

Abstract
Objective
The objective of this study was to employ ensemble clustering and tree-based risk model approaches to identify interactions between clinicogenomic features for colorectal cancer using the 100,000 Genomes Project.
Results
Among the 2211 patients with colorectal cancer (mean age of diagnosis: 67.7; 59.7% male), 16.3%, 36.3%, 39.0% and 8.4% had stage 1, 2, 3 and 4 cancers, respectively. Almost every patient had surgery (99.7%), 47.4% had chemotherapy, 7.6% had radiotherapy and 1.4% had immunotherapy. On average, tumour mutational burden (TMB) was 18 mutations/Mb and 34.4%, 31.3% and 25.7% of patients had structural or copy number mutations in KRAS, BRAF and NRAS, respectively. In the fully adjusted Cox model, patients with advanced cancer [stage 3 hazard ratio (HR) = 3.2; p < 0.001; stage 4 HR = 10.2; p < 0.001] and those who had immunotherapy (HR = 1.8; p < 0.04) or radiotherapy (HR = 1.5; p < 0.02) treatment had a higher risk of dying. The ensemble clustering approach generated four distinct clusters where patients in cluster 2 had the best survival outcomes (1-year: 98.7%; 2-year: 96.7%; 3-year: 93.0%) while patients in cluster 3 (1-year: 87.9; 2-year: 70.0%; 3-year: 53.1%) had the worst outcomes. Kaplan–Meier analysis and log rank test revealed that the clusters were separated into distinct prognostic groups (p < 0.0001). Survival tree or recursive partitioning analyses were performed to further explore risk groups within each cluster. Among patients in cluster 2, for example, interactions between cancer stage, grade, radiotherapy, TMB, BRAF mutation status were identified. Patients with stage 4 cancer and TMB ≥ 1.6 mutations/Mb had 4 times higher risk of dying relative to the baseline hazard in that cluster.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13104-021-05789-0.
Introduction
An evaluation of cancer drug approvals by the European Medicines Agency (EMA) found that 57% of drugs entered the market with limited evidence of survival benefits where at 3 years after market entry, survival gains in patients receiving 33 of the 39 cancer drugs were marginal [1]. Long-term follow-up analyses of data from trials in the post-market entry period are rare. Moreover, only 26% of randomised controlled trials (RCTs) investigated extension of life as the primary outcome [1] despite the EMA’s recommendation that overall survival is the most crucial outcome for investigating efficacy and safety of oncology drugs [2].
In light of these issues, population-based health records may help advance the long-term post-market evaluation of cancer drugs and support the identification and approval of new indications. Since the inception of the 21st Century Cures Act that aims to accelerate the development and innovation of medicines, the Food and Drug Administration created a framework for evaluating the use of real-world data (RWD) to support drug trials [3]. Overall survival in patients with cancer is well studied, particularly in the context of how genetic and transcriptomic alterations in tumours affect patient outcomes. Harnessing data from the International Cancer Genome Consortium and The Cancer Genome Atlas, there have been concerted efforts by the cancer community to develop genetic signatures that are predictive of overall survival rates [4–10]. Linking genetic data with electronic health records (EHRs) may allow further exploration of associations between clinical characteristics and tumour genetic profiles.
We seek to evaluate whether a clinicogenomic dataset from Genomics England could be useful for predicting survival outcomes based on clinical and genetic features in patients with colorectal cancer. We employed an ensemble clustering approach and survival tree analysis to identify clinicogenomic features that are indicative of prognosis.
Main text
Materials and methods
Dataset
Clinical and genome sequence data from 2211 patients with colorectal cancer was used.
Clinical data
Clinical data consisted of data from secondary care, the National Cancer Registration and Analysis Service and the Office for National Statistics (ONS). We obtained patient demographic details, age at cancer diagnosis, sex, tumour grade, tumour, nodes metastasis (TNM) stage and information on cancer therapy. Cancer type was defined based on site and morphology of cancer coded in ICD-O2 and ICD-10. Tumour grade was defined as G1 (well differentiated), G2 (moderately differentiated) and G3 (poorly differentiated). Cancer treatment was categorised as surgery, chemotherapy, radiotherapy and immunotherapy. Mortality data were obtained from the ONS registry.
