

Abstract
Purpose
Growth differentiation factor 11 (GDF11) is a key signaling protein required for proper development of many organ systems. Only one prior study has associated an inherited GDF11 variant with a dominant human disease in a family with variable craniofacial and vertebral abnormalities. Here, we expand the phenotypic spectrum associated with GDF11 variants and document the nature of the variants.
Methods
We present a cohort of six probands with de novo and inherited nonsense/frameshift (4/6 patients) and missense (2/6) variants in GDF11. We generated gdf11 mutant zebrafish to model loss of gdf11 phenotypes and used an overexpression screen in Drosophila to test variant functionality.
Results
Patients with variants in GDF11 presented with craniofacial (5/6) , vertebral (5/6), neurological (6/6), visual (4/6), cardiac (3/6), auditory (3/6) and connective tissue abnormalities (3/6). gdf11 mutant zebrafish show craniofacial abnormalities and body segmentation defects that match some patient phenotypes. Expression of the patients’ variants in the fly showed that one nonsense variant in GDF11 is a severe loss-of-function (LOF) alleles whereas the missense variants in our cohort are partial LOF variants.
Conclusion
GDF11 is needed for human development, particularly neuronal development, and LOF GDF11 alleles can affect the development of numerous organs and tissues.
Introduction
Growth Differentiation Factor (GDF) proteins are members of the Bone Morphogenetic Proteins (BMP) subfamily of transforming growth factor-beta (TGF-β) ligands and are key signaling proteins for development1,2. Loss-of-function (LOF) variants in GDF genes are associated with disorders affecting many different organs and tissues (Supplementary Table 1). Additionally, individual LOF variants within the same GDF gene can lead to pleiotropic effects3,4. Pleiotropy of individual GDF genes is likely due to the complex role of these genes in the development of multiple tissues5,6 and functional redundancies among GDF/BMP genes7–9.
GDF11 has three domains: a signal peptide (Amino Acid (AA)1-24), a mature proprotein (AA25-298), and the TGF-β domain (AA299-407) (Figure 2C)10. The signal peptide localizes the protein to the plasma membrane, where Furin proteases cleave the TGF-β domain at an RXXR motif (AA295-298) allowing secretion of the mature protein containing TGF-β domain while the cleaved propeptide is retained in the membrane11. Secreted GDF11 binds to Activin receptors, which triggers phosphorylation of SMAD2 and subsequent translocation to the nucleus, upregulating genes required for cell differentiation and tissue patterning12–15. GDF11 is broadly expressed, with expression highest in skeletal muscle, pancreas, kidney, retina, and the brain10,16–18. GDF11 is expressed ubiquitously within the brain with expression highest in oligodendrocytes, oligodendrocyte precursors, and astrocytes, followed by neurons19. GDF11 is most highly expressed during development and early life and its levels decline with aging20,21. The breadth of GDF11 expression, coupled with high levels during pre- and post-natal developmental stages, indicates that GDF11 may be required for proper organogenesis and homeostasis after birth.
A GDF11 variant (NP_005802.1:p.(R298Q)) with a dominant inheritance pattern and variable penetrance and expressivity has been documented in a large family whose members presented with cleft lip/palate as well as rib and vertebral hypersegmentation22. The affected arginine (R) is the second arginine in the RXXR motif essential for TGF-β domain cleavage11. When this arginine is replaced with glutamine, the TGF-β domain is not cleaved by Furin proteases22. The biochemical data, coupled with the dominant inheritance pattern, suggest that this allele behaves as a dominant LOF variant.
Model organism studies have defined a developmental role for GDF1110,23–27. Gdf11-deficient (Gdf11−/−) mice die within 24 hours of birth with renal and palate abnormalities10. The skeleton of Gdf11−/− mice exhibits an increased number of ribs, anteriorly directed homeotic transformations, posterior displacement of hindlimbs and defective inner ear structure10,28. Gdf11 is a haploinsufficient locus in mice and skeletal abnormalities are seen in heterozygous animals; Gdf11+/− mice present fewer additional ribs and less severe craniofacial abnormalities than Gdf11−/− mice indicating that the effect of GDF11 function on skeletal development is dose-dependent10. Gdf11 is also required for the timing and progression of neurogenesis during the development of the spinal cord, retina, and olfactory epithelium23,26,29. Gdf11-related defects are typically attributed to aberrant Hox gene expression downstream of Gdf11 signaling, which in turn causes major tissue patterning defects in development10.
We have identified a cohort of patients with both de novo and inherited variants in GDF11 presenting with complex neurological, cardiovascular, connective tissue, ocular, and auditory phenotypes, in addition to the craniofacial and skeletal abnormalities previously described. Additionally, we generated a gdf11 LOF Zebrafish model and we used Drosophila to evaluate the function of three of the patients’ GDF11 variants.
Materials and Methods
Human genetics
All probands were exome or genome sequenced (Supplementary Methods (SM)). All GDF11 variants were sanger confirmed. GDF11 variants are mapped onto the NM_005811.5 RefSeq transcript.
Sequence alignment
Protein sequences from human GDF11 (NP_005802.1), mouse Gdf11 (NP_034402.1), zebrafish gdf11 (NP_998140.1), and Drosophila myo (NP_726604.1) were obtained from NCBI and aligned using BoxShade (https://embnet.vital-it.ch/software/BOX_form.html).
Quantification of GDF11 gene and protein levels from peripheral blood mononuclear cells (PBMC)
PBMC samples were quickly thawed at room temperature and centrifuged at 500xg for 5 minutes at room temperature. RNA and protein were isolated and analyzed using separate protocols described in SM. The primers used to quantify gene expression are provided in SM. For Western blotting standard protocols were used and are described in the SM alongside antibodies used. For ELISA circulating GDF11 levels in plasma were quantified using the human GDF11/GDF-11 Sandwich ELISA kit (LSBio #LS-F11519) according to the manufacturer’s recommendations. Plasma samples were diluted 1:1 in sample diluent before processing. The qPCR was performed with one technical replicate and the ELISA was performed with 3 technical replicates. Center values represented in Figure 1B–C represent mean.
Figure 1 -. Overview of patients with GDF11 variants.
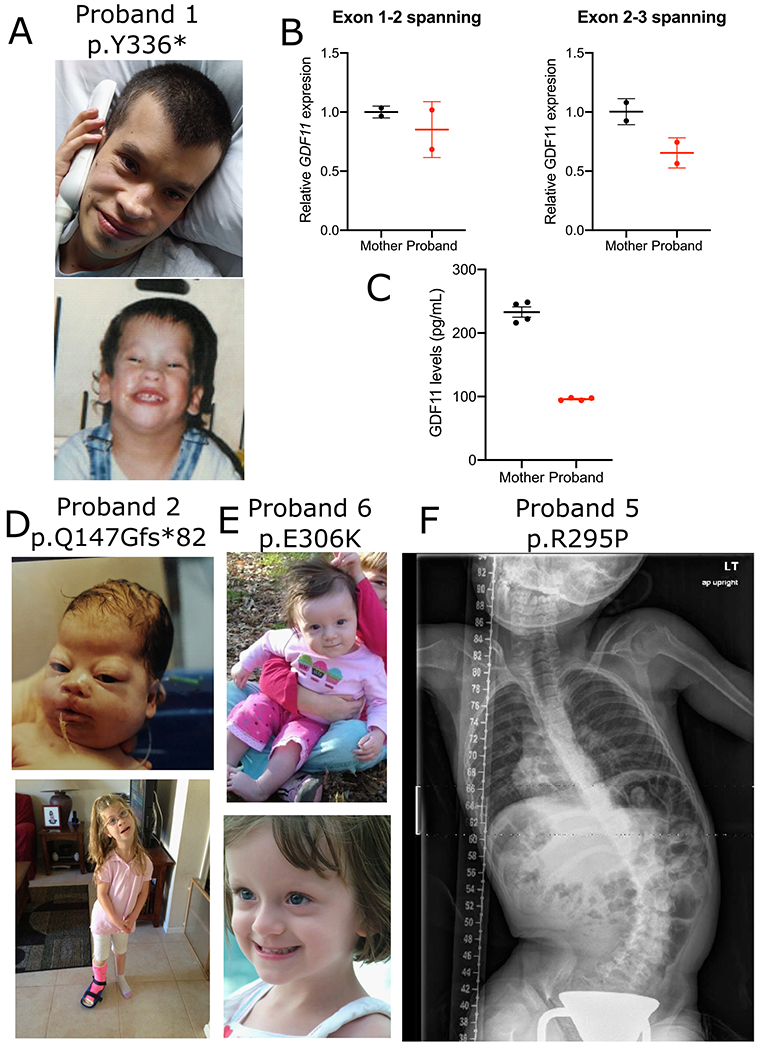
(A) Pictures of Proband 1 (B) GDF11 expression was measured in PBMCs derived from the proband or unaffected mother by qPCR using primer sets spanning exons 1 and 2 (left) or 2 and 3 (right) normalized to GUSB loading control expression. RNA was collected from n = 2 technical replicates from N = 1 blood draws per patient. Error bars = SD. (C) GDF11 expression was measured in plasma derived from the proband or unaffected mother using a commercial GDF11 ELISA kit (LSBio #LS-F11519) Error bars = SEM. Quantification was performed in n = 4 technical replicates from N = 1 blood draw per patient. Pictures of proband 2 (D) and proband 6 (E). X-ray of proband 5 (F).
