fold reduction in Vcam1 mRNA in post-ischemic kidneys of kynurenine-treated mice, but Kim1 and Tnfa transcripts, injury scores, and Ly6.B2+ve infiltration were comparable to vehicle-treated mice (Figure 3B). While these findings suggest that an increase in serum kynurenine by exogenous administration is not sufficient to mimic HP, we also examined whether kynurenine administration enhances the renoprotection generated by PHI. For this purpose, animals were pretreated with a PHI (same as in Figure 2B, experimental protocol) and received either one single injection of kynurenine or normal saline 2 h prior to IRI. Based on the analysis of Kim1, Vcam1, and Tnfa mRNA, kynurenine administration failed to enhance the cytoprotection generated by PHD inhibition (Figure S3C). Because these findings suggest that an increase in serum kynurenine by exogenous administration is not sufficient to mimic or enhance the cytoprotective effects induced by activation of hypoxia signaling, the renoprotective action of kynurenine may require additional upstream hypoxia-induced alterations. In summary, our results demonstrate that the increase in serum kynurenine is required in the concerted molecular response by which HP provides protection against post-ischemic kidney injury and inflammation.
HP preserves NAD+ levels in the post-ischemic kidney to promote favorable metabolic reprogramming
To understand how HP provides protection against post-ischemic renal injury, we performed RNA sequencing (RNA-seq) analysis on the post-IRI kidneys of wild-type mice subjected to HP compared to normoxic controls. RNA-seq identified 2,633 differentially expressed genes (DEGs) in the context of HP, and heatmap of the top100 most DEGs showed a distinct transcriptional profile in HP compared to normoxia (Figure S4A). Following Gene Ontology (GO) term enrichment analysis, we detected immune system process and inflammatory response among the key pathways regulated by HP (Figure S4B). Consistent with the blunted inflammation by HP was the suppression of several genes encoding inflammatory molecules, including chemokine (C-C motif) ligand 2 (Ccl2), chemokine (C-X-C motif) ligand 2 (Cxcl2), and chitinase-like 3 (Chil3) (Figure 4A). On the other hand, among the upregulated genes, Cxcl12 promotes kidney repair by facilitating glycolysis (Yakulov et al., 2018), while Ccl28 has been implicated in dampening inflammation in hypoxic tumors by promoting the recruitment of regulatory T (Treg) cells (Facciabene et al., 2011).
Figure 4. HP suppresses proinflammatory pathways and preserves NAD+ levels in the post-ischemic kidney to promote favorable metabolic reprogramming.
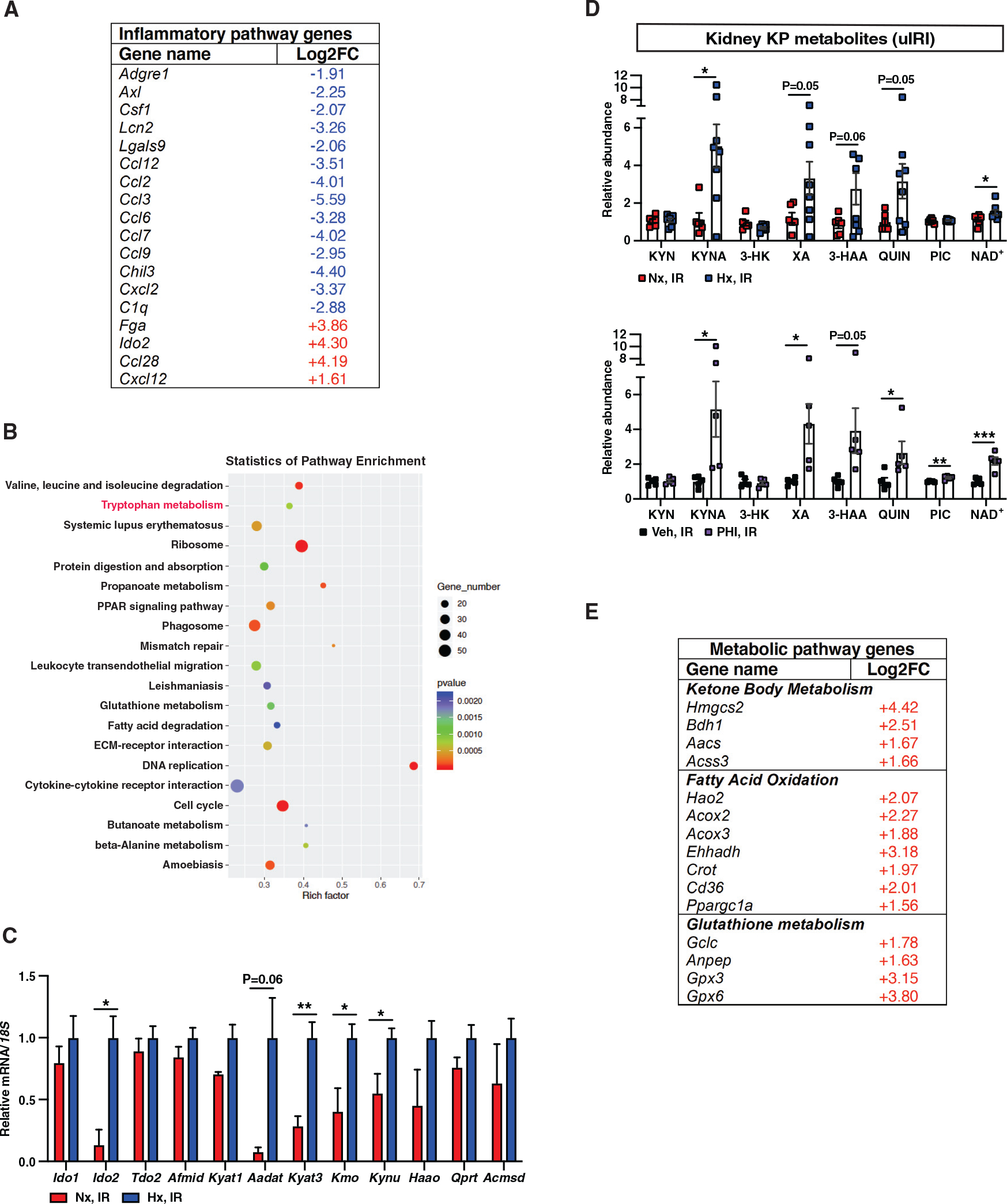
(A) Shown is a list of selected genes with ontologies of inflammatory response and immune system process among DEGs in response to HP with a log2FC greater than 1 or less than −1 and false discovery rate (FDR)-adjusted p < 0.05.
