

Abstract
Background
Recent studies have found that programmed death ligand 1 (PD‐L1) might be involved in chemotherapy resistance in non‐small cell lung cancer (NSCLC). Arsenic sulfide (As4S4) has been recognized to have antitumor activities and enhance the cytotoxic effect of chemotherapy drugs. In this study, we aimed to verify the relationship between PD‐L1 and cisplatin (DDP) resistance and identify whether As4S4 could reverse DDP resistance through targeting PD‐L1 in NSCLC.
Methods
The effect of As4S4 and DDP on cell proliferation and apoptosis was investigated in NSCLC cell lines. The expression of p53 and PD‐L1 proteins was measured by western blotting analysis. The levels of miR‐34a‐5p, miR‐34a‐3p and PD‐L1 in cells were measured by real‐time qPCR analysis. Mouse xenograft models were established by inoculation with A549/DDP (DDP‐resistant) cells.
Results
Depletion of PD‐L1 inhibited DDP resistance in A549/DDP and H1299/DDP cells. As4S4 was capable of sensitizing A549/DDP cells to DDP by enhancing apoptosis. As4S4 upregulated p53 expression and downregulated PD‐L1 expression in A549/DDP cells. As4S4 increased miR‐34a‐5p level in A549/DDP cells. Inhibition of p53 by PFT‐α partially restored the levels of PD‐L1 and miR‐34a‐5p. Pretreatment with PFT‐α suppressed the apoptosis rate induced by cotreatment of As4S4 and DDP in A549/DDP cells. Cotreatment of DDP and As4S4 notably reduced the tumor size when compared with DDP treatment alone in vivo.
Conclusions
Upregulation of PD‐L1 was correlated with DDP resistance in NSCLC cells. Mechanistic analyses indicated that As4S4 might sensitize NSCLC cells to DDP through targeting p53/miR‐34a‐5p/PD‐L1 axis.
Keywords: As4S4 , chemoresistance, cisplatin, NSCLC, PD‐L1
PD‐L1 is correlated with DDP resistance in non‐small cell lung cancer. As4S4 reverses DDP resistance through targeting PD‐L1 in NSCLC in vitro and in vivo.
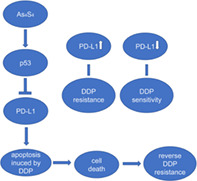
INTRODUCTION
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer death worldwide, with 1 761 007 deaths reported in 2018.1 Non‐small cell lung cancer (NSCLC) accounts for nearly 85% of all lung cancer diagnoses, and constitutes a heterogeneous population of adenocarcinoma, squamous and large cell carcinomas.2 Platinum‐based chemotherapy, particularly cisplatin (DDP), has been demonstrated to be efficient therapeutic treatment for NSCLC. However, acquired drug resistance, reported to develop during clinical treatment, is a large barrier that negatively impacts the survival rate of patients.3
Programmed cell death ligand 1 (PD‐L1), also known as B7‐H1 or CD274, has been found to be widely overexpressed in various types of cancer cells, including NSCLC, and is believed to play an important role for cancer cells to escape from immune surveillance.4 Programmed cell death 1 (PD‐1), also known as CD279, is predominantly expressed on activated T cells. Binding of PD‐L1 to PD‐1 inhibits T cell effector function by inducing exhaustion of T cells, resulting in an immunosuppressive state.5 There have been several monoclonal antibodies targeting PD‐1/PD‐L1 axis approved by the US Food and Drug Administration (FDA) which have achieved great success in treating NSCLC.6, 7 Recent studies have found that PD‐L1 is involved in chemotherapy resistance in several cancer cell lines, including ovarian cancer, myeloma and NSCLC. Upregulation of PD‐L1 after chemotherapy has also been reported to be correlated with poor prognosis in patients with NSCLC.8, 9, 10, 11
Arsenic sulfide (As4S4), the active ingredient of the traditional Chinese medicine realgar, has been used for more than 2400 years and has attracted much research attention in recent years. Several studies have reported that As4S4 has antitumor activities in several cancers, including acute promyelocytic leukemia (APL), melanoma and gastric cancer,12, 13, 14, 15 and the antitumor activities are correlated with its ability to activate the p53‐dependent pathway.15, 16 A recent study has revealed that p53 regulates PD‐L1 via miR‐34, which directly binds to the PD‐L1 3′ untranslated region in models of NSCLC.17 Based on the above research, we conducted this study to verify the relationship between PD‐L1 and DDP resistance and identify whether As4S4 could decrease the PD‐L1 expression and reverse the DDP resistance in NSCLC.
METHODS
Chemicals, solutions, and antibodies
Highly purified realgar supplied by the Sanmenxia Yuhuangshan Pharmaceutical Co., Ltd was prepared from mined natural realgar. The purity of As4S4 in the realgar preparation was greater than 95.0%, confirmed by repeated X‐ray powder diffraction analyses. Realgar was dissolved in Dulbecco's phosphate‐buffered saline (DPBS) (Thermo Fisher Scientific) and sterilized by filtration. The content of As in DPBS solution was determined by inductively coupled plasma atomic emission spectrometry at the Instrumental Analysis Center of Shanghai Jiao Tong University (Shanghai, People's Republic of China). As4S4 stock solution of 766.36 μM was stored at 4°C. DDP was supplied by Qilu Pharmaceutical. Anti‐p53 (cat: 21891‐1‐AP), anti‐PD‐L1 (cat: 17952‐1‐AP), and anti‐GAPDH (cat: 10494‐1‐AP) antibodies were purchased from Proteintech. Pifithrin‐α (PFT‐α) (cat: S1816‐25 mg), methyl‐thiazolyl‐tetrazolium (MTT) (cat: ST316), interferon‐γ (IFN‐γ) (cat: P5664‐100 ug) and sodium dodecyl sulfate (SDS) powder (cat: ST626) were purchased from Beyotime Institute of Biotechnology. MTT powder was dissolved in DPBS at a concentration of 5 mg/ml. SDS powder was dissolved in water at aconcentration of 10%.
Cells and cell culture
The human lung adenocarcinoma cell lines A549 (p53 wild‐type, cat: 3111C0001CCC000002), A549/DDP (p53 wild‐type, DDP‐resistant cells, cat: 3111C0001CCC000519) and H1299 (p53 deficient, cat: 1101HUM‐PUMC000469) were obtained from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, People's Republic of China). A549 and A549/DDP cells were cultured in McCoy's 5A Media (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), 100 U/ml penicillin (Thermo Fisher Scientific) and 100 μg/ml streptomycin (Thermo Fisher Scientific) in a humid atmosphere incubator with 5% CO2 at 37°C.