Generation of Zebrafish gdf11 mutants
Three zebrafish indel alleles were generated using CRISPR-Cas9 (SM). We generated three different frameshift deletions: b1407, a 2bp deletion in exon 1, c.374-5, resulting in an E125Vfs*15 truncation; b1408 a 7bp deletion in exon 3, c.922-28, creating an F308Gfs*53 truncation, and b1396, which has a 703bp deletion removing the 5’UTR and most of the first exon. All alleles were confirmed by sequencing aligned to the GRCz11 reference transcript ENSDART00000066033.8. Surviving F1s for each allele were raised to adulthood and genotyped to identify heterozygotes that were then increased. Homozygous viable F2 mutants were raised to adulthood and increased to obtain larvae for the described experiments, alongside control larvae from homozygous wild-type F2 siblings.
Analysis of gdf11 expression in Zebrafish
In situ hybridization was performed as described30. Primers used are described in SM. Image acquisition detailed in SM.
Single-cell RNA-seq expression for gdf11 was retrieved from the Zebrafish single-cell transcriptome atlas (http://cells.ucsc.edu/?ds=zebrafish-dev). Tissue-specific assignments of cell-type identities are those previously annotated 31.
Analysis of zebrafish craniofacial structures.
Zebrafish skeletal elements were fixed and stained with Alcian blue and Alizarin red as previously described 32. Image acquisition and statistical analysis are detailed in SM.
Fly stocks and maintenance
All fly stocks used in this study were either generated in-house or were obtained from the Bloomington Drosophila Stock Center (BDSC). All flies were reared on standard fly food and maintained at room temperature unless specified. Fly lines used are listed in SM.
Generation of UAS-myo and myo-T2A-GAL4 flies
The Drosophila melanogaster cDNA for myo (isoform myo-PA, FlyBase ID: FBal0267088) was generously provided by Michael O’Connor33. Identification of conserved amino acids corresponding to variants in human GDF11 (fly variant in myo in parenthesis): p.E306K (p.E500K), p.Y336* (P.F530*), and p.R295P (p.R489P) was done using multiple protein alignment DIOPT v6 34 via Marrvel1.2 (www.marrvel.org)35. Mutagenesis and transgene injection were done as previously described36. Two independent lines were made for each injected construct, and both constructs were used in all future studies. The myo-T2A-GAL4 allele was made as previously described37. Detailed reagents are available in the SM.
Overexpression of myo assay
To determine the viability of each myo variant when overexpressed, UAS-myo-WT and variant flies, as well as UAS-empty, were crossed to various GAL4 driving lines (Act-GAL4, repo-GAL4, mef2-GAL4, and myo-T2A-GAL4) at 18°C, 22°C, 25°C, and 29°C. Following standard practice in the fly community, two biological replicates of each cross were performed (unblinded) from each cross to determine the percentage of viable flies (N>150: exact numbers are provided in supplementary data file 1). A chi-squared test, with expected totals derived from the number of viable GAL4>UAS-empty (pUAST-attB without any insert injected into VK00033) animals with the respective GAL4, was performed to determine if differences in viability were significant. No variation was estimated.
Results
Patients with variants in GDF11 exhibit multisystemic phenotypes
Probands 1-6, with both de novo and inherited variants in GDF11 (NM_005811.4, NP_005802.1), present with complex neurological, craniofacial, skeletal, cardiovascular, connective tissue, ocular, and auditory phenotypes (Figure 1, Table 1)22. Of the six patients in our cohort, four have predicted nonsense or frameshift variants (p.N94Rfs*47, p.Q147Gfs*82, p.T319Nfs*5, p.Y336*), and two have missense variants (p.R295P, p.E306K) (Supplementary Table 2). One missense variant perturbs the first arginine in the RXXR motif (p.R295P) and the other missense variant reverses the charge of a conserved residue in the TGF-β domain (p.E306K) (Table 1) (Figure 2B, 2C). RNA expression in PBMCs from proband 1 (p.Y336*) showed GDF11 levels comparable to the patient’s unaffected mother (Figure 1B), suggesting that this variant does not undergo nonsense-mediated decay (NMD) which is expected as this variant lies in the final coding exon (Figure 2C). However, quantification of GDF11 protein levels in blood plasma using ELISA showed 50% less GDF11 protein when compared to an unaffected relative (Figure 1C). This is expected as the truncating mutant protein does not contain the antibody epitope in the TGFβ domain (Figure 1B). The frameshift variants are not documented in gnomAD2.1.138 and are expected to produce a protein that lacks the functional TGF-β domain (Figure 2B, 2C). Additionally, the pLI score for GDF11 is 0.98 with an observed/expected (o/e) score of 0.06 in gnomAD indicating a high intolerance for LOF variants in GDF1138. A query of missense variants in GDF11 in MARRVEL35 revealed that p.R295P has a high CADD score39 of 34 and is not seen in the gnomAD database (Supplementary Table 2)38. Although the p.E306K variant is observed once in gnomAD, the variant also has a high CADD score of 27 (Supplementary Table 2). Both missense variants are predicted to be damaging by various in silico prediction algorithms40. Additionally, the missense Z-score for GDF11 is 2.98, with an o/e score of 0.45 which indicates that GDF11 is intolerant of missense variants38. Table 1 lists clinical presentations, which are summarized in the following paragraphs (more information is available in the supplementary information).
Table 1.
Summary of clinical information from each proband
| Proband 1 | Proband 2 | Proband 3 | Proband 4 | Proband 5 | Proband 6 | ||
|---|---|---|---|---|---|---|---|
| Human Variant | Y336* | Q147Gfs*82 | T319Nfs*5 | N94Rfs*47 | R295P | E306K | |
| Inheritance Pattern | De novo | Autosomal Dominant | De novo | Autosomal Dominant | De novo | De Novo | |
| Age of Onset (y/o) | 1 month | 0 | 3 | 0 | 0 | 2 months | |
| Current Age (y/o) | 32 | 17 | 8 | 15 months | 11 | 12 | |
| Sex | Male | Female | Male | Male | Male | Female | |
| Intellectual Disability | + | − | + | NA | + | − | 3/5 |
| Developmental Delay | + | + | + | + | − | + | 5/6 |
| Seizures | + | − | +b | + | +c | − | 4/6 |
| Neurological Abnormalities | + | + | + | + | + | + | 6/6 |
| Visual Disorders | + | + | − | + | − | + | 4/6 |
| Hearing Disorders | + | + | − | − | + | − | 3/6 |
| Craniofacial Abnormalities | + | +a | − | + | + | + | 5/6 |
| Palate Abnormalities | + | +a | − | − | − | + | 3/6 |
| Vertebral Abnormalities | + | + | + | − | + | + | 5/6 |
| Scoliosis | + | − | − | − | + | + | 3/6 |
| Toe Abnormalities | + | +a | − | − | − | + | 3/6 |
| Connective Tissue Abnormalities | + | − | − | − | + | + | 3/6 |
| Cardiac Abnormalities | + | +a | − | − | + | − | 3/6 |
| Aortic Dilation | + | − | − | − | + | − | 2/6 |
Summary of clinical information from each proband; detailed reports can be found in the supplemental materials. Proband 2 inherited the variant from her mother who has a milder phenotypic presentation. These phenotypes are indicated with an a. Proband 4 inherited his variant from his father, the father did not report any shared phenotypes. It is not known if the mother of proband 2 or father of proband 4 is mosaic.