Genetic data
Tumour mutational burden (TMB) was computed as the number of somatic non-synonymous small variants per megabase (Mb) of coding sequence. Only variants meeting the quality threshold criteria were included in TMB ascertainment (detailed calculations of variant quality metrics have been described previously) [11]. Somatic structural variants (SVs) and copy number variants (CNVs) for BRAF, KRAS and NRAS were obtained using the R package ‘getSVCNVperGene’. SVs and long indel (> 50 bp) calling were performed using the Manta Structural Variant Caller [12]. CNVs were called using the Canvas algorithm that identified genomic regions that had been lost or gained and investigated minor allele frequencies and coverage to determine copy number [13].
Ensemble clustering and validation
To perform clustering on mixed data types (i.e., numerical and categorical data), we first analysed dissimilarity between observations using the Gower distance. We employed four clustering algorithms: partitioning around medoids (PAM), hierarchical clustering (i.e., divisive analysis or DIANA), Fuzzy C-means (FCM) and k-means. We employed ensemble clustering (consensus clustering) [14] to merge results from multiple clustering algorithms above using the diceR package. The ‘dice’ function was used to perform consensus clustering across subsamples and algorithms for a different number of clusters (k). The number of subsamples was specified as five and the consensus function to use was specified as the cluster-based similarity partitioning algorithm. Internal cluster validation indices were used to assess the performance by considering the separability and compactness of the clusters. We selected the C-index, silhouette coefficient, compactness and connectivity indices for validation. The relative ranks of each algorithm across the internal indices were considered and their sum was computed. Algorithms below 75% for the sum rank were trimmed. Post trimming, algorithms were reweighted based on their internal index magnitudes and fed into the consensus function.
Survival analyses
Prior to clustering, we applied the Cox proportional hazards regression analysis to estimate overall survival outcomes for each clinicogenomic feature. All hazard ratios were fully adjusted for all other features investigated. To examine potential interactions between clinicogenomic predictors, we performed survival tree (recursive partitioning) analyses [15] for each cluster using the rpart package. Tree-based models allowed the visualisation of decision rules for predicting an outcome for different patient groups within each cluster. Survival data were pre-scaled to fit an exponential model in that the predicted risk in the root node is fixed to 1.0. Relative risk estimates in other nodes were reported as relative to the survival in the root node (i.e., relative to the baseline hazard). Mean deviance was ascertained to measure the variability among all observations that reached a specified node.
Results
Clinicogenomic features of the colorectal cancer cohort
The Genomics England cohort consisted of 2211 patients diagnosed with colorectal cancer (see Fig. 1 for the study design). Of these patients, 59.7% were men and the mean age of diagnosis was 67.7 (Additional file 1). The proportions of patients with colorectal cancer diagnosed at different stages were as follow: stage 1 (16.3%), stage 2 (36.3%), stage 3 (39.0%) and stage 4 (8.4%). Patients were also classified according to tumour: grade 1 (3.0%), grade 2 (81.9%) and grade 3 (15.1%). Almost all patients underwent surgical intervention (99.7%), while 47.4% of patients underwent chemotherapy, 7.6% had radiotherapy and 1.4% had immunotherapy. With regards to genetic features, we explored tumour mutational burden (TMB) and observed that patients had 18 mutations/Mb on average (Additional file 1). We also explored structural variants and copy number variants and found that 34.4%, 31.3% and 25.7% of patients had mutations in KRAS, BRAF and NRAS respectively (Additional file 1). We performed Cox regression analyses where all hazard ratios (HRs) were fully adjusted for all other features considered. In the fully adjusted model, when considering cancer stage (with stage 1 as the reference), patients who were at stage 3 (HR = 3.2; p < 0.001) and stage 4 (HR = 10.2; p < 0.001) had a significantly higher risk of death, while stage 2 patients did not show any increase in risk (HR = 1.3; p = 0.29). Additionally, immunotherapy (HR = 1.8; p < 0.04) and radiotherapy (HR = 1.5; p < 0.02) were significantly associated with poorer survival outcomes (Additional file 2).