(B) KEGG enrichment graph displays the top 20 pathways. The number of DEGs enriched in KEGG terms, p value, and rich factor are shown in scatterplot. Rich factor = (number of DEGs in KEGG term)/(total number of genes in KEGG term).
(C and D) Relative mRNA levels of genes associated with tryptophan metabolism (n = 3 [Nx, IR] and 4 [Hx, IR]) (C) and KP metabolites (D) in day 3 post-IRI kidneys from mice subjected to HP (upper panel) compared to normoxia (n = 6 [Nx, IR] and 8 [Hx, IR]) or PHI-treated mice compared to vehicle (n = 5) (lower panel).
(E) List of selected metabolic pathway genes among DEGs in response to HP with FDR-adjusted p < 0.05.
Error bars represent SEM. For (C) and (D), statistics were determined by two-tailed t test; *p < 0.05. Afmid, arylformamidase; Kyat, kynurenine aminotransferase; Aadat, aminoadipate aminotransferase; Kmo, kynurenine 3-monooxygenase; Kynu, kynurerinase; Haao, 3-hydroxyanthranilate 3,4-dioxygenase; Qprt, quinolinate phosphoribosyltransferase; Acmsd, alpha-amino-beta-carboxy-muconate-semialdehyde decarboxylase. KYN, kynurenine; KYNA, kynurenic acid; 3-HK, 3-hydroxykynurenine; XA, xanthurenic acid; 3-HAA, 3-hydroxyanthranilic acid; QUIN; quinolinic acid, PIC; picolinic acid.
KEGG pathway analysis of DEGs revealed significant enrichment in several metabolic and signaling pathways (Figure 4B). Remarkably, tryptophan metabolism was identified among the most enriched metabolic pathways in post-ischemic kidneys subjected to HP. To validate this finding, we quantified the transcripts encoding enzymes in the KP by RT-PCR. As shown in Figure 4C, we found significant upregulation of Ido2, kynurenine aminotransferase 3 (Kyat3), kynurenine 3-monooxygenase (Kmo), and kynureninase (Kynu) (Figure 4C). These findings motivated us to further investigate the impact of HP on generation of KP metabolites and NAD+. We observed significant increases in KYNA (by 345%), XA (by 171%), 3-HAA (by 219%), QUIN (by 216%), and NAD+ (by 35%) in post-IRI kidneys of HP-subjected mice compared to normoxic controls (Figure 4D). The increase in NAD+ was also confirmed by a biochemical assay (Figure S4C). Importantly, the analysis of PHI-treated post-IRI kidneys yielded consistent results, with even more prominent increase in NAD+ levels by 118% (Figure 4D). NAD+ has emerged as a key metabolite exerting cytoprotection through an array of actions that include regulation of cellular metabolism and antioxidant defense (Cantó et al., 2015; Ralto et al., 2020). As shown in Figure 4E, we found significant upregulation of genes involved in ketone body metabolism, fatty acid oxidation, and glutathione metabolism, a coordinated metabolic response that may protect from bioenergetic failure in AKI. For instance, upregulation of glutathione metabolism related genes enhances antioxidant defense, while tissue ketogenesis has been implicated in the renoprotective effects mediated by SGLT2 inhibition (Tomita et al., 2020). Also, among the upregulated genes involved in fatty acid oxidation, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a) protects against tubular injury from diverse insults such as IRI, toxins, and sepsis (Kang et al., 2015; Tran et al., 2011, 2016). Of interest, skeletal muscle Ppargc1a ameliorates stress-induced depression by enhancing the conversion of kynurenine into kynurenic acid through induction of Kyats (Agudelo et al., 2014). Therefore, our data suggest that HP protects against ischemic kidney injury by increasing circulating kynurenine and boosting the generation of kynurenine metabolites and NAD+ through activation of the KP in the post-ischemic kidney.
DISCUSSION
Besides their role in energy conversion, metabolites have a myriad of functions in dictating responses to cellular injury. Here, we show that two potent cytoprotective strategies, HP and pharmacologic inhibition of PHDs, lead to systemic accumulation of the tryptophan metabolite, kynurenine. The increase in serum kynurenine represents an essential metabolic adaptation in the context of renoprotection generated by both HP and PHD inhibition. Furthermore, by enhancing the kynurenine biotransformation in the post-ischemic kidney, HP generates kynurenine metabolites and NAD+, a response that augments kidney tissue resilience against ischemic injury.
