

Abstract
Motivation
Despite widespread prevalence of somatic structural variations (SVs) across most tumor types, understanding of their molecular implications often remains poor. SVs are extremely heterogeneous in size and complexity, hindering the interpretation of their pathogenic role. Tools integrating large SV datasets across platforms are required to fully characterize the cancer’s somatic landscape.
Results
svpluscnv R package is a swiss army knife for the integration and interpretation of orthogonal datasets including copy number variant segmentation profiles and sequencing-based structural variant calls. The package implements analysis and visualization tools to evaluate chromosomal instability and ploidy, identify genes harboring recurrent SVs and detects complex rearrangements such as chromothripsis and chromoplexia. Further, it allows systematic identification of hot-spot shattered genomic regions, showing reproducibility across alternative detection methods and datasets.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Somatic structural variations (SVs) are a fundamental contributor to cancer genetics and pathogenesis (Futreal et al., 2004). Historically, copy number variants (CNVs) have been instrumental in cancer diagnosis and classification. Genome-wide profiling via genotyping or comparative genomic hybridization (CGH) arrays coupled with tools such as GISTIC (Mermel et al., 2011) have facilitated identification of oncogenic events. However, the surge of available large whole genome sequencing (WGS) cancer datasets is expanding our understanding of the role of SVs (Consortium, 2020; Grobner et al., 2018; Ma et al., 2018) and has revealed widespread prevalence of complex catastrophic events such as chromothripsis and chromoplexia (Cortes-Ciriano et al., 2020).
Recently, we integrated CNVs from genotyping arrays with structural variant calls (SVCs) from WGS in a neuroblastoma cohort, showing that orthogonal data types allowed identification of novel oncogenic alterations and revealed insights about the pathogenic role of chromothripsis (Lopez et al., 2020). Here, we implement the methodological framework into an R package incorporating multiple functionalities to integrate orthogonal data types and characterize SVs in large cancer datasets.
2 Materials and methods
The svpluscnv R package operates with two input data types: (i) CNV: segmentation profiles including logR (E.g. log2 of the ratio of the signal between paired samples; e.g. tumor/normal). CNVs derive from arrays (SNP, CGH) as well as sequencing read-depth data and provide genome-wide gain/loss dosage information. (ii) SVC: derived from WGS discordantly aligned reads. A plethora of available algorithms use different approaches to identify SVC (reviewed in Kosugi et al., 2019). Variant classes include: duplication (DUP), deletion (DEL), inversion (INV), insertion (INS), translocation (TRA) and break-end (BND) for undefined SVCs.
2.1 Integrated analyses of CNV and SV datasets
Aneuploidy and CNV visualization: (i)‘cnv.freq.plot’ maps segments with copy number logR ≠ 0 across samples and plots a genome wide map of gain/loss frequencies (Supplementary Fig. S1); (ii) ‘chr.arm.cnv’ retrieves chromosome arm median CNV logR values.
Chromosomal instability measurements: (i) ‘percent.genome.changed’ measures the percentage of the genome’s CN with logR ≠ 0; (ii) ‘cnv.breaks’ identifies CNV breakpoints based on fold change threshold of CN between contiguous segments and (iii) ‘svc.breaks’ reports per sample SVC breakpoints. Finally, ‘match.breaks’ identifies colocalizing breakpoints between CNV and SVC for a set of common samples.
Gene annotation functionalities: (i) ‘gene.cnv’ transforms segmentation data into a gene-level CN matrix from which amplifications and deletions can be queried. (ii) ‘cnv.break.annot’ and ‘svc.break.annot’ provides genomic feature annotation tools for CNV and SVC breakpoints such as overlapping genes and their upstream regions (Supplementary Fig. S2). Finally, ‘sv.model.view’ and ‘gene.track.view’ display genomic track visualizations overlaying CNV and SVC for defined regions (Supplementary Fig. S2).