Fig. 1.

Schematic diagram depicting the study design
Ensemble clustering and validation
Employing resampling of the four clustering algorithms (K-means, PAM, DIANA and fuzzy C-means) on five replicates of the dataset, the optimal number of clusters was found to be between four and six. Internal cluster validity indices (i.e., C-index, silhouette coefficient, compactness and connectivity) for cluster validation were assessed to compare results from the varying number of clusters (k) (Additional file 3). Based on the cluster validity indices, k = 4 was found to be optimal based on compactness and connectivity (Additional file 4). We employed the ensemble clustering approach to consider the relative ranks of each of the four clustering algorithms across all internal validity indices to compute the overall rank sum to generate a final ensemble with the largest silhouette coefficient and lowest connectivity (Additional file 4).
Descriptive features of the clusters and overall survival outcomes
Descriptive statistics of the clusters were provided in Additional file 5. Cluster 1 featured patients with high TMB (mean = 32.3 non-synonymous somatic mutations per Mb). The proportion of patients having stage 1 or stage 2 tumours were as follow: cluster 1 (69.2%), cluster 2 (32.2%), cluster 3 (8.7%) and cluster 4 (80.9%) (Additional file 5). Cluster 2 consisted of the highest proportion of patients who had chemotherapy (75.2%) while cluster 4 had the least number of patients who had chemotherapy (27.7%) (Additional file 5). Cluster 4 had the highest proportion of patients with mutations in KRAS (60.8%), BRAF (55.8%) and NRAS (54.7%). We estimated overall survival rates at 1 year, 2 years and 3 years post-diagnosis for each cluster. Patients in cluster 2 had the best survival outcomes (1-year: 98.7%; 2-year: 96.7%; 3-year: 93.0%) while patients in cluster 3 (1-year: 87.9; 2-year: 70.0%; 3-year: 53.1%) had the worst outcomes (Additional file 5). We generated a Kaplan–Meier plot and to illustrate the overall survival outcomes stratified by the clusters generated from ensemble clustering. Log rank test revealed that the clusters were separated into distinct prognostic groups (p < 0.0001) (Fig. 2).
Fig. 2.
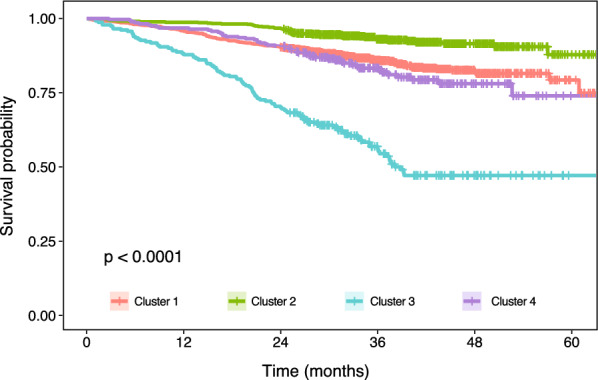
Overall survival outcomes in patients with colorectal cancer stratified using the ensemble clustering approach
Survival tree analyses within each prognostic cluster offered an additional resolution to stratify patients into risk group by clinicogenomic features
We performed survival tree (recursive partitioning) analyses to garner additional insights into the potential interactions between clinicogenomic predictors. Survival trees allowed the identification of interactions between clinicogenomic predictors by grouping subjects according to their survival profiles. Survival trees for each cluster were shown in Fig. 3. For cluster 1, cancer stage was found to be a significant predictor of overall survival (Fig. 3A). For cluster 2, cancer stage, grade, radiotherapy, TMB, BRAF mutation status were important predictors (Fig. 3B). Survival tree for cluster 3 featured TMB, BRAF and KRAS mutation status and cancer stage as predictors (Fig. 3C). For cluster 4, stage, radiotherapy and TMB were found to be important (Fig. 3D).
Fig. 3.

