

Abstract
Background
Src homology region 2 domain‐containing phosphatase 2 (SHP2) is a novel target for Kirsten rat sarcoma oncogene (KRAS) mutant cancer. We retrospectively studied the significance of SHP2 in KRAS mutant non‐small cell lung cancer (NSCLC) treated with immunotherapy and its relationship with tumor microenvironment (TME).
Methods
Sixty‐one advanced KRAS mutant NSCLC patients who underwent immunotherapy were enrolled. Next‐generation sequencing (NGS) was used to profile mutation status. The expression of SHP2, phospho‐SHP2 (pSHP2), and programmed death ligand 1 (PD‐L1) were analyzed by immunohistochemistry (IHC). Quantitative multiplexed immunofluorescence cytochemistry (mIFC) analysis was conducted to describe the TME.
Results
SHP2 was heterogeneously expressed in 32 samples in both tumor cells and immune cells and highly expressed (H‐score >10) in 25 (78.1%) samples. The expression levels of SHP2 and pSHP2 were positively correlated. Stromal SHP2 (s‐SHP2) was higher in tumors with PD‐L1 ≥50% versus PD‐L1 <50% (p = 0.039). By quantitative mIFC analysis, the expression of s‐SHP2 had positive correlation with CD8, CD4, CD68, and PD‐L1 levels in stromal area. Patients with high SHP2 expression made up 100.0% of the partial respond (PR) and 80.0% of the stable disease (SD), whereas 50.0% of the progress disease (PD). High SHP2 expression was associated with longer progression‐free survival (PFS) and overall survival (OS) (p < 0.001, p = 0.013). Patients with high expression of both SHP2 and PD‐L1 had longer PFS (p < 0.001).
Conclusion
High SHP2 expression could predict the efficacy of immunotherapy and better survival in advanced KRAS mutant NSCLC. SHP2 may function in both tumor cells and immune cells, warranting further study on the potential diverse effects of SHP2 inhibition in TME.
Keywords: immunotherapy, KRAS, non‐small cell lung cancer, SHP2
SHP2 was heterogeneously expressed in both tumor cells and immune cells in KRAS mutant NSCLC. ICI can benefit more in patients with high SHP2‐expressed NSCLC. High level of SHP2 was associated with better PFS and OS, suggesting SHP2 is a promising marker to predict the efficacy of ICI and better survival for KRAS mutant NSCLC.
INTRODUCTION
Non‐small cell lung cancer (NSCLC) is the leading cause of cancer‐related death in China and primarily diagnosed as stage IV disease.1 Kirsten rat sarcoma oncogene (KRAS) is one of the most prevalent oncogenic mutated genes in NSCLC, making up ~ 7.2%–8.0% of Chinese adenocarcinoma patients and 5% squamous carcinoma, whereas ~30% in Caucasian patients.2, 3 Treatment of KRAS mutant lung cancer has been difficult until recently. Great breakthroughs have been made showing that KRAS G12C kinase inhibitors AMG510 and MRTX849 show a significant effect on NSCLC patients with KRAS G12C mutation.4, 5, 6 Recently, the combination of MEK and Src homology region 2 domain‐containing phosphatase‐2 (SHP2) inhibitors is highlighted for its synthetic lethality in KRAS mutant cancer models, which provides another therapeutic potential.7, 8, 9
SHP2, composed of two SH2 domains (N‐SH2 and C‐SH2), a protein‐tyrosine phosphatases (PTP) domain and a C‐terminal tail with two tyrosine phosphorylation sites, is one of the two SH2 domain‐containing PTPs.10 SHP2 is a molecular switch that the N‐SH2 is wedged into the PTP domain, which blocks substrate access to keep the closed state and is activated by phosphotyrosyl (pTyr) peptide that disrupts the auto‐inhibition state.11 SHP2 plays multiple roles in tumor formation and progression, especially in KRAS mutant‐driven cancers.7, 8, 9 It is located downstream of growth factor receptors and upstream of RAS that is critical for the mitogen‐activated protein kinases (MAPK)/extracellular‐signal‐regulated‐kinase (ERK) pathway, and the mechanism of how SHP2 mediates the activation of RAS is multiple.12 SHP2 also participates in the phosphoinositide 3‐kinase (PI3K)/protein kinase B (AKT), JAK/STAT, JNK, and nuclear factor κB (NF‐κB) signaling pathways, which are associated with tumor formation.13 SHP2 plays an important role in immune cells (e.g., T cells, B cells, and macrophages). When the two tyrosine‐containing motifs located in its cytoplasmic tail of programmed cell death protein 1 (PD‐1) become phosphorylated, SHP2 is recruited and then promotes immunosuppression in immune cells.14, 15
Previous studies showed that SHP2 was an independent prognostic factor for NSCLC as well as thyroid carcinoma. Patients with high level of SHP2 expression had shorter overall survival (OS) in comparison to those with low level of SHP2 expression.16, 17, 18 Receptor tyrosine kinase (RTK)‐driven malignant cells depended on SHP2 for survival. In epidermal growth factor receptor (EGFR) mutant NSCLC patients, SHP2 was associated with tyrosine kinase inhibitor (TKI) resistance and those with a high level of SHP2 messenger RNA (mRNA) had a shorter progression‐free survival (PFS) and shorter OS.19 Notably, SHP2 inhibitor could enhance the activity of T cells in the mouse model, and the combination of SHP2 inhibitor SHP099 and anti‐PD‐1 could kill tumor cells more effectively, which showed that SHP2 was a potentially robust therapeutic strategy for cancer treatment.20 Recently, some clinical trials of combination treatments (e.g., SHP2 inhibitor TNO155 combined with PD‐1 inhibitor/KRAS inhibitor and RMC‐4630 combined with KRAS inhibitor/MEK inhibitor) have been carried out.21, 22, 23, 24
SHP2 is required for growth of KRAS mutant NSCLC.7, 8, 9 However, studies regarding the relationship of SHP2 in KRAS mutant NSCLC with its genomic profile as well as tumor immune microenvironment are lacking. Considering the function of SHP2 in both epithelial cancer cells and immune cells, SHP2 could be a key player and affect the clinical efficacy of immunotherapy in KRAS mutant cancers. In this study, we focus on the expression of SHP2 and its predictive and prognostic significance in immune checkpoint inhibitor (ICI)‐treated KRAS mutant NSCLC patients. We further explore the tumor immune environment and analyze the correlation of SHP2 with immune cells.