Our findings, together with recently published studies, suggest that the KP may be one of the key pathways mediating organ protection by HP. First, the fact that the KP emerged from non-biased metabolomic screening in our study and in the cardioprotection study by Olenchock et al. (2016) suggests that this metabolic pathway plays a central and perhaps unifying role in organ protection provided by HP. Second, kynurenine interacts with the arylhydrocarbon receptor (Ahr) with downstream effects that include, among others, induction of Treg cells (Mezrich et al., 2010) and inhibition of the nucleotide-binding domain, leucine-rich repeat, and pyrin domain containing protein 3 (Nlrp3) transcription and inflammasome activation (Huai et al., 2014), effects known to suppress post-ischemic inflammation (Anders and Muruve, 2011; Kinsey et al., 2009; Nazir et al., 2017). Consistently, we found downregulation of Nlrp3 in wild-type mice subjected to HP, an effect that was lost in Ido1 KO mice but could be restored upon injection of kynurenine (Figure S4D). Third, kynurenine is converted to other metabolites with immunosuppressant functions. Specifically, kynurenic acid reduces the inflammatory response triggered by lipopolysaccharide in macrophages (Tiszlavicz et al., 2011) and controls cytokine release from invariant natural killer T (iNKT) cells by interacting with G-protein-coupled receptor 35 (GPR35) (Fallarini et al., 2010). Other examples include 3-hydroxyanthranilic acid and quinolinic acid, which induce apoptosis of type 1 T helper (Th1) cells, while promoting proliferation of type 2 T helper (Th2) cells, a switch that protects against hyperinflammation (Fallarino et al., 2002). Finally, KP enzymes have been shown to play critical roles in preserving tissue homeostasis against injury and inflammation. Specifically, IDO1-driven activation of general control nonderepressible 2 (GCN2)-dependent signals induces autophagy and reduces inflammatory pathology in a mouse model of glomerulonephritis (Chaudhary et al., 2015). Similarly, IDO1 limits vascular inflammation and atherosclerosis in ApoE−/− mice (Cole et al., 2015; Polyzos et al., 2015). Furthermore, inhibition of kynurenine 3-monooxygenase (KMO), an intervention that raises serum kynurenine, protects against tissue injury in experimental multiorgan dysfunction syndrome (Mole et al., 2016).
NAD+ has emerged as a guardian promoting renal resistance against insults by connecting oxidative metabolism in the epithelium to overall organ function. Augmentation of NAD+ levels by increasing PGC1a levels (Kang et al., 2015; Tran et al., 2011, 2016), providing the precursor nicotinamide (Poyan Mehr et al., 2018), or α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD) inhibition (Katsyuba et al., 2018) protects against AKI. Our study now demonstrates that HP increased circulating kynurenine and preserved NAD+ levels following renal IRI, perhaps by promoting kynurenine degradation to NAD+ in the post-ischemic kidney. Our findings have implications on the interpretation of two recent negative trials on ischemic preconditioning (Hausenloy et al., 2015; Meybohm et al., 2015). Defects in hypoxic response and tryptophan metabolism due to aging, comorbidities, and medications (Cho et al., 2021; Kartika et al., 2020; Orabona et al., 2018; Takabuchi et al., 2004) may have blunted the beneficial effects of remote ischemic preconditioning, issues that warrant further investigation.
Collectively, our findings indicate that HP and PHD inhibition increase circulating kynurenine, which functions as a critical metabolic signal in mediating renoprotection by these approaches. It could therefore represent a valuable target to protect against ischemic AKI and potentially synergize with emerging therapies focused on NAD+ preservation.
LIMITATIONS OF STUDY
First, our study identified kynurenine as an essential metabolite in renoprotective responses generated by HP and PHI, but it cannot be assumed that it is the sole mediator. Second, while HP and PHI preserve NAD+ levels in the post-ischemic kidney, probably through a combination of increased circulating kynurenine and induction of genes encoding enzymes in tryptophan metabolism, the exact pathway by which kynurenine acts on kidney tissue remains to be explored. Third, IDO1 is required for the increase in serum kynurenine observed following HP and PHD inhibition, but it remains unclear which cells drive this response and how alterations in other enzymatic steps contribute. Finally, it remains to be determined whether the systemic alteration in tryptophan metabolism represents a broader mechanism for homeostatic regulation in the context of hypoxia.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pinelopi P. Kapitsinou (pinelopi.kapitsinou@northwestern.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq generated during this study are available on GEO: GSE156516. The metabolomic data has been deposited at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001178. The data can be accessed directly via the associated DOI: 10.21228/M8F99F.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J mice (WT) and Ido1 KO mice (stock#005867, C57BL/6J background) were purchased from the Jackson Laboratory (Bar Harbor, ME) and then bred in the Laboratory Research Animal Center of University of Kansas Medical Center. Mice were maintained in a specific pathogen-free facility on a 12-hour light/12-hour dark cycle. Littermates were randomly assigned to experimental groups.
Animal experiments
For the unilateral renal IRI model, anesthesia was induced with xylazine (10 mg/kg i.p.) and ketamine (90–120 mg/kg i.p.). After making a small midline abdominal incision, the left renal pedicle was occluded with a microaneurysm clamp while the right kidney was used as an internal control. The abdomen was partially closed, temporarily, with sutures, and body temperature was monitored by rectal probe and controlled with a heating pad at 37°C. After 25 minutes, the clamp was removed, and reperfusion was visually confirmed. The abdomen was closed with a 6–0 suture and the skin was closed with Michel miniature clips. Male mice 8–12 weeks of age were used for the renal IRI studies. For HP, mice were subjected to 8% O2 in an animal hypoxia chamber (Biospherix Ltd) for 2 days prior to IRI. For pharmacologic PHD inhibition, HIF prolyl-4-hydroxylase inhibitor IOX2 (Tocris) was dissolved in 1% methylcellulose and administered by oral gavage at a dose of 60 mg/kg. Mice received a total of 2 doses of IOX2 6 hours and 2 days prior to IRI. Mice in the control group were treated with 1% methylcellulose (vehicle) at the same time points prior to IRI. For the kynurenine repletion experiments, mice were i.p. injected with L-kynurenine (50 mg.kg or 200 mg/kg) or normal saline 2hrs prior to renal IRI.