Survival tree analyses for each of the four prognostic clusters. A Cluster 1, B cluster 2, C cluster 3 and D cluster 4. Proportion of patients within each node are indicated as a percentage. Relative risk (RR) for each node is indicated, along with the mean deviance (md) value. Nodes described in the results section are highlighted in blue boxes
Relative risk estimates were shown in the survival trees for each node as relative to the survival in the root node. In the survival tree for cluster 2, the rightmost terminal node had a relative death rate of 4 times the overall rate and was defined by stage 4 and TMB ≥ 1.6 (Fig. 3B). For cluster 3, TMB was found to be the most important predictor of overall survival with a cut-off value of 3.9 mutations/Mb. Patients with TMB < 3.9 mutations/Mb were further split by TMB and BRAF mutation status, in which the group with TMB < 3.6 mutations/Mb, no BRAF mutation and TMB ≥ 3.3 mutations/Mb had a relative death rate of 2.5 times the overall rate (Fig. 3C). In contrast, we observed a low relative risk for death of 0.13 in patients with TMB < 3.9 but TMB ≥ 3.6 mutations/Mb (Fig. 3C). Similarly, a relative risk of 0.13 is also observed in another group of patients having TMB < 3.9 and also TMB < 3.6 mutations/Mb, followed by having BRAF mutation and no KRAS mutation (Fig. 3C). Survival tree for cluster 4 revealed that patients were initially split by cancer stage, where the left side of the tree consisted of patients at stage 1 or 2, while the right side was for patients at stage 3 or 4 (Fig. 3D). Then, patients at stage 3 or 4 were split by TMB with a cut-off value of 3.8 mutations/Mb. Patients with TMB ≥ 3.8 had a relative death rate of 1.9 times. Patients with TMB < 3.8 were further split by TMB with a cut-off of 3.0, and patients with TMB ≥ 3.0 but < 3.8 had a relative death rate of 6.4 times the overall rate (Fig. 3D).
Discussion
We demonstrated the feasibility of predicting overall survival outcomes in patients with colorectal cancer using ensemble clustering and survival tree analyses on a clinicogenomic dataset. Large-scale national estimates of colorectal cancer survival rates have mostly focused on cancer survival by stage using staging information collected from population-based cancer registries [16]. Independently, other studies have investigated the role of genomic biomarkers on cancer survival outcomes [17–23]. Our study demonstrated that clinicogenomic features can be employed to provide additional resolution for stratifying patients into risk groups not currently afforded by staging information alone. We have identified four prognostic clusters using ensemble clustering. As each cluster consists of a heterogeneous group of patients, subsequent survival tree analyses within the clusters revealed the different contributions of cancer stage/grade, radiotherapy treatment, TMB, BRAF or KRAS mutation status in predicting the relative risk of death in patients with colorectal cancer.
Limitations
First, the study is underpowered to investigate prognosis in relation to specific cancer therapy. Second, we have only investigated mutation profiles of KRAS, BRAF and NRAS as an initial proof of concept. Third, analyses were performed only in patients with complete staging information. Future work may explore imputation methods to address missing data. Fourth, the selection of patients for recruitment into the 100,000 Genomes Project may introduce a selection bias for individuals with access to specific healthcare services.
Supplementary Information
Additional file 1: Baseline characteristics of patients with colorectal cancer in the 100,000 Genomes Project cohort.
Additional file 2: Hazard ratios and 95% confidence intervals for overall survival.
Additional file 3: Internal cluster validity indices for k = 5 and 6.
Additional file 4: Internal cluster validity indices for k = 4.
Additional file 5: Clinicogenomic features after ensemble clustering.