METHODS
Patients and clinical management
We retrospectively reviewed 217 KRAS mutant NSCLC patients at the Guangdong Lung Cancer institute (GLCI) from January 27, 2016, to October 1, 2019, and 61 of them were treated with ICI. This study was approved by the Guangdong Provincial People's Hospital's Research Ethics Committee, and all patients provided written informed consent. Patients’ baseline characteristics, including age at diagnosis, gender, histologic type, stage, smoking history, Eastern Cooperative Oncology Group (ECOG) performance status (PS) at diagnosis, treatment history, best overall response (BOR), PFS, and OS, were obtained.
All the patients had received at least 2 cycles of immunotherapy until disease progression or unacceptable toxicity occurred. Treatment regimens included ICI only, ICI plus chemotherapy, and ICI plus ICI. Treatment responses were assessed by specialists according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. PFS was defined from the start of immunotherapy to disease progression. OS was defined from the start of immunotherapy to death or last follow‐up. The final follow‐up was June 31, 2020.
Genetic profiling of tumors by next‐generation sequencing
All the patients were tested by next‐generation sequencing (NGS) (Burning Rock Biotech) using tissue DNA or circulating cell‐free DNA (cfDNA) obtained from lung biopsy, surgery resection, or blood. Tumor tissue or 10 mL of whole blood were collected, and DNA was extracted. Circulating cfDNA was extracted from plasma samples using the QIAamp circulating nucleic acid kit (Qiagen) according to the manufacturer's instructions. Quantification of cfDNA was performed using the Qubit 2.0 Fluorometer with the double stranded DNA (dsDNA) HS assay kits (Life Technologies). After quantification, ~50 ng of DNA were used to construct NGS library and sequenced on Nextseq500 sequencer (Illumina). Targeted gene panels covering 168 or 520 tumor‐related genes were sequenced with target sequencing depths of 1000× for the tissue samples and 10 000× for the liquid biopsy samples. Sequence data was obtained by high‐flux sequencing technology and mapped to the human genome (hg19) using BWA aligner 0.7.10. Local alignment optimization, variant calling, and annotation were performed using GATK 3.2, MuTect, and VarScan. Alterations of SNP, Indel, Rearrangement, and CNV were analyzed and reported. At least two supporting reads were required for INDELs in plasma samples and five supporting reads were required in tissue samples. Eight supporting reads were sufficient for SNVs in plasma as well as tissue samples.
Immunohistochemistry
The expression levels of SHP2 (6D9, 1:500, NOVUS), phospho‐SHP2 (pSHP2) (Y542, 1:150, ABCAM), and PD‐L1 (E1L3N, 1:200, CST) were assessed using immunohistochemistry (IHC). The procedures were carried out according to manufacturer's instructions, and IHC staining was scored based on semiquantitative analyses by two independent pathologists. Areas of tumor cell aggregates and stroma were evaluated, respectively. The evaluation of SHP2 and pSHP2 was defined by the percentage of positive cells multiplying the staining intensity. The staining intensity was ruled as +3 (strong), +2 (moderate), +1 (weak), and 0 (negative). The scoring formula was: H‐score = 3× (+3)% + 2× (+2)% + 1× (+1)%. The evaluation of PD‐L1 was defined according to the percentage of positive tumor cells.
Multiplexed immunofluorescence cytochemistry
Multiplexed immunofluorescence cytochemistry (mIFC) was conducted at Genecast Biotechnology. Biomarker panel 1 of SHP2, CD4, CD8, CD68, and CK, and panel 2 of CD8, PD‐L1, PD‐1, TIM3, and CK were sequentially detected. The slides cut from FFEP NSCLC tissues sequentially went through epitope retrieval, protein blocking and were then incubated sequentially with primary antibody, secondary antibody, and tyramine signal amplification (TSA) visualization. Primary antibodies for CK (AE1/AE3, 1:100, Zsbio), CD68 (KP1, 1:1000, Zsbio), CD4 (EP204, 1:100, Zsbio), SHP2(6D9, 1:500, NOVUS), PD‐L1 (E1L3N, 1:100, CST), PD‐1 (UMAB199, 1:100, Zsbio), and TIM3 (D5D5R, 1:100, CST) were incubated for 1 hour at room temperature, and CD8 antibody (SP16, 1:100, Zsbio) was incubated for overnight at 4°C. TSA visualization was performed with the opal seven‐color multiplex IHC Kit (NEL797B001KT, PerkinElmer), containing fluorophores (DAPI), Opal 520 (CK), Opal 650 (CD68, PD‐1), Opal 620 (CD8), Opal 690 (CD4, TIM3), Opal 570 (SHP2, PD‐L1), and TSA coumarin system (NEL703001KT, PerkinElmer). Slides were scanned using the PerkinElmer Vectra (Vectra 3.0.5; PerkinElmer), and the inForm Advanced Image Analysis software (inForm 2.3.0; PerkinElmer) was used to unmix multispectral images with spectral libraries. An algorithm was trained for batch analysis. An experienced pathologist determined appropriate positive threshold X for each biomarker. Corresponding fluorescent staining intensity of X, 2X, 3X, or more were determined as 1+, 2+, 3+, respectively, for each biomarker. The scoring formula was H‐score = (3+)% × 3+ (2+)% × 2+ (1+)% × 1.