Induction of DDP‐resistant cells
H1299 cells were treated with gradually increasing concentrations of DDP. Cells were incubated with 1 μM DDP for 48 h. After that, cells were digested, passed on and cultured in the fresh cell culture medium. After cells had adhered to the wall, DDP was added, and the final concentration of DDP in the medium was 2 μM. Repeat culture and passage of cells cultured in gradually increased concentrations of DDP induced the production of DDP‐resistant cells. Finally, H1299/DDP cells that could grow in the environment with 40 μM DDP were established. In addition, 40 μM DDP was used for continuous induction for 1 month to maintain its resistance.
Cell viability assay
The effect of DDP and As4S4 on cell proliferation was measured using MTT assay. Cells were seeded into 96‐well plates at a density of 5 × 103 cells/well and incubated overnight for adherence. Cells were exposed to different concentrations of cisplatin (8, 16, 32, 64, 128, 256 μM), As4S4 (0.5, 1.0, 1.5, 2.0, 2.5 and 3.0 μM), or their combinations. After treatment for 48 h, 10 μl MTT solutions (5 mg/ml) were added to each well and cells were incubated at 37°C for another 4 h. Following 4 h incubation, 150 μl SDS solutions (10% concentration) were added to each well to dissolve the formazan at 37°C overnight. Absorbance of each sample was measured at 570 nm. Data were analyzed based on three independent experiments.
Apoptosis analysis
Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining was used to quantify the apoptotic rate of A549/DDP cells. Cells were seeded at 1 × 106 cells/dish in 10 cm dishes and incubated overnight, then treated with As4S4, DDP or a combination of As4S4 and DDP for 48 h. Cells were then harvested and stained with annexin V–FITC/PI (cat: KGA 105‐DGA 108, KeyGEN BioTECH) following the manufacturer's instructions. After incubation for 30 min at 4°C, cells were analyzed using flow cytometry (FACS Canto; BD Biosciences).
siRNA and transfection
siRNA was transfected into cells using RFect (cat: 11013, Bio‐generating Biotechnology Corp.). Cells were seeded at 5 × 105 cells/well in 6 well plates and incubated overnight, then treated with a combination of siRNA and Rfect for 24 h following the manufacturer's instructions. The siRNA target sequences synthesized by Sangon Biotech Co., Ltd were as follows. Negative control (siRNA NC): sense ‐ UUC UCC GAA CGU GUC ACG UTT; antisense ‐ ACG UGA CAC GUU CGG AGA ATT; siRNA PD‐L1: sense ‐ AG GAA GAC CUG AAG GUU CAG CAU A; antisense ‐ AU GCU GAA CCU UCA GGU CUU CCU C.
Western blotting analysis
Total protein from cells or tumor tissues of animals was extracted using radioimmunoprecipitation assay lysis buffer (cat: P0013B, Beyotime Institute of Biotechnology). Protein concentrations were analyzed using a bicinchoninic acid protein assay kit (cat: P0010S, Beyotime Institute of Biotechnology) according to the manufacturer's protocol. Equal amounts of protein (60 μg) were loaded, separated by SDS polyacrylamide gel electrophoresis gels, and transferred onto the polyvinylidene fluoride membrane (cat: FFP24, Beyotime Institute of Biotechnology). The membrane was incubated with specific primary antibodies (1:1000) at 4°C overnight after blocking nonspecific binding sites with 5% nonfat milk. Membranes were then incubated with anti‐rat antibody labeled with horseradish peroxidase (cat: SA00001‐2, Proteintech) for 1 h at room temperature. Proteins were detected with an enhanced chemiluminescence system using the Beyo ECL Plus kit (cat: P0018S, Beyotime Institute of Biotechnology) and were semi‐quantified using Image Lab software Version 2.0.1 (Bio‐Rad Laboratories, Inc).
Real‐time qPCR analysis
Total RNAs were extracted from cells using RNA extraction kit (cat: G3013, Servicebio) and reverse‐transcribed to complementary DNA using RevertAid First Strand cDNA Synthesis Kit (cat: K1622, Thermo Fisher Scientific). The PCR detection was performed using FastStart Universal SYBR Green Master Kit (cat: G3008, Servicebio). The sequences of miR‐34a‐5p, miR‐34a‐3p and PD‐L1 were as follows: miR‐34a‐5p‐forward: ACACTCCAGCTGGGTGGCAGTGTCTTAGCT; miR‐34a‐5p‐reverse: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACAACCAG; miR‐34a‐3p‐forward: ACACTCCAGCTGGGCAATCAGCAAGTATAC; miR‐34a‐3p‐reverse: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGGGCAGT; PD‐L1‐forward: AGAACTACCTCTGGCACAT; PD‐L1‐reverse: ATCCATCATTCTCCCTTT.
U6 was the internal control of miR‐34a‐5p and miR‐34a‐3p. U6‐forward: CTCGCTTCGGCAGCACA; U6‐reverse: AACGCTTCACGAATTTGCGT. GAPDH was the internal control of PD‐L1. GAPDH‐forward: GGCACAGTCAAGGCTGAGAATG; GAPDH‐reverse: ATGGTGGTGAAGACGCCAGTA. The relative expression of miR‐34a‐5p, miR‐34a‐3p and PD‐L1 was calculated using the 2‐ΔΔCt method.
In vivo treatment
The animal experimental protocol was established according to the ethical guidelines of the Shandong Cancer Hospital and Institute, Cheeloo College of Medicine, Shandong University. The mice were housed in the animal center of Shandong Cancer Hospital and Institute in a specific pathogen free environment. A549/DDP cells (4 × 106) were subcutaneously injected into the right flank of 6‐week‐old female BALB/c nude mice, which were purchased from Yongteng Biotech Co., Ltd. At day 7 after tumor inoculation, when established tumors of 0.2 to 0.3 cm3 in diameter were detectable, drug administration was commenced. Animals with tumors less than 0.2 cm3 or more than 0.3 cm3 were excluded. Animals were randomly divided into four groups consisting of six animals each: (1) Normal saline (NS) group (20 ml/kg, once‐a‐day); (2) As4S4 group (As4S4 1 mg/kg in 0.4 ml, once‐a‐day); (3) DDP group (DDP 5 mg/kg in 0.4 ml, every 3 days); and (4) As4S4 + DDP group (As4S4 1 mg/kg in 0.4 ml, once‐a‐day; DDP 5 mg/kg in 0.4 ml, every 3 days). The randomization method was online random number generators. DDP was diluted using NS for the certain dosage, and As4S4 was dissolved in DPBS. The method of injection was intraperitoneal injection for 3 weeks. The mice were sacrificed after treatment for 3 weeks. Tumor volumes were calculated according to the following formula: (length × width2)/2.
Statistical analysis
SPSS software (v 13.0; SPSS Inc.) was used to perform statistical analysis. All data were expressed as mean ± SD. Data were presented as mean ± SD as stated. Student's t‐test was used to calculate p‐values. A two‐sided p < 0.05 was considered statistically significant.
