

Abstract
The Polycomb group (PcG) protein Enhancer of Zeste Homolog 2 (EZH2) is one of the three core subunits of the Polycomb Repressive Complex 2 (PRC2). It harbors histone methyltransferase activity (MTase) that specifically catalyze histone 3 lysine 27 (H3K27) methylation on target gene promoters. As such, PRC2 are epigenetic silencers that play important roles in cellular identity and embryonic stem cell maintenance. In the past two decades, mounting evidence supports EZH2 mutations and/or over-expression in a wide array of hematological cancers and solid tumors, including prostate cancer. Further, EZH2 is among the most up-regulated genes in neuroendocrine prostate cancers, which become abundant due to the clinical use of high-affinity androgen receptor pathway inhibitors. While numerous studies have reported epigenetic functions of EZH2 that inhibit tumor suppressor genes and promote tumorigenesis, discordance between EZH2 and H3K27 methylation has been reported. Further, enzymatic EZH2 inhibitors have shown limited efficacy in prostate cancer, warranting a more comprehensive understanding of EZH2 functions. Here we first review how canonical functions of EZH2 as a histone MTase are regulated and describe the various mechanisms of PRC2 recruitment to the chromatin. We further outline non-histone substrates of EZH2 and discuss post-translational modifications to EZH2 itself that may affect substrate preference. Lastly, we summarize non-canonical functions of EZH2, beyond its MTase activity and/or PRC2, as a transcriptional cofactor and discuss prospects of its therapeutic targeting in prostate cancer.
Keywords: histone methylation, non-histone substrate, PRC2, post-translational modifications, SUZ12, lncRNA, neuroendocrine prostate cancer, FOXA1, androgen receptor, EZH2 inhibitors, protein degradation, DNA methylation
INTRODUCTION
Polycomb Group (PcG) proteins are an important family of epigenetic regulators that modify histones for epigenetic silencing of gene transcription. PcG proteins were originally discovered in Drosophila melanogaster where they function as negative regulators of homeotic gene expression during development [1]. In mammals, PcG proteins form two major multiprotein complexes termed Polycomb Repressive Complex 1 (PRC1) and Polycomb Repressive Complex 2 (PRC2) [2–4], the latter of which is the focus of this present review. Enhancer of Zeste Homolog 2 (EZH2), the catalytic subunit of PRC2 that mediates histone H3 lysine 27 trimethylation (H3K27me3), has been shown to be frequently mutated in hematological cancers but overexpressed in human solid tumors, where its expression and activity correlate with disease progression [5]. The initial discovery of EZH2 overexpression in prostate cancer (PCa) has fueled a plethora of studies on the roles of PRC2-dependent gene silencing in driving cancer progression, which we have previously reviewed [6]. EZH2 has thus become an attractive target for cancer therapy and a number of small molecule inhibitors have been developed to block its enzymatic activity [5]. However, while these enzymatic inhibitors of EZH2 have proven particularly effective in lymphoma [5], they have shown limited efficacy in PCa. Increasing evidence suggest that EZH2 may harbor context-dependent multifunctionalities beyond H3K27me3, introducing complexity into our current understanding of its role in cancer progression. The purpose of this review is to provide a general discussion of the canonical and non-canonical functions of EZH2 and then focus on the diverse roles of EZH2 specifically in PCa and outline strategies to target EZH2 and combat PCa progression.
I. CANONICAL FUNCTION OF EZH2 AS A HISTONE METHYLTRANSFERASE
Regulation of EZH2 histone methyltransferase activity
PcG proteins EZH2, Suppressor of Zeste 12 Protein Homolog (SUZ12), and Embryonic Ectoderm Development (EED), along with histone-binding protein RBBP4/7, are core subunits of the PRC2 [7]. EZH2 contains a C-terminal SET domain, a conserved feature of histone methyltransferases (HMTase), and carries out the catalytic activities of PRC2 specifically towards H3K27, leading to chromatin compaction and epigenetic silencing of target genes [7–11]. A large portion of the genome is methylated on H3K27 by PRC2 in a step-wise process involving the transfer of a methyl group to form mono- (me1), di- (me2), and ultimately trimethylated H3K27 (H3K27me3) [12, 13]. Of these methylated H3, roughly 20% harbor H3K27me1, 50% harbor H3K27me2, and 10%–20% harbor H3K27me3 in embryonic stem cells (ESC) [14, 15]. Though EZH2 contains a SET domain that possesses HMTase activity, EZH2 protein by itself has low HMTase activity, at least when probed in vitro [9, 11]. Multiple studies in Drosophila melanogaster and mammalian systems have focused on delineating the functions of other PRC2 subunits in facilitating EZH2 HMTase activity. EZH2 catalytic SET domain adapts an auto-inhibitory conformation, which is relieved upon EZH2 binding with EED and SUZ12 [16, 17]. Initial H3K27me3 deposition generates a feedforward loop by binding to the aromatic cage of EED, resulting in allosteric activation of the PRC2 complex, which further leads to additional H3K27me3 deposition and propagation of repressive chromatin domains (Fig. 1A) [18–21]. Moreover, SUZ12 is indispensable for protein stability and catalytic activity of the PRC2 complex (Fig. 1B) [7, 22, 23]. Mechanistically, the C-terminal VEFS domain of SUZ12 forms a stable complex with EZH2 and EED that is catalytically competent to establish normal global H3K27 methylation in vivo [15]. In addition, PRC2 core subunits RBBP4 and RBBP7 are required for PRC2 binding to unmodified nucleosomes and are necessary for full HMTase activity [7, 24].