Statistical analyses
All statistical analyses were performed in R software (version 3.6.3). Associations between variables were examined with either Wilcoxon rank‐sum test, Kruskal–Wallis test, χ2 test, or Spearman correlation test, as appropriate. Survival analysis was performed by Kaplan–Meier plot methods, and p value was calculated using log‐rank test. p < 0.05 was considered statistically significant.
RESULTS
Clinical characteristics of ICI‐treated KRAS mutant NSCLC patients
We screened 217 KRAS mutant NSCLC patients in our hospital, 61 of which had ICI treatment, and the main characteristics were summarized in Table 1. Of all the KRAS mutant patients, 5 (8.2%) were female, 56 (91.8%) were male, and the median age was 62.6 (ranging from 47.3–78.3 years old). Forty‐one (67.2%) patients were former or current smokers and median pack‐year was 47.3 (ranging from 10.0–100.0). Fifty‐nine (96.7%) patients were adenocarcinoma, and the remaining two patients were small cell lung cancer compounded with adenocarcinoma and pulmonary sarcomatoid carcinoma compounded with adenocarcinoma. Fifty‐seven (93.4%) of the patients were stage IV, the ECOG PS for most of the patients (n = 59) was 0–1, and only 2 patients were 3. Twenty‐five (41.0%) of the patients accepted immunotherapy as first‐line treatment and the rest of them were the second line or more. The main immunotherapy regimens were anti‐PD‐1/PD‐L1 only (n = 41) or anti‐PD‐1/PD‐L1 plus chemotherapy (n = 18). Eleven patients had partial respond (PR), 19 patients had stable disease (SD), and 9 patients had progressive disease (PD) according to the RECIST version 1.1. NGS showed that 25 (41.0%) patients had KRAS G12C, the most common mutation subtype, 9 (14.8%) patients had G12V, 7 (11.5%) patients had G13D, and 6 (9.8%) patients had G12D. Thirty‐seven (60.7%) of the patients harbored TP53, 16 (26.2%) patients harbored STK11/LKB1, and 9 patients harbored (14.8%) KEAP1, whereas only 2 patients had EGFR mutation and 3 patients had MET amplification (Table 2).
TABLE 1.
The clinical characteristics of 61 ICI‐treated KRAS mutant NSCLC patients
| Characteristics | No. (n = 61) (%) | SHP2 evaluation (n = 34) | p value | |
|---|---|---|---|---|
| High | Low | |||
| Age (y) | ||||
| ≥60 | 37 (60.7) | 14 | 5 | 0.640 |
| ˂60 | 24 (39.3) | 11 | 4 | |
| Median | 62.6 | |||
| Range | 47.3–78.3 | |||
| Gender | ||||
| Female | 5 (8.2) | 2 | 1 | 0.616 |
| Male | 56 (91.8) | 23 | 8 | |
| Histologic type | ||||
| ADC | 59 (96.7) | 24 | 9 | |
| ADC/SCLC | 1 (1.6) | 0 | 0 | |
| ADC/sarcomatoid | 1 (1.6) | 1 | 0 | |
| Smoking status | ||||
| Former/current | 41 (67.2) | 18 | 6 | 0.538 |
| Never | 20 (32.8) | 7 | 3 | |
| Pack‐years of smoking | ||||
| Median | 47.3 | |||
| Range | 10.0–100.0 | |||
| Stage | ||||
| IIB | 1 (1.6) | 0 | 0 | |
| IIIB | 3 (4.9) | 0 | 0 | |
| IV | 57 (93.4) | 25 | 9 | |
| ECOG PS at diagnosis | ||||
| 0–1 | 59 (96.7) | 24 | 8 | |
| 2–3 | 2 (3.3) | 1 | 1 | |
| Type of ICI | ||||
| Anti‐PD‐1/PD‐L1 | 41 (67.2) | 19 | 5 | |
| Anti‐PD‐1/PD‐L1 plus chemotherapy | 18 (29.5) | 5 | 3 | |
| Anti‐PD‐1/PD‐L1 plus targeted therapy | 1 (1.6) | 1 | 0 | |
| Anti‐PD‐1/PD‐L1 plus anti‐CTLA‐4 | 1 (1.6) | 0 | 1 | |
| Line of ICI | ||||
| First line | 25 (41.0) | 10 | 3 | |
| Second line | 16 (26.2) | 8 | 3 | |
| Third line or more | 20 (32.8) | 7 | 3 | |
| Best overall response | ||||
| PR | 11 (18.0) | 6 | 0 | |
| SD | 19 (31.1) | 8 | 3 | |
| PD | 9 (14.8) | 4 | 2 | |
| NA | 22 (36.1) | 7 | 4 | |
Abbreviations: ADC, adenocarcinoma; CTLA‐4, cytotoxic T lymphocyte–associated protein 4; ECOG, Eastern Cooperative Oncology Group; ICI, immune checkpoint inhibitor; NA, not available; NSCLC, non‐small cell lung cancer; PD, progress disease; PD‐1, programmed cell death protein 1; PD‐L1, programmed death ligand 1; PR, partial respond; PS, performance status; SCLC, small cell lung cancer; SD, stable disease.
TABLE 2.
