

Abstract
APOBEC3 enzymes are cytosine deaminases implicated in cancer. Precisely when APOBEC3 expression is induced during cancer development remains to be defined. Here we show that specific APOBEC3 genes are upregulated in breast DCIS, and in pre-invasive lung cancer lesions coincident with cellular proliferation. We observe evidence of APOBEC3-mediated subclonal mutagenesis propagated from TRACERx pre-invasive to invasive NSCLC lesions. We find that APOBEC3B exacerbates DNA replication stress and chromosomal instability through incomplete replication of genomic DNA, manifested by accumulation of mitotic ultrafine bridges and 53BP1 nuclear bodies in the G1 phase of the cell cycle. Analysis of TRACERx NSCLC clinical samples and mouse lung cancer models, revealed APOBEC3B expression driving replication stress and chromosome missegregation. We propose that APOBEC3 is functionally implicated in the onset of chromosomal instability and somatic mutational heterogeneity in pre-invasive disease, providing fuel for selection early in cancer evolution.
Keywords: APOBEC, non-small cell lung cancer, breast cancer, intratumor heterogeneity, chromosomal instability, pre-invasive
Statement of Significance
This study reveals the dynamics and drivers of APOBEC3 gene expression in pre-invasive disease and the exacerbation of cellular diversity by APOBEC3B through DNA replication stress to promote chromosomal instability early in cancer evolution.
INTRODUCTION
Non-small cell lung cancer (NSCLC) is characterized by somatic copy number and point mutation intratumor heterogeneity, whereby chromosomal instability (CIN) is associated with an increased risk of recurrence or death (1). Large-scale genomic sequencing studies have implicated APOBEC3 enzymes in somatic mutagenesis, mediating the mutational signatures single base substitutions (SBS) 2 and 13 in cancer genomes (2). These enzymes form a barrier against viral and transposon replication through cytosine deaminase-dependent and -independent mechanisms (3). However, APOBEC3 family members have also been implicated in the generation of C>T and C>G DNA point mutations in 5’-TCA-3’ and 5’-TCT-3’ trinucleotide motifs during cancer evolution (4,5). The clinical importance of APOBEC3 in cancer is of broad interest because of its associations with treatment outcome (6), patient outcome (7,8), point mutation heterogeneity (9, 10), enrichment of APOBEC3 signature mutations in metastases (9, 11, 12), the genesis of oncogenic driver mutations (4, 13, 14) and the immune response (15, 16). Currently, APOBEC3A (A3A) and APOBEC3B (A3B) are thought to be the principal family members involved in APOBEC3-mediated cancer mutagenesis (17–19).
Several pathways have been identified that drive A3A and A3B gene expression in cancer, including the DNA damage response (20,21), the PKC-NFκB pathway (22) and the interferon signaling pathway (9). In NSCLC, APOBEC3-mediated mutations are widespread and are enriched as subclonal mutations (1). As a consequence, APOBEC3-mediated mutagenesis is thought to be a late mutagenic process, enriched in tumor subclones in NSCLC (1,9). How early branched evolution and subclone dispersion occurs in NSCLC is unclear. In breast cancer this process can already occur in pre-invasive ductal carcinoma in situ (DCIS) lesions (23,24). Although some sequencing studies have reported APOBEC3 signature mutations in pre-invasive lung disease (25–28), the timing and consequences of APOBEC3 induction during cancer evolution are currently unclear.
Using a combination of integrated molecular and clinical analysis of a comprehensively annotated prospective cohort study of patients with NSCLC and pre-invasive disease in the TRACERx (TRAcking Cancer Evolution through therapy (Rx)) study (1) and pre-invasive lung squamous cell carcinoma (LUSC) lesions (29), as well as published datasets, in vitro and in vivo experimentation, we characterize APOBEC3 family mRNA and protein expression during NSCLC and breast cancer evolution and investigate the underlying causes and consequences of APOBEC3 expression in cancer evolution.
RESULTS
APOBEC3 expression increases in pre-invasive lung and breast cancer lesions
We initially investigated APOBEC3 expression during NSCLC evolution by immunohistochemical staining of sections from different tumor morphology-defined progression stages with a monoclonal antibody that detects a shared epitope in A3A, A3B and A3G (30). Control experiments demonstrate that this protocol can detect A3B protein in formalin-fixed paraffin-embedded (FFPE) samples, since nuclear APOBEC3 staining was absent in SKBR3 cells (A3B-null) but was strong in HCC1954 cells (A3B-high) (Supplementary Fig. 1A). Weak or no nuclear immunoreactivity was observed in histologically normal bronchial (Fig. 1A) and alveolar epithelium (Fig. 1B). The presence of stromal cells with cytoplasmic APOBEC3 staining below the epithelial lining (Fig. 1A; orange arrow) is indicative of A3G and A3A protein, but not A3B, which is reported to be extensively nuclear (30–32). We observed over 10% nuclear positivity in 1% (1 out of 94) of normal tissue sections, in 93% (62 out of 67) of pre-invasive samples and in 52% (47 out of 90) of NSCLC samples, respectively (Fig. 1C). The pre-invasive lesions showed the highest APOBEC3 immunoreactivity (Fig. 1A–C; two-tailed Fisher’s exact test, P ≤ 0.0001), indicating that APOBEC3 expression varies during NSCLC evolution and may peak in pre-invasive lesions. Among the 67 pre-invasive lesions, 7 were classified as severe dysplasia, 16 as less severe dysplasia, 18 as carcinoma in situ (CIS), 7 as atypical adenomatous hyperplasia (AAH), 13 as adenocarcinoma in situ (AIS) and 6 as minimally invasive adenocarcinoma (MIA). All 7 severely dysplastic lesions showed 60–75% nuclear APOBEC3 positivity, whereas the less severe dysplastic lesions showed anywhere between 2–50% nuclear APOBEC3 positivity. All 18 CIS lesions showed >75%, 4 out of 7 AAH lesions exhibited 10–20% and the remaining 3 AAH lesions showed 2–10% nuclear APOBEC3 positivity, while both the AIS (N = 13) and MIA (N = 6) lesions showed a heterogeneous pattern with overall 10–20% nuclear positivity.
Figure 1. APOBEC3 expression increases during early NSCLC evolution.
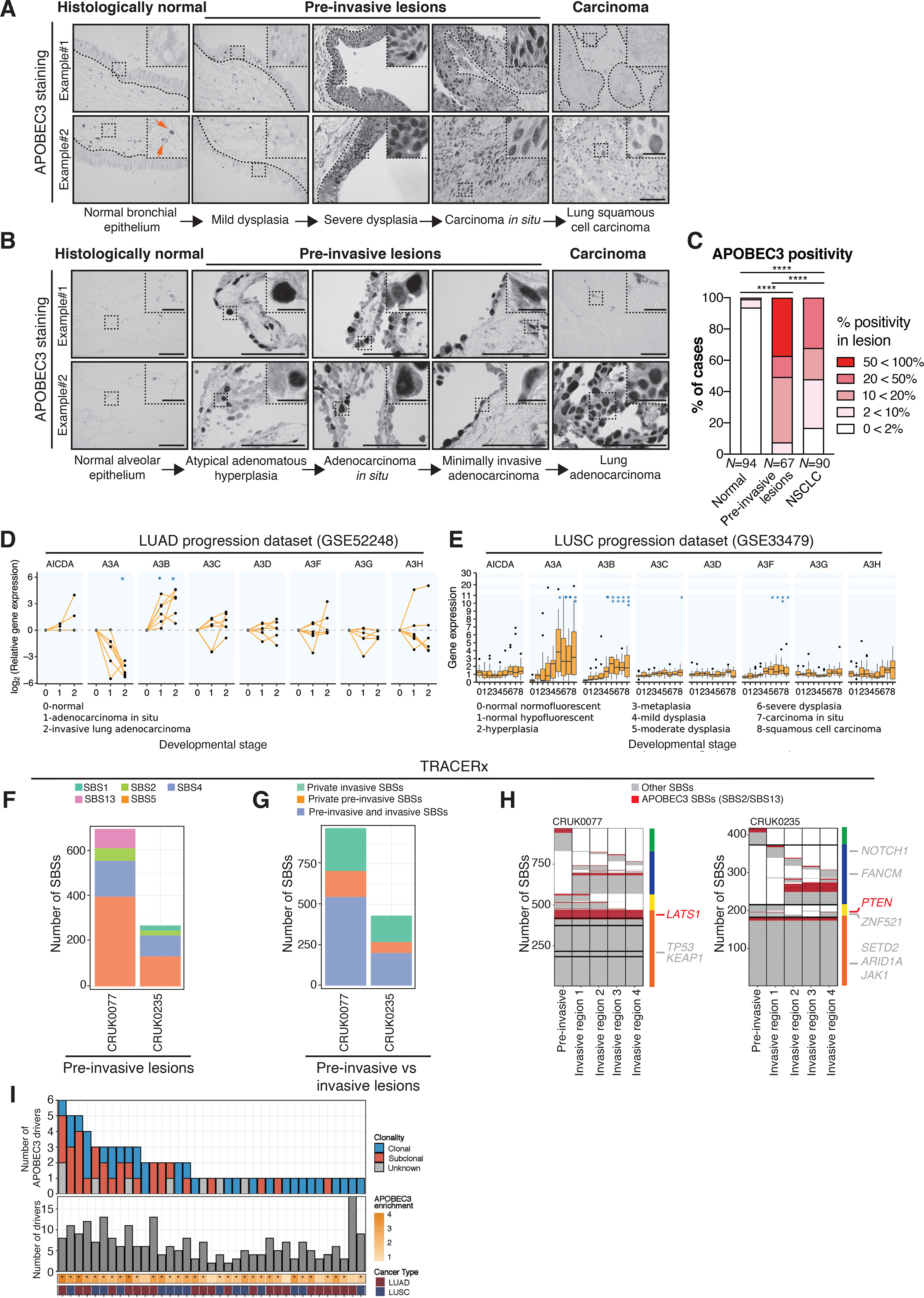
A-B, Representative images from 2 different samples with APOBEC3 immunohistochemistry in normal lung epithelium, pre-invasive lesions, LUSC and LUAD. Note the nuclear APOBEC3 staining in epithelial cells, which is indicative of A3B and potentially also A3A staining. The samples originate from NSCLC patients from the TRACERx 100 cohort (1), the UCLH Surveillance Study (29) and a Danish cohort of NSCLC patients. The orange arrows indicate stromal cells that have cytoplasmic APOBEC3 staining, which is indicative of A3G and A3A. The black dotted lines indicate the epithelial lining. C, Summary of APOBEC3 nuclear positivity in NSCLC development as assessed by immunohistochemistry. Percentage of cases scoring between 0<2%, 2<10%, 10<20%, 20<50% and 50<100% APOBEC3 positivity in each category are shown (N = 251 cases in total). The number of cases scoring >10% nuclear positivity was compared between each group (two-tailed Fisher’s exact test, ****P ≤ 0.0001). D, APOBEC3 gene expression during different stages of LUAD relative to normal tissue (N = 18 samples, linear mixed-effects model, •(blue dot)FDR ≤ 0.1, *FDR ≤ 0.05). Each dot represents a sample. The connected dots represent samples from the same patient. E, APOBEC3 gene expression during different stages of LUSC carcinogenesis. The boxplots represent median ± 1st and 3rd quartiles, P-values are FDR-adjusted for multiple comparison (N = 122 samples from 77 patients, linear mixed-effects model, •(blue dot)FDR ≤ 0.1, *FDR ≤ 0.05, **FDR ≤ 0.01, ***FDR ≤ 0.001). F, Number of SBSs as detected by whole-exome sequencing (WES) in 2 pre-invasive NSCLC lesions. Each color represents a different SBS signature. G, Number of SBSs in the 2 synchronous patient-matched pre-invasive and invasive NSCLCs that are ubiquitous between the pre-invasive and invasive lesion or private (i.e. exclusive) to the pre-invasive or invasive lesion, respectively. H, Heat maps show the distribution of APOBEC3- (red) and non-APOBEC3-mediated (grey) SBSs. Columns next to the heat maps show the SBS distribution, which are either present in both the pre-invasive and all invasive regions (orange), present in both the pre-invasive but not all invasive regions (yellow), private to the invasive lesion (blue) or private to the pre-invasive lesion (green). Driver mutations are shown next to the heat map, indicating in which region they are present (in non-APOBEC3 context, grey; in APOBEC3 context, red). I, Overview of the total number of driver mutations (bottom panel) and the number of driver mutations in the APOBEC3 context (top panel). Asterisks indicate significant enrichment for APOBEC3 signature mutations (two-tailed Fisher’s exact test, P ≤ 0.05). Scale bars, 100 μm. Scale bar in inset magnification, 25 μm.