Study Approval
All animal studies were conducted in accordance with NIH guidelines for the use and care of live animal and were approved by the Institutional Animal Care and Use Committees at University of Kansas and at Northwestern University.
METHOD DETAILS
Histology
For the morphologic analysis, kidneys were fixed with 10% buffered formalin. Tubular injury was semiquantitatively graded based on the percentage of affected tubules (0, unaffected; 1, 1%–25%; 2, 26%–50%; 3, 51%–75%; 4, 76%–100%) in at least 5 random high-power fields in each section. Neutrophil infiltration was examined with a rat anti–Ly-6B.2 antibody (Bio-Rad, Hercules, CA), and for quantification, five random visual fields were analyzed per kidney section at a magnification of 200x using ImageJ software.
RNA isolation and Real-Time PCR
Total RNA was extracted with Trizol reagent (Invitrogen) according to the manufacturer’s instructions. First-strand cDNA synthesis was performed with 1 μg of RNA using a High Capacity cDNA Reverse Transcription Kit (Applied BioSystems). For real-time PCR analysis, cDNA was subjected to PCR amplification using either SYBR Green PCR Master Mix or TaqMan Universal PCR Master Mix on the Step-One Plus Real-Time PCR system (Applied Biosystems). The sequences of the primers used in this study are reported in Table S1. 18S rRNA was used to normalize mRNA.
RNA sequencing
RNA integrity was checked with Agilent Technologies 2100 Bioanalyzer. Poly(A) RNA sequencing library was prepared by LC Sciences following Illumina’s TruSeq-stranded-mRNA sample preparation protocol. Quality control analysis and quantification of the sequencing library were performed using Agilent Technologies 2100 Bioanalyzer High Sensitivity DNA Chip. Paired-ended sequencing was performed on Illumina’s NovaSeq 6000 sequencing system. First, Cutadapt (Martin, 2011) and perl scripts in house were used to remove the reads that contained adaptor contamination, low quality bases and undetermined bases. Then sequence quality was verified using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). HISAT2 (Kim et al., 2015) was used to map reads to the genome of ftp://ftp.ensembl.org/pub/release-96/fasta/mus_musculus/dna/. The mapped reads of each sample were assembled using StringTie (Pertea et al., 2015). Then, all transcriptomes were merged to reconstruct a comprehensive transcriptome using a proprietary Perl script of LC Sciences (Houston, Texas, USA). After the final transcriptome was generated, FPKM reads were evaluated by StringTie, and differential expressed genes were evaluated by edgeR (Robinson et al., 2010). The differentially expressed mRNAs and genes were selected with log2 (fold change) ≥ 1 or log2 (fold change) ≤ −1, and with p values < 0.05.
Metabolomic profiling
The metabolomic screening was conducted by Metabolon, Inc (Durham, NC). The sample preparation process was carried out using the automated MicroLab STAR® system from Hamilton Company. Sample preparation was conducted using a proprietary series of organic and aqueous extractions to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into two fractions; one for analysis by LC and one for analysis by GC. Samples were placed briefly on a TurboVap® (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum. Samples were then prepared for the appropriate instrument, either LC/MS or GC/MS.
Technical replicate samples created from a homogeneous pool containing a small amount of all study samples (“Client Matrix”) were also included. The instrument variability was determined by calculating the median relative standard deviation for the internal standards that were added to each sample prior to injection into the mass spectrometers. The overall process variability was determined by calculating the median relative standard deviation for all endogenous metabolites present in 100% of the Client Matrix samples, which were technical replicates of pooled client samples.
The informatics system consisted of four major components, the Laboratory Information Management System (LIMS), the data extraction and peak-identification software, data processing tools for QC and compound identification, and a collection of information interpretation and visualization tools for use by data analysts. The hardware and software foundations for these informatics components were the LAN backbone, and a database server running Oracle 10.2.0.1 Enterprise Edition.
The data extraction of the raw mass spec data files yielded information that could be loaded into a relational database and manipulated without resorting to BLOB manipulation. Once in the database, the information was examined and appropriate QC limits were imposed. Peaks were identified using Metabolon’s proprietary peak integration software, and component parts were stored in a separate, specifically designed complex data structure.
Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Identification of known chemical entities was based on comparison to metabolomic library entries of purified standards. At the time of analysis, more than 1000 commercially available purified standard compounds had been acquired and registered into LIMS for distribution to both the LC and GC platforms for determination of their analytical characteristics. The combination of chromatographic properties and mass spectra gave an indication of a match to the specific compound or an isobaric entity.