RESULTS
Expression of PD‐L1 in A549/DDP and H1299/DDP cells was significantly higher than that of A549 and H1299 cells
We first used the MTT assay to assess the effects of serial concentrations of DDP on cell proliferation in A549, A549/DDP, H1299 and H1299/DDP cells. A549/DDP and H1299/DDP showed high resistance to the DDP challenge compared to their parental cells (Figure 1a,f). There was an increase of the DDP half‐maximal inhibitory concentration values (IC50) in DDP‐resistant cells (Figure 1b,g). Higher protein expression of PD‐L1 was observed in A549/DDP and H1299/DDP cells compared with A549 and H1299 cells using western blot assay (Figure 1c,d,h,i). Increased mRNA level of PD‐L1 was detected using real‐time qPCR in DDP‐resistant cells (Figure 1e,j).
FIGURE 1.
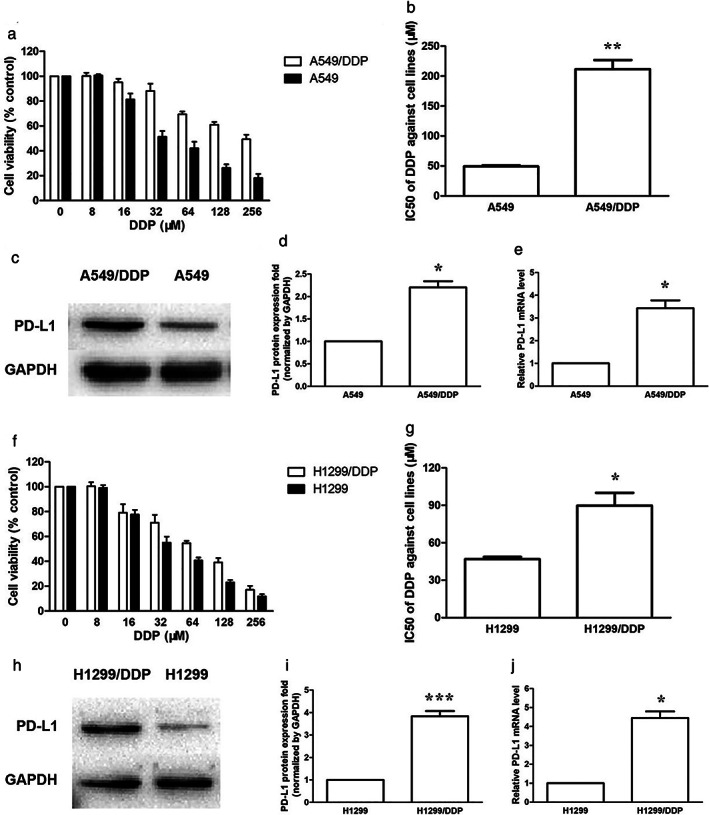
Expression of PD‐L1 was significantly higher in DDP‐resistant cells. (a) The cytotoxicity of DDP against A549 and A549/DDP cell lines when treated for 48 h. (b) The IC50 of DDP against A549 and A549/DDP cells when treated for 48 h. (c) The basic PD‐L1 protein expression of A549 and A549/DDP cells. (d) PD‐L1 protein quantification of the western blot results in A549 and A549/DDP cells. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (e) The basic PD‐L1 mRNA level of A549 and A549/DDP cells. (f) The cytotoxicity of DDP against H1299 and H1299/DDP cell lines when treated for 48 h. (g) The IC50 of DDP against H1299 and H1299/DDP cells when treated for 48 h. (h) The basic PD‐L1 protein expression of H1299 and H1299/DDP cells. (i) PD‐L1 protein quantification of the western blot results in H1299 and H1299/DDP cells. (j) The basic PD‐L1 mRNA level of H1299 and H1299/DDP cells. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
Increase of PD‐L1 enhanced DDP resistance in A549 and H1299 cells
It is well known that IFN‐γ, produced by activated T cells, stimulates PD‐L1 expression in the tumor microenvironment.18 Treatment with IFN‐γ (10 ng/ml) increased the protein expression of PD‐L1 in a time‐dependent manner in A549 and H1299 cells (Figure 2a,b,e,f). Cotreatment of IFN‐γ and DDP for 48 h significantly enhanced DDP resistance in A549 and H1299 cells (Figure 2c,g). In the presence of IFN‐γ, the IC50 values of DDP were increased in A549 and H1299 cells (Figure 2d,h).
FIGURE 2.
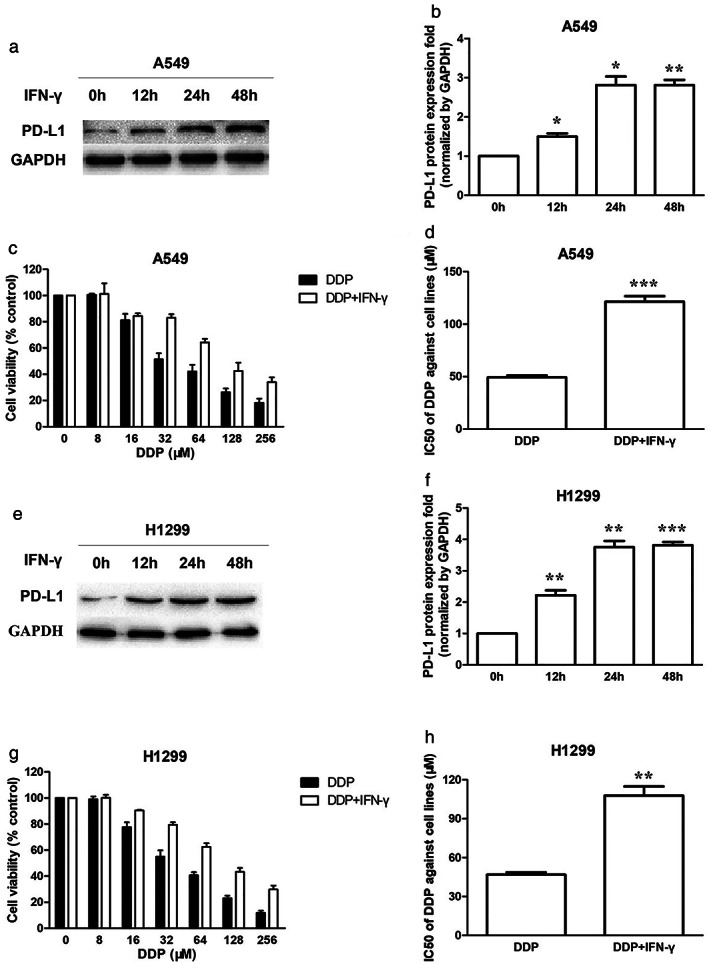
Increase of PD‐L1 enhanced DDP resistance in A549 and H1299 cells. (a) PD‐L1 protein expression in A549 cells when treated with 10 ng/ml IFN‐γ for 0, 12, 24, 48 h. (b) PD‐L1 protein quantification of the western blot results in A549 cells treated with IFN‐γ. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (c) The cytotoxicity of DDP against A549 cells in the absence or presence of IFN‐γ (10 ng/ml) when treated for 48 h. (d) The IC50 of DDP against A549 cells in the absence or presence of IFN‐γ (10 ng/ml) when treated for 48 h. (e) PD‐L1 protein expression in H1299 cells when treated with 10 ng/ml IFN‐γ for 0, 12, 24, 48 h. (f) PD‐L1 protein quantification of the western blot results in H1299 cells treated with IFN‐γ. (g) The cytotoxicity of DDP against H1299 cells in the absence or presence of IFN‐γ (10 ng/ml) when treated for 48 h. (h) The IC50 of DDP against H1299 cells in the absence or presence of IFN‐γ (10 ng/ml) when treated for 48 h. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001). IFN‐γ, interferon‐γ
Depletion of PD‐L1 inhibited DDP resistance in A549/DDP and H1299/DDP cells
Western blot assay indicated that PD‐L1 was successfully downregulated in A549/DDP and H1299/DDP cells by specific siRNA (Figure 3a,b,e,f). Furthermore, the serial concentrations of DDP were used to treat the PD‐L1 silenced A549/DDP and H1299/DDP cells. The depletion of PD‐L1 inhibited DDP resistance in A549/DDP and H1299/DDP cells (Figure 3c,g). The IC50 values of DDP in PD‐L1 silenced A549/DDP and PD‐L1 silenced H1299/DDP cells treated for 48 h were decreased with downregulation of PD‐L1 (Figure 3d,h).