We sought to confirm our immunohistochemical analysis by deciphering the timing of the full repertoire of APOBEC3 gene expression changes during cancer evolution in expression studies (see Supplementary Table 1) that assayed multiple morphological stages in the progression from normal tissue to established lung adenocarcinoma (LUAD) (33) or LUSC (34). Within the LUAD dataset (33), we observed increased expression of A3B in AIS and LUAD relative to normal lung tissue, whereas A3A expression decreased in the invasive stage relative to normal lung tissue (Fig. 1D; linear mixed-effects model, FDR ≤ 0.1; LUAD, FDR ≤ 0.05). Within the LUSC dataset (34), significant increases in A3A, A3B and A3F expression were detected in the stages from moderate dysplasia to invasive carcinoma compared to normal tissue samples (Fig. 1E; linear mixed-effects model A3A, FDR ≤ 0.05; A3B, FDR ≤ 0.01; A3F, FDR ≤ 0.05). Immunohistochemical quantification of A3A and A3B transcripts using BaseScope in a small set of samples, including normal, dysplastic, CIS and NSCLC tissue, confirmed an increase in A3A and A3B expression in CIS and NSCLC samples relative to normal lung tissue (Supplementary Fig. 1B-C). We then sought to investigate whether early APOBEC3 expression changes are specific to NSCLC. We re-analyzed multiple independent datasets of gene expression changes during breast cancer progression (35–37). Similar to LUSC, data from breast cancer progression studies, including analyses from normal to either DCIS or invasive ductal carcinoma (IDC), demonstrate an increased expression of A3B at the DCIS and IDC stages in 2 out of 3 datasets relative to normal breast tissue (Supplementary Fig. 1D–F; linear mixed-effects model, FDR ≤ 0.1; FDR ≤ 0.05; FDR ≤ 0.01). Taken together, these data suggest that APOBEC3 expression is dynamic and is upregulated during early lung and breast cancer development.
Next, we investigated the mutational signatures of APOBEC3-mediated mutagenesis in pre-invasive and invasive NSCLC (Fig. 1F and Supplementary Fig. 1G). In 2 patients from the TRACERx study in whom rare synchronous pre-invasive lesions and invasive tumors are clonally related (Fig. 1G), truncal mutations were found in the APOBEC3 context (Fig. 1H). A LATS1 driver event in an APOBEC3 context was ubiquitously found in the pre-invasive and invasive lesions of patient CRUK0077. Patient CRUK0235 had a PTEN driver in an APOBEC3 context detectable in the pre-invasive lesion and regions 1 and 4 of the invasive lesion, indicating early subclonal seeding of the primary tumor. However, we cannot exclude later subclonal diversification and seeding of the pre-invasive lesion with a PTEN mutant subclone. In addition, we observed subclonal APOBEC3 signature mutations that were unique (i.e. private) to the matched pre-invasive or invasive lesions in both patients (Fig. 1H), indicating that APOBEC3-mediated polyclonal diversification can occur at the pre-invasive stage prior to malignant transformation. Within the first 100 patients of the TRACERx cohort (1) we identified 487 driver mutations of which 71 (15%) were in an APOBEC3 context. In total 37 out of 100 tumors harbored at least 1 driver mutation in an APOBEC3 context, of which 26 tumors have clonal APOBEC3 driver events (Fig. 1I). These data strengthen our observations that APOBEC3 plays an important role in pre-invasive lung cancer mutagenesis.
To further investigate which APOBEC3 family members might be impacting NSCLC progression, we studied the association between APOBEC3 gene expression and progression-free interval (PFI) in the TCGA LUAD and LUSC datasets (Supplementary Fig. 1H–K). In earlier stage (I and IA) LUAD patients, we observed that higher A3B expression was associated with shorter PFI (Supplementary Fig. 1H; Wald test, A3B, P = 0.012), whereas this was not the case in later stage (IIIA, IIIB and IV) LUAD patients (Supplementary Fig. 1I). Within LUSC patients, higher A3A and A3F expression was associated with shorter PFI (Supplementary Fig. 1J; Wald test, A3A, P = 0.018; A3F, P = 0.012) but not in later stage (IIIA, IIIB and IV) LUSC patients (Supplementary Fig. 1K). No other APOBEC3 genes were associated with significantly worse PFI (Supplementary Fig. 1H–K). Altogether, our data suggest that APOBEC3 plays an important role in early lung and breast cancer development and progression.
APOBEC3 expression increases during either replication stress-associated senescence or proliferation
Previously, we have demonstrated that DNA replication stress can drive A3B expression (20). To explore the underlying basis of the high APOBEC3 expression observed in the pre-invasive NSCLC (Fig. 1), we investigated potential mechanisms that induce APOBEC3 expression prior to the establishment of invasive cancer. Pre-invasive lesions contain epithelial cells undergoing replication stress, and this could result in either senescence (38) or transformation and unchecked proliferation. Indeed, normal bronchial epithelium was negative for (phosphorylated) pRPA(S33), a common marker of replication stress, whereas > 75% of epithelial cells in all the examined pre-invasive lesions (N = 5 AAH, N = 13 AIS, N = 6 MIA, N = 15 CIS) were pRPA(S33)-positive (Fig. 2A). Furthermore, APOBEC3-staining of adjacent sections, revealed that almost all APOBEC3-positive cells were pRPA(S33)-positive (> 95%; Fig. 2B). We next sought to clarify a potential link between APOBEC3 with either senescence or proliferation.
Figure 2. APOBEC3 expression increases during either replication stress-associated senescence or proliferation.
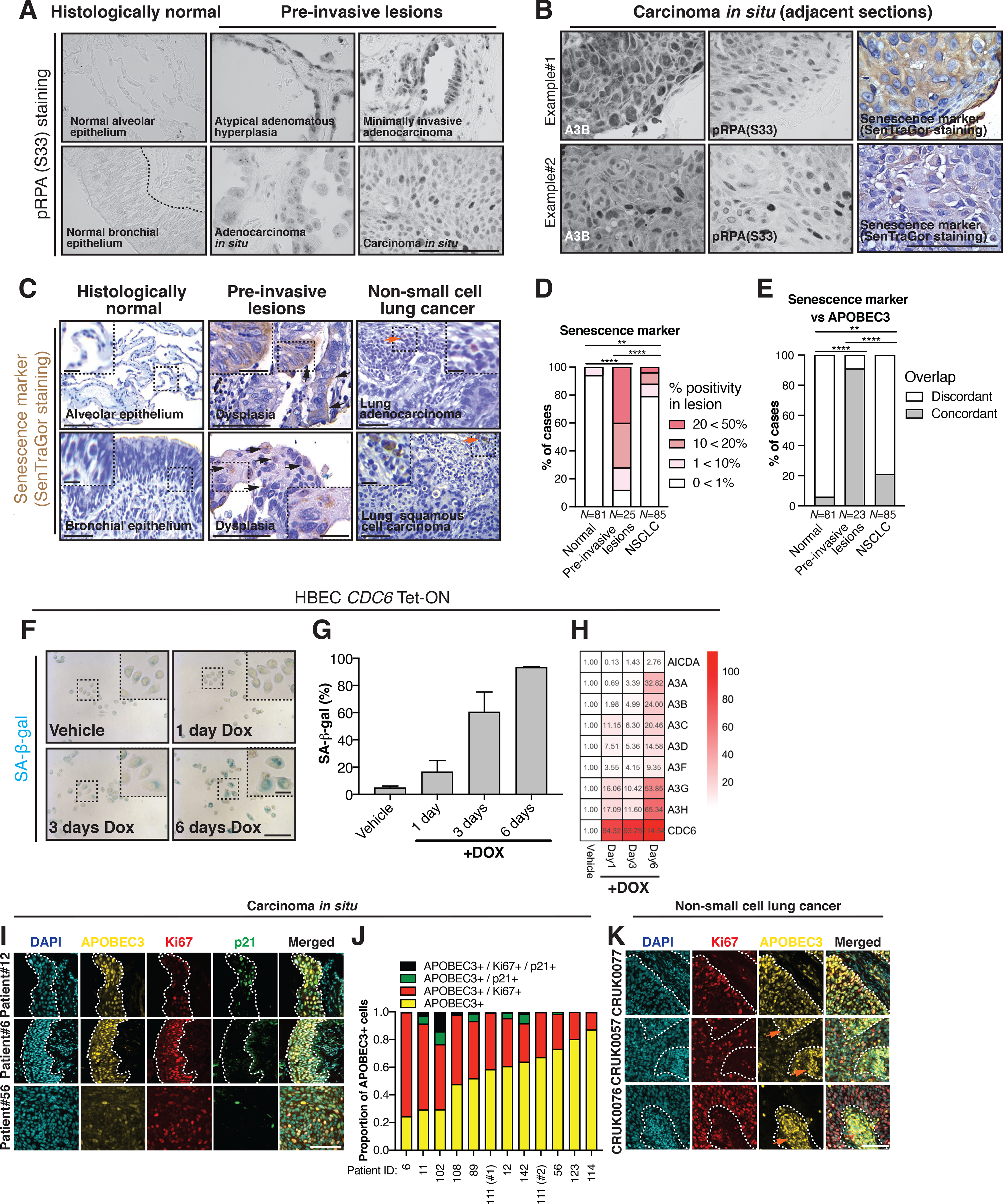
A, Representative examples of pRPA(S33) staining in different types of pre-invasive lesions. The following pre-invasive lesions were stained: N = 5 AAH, N = 13 AIS, N = 6 MIA and N = 15 CIS samples. B, Representative example of adjacent 4 μm thick sections of a CIS with pRPA(S33), SenTraGor and APOBEC3 immunohistochemistry. Immunohistochemistry was performed on a subset of pre-invasive lesions that contained enough tissue (N = 15 CIS samples). C, Representative examples of senescent cell detection by SenTraGor staining in normal lung epithelium, pre-invasive lesions and NSCLC. Black and orange arrowheads indicate SenTraGor-positive epithelial and stromal cells, respectively. D, Percentage of cases scoring 0<1%, 1<10%, 10<20%, 20<50% in each category. N = 191 samples (two-tailed Fisher’s exact test, **P ≤ 0.01; ****P ≤ 0.0001). E, APOBEC3-positive lesions were assessed for the presence of SenTraGor-positive cells. Concordance between SenTraGor and APOBEC3 staining (N = 189 samples, two-tailed Fisher’s exact test, **P ≤ 0.01; ****P ≤ 0.0001). Within the adjacent anti-APOBEC3 stained sections, 2 pre-invasive lesions were missing and thus were unevaluable. F, Representative images of SA-β-gal staining of HBEC CDC6 Tet-ON cells. G, Quantification of SA-β-gal positive cells. Results represent mean ± SD, N = 3 independent experiments. H, Relative changes in APOBEC3 gene expression compared to vehicle-treated condition as assessed by qRT-PCR. Results represent mean, N = 3 independent experiments. I, Representative examples of a triple immunofluorescence stain for APOBEC3, Ki67 and p21 in lung CIS samples. The dashed white line is drawn around the epithelium. J, Proportion of APOBEC3-positive cells that also stained positive for Ki67 and/or p21. Note patient 111 has been biopsied twice with an interval of 2 years. K, Representative examples of a double immunofluorescence staining for APOBEC3 and Ki67 (p21 was not evaluable) in NSCLC. Example regions within sections are shown originating from different patients with relatively high (CRUK077), moderate (CRUK0057) and low (CRUK0076) APOBEC3 immunofluorescence. The dashed white line separates the tumor cells from the tumor stroma. Note the nuclear APOBEC3 staining in epithelial cells, which is indicative of A3B and A3A staining. The orange arrows indicate stromal cells with cytoplasmic APOBEC3 staining, which is derived from A3G and A3A. Scale bars, 100 μm. Scale bars in inset magnification, 25 μm.