For proband 3 absence seizures were also reported in a sister who did not carry a variant in GDF11.
For proband 5 seizures are likely due to Aicardi-Goutieres type 6.
Figure 2 -. GDF11 is conserved across species –
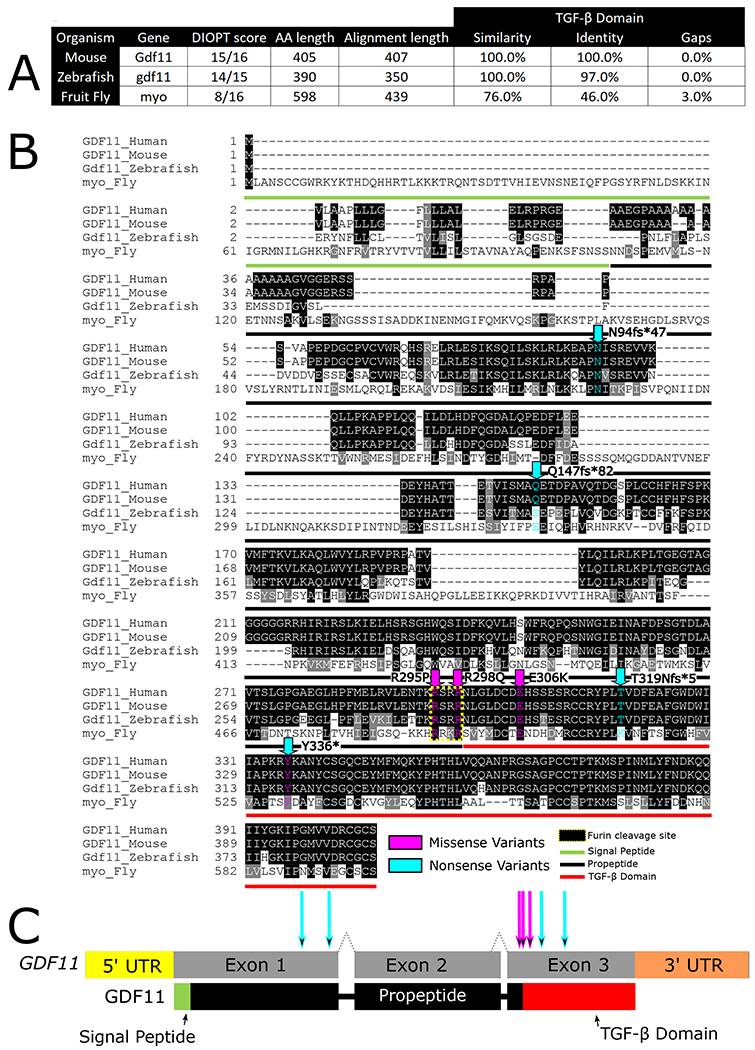
(A)GDF11 is highly conserved, sharing very high DIOPT scores with mice, fish, and flies. (B) Both the missense variants (p.R298P and p.E306K) modeled in this study affect conserved amino acids in Drosophila. (C) Both missense variants lie within the Furin cleavage site or the TGF-β signaling domain of GDF11 and its homologs.
Proband 1 has a de novo p.Y336* (NP_005802.1) (NM_005811.4:c.1008C>G) variant in GDF11. The patient was born with breathing problems, hypotonia, poor suck, and many craniofacial abnormalities including a high palate, wide nose, and a broad forehead. He displayed overlapping toes and vertebral abnormalities including a spinal fusion which led to scoliosis (Figure 1A). He had profoundly delayed motor milestones, global developmental delay (DD), and intellectual disability (ID). Additionally, he has a dilated aortic root, macrocephaly, brain anomalies including agenesis of the corpus callosum, seizures, pronounced visual problems including congenital cataracts, bilateral central lens opacities, and myopia, and bilateral hearing loss.
Proband 2 has a maternally inherited heterozygous p.Q147Gfs*82 (NP_005802.1) (NM_005811.4:c.434_437dup) variant in GDF11. She presented with respiratory problems secondary to tracheomalacia at birth as well as a cleft lip and cleft palate (Figure 1B). She has mild DD and mild bilateral hearing loss with receptive and expressive speech delays that improved greatly over time. She has craniofacial abnormalities including a large and mildly dolichocephalic head with a narrow forehead. She displays vertebral abnormalities (a long neck) and additional skeletal abnormalities with short fingers, small feet, and syndactyly of the fourth and fifth toes bilaterally. She is mildly hypotonic but otherwise normal neurologically and has no observed cardiac phenotype. The proband’s mother also carries the variant and presented with similar but milder symptoms. The mother has cleft lip and palate and dolichocephaly and a long neck, missing wisdom teeth, and has narrow feet and toe abnormalities. Neurologically, the mother is normal with no ID or DD. It is not known if the mother is mosaic for the GDF11 variant.
Proband 3 has a de novo p.T319Nfs*5 (NP_005802.1) (NM_005811.4:c.955dup) variant in GDF11. He has ID and DD with delayed speech and language development. Besides a pectus excavatum and mild scapula alata, he had no craniofacial or vertebral abnormalities. This individual also presented with absence seizures; however, seizures were also observed in a sister who does not have the T319Nfs*5 variant in GDF11.
Proband 4 has a paternally inherited heterozygous p.N94Rfs*47 (NP_005802.1 ) (NM_005811.4:c.279_289del) variant in GDF11. She presented with hypoglycemia and neonatal seizures. The individual has significant DD, microcephaly, and cerebral atrophy in addition to a lack of visual fixation. This proband has no skeletal abnormalities. The father of this proband has no reported phenotypes. It is not known if the father is mosaic for the GDF11 variant.
Proband 5 has a de novo p.R295P (NP_005802.1) (NM_005811.4:c.884G>C) variant in GDF11. He has craniofacial abnormalities with marked brachycephaly and bilateral ptosis, prominent ears, and short stature with preservation of head circumference. He has additional skeletal abnormalities with marked scoliosis with hypersegmentation of his vertebrae (Figure 1F) and has a mildly dilated aortic root. He presented with a history of regression at 18 months of age following scarlet fever with a loss of speech and language skills and delayed motor milestones. He developed spasticity, episodes of dystonia, small joint hypermobility, and contractures to hips, knees, and elbows. Prior sequencing identified a p.P193A (maternal) and a p.W1211C (paternal) variant in Adenosine deaminase RNA specific (ADAR) (NM_001111.4), that has been associated with a diagnosis of Aicardi-Goutieres type 6 (AGS6, MIM#615010)42–44. His seizures, dystonia, and spasticity can probably be attributed to ADAR, however, the remaining phenotypes have not been previously associated with AGS6.
Proband 6 has a de novo p.E306K (NP_005802.1) (NM_005811.4:c.916G>A) variant in GDF11. She presented with proximal weakness and myasthenic syndrome in addition to recurrent retinal vasculitis (Figure 1G) and recurrent abdominal adhesions and hepatitis with an unclear etiology. She has mild dysmorphic facial features including a slender nasal bridge with prominent columella, significant malar flattening, a prominent forehead, flat midface, and mildly high-arched palate in addition to scoliosis, pectus carinatum, spina bifida occulta, Bertalotti Syndrome and hypermobile joints. This individual has DD but no ID or cardiac abnormalities.