FIGURE 3.
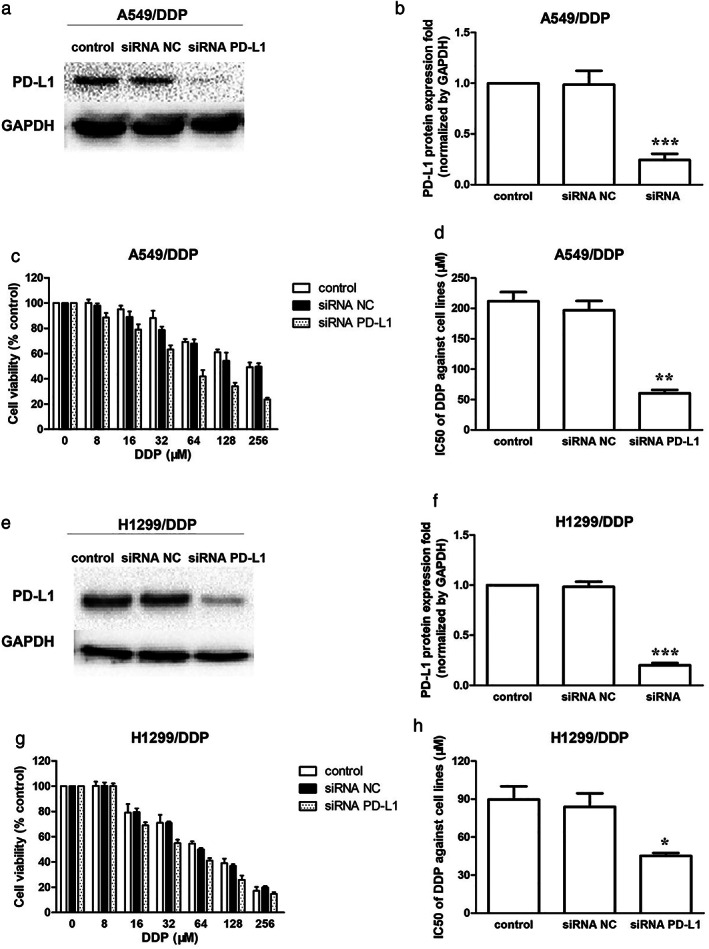
Depletion of PD‐L1 inhibited DDP resistance in A549/DDP and H1299/DDP cells. (a) Effect of PD‐L1 siRNA on PD‐L1 protein expression in A549/DDP cells. A549/DDP cells were transfected with or without PD‐L1 siRNA for 24 h. (b) PD‐L1 protein quantification of the western blot results in A549/DDP cells transfected with or without PD‐L1 siRNA for 24 h. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (c) The cytotoxicity of DDP against A549/DDP cells and PD‐L1 silenced A549/DDP cells when treated for 48 h. (d) The IC50 of DDP against A549/DDP cells and PD‐L1 silenced A549/DDP cells when treated for 48 h. (e) Effect of PD‐L1 siRNA on PD‐L1 protein expression in H1299/DDP cells. (f) PD‐L1 protein quantification of the western blot results in H1299/DDP cells transfected with or without PD‐L1 siRNA for 24 h. (g) The cytotoxicity of DDP against H1299/DDP cells and PD‐L1 silenced H1299/DDP cells when treated for 48 h. (h) The IC50 of DDP against H1299/DDP cells and PD‐L1 silenced H1299/DDP cells when treated for 48 h. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001). NC, negative control
As4S4 enhanced drug susceptibility of DDP in A549/DDP cells
A549/DDP and H1299/DDP cells were treated with different concentrations of As4S4 (0, 0.5, 1.0,1.5, 2.0, 2.5, 3.0 μM) for 48 h. In order to investigate whether As4S4 synergistically facilitated the sensitivity of A549/DDP and H1299/DDP to DDP, As4S4 at a nonlethal dose was used. In particular, 1.5 μM As4S4 and 2.0 μM As4S4 did not yield a measurable impact on cell viability in A549/DDP and H1299/DDP cells, respectively (Figure 4a,f). However, 1.5 μM As4S4 clearly enhanced the sensitivity of A549/DDP cells to DDP when treated for 48 h. The combination of 1.5 μM As4S4 and 8 μM DDP showed an obvious synergism (Figure 4b). Thus, 1.5 μM As4S4 and 8 μM DDP were used for further study. Cotreatment of As4S4 (1.5 μM) and DDP decreased the IC50 value of DDP in A549/DDP cells, when compared with DDP alone (Figure 4e). We next examined whether As4S4 could enhance DDP‐induced apoptosis using flow cytometry. As4S4 increased DDP‐induced apoptosis in A549/DDP cells (Figure 4c,d). However, cotreatment of As4S4 (2 μM) and DDP did not exhibit synergistic inhibition effect in H1299/DDP cells (Figure 4g).
FIGURE 4.