KRAS mutation subtypes and co‐occurring genomic alterations
| Number (n = 61) | % | |
|---|---|---|
| KRAS mutation subtypes | ||
| G12C | 25 | 41.0 |
| G12V | 9 | 14.8 |
| G13D | 7 | 11.5 |
| G12D | 6 | 9.8 |
| G12A | 3 | 4.9 |
| G12S | 2 | 3.3 |
| G12C + Q61H | 1 | 1.6 |
| G12C + Q61L | 1 | 1.6 |
| G12F | 1 | 1.6 |
| G12R | 1 | 1.6 |
| G12V + G12R | 1 | 1.6 |
| G13C | 1 | 1.6 |
| G13R | 1 | 1.6 |
| G13V | 1 | 1.6 |
| Q61H | 1 | 1.6 |
| Co‐occurring genomic alterations | ||
| TP53 mutation | 37 | 60.7 |
| EGFR mutation | 2 | 3.3 |
| STK11/LKB1 mutation | 16 | 26.2 |
| KEAP1 mutation | 9 | 14.8 |
| MET amplification | 2 | 3.3 |
The expression of SHP2 was not significantly different between subtypes of KRAS mutant NSCLC
Of the 61 KRAS mutant patients, 34 had available tumor tissue to detect the expression of SHP2 (Figure 1). IHC was performed to evaluate the expression level of SHP2 and pSHP2 in intratumoral area (it‐SHP2, it‐pSHP2) and stromal area (s‐SHP2, s‐pSHP2) shown in Figure 2(a). Tumors of 32 (94.1%) patients positively expressed SHP2, and the expression level of SHP2 varies in different patients (Figure S1). SHP2 expression was higher in intratumoral area than in stromal area and there is a correlation between them. The overall expression level of SHP2 was correlated with pSHP2 as well as in both intratumoral area and stromal area (Figure 2(b)). Patients with PD‐L1 ≥50% had higher level of s‐SHP2 than patients with PD‐L1 <50% (p = 0.039) (Figure 2(c)). There is no significant difference in it‐SHP2 between diverse levels of PD‐L1. Whether the patient did or did not smoke did not influence the expression of it‐SHP2 and s‐SHP2 (Figure 2(d)). There was no significant difference in the expression level of SHP2 among subgroups of different KRAS mutations as well as subgroups of different co‐occurring mutated genes like TP53, STK11/LKB1, and KEAP1 (Figure 2(e),(f)).
FIGURE 1.
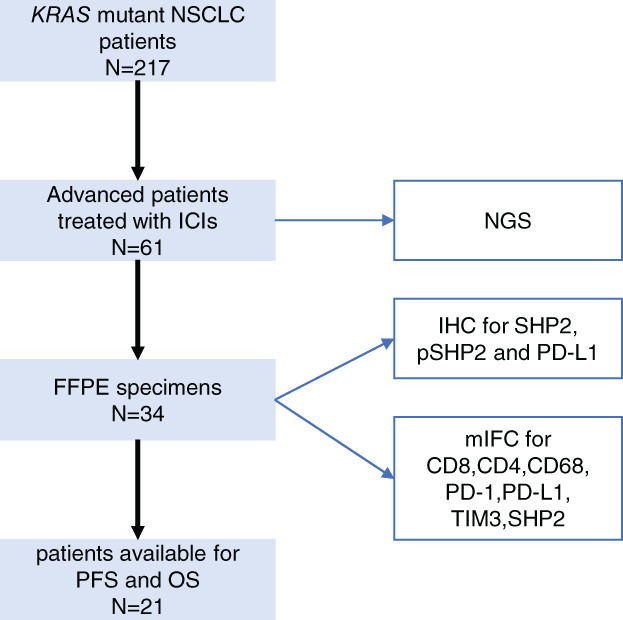
Flow diagram of the specimen included in the study. NSCLC, non‐small cell lung cancer; ICI, immune checkpoint inhibitor; NGS, next‐generation sequencing; FFPE, formalin‐fixed paraffin‐embedded; IHC, immunohistochemistry; pSHP2, phospho‐SHP2; PD‐L1, programmed death ligand 1; PFS, progression‐free survival; OS, overall survival; mIFC, multiplexed immunofluorescence cytochemistry
FIGURE 2.
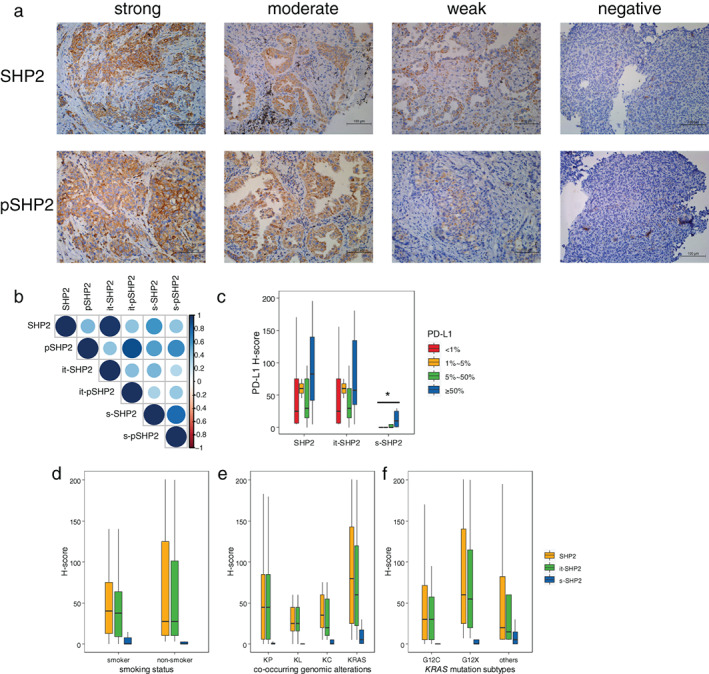