We identified senescent cells using a biotin-linked Sudan Black B analogue (SenTraGor™), which stains for lipofuscin-containing senescent cells (39) (N = 81 normal lung samples, N = 25 pre-invasive lesions, N = 85 NSCLC; Fig. 2C–D). In addition, we stained for APOBEC3 protein expression on consecutive sections of FFPE clinical samples (N = 81 normal lung samples, N = 23 pre-invasive lesions, N = 85 NSCLC; Fig. 2E). As expected, the SenTraGor staining was more prevalent in pre-invasive lesions (> 10% positivity in 18 out of 25 cases) relative to normal lung epithelium (> 10% positivity in 0 out of 81 cases) and invasive carcinoma (> 10% positivity in 10 out of 85 cases) (Fig. 2B and D; two-tailed Fisher’s exact test, P ≤ 0.0001). The APOBEC3-positive lesions were enriched with patches of overlapping SenTraGor-positive cells in 5 out of 81 histologically normal regions (6%), 21 out of 23 evaluable pre-invasive lesions (91%) and 18 out of 85 carcinomas (21%) as evaluated by the adjacent sections (Fig. 2E; two-tailed Fisher’s exact test, P ≤ 0.01, P ≤ 0.0001).
We hypothesized that replication stress-associated senescence may underlie this observed APOBEC3 upregulation, so we next monitored APOBEC3 expression using a previously described epithelial cell line model of oncogene-induced senescence (40). In the human bronchial epithelial cell (HBEC) CDC6 Tet-ON cell line, induction of CDC6 overexpression through prolonged doxycycline treatment, triggers replication stress and subsequent senescence (40). As expected upon doxycycline exposure, HBEC CDC6 Tet-ON cells flattened, contained more vacuoles and exhibited increased senescence-associated β-gal (SA-β-gal) staining (Supplementary Fig. 2A; Fig. 2F–G). Quantitative reverse transcription PCR (qRT-PCR) of APOBEC3 genes in HBEC CDC6 Tet-ON cells, revealed an over 15-fold increase in several APOBEC3 transcripts including A3A, A3B, A3C, A3G and A3H mRNA expression by day 6 (Fig. 2H). The CDC6-induced increase in A3B transcript and protein levels were abrogated after 24-hour Chk1 kinase inhibition (Supplementary Fig. 2B–C), indicating that the increase in gene expression is dependent on the activation of a Chk1-mediated replication stress checkpoint (41). Interestingly, A3B protein levels in senescence-escaped HBEC-CDC6 cells returned to baseline (Supplementary Fig. 2D–E), potentially indicating adaptation.
To further assess whether senescence drives or is merely coincident with APOBEC3 expression, we utilized a recent study showing that microRNA146a (miR146a) is strongly upregulated in senescent cells (42). Ectopic expression of miR146a-EGFP, a reporter of senescence (42), in H2122 and H1944 lung cancer cell lines was carried out to investigate the relationship between senescence and APOBEC3 expression. We treated these cell lines with a high dose of hydroxyurea (HU; 1 mM, 1–4 days) to induce replication stress and prolonged fork arrest in order to investigate the dynamics of senescence and APOBEC3 gene expression. In addition, we used a high dose of irradiation (IR; 8 Gy IR, 4 days recovery) as a positive control in order to initiate senescence through an excess of acute DNA double strand breaks rather than DNA replication stress. Both HU- and IR-induced senescence-associated cell morphological changes and increased SA-β-gal staining (Supplementary Fig. 2F–I) increased the miR146a-EGFP signal (Supplementary Fig. 2J–K) and reduced EdU incorporation (Supplementary Fig. 2L–M). Interestingly, HU but not IR increased APOBEC3 expression, despite both conditions inducing senescence at day 4 (Supplementary Fig. 2N–O). These data suggest that the induction of APOBEC3 expression and senescence are parallel pathways driven by replication stress.
The percentage of APOBEC3-positive cells was greater than that of senescent cells in pre-invasive NSCLC (compare Fig. 1A–C and Fig. 2B–D), implying that despite overlapping in many cases (Fig. 2E), most APOBEC3-positive cells were non-senescent. To elucidate whether proliferation could instead be implicated in APOBEC3 expression, a triple immunofluorescence stain for APOBEC3, Ki67 (indicative of proliferation) and the cyclin-dependent kinase inhibitor p21 (indicative of cell cycle arrest) in 12 CIS samples with available tissue (from (29)) showed that on average 39% (range 13%−75%) of APOBEC3-positive cells were also positive for Ki67, whereas only 4.8% (range 0.2%−23.6%) were positive for p21 (Fig. 2I–J). For 79 NSCLC samples with available tissue from the TRACERx 100 cohort (1), a double immunofluorescence stain for APOBEC3 and Ki67 (p21 was not evaluable) was performed (Fig. 2K). Similar to the CIS samples, a large proportion of APOBEC3-positive cells in NSCLC were also Ki67-positive (Fig. 2K; average 30% of A3B-positive cells were Ki67-positive, range 5%−70%). Concordant with our earlier observation (Fig. 1C), pre-invasive lesions contained more APOBEC3-positive epithelial cells than NSCLCs (Supplementary Fig. 2P; N = 12 CIS, N = 79 NSCLC, two-tailed Mann-Whitney test, P ≤ 0.0001).
To clarify which APOBEC3 gene family member best correlates with nuclear APOBEC3 immunofluorescence, we explored matched RNAseq-derived APOBEC3 expression within the TRACERx 100 cohort (1). We observed a stronger correlation with A3B (Supplementary Fig. 2Q; Spearman correlation, N = 54, rho = 0.41; P = 0.0023) relative to A3A mRNA expression (Supplementary Fig. 2R; Spearman correlation, N = 54, rho = 0.16; P = 0.24). There was no significant correlation between the percentage APOBEC3-positive tumor cells and the subclonal, clonal nor total number of APOBEC3 signature mutations (SBS2/SBS13) in either the CIS (from (29)) or NSCLC samples (from (1)) (Supplementary Fig. 2S; Spearman correlation, N = 12 CIS and N = 79 NSCLC, rho ≤ 0.17, P ≥ 0.14).
Next, to test whether cell proliferation was required for APOBEC3 expression in the absence of DNA damage, we densely seeded RPE-1 cells to facilitate contact inhibition. Contact inhibition in RPE-1 cells strongly reduced A3B mRNA expression, whereas subconfluent re-seeding and cell cycle progression elicited a rebound of A3B mRNA expression towards baseline (Supplementary Fig. 2T–U). Taken together, these data suggest that replication stress and cell cycle progression drive A3B expression. In contrast, the senescence program does not induce A3B expression and is a parallel pathway downstream of replication stress. Thus, our data suggest that APOBEC3 is upregulated early during breast cancer and NSCLC development with DNA replication stress being a likely driver of APOBEC3 expression (Fig. 1 and 2).
A3B exacerbates DNA replication stress
We next reasoned that, since APOBEC3 deaminates cytosines on ssDNA, it could itself induce replication stress as previously described (43–46), pointing towards a possible feed-forward loop driving CIN. Since A3A and A3B are the prime candidate APOBEC3 genes implicated in cancer (2,17–19,47), we generated A3A and A3B single-gene knockouts (KO) in the immortalized human type II pneumocyte cell line stably expressing a 4-hydroxytamoxifen (4-OHT)-regulatable oncogenic RAS chimeric protein (48) (from now on referred to as TIIP; Supplementary Fig. 3A–F). We investigated whether A3A and A3B could contribute to replication stress and ensuing accumulation of under-replicated DNA. Consistent with this hypothesis, in unperturbed proliferating cells, TIIP A3B-KO cells showed a strong reduction of pRPA(S33) and pChk1 (S345) compared to TIIP wild type (WT) cells, confirming previous observations (43–46), whereas this reduction was not observed in TIIP A3A-KO cells (Supplementary Fig. 3E). Furthermore, TIIP A3B-KO cells showed higher fork extension rates, suggesting that A3B might promote fork slowing, exacerbating replication stress (Fig. 3A–B; two-tailed Mann-Whitney test, P ≤ 0.0001). Concordant with these data, we detected an increased fork extension rate also in the LUSC cell line H520, using siRNA-mediated knockdown of A3B (Fig. 3C; two-tailed Mann-Whitney test, P ≤ 0.01; Supplementary Fig. 3G) and a reduction in fork extension rate upon A3B overexpression in HEK293 cells (Fig 3D; two-tailed Mann-Whitney test, P ≤ 0.0001; Supplementary Fig. 3H–I). In addition, we found that in the presence of mild replication stress (0.2 μM aphidicolin, 24 hours), TIIP A3B-KO cells accumulated fewer FANCD2 foci in prometaphase than TIIP WT or TIIP A3A-KO cells (Fig. 3E–F; two-tailed Mann-Whitney test, P ≤ 0.001) (49). In contrast, A3B overexpression increased the number of FANCD2 foci in prometaphase (Supplementary Fig. 3J; two-tailed Mann-Whitney test, P ≤ 0.0001).
Figure 3. APOBEC3B contributes to the accumulation of under-replicated DNA through replication stress.
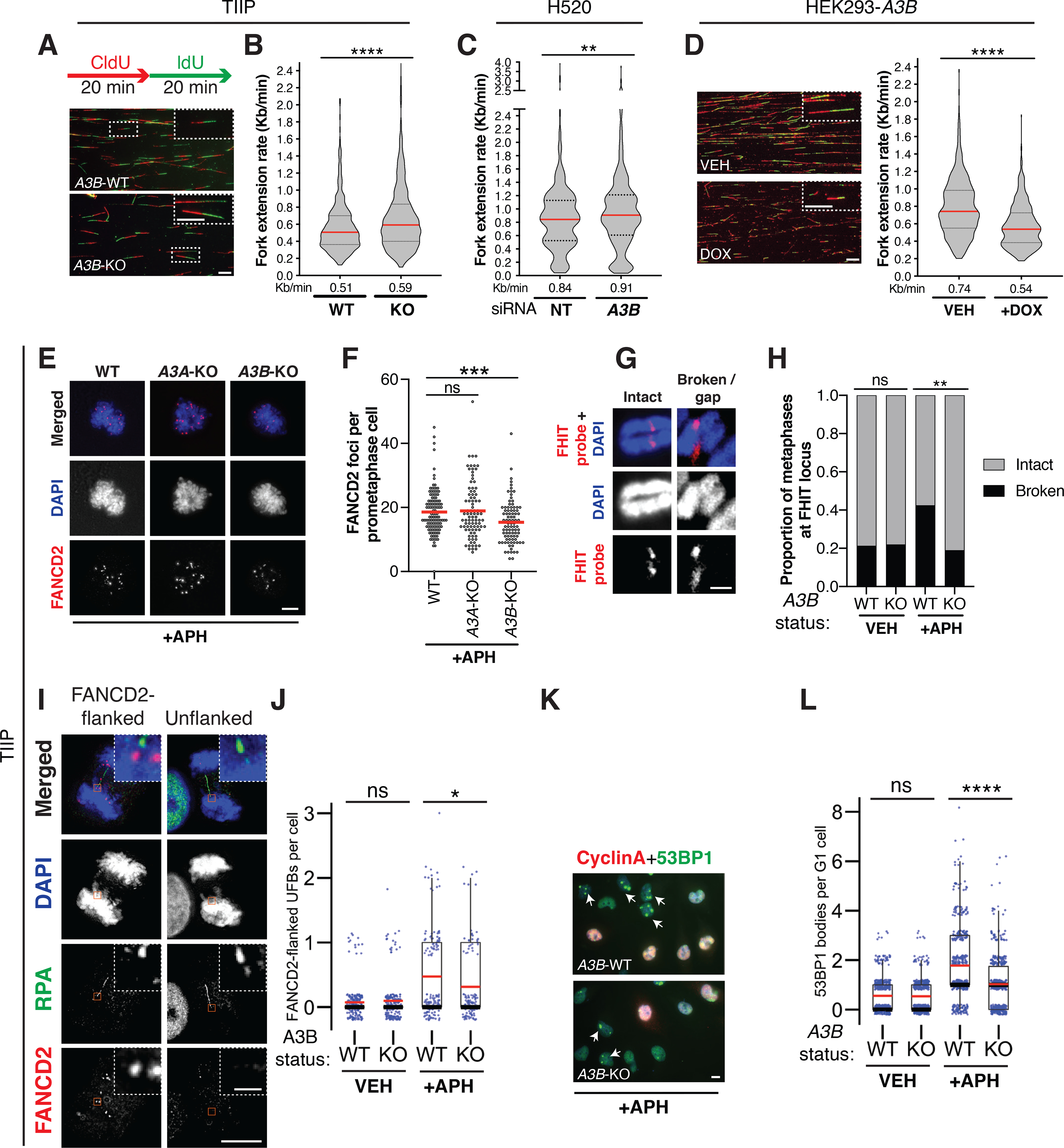
A-D, 20 min sequential labeling pulses of CIdU (red) and IdU (green) were subjected to DNA fiber stretching analysis. Representative images of DNA fibers are shown. DNA fiber stretching analysis of TIIP (WT and A3B-KO#clone13), H520 and HEK293-A3B cells. A minimum of 530 tracts were measured per condition. Results represent median (red) ± 1st and 3rd quartiles (black), N = 3 independent experiments (two-tailed Mann-Whitney test, **P ≤ 0.01; ****P ≤ 0.0001). Scale bars, 10 μm and in inset magnification, 100 μm. E, Representative images of FANCD2 foci in TIIP prometaphase cells. F, A minimum of 75 prometaphase cells were assessed per condition. Results represent mean, N = 3 independent experiments (two-tailed Mann-Whitney test, ***P ≤ 0.001). Scale bar, 10 μm. G, Representative images of chromosome 3 without and with a break/gap at the FHIT locus. Scale bar, 1 μm. H, Proportion of cells with an intact or broken chromosome 3 at the FHIT locus. At least 68 metaphases were evaluated per condition. N = 2 independent experiments (two-tailed Fisher’s exact test, **P ≤ 0.01). I, Representative images of RPA-positive ultrafine bridges (UFBs) either unflanked or flanked by FANCD2-positive foci. Scale bar, 10 μm and in inset magnification, 1 μm. J, Number of FANCD2-flanked UFBs per anaphase cell. A minimum of 115 anaphase cells were assessed per condition. Boxplots represent mean (red), median (black) ± 1st and 3rd quartiles (box), N = 3 independent experiments (two-tailed Mann-Whitney test, *P ≤ 0.05). K, Representative images of 53BP1 nuclear bodies in the G1 cell cycle phase (53BP1-positive foci in Cyclin A-negative cells) in TIIP cells. Scale bar, 10 μm. L, A minimum of 400 cells were assessed per condition. Boxplots represent mean (red), median (black) ± 1st and 3rd quartiles (box), N = 3 independent experiments (two-tailed Mann-Whitney test, ****P ≤ 0.0001).