In summary, most patients presented with craniofacial (5/6) and vertebral (5/6) abnormalities, in agreement with previously reported phenotypes 22. However, additional shared neurological phenotypes were present, with ID identified in 3/5 individuals, DD in 5/6, and some form of abnormal neurological presentations were identified in all probands. Other phenotypes shared amongst probands are visual disorders (4/6), hearing disorders (3/6), toe abnormalities (3/6), cardiac disorders (3/6) (with two individuals exhibiting aortic dilations), and connective tissue disorders (3/6). Additional individuals with copy number variants (CNVs) in GDF11 were identified using the DECIPHER database45. Of the eight patients with a CNV involving GDF11, three were deletions (1.28 Mb, 2.94 Mb, and 101.3Mb) and five were duplications (2.18 Mb, 3.16 Mb, 3.42 Mb, 8.80 Mb, and 9.15 Mb). These individuals are reported to have craniofacial (4/8), vertebral (4/8), and neurological abnormalities including DD (5/8) and ID (5/8). The CNVs in the DECIPHER database include many genes neighboring GDF11 (70 total genes in the smallest deletion (1.28Mb) and 1305 genes in the largest deletion (101.3Mb)) which may influence the phenotypes in each patient. Given that GDF11 is an established key signaling protein required in the development of multiple tissues in mice10,11, the diverse array of phenotypes presented in this cohort and the DECIPHER database, is consistent with these observations.
gdf11 expression in Zebrafish is analogous to GDF11 expression in humans
In mice and zebrafish, the orthologs of human GDF11 are highly conserved at the protein level (Figure 2A). The conservation of the structure of GDF11 across species predicts that the functions of GDF11 may be conserved. In zebrafish, gdf11 is expressed in numerous tissues throughout embryonic and larval development. Strong gene expression in the tailbud region at the end of gastrulation27 is consistent with a role in posterior body axis patterning noted in avian and mammalian studies10,25, and expression in the brain and pharyngeal arches was noted at later larval stages46. Using in situ hybridization and analysis of a recently published single-cell transcriptomics dataset we show that gdf11 is expressed in organs and cell types that are affected in the probands (Supplementary results, Figure S1, Figure S2).
gdf11 loss-of-function in Zebrafish phenocopies some patient phenotypes
Published functional analyses of gdf11 in Zebrafish are limited in scope and reported only for transient knockdown by morpholino oligonucleotide (MO) injection. In the initial analysis, gdf11 was knocked down to evaluate the histone deacetylase regulation of liver growth46. In a second report, gdf11 depletion by MO resulted in a caudal shift of hoxc10a expression and a corresponding caudal displacement of the pelvic fin27, similar to mouse mutant phenotypes10. To determine the role of gdf11 in additional organ systems in fish using clean genetic tools, we used CRISPR/Cas9 gene editing to generate gdf11 variants predicted to be LOF alleles (Figure 3A): one allele, b1407, contains a truncating frameshift variant in the first exon, abrogating most of the open reading frame. The second, b1408, is a truncating frameshift in the third exon, removing the region that encodes the C-terminal TGF-β domain at the region similar to the truncating variant documented in proband 1. The third, b1396, is a 703bp deletion removing the 5’UTR and most of the first exon to eliminate transcription and hence avoid genetic compensation47. Homozygotes for all three gdf11 alleles are viable but display notable abnormalities in larval and adult stages. Alcian blue and Alizarin red staining to label cartilage and bone, respectively, in 7 dpf larval zebrafish revealed a disrupted arrangement of craniofacial elements in mutants compared to wild-type siblings (Figure 3B–D). Mutants displayed an increased angle of articulation between the ceratohyal cartilage elements in young fish homozygous for the early and late truncating variants of 60.1± 4.9° and 73.3±11.2°, respectively, compared to 54.4±1.1° in wild-type fish (p = 0.014 and 0.0006). Although both statistically significant, the defects in the later truncating b1408 mutant were more severe and extended throughout the other cartilage elements of the jaw and face, including a morphological defect in the shape of the opercular bone (Figure 3D). The opercular bone is one of the first ossified bone structures formed in developing fish and provides an effective model of morphogenic variations48,49. In 7 dpf wild-type larvae, the opercular bone had a distinctive shape, narrow medially with a fan-shaped expansion of the distal end. The wild type opercular bone had a measured mean area of 1950 ± 92 μm2. By contrast, opercular bones of gdf11b1396 and gdf111408 homozygous larvae were narrow and stick-like, lacking the distal fan, with mean areas reduced by 38% and 32% (1207 ± 82 μm2 ; p < 0.0001 and 1323 ± 73.17 μm2 ; p < 0.0001), respectively. The gdf11b1407 allele had a slightly reduced operculum (1719 ± 62.7 μm2), but the 12% reduction is not statistically significant (p = 0.072). Other signs of facial dysmorphia were apparent in animals homozygous for the b1396 large deletion allele, where sagittal sections of the larval head revealed an abnormal rostral protrusion of the upper jaw element (Fig. 3F). This phenotype persisted in mutant adult fish (Fig. 3G–H) in which the rostral portion of the face was elongated, and the dorsoventral head width diminished relative to wild types. While we were unable to examine adult skeletal elements, measurements of live fish revealed that the body axis of young adult b1396 homozygotes was also abnormal; the pelvic fin was posteriorized by one body segment (Fig. 3I–J), consistent both with the earlier MO study in zebrafish27 and the mouse model in which homeotic transformations in the anterior-posterior axis were noted10. We conclude that Zebrafish lacking gdf11 function have several phenotypes similar to those observed in human probands.