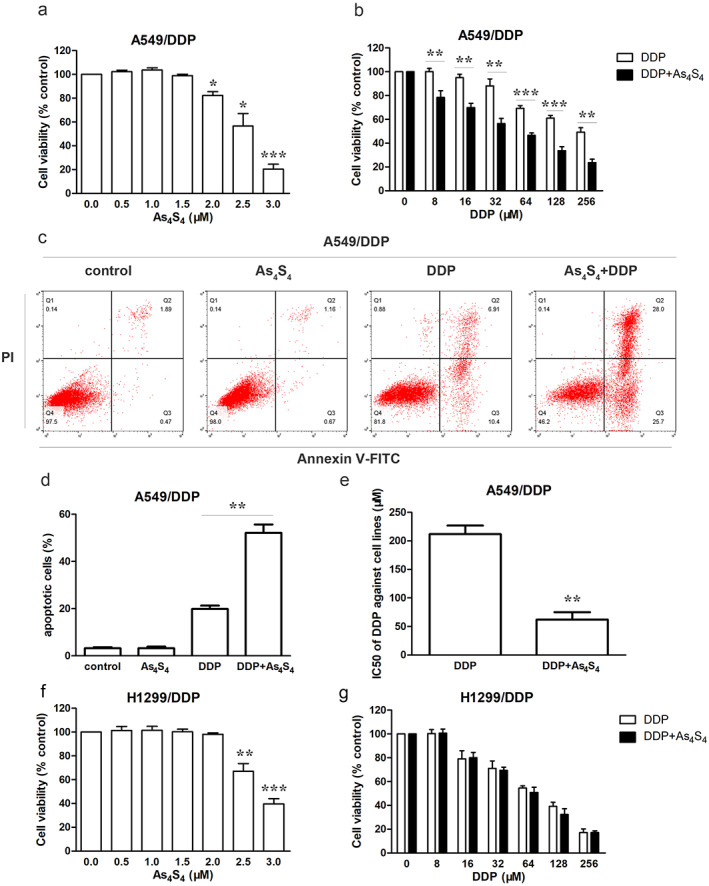
As4S4 enhanced drug susceptibility of DDP in A549/DDP cells. (a) The cytotoxicity of As4S4 against A549/DDP cells when treated for 48 h. (b) The cytotoxicity of DDP against A549/DDP cells in the absence or presence of As4S4 (1.5 μM) when treated for 48 h. (c) Flow cytometric analysis of A549/DDP cells treated with As4S4 (1.5 μM), DDP (8 μM), or the combination of As4S4 and DDP for 48 h. (d) Apoptotic rate in each group. (e) The IC50 of DDP against A549/DDP cells in the absence or presence of As4S4 (1.5 μM) when treated for 48 h. (f) The cytotoxicity of As4S4 against H1299/DDP cells when treated for 48 h. (g) The cytotoxicity of DDP against H1299/DDP cells in the absence or presence of As4S4 (2.0 μM) when treated for 48 h. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
As4S4 changed the levels of p53, PD‐L1 and miR‐34a‐5p in A549/DDP cells
Western blot analysis showed that treatment of As4S4 (1.5 μM) upregulated p53 expression and downregulated PD‐L1 expression in A549/DDP cells in a time‐dependent manner (Figure 5a–c). Real‐time qPCR analysis showed that As4S4 (1.5 μM) increased miR‐34a‐5p level in a time‐dependent manner and had no influence on miR‐34a‐3p level in A549/DDP cells (Figure 5d,e). To determine whether As4S4 could modify the levels of p53, PD‐L1 and miR‐34a in H1299/DDP cells, different concentrations of As4S4 (0, 2, 2.5, 3 μM) were used. We observed that As4S4 had no influence on expression of p53 and PD‐L1, as well as miR‐34a level in H1299/DDP cells when treated for 48 h (Figure 5f–i).
FIGURE 5.
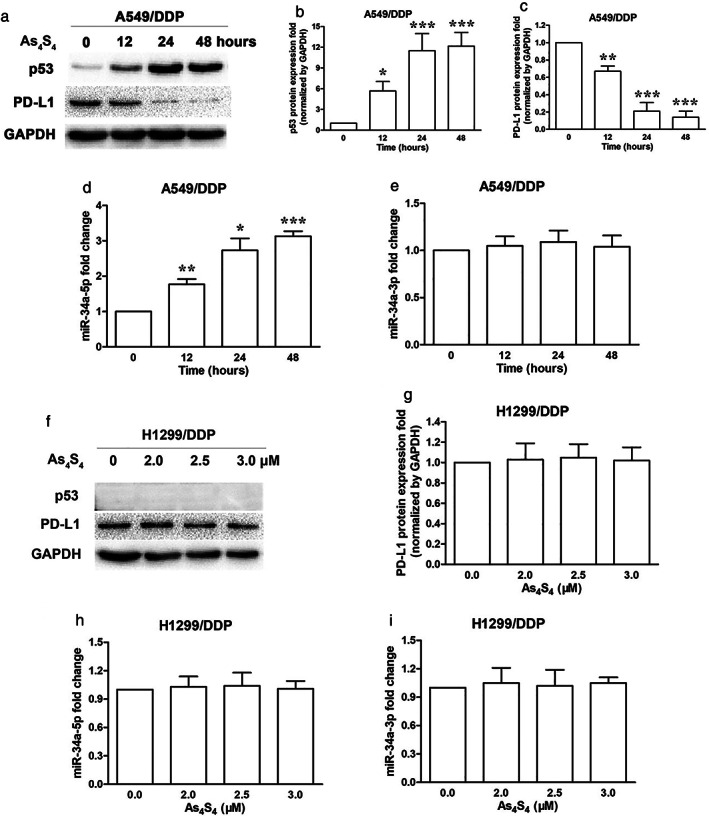
As4S4 changed the levels of p53, PD‐L1 and miR‐34a‐5p in A549/DDP cells. (a) Effect of As4S4 (1.5 μM) on p53 and PD‐L1 protein expression in A549/DDP cells treated for 0, 12, 24 and 48 h. (b) p53 protein quantification of the western blot results in A549/DDP cells treated with As4S4 (1.5 μM) for 0, 12, 24 and 48 h. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (c) PD‐L1 protein quantification of the western blot results in A549/DDP cells treated with As4S4 (1.5 μM) for 0, 12, 24 and 48 h. (d) miR‐34a‐5p levels of the real‐time qPCR results in A549/DDP cells treated with As4S4 (1.5 μM) for 0, 12, 24 and 48 h. (e) miR‐34a‐3p levels of the real‐time qPCR results in A549/DDP cells treated with As4S4 (1.5 μM) for 0, 12, 24 and 48 h. (f) Effect of As4S4 (0, 2.0, 2.5, 3.0 μM) on p53 and PD‐L1 protein expression in H1299/DDP cells treated for 48 h. (g) PD‐L1 protein quantification of the western blot results in H1299/DDP cells treated with As4S4 (0, 2.0, 2.5, 3.0 μM) for 48 h. (h) miR‐34a‐5p levels of the real‐time qPCR results in H1299/DDP cells treated with As4S4 (0, 2.0, 2.5, 3.0 μM) for 48 h. (i) miR‐34a‐3p levels of the real‐time qPCR results in H1299/DDP cells treated with As4S4 (0, 2.0, 2.5, 3.0 μM) for 48 h. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
As4S4 downregulated the PD‐L1 expression through p53/miR‐34a‐5p axis
To further understand the role of p53 in PD‐L1 regulation, we used As4S4 with or without PFT‐α (a p53 specific inhibitor) and determined whether p53 inhibiting could influence PD‐L1 expression in A549/DDP cells. A549/DDP cells were pretreated with 30 μM PFT‐α (a concentration that did not cause significant cytotoxicity in cells) for 30 min prior to addition of As4S4. Inhibition of p53 by PFT‐α partially restored the protein levels of p53 and PD‐L1, which were changed by As4S4 treatment (Figure 6a–c). Flow cytometry analysis revealed that PFT‐α alone did not cause apoptosis and had no influence on the apoptosis rate induced by DDP in A549/DDP cells. However, pretreatment with PFT‐α suppressed the apoptosis rate induced by cotreatment of As4S4 and DDP compared to the nontreated control in A549/DDP cells (Figure 6d,e). Furthermore, the upregulation of miR‐34a‐5p level induced by As4S4 was significantly decreased by additional PFT‐α (Figure 6f).