Figure 1. Cooperation between PRC2 and other proteins regulates gene transcription.
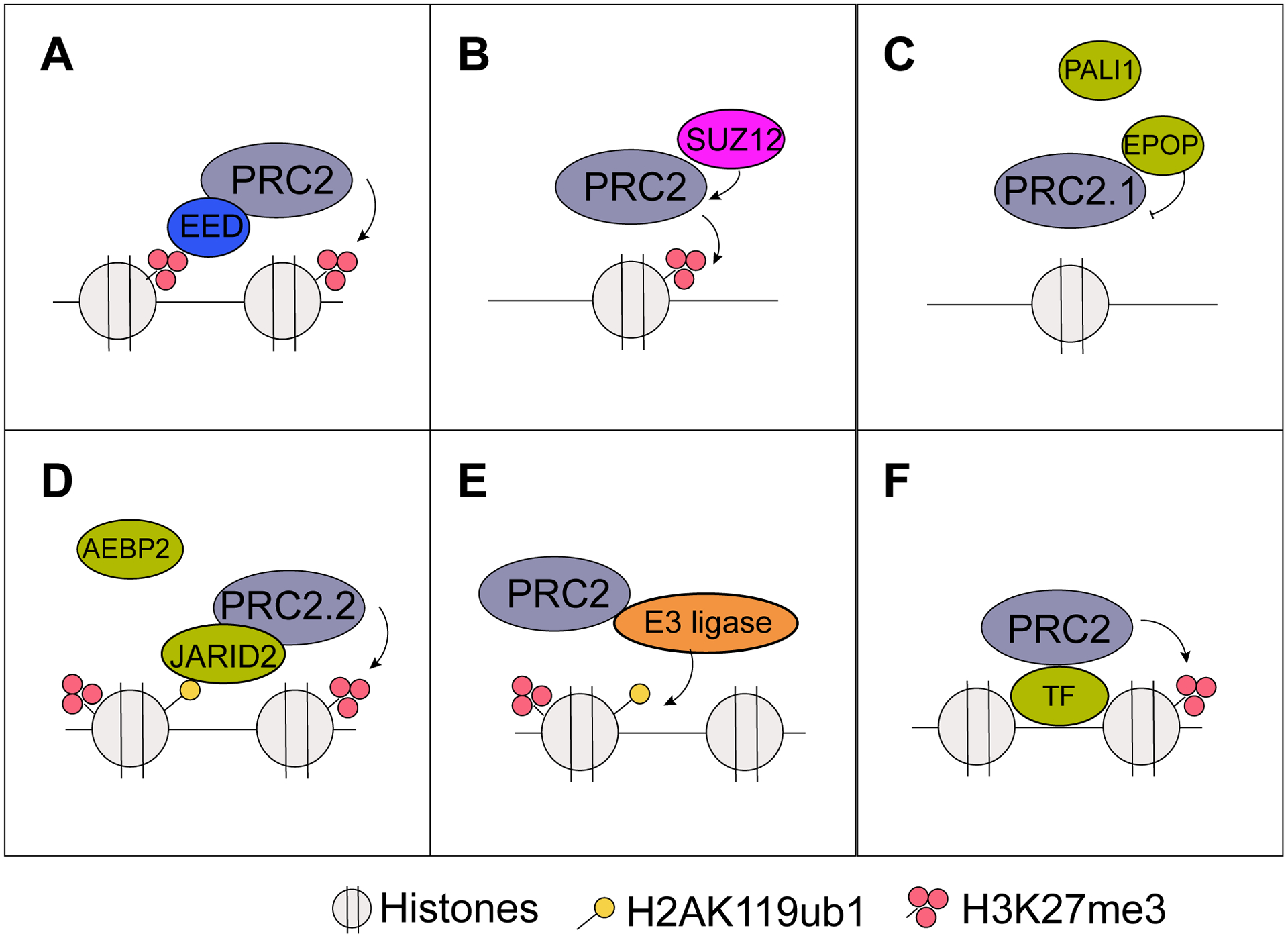
(A) EED binds H3K27me3 and recruits the PRC2 complex. (B) SUZ12 regulates the activity and stability of the PRC2 complex. (C) EPOP inhibits excessive activity and chromatin binding of the PRC2.1 complex. (D) JARID2 binds H2AK119ub1 and recruits the PRC2.2 complex. (E) The interplay between PRC2 and different E3 ligases. (F) Different DNA-binding proteins regulate PRC2 actions.