LC-MS/MS-based quantification of kynurenine and tryptophan
Instrumentation
Sample analyses were carried out using a Waters Acquity UPLC system (Waters, Milford, MA), made up of a binary solvent manager, refrigerated sample manager, and a heated column manager. Tandem mass spectrometric detection was performed using a TSQ Quantum triple-stage quadrupole mass spectrometer (Thermo-Electron, San Jose, CA) equipped with a standard API-1 electrospray source and a 50 μm I.D. stainless steel capillary
Liquid chromatographic conditions
A BEH-C18 Acquity UPLC column (2.1 mm × 100 mm, 1.7 μm) (Waters, Milford MA) equipped with an Acquity UPLC in-line stainless steel filter unit (0.2 μm, Waters) was used for all chromatographic separations. The column and autosampler tray temperatures were set to 50°C and 10°C, respectively. Mobile phases were made up of 20 mM ammonium acetate pH 5.5 in (A) H2O/CH3CN (95:5) and in (B) H2O/CH3CN (5:95). Gradient conditions were as follows: 0–2 min, B = 5%; 2–7 min, B = 5%–90%; 7–9 min, B = 90%; 9–10 min, B = 90%–5%; 10–15 min, B = 5%. The flow rate was maintained at 350 μL/min. A software-controlled divert valve was used to transfer eluent from 0–4.8 min and from 6.0–15 min of each chromatographic run to waste. The total chromatographic run time was 15 min. The sample injection volume was 10 μL. The autosampler injection valve and the sample injection needle were flushed and washed sequentially with mobile phase B (1 mL) and mobile phase A (2 mL) between each injection
Tandem mass spectrometric detection
The mass spectrometer was operated in positive ion mode. Quantitation was based on multiple reaction monitoring (MRM) detection of dansyl derivatives (tryptophan-d0: m/z 438 → 170, 28 V; tryptophan-d5: m/z 443 → 170, 24 V; kynurenine-d0: m/z 442 → 170, 36 V; kynurenine-d4: m/z 446 → 170, 32 V). The following optimized parameters were used for the detection of analytes and internal standards: N2 sheath gas 45 psi; N2 auxiliary gas 25 psi; spray voltage 5.0 kV; capillary temperature 280°C; capillary offset 35 V, tube lense offset 150 V, Ar collision gas 1.5 mtorr; scan time 50 ms; Q1/Q3 peak width at half-maximum 0.7 m/z.
Sample processing
Aliquots of thawed serum (50 μL) were transferred to clean microcentrifuge tubes and spiked with internal standards tryptophan-d5 (5 μL) and kynurenine-d4 (5 μL). Spiked serum was lightly vortexed, allowed to stand at room temperature for 15–20 min, and deproteinized with 75 μL of 2-propanol. Samples were then cooled to −20°C for 45–60 min, and precipitated proteins were removed by centrifugation (18,000 × g, 30 min, 5°C). The clear supernatant of each sample was transferred to a membrane dialysis cartridge (5000 MWCO) and filtered at 10°C for 6–8 h (10,000 × g). The filtrate was evaporated under a gentle stream of N2 gas. The residue was reconstituted in 80 μL of H2O/CH3CN (1:1) containing 20 mM dansyl chloride and 100 mM NaHCO3 (pH 8.0), and lightly vortexed for 20 min at room temperature. Following derivatization, the samples were diluted with 120 μL H2O and centrifuged to remove particulates (18,000 × g, 22°C, 20 min). The clear supernatant was transferred to 200 μL silanized autosampler vials equipped with Teflon-lined bonded rubber septa.
LC/UV based quantification of kynurenine
The LC/UV method was used for the quantification of serum kynurenine following exogenous L-kynurenine administration as reported by Veres G et al. (Veres et al., 2015). Briefly, 3-nitro-L-tyrosine (3-NLT) was used as an internal standard and added to a final concentration of 3 μM to 80 μl of serum. Serum samples were acidified with an equal volume of 5% perchloric acid and centrifuged at 12,000 ×g for 10 min at 4°C. The supernatant was removed and kynurenine was separated by high performance liquid chromatography (HPLC) coupled with UV detection on a 4.6 × 150 mm, 3 μm C-18 column (Acclaim 120) kept at 30°C. Elution was performed with 15 mM ammonium formate, 10% acetonitrile, pH 4.0, at flow rate of 0.7 ml/min. Both kynurenine and 3-NLT were detected by measuring the absorbance at 360 nm. Serum kynurenine was quantified using standard curves built from dilutions of a kynurenine standard that were treated like the serum samples.
HPLC-MS/MS based measurement of kynurenine pathway metabolites
For extraction of metabolites from serum, each sample was mixed with extraction solvent (methanol-water, 80:20; v/v; cooled to −80°C) at 1:10 ratio and vortexed for 1 min. Samples were further incubated at −80°C overnight and then spun at 20,000 ×g for 30 min. The supernatant was collected in a new tube and dried down using a SpeedVac concentrator (Thermo Savant). For extraction of metabolites from tissue, samples were homogenized in extraction solvent at 20 μL/1mg of tissue. A fifth of the homogenate was further diluted five times with extraction solvent in a new tube and vortexed for 1 min. Samples were incubated at −80°C overnight and then spun at 20,000 ×g for 30 min. The supernatant was collected in a new tube and dried down using a SpeedVac concentrator (Thermo Savant). After reconstitution in 60% acetonitrile, samples were analyzed by High-Performance Liquid Chromatography and Triple-quadruple Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, the system consisted of a Thermo TSQ in line with an electrospray source and a Vanquish (Thermo) UHPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a XBridge C18 column (Waters, dimensions of 2.1 mm × 50 mm and a 3.5 μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 3 min, 45% A; 10 min, 60% A; 10.1–11 min, 75% A; 11.1 min, 15% A; 11.1–15 min, 15% A with a flow rate of 400 μL/min. In positive mode, the capillary of ESI source was set to 325°C, with sheath gas at 35 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 3.5 kV. A selective reaction monitoring (SRM) of the protonated precursor ion and the related product ions were monitored. The transitions are listed as following: 3-hydroxy-anthranilic acid 154→136, 3-hydroxykynurenine 225→208, kynurenic acid 190→144, kynurenine 209→192, NAD+ 664.078→428, picolinic acid 124→83, quinolinic acid 168.03→127, tryptophan 205→188, xanthurenic acid 206→160. Peak area was integrated, and data acquisition and analysis were carried out by Xcalibur 4.1 software and TraceFinder 4.1 software, respectively (both from Thermo Fisher Scientific).