Figure 3 – Zebrafish models of gdf11 loss of function exhibit craniofacial and body axis patterning defects—
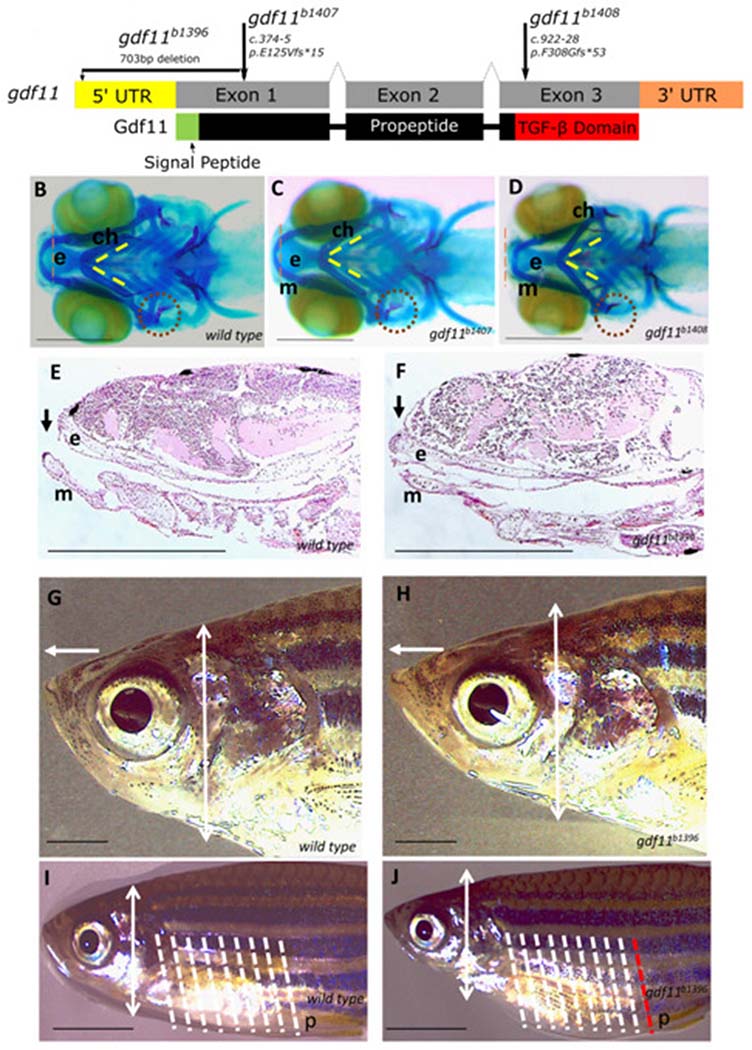
(A) Overview of the gdf11 mutants generated via CRISPR/Cas9 gene editing (B-D) Alcian and Alizarin staining of the 7dfp larval head skeleton labels cartilage (blue) and bone (red) elements. From the ventral aspect, Meckel’s cartilage (m) in the wild type larval fish (B) extends rostrally beyond the ethmoid plate of the upper jaw (e, red dotted line delineates the rostral-most edge), the bilateral ceratohyal elements (ch) meet at the midline in a constrained angle of articulation (yellow dotted lines), and the opercular bone (op), red dotted circle) is ossified in with a broadening flare at its distal end. gdf11 mutants (C, D) exhibit defects in the alignment of upper and jaw elements, in the angle of ch articulation, and the morphology of the op with a more severe phenotype observed in the late truncating allele (D). (E-F) Upper and lower jaw element alignment are visualized again in sagittal sections of H & E stained 7dfp wild type (E) and gdf11 mutant (F) larvae, in which the ethmoid plate protrudes beyond the rostral limit of Meckel’s cartilage. (G-H) 6 month gdf11 mutant (H) rostral length measured from the anterior edge of the eye to the tip of the nose (white arrow) is 15% longer than in stage-matched wild type (G; p = 0.0007) while the dorsoventral thickness of the head posterior to the eye (white double arrowhead, also marked in panels I & J) is an average of 15% less (p = 0.001) than in wild type. (I-J) Regular anterior-posterior arrangements of body segments are visible on the lateral exterior or the juvenile fish (shown at 2 months in I and J), with eight such segments (white dotted lines) falling between the pectoral and pelvic (p) fins. One additional segment is noted in gdf11 mutants (J, white, and red dotted lines). N ≥ 8 for each group; scale bars: B-F 250μm; G-J 1mm.
Overexpression based assays of GDF11 variants in Drosophila indicates that they are LOF variants
Variant pathogenicity prediction programs suggest that the human GDF11 variants are damaging. To test this hypothesis, we used the fruit fly Drosophila melanogaster. Flies have been used effectively to identify LOF variants in human genes, elucidate mechanisms, and identify therapeutic drugs50. In Drosophila, the closest homolog to GDF11 is myoglianin (myo) (Figure 2A)51. The fly myo gene is the only orthologue of both GDF11 (DIOPT 7/15) and GDF8/MSTN (myostatin, DIOPT 8/15)34. myo encodes a larger protein than human GDF11 (598 vs 405 AA) which affects protein similarity and identity scores. However, the amino acid similarity of the secreted TGF-β domain is 76%, indicating that the key signaling domain of GDF11 is highly conserved in flies (Figure 2A). LOF alleles in myo have been reported to cause pupal lethality before head eversion33.
To determine the functionality of the probands’ variants, we generated constructs containing the wild-type myo gene (myo-WT) with an upstream activation sequence (UAS). We also generated UAS-myo constructs with variants in the location homologous to three of the probands in this cohort, one nonsense variant p.Y336* from proband 1 (myo-F530*), and two missense variants, p.R298P from proband 5 (myo-R489P) and p.E306K from proband 6 (myo-E500K) (Figure S4C). We used site-directed mutagenesis and injected each construct into the VK00033 landing site via phiC31 integrase mediated transgenesis to ensure constant transgene expression across constructs (Figure S4B)52,53. To assess the function of each myo variant, we first replaced the endogenous myo by inserting a T2A-GAL4 CRISPR-Mediated Integration Cassette (CRIMIC) cassette into the first coding intron of myo54, creating a myo-T2A-GAL4 allele (Figure S4A). Unfortunately, we were not able to rescue myo null induced homozygous lethality (supplemental results).
Ubiquitous overexpression of myo has been shown to cause pupal lethality when driven with Actin-Gal4 (Act-GAL4)33. To detect differences in functionality of the myo variants, we overexpress myo-WT, myo-F530*, myo-E500K, or myo-R489P using Act-GAL4 to assess the lethality of each of the variants (Figure 4A). As a control, we use animals containing an empty UAS promoter (UAS-empty) inserted into the same docking site. When UAS-myo-WT is driven ubiquitously we observe lethality at 22°C or higher. However ubiquitous expression of myo is toxic even at low levels, as only 1.91% of Act-GAL4>UAS-myo-WT eclose as adults compared to Act-GAL4>UAS-empty at 18°C (Figure 4B). We observe no toxicity when overexpressing UAS-myo-F530X with Act-GAL4, suggesting that this truncation is indeed a LOF allele. In contrast, the two missense (p.E500K, p.R489P) alleles do cause lethality when overexpressed, but to different degrees when compared to WT. UAS-myo-E500K had similar toxicity as UAS-myo-WT (lethal at all temperatures). However, the number of Act-GAL4>UAS-myo-E500K animals that eclose at 18°C (9.00%) is significantly greater (χ2, p-value = 0.0003) than the number of Act-GAL4>UAS-myo-WT animals that eclose (1.91%), indicating a possible minor loss of myo toxicity (Figure 4B, C). UAS-myo-R489P is viable at low temperatures (18°C and 22°C), but the viability decreased at temperatures >25°C, suggesting the impaired function of this variant. In addition to lethality, we also find that ectopic expression of myo variants causes morphological phenotypes in the eye (Figure 4E). Act-GAL4 driving UAS-myo-E500K or UAS-myo-R489P at 18°C causes a rough eye phenotype. This phenotype was not seen with Act-GAL4>UAS-myo-F530* again suggesting residual functions of the two missense variants. We did not obtain enough UAS-myo-WT animals to analyze whether this transgene causes a rough eye phenotype or not.
Figure 4 – Patient variants behave as strong or mild loss-of-function alleles in flies.