Consistent with the attenuation of DNA replication stress following A3B deletion, after treatment with 0.2 μM aphidicolin TIIP A3B-KO cells presented with fewer metaphase breaks at the FHIT common fragile site locus (Fig. 3G–H, two-tailed Fisher’s exact test, P ≤ 0.01), fewer FANCD2-flanked ultrafine bridges (UFBs) (Fig. 3I–J; two-tailed Mann-Whitney test, P ≤ 0.05) and fewer 53BP1 nuclear bodies in the G1 cell cycle phase relative to TIIP WT cells (Fig. 3K–L and Supplementary Fig. 3K; two-tailed Mann-Whitney test, P ≤ 0.0001). After 24 hours low dose (0.2 μM) aphidicolin treatment, the percentage of TIIP A3B-KO cells in the G1 cell cycle phase was greater than that of TIIP WT cells (Supplementary Fig. 3L; two-tailed unpaired t-test, P ≤ 0.05), suggesting cells might cope better without A3B-induced genome instability, allowing more cells to complete the cell cycle. Similarly, confirming the findings in TIIP cells, we observed that U2OS A3B-KO cells (Supplementary Fig. 3M–O) also had fewer 53BP1 nuclear bodies in the G1 cell cycle phase relative to U2OS WT cells after 24h low dose (0.2 μM) aphidicolin treatment (Supplementary Fig. 3P; two-tailed Mann-Whitney test, P ≤ 0.0001). Since TIIP cells were immortalised and therefore do not properly senesce (see Materials and Methods), we also used the U2OS A3B-KO cells to investigate whether A3B-KO cells were still capable of senescing. Many U2OS A3B-KO cells harbored SA-β-gal-positive signals after 4 days HU treatment (0.2mM and 1mM), suggesting senescence can occur in the absence of A3B (Supplementary Fig. 3Q–R; two-tailed unpaired t-test, P ≤ 0.05).
Analogous to aphidicolin, 4OHT-mediated RAS induction (3 days) resulted in fewer DNA damage related foci in TIIP A3B-KO relative to TIIP WT cells (Supplementary Fig. 3S–T, two-tailed Mann-Whitney test). Taken together, these results suggest A3B exacerbates DNA replication stress, likely contributing to CIN.
A3B exacerbates chromosomal instability and promotes aneuploidy
Since under-replicated regions of DNA contribute to CIN (49), we investigated whether A3B might contribute to CIN. We employed the ImageStream cytometer together with centromere-specific FISH probes (hereafter ImageStream FISH) to quantify aneuploidy frequencies after 24 hours of low dose (0.2 μM) aphidicolin exposure. A centromeric chromosome 15 probe was used, overcoming the bias of the elevated missegregation observed with some larger chromosomes (50). A significant decrease in cells deviating from the modal chromosome 15 signal was detected in TIIP A3B-KO cells compared to TIIP WT cells (Fig. 4A; two-tailed unpaired t-test, P ≤ 0.05).
Figure 4. APOBEC3B exacerbates chromosomal instability and promotes aneuploidy.
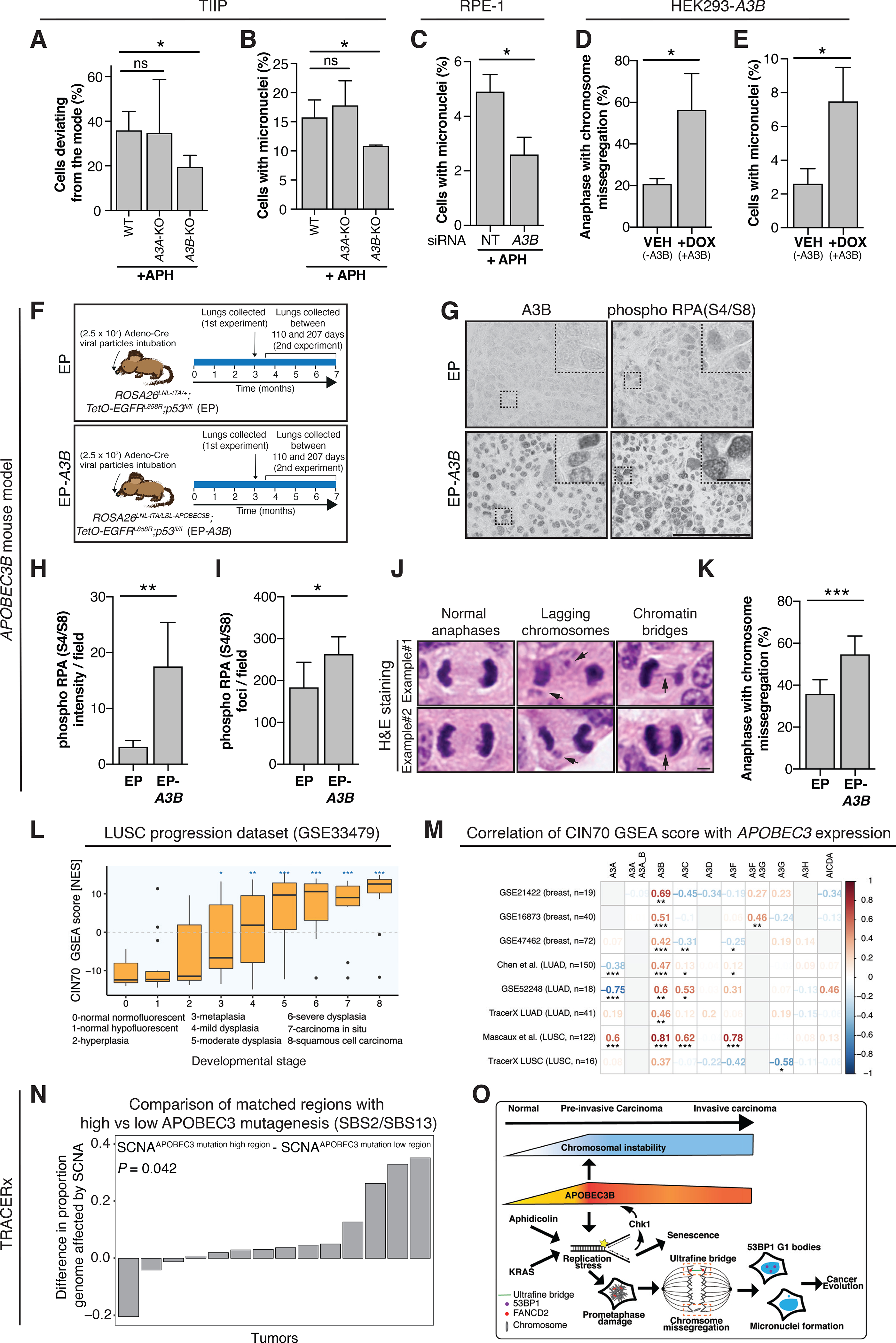
A, Percentage of cells deviating from modal chromosome 15 as assessed by ImageStream-FISH. Approximately 4000 cells were assessed per condition. Results represent mean ± SD, N = 4 independent experiments (two-tailed unpaired t-test, *P ≤ 0.05). B-C, Percentage of cells with micronuclei. A minimum of 450 cells per condition were assessed. Results represent mean ± SD, N = 3 independent experiments (two-tailed unpaired t-test, *P ≤ 0.05). D, A minimum of 130 anaphases were assessed per condition. Results represent mean ± SD, N = 3 independent experiments (two-tailed unpaired t-test, *P ≤ 0.05). E, Percentage of cells with micronuclei. A minimum of 450 cells per condition were assessed. Results represent mean ± SD, N = 3 independent experiments (two-tailed unpaired t-test, *P ≤ 0.05). F, Tumors were induced in the lungs of EGFRL858R;Tp53flox/flox (EP; N = 7) or EGFRL858R;Tp53flox/flox;R26LSL-A3B/LSL-tTA mice (EP-A3B; N = 8, 2 combined experiments) (Fig. 4F). Lungs were harvested either at 3 months in 1 experiment or at termination in an additional experiment (between 110 and 207 days). G, Representative examples of A3B and pRPA(S4/S8) immunohistochemistry in lung cancers derived from EP and EP-A3B mice. Scale bar, 100 μm and in inset magnification, 25 μm. H, Intensity of pRPA(S4/S8) staining. Results represent mean ± SD, N = 6 mice per group (two-tailed unpaired t-test, **P ≤ 0.01). I, Number of pRPA(S4/S8) foci per field. Results represent mean ± SD, N = 6 mice per group (two-tailed unpaired t-test, *P ≤ 0.05). J, Representative images of anaphase cells that were assessed for the presence of lagging chromosomes and chromatin bridges within H&E sections of EP and EP-A3B lung cancers. Scale bar, 5 μm. K, Results represent mean ± SD, N = 7 EP and N = 8 EP-A3B mice (Mann-Whitney test, ***P ≤ 0.001). L, CIN70 GSEA scores at different stages of LUSC. Boxplots represent median ± 1st and 3rd quartiles, (linear mixed-effects model, *FDR ≤ 0.05, **FDR ≤ 0.01, ***FDR ≤ 0.001). M, Heatmap of the Spearman correlation coefficients between the CIN70 GSEA score and APOBEC3 mRNA expression per dataset. (Spearman correlation, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). N, For tumors of the lung TRACERx cohort, the proportion of the genome affected by SCNAs of regions with a higher number of APOBEC3 signature mutations were compared to those with a relatively lower number of APOBEC3 signature mutations (two-tailed paired Wilcoxon test). Only tumors with a significant difference in APOBEC3 mutations between the 2 tumor regions were considered in this analysis (N = 14). Each comparison is confined to within-tumor regions and is represented by a bar. O, Model of A3B contributing to CIN early in lung cancer development. A3B exacerbates aphidicolin and KRAS-induced replication stress, hindering the complete replication of the genome. Persistent regions of under-replicated DNA lead to the formation of UFBs during mitosis. After mitosis the resolved UFBs contribute to the formation of 53BP1 nuclear bodies and missegregated chromosomes contribute to the formation of micronuclei.