Extraction and measurement of NAD+
Intracellular NAD+ and NADH levels in the kidney were determined by using the NAD/NADH Quantitation Colorimetric Kit (BioVision incorporation, San Francisco, CA, Catalog #K337) following the manufacturer’s instruments. Briefly, 20–30 mg mouse kidneys were extracted in 500 μL NAD/NADH extraction buffer on ice. Protein concentration was measured with a BioRad protein assay (BioRad). The homogenate was filtered through 10 kDa molecular weight cut-off filters (Amicon Ultra, EMD Millipore) to remove NADH or NADPH consuming enzymes and subsequently assayed following manufacturer protocol. Finally, the results were obtained using a 96 well plate reader (BioTek) at the optical density of 450 nm.
Measurement of serum Epo
Serum Epo concentration was determined with a commercially available ELISA kit following the manufacturer provided instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the program “R” (https://cran.rproject.org/) or Prism 8 (GraphPad Software, La Jolla, CA). Two-tailed t test was used to calculate between-group differences while multiple comparisons were calculated by using one-way ANOVA with Sidak correction. For metabolites, statistical comparisons were performed by Welch’s two sample t tests and random effects ANOVA. For RNA-seq, differential expression analysis was done by the R package edgeR. Statistical tests are referenced in the figure legends. A two-sided significance level of 5% was considered statistically significant. Results were reported as mean values ± SEM. n indicates biological replicates per experimental group.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rat monoclonal anti Mouse Ly-6B.2 | Bio-rad | Cat#MCA771G; RRID: AB_322950 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| IOX 2 | Tocris | Cat# 4451 |
| L-Kynurenine | Sigma-Aldrich | Cat#K8625 |
| L-Tryptophan | Sigma-Aldrich | Cat#T0254 |
| Water, BAKER ANALYZED LC/MS Reagent Grade | J.T. Baker | Cat#9831-02 |
| Methylcellulose | ACROS Organics | 182311000 |
| TRIzol™ reagent | Invitrogen | Cat#15596026 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat#4368706 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat#4305719 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NAD/NADH Assay | Biovision | Cat#K337 |
| Mouse EPO Quantakine ELISA Kit | R&D systems | Cat#MEP00B |
| Standard for Tryptophan-d5 | Cambridge Isotope Laboratories | Cat#DLM-1092-PK |
| Standard for Kynurenine-d4 | Cambridge Isotope Laboratories | Cat#DLM-7842-PK |
| 3-Nitro-L-tyrosine | Sigma-Aldrich | Cat#851914 |
| 2-propanolol (LC-MS) | MilliporeSigma | Cat#1027814000 |
| Acetonitrile (LC-MS) | MilliporeSigma | Cat# 1000294000 |
| Bio-Rad Protein Assay | BioRad | Cat# 5000001 |
|
| ||
| Deposited data | ||
|
| ||
| RNaseq data | This manuscript-GEO | GEO: GSE156516 |
| Metabolomic data | This data has been deposited at the NIH Common Fund’s NMDR website, https://www.metabolomicsworkbench.org | Project id PR001178; https://doi.org/10.21228/M8F99F |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mus musculus (C57BL/6J) | The Jackson Laboratory | stock#000664 |
| Mus musculus Ido1 KO | The Jackson Laboratory | stock#005867 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Ido1 Mutant Forward 5′-CGTGCAATCCATCTTGTTCA-3′ | Integrated DNA Technologies | N/A |
| Ido1 Wild type Forward 5′-TATTGAAAGGGGAATCCAGA –3′ | Integrated DNA Technologies | N/A |
| Ido1 Reverse 5′- GTGTCAGAAAGCTCACTGCTT –3′ | Integrated DNA Technologies | N/A |
| RT-PCR Tnfα forward 5′-GCTGAGCTCAAACCCTGGTA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Tnfα reverse 5′-CGGACTCCGCAAAGTCTAAG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Vcam1 forward 5′- TAGAGTGCAAGGAGTTCGGG –3′ | Integrated DNA Technologies | N/A |
| RT-PCR Vcam1 reverse 5′-CCGGCATATACGAGTGTGAA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kim1 forward 5′-AAACCAGAGATTCCCACACG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kim1 reverse 5′-GTCGTGGGTCTTCCTGTAGC-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Ido1 forward 5′-ACTGTGTCCTGGCAAACTGGAAG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Ido1 reverse 5′-AAGCTGCGATTTCCACCAATAGAG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Ido2 forward 5′-CTTGGCCCAAATGCATGATT-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Ido2 reverse 5′-AAGATCCGGATGACCGAGTAAA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Tdo2 forward 5′-GGCAGAGTTCCGGAAGCA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Tdo2 reverse 5′-CATGACGCTTCTCATCAAACAAG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Afmid forward 5′-CCAGCAGATGGGTCATCCA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Afmid reverse 5′-TCTGCACGAAGTTCCCAACA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kmo forward 5′-AGCTGAGCGAGGGAAGAATCT-3 | Integrated DNA Technologies | N/A |
| RT-PCR Kmo reverse 5′-CAGCCCAAGCACGCAAAC-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kynu forward 5′-GAAGTAATCGAGGAGGAAGGAGACT-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kynu reverse 5′-AGTATAAAAGTGCAGCCCACTGAA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Haao forward 5′- ACATCCCTAAGCCTGTTTGG –3′ | Integrated DNA Technologies | N/A |