The expression of SHP2 in different subtypes of KRAS mutant NSCLC. (a) Representative immunohistochemistry (IHC) images of SHP2 and pSHP2. IHC staining intensity was +3 (strong), +2 (moderate), +1 (weak), and 0 (negative), respectively. pSHP2, phospho‐SHP2. (b) Heatmap of correlation analysis including SHP2, it‐SHP2, s‐SHP2, pSHP2, it‐pSHP2, and s‐pSHP2. it‐SHP2, intratumoral SHP2; s‐SHP2, stromal SHP2; it‐pSHP2, intratumoral pSHP2; s‐pSHP2, stromal pSHP2. (c)–(f) The expression of SHP2, it‐SHP2 and s‐SHP2 in different PD‐L1 status (c), smoking status (d), KRAS mutation subtypes (e) and co‐occurring genomic alterations (f). PD‐L1, programmed death ligand 1; KP, KRAS + TP53; KL, KRAS + LKB1; KC, KRAS + CDKN2A/B; KRAS, KRAS alone. *p < 0.05
s‐SHP2 is correlated with PD‐L1 expression, T cells, and macrophages infiltration in stromal area
We performed mIFC for SHP2, CD8, CD4, CD68, PD‐L1, PD‐1, TIM3, and CK presented in Figure 3(a). It showed that SHP2 was widely expressed in 43.2% of CD4+ T cells, 34.4% of CD8+ T cells, and 62.3% of CD68+ macrophage cells in addition to tumor cells. Macrophages had the highest percentage of SHP2 expression positivity. There was significant difference between the SHP2‐positive percentages of tumor cells and CD68+ macrophage cells in intratumoral area (64.6% vs. 28.1%, p = 0.012) (Figure 3(b)). The expression of s‐SHP2 was positively correlated with it‐PD‐L1, it‐CD8, it‐CD4, s‐PD‐L1, s‐CD8, s‐CD4, and s‐CD68 (Figure 3(c)). Significantly higher percentages of CD8+ T cells, CD4+ T cells, and CD68+ macrophages in stromal area were observed in patients with high versus low s‐SHP2 expression (4.0% vs. 0.6%, p = 0.014; 10.9% vs. 3.6%, p = 0.005; 2.9% vs. 0.6%, p = 0.007) (Figure 3(d)).
FIGURE 3.
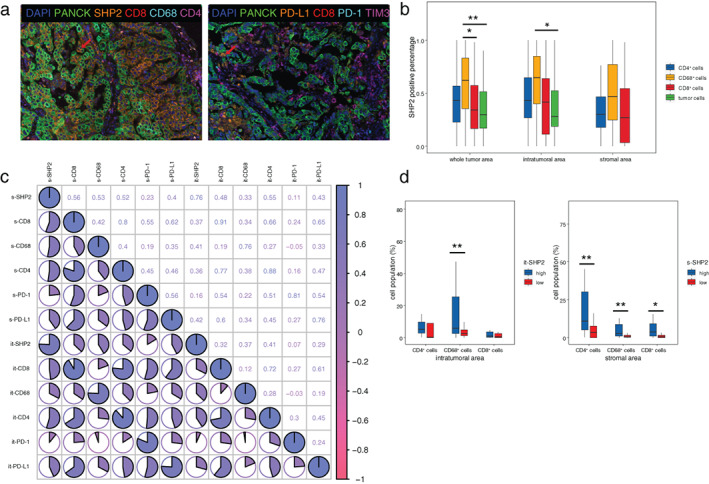
The correlation between SHP2 and immune cells. (a) Representative multiplexed immunofluorescence cytochemistry (mIFC) staining of SHP2, CD4, CD8, CD68, and CKs (panel 1) (left), and CD8, PD‐L1, PD‐1, TIM3, and CKs (panel 2) (right). (b) The percentage of SHP2+/CD8+ T cells, SHP2+/CD4+ T cells, and SHP2+/CD68+ macrophages in CD8+ T cells, CD4+ T cells, and CD68+ macrophages, respectively. (c) Heatmap of correlation analysis including SHP2, CD8, CD4, CD68, PD‐1, and PD‐L1 in intratumoral and stromal area. “X” in the circle means there is no significance. PD‐L1, programmed death ligand 1. (d) Quantification of CD8+, CD4+, and CD68+ cells in high and low expression level of it‐SHP2 and s‐SHP2. *p < 0.05, **p < 0.01
High level of SHP2 is related with better survival in ICI‐treated patients
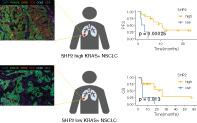
Survival analysis was assessable for 21 patients, and the median follow‐up was 3.7 years with 6 patients continuing immunotherapy at the cut‐off time of the analysis. We assessed the BOR during immunotherapy. It showed that all the patients with PR, and 80% of patients with SD, had high levels of SHP2, whereas half of the patients with PD had low levels of SHP2 (Figure 4(a)). In 16 patients with high SHP2, 6 had PR, and 8 had SD, whereas in 4 patients with low SHP2, 2 had SD and 2 had PD.
FIGURE 4.