FIGURE 6.
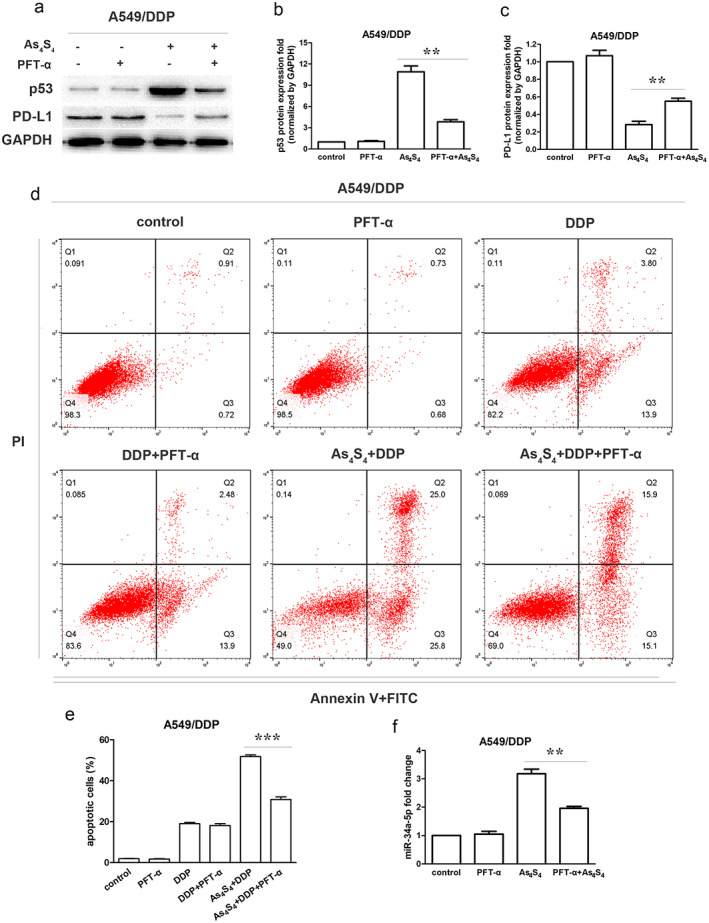
As4S4 downregulated the PD‐L1 expression through p53/miR‐34a‐5p axis. (a) Effect of As4S4 (1.5 μM) on p53 and PD‐L1 protein expression in A549/DDP cells in the presence or absence of PFT‐α (30 μM) treated for 48 h. (b) p53 protein quantification of the western blot results in A549/DDP cells treated with As4S4, PFT‐α or their combinations for 48 h. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (c) PD‐L1 protein quantification of the western blot results in A549/DDP cells treated with As4S4, PFT‐α or their combination for 48 h. (d) Flow cytometric analysis showing apoptosis in A549/DDP cells treated by DDP (8 μM), DDP plus As4S4 (1.5 μM), or PFT (30 μM), or the combinations. (e) Apoptotic rate in each group. (f) miR‐34a‐5p levels in A549/DDP cells treated with As4S4, PFT‐α or their combination for 48 h. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001). PFT‐α, Pifithrin‐α
As4S4 enhanced antitumor efficacy of DDP in vivo
To verify the synergistic effect of As4S4 and DDP, we established xenograft mouse models. A549/DDP cells were subcutaneously implanted into BALB/c nude mice, and the mice were treated with DDP with or without As4S4. DDP significantly inhibited tumor growth, and the efficacy was significantly enhanced by additional As4S4 treatment, whereas the antitumor activity of As4S4 alone was dismal (Figure 7a–c). DDP treatment alone had no apparent influence on expression of p53 in tumor tissue. The expression of p53 was significantly increased after treatment with As4S4. DDP treatment upregulated PD‐L1 expression in xenograft mouse models. Upregulation of PD‐L1 induced by DDP in the xenograft models was inhibited by treatment of As4S4 (Figure 7d–f).
FIGURE 7.