| RT-PCR Haao reverse 5′- CTATACACTGCCCTCCCATTG –3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kyat1 forward 5′-AGCGCGTCCCTTTCCATAC-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kyat1 reverse 5′-GCGGAGACCTAAGAAGCGG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Aadat forward 5′-ATGAATTACTCACGGTTCCTCAC-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Aadat reverse 5′-AACATGCTCGGGTTTGGAGAT-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kyat3 forward 5′-TTCAAAAACGCCAAACGAATCG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Kyat3 reverse 5′-GATGACCAAAGCCTCTTGTGT-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Nlrp3 forward 5′-ATTACCCGCCCGAGAAAGG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Nlrp3 reverse 5′-TCGCAGCAAAGATCCACACAG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Qprt forward 5′-CCGGTTCTAAAGCCGAAGAA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Qprt reverse 5′-GCCCAACAAGCCGCAGTA-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Acmsd forward 5′-TCCACACTCATATTCTACCAAAGG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Acmsd reverse 5′-AGCAGTTCTGTTGGATCACTC-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Epo forward 5′-CATCTGCGACAGTCGAGTTCTG-3′ | Integrated DNA Technologies | N/A |
| RT-PCR Epo reverse 5′-CACAACCCATCGTGACATTTTC-3′ | Integrated DNA Technologies | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| edgeR | Bioconductor | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Xcalibur 4.1 | Thermo Fisher Scientific | https://assets.thermofisher.com/TFS-Assets/CMD/manuals/man-xcali-97928-xcalibur-41-quan-start-manxcali97928-en.pdf |
| TraceFinder 4.1 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/tracefinder-software.html |
|
| ||
| Other | ||
|
| ||
| Amicon Ultra-0.5 Centrifugal Filter Unit | Millipore | UFC501024 |
Highlights.
Hypoxia and PHD inhibition increase serum kynurenine
Ido1 deficiency blunts the increase in serum kynurenine by hypoxia and PHD inhibition
The IDO1-kynurenine axis is required in renoprotection by hypoxic preconditioning
Hypoxia and PHD inhibition activate the kynurenine pathway in the post-ischemic kidney
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants P20 GM104936 (P.P.K.), R01DK115850 (P.P.K.), R35GM119528 (R.L.), T32DK071496 (R. Torosyan). Metabolomics services were performed by the Metabolomics Core Facility at Robert H. Lurie Comprehensive Cancer Center of Northwestern University. We acknowledge the Northwestern University George M. O’Brien Kidney Research Core Center (NU GoKidney), an NIH/NIDDK-funded program (P30 DK114857), for their core services and support. We thank Breeanna Mintmier (West Virginia University) for performing LC/UV-based quantification of serum kynurenine. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109547.
REFERENCES
- Agudelo LZ, Femenía T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, et al. (2014). Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159, 33–45. [DOI] [PubMed] [Google Scholar]
- Anders H-J, and Muruve DA (2011). The inflammasomes in kidney disease. J. Am. Soc. Nephrol. 22, 1007–1018. [DOI] [PubMed] [Google Scholar]
- Baban B, Chandler P, McCool D, Marshall B, Munn DH, and Mellor AL (2004). Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J. Reprod. Immunol. 61, 67–77. [DOI] [PubMed] [Google Scholar]
- Bernhardt WM, Câmpean V, Kany S, Jürgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Günzler V, Amann K, et al. (2006). Pre-conditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 17, 1970–1978. [DOI] [PubMed] [Google Scholar]
- Cantó C, Menzies KJ, and Auwerx J (2015). NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka I, Agudelo LZ, and Ruas JL (2017). Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 357, eaaf9794. [DOI] [PubMed] [Google Scholar]
- Chaudhary K, Shinde R, Liu H, Gnana-Prakasam JP, Veeranan-Karmegam R, Huang L, Ravishankar B, Bradley J, Kvirkvelia N, McMenamin M, et al. (2015). Amino acid metabolism inhibits antibody-driven kidney injury by inducing autophagy. J. Immunol. 194, 5713–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SJ, Hong KS, Schenck E, Lee S, Harris R, Yang J, Choi AMK, and Stout-Delgado H (2021). Decreased IDO1-dependent tryptophan metabolism in aged lung during influenza. Eur. Respir. J. 57, 2000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JE, Astola N, Cribbs AP, Goddard ME, Park I, Green P, Davies AH, Williams RO, Feldmann M, and Monaco C (2015). Indoleamine 2,3-dioxygenase-1 is protective in atherosclerosis and its metabolites provide new opportunities for drug development. Proc. Natl. Acad. Sci. USA 112, 13033–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Bratton DL, and Colgan SP (2014). Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 13, 852–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. [DOI] [PubMed] [Google Scholar]
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L-P, Gimotty PA, Gilks CB, Lal P, Zhang L, and Coukos G (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230. [DOI] [PubMed] [Google Scholar]
- Fallarini S, Magliulo L, Paoletti T, de Lalla C, and Lombardi G (2010). Expression of functional GPR35 in human iNKT cells. Biochem. Biophys. Res. Commun. 398, 420–425. [DOI] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, Fioretti MC, and Puccetti P (2002). T cell apoptosis by tryptophan catabolism. Cell Death Differ. 9, 1069–1077. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, et al. ; ERICCA Trial Investigators (2015). Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 373, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Hill P, Shukla D, Tran MGB, Aragones J, Cook HT, Carmeliet P, and Maxwell PH (2008). Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 19, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai W, Zhao R, Song H, Zhao J, Zhang L, Zhang L, Gao C, Han L, and Zhao W (2014). Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat. Commun. 5, 4738. [DOI] [PubMed] [Google Scholar]