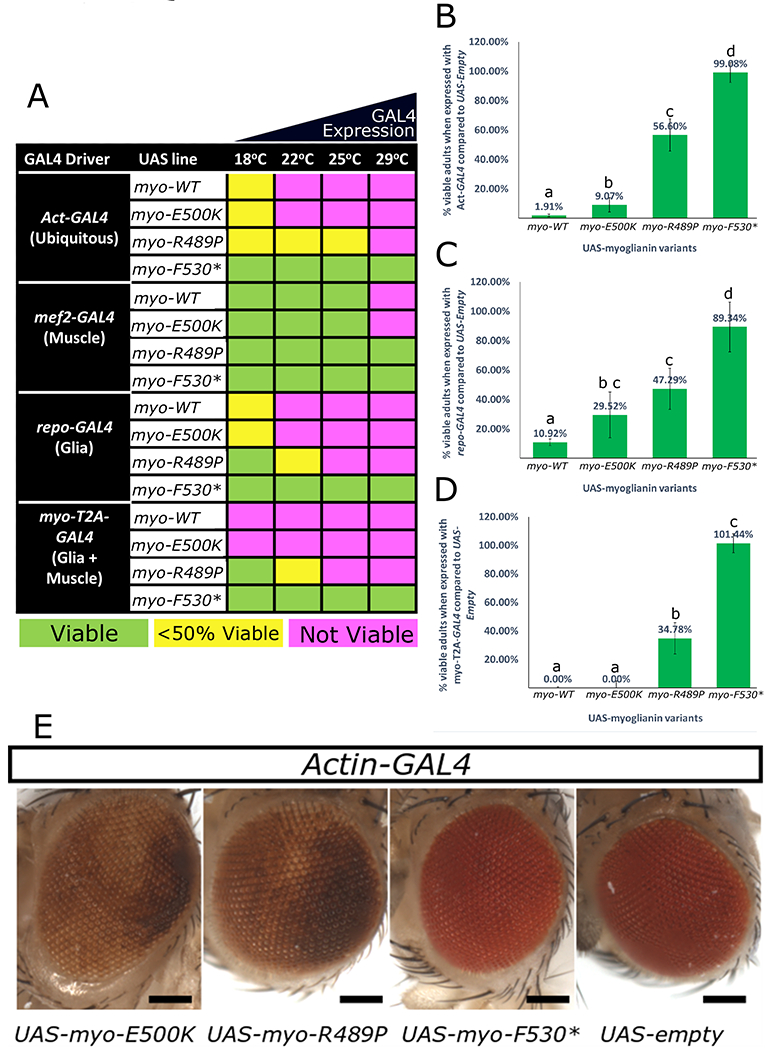
A mutant form of myo that corresponds to 3 of the proband’s variants (p.R295P, p.E306K, and p.Y336*) along with a wild type myo construct (WT) and an empty UAS-vector (negative control) were expressed with various GAL4 drivers to determine their effect when overexpressed. (A) Ubiquitous overexpression of myo-WT and overexpression with myo-T2A-GAL4 allele is lethal except at low temperatures (18°C) when GAL4 is less abundant. Ubiquitous overexpression of myo-E500K mirrors the lethality of myo-WT, myo-R498P is viable at higher temperatures and no lethality is observed when myo-F530* is expressed at any temperature. When overexpressed specifically in muscles, myo-WT and myo-E500K are only lethal at 29°C while myo-R498P and myo-F530X are viable. When overexpressed specifically in glial cells, the toxicity mirrors that seen with ubiquitous overexpression. The numbers of viable animals were quantified for ubiquitous expression (B), glial expression (C), and with myo-T2A-GAL4 expression (D). These data indicate a decreasing scale of toxicity of myo-WT>myo-E500K>myo-R489P>myo-F530X. This trend is also seen with repo-GAL4 and myo-T2A-GAL4 at 18°C. (B-D) Lower case letters represent groups significantly different (χ2, p <0.05) from each other. (E) When myo-E500K and myo-R489P variants are expressed ubiquitously at 18°C a rough eye phenotype is observed indicating a developmental issue. All eye pictures are taken under the same magnification and were processed identically. Scale bar = 200μm. Error bars = SD.
To assess the consequences of overexpression of the myo WT and variant alleles in the cells where myo is normally expressed we used myo-T2A-GAL4 (muscle and glia), mef2-GAL4 (muscle) and repo-GAL4 (glia) to drive various myo transgenes at different temperatures. The same trend for toxicity was seen for each driver with UAS-myo-WT showing the strongest toxicity, followed by UAS-myo-E500K then UAS-myo-R489P, and finally UAS-myo-F530* and UAS-empty causing no lethality (Supplementary results, Figure 4A, 4C, 4D). The absence of increased lethality at any temperature when the myo-F530* allele is expressed with any GAL4 driver indicates that the allele is unlikely to have a dominant negative effect. These data indicate that myo-F530* is a strong LOF allele, myo-R489P a partial LOF allele, and myo-E500K a milder LOF allele.
Discussion
Craniofacial and vertebral abnormalities are related to LOF variants in GDF11 in human patients22 and rodent knockout models10. Here, we report four patients with strong LOF variants in GDF11, with only one patient having severe craniofacial and vertebrae abnormalities. Patients with truncation alleles in GDF11 present with a higher prevalence of neurological abnormalities, developmental delays, and visual problems. Additionally, neurological, developmental, and ocular abnormalities have a stronger correlation with the degree of GDF11 LOF than do vertebral and craniofacial abnormalities, indicating GDF11 dosage may have a greater influence on nervous system development than on the development of other tissues.
In Zebrafish, craniofacial abnormalities vary in severity among LOF alleles. Variants that result in NMD have been found to trigger genetic compensation through the activation of related genes47. Thus, the milder phenotype observed in the early truncating allele (b1407) may be due to this transcriptional switch, whereas the later truncation (b1408), would be presumed to escape genetic compensation. The large deletion (b1396), which was designed to block transcription altogether, is predicted to be immune from genetic compensation and thus a complete LOF. The viability and somewhat milder phenotypes of these Zebrafish mutant alleles, compared to the mouse and fly models, suggest some functional redundancy, which may mirror some of the clinical phenotypes of the probands in this study.
Interestingly, the severity of the LOF alleles reported from the fly experiments correlates with the severity of the neurological phenotypes seen in our patient cohort. The four probands with nonsense variants all show profound DD and 3/4 probands have associated ID. The patient with a partial LOF allele (proband 5 – p.R295P) presents with ID but not DD, a milder presentation than the complete LOF variant patients but more severe than the milder LOF patient (proband 6 – p.E306K). This gradient of symptom severity indicates that the degree of GDF11 function loss in patients reflects the severity of the neurological disorder. In agreement with this observation, LOF alleles in Drosophila myo33 and mice Gdf1110 have severe nervous system defects. Additionally, overexpression of myo variants causes a rough eye phenotype in Drosophila, indicative of a neurodevelopmental defect in the fly visual system. Although the severity of craniofacial and vertebral dysmorphism in probands is variable, genotype-phenotype correlation can be seen in these organ systems. Probands with full cleft lip/palate have a complete LOF nonsense variant and those with minor craniofacial phenotypes have partial/milder LOF alleles. However, the minor phenotypic presentation in the mother of proband 2 and the lack of any reported phenotypes in the father of proband 4 is an indicator of the variable expressivity and incomplete penetrance associated with GDF11 LOF variants. In agreement with this is the lack of vertebral phenotypes in probands 3 and 4, the lack of craniofacial dysmorphism in proband 3 and the variability of phenotypes in a previously reported family with a GDF11 variant22. These phenotypes are likely more influenced by other genetic or environmental factors than the neurological phenotypes, which more closely correlate with the severity of the GDF11 LOF variants.
How loss of GDF11 disrupts neuronal development is unclear. In mouse olfactory epithelium, Gdf11 negatively regulates neurogenesis by promoting cell cycle arrest in neuronal progenitors via 27Kip1 and/or p21Cip1 and inactivation of Foxg123,55. Also in the brain, Gdf11 acts as a negative regulator of gliogenesis, favoring stem cell differentiation into neuronal precursor cells56. In contrast, in the spinal cord, loss of Gdf11 causes a decrease in proliferation of spinal cord motoneurons in addition to aberrant rostral/caudal patterning of motoneurons as a result of expanded Hoxc expression25,29. In the retina, Gdf11 is a negative regulator of retinal ganglion cell proliferation. Interestingly the latter is not via cell cycle arrest as in the olfactory epithelium, but instead via downregulation of Math526. Hence, although Gdf11 is a key player in neuronal development, predicting how these disruptions manifest in a phenotype in humans, is not yet obvious.
The impact on the cardiovascular system is also seen in patients with GDF11 LOF variants. GDF11 is expressed in cardiac muscle in adults and is expressed in neural crest cells that signal the development of cardiac structures such as the aorta in mammals and zebrafish. In both adult mice and humans, the role of GDF11 is controversial with debate on whether increasing circulating GDF11 helps cardiac health20,57–59, and the role of GDF11 in the developing heart has not been well studied in vivo in model organisms. Cardiomyocyte Gdf11 knockout mice have left ventricular dilation60, indicating a potential association between a loss of GDF11 and cardiovascular abnormalities, which is consistent with the two patients in our cohort with aortic dilation. Gdf11 initiates intracellular Smad2 activation by binding to the Activin receptors TGFBR1 and ACVR2B12–14. LOF variants in human TGFBR1 and ACVR2B are associated with defects in cardiac development61,62. Among our cohort of patients with GDF11 LOF variants, 3/6 patients have cardiac abnormalities and two have aortic dilations. The influence of GDF11 specifically on the developing human heart is likely to be complex due to the compensatory roles of MSTN and its ability to bind the same receptors as GDF1163. The expression of these different GDF paralogs, the diversity of the receptors, and modulators, such as follistatin, may impact how cardiac malformations present in GDF11 LOF variants. However, cardiac abnormalities, particularly aortic dilations, should be screened for in patients with variants in GDF11.