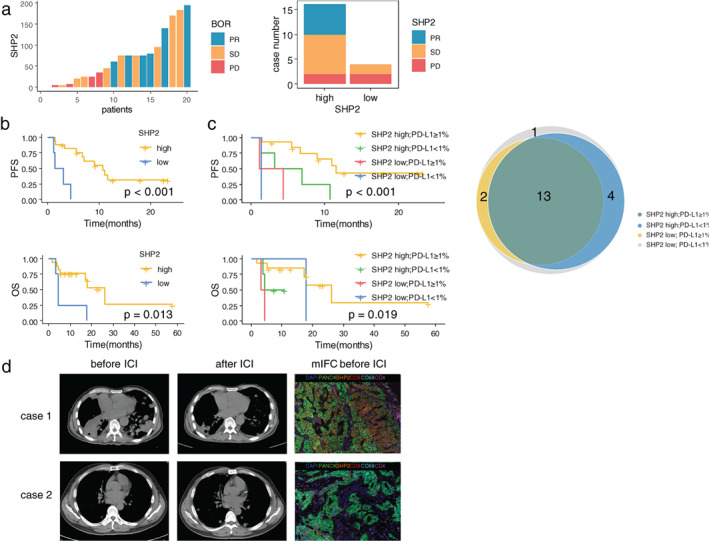
Kaplan–Meier curves for PFS and OS according to the expression level of SHP2 and PD‐L1. (a) Best overall response (BOR) and the expression level of SHP2 in patients available for survival analysis. PR, partial respond; SD, stable disease; PD, progress disease. (b) PFS and OS in patients with high SHP2 expression versus low SHP2 expression. PFS, progression‐free survival; OS, overall survival. (c) PFS and OS in patients with high/low SHP2 and PD‐L1. Venn diagram depicting the number of patients with different SHP2 and PD‐L1 expression state. PD‐L1, programmed death ligand 1. (d) Radiological evaluation before and after ICI and mIFC analysis of two patients with high (case 1) and low (case 2) expression of SHP2. ICI, immune checkpoint inhibitor; mIFC, multiplexed immunofluorescence cytochemistry
Survival analysis demonstrated that patients with high level of SHP2 as well as it‐SHP2 showed better PFS and OS (p < 0.001, p < 0.001) (Figure 4(b), Figure S2(a),(b)). Patients with high level of s‐SHP2 also showed better PFS (p = 0.014). Patients with PD‐L1 ≥1% had a longer PFS (p = 0.023) than patients with PD‐L1 <1%, and there was a tendency of benefit when the cut‐off point was 5% (Figure S2(c)). Notably, a special group of patients with both high level SHP2 and PD‐L1 ≥1% survived better than the rest (p < 0.001) (Figure 4(c)). Patients with high level of s‐CD8 and s‐TIM3 and low level of s‐CD68 were associated with better PFS (p = 0.017, p < 0.001, p = 0.049) (Figure S2(d)). There was no difference of survival among KRAS sub‐genotypes or subgroups of KRAS mutations with different co‐occurring mutated genes.
DISCUSSION
KRAS mutation makes up nearly 10% of Chinese and 30% of Caucasian NSCLC. Mutant KRAS has been deemed undruggable since it was discovered. The survival rate of KRAS mutant NSCLC patients was inferior to patients with EGFR alteration, which makes it urgent for further research on novel therapeutics.25 Over the past several years, ICI had transformed the treatment pattern of tumors, and a fraction of patients with lung cancer could benefit.26 However, the therapeutic effects varied in distinct common driver genes such as KRAS, EGFR, and anaplastic lymphoma kinase (ALK). Increasing attention has been paid to the immunotherapy of KRAS mutant NSCLC for its high tumor mutation burden (TMB), increasing tumor‐infiltrating lymphocytes (TILs), elevated expression of PD‐L1, and patient's smoking history.27, 28 In KRAS mutant lung cancer, there is also a great need to further refine the efficient biomarker to predict the efficacy of ICI treatment.
SHP2 is one of the newly emerging anticancer targets that regulates RTK‐RAS‐ERK signaling pathway and also participates in PD‐1/PD‐L1 signaling pathway.12, 14 In the tumor microenvironment, SHP2 may play diversified roles in different cell types, especially epithelial cancer cells, immune T cells, macrophages, etc. It has been reported that mutant cancer cells SHP2 played critical roles in survival and proliferation in KRAS. However, it remains unclear whether SHP2 mediates differential roles in T cells, macrophages, and cancer cells in clinical KRAS mutant NSCLC, and whether SHP2 expression predicts the efficacy of ICI in KRAS mutant cancers. Our study explored the expression and potentially predictive role of pSHP2 in tumor microenvironment of KRAS mutant ICI‐treated NSCLC.
We assessed the expression of SHP2 in both intratumoral and stromal areas by routine single plex IHC. SHP2 was expressed in 32 (94.1%) samples. In these KRAS mutant lung cancer specimens, SHP2 was heterogeneously expressed in both tumor cells and immune cells. SHP2 expression level in intratumoral and stromal areas was positively correlated, but higher in intratumoral area. mIFC was used to present the expression level of SHP2 in CD4+ T cells, CD8+ T cells, CD68+ macrophage cells, and tumor cells. Again, we found SHP2 is commonly expressed in KRAS mutated tumor cells and is also expressed in immune cells including T cells and macrophages. Expression of s‐SHP2 is correlated with s‐PD‐L1 expression, it/s‐CD8+ T cells, and it/s‐CD4+ T cells. Expression of SHP2 in different types of immune cells strongly suggested it played key roles in orchestrating the formation of immunosuppressive tumor microenvironment.
Previous studies showed that KRAS G12D, G12V, and G13C mutation groups tended to have higher PD‐L1 expression whereas KRAS G12A and G12C mutation groups tended to lack PD‐L1 expression, which may affect the clinical efficacy of ICI.27 TP53 co‐mutation group exhibited better objective response rate with ICI, whereas LKB1 co‐mutation group promoted the resistance to ICI.29 To explore if SHP2 expression varies in different subgroups of KRAS mutant lung cancer, we compared SHP2 levels among them. In our study, however, there is no discrepancy of SHP2 expression among subgroups of different KRAS mutation subtypes or of different co‐occurring mutated genes, suggesting SHP2 may play a similar role in all sub‐genotypes of KRAS mutant lung cancers. It is has been reported that only part of KRAS mutant lung cancers could benefit from ICI. Our finding displayed that ICI can benefit more in patients with high SHP2‐expressed NSCLC. A total of 100% of the PR patients belong to the high SHP2 group, whereas 50% of the PD patients belong to the low SHP2 group. We also observed that a high level of SHP2 was associated with better PFS and OS, suggesting the survival benefit in high SHP2‐expressing KRAS mutant cases may result from the SHP2‐mediated modification of immune microenvironment. Therefore, SHP2 seems to be a significant biomarker to predict the efficacy of immunotherapy. We only detect SHP2 in ICI‐treated tumors, so it remains to be further investigated whether SHP2 is related with longer OS in non‐ICI‐treated KRAS mutant patients. We also note that the differential expression of SHP2 between KRAS mutant NSCLC and KRAS wild‐type NSCLC, or whether it plays the same role as a predictive marker for the immunotherapy in KRAS wild‐type NSCLC, remains unclear. ICI has already been approved as standard care for NSCLC, especially without driver gene alterations. Therefore, the expression patterns and the potentially predictive value of SHP2 in KRAS wild‐type NSCLC need to be confirmed clinically.