2.2 Chromosome shattering and hot-spot analyses
‘shattered.regions’ combines CNVs and SVCs to identify complex rearrangements. The algorithm is highly parametrizable and follows two major steps: (i) identification of High Breakpoint Density (HBD) genomic bins: breakpoints are mapped into a default 10 Mb sliding genomic windows calculated every 2 Mb, and (ii) shattered regions are defined by collapsing contiguous HBDs. Next, further testing for true likely catastrophic events uses information about interleaved SVCs, links between distant regions and the dispersion of breakpoints within regions (Supplementary Date). A simplified ‘shattered.regions.cnv’ algorithm, identifies catastrophic events using CNV segmentation data only. It follows same approach as shattered.regions, using only parameters derived from CNV. A ‘circ.chromo.plot’ function wraps circlize (Gu et al., 2014) functionalities for circus plotting zooming into shattered regions (Fig. 1A).
Fig 1.
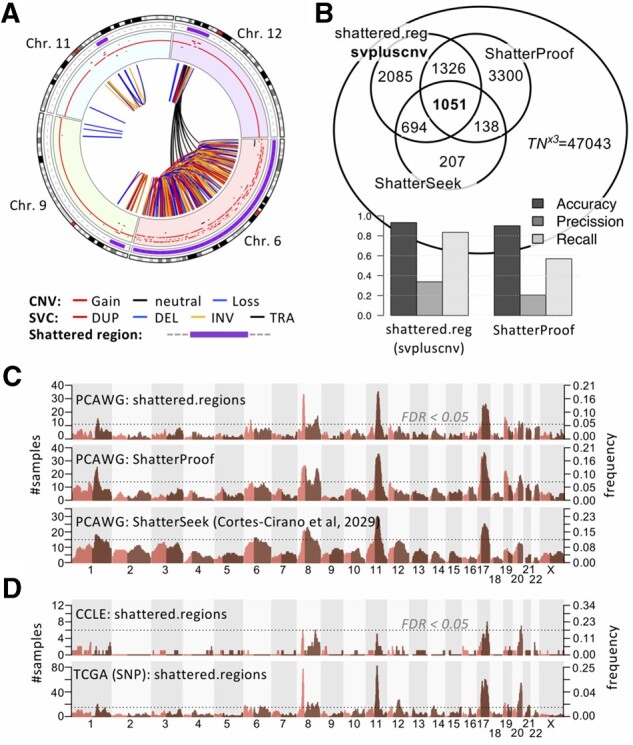
Identification of shattered chromosome regions. (A) Circos plot representing a breast cancer sample (BRCA-UK: SA541850) shows shattered regions at 6, 9, 11 and 12 Chromosomes (purple band), CNVs (outer) and SVCs (inner) tracks. (B) Venn diagram representing the overlap of shattered chromosomes detected by three different methods across all PCAWG samples (top) and prediction performance(bottom); (C, D) Genome wide shattered region frequency maps and their FDR < 0.05 threshold (Supplementary Fig. S4) of (C) PCAWG breast cancer samples derived from shattered.regions (top), ShatterProof (middle) and ShatterSeek (bottom); (D) shattered.regions identified in breast cancer cell lines (top) and TCGA samples (bottom)
‘shattered.map.plot’ generates a genome wide map of regions with chromosome shattering and their frequencies (Fig. 1C and D). Using a permutation test ‘freq.p.test’ assigns corrected P-values to observed frequencies defining hot-spots regions above significance threshold; such regions are deemed under selection pressure for chromosome shattering.
3 Results
3.1 Integrated SV analysis of cancer datasets
In order to test svpluscnv tools, we compared the results obtained from analyzing different breast cancer cell line and primary tumor datasets including: 59 CCLE cell lines (Ghandi et al., 2019), 1088 primary tumors from TCGA and 198 breast adenocarcinomas from PCAWG (Consortium, 2020). The three datasets presented similar CNV gain/loss frequency profiles (Supplementary Fig. S1). We mapped breakpoints to known genes and ranked altered genes, again showing concordance across the three datasets (Supplementary Fig. S2A–C); 35 of 59 breast lines had orthogonal data from SNP (CNV) and WGS (SVC) profiles. We observed complete overlap of CNV and SVC breakpoints in the most frequently altered gene and fragile site, FHIT (Supplementary Fig. S2D). Overall, 30.2–28.3% of all breakpoints colocalized across CNV and SVC data types (Supplementary Fig. S3).