Furthermore, after 24 hours low dose (0.2 μM) aphidicolin exposure, A3B depleted cells had fewer micronuclei (Fig. 4B; two-tailed unpaired t-test, P ≤ 0.05), in both RPE-1 cells through siRNA-mediated knockdown of A3B (Fig. 4C, two-tailed unpaired t-test, P ≤ 0.05; Supplementary Fig. 4A) and in TIIP A3B-KO cells in the presence of 40HT (3 days RAS) (Supplementary Fig. 4B, two-tailed Mann-Whitney test, P ≤ 0.0001). In contrast, A3B overexpression increased the percentage of cells with chromosome missegregation and micronuclei (Fig. 4D–E, two-tailed unpaired t-test, P ≤ 0.05).
To test whether A3B overexpression could contribute to CIN in vivo, we combined a Cre-inducible model for human A3B expression (Rosa26::LSL-A3B/LSL-A3B) with an EGFRL858R;Tp53flox/flox driven lung cancer mouse model and a Cre-inducible tetracycline-controlled transactivator (R26LNL-tTA) (see Materials and Methods and (51,52)). Tumors were induced in the lungs of EGFRL858R;Tp53flox/flox (EP; N = 7) or EGFRL858R;Tp53flox/flox;R26LSL-A3B/LSL-tTA (EP-A3B; N = 8, 2 combined experiments) (Fig. 4F). Lungs were harvested either at 3 months in 1 experiment or at termination in an independent experiment (between 110 and 207 days). As expected, lung cancers from EP-A3B mice stained positive for A3B in contrast to those from EP mice (Fig. 4G). There was significantly more pRPA(S4/S8) staining and foci (indicative of DNA damage) in lung cancers from EP-A3B mice relative to EP mice (Fig. 4G–I, two-tailed unpaired t-test, P ≤ 0.01; P ≤ 0.05). We examined haematoxylin and eosin-stained (H&E) sections obtained from the mouse lung cancers for chromosome missegregation events (Fig. 4J). The EP lung cancer cells missegregated in 36%, whereas the EP-A3B lung cancer cells missegregated in 55% (Fig. 4K; Mann-Whitney test, P ≤ 0.001). These data suggest that A3B can contribute to CIN in developing lung cancers in vivo.
To assess the clinical relevance of our findings, we examined published gene expression datasets of pre-invasive lung and breast cancer in addition to the TRACERx lung study involving the first 100 patients (1). The enrichment of the CIN70 gene signature, a surrogate measure of CIN (53), increased during pre-invasive LUSC evolution (Fig. 4L; two-tailed Mann-Whitney test, FDR ≤ 0.05). Unlike other APOBEC3 family members, A3B expression correlated significantly with the CIN70 enrichment score in almost all examined datasets (Fig. 4M; Spearman correlation, P ≤ 0.05, P ≤ 0.01, P ≤ 0.001). A3A, A3C and A3F expression significantly positively correlated with the CIN70 gene signature in only a few datasets (Fig. 4M; Spearman correlation, P ≤ 0.05, P ≤ 0.01, P ≤ 0.001).
Additionally, we examined diagnostic H&E samples from the TRACERx 100 cohort (1) with microscopy for chromosome missegregation events (Supplementary Fig. 4C–D) and investigated potential associations with patient-matched APOBEC3 expression profiles. Only H&E sections with ≥ 10 anaphases were considered (9 diagnostic H&E sections had <10 anaphases and were not considered). Unlike any other APOBEC3 family members, only A3B expression significantly, albeit moderately, correlated with the percentage of anaphases with chromosome missegregation (Supplementary Fig. 4D; Spearman correlation, N = 58, rho = 0.27; P = 0.038). Within the same cohort of patients, again only A3B expression was significantly associated with the proportion of the genome affected by somatic copy number alterations (SCNAs) (Supplementary Fig. 4E; Spearman correlation, N = 58, rho=0.38; P = 0.0030). However, the percentage of APOBEC3-positive tumor cells did not correlate with the percentage of anaphases with chromosome missegregations (Supplementary Fig. 4F; Spearman correlation, N = 70, rho = 0.047; P = 0.70) nor the proportion of the genome affected by SCNAs (both clonal and subclonal) (Supplementary Fig. 4G; Spearman correlation, N = 72 (for 7 patients we could not derive SCNA measures), rho = 0.11; P = 0.35). This might be explained by the antibody detecting both nuclear A3A and A3B, whereas RNAseq enables reliable separation of A3A and A3B transcripts.
Finally, we examined the relationship between the burden of APOBEC3-mediated mutagenesis at a regional level within the TRACERx 100 cohort (1). Only tumors with a significant difference in APOBEC3 mutations between the 2 tumor regions have been considered in this analysis with each comparison being confined to within-tumor regions (N = 14). We found that regions with higher numbers of APOBEC3 signature mutations had a significantly higher proportion of their genomes affected by SCNAs relative to tumor regions from the same patient with lower numbers of APOBEC3 signature mutations (Fig. 4N; two-tailed paired Wilcoxon test, N = 14, P = 0.042). Taken together, these data support the role of A3B induction in pre-invasive disease due to DNA replication stress, exacerbating replication stress and driving CIN (Fig. 4O).
DISCUSSION
APOBEC3 expression is upregulated in a wide range of cancers including breast cancer and NSCLC (4,5). A3A and A3B are 2 prominent candidates thought to be responsible for the APOBEC3-mediated mutational signature in numerous cancer types (2,4,5). While recent next-generation sequencing efforts have detected APOBEC3 signature mutations within pre-invasive LUAD (25–27), these studies have not analyzed the basis for APOBEC3 expression dynamics and the specific contribution of APOBEC3 to genome instability from pre-invasive to invasive disease. Recent studies in breast cancer identified that A3B mRNA is upregulated at the DCIS stage (24,54), but these studies did not investigate the entire repertoire of APOBEC3 genes. Here, we took an unbiased approach to investigate mRNA expression changes of all APOBEC3 genes during breast cancer and NSCLC evolution. Our immunohistochemistry data confirmed overexpression of APOBEC3 in pre-invasive lesions relative to normal lung epithelium and strongly suggest that APOBEC3 protein expression peaks during the pre-invasive stage of NSCLC development. A part of the heterogeneity in APOBEC3 protein expression found in cancer could potentially be explained by SCNAs driving downstream changes in protein abundance (9) or associations with the cell cycle (55). The APOBEC3 nuclear staining likely originates from both A3B and A3A proteins, in contrast the cytoplasmic signal has been reported to derive from A3A and A3G (30,32). Furthermore, through sequencing of synchronous pre-invasive and invasive lesions in 2 TRACERx patients, we observed both ubiquitous and private APOBEC3 signature mutations between the matched lesions. Additionally, both patients had a driver event in an APOBEC3 context that was present in the pre-invasive and invasive lesions. Given our findings that APOBEC3 expression occurs early in pre-invasive breast and NSCLC evolution and the role of APOBEC3 in catalyzing CIN, combined with our recent findings that tumor suppressor gene (TSG) losses are commonly early truncal events in tumor evolution (56), we suggest that APOBEC3 induction following DNA replication stress may drive the early onset of CIN, fuelling clonal TSG copy number loss events. In either scenario, our data suggest that APOBEC3 plays a role in driving genome instability and diversification in pre-invasive disease, contributing to cancer evolution.
Previously, we have demonstrated that replication stress can drive APOBEC3 expression in breast cancer (20). We now demonstrate that APOBEC3-positive pre-invasive lesions are enriched for a marker of senescence (SenTraGor) as wells as a marker of replication stress (pRPA(S33)). Our in vitro experiments suggest that replication stress drives APOBEC3 expression and senescence independently. Interestingly, U2OS A3B-KO cells had more SA-β-gal staining after prolonged replication fork arrest, suggesting A3B may support senescence bypass. At the very least, A3B does not appear to be required for senescence induction.
Consistent with a functional role for replication stress in driving A3B expression, Chk1 kinase inhibition reversed CDC6-induced A3B protein levels. Note that the profile of the induced APOBEC3 genes and the dynamics of APOBEC3 induction can differ between experimental conditions. Despite APOBEC3-positive cells having patches of SenTraGor staining, the majority of these cells were in fact SenTraGor-negative. A triple immunofluorescence staining for APOBEC3, Ki67 and p21 in lung CIS samples, showed that APOBEC3-positive cells were more often Ki67-positive than p21-positive. A strong association between Ki67 and A3B was recently also reported in HPV-negative oral epithelial dysplasias and head and neck cancers (57). Interestingly, here we found that APOBEC3 protein levels did not correlate with the number of APOBEC3 signature mutations. These data indicate that the sum of A3A and A3B protein levels may not fully reflect the history of APOBEC3-mediated mutagenesis. This is in line with observations that APOBEC3-mediated mutagenesis can occur in bursts (58). In summary, our immunohistochemistry data together with in vitro experiments point towards replication stress and cell cycle progression driving A3B expression. In contrast, the senescence program does not induce A3B expression and is a separate pathway downstream of replication stress (Fig. 4O).
Interestingly, our data supports a model whereby A3B promotes the accumulation of under-replicated DNA through replication stress. It appears that A3B and likely other APOBEC3 genes as well, including A3A (59), exacerbate replication stress in the presence of mildly elevated levels of replication stress, hinting towards a positive feed-forward loop (Fig. 4O). Similar to our observations of A3B inducing replication stress, Mehta and colleagues have described that A3A can induce replication fork slowing. The initiating factor for this feed-forward loop might be caused by endogenous factors, such as oncogene-induced replication stress (60) or exogenous factors such as tobacco smoking (61). These observations are particularly intriguing in light of a recent study in yeast, showing that A3B causes 9- to 28-fold more mutations in engineered yeast strains with low levels of replicative DNA polymerases (62).
In conclusion, our in vitro and in vivo experimental data together with clinical evidence, suggest that A3B induction early in tumorigenesis, exacerbates both mutational as well as large-scale chromosomal copy number diversity, providing a potent evolutionary fuel for selection and cancer adaptation.
MATERIALS AND METHODS
Ethical approval
Approval for study (1) was provided by the London Camden & Kings Cross Research Ethics Committee (13/LO/1546). Approval for study (29) was provided by the UCL/UCLH Local Ethics Committee (06/Q0505/12 and 01/0148). Approval for using tissue specimens from a Danish cohort of NSCLC was provided by the local ethical committee of Rigshospitalet, Copenhagen University Hospital and by the Danish Capital Region’s Committee for Health Research Ethics (H-15008619).
All patients provided written informed consent and the studies were performed and all relevant ethical regulations were followed. These studies were conducted in accordance with recognized ethical guidelines in accordance with the Declaration of Helsinki.
Hybrid histochemistry / Immunohistochemistry
Sections were stained from previously initiated cohorts of NSCLC patients, namely from TRACERx (1), patients with pre-invasive LUSC lesions undergoing surveillance (29), and a Danish cohort of NSCLC.
For APOBEC3 and pRPA(S33) staining of deparaffinized sections, we employed our established sensitive immunohistochemical staining protocol including antigen unmasking in citrate buffer (pH 6) using microwave (15 min), followed by overnight incubation with the primary rabbit monoclonal anti-APOBEC3 antibody (5210–87-13) (30) and the primary rabbit polyclonal anti-RPA2(S33) antibody (NB100–544, Novus Biologicals), and the ensuing processing by the indirect streptavidin-biotin-peroxidase method using the Vectastain Elite kit (Vector Laboratories, Burlingame, CA, USA) and nickel-sulphate-based chromogen enhancement detection as previously described, without nuclear counterstaining (63). The primary rabbit polyclonal anti-RPA2(S33) antibody was not reactive against murine tissue, therefore the rabbit anti-RPA2 S4/S8 (NBP123017, Novus Biologicals) was used instead.