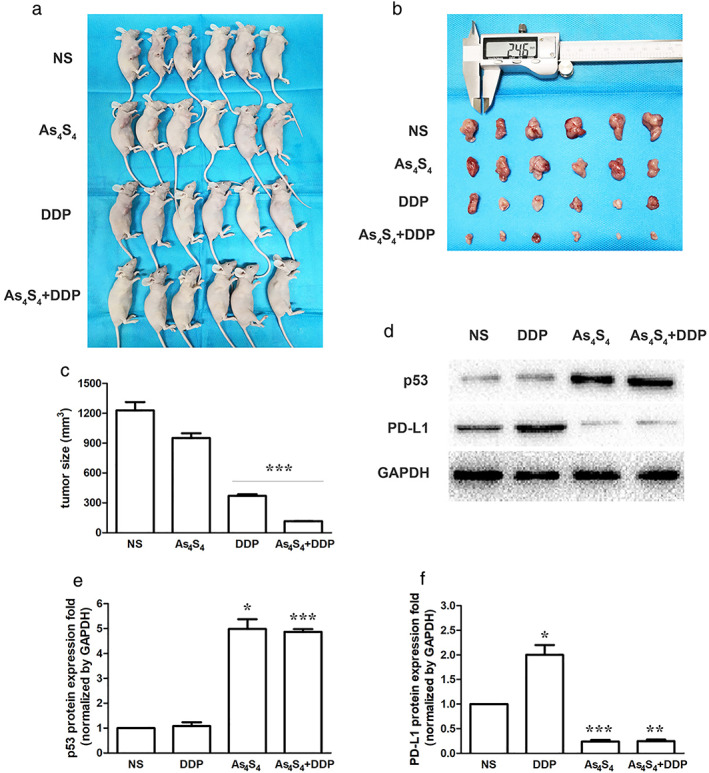
As4S4 enhanced antitumor efficacy of DDP in vivo. (a) Representative photograph of mice in each group after treatment for 21 days (n = 6). NS group: NS 20 ml/kg, once‐a‐day; As4S4 group: As4S4 1 mg/kg in 0.4 ml, once‐a‐day; DDP group: DDP 5 mg/kg in 0.4 ml, every three days; As4S4 + DDP group: As4S4 1 mg/kg in 0.4 ml, once‐a‐day, DDP 5 mg/kg in 0.4 ml, every three days. (b) Representative photograph of tumor tissues from xenograft mice in each group (n = 6). Tumor dimensions were measured by a digital caliper. (c) Tumor sizes in each group (n = 6). (d) Effect of DDP, As4S4 and their combinations on p53 and PD‐L1 protein expression in xenograft models. (e) p53 protein quantification of the western blot results in each group. Protein levels were normalized to the GAPDH levels and are shown as fold increase or decrease relative to the levels for the control strain. (f) PD‐L1 protein quantification of the western blot results in each group. Data represents the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001). NS, normal saline
DISCUSSION
It is important to note that PD‐L1 in tumor cells has functions other than as an immune checkpoint ligand, including stimulation of cancer progression, promotion of epithelial‐mesenchymal transition (EMT) and resistance to chemotherapy drugs.19, 20 In this study, we demonstrated that increase of PD‐L1 was correlated with DDP resistance in NSCLC cells, as evidenced by cell proliferation assay. Furthermore, our study revealed that As4S4 could block PD‐L1 overexpression in A549/DDP cells, and increase cell apoptosis induced by DDP.
Tumor suppressor p53 is a transcription factor and plays pivotal roles in cell cycle, DNA damage repair, and apoptosis.21 Wild‐type p53 protein is frequently downregulated because of its function of tumor suppression in many cancer cells. MicroRNAs (miRNAs), well‐known as small non‐coding RNAs, can interact with the 3′‐untranslated region (3′‐UTR) of multiple target messenger RNAs (mRNAs) and regulate gene expression at the post‐transcriptional levels.22 Transcription of miR‐34a is regulated dominantly by the crucial tumor suppressor p53.23 A recent study has revealed that p53 can augment the expression of miR‐34a, which leads to the decreased expression level of PD‐L1 in NSCLC cells.17, 24 In our study, we found that As4S4 decreased the protein level of PD‐L1, and the expression of p53 and miR‐34a‐5p level were upregulated by As4S4 in A549/DDP cells (p53 wild‐type). No such decrease of PD‐L1 was found in H1299/DDP cells (p53 deficient), which was probably due to the lack of expression of p53. Cotreatment of DDP and As4S4 had a synergistic inhibitory effect on A549/DDP cell viability, but not H1299/DDP cell viability. These results implied that p53 might play an important role in the reversal effect of chemoresistance and downregulation of PD‐L1 induced by As4S4 in NSCLC cells.
To further understand the role of p53 in PD‐L1 regulation, we used PFT‐α to inhibit the transcriptional activity of p53. PFT‐α partially reversed the synergistic inhibition effect of As4S4 and DDP. Moreover, blocking the activation of p53 by PFT‐α could partially restore the levels of p53, PD‐L1 and miR‐34a‐5p. These results implied that the upregulation of p53 by As4S4 might induce various p53‐mediated antioncogenic factors, such as the regulation of miRNA. Based on the study which revealed that p53 regulates PD‐L1 via miR‐34a in NSCLC, our results proposed that As4S4 might control the expression of PD‐L1 by regulating the p53/miR‐34a pathway in NSCLC cells.
Early reports showed that PD‐L1 expression was increased in breast cancer and melanoma cells with cytotoxic drug treatment.25, 26 DDP treatment also upregulated PD‐L1 expression in xenograft mouse models implanted with NSCLC cells.10 Our results showed that PD‐L1 expression was elevated by DDP treatment in vivo. Higher expression of this protein was also observed in DDP‐resistant sublines. Furthermore, As4S4 significantly upregulated p53 protein expression and downregulated PD‐L1 protein expression in tumor tissues in vivo, and inhibited the upregulation of PD‐L1 induced by DDP treatment. It has been reported that doxorubicin treatment could upregulate the total PD‐L1 protein expression in breast cancer cells, with a decrease in the membrane and cytoplasmic fraction and increase in the nuclear fraction.25 In our study, we explored the changes in the total expression of PD‐L1 protein in NSCLC cells. Whether nuclear translocation participates in the changes of PD‐L1 expression induced by As4S4 remains to be further investigated. Moreover, no study has so far demonstrated the exact mechanism of how DDP treatment upregulates the expression of PD‐L1 in vivo. Variations of PD‐L1 expression after DDP based chemotherapy in NSCLC patients and its possible mechanisms are being investigated in our laboratory for this reason.
Overexpression of the ATP‐dependent efflux pump, known as P‐glycoprotein (P‐gp), is the main mechanism of drug resistance. P‐gp is the first known member of the ABC transporter superfamily, and is encoded by the multidrug resistance 1 (MDR1) gene.27 It has been reported that PD‐1/PD‐L1 interaction increased MDR1/P‐gp expression in breast cancer cells.28 As we did not investigate the association between PD‐L1 and the MDR1 gene in NSCLC cells, we will investigate this further in future studies.
Phase III clinical trials reported that the combination of chemotherapy and immune checkpoint‐targeted antibodies showed superiority to chemotherapy alone in patients with advanced NSCLC.29, 30 It has also been reported that the combination of As4S4 and anticancer compounds showed synergistic enhancement of anticancer efficacy in numerous human cancer cell lines and xenograft mouse models.16 Our results indicated that one possible mechanism of As4S4 was to break down the drug resistance of tumor cells through downregulation of PD‐L1. In vitro inoculation and in vivo injection of As4S4 could reverse chemoresistance of DDP in NSCLC. These findings suggest that the combination of DDP treatment with As4S4 will further increase the benefits of cancer treatment of NSCLC.