Both partial LOF variants present in this cohort, in addition to a family member in the previously reported family22, present with connective tissue abnormalities resulting in hypermobile joints. Because the most common cause of joint hypermobility is a lack of collagen and GDF11 induces the expression of collagen I and III, the connective tissue disorders are seen in patients may also be due to partial LOF variants in GDF1164, which will require further biological studies.
In conclusion, we have identified a cohort of six patients from six families with LOF variants in GDF11. The cohort has complex clinical presentations significantly expanding the phenotypes linked to variants in this gene. We have generated gdf11 Zebrafish mutants that exhibit craniofacial and body axis patterning abnormalities that reflect gdf11 expression patterns and some of the key clinical presentations of the human subjects. Using Drosophila, we have been able to determine the degree of GDF11 functional loss for a subset of variants, showing that LOF severity measured in flies correlates with the severity of neurological phenotypes in humans. The variable expressivity of GDF11-associated phenotypes is likely a result of the complexities and redundancies of GDF signaling throughout development as well as other genetic and environmental factors. To further elucidate these additional factors, we will need an expanded cohort of patients with LOF variants in GDF11. This study provides the resources for modeling and evaluating GDF11 LOF variants in model organisms and the potential phenotypes caused by GDF11 variants.
Supplementary Material
Acknowledgments
We would like to thank the patients and their families who participated in this study. The research reported in this manuscript was supported by the NIH Common Fund, the Office of Strategic Coordination and Office of the NIH Director under Award Numbers U01HG007942 (BCM sequencing core), U54NS093793 (Model Organism Screening Center of the Undiagnosed Diseases Network), R24OD026591 (J.H.P and M.W) and U01HG007690 (BWH clinical site). The Care4Rare Research Consortium performed the re-analysis of the exome data for patient 5 and is funded by Genome Canada and the Ontario Genomics Institute (OGI-147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Q8 Quebec, and Children’s Hospital of Eastern Ontario Foundation. H.J.B. and S.Y. are supported by R24OD022005 from ORIP at NIH. myo-T2A-GAL4 generation was funded as part of the genome disruption project (NIGMS GM132087). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. T.A.R. has been supported by The Cullen Foundation. S.S.B. is supported by F32HD100048 from NICHD at NIH. H.J.B. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of interests
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories. The authors have no other conflicts of interest.
Description of supplemental data
Supplemental data include 4 figures, 2 tables, patient clinical reports, supplemental materials, methods, and results.
Data and code availability
The manuscript includes all [datasets/code] generated or analyzed during this study.
Ethics Declaration
Written informed consent for genetic testing and publication of relevant findings and photographs was obtained from all patients or their parents. Research using patient cells is approved by the Institutional Review Board for Human Subject Research for Baylor College of Medicine and Affiliated Hospitals (BCM IRB) for translational models of neurological disease at the neurological research institute (Human Subjects Assurance Number: 00000286). The BCM IRB is organized, operates, and is registered with the United States Office for Human Research Protections according to the regulations codified in the United States Code of Federal Regulations at 45 CFR 46 and 21 CFR 56. The BCM IRB operates under the BCM Federal Wide Assurance No. 00000286, as well as those of hospitals and institutions affiliated with the College. Zebrafish were raised and all experiments were conducted according to standard protocols approved by the University of Oregon IACUC.
References
- 1.Lee SJ (1990). Identification of a novel member (GDF-1) of the transforming growth factor-β superfamily. [DOI] [PubMed] [Google Scholar]
- 2.Akhurst RJ, and Hata A (2012). Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frikha R (2020). Klippel-Feil syndrome: a review of the literature. Clin. Dysmorphol 29, 35–37. [DOI] [PubMed] [Google Scholar]
- 4.Naikmasur VG, Sattur AP, Kirty RN, and Thakur AR (2011). Type III Klippel-Feil syndrome: Case report and review of associated craniofacial anomalies. Odontology 99, 197–202. [DOI] [PubMed] [Google Scholar]
- 5.Asai-Coakwell M, French CR, Ye M, Garcha K, Bigot K, Perera AG, Staehling-Hampton K, Mema SC, Chanda B, Mushegian A, et al. (2009). Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum. Mol. Genet 18, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otsuka F, McTavish KJ, and Shimasaki S (2011). Integral role of GDF-9 and BMP-15 in ovarian function. Mol. Reprod. Dev 78, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McPherron AC, Huynh TV, and Lee SJ (2009). Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev. Biol 9,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, and Tabin CJ (2006). Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, 2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao R, Lawler AM, and Lee SJ (1999). Characterization of GDF-10 expression patterns and null mice. Dev. Biol 212, 68–79. [DOI] [PubMed] [Google Scholar]
- 10.McPherron AC, Lawler AM, and Lee SJ (1999). Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat. Genet 22, 260–264. [DOI] [PubMed] [Google Scholar]
- 11.Walker RG, Poggioli T, Katsimpardi L, Buchanan SM, Oh J, Wattrus S, Heidecker B, Fong YW, Rubin LL, Ganz P, et al. (2016). Biochemistry and Biology of GDF11 and Myostatin: similarities, differences and questions for future investigation HHS Public Access. Circ Res 118, 1125–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul Oh S, Yeo CY, Lee Y, Schrewe H, Whitman M, and Li E (2002). Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev. 16, 2749–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, and Attisano L (2003). Myostatin Signals through a Transforming Growth Factor β-Like Signaling Pathway To Block Adipogenesis. Mol. Cell. Biol 23, 7230–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson O, Reissmann E, Jörnvall H, and Ibáñez CF (2006). Synergistic interaction between Gdf1 and Nodal during anterior axis development. Dev. Biol 293, 370–381. [DOI] [PubMed] [Google Scholar]
- 15.Bajikar SS, Wang CC, Borten MA, Pereira EJ, Atkins KA, and Janes KA (2017). Tumor-Suppressor Inactivation of GDF11 Occurs by Precursor Sequestration in Triple-Negative Breast Cancer. Dev. Cell 43, 418–435.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A. C. McPherron (2012). Metabolic Functions of Myostatin and GDF11. Immunol. Endocr. Metab. Agents Med. Chem 10, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamer LW, Wolfman NM, Celeste AJ, Hattersley G, Hewick R, and Rosen V (1999). A novel BMP expressed in developing mouse limb, spinal cord, and tail bud is a potent mesoderm inducer in Xenopus embryos. Dev. Biol 208, 222–232. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima M, Toyono T, Akamine A, and Joyner A (1999). Expression of growth/differentiation factor 11, a new member of the BMP/TGFβ superfamily during mouse embryogenesis. Mech. Dev 80, 185–189. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Shao J, Wang Z, Yang T, Liu S, Liu Y, Fan X, and Ye W (2015). Growth differentiation factor 11 is a protective factor for osteoblastogenesis by targeting PPARgamma. Gene 557, 209–214. [DOI] [PubMed] [Google Scholar]
- 20.Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’Osso C, Khong D, Shadrach JL, et al. (2013). Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153, 828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeanplong F, Falconer SJ, Thomas M, Matthews KG, Oldham JM, Watson T, and Mcmahon CD (2012). Growth and differentiation factor-11 is developmentally regulated in skeletal muscle and inhibits myoblast differentiation. Open J. Mol. Integr. Physiol 2, 127–138. [Google Scholar]