For senescence detection by SenTraGor™, a GL13-based (a biotinylated Sudan Black-B chemical analogue that specifically reacts against lipofuscin) hybrid histochemical/immunohistochemical method was employed, performed as previously published (39). Results were evaluated independently by two experienced oncopathologists, and scored on at least 400 cells per section, based on percentage of cells with nuclear positivity of APOBEC3, or cytoplasmic positivity by the SenTraGor method, respectively. A similar quantitative method has been used and described in previous publications (20,63). Representative images are shown in Figures 1 (APOBEC3) and 2 (senescence, pRPA2(S33), APOBEC3), and scores categorized as indicated in their respective legends.
The pRPA2(S4/S8) immunohistochemistry images from the mouse lung cancers were analyzed using Image J. The signal was assessed on the representative images derived from 6 tumors without inducing the engineered human A3B and 6 tumors expressing A3B, compared with the negative control staining (the sections from tumors without induced A3B, stained with the anti-APOBEC3 antibody: see upper left panel in Fig. 4G for an example of such negative control) as a background staining. The immunohistochemistry signals were inverted and the threshold adjusted equally for each image (64). Both the total intensity of the signal was measured, and the number of foci detected (particle size above 20 pixels).
BaseScope
BaseScope was performed according to the manufacturers’ instructions (Biotechne). BaseScope™ probe Hs-APOBEC3A-2zz-st1 (701261) was used for detecting A3A transcripts and probe Hs-APOBEC3B-1zz-st1-C2 (701271-C2) was used for detecting A3B transcripts.
Whole exome sequencing of patient-matched synchronous pre-invasive lesions and invasive NSCLC
Two patients, enrolled in the TRACERx study, presented with driver mutations in the APOBEC3 context that were both present in the pre-invasive and invasive NSCLC lesions (1). Pre-invasive and invasive lesions were considered to be clonally related if there were ≥ 20 mutations shared. Pre-invasive lesions were identified from archival formalin-fixed paraffin-embedded blocks. These 2 TRACERx cases were each diagnosed with synchronous CIS and LUSC. Laser capture microdissection was performed to sample pre-invasive lesions, followed by DNA extraction using GeneRead DNA FFPE Kit (QIAGEN). A similar strategy for DNA quality control and WES protocols was used as previously described (1). SBS mutational signatures were called in the pre-invasive and invasive samples as described previously (1). To account for FFPE induced artefacts, we removed C>T SBSs at CpG sites that had a variant count less than 10.
TRACERx RNAseq
The RNAseq data has been described elsewhere (65). Median values from the regional RNAseq data were used per patient.
Cell culture
The H2122, H1944, H520 lung cancer cell lines and the U2OS and RPE-1 cell lines were obtained from Cell Services at The Francis Crick Institute, UK. H2122, H1944 and H520 cells were cultured in RPMI 1640 media (Thermo Fisher Scientific), supplemented with 10% foetal bovine serum and 1/10,000 units of penicillin-streptomycin (Sigma Aldrich) and with L-glutamine (Thermo Fisher Scientific). RPE-1 and U2OS cells were cultured in DMEM media (Thermo Fisher Scientific), supplemented with 10% foetal bovine serum and 1/10,000 units of penicillin-streptomycin (Sigma Aldrich) and L-glutamine (Thermo Fisher Scientific). TIIP-ER-KRAS V12 (referred to as TIIP) cells were kindly provided by J. Downward (The Francis Crick Institute, London, UK) (48). The process of TIIP cell derivation and immortalisation has been described elsewhere (66). TIIP cells were cultured in DMEM/F-12 without phenol red (Thermo Fisher Scientific), supplemented with charcoal-stripped 10% foetal bovine serum and 1/10,000 units of penicillin-streptomycin (Sigma Aldrich) and with L-glutamine (Thermo Fisher Scientific). The generation of HBEC CDC6 Tet-ON cells is described elsewhere (40). These cells were maintained in keratinocyte serum-free medium (#17005042, Thermo Fisher Scientific) supplemented with 25 mg Bovine Pituitary Extract and 2.5 μg EGF. All the cell lines have been validated by short tandem repeat (STR) profiling and regularly tested for the presence of mycoplasma. All cell culture experiments were performed within 15 passages from thawing.
Generation of knockout cells
A3A knockout and A3B knockout cells were generated using a similar strategy as described elsewhere (30) (Supplementary Fig. 3A). The TIIP and U2OS bulk cell lines were single cell cloned to create a parental (i.e. wild type) cell line (Supplementary Fig. 3B and 3M). The parental clone was transiently transfected with PX458(193–194) and PX458(199–200) and single cell cloned to create the TIIP A3A#4 knockout cell line, or the parental clone was transiently transfected with PX458(197–198) and PX458(199–200) and single cell cloned to create the U2OS A3B#38, TIIP A3B#8 and TIIP A3B#13 knockout cell lines. Confirmation of knockout was performed through PCR and western blotting (Supplementary Fig. 3C–E and 3N–O).
Treatments
For oncogene induction in the TIIP cell line, cells were incubated with 500 nM 4-hydroxytamoxifen (4-OHT) (Sigma Aldrich) and this was replenished every day. For CDC6 induction in the HBEC-CDC6 cell line, cells were incubated in 1 μg/ml doxycycline and this was replenished every second day. Senescence was induced with 8 Gy ionizing radiation or 1 mM hydroxyurea. Low levels of replication stress were induced by exposing cells to 24 hours 0.2 μM aphidicolin (Sigma Aldrich) after which they were processed accordingly. Chk1 inhibition in HBEC-CDC6 cells was performed using 2 μM LY2603618 (Selleckchem) for 24 hours prior to harvesting.
RNA interference
Reverse transfections were performed with siRNA (Dharmacon, GE Healthcare) at a final concentration of 40 nM using lipofectamine® RNAiMax (Thermo Fisher Scientific). Non-targeting (NT) control siRNA was used as control. For specific APOBEC3B knockdown the following was used: ON-TARGETplus APOBEC3B siRNA Targeted Region (3’UTR, J-017322–08-0005).
Gel-based deamination assay
The gel-based deamination assay was performed as described elsewhere (20). We used the following probe: 5′-(6-FAM)-AAAAAAAAATCGGGAAAAAAA-3′.
Immunofluorescence
Immunofluorescence was performed as described elsewhere (67). Primary antibodies are described in Supplementary Table 2. Secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 555 (1:2000, Thermo Fisher) were used for detection. DNA was stained with DAPI (Thermo Fisher Scientific). Images of DNA fibers were acquired using a Zeiss AXIO Imager M2 microscope 40× 1.3 oil immersion objective (Zeiss) equipped with a Hamamatsu photonics camera. Images of anaphase cells and ultrafine bridges were acquired using an Olympus DeltaVision RT microscope (Applied Precision, LLC) with a 60× 1.3 oil immersion objective (Olympus) equipped with a Coolsnap HQ camera. Z stacks were acquired at 0.2 μm intervals at 12 μm thick sections. Images of interphase cells were acquired using a 40× 1.4 oil immersion objective (Olympus). Z stacks of interphase cells were acquired at 0.8 μm intervals at 12 μm thick sections. Deconvolution (Iterative restoration with 8 iterations) was performed using the softWoRx deconvolution tool.
Tissue sections were stained from 12 CIS lesions derived from patients with pre-invasive LUSC lesions undergoing surveillance (29). The APOBEC3 and Ki67 double immunofluorescence was performed on a diagnostic tissue microarray prepared from 79 NSCLC from the TRACERx lung study involving the first 100 patients (1). The immunofluorescence images were analyzed using Qpath software (68). Qpath was used to distinguish epithelial cells from stromal cells and manually reviewed thresholds were used to classify epithelial cells as positive or negative for the respective antibody staining.
Preparation of metaphase spreads and FISH
Cells were incubated with colcemid (Thermo Fisher Scientific) at 0.1 μg/mL for 3 hours, trypsinized, washed and spun down. Cells were incubated at 37 °C for 7 min in 2 mL pre-warmed hypotonic solution (1:1, 0.4% KCl + 0.4% sodium citrate). Hereafter, 2 mL fixative was added (3:1, MeOH + acetic acid) and cells were spun. This process was repeated 3 times, after which cells were dropped. Samples were denatured, ethanol dehydrated and subsequently incubated overnight at 37 °C in a humidified chamber with FISH probe against the FHIT locus (FHIT-20-RE, Pisces Scientific). The next day, slides were plunged in wash buffer at room temperature and again at 65 °C for 10 min, ethanol dehydrated, counterstained with DAPI and finally mounted.
Mouse strains and tumor induction
The Cre-inducible Rosa26::LSL-APOBEC3Bi mice are described elsewhere (51,52) (manuscripts under consideration). Mouse lung tumors were initiated in EGFRL858R;Tp53flox/flox (EP; N = 7) and EGFRL858R;Tp53flox/flox;R26LSL-A3B/LSL-tTA (EP-A3B; N = 8, 2 combined experiments) mice by intratracheal infection with adenoviral vectors expressing Cre recombinase (2.5×107 adenoviral particles per mouse). Adenoviral-Cre (Ad-Cre-GFP) was obtained from the University of Iowa Gene Transfer Core. All animal regulated procedures were approved by the Francis Crick Institute BRF Strategic Oversight Committee that incorporates the Animal Welfare and Ethical Review Body and conformed with the UK Home Office guidelines and regulations under the Animals (Scientific Procedures) Act 1986 including Amendment Regulations 2012.
Evaluation of chromosome missegregation errors in H&E samples
Diagnostic H&E samples from NSCLC samples in the TRACERx 100 cohort (1) were evaluated for anaphases with chromosome missegregation events using a 100x objective light microscope to evaluate cells undergoing anaphase. Only H&E sections with ≥ 10 anaphases were considered.
H&E samples from lung cancer samples from the EP (N = 7) and EP-A3B (N = 8) mouse models were evaluated for anaphases with chromosome missegregation events using a 100x objective light microscope. Depending on availability 1–2 sections per mouse were evaluated. For each mouse we observed at least 17 anaphases.
Immunoblot assays
Immunoblotting was carried out according to standard procedures. Primary antibodies are described in Supplementary Table 3.
RNA extraction and quantitative reverse transcription PCR
RNA extraction was performed using a Qiagen RNeasy kit and reverse transcription using an AffinityScript cDNA synthesis kit (Agilent Technologies) according to the manufacturers’ instructions. Quantitative reverse transcription PCR was performed on a QuantStudio™ 7 Flex Real-Time PCR System (Thermo Fisher Scientific). Previously described custom primers were used (69) (Sigma Aldrich) and are shown in Supplementary Table 4.
Flow cytometry - Cell cycle analysis
Cells were fixed in ice cold 70% ethanol overnight at −20 °C. Afterwards, cells were washed with PBS and labelled for 30 min (RT) with 1 μg/mL of DAPI (Thermo Fisher Scientific) in PBS, containing 50 μg/mL RNase A (Sigma Aldrich). Cells were analyzed on a BD LSR Fortessa X-20 (BD Biosciences) and acquired data were analyzed using the Cell Cycle platform of FlowJo software.
DNA fiber stretching assay
The DNA fiber stretching assay was performed as described in (67). 20 min sequential labelling pulses of CIdU (red) and IdU (green) in TIIP, H520 and HEK293-A3B cell lines were subjected to DNA fiber stretching analysis.
EdU incorporation assay
The Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific) was used. Cells were supplemented with 10 μM EdU for 30 min after which they were processed according to the manufacturer’s instructions.
ImageStream-FISH
Cells were exposed for 24 hours to vehicle or 0.2 μM aphidicolin (Sigma Aldrich), after which they were processed as described elsewhere (50). Briefly, 1.5 million cells were hybridized with chromosome 15 satellite enumeration probe (LPE015G, Cytocell) prior to analysis on an ImageStream X Mk II (Amnis).
Senescence-associated β-galactosidase assay (Chromogenic assay)
Senescence within cell culture was detected using the SA-β-gal assay (Sigma Aldrich) as described elsewhere (70).
Recombinant DNA
Senescence reporter cell lines were created using the miR146a-EGFP plasmid (kind gift from S. Elledge, Harvard University, USA) (42). The pSpCas9(BB)-2A-GFP (PX458, Addgene plasmid #48138) plasmid was used for the generation of knockout cell lines (kind gift from F. Zhang, MIT, USA).