There is a limitation in this study. Although blocking the activation of p53 by PFT‐α can partially restore the levels of miR‐34a‐5p and PD‐L1, which are changed by As4S4 treatment, the relationship between miR‐34a‐5p and PD‐L1 remains uncertain. Therefore, the current results are insufficient to support the conclusion that As4S4 downregulates PD‐L1 expression through the p53/miR‐34a‐5p axis. To explore whether miR‐34a‐5p could directly regulate PD‐L1 expression in NSCLC, we should conduct miR‐34a‐5p overexpression and knockdown. Studies are currently underway to specifically address this issue.
In conclusion, the present study indicates that increase of PD‐L1 is correlated with DDP resistance in NSCLC. As4S4 can block PD‐L1 overexpression in DDP resistant NSCLC cells with wild‐type p53 to break down the drug resistance, probably via targeting the p53/miR‐34a‐5p axis. These findings suggest that As4S4 may be a potential agent to treat chemotherapy refractory NSCLC.
CONFLICT OF INTEREST
The authors reported no conflicts of interest in this study.
ACKNOWLEDGMENTS
This work was supported by the Shandong Key Research and Development Program (2019GSF108251). The authors wish to acknowledgment Yinghong Zhou for his help in experiment recommendations and animal feeding.
Tian W, Sun Y, Cheng Y, Ma X, Du W, Shi W, et al. Arsenic sulfide reverses cisplatin resistance in non‐small cell lung cancer in vitro and in vivo through targeting PD‐L1 . Thorac Cancer. 2021;12:2551–2563. 10.1111/1759-7714.14136
Wenna Shi and Qisen Guo contributed equally to this work.
Funding information Shandong Key Research and Development Program, Grant/Award Number: 2019GSF108251
Contributor Information
Wenna Shi, Email: nanazaici312@qq.com.
Qisen Guo, Email: guoqs369@163.com.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Parsons A, Daley A, Begh R, Aveyard P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: systematic review of observational studies with meta‐analysis. BMJ. 2010;340:b5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel‐Bellan A, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, et al. Structure of the complex of human programmed death 1, PD‐1, and its ligand PD‐L1. Structure. 2015;23:2341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Huang S, Zhang Y, Zhu L, Wu X. The application and mechanism of PD pathway blockade for cancer therapy. Postgrad Med J. 2018;94:53–60. [DOI] [PubMed] [Google Scholar]
- 7.Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD‐L1 as cancer therapeutics. J Hematol Oncol. 2019;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7‐H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. 2013;27:464–72. [DOI] [PubMed] [Google Scholar]
- 9.Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR‐197/CKS1B/STAT3‐mediated PD‐L1 network in chemoresistant non‐small‐cell lung cancer. Mol Ther. 2015;23:717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang P, Ma Y, Lv C, Huang M, Li M, Dong B, et al. Upregulation of programmed cell death ligand 1 promotes resistance response in non‐small‐cell lung cancer patients treated with neo‐adjuvant chemotherapy. Cancer Sci. 2016;107:1563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo Y, Zheng W, Liu J, Tang Q, Wang SS, Yang XS. MiR‐34a‐5p/PD‐L1 axis regulates cisplatin chemoresistance of ovarian cancer cells. Neoplasma. 2020;67:93–101. [DOI] [PubMed] [Google Scholar]
- 12.Lu DP, Qiu JY, Jiang B, Wang Q, Liu KY, Liu YR, et al. Tetra‐arsenic tetra‐sulfide for the treatment of acute promyelocytic leukemia: a pilot report. Blood. 2002;99:3136–43. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Fang Y, Ma L, Liu S, Li X. Realgar‐induced apoptosis and differentiation in all‐trans retinoic acid (ATRA)‐sensitive NB4 and ATRA‐resistant MR2 cells. Int J Oncol. 2012;40:1089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pastorek M, Gronesova P, Cholujova D, Hunakova L, Bujnakova Z, Balaz P, et al. Realgar (As4S4) nanoparticles and arsenic trioxide (As2O3) induced autophagy and apoptosis in human melanoma cells in vitro. Neoplasma. 2014;61:700–9. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Tian W, Kim S, Ding W, Tong Y, Chen S. Arsenic sulfide, the main component of realgar, a traditional Chinese medicine, induces apoptosis of gastric cancer cells in vitro and in vivo. Drug Des Devel Ther. 2015;9:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Tong Y, Zhang X, Pan M, Chen S. Arsenic sulfide combined with JQ1, chemotherapy agents, or celecoxib inhibit gastric and colon cancer cell growth. Drug Des Devel Ther. 2015;9:5851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR‐34. J Natl Cancer Inst. 2016;108:djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishibashi M, Tamura H, Sunakawa M, Kondo‐Onodera A, Okuyama N, Hamada Y, et al. Myeloma drug resistance induced by binding of myeloma B7‐H1 (PD‐L1) to PD‐1. Cancer Immunol Res. 2016;4:779–88. [DOI] [PubMed] [Google Scholar]
- 20.Marcucci F, Rumio C, Corti A. Tumor cell‐associated immune checkpoint molecules ‐ drivers of malignancy and stemness. Biochim Biophys Acta Rev Cancer. 2017;1868:571–83. [DOI] [PubMed] [Google Scholar]
- 21.Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18:6145–57. [DOI] [PubMed] [Google Scholar]
- 22.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA. RNA. 2005;11:1753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slabáková E, Culig Z, Remšík J, Souček K. Alternative mechanisms of miR‐34a regulation in cancer. Cell Death Dis. 2017;8:e3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang DY, Sp N, Jo ES, Rugamba A, Hong DY, Lee HG, et al. The inhibitory mechanisms of tumor PD‐L1 expression by natural bioactive gallic acid in non‐small‐cell lung cancer (NSCLC) cells. Cancers (Basel). 2020;12(3):727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghebeh H, Lehe C, Barhoush E, al‐Romaih K, Tulbah A, al‐Alwan M, et al. Doxorubicin downregulates cell surface B7‐H1 expression and upregulates its nuclear expression in breast cancer cells: role of B7‐H1 as an anti‐apoptotic molecule. Breast Cancer Res. 2010;12:R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Zhou J, Giobbie‐Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD‐L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609. [DOI] [PubMed] [Google Scholar]
- 27.Raaijmakers MH. ATP‐binding‐cassette transporters in hematopoietic stem cells and their utility as therapeutical targets in acute and chronic myeloid leukemia. Leukemia. 2007;21:2094–102. [DOI] [PubMed] [Google Scholar]
- 28.Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD‐1/PD‐L1 interaction up‐regulates MDR1/P‐gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. 2017;8:99901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first‐line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301. [DOI] [PubMed] [Google Scholar]
- 30.Melosky B, Juergens R, Hirsh V, McLeod D, Leighl N, Tsao MS, et al. Amplifying outcomes: checkpoint inhibitor combinations in first‐line non‐small cell lung cancer. Oncologist. 2020;25:64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]