- Johnsen M, Kubacki T, Yeroslaviz A, Späth MR, Mörsdorf J, Göbel H, Bohl K, Ignarski M, Meharg C, Habermann B, et al. (2020). The Integrated RNA Landscape of Renal Preconditioning against Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 31, 716–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitsinou PP, and Haase VH (2015). Molecular mechanisms of ischemic preconditioning in the kidney. Am. J. Physiol. Renal Physiol. 309, F821–F834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitsinou PP, Jaffe J, Michael M, Swan CE, Duffy KJ, Erickson-Miller CL, and Haase VH (2012). Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Renal Physiol. 302, F1172–F1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, Yao B, Zhang MZ, Harris RC, Duffy KJ, et al. (2014). Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest. 124, 2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, Shay S, French JL, West J, and Haase VH (2016). The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell. Biol. 36, 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartika R, Wibowo H, Purnamasari D, Pradipta S, and Larasati RA (2020). Altered Indoleamine 2,3-Dioxygenase Production and Its Association to Inflammatory Cytokines in Peripheral Blood Mononuclear Cells Culture of Type 2 Diabetes Mellitus. Int. J. Tryptophan Res. 13, 1178646920978236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, Ryu D, Cialabrini L, Matilainen O, Liscio P, et al. (2018). De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, Ju S-T, and Okusa MD (2009). Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 20, 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, and Simon MC (2010). Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 3. [Google Scholar]
- Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Böning A, Niemann B, et al. ; RIPHeart Study Collaborators (2015). A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 373, 1397–1407. [DOI] [PubMed] [Google Scholar]
- Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, and Bradfield CA (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole DJ, Webster SP, Uings I, Zheng X, Binnie M, Wilson K, Hutchinson JP, Mirguet O, Walker A, Beaufils B, et al. (2016). Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 22, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, and Reimer KA (1986). Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC, et al. (2017). Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130, 2664–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ, Chakraborty AA, Pierce KA, Miller CM, Hanse EA, et al. (2016). EGLN1 Inhibition and Rerouting of α-Ketoglutarate Suffice for Remote Ischemic Protection. Cell 164, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orabona C, Mondanelli G, Pallotta MT, Carvalho A, Albini E, Fallarino F, Vacca C, Volpi C, Belladonna ML, Berioli MG, et al. (2018). Deficiency of immunoregulatory indoleamine 2,3-dioxygenase 1in juvenile diabetes. JCI Insight 3, e96244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyzos KA, Ovchinnikova O, Berg M, Baumgartner R, Agardh H, Pirault J, Gisterå A, Assinger A, Laguna-Fernandez A, Bäck M, et al. (2015). Inhibition of indoleamine 2,3-dioxygenase promotes vascular inflammation and increases atherosclerosis in Apoe−/− mice. Cardiovasc. Res. 106, 295–302. [DOI] [PubMed] [Google Scholar]
- Poyan Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J, Lerner A, Kher A, Kim SH, Khoury CC, et al. (2018). De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 24, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralto KM, Rhee EP, and Parikh SM (2020). NAD+ homeostasis in renal health and disease. Nat. Rev. Nephrol. 16, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, and Ratcliffe PJ (2004). Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabuchi S, Hirota K, Nishi K, Oda S, Oda T, Shingu K, Takabayashi A, Adachi T, Semenza GL, and Fukuda K (2004). The intravenous anesthetic propofol inhibits hypoxia-inducible factor 1 activity in an oxygen tension-dependent manner. FEBS Lett. 577, 434–438. [DOI] [PubMed] [Google Scholar]
- Tiszlavicz Z, Németh B, Fülöp F, Vécsei L, Tápai K, Ocsovszky I, and Mándi Y (2011). Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-α (TNF-α) production by mononuclear cells, HMGB1 production by monocytes and HNP1–3 secretion by neutrophils. Naunyn Schmiedebergs Arch. Pharmacol. 383, 447–455. [DOI] [PubMed] [Google Scholar]
- Tomita I, Kume S, Sugahara S, Osawa N, Yamahara K, Yasuda-Yamahara M, Takeda N, Chin-Kanasaki M, Kaneko T, Mayoux E, et al. (2020). SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 32, 404–419. [DOI] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al. (2011). PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121, 4003–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, and Parikh SM (2016). PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres G, Molnár M, Zádori D, Szentirmai M, Szalárdy L, Török R, Fazekas E, Ilisz I, Vécsei L, and Klivényi P (2015). Central nervous system-specific alterations in the tryptophan metabolism in the 3-nitropropionic acid model of Huntington’s disease. Pharmacol. Biochem. Behav. 132, 115–124. [DOI] [PubMed] [Google Scholar]
- Yakulov TA, Todkar AP, Slanchev K, Wiegel J, Bona A, Groß M, Scholz A, Hess I, Wurditsch A, Grahammer F, et al. (2018). CXCL12 and MYC control energy metabolism to support adaptive responses after kidney injury. Nat. Commun. 9, 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq generated during this study are available on GEO: GSE156516. The metabolomic data has been deposited at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001178. The data can be accessed directly via the associated DOI: 10.21228/M8F99F.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.