- 22.Cox TC, Lidral AC, McCoy JC, Liu H, Cox LL, Zhu Y, Anderson RD, Moreno Uribe LM, Anand D, Deng M, et al. (2019). Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial clefting in humans. Hum. Mutat 40, 1813–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, and Calof AL (2003). Autoregulation of neurogenesis by GDF11. Neuron 37, 197–207. [DOI] [PubMed] [Google Scholar]
- 24.Suh J, Kim NK, Lee SH, Eom JH, Lee Y, Park JC, Woo KM, Baek JH, Kim JE, Ryoo HM, et al. (2020). GDF11 promotes osteogenesis as opposed to MSTN, and follistatin, a MSTN/GDF11 inhibitor, increases muscle mass but weakens bone. Proc. Natl. Acad. Sci. U. S. A 117, 4910–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JP (2006). The function of growth/differentiation factor 11 (Gdf11) in rostrocaudal patterning of the developing spinal cord. Development 133, 2865–2874. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, and Calof AL (2005). Developmental biology: GDF11 controls the timing of progenitor cell competence in developing retina. Science (80-.). 308, 1927–1930. [DOI] [PubMed] [Google Scholar]
- 27.Murata Y, Tamura M, Aita Y, Fujimura K, Murakami Y, Okabe M, Okada N, and Tanaka M (2010). Allometric growth of the trunk leads to the rostral shift of the pelvic fin in teleost fishes. Dev. Biol 347, 236–245. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein JM, Valido A, Lewandowski JP, Walker RG, Mills MJ, Messemer KA, Besseling P, Lee KH, Wattrus SJ, Cho M, et al. (2019). Variation in zygotic CRISPR/Cas9 gene editing outcomes generates novel reporter and deletion alleles at the Gdf11 locus. Sci. Rep 9, 18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Y, and Liu JP (2011). Gdf11 facilitates temporal progression of neurogenesis in the developing spinal cord. J. Neurosci 31, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodríguez-Marí A, Yan YL, BreMiller RA, Wilson C, Cañestro C, and Postlethwait JH (2005). Characterization and expression pattern of zebrafish anti-Müllerian hormone (amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development. Gene Expr. Patterns 5, 655–667. [DOI] [PubMed] [Google Scholar]
- 31.Farnsworth DR, Saunders LM, and Miller AC (2020). A single-cell transcriptome atlas for zebrafish development. Dev. Biol 459, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker MB, and Kimmel CB (2007). A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem 82, 23–28. [DOI] [PubMed] [Google Scholar]
- 33.Awasaki T, Huang Y, O’Connor MB, and Lee T (2011). Glia instruct developmental neuronal remodeling through TGF-Î 2 signaling. Nat. Neurosci 14, 821–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Al-Ouran R, Hu Y, Kim SY, Wan YW, Wangler MF, Yamamoto S, Chao HT, Comjean A, Mohr SE, et al. (2017). MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung H. lok, Wangler MF, Marcogliese PC, Jo J, Ravenscroft TA, Zuo Z, Duraine L, Sadeghzadeh S, Li-Kroeger D, Schmidt RE, et al. (2020). Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms. Neuron 106, 589–606.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanca O, Zirin J, Garcia-Marques J, Knight SM, Yang-Zhou D, Amador G, Chung H, Zuo Z, Ma L, He Y, et al. (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. Elife 8,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rentzsch P, Witten D, Cooper GM, Shendure J, and Kircher M (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H, and Wang K (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJV, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, et al. (2014). Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics 45, 386–391. [DOI] [PubMed] [Google Scholar]
- 43.Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CGEL, Falconer A, et al. (2014). A type i interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J. Med. Genet 51, 76–82. [DOI] [PubMed] [Google Scholar]
- 44.Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012). Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type i interferon signature. Nat. Genet 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Vooren S. Van, Moreau Y, Pettett RM, and Carter NP (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farooq M, Sulochana KN, Pan X, To J, Sheng D, Gong Z, and Ge R (2008). Histone deacetylase 3 (hdac3) is specifically required for liver development in zebrafish. Dev. Biol 317, 336–353. [DOI] [PubMed] [Google Scholar]
- 47.El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huycke TR, Frank Eames B, and Kimmel CB (2012). Hedgehog-dependent proliferation drives modular growth during morphogenesis of a dermal bone. Dev. 139, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tarasco M, Laizé V, Cardeira J, Cancela ML, and Gavaia PJ (2017). The zebrafish operculum: A powerful system to assess osteogenic bioactivities of molecules with pharmacological and toxicological relevance. Comp. Biochem. Physiol. Part - C Toxicol. Pharmacol 197, 45–52. [DOI] [PubMed] [Google Scholar]
- 50.Bellen HJ, Wangler MF, and Yamamoto S (2019). The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet 28, R207–R214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo PCH, and Frasch M (1999). Sequence and expression of myoglianin, a novel Drosophila gene of the TGF-β superfamily. Mech. Dev 86, 171–175. [DOI] [PubMed] [Google Scholar]
- 52.Groth AC, Fish M, Nusse R, and Calos MP (2004). Construction of Transgenic Drosophila by Using the Site-Specific Integrase from Phage ϕC31. Genetics 166, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venken KJT, He Y, Hoskins RA, and Bellen HJ (2006). P[acman]: A BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science (80-.). 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- 54.Lee PT, Zirin J, Kanca O, Lin WW, Schulze KL, Li-Kroeger D, Tao R, Devereaux C, Hu Y, Chung V, et al. (2018). A gene-specific T2A-GAL4 library for drosophila. Elife 7,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawauchi S, Kim J, Santos R, Wu HH, Lander AD, and Calof AL (2009). Foxg1 promotes olfactory neurogenesis by antagonizing Gdf11. Development 136, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gokoffski KK, Wu HH, Beites CL, Kim J, Kim EJ, Matzuk MM, Johnson JE, Lander AD, and Calof AL (2011). Activin and GDF11 collaborate in feedback control of neuroepithelial stem cell proliferation and fate. Development 138, 4131–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schafer MJ, Atkinson EJ, Vanderboom PM, Kotajarvi B, White TA, Moore MM, Bruce CJ, Greason KL, Suri RM, Khosla S, et al. (2016). Quantification of GDF11 and Myostatin in Human Aging and Cardiovascular Disease. Cell Metab. 23, 1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith SC, Zhang X, Zhang X, Gross P, Starosta T, Mohsin S, Franti M, Gupta P, Hayes D, Myzithras M, et al. (2015). GDF11 Does Not Rescue Aging-Related Pathological Hypertrophy. Circ. Res 117, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olson KA, Beatty AL, Heidecker B, Regan MC, Brody EN, Foreman T, Kato S, Mehler RE, Singer BS, Hveem K, et al. (2015). Association of growth differentiation factor 11/8, putative anti-ageing factor, with cardiovascular outcomes and overall mortality in humans: Analysis of the Heart and Soul and HUNT3 cohorts. Eur. Heart J 36, 3426–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garbern J, Kristl AC, Bassaneze V, Vujic A, Schoemaker H, Sereda R, Peng L, Ricci-Blair EM, Goldstein JM, Walker RG, et al. (2019). Analysis of Cre-mediated genetic deletion of Gdf11 in cardiomyocytes of young mice. Am. J. Physiol. - Hear. Circ. Physiol 317, H201–H212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al. (2005). A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet 37, 275–281. [DOI] [PubMed] [Google Scholar]
- 62.Kosaki R, Gebbia M, Kosaki K, Lewin M, Bowers P, Towbin JA, and Casey B (1999). Left-right axis malformations associated with mutations in ACVR2B, the gene for human activin receptor type IIB. Am. J. Med. Genet 82, 70–76. [DOI] [PubMed] [Google Scholar]
- 63.Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE, Mallozzi C, Jacobi C, Jennings LL, Clay I, et al. (2015). GDF11 Increases with Age and Inhibits Skeletal Muscle Regeneration. Cell Metab. 22, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tito A, Barbulova A, Zappelli C, Leone M, Ruvo M, Mercurio FA, Chambery A, Russo R, Colucci MG, and Apone F (2019). The Growth Differentiation Factor 11 is Involved in Skin Fibroblast Ageing and is Induced by a Preparation of Peptides and Sugars Derived from Plant Cell Cultures. Mol. Biotechnol 61, 209–220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.