High throughput imaging of DNA repair foci and micronuclei
Cells were seeded, treated, fixed and stained in a black and clear bottom 96-well plate (Greiner μClear® 655090). The antibody-stained cells were imaged on a Perkin Elmer Opera Phenix using a water immersion 63x lens to capture confocal stacks of 7 planes. The images were projected and analyzed using the associated Phenix software, Harmony. For these high throughput imaging experiments, all values above and below the median of the WT vehicle condition are shown as red and blue dots, respectively.
Analysis of gene expression in cancer progression datasets
Cross-platform gene-expression profiles from different stages of cancer development were compiled from Gene Expression Omnibus (GEO) for breast (GSE16873, N = 40 samples from 12 patients; GSE21422, N = 19 samples; GSE47462, N = 72 samples from 25 patients) and lung tissue (GSE52248, N = 18; Mascaux et al. (34)/GSE33479, N = 122 samples from 77 patients) and Chen et al. (25). Differential gene expression in developmental stages relative to normal/healthy tissue was determined for each gene in the expression dataset, using linear mixed-effects model with patient as random effect. Fixed effects were applied only for Mascaux et al. (GSE33479), as reported previously (34). P-values were adjusted for multiple testing across all genes in each expression profile (FDR).
Chromosomal instability (CIN70) gene signature enrichment
We evaluated an enrichment score for a previously published set of genes associated with CIN, CIN70 (53). The method single-sample Gene Set Enrichment (ssGSEA) (71) was applied on the gene expression profile of each sample individually to calculate a normalized enrichment score (NES) and to determine whether the CIN70 gene signature was enriched among the up-regulated (positive NES) or downregulated genes (negative NES). The parameters sample and gene normalization were set to rank and z-score, respectively, and a NES was calculated for each log2-transformed gene expression profile (GSE16873, GSE21422, GSE47462, GSE52248, Mascaux et al. (34), Biswas et al. (65) and Chen et al. (25)). A Spearman correlation test between CIN70 NES score and APOBEC3 gene expression was used to calculate the reported correlation coefficients and associated P-values.
Determining the progression-free interval hazard ratio
Data generated by The Cancer Genome Atlas (TCGA) pilot project established by the NCI and the National Human Genome Research Institute were downloaded for survival analysis in LUAD and LUSC patients. The data were retrieved through database of Genotypes and Phenotypes (dbGaP) authorization (accession no. phs000178.v9.p8). Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at https://cancergenome.nih.gov/.
In order to estimate the influence of expression of APOBEC3 genes on survival, a Cox proportional-hazards model with PFI as end point estimate was used. The definition of PFI has been used as described elsewhere (72). The expression of different APOBEC3 genes was measured as normalized expression counts. Before adding the different genes expressed as continuous predictor variables to the model, each variable has been z-transformed to make sure the predictor estimates are comparable between genes. The Cox model was calculated for early-stage tumor samples (stage I and stage IA) and later stage tumor samples (stage IIIA, IIIB and IV) separately.
Analysis of genome instability in APOBEC3 heterogeneous tumors
The multi-region WES data of the TRACERx100 cohort (1) was used to analyze differences in genome instability between tumor regions with a high and low number of APOBEC3 signature mutations within the same tumor. Only patients with a significant enrichment of the APOBEC3 mutation signature, as defined by Roberts et al. (4), have been considered in this analysis. The tumor regions with the highest and lowest ratio of APOBEC3 to non-APOBEC3 signature mutations have been identified for each tumor. Only patients with a significant difference in APOBEC3 mutation ratio between the high and low region have been considered for further analysis (N = 14). The proportion of the genome altered by SCNAs was calculated as the sum of the segment sizes with a copy number gain or loss relative to the ploidy of the tumor region divided by the sum of all segments within the tumor region (1). A two-tailed paired Wilcoxon test was used to analyze the difference in the proportion of the genome altered between regions with high and low number of APOBEC3 signature mutations.
Data availability
The TRACERx whole exome sequencing data generated, used or analyzed during this study are not publicly available and restrictions apply to the availability of these data. Such TRACERx whole exome sequencing data are available through the Cancer Research UK & University College London Cancer Trials Centre (ctc.tracerx@ucl.ac.uk) for academic non-commercial research purposes upon reasonable request, and subject to review of a project proposal that will be evaluated by a TRACERx data access committee, entering into an appropriate data access agreement and any applicable ethical approvals.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the TRACERx consortium for participating in this study.
The results published here are in part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and the National Human Genome Research Institute.
R.E. Hynds is a Wellcome Trust Sir Henry Wellcome Fellow (WT209199/Z/17/Z) and received grant funding from the Roy Castle Lung Cancer Foundation that supported this work. P.G. is funded by KBVU grant R167-A11068. The work in the Boulton lab is supported by a European Research Council (ERC) Advanced Investigator Grant (TelMetab); and Wellcome Trust Senior Investigator and Collaborative Grants. T. Marafioti is supported by the UK National Institute of Health Research University College London Hospital Biomedical Research Centre and A.U. Akarca is supported by Cancer Research UK-UCL Centre Cancer Immuno-therapy Accelerator Award. R.A.M. de Bruin and C. Bertoli are supported by core funding to the MRC-UCL University Unit (Ref. MC_EX_G0800785) and funded by R.A.M. de Bruin’s Cancer Research UK Programme Foundation Award. Cancer studies in the Harris lab are supported by NCI P01-CA234228. R.S. Harris is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished McKnight University Professor, and an Investigator of the Howard Hughes Medical Institute. K. Evangelou and V.G. Gorgoulis were financially supported by the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grants agreement no. 722729 (SYNTRAIN); the National Public Investment Program of the Ministry of Development and Investment / General Secretariat for Research and Technology, in the framework of the Flagship Initiative to address SARS-CoV-2 (2020ΣE01300001); the Welfare Foundation for Social & Cultural Sciences (KIKPE), Greece; H. Pappas donation; grant no. 775 from the Hellenic Foundation for Research and Innovation (HFRI); and NKUA-SARG grants 70/3/9816, 70/3/12128, and 70/3/15603 S.F. Bakhoum is supported by the Office Of The Director, the National Institutes of Health under Award Number DP5OD026395 High-Risk High-Reward Program, the NCI Breast Cancer SPORE (P50CA247749) and R01 (R01CA256188–01), the Burroughs Wellcome Fund Career Award for Medical Scientists, the Parker Institute for Immunotherapy at MSKCC, the Josie Robertson Foundation, and the MSKCC core grant P30-CA008748. J. Bartek and his team were funded by grants from the Danish Cancer Society (R1123-A7785–15-S2, R167-A11068), the Novo Nordisk Foundation (16854, 0060590), the Lundbeck foundation (R266–2017-4289, R322–2019-2577), the Swedish Research council (VR-MH 2014–46602-117891–30), The Swedish Cancer Foundation/Cancerfonden (170176), and the Danish national research foundation (project CARD, DNRF 125). N. Kanu receives funding from Cancer Research UK. C. Swanton is a Royal Society Napier Research Professor (RP150154). This work was supported by the Francis Crick Institute that receives its core funding from Cancer Research UK (FC001169), the UK Medical Research Council (FC001169), and the Wellcome Trust (FC001169). This research was funded in whole, or in part, by the Wellcome Trust (FC001169). For the purpose of open access, the authors have applied a CC by copyright licence to any author-accepted manuscript version arising from this submission. This work was supported by Breast Cancer Research Foundation, USA (BCRF), the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007–2013) Consolidator Grant (FP7-THESEUS-617844), an ERC Advanced Grant (PROTEUS) from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (835297), Novo Nordisk Foundation (ID16584), and the National Institute for Health Research (NIHR) Biomedical Research Centre at University College London Hospitals.
The authors thank Dr. Silvestro Conticello and Dr. Uday Munagala (ISPRO, Italy) for critical discussions. The authors thank Dr. Stephen Elledge (Harvard University, USA) for providing miR-146a-EGFP plasmid, Dr. Julian Downward and Dr. Miriam Molina (The Francis Crick Institute, UK) for providing TIIP-ER-KRAS V12 cells. We also thank Dr. Sarah Clarke (University College London, UK) for their assistance with normal lung and CIS FFPE blocks. We also thank Laura Tovini and Dr. Sarah McClelland (Barts Cancer Institute, London) for their helpful advice on the ImageStream. We also thank Dr. Christoffer L. Halvorsen (Danish Cancer Society Research Center, Denmark), Dr. Robert Strauss (Danish Cancer Society Research Center, Denmark), Dr. Agostina Bertolin (The Francis Crick Institute, UK) and Dr. David Moore (The Francis Crick Institute, UK) for helpful suggestions. Finally, the authors gratefully acknowledge members of Experimental Histopathology, Light Microscopy, Flow Cytometry, Cell Services and Genomics Equipment Park at the Francis Crick Institute (The Francis Crick Institute, UK).
AUTHORS’ CONTRIBUTIONS
Conception and design: S. Venkatesan, S.F. Bakhoum, J. Bartkova, J. Bartek, N. Kanu and C. Swanton
Development of methodology: S. Venkatesan., M. Angelova, H. Zhai, C. Puttick, W-T. Lu, R.E. Hynds, M. Dietzen, P. Galanos, K. Evangelou, R. Bellelli, E.L. Lim, T.B.K. Watkins, A.U. Akarca, T. Marafioti, M.Y. Wu, M. Howell, S.J. Boulton, S.M. Janes, R.A.M. de Bruin, W.L. Brown, R.S. Harris, V.G., Gorgoulis, J. Bartkova, N. McGranahan, S.F. Bakhoum, J. Bartek, N. Kanu and C. Swanton
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Venkatesan, M. Angelova, C. Puttick, H. Zhai, D. Caswell, W-T Lu, M. Dietzen, P. Galanos, K. Evangelou, R. Bellelli, E.L. Lim, T.B.K. Watkins, A. Rowan, V.H. Teixeira, Y. Zhao, H. Chen, L-P Zalmas, M.A. Bakir, S. Hobor, E. Grönroos, A. Pennycuick, E. Nigro, B.B. Campbell, W.L. Brown, M.Y. Wu, M. Howell, S.J. Boulton, C. Bertoli, T.R. Fenton, R.A.M. de Bruin, Apolinar Maya-Mendoza, E. Santoni-Rugiu, R.E. Hynds, S.M. Janes, V.G. Gorgoulis, M. Jamal-Hanjani, N. McGranahan, R.S. Harris, J. Bartkova, S.F. Bakhoum, J. Bartek, N. Kanu and C. Swanton
Writing, review, and/or revision of the manuscript: S. Venkatesan, R.E. Hynds, E. Grönroos, R.S. Harris, J. Bartkova, S.F. Bakhoum, J. Bartek, N. Kanu and C. Swanton
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S. Venkatesan, M. Jamal-Hanjani, A. Rowan, V.H. Teixeira, J. Bartkova, S.F. Bakhoum, J. Bartek, N. Kanu and C. Swanton
Study supervision: S.F. Bakhoum, J. Bartek, N. Kanu and C. Swanton
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
C. Swanton acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Archer Dx Inc (collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical, is an AstraZeneca Advisory Board member and Chief Investigator for the MeRmaiD1 clinical trial, has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Bicycle Therapeutics, and the Sarah Cannon Research Institute, has stock options in Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options and is co-founder of Achilles Therapeutics. C. Swanton is editorial board member for Cell and PLOS medicine, associate editor and editorial board member for Annals of Oncology, scientific editor for Cancer Discovery and advisory board member for Nature Reviews Clinical Oncology and Cancer Cell. C. Swanton holds European patents relating to assay technology to detect tumor recurrence (PCT/GB2017/053289); to targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), a US patent relating to detecting tumor mutations (PCT/US2017/28013) and both a European and US patent related to identifying insertion/deletion mutation targets (PCT/GB2018/051892). S.M. Janes receives grant funding from GRAIL, and has consulted for JnJ, GSK, AZ and Bard1. T.R. Fenton is on the clinical and scientific advisory board of and holds stock options in APOBEC Discovery Ltd. R.S. Harris is a co-founder, shareholder, and consultant of ApoGen Biotechnologies. S.F. Bakhoum holds a patent related to some of the work described targeting CIN and the cGAS-STING pathway in advanced cancer. He owns equity in, receives compensation from, and serves as a consultant and the Scientific Advisory Board and Board of Directors of Volastra Therapeutics Inc. He has also consulted for Sanofi, received sponsored travel from the Prostate Cancer Foundation, and both travel and compensation from Cancer Research UK. No potential conflicts of interest were disclosed by the other authors.