It is reported that phosphorylation of intracellular ITIM and/or ITSM of PD‐1 receptor is required for activation of SHP2 phosphatase.15 In infiltrating T cells, PD‐1 blockade by ICI may reduce the ITIM/ITSM phosphorylation, downregulate the SHP2 signaling, and alleviate its inhibitive effects on T cell activation. Therefore, antitumor immune response can be promoted. In KRAS mutant cancer cells, however, how the ICI treatment affects the SHP2 action of mechanism remains unclear.
Previous studies demonstrated that the application of SHP2 inhibitor was able to transform the immunosuppressive environment through upregulating T cells and increasing M1 macrophages.30 Zhao et al.20 illustrated that in a CT‐26 colon cancer xenograft model, SHP2 inhibitor could elevate proportion of CD8+IFN‐γ+ T cells, and the combination of SHP2 and PD‐1 inhibitor synergized the tumor killing effect. We assume that immunotherapy can transform the depressed state of T cells caused by SHP2 and, therefore, exert enhanced antitumor effect in some degree. Mainardi et al.7 reported that the combination of SHP2 and MEK inhibitors decreased the level of KRAS‐GTP and pERK, and decelerated tumor progression generated synthetic lethality in KRAS mutant lung cancer cell lines and organoid models. However, it is hard to explain how SHP2 in tumor cell worked with immunotherapy and the reason why high level of SHP2 showed better survival. Hence, it is worth investigating the effects of immune checkpoint inhibitors on SHP2‐signaling in tumor cells.
There are a few limitations in our study. First, the number of KRAS mutant NSCLC patients treated with immunotherapy is not large. More patients will be recruited in the future to further certify the accuracy of SHP2 to predict the effect of immunotherapy on KRAS mutant NSCLC patients. Treg cells were not included for detailed analysis in our study. We have indicated that SHP2 had close relation with CD8+ T cells and CD4+ T cells, so we wonder whether SHP2 modifies tumor immune microenvironment through regulatory T cells. In the next step, we plan to use in vitro cell models or organoid models to investigate how SHP2 plays a master role in orchestrating the interaction between tumor cells and immune cells. There is also a need to search effective treatment by targeted inhibition of SHP2 and to identify the detailed effects of SHP2 inhibition on cytotoxic T cells, Tregs, macrophages, and other immune cells in clinical samples.
In summary, our findings showed that SHP2 is a promising predictive marker for ICI treatment in KRAS mutant NSCLC. In KRAS mutant lung cancers, SHP2 expression was correlated with PD‐L1 and may be complementary to PD‐L1 as a routine biomarker testing for ICI. We also observed that SHP2 was expressed in both tumor cells and immune cells in clinical samples. Intratumoral or stromal SHP2 was associated with CD8+ T cells and macrophages infiltration. Together, these data suggest a rationale for conditional targeting SHP2 to improve the TME and the efficacy of ICI in NSCLC. Further studies are needed to validate the predictive role of SHP2 protein expression. Studies on the roles of SHP2 in tumor cells, cytotoxic T cells, Tregs, macrophages, and other stromal cells in TME are also warranted.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
FIGURE S1. The expression level of it‐SHP2 and s‐SHP2 in 34 patients. The expression level was evaluated by H‐score method. it‐SHP2, intratumoral SHP2; s‐SHP2, stromal SHP2.
FIGURE S2. Kaplan–Meier curves for PFS and OS of it‐SHP2, s‐SHP2 and immune markers. (a) PFS and OS in patients with different it‐SHP2 and s‐SHP2 levels. it‐SHP2, intratumoral SHP2; s‐SHP2, stromal SHP2; PFS, progression‐free survival; OS, overall survival. (b) PFS and OS in patients with different pSHP2, it‐pSHP2, and s‐pSHP2 levels. pSHP2, phospho‐SHP2; it‐pSHP2, intratumoral pSHP2; s‐pSHP2, stromal pSHP2. (c) PFS and OS in patients with different PD‐L1 status. The cut‐off points were 1%, 5%, and 50% respectively. PD‐L1, programmed death ligand 1. (d) PFS and OS in patients with high/low level of s‐CD8, s‐CD68 and s‐TIM3.
ACKNOWLEDGMENTS
This work was supported by following grants: Guangdong Provincial Natural Science Program (No.2019A1515010900, X.Z.); GDPH Dengfeng Program (No. DFJH201903 and KJ012019444 and 8197103306, X.Z.); Guangzhou Health S and T Project (No. 20191A011097, H.L.L.), Guangzhou S and T Project (No. n201904010331, J.Q.H.); Guangdong Provincial Key Lab of Translational Medicine in Lung Cancer (2017B030314120, Y.L.W.).