Recent studies have further stratified PRC2 into two subcomplexes, namely PRC2.1 and PRC2.2, each with distinct non-core PRC2 subunits. PRC2.1 contains Polycomb-like (PCL) proteins (PCL1/PHF1, PCL2/MTF2, and PCL3/PHF19), EPOP, and PALI1, whereas PRC2.2 utilizes accessory proteins JARID2 and AEBP2 [25, 26]. Although all of these PRC2 accessory proteins have been shown to stimulate PRC2 HMTase activity in in vitro assays [7, 27–31], some discrepancies have been observed in vivo. PCL family member PCL1/PHF1 was first reported to enhance H3K27me3, but not H3K27me1 or H3K27me2 [27, 28]. However, PCL1 was also shown to bind to H3K36me3 through its Tudor domain and inhibit PRC2 HMTase activity [32, 33]. PCL3, on the other hand, represses gene transcription by recruiting the H3K36me3 demethylase NO66 [34]. EPOP is another important accessory protein of PRC2.1. In mouse embryonic stem cells (mESCs), both PRC2 recruitment and H3K27me3 were increased in EPOP-targeted regions upon EPOP knockout, indicating that EPOP may inhibit PRC2 methyltransferase activity in vivo (Fig. 1C) [35, 36]. The exact mechanism by which EPOP suppresses PRC2 catalytic activity warrants further investigation. PALI1 is the most recently characterized PRC2 non-core subunit. Although PALI1 has been consistently identified as part of the PRC2.1 complex using mass spectrometry analyses [31, 37], its presence in PRC2.1 is mutually exclusive with EPOP. PALI1 was also shown to compete with AEBP2 at similar genomic targets during mESCs differentiation, which is essential for balancing the activities of both PRC2.1 and PRC2.2 during developmental processes [31].
As a PRC2.2 sub-unit, JARID2 enhances PRC2 activity through two distinct mechanisms. First, PRC2 trimethylates JARID2 at lysine 116, which is recognized by the aromatic cage of EED, and subsequently leads to allosteric activation of PRC2, reminiscent of allosteric activation of PRC2 by H3K27me3 [17, 38]. Second, JARID2 was shown to exhibit nucleosome-binding ability, which stimulates PRC2 activity through tethering PRC2 to nucleosome substrate [39]. On the contrary, AEBP2, another accessory member of PRC2.2, stimulates PRC2 through a mechanism distinct from allosteric activation by the methylated substrate. The KR-motif within AEBP2 binds to nucleosome, and both nucleosome binding by AEBP2 and PRC2 interaction with AEBP2 are required to enhance PRC2 activity [19].
Mechanisms of PRC2 recruitment to the chromatin
Although EZH2 is a major chromatin regulator, it is not a DNA-binding protein. Therefore, its recruitment to target genomic loci is largely dependent on multiple cofactors and chromatin features, which are reviewed in this section.
1). Histone Modifiers
Polycomb Repressive Complex 1 (PRC1) is the other one of the two classes of PRCs that are essential for polycomb-mediated gene silencing and epigenetic inheritance of cell fate [40]. The core subunits of PRC1 include a RING1 protein (RING1A or RING1B) and one of the six polycomb group ring finger proteins (PCGF1–6), corresponding to PRC1.1–1.6 sub-complex, respectively. Out of these, PRC1.2 and PRC1.4 complexes are named as canonical PRC1 (cPRC1) due to the presence of CBX subunits that bind H3K27me3. In contrast, PRC1.1, PRC1.3, PRC1.5, and PRC1.6 are defined as variant PRC1 (vPRC1) that exclusively associate with RYBP, RING1, and yin yang 1 (YY1) binding proteins, proteins that do not recognize H3K27me3. Through RING1 proteins, PRC1 catalyzes the mono-ubiquitination of histone H2A at lysine 119 (H2AK119ub1) [41–43], which is recognized by JARID2 for subsequent recruitment of PRC2 to the chromatin (Fig. 1D) [43–45]. To substantiate these findings, two recent studies reported that it is the DNA-binding vPRC1 that catalyzes H2AK119ub1, which serves as an upstream signal to stabilize PRC2.2 recruitment through JARID2 binding for subsequent methylation of H3K27. Accumulation of H3K27me3 eventually recruits cPRC1 through CBX proteins, which serve to amplify H3K27me3 over large genomic regions for epigenetic silencing [46, 47]. Interestingly, in addition to RING1A/B, multiple other E3 ubiquitin ligases, such as MDM2, TRIM37, and CRL4B, have also been shown to modify H2AK119ub1 and interact with PRC2, thereby further enhancing H3K27me3 modifications (Fig. 1E) [48–50]. Thus, PRC2-interacting E3 ligases provide effective means to regulate dynamic PRC2 recruitment to specific genomic regions [8, 51–53].