3.2 Analysis of shattered regions
In order to test the shattered.regions algorithm with other available tools, we evaluated 2658 human cancer whole genomes, previously analyzed with the algorithm ShatterSeek and manually curated (Cortes-Ciriano et al., 2020). In addition, we obtained predictions from ShatterProof (Govind et al., 2014) for the same set of samples using default parameters. The three sets of predictions significantly overlapped (P-value < 2.2e-16, Fig. 1B, Supplementary Data). We then used the curated set (ShatterSeek) as the gold standard to test for the precision/recall performance: shattered.regions achieved superior results (pre = 0.34; rec = 0.83) compared to ShatterProof (pre = 0.20; rec = 0.57) although the later allows additional input data types (i.e. Loss of Heterozygosity) not included here (Fig. 1B).
svpluscnv introduces a novel tool to identify recurrently shattered regions that could be under selection pressure in cancer histotypes. We evaluated PCAWG breast dataset shattered regions calculated using alternative algorithms (Fig. 1C); the three methods returned a strongly similar landscape of genome-wide frequencies (Pearson’s correlation P-value < 2.2×10−16, Supplementary Fig. S5A–C); The same landscape was reproduced when tested across two additional datasets including breast cancer derived cell lines (CCLE) and primary tumors from TCGA (based on CNV data only) (Fig. 1D, Supplementary Fig. S5D). Recurrently shattered regions (FDR<0.05) were identified in chr8p11, chr8q24 (MYC locus), chr11q13 (CCND1 locus), chr17q and chr20q in all three datasets; highlighting their reproducibility and likely biological relevance (Fig. 1C and D, Supplementary Fig. S4).
4 Conclusion
svpluscnv aims to become instrumental in the study of SVs in cancer genomics, enabling the identification of recurrent complex rearrangements that may pinpoint disease driver events. This toolset allows the research community to easily perform complex analysis of high throughput SV profiling data and will support extensions to further integrate analyses of other types of omics data.
Financial Support: This work was supported by the following NIH grant to SJD: [R01-CA204974].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Gonzalo Lopez, Department of Genetics and Genomics Sciences, Icahn School of Medicine, New York, NY, 10029, USA.
Laura E Egolf, Division of Oncology and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, Philadelphia, PA, 19104, USA; Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Federico M Giorgi, Department of Pharmacy and Biotechnology, University of Bologna, Bologna, 40126, Italy.
Sharon J Diskin, Division of Oncology and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, Philadelphia, PA, 19104, USA; Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Adam A Margolin, Department of Genetics and Genomics Sciences, Icahn School of Medicine, New York, NY, 10029, USA.
Data Availability
No new data were generated or analysed in support of this research.
References
- Consortium,ITP-CAoWG. (2020) Pan-cancer analysis of whole genomes. Nature, 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ciriano I. et al. (2020) Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet, 52, 331–341. [doi: 10.1038/s41588-019-0576-7, Epub February 5 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal P.A. et al. (2004) A census of human cancer genes. Nat. Rev. Cancer, 4, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M. et al. (2019) Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature, 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind S.K. et al. (2014) ShatterProof: operational detection and quantification of chromothripsis. BMC Bioinformatics, 15, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobner S.N. et al. (2018) The landscape of genomic alterations across childhood cancers. Nature, 555, 321–327. [DOI] [PubMed] [Google Scholar]
- Gu Z. et al. (2014) circlize Implements and enhances circular visualization in R. Bioinformatics, 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- Kosugi S. et al. (2019) Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol., 20, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez G. et al. (2020) Somatic structural variation targets neurodevelopmental genes and identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome Res, 30, 1228–1242. [doi: 10.1101/gr.252106.119, Epub 13 August 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X. et al. (2018) Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature, 555, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C.H. et al. (2011) GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol., 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data were generated or analysed in support of this research.