Acknowledgements
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. Genomics England Research Consortium: Ambrose, J. C.; Arumugam, P.; Bleda, M.; Boardman-Pretty, F.; Boustred, C. R.; Brittain, H.; Caulfield, M. J.; Chan, G. C.; Fowler, T.; Giess A.; Hamblin, A.; Henderson, S.; Hubbard, T. J. P.; Jackson, R.; Jones, L. J.; Kasperaviciute, D.; Kayikci, M.; Kousathanas, A.; Lahnstein, L.; Leigh, S. E. A.; Leong, I. U. S.; Lopez, F. J.; Maleady-Crowe, F.; Moutsianas, L.; Mueller, M.; Murugaesu, N.; Need, A. C.; O‘Donovan P.; Odhams, C. A.; Patch, C.; Perez-Gil, D.; Pereira, M. B.; Pullinger, J.; Rahim, T.; Rendon, A.; Rogers, T.; Savage, K.; Sawant, K.; Scott, R. H.; Siddiq, A.; Sieghart, A.; Smith, S. C.; Sosinsky, A.; Stuckey, A.; Tanguy M.; Thomas, E. R. A.; Thompson, S. R.; Tucci, A.; Walsh, E.; Welland, M. J.; Williams, E.; Witkowska, K.; Wood, S. M.
Abbreviations
- RWD
Real-world data
- RCT
Randomised control trial
- EMA
European Medicines Agency
- HER
Electronic health record
- TMB
Tumour mutational burden
- HR
Hazard ratio
- TNM
Tumour, nodes, metastasis
- SV
Structural variant
- CNV
Copy number variant
- PAM
Partition around medoids
- DIANA
Divisive analysis
- FCM
Fuzzy C-means
Authors’ contributions
YW analysed the data, interpreted the results and wrote the initial manuscript draft. NP contributed to the study design and supervision. SM contributed to the interpretation of the results. WHC contributed to the interpretation of the results and writing of the final manuscript. AGL designed the study, supervised the research and wrote the final manuscript. All authors read and approved the final manuscript.
Funding
AGL is supported by funding from the Wellcome Trust (204841/Z/16/Z), National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre (BRC714/HI/RW/101440), NIHR Great Ormond Street Hospital Biomedical Research Centre (19RX02), the Health Data Research UK Better Care Catalyst Award (CFC0125) and the Academy of Medical Sciences (SBF006\1084). The funders have no role in the writing of the manuscript or the decision to submit it for publication.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Access to the 100,000 Genomes Project database (https://www.genomicsengland.co.uk/) is subjected to approval.
Declarations
Ethics approval and consent to participate
The 100,000 Genomes Project received ethics approval from the Health Research Authority Committee East of England, Cambridge South (Reference: 14/EE/1112). This project is covered by the aforementioned ethics approval and as such a separate informed consent is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alvina G. Lai, Email: alvina.lai@ucl.ac.uk
Genomics England Research Consortium:
J. C. Ambrose, P. Arumugam, M. Bleda, F. Boardman-Pretty, C. R. Boustred, H. Brittain, M. J. Caulfield, G. C. Chan, T. Fowler, A. Giess, A. Hamblin, S. Henderson, T. J. P. Hubbard, R. Jackson, L. J. Jones, D. Kasperaviciute, M. Kayikci, A. Kousathanas, L. Lahnstein, S. E. A. Leigh, I. U. S. Leong, F. J. Lopez, F. Maleady-Crowe, L. Moutsianas, M. Mueller, N. Murugaesu, A. C. Need, P. O’Donovan, C. A. Odhams, C. Patch, D. Perez-Gil, M. B. Pereira, J. Pullinger, T. Rahim, A. Rendon, T. Rogers, K. Savage, K. Sawant, R. H. Scott, A. Siddiq, A. Sieghart, S. C. Smith, A. Sosinsky, A. Stuckey, M. Tanguy, E. R. A. Thomas, S. R. Thompson, A. Tucci, E. Walsh, M. J. Welland, E. Williams, K. Witkowska, and S. M. Wood
References
- 1.Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009–13. BMJ. 2017;359:4530. doi: 10.1136/bmj.j4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Medicines Agency. Evaluation of anticancer medicinal products in man. https://www.ema.europa.eu/en/evaluation-anticancer-medicinal-products-man. Accessed 1 April 2021.
- 3.US Food and Drug Administration. Framework for FDA’s real-world evidence program. https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence. Accessed 16 Nov 2020.