Feng H‐B, Chen Y, Xie Z, Jiang J, Zhong Y‐M, Guo W‐B, et al. High SHP2 expression determines the efficacy of PD‐1/PD‐L1 inhibitors in advanced KRAS mutant non‐small cell lung cancer. Thorac Cancer. 2021;12:2564–2573. 10.1111/1759-7714.14137
Funding information GDPH Dengfeng Program, Grant/Award Number: DFJH201903 & KJ012019444 & 8197103306; Guangdong Provincial Key Lab of Translational Medicine in Lung Cancer, Grant/Award Number: 2017B030314120; Guangdong Provincial Natural Science Program, Grant/Award Number: 2019A1515010900; Guangzhou Health S&T Project, Grant/Award Number: 20191A011097; Guangzhou S&T Project, Grant/Award Number: 201904010331
REFERENCES
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. [DOI] [PubMed] [Google Scholar]
- 2.Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small‐cell lung cancer: meta‐analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24:2371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aviel‐Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K‐ras mutations in non‐small‐cell lung carcinoma: a review. Clin Lung Cancer. 2006;8:30–8. [DOI] [PubMed] [Google Scholar]
- 4.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575:217–23. [DOI] [PubMed] [Google Scholar]
- 5.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS‐mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ClinicalTrials.gov. NCT03600883. [cited Feb 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT03600883?term=AMG510&draw=2&rank=3.
- 7.Mainardi S, Mulero‐Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS‐mutant non‐small‐cell lung cancer in vivo. Nat Med. 2018;24:961–7. [DOI] [PubMed] [Google Scholar]
- 8.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS‐driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24:954–60. [DOI] [PubMed] [Google Scholar]
- 9.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild‐type KRAS‐amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neel BG, Gu H, Pao L. The 'Shp'ing news: SH2 domain‐containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93. [DOI] [PubMed] [Google Scholar]
- 11.Ran H, Tsutsumi R, Araki T, Neel BG. Sticking it to Cancer with molecular glue for SHP2. Cancer Cell. 2016;30:194–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/mitogen‐activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453–9. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q, Qu J, Zhao M, Xu Q, Sun Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol Res. 2020;152:104595. [DOI] [PubMed] [Google Scholar]
- 14.Boussiotis VA. Molecular and biochemical aspects of the PD‐1 checkpoint pathway. N Engl J Med. 2016;375:1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD‐1 stimulation. Sci Adv. 2020;6:eaay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao J, Huang YQ, Jiao S, Lan XB, Ge MH. Clinicopathological and prognostic significance of SHP2 and Hook1 expression in patients with thyroid carcinoma. Hum Pathol. 2018;81:105–12. [DOI] [PubMed] [Google Scholar]
- 17.Chen MJ, Wang YC, Wu DW, Chen CY, Lee H. Association of nuclear localization of SHP2 and YAP1 with unfavorable prognosis in non‐small cell lung cancer. Pathol Res Pract. 2019;215:801–6. [DOI] [PubMed] [Google Scholar]
- 18.He L, Li Y, Huang X, Cheng H, Ke Y, Wang L. The prognostic significance of SHP2 and its binding protein Hook1 in non‐small cell lung cancer. Onco Targets Ther. 2019;12:5897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karachaliou N, Cardona AF, Bracht JWP, Aldeguer E, Drozdowskyj A, Fernandez‐Bruno M, et al. Integrin‐linked kinase (ILK) and src homology 2 domain‐containing phosphatase 2 (SHP2): novel targets in EGFR‐mutation positive non‐small cell lung cancer (NSCLC). EBioMedicine. 2019;39:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti‐tumor immunity and synergizes with PD‐1 blockade. Acta Pharm Sin B. 2019;9:304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ClinicalTrials.gov. NCT04000529. [cited 2021 Feb 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT04000529?term=SHP2&draw=4&rank=10.
- 22.ClinicalTrials.gov. NCT04330664. [cited 2021 Feb 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT04330664?term=SHP2&draw=4&rank=12.
- 23.ClinicalTrials.gov. NCT04185883. [cited 2021 Feb 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT04185883?term=SHP2&draw=4&rank=15
- 24.ClinicalTrials.gov. NCT03989115. [cited Feb 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT03989115?term=SHP2&draw=4&rank=20
- 25.Johnson ML, Sima CS, Chaft J, Paik PK, Pao W, Kris MG, et al. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer. 2013;119:356–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): a randomised, open‐label, controlled, phase 3 trial. Lancet. 2019;393:1819–30. [DOI] [PubMed] [Google Scholar]
- 27.Jeanson A, Tomasini P, Souquet‐Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of immune checkpoint inhibitors in KRAS‐mutant non‐small cell lung Cancer (NSCLC). J Thorac Oncol. 2019;14:1095–101. [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti‐PD‐1/PD‐L1 immunotherapy in KRAS‐mutant non‐small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105. [DOI] [PubMed] [Google Scholar]
- 29.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintana E, Schulze CJ, Myers DR, Choy TJ, Mordec K, Wildes D, et al. Allosteric inhibition of SHP2 stimulates antitumor immunity by transforming the immunosuppressive environment. Cancer Res. 2020;80:2889–902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. The expression level of it‐SHP2 and s‐SHP2 in 34 patients. The expression level was evaluated by H‐score method. it‐SHP2, intratumoral SHP2; s‐SHP2, stromal SHP2.
FIGURE S2. Kaplan–Meier curves for PFS and OS of it‐SHP2, s‐SHP2 and immune markers. (a) PFS and OS in patients with different it‐SHP2 and s‐SHP2 levels. it‐SHP2, intratumoral SHP2; s‐SHP2, stromal SHP2; PFS, progression‐free survival; OS, overall survival. (b) PFS and OS in patients with different pSHP2, it‐pSHP2, and s‐pSHP2 levels. pSHP2, phospho‐SHP2; it‐pSHP2, intratumoral pSHP2; s‐pSHP2, stromal pSHP2. (c) PFS and OS in patients with different PD‐L1 status. The cut‐off points were 1%, 5%, and 50% respectively. PD‐L1, programmed death ligand 1. (d) PFS and OS in patients with high/low level of s‐CD8, s‐CD68 and s‐TIM3.