REFERENCES
- 1.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017;376(22):2109–21 doi 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salter JD, Bennett RP, Smith HC. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem Sci 2016;41(7):578–94 doi 10.1016/j.tibs.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45(9):970–6 doi 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013;494(7437):366–70 doi 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv 2016;2(10):e1601737 doi 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Cancer 2014;5(6):405–13 doi 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen TW, Lee CC, Liu H, Wu CS, Pickering CR, Huang PJ, et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat Commun 2017;8(1):46S doi 10.1038/s41467-017-00493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roper N, Gao S, Maity TK, Banday AR, Zhang X, Venugopalan A, et al. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep 2019;26(10):26S1–66 e6 doi 10.1016/j.celrep.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabbie R, Ansari-Pour N, Cast O, Lau D, Scott F, Welsh SJ, et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nature Communications 2020;11(1):4306 doi 10.1038/s41467-020-18060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nature Genetics 2019;51(10):1450–8 doi 10.1038/s41588-019-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569(77S7):560–4 doi 10.1038/s41586-019-1056-z. [DOI] [PubMed] [Google Scholar]
- 13.Cannataro VL, Gaffney SG, Sasaki T, Issaeva N, Grewal NKS, Grandis JR, et al. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019;38(18):3475–87 doi 10.1038/s41388-018-0657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep 2014;7(6):1833–41 doi 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 15.Smid M, Rodríguez-González FG, Sieuwerts AM, Salgado R, Prager-Van der Smissen WJC, Vlugt-Daane Mvd, et al. Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration. Nature Communications 2016;7(1):12910 doi 10.1038/ncomms12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Driscoll CB, Schuelke MR, Kottke T, Thompson JM, Wongthida P, Tonne JM, et al. APOBEC3B-mediated corruption of the tumor cell immunopeptidome induces heteroclitic neoepitopes for cancer immunotherapy. Nature Communications 2020;11(1):790 doi 10.1038/s41467-020-14S68-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet 2020;52(9):884–90 doi 10.1038/s41588-020-0667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortez LM, Brown AL, Dennis MA, Collins CD, Brown AJ, Mitchell D, et al. APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet 2019;15(12):e1008S45 doi 10.1371/journal.pgem1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng JCF, Quist J, Grigoriadis A, Malim MH, Fraternali F. Pan-cancer transcriptomic analysis dissects immune and proliferative functions of APOBEC3 cytidine deaminases. Nucleic Acids Res 2019;47(3):1178–94 doi 10.1093/nar/gky1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanu N, Cerone MA, Goh G, Zalmas LP, Bartkova J, Dietzen M, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol 2016;17(1):185 doi 10.1186/s13059-016-1042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Periyasamy M, Singh AK, Gemma C, Kranjec C, Farzan R, Leach DA, et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res 2017;45(19):11056–69 doi 10.1093/nar/gkx721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, et al. The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res 2015;75(21):4538–47 doi 10.1158/0008-5472.CAN-15-2171-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018;172(1–2):205–17 e12 doi 10.1016/j.cell.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular Carcinomas In Situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin Cancer Res 2019;25(2):674–86 doi 10.1158/1078-0432.CCR-18-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, et al. Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nature Communications 2019;10(1):5472 doi 10.1038/s41467-019-13460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu X, Fujimoto J, Ying L, Fukuoka J, Ashizawa K, Sun W, et al. Multi-region exome sequencing reveals genomic evolution from preneoplasia to lung adenocarcinoma. Nature Communications 2019;10(1):2978 doi 10.1038/s41467-019-10877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vinayanuwattikun C, Le Calvez-Kelm F, Abedi-Ardekani B, Zaridze D, Mukeria A, Voegele C, et al. Elucidating Genomic Characteristics of Lung Cancer Progression from In Situ to Invasive Adenocarcinoma. Sci Rep 2016;6:31628 doi 10.1038/srep31628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian J, Zhao S, Zou Y, Rahman SMJ, Senosain MF, Stricker T, et al. Genomic Underpinnings of Tumor Behavior in In Situ and Early Lung Adenocarcinoma. Am J Respir Crit Care Med 2020;201(6):697–706 doi 10.1164/rccm.201902-02940C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D, Beane J, et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nature Medicine 2019;25(3):517–25 doi 10.1038/s41591-018-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown WL, Law EK, Argyris PP, Carpenter MA, Levin-Klein R, Ranum AN, et al. A Rabbit Monoclonal Antibody against the Antiviral and Cancer Genomic DNA Mutating Enzyme APOBEC3B. Antibodies (Basel) 2019;8(3) doi 10.3390/antib8030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leonard B, Starrett GJ, Maurer MJ, Oberg AL, Van Bockstal M, Van Dorpe J, et al. APOBEC3G Expression Correlates with T-Cell Infiltration and Improved Clinical Outcomes in High-grade Serous Ovarian Carcinoma. Clin Cancer Res 2016;22(18):4746–55 doi 10.1158/1078-0432.CCR-15-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J Biol Chem 2013;288(24):17253–60 doi 10.1074/jbc.M113.458661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morton ML, Bai X, Merry CR, Linden PA, Khalil AM, Leidner RS, et al. Identification of mRNAs and lincRNAs associated with lung cancer progression using next-generation RNA sequencing from laser micro-dissected archival FFPE tissue specimens. Lung Cancer 2014;85(1):31–9 doi 10.1016/j.lungcam2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mascaux C, Angelova M, Vasaturo A, Beane J, Hijazi K, Anthoine G, et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 2019;571(7766):570–5 doi 10.1038/s41586-019-1330-0. [DOI] [PubMed] [Google Scholar]
- 35.Emery LA, Tripathi A, King C, Kavanah M, Mendez J, Stone MD, et al. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. Am J Pathol 2009;175(3):1292–302 doi 10.2353/ajpath.2009.090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunner AL, Li J, Guo X, Sweeney RT, Varma S, Zhu SX, et al. A shared transcriptional program in early breast neoplasias despite genetic and clinical distinctions. Genome Biol 2014;15(5):R71 doi 10.1186/gb-2014-15-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kretschmer C, Sterner-Kock A, Siedentopf F, Schoenegg W, Schlag PM, Kemmner W. Identification of early molecular markers for breast cancer. Mol Cancer 2011;10(1):15 doi 10A186/1476-4598-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006;444(7119):633–7 doi 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 39.Evangelou K, Lougiakis N, Rizou SV, Kotsinas A, Kletsas D, Munoz-Espin D, et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017;16(1):192–7 doi 10.1111/acel.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komseli ES, Pateras IS, Krejsgaard T, Stawiski K, Rizou SV, Polyzos A, et al. A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genomics 2018;19(1):37 doi 10.1186/s12864-017-4375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma CX, Janetka JW, Piwnica-Worms H Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011;17(2):88–96 doi 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015;349(6255):aaa5612 doi 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buisson R, Lawrence MS, Benes CH, Zou L. APOBEC3A and 3B activities render cancer cells susceptible to ATR inhibition. Cancer Research 2017:canres.3389.2016 doi 10.1158/0008-5472.Can-16-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikkila J, Kumar R, Campbell J, Brandsma I, Pemberton HN, Wallberg F, et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br J Cancer 2017;117(1):113–23 doi 10.1038/bjc.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, et al. Cytosine Deaminase APOBEC3A Sensitizes Leukemia Cells to Inhibition of the DNA Replication Checkpoint. Cancer Research 2017;77(17):4579–88 doi 10.1158/0008-5472.Can-16-3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep 2011;12(5):444–50 doi 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. Journal of Experimental Medicine 2020;217(12) doi 10.1084/jem.20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov 2013;3(5):548–63 doi 10.1158/2159-8290.CD-12-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nature Cell Biology 2009;11(6):753–60 doi 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 50.Worrall JT, Tamura N, Mazzagatti A, Shaikh N, van Lingen T, Bakker B, et al. Non-random Mis-segregation of Human Chromosomes. Cell Rep 2018;23(11):3366–80 doi 10.1016/j.celrep.2018.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayekar MK, Caswell DR, Vokes NI, Law EK, Wu W, Hill W, et al. Targeted cancer therapy induces APOBEC fuelling the evolution of drug resistance. bioRxiv 2020:2020.12.18.423280 doi 10.1101/2020.12.18.423280. [DOI] [Google Scholar]
- 52.de Carné Trécesson S, Boumelha J, Law EK, Romero-Clavijo P, Mugarza E, Coelho MA, et al. APOBEC3B expression generates an immunogenic model of Kras mutant lung cancer. bioRxiv 2020:2020.12.22.423126 doi 10.1101/2020.12.22.423126. [DOI] [Google Scholar]
- 53.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 2006;38(9):1043–8 doi 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 54.Sieuwerts AM, Doebar SC, de Weerd V, Verhoef EI, Beauford CM, Agahozo MC, et al. APOBEC3B Gene Expression in Ductal Carcinoma In Situ and Synchronous Invasive Breast Cancer. Cancers (Basel) 2019;11(8) doi 10.3390/cancers11081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roelofs PA, Goh CY, Chua BH, Jarvis MC, Stewart TA, McCann JL, et al. Characterization of the mechanism by which the RB/E2F pathway controls expression of the cancer genomic DNA deaminase APOBEC3B. eLife 2020;9:e61287 doi 10.7554/eLife.61287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020;587(7832):126–32 doi 10.1038/s41586-020-2698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Argyris PP, Wilkinson PE, Jarvis MC, Magliocca KR, Patel MR, Vogel RI, et al. Endogenous APOBEC3B overexpression characterizes HPV-positive and HPV-negative oral epithelial dysplasias and head and neck cancers. Mod Pathol 2021;34(2):280–90 doi 10.1038/s41379-020-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019;176(6):1282–94 e20 doi 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, Cortez D. HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell Rep 2020;31(9):107705 doi 10.1016/j.celrep.2020.107705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008;319(5868):1352–5 doi 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 61.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354(6312):618–22 doi 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sui Y, Qi L, Zhang K, Saini N, Klimczak LJ, Sakofsky CJ, et al. Analysis of APOBEC-induced mutations in yeast strains with low levels of replicative DNA polymerases. Proceedings of the National Academy of Sciences 2020;117(17):9440–50 doi 10.1073/pnas.1922472117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bartkova J, Hořejší Z, Koed K, Krämer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434(7035):864–70 doi 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 64.Russ JC, Russ JC. Automatic discrimination of features in grey-scale images. Journal of Microscopy 1987;148(3):263–77 doi 10.1111/j.1365-2818.1987.tb02872.x. [DOI] [Google Scholar]
- 65.Biswas D, Birkbak NJ, Rosenthal R, Hiley CT, Lim EL, Papp K, et al. A clonal expression biomarker associates with lung cancer mortality. Nat Med 2019;25(10):1540–8 doi 10.1038/s41591-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kemp SJ, Thorley AJ, Gorelik J, Seckl MJ, O’Hare MJ, Arcaro A, et al. Immortalization of human alveolar epithelial cells to investigate nanoparticle uptake. Am J Respir Cell Mol Biol 2008;39(5):591–7 doi 10.1165/rcmb.2007-03340C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan YW, Fugger K, West SC. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat Cell Biol 2018;20(1):92–103 doi 10.1038/s41556-017-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Scientific Reports 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 2010;38(13):4274–84 doi 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint 0. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009;4(12):1798–806 doi 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 71.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173(2):400–16 e11 doi 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TRACERx whole exome sequencing data generated, used or analyzed during this study are not publicly available and restrictions apply to the availability of these data. Such TRACERx whole exome sequencing data are available through the Cancer Research UK & University College London Cancer Trials Centre (ctc.tracerx@ucl.ac.uk) for academic non-commercial research purposes upon reasonable request, and subject to review of a project proposal that will be evaluated by a TRACERx data access committee, entering into an appropriate data access agreement and any applicable ethical approvals.