- 4.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang WH, Lai AG. An integrative pan-cancer investigation reveals common genetic and transcriptional alterations of AMPK pathway genes as important predictors of clinical outcomes across major cancer types. BMC Cancer. 2020;20:773. doi: 10.1186/s12885-020-07286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang WH, Forde D, Lai AG. Dual prognostic role of 2-oxoglutarate-dependent oxygenases in ten cancer types: implications for cell cycle regulation and cell adhesion maintenance. Cancer Commun. 2019;39:23. doi: 10.1186/s40880-019-0369-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang WH, Lai AG. An immunoevasive strategy through clinically-relevant pan-cancer genomic and transcriptomic alterations of JAK-STAT signaling components. Mol Med. 2019;25:1–14. doi: 10.1186/s10020-019-0114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang WH, Lai AG. Pan-cancer genomic amplifications underlie a Wnt hyperactivation phenotype associated with stem cell-like features leading to poor prognosis. Transl Res. 2019 doi: 10.1016/j.trsl.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Korkut A, Zaidi S, Kanchi RS, Rao S, Gough NR, Schultz A, et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-β superfamily. Cell Syst. 2018;7:422–437. doi: 10.1016/j.cels.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genomics England. Site QC, FILTER and INFO Fields. https://research-help.genomicsengland.co.uk/display/GERE/Site+QC%2C+FILTER+and+INFO+Fields. Accessed 1 April 2021.
- 12.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 13.Roller E, Ivakhno S, Lee S, Royce T, Tanner S. Canvas: versatile and scalable detection of copy number variants. Bioinformatics. 2016;32:2375–2377. doi: 10.1093/bioinformatics/btw163. [DOI] [PubMed] [Google Scholar]
- 14.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52:91–118. doi: 10.1023/A:1023949509487. [DOI] [Google Scholar]
- 15.Ramezankhani A, Tohidi M, Azizi F, Hadaegh F. Application of survival tree analysis for exploration of potential interactions between predictors of incident chronic kidney disease: a 15-year follow-up study. J Transl Med. 2017;15:1–17. doi: 10.1186/s12967-017-1346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surveillance, epidemiology, and end results (SEER) program. Overview of the SEER program. https://seer.cancer.gov/about/overview.html. Accessed 19 April 2020.
- 17.Chang WH, Lai AG. Timing gone awry: distinct tumour suppressive and oncogenic roles of the circadian clock and crosstalk with hypoxia signalling in diverse malignancies. J Transl Med. 2019;17:132. doi: 10.1186/s12967-019-1880-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang WH, Lai AG. Transcriptional landscape of DNA repair genes underpins a pan-cancer prognostic signature associated with cell cycle dysregulation and tumor hypoxia. DNA Repair. 2019;78:142–153. doi: 10.1016/j.dnarep.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Chang WH, Forde D, Lai AG. A novel signature derived from immunoregulatory and hypoxia genes predicts prognosis in liver and five other cancers. J Transl Med. 2019;17:14. doi: 10.1186/s12967-019-1775-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge Z, Leighton JS, Wang Y, Peng X, Chen Z, Chen H, et al. Integrated genomic analysis of the ubiquitin pathway across cancer types. Cell Rep. 2018;23:213–226.e3. doi: 10.1016/j.celrep.2018.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang WH, Lai AG. The pan-cancer mutational landscape of the PPAR pathway reveals universal patterns of dysregulated metabolism and interactions with tumor immunity and hypoxia. Ann NY Acad Sci. 2019;1448:65–82. doi: 10.1111/nyas.14170. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Caesar-Johnson SJ, et al. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173:386–399.e12. doi: 10.1016/j.cell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang WH, Lai AG. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br J Cancer. 2019 doi: 10.1038/s41416-019-0572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Baseline characteristics of patients with colorectal cancer in the 100,000 Genomes Project cohort.
Additional file 2: Hazard ratios and 95% confidence intervals for overall survival.
Additional file 3: Internal cluster validity indices for k = 5 and 6.
Additional file 4: Internal cluster validity indices for k = 4.
Additional file 5: Clinicogenomic features after ensemble clustering.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files. Access to the 100,000 Genomes Project database (https://www.genomicsengland.co.uk/) is subjected to approval.