2). Site-specific DNA-binding proteins
Specific genetic information stored in DNA sequences is another important signal for PRC2 recruitment to the chromatin. This is mediated by EZH2-interacting transcription factors that recognize DNA sequence motifs and subsequently recruit PRC2. For example, PRC2 accessory proteins AEBP2 and JARID2 contain a DNA-binding Zinc-finger domain and A/T-rich interaction domain (ARID) that bind to specific DNA regions [54, 55] (Fig. 1F). Other transcription factors that have been shown to regulate PRC2 recruitment to specific DNA sequences include hypermethylated in cancer 1 (HIC1), circadian clock gene Period2 (PER2), runt-related transcription factor 3 (RUNX3), CCCTC-binding factor (CTCF), PML-RARα, scaffold attachment factor B1 (SAFB1), and YY1 [56–63]. In PCa, the chromatin scaffold protein SAFB1 has been shown to repress AR transcriptional activity [62]. This is achieved by SAFB1 forming a complex with EZH2 at AR-interacting chromatin sites, such as PSA enhancers, in association with other PRC2 proteins to catalyze H3K27me3 and epigenetic silencing.
3). RNA molecules
Several long noncoding RNAs (lnRNAs) have also been shown to mediate the recruitment of PRC2. XIST RNA was implicated in tethering PRC2 complex to the X chromosome during X chromosome inactivation [64]. The lncRNA HOTAIR, transcribed from HOX gene loci, interacts with PRC2 for its local chromatin recruitment and transcriptional silencing of neighboring HOX genes [65]. In PCa cells, EZH2 was shown to bind to MALAT1, a lncRNA that is overexpressed in castration-resistant PCa (CRPC). MALAT1 plays a vital role in recruiting EZH2 to target genomic loci for tumor suppressor gene repression and in mediating EZH2-driven PCa cell migration and invasion [66]. Further, a recent study addressed the mechanisms underlying the requirement of RNA for PRC2 localization in human induced pluripotent stem cells. The authors demonstrated that genome-wide PRC2 occupancy on the chromatin is abolished by either RNA degradation, inhibitors of transcription, or EZH2 mutants defective in RNA binding, suggesting the importance of RNA-EZH2 interaction for chromatin recruitment of PRC2 [67]. However, other studies have shown that the interaction of PRC2 with RNA antagonizes its binding to the chromatin through a competitive mechanism. It has been suggested that RNA prevents PRC2 recruitment to actively transcribed genes, underlying the global pattern of PRC2 association with compact chromatin [68, 69]. Despite these controversies, it is clear that EZH2 is an RNA-binding protein and that RNA is a critical regulator of genome-wide localization of EZH2 and PRC2.
4). CpG-Islands (CGIs)
CGIs are stretches of DNA sequences of several hundred base pairs that have a high frequency of CpG dinucleotides. The human genome contains approximately 30,000 CGIs, many of which are located at gene promoters. They are target genomic regions for DNA methylation, which mediates transcriptional silencing of corresponding genes [70]. PRC2 mainly enriches in CGI-containing promoters of the non-transcribed, inactive genes [70, 71]. Evolutionary hyper-conserved CpG domains largely coincide with PRC2 occupancy in human ESCs, indicating a role of distinct sequence properties in PRC2 recruitment. In addition, studies have shown that a high density of unmethylated CpG dinucleotides is sufficient to recruit PRC2, which subsequently deposits H3K27me3 and ultimately leads to DNA methylation through interaction with DNMTs [72–75]. This recruitment is obstructed by DNA methylation and transcriptional activity. Accordingly, inhibition of transcription induced global recruitment of PRC2 to bona fide PRC2 targets [76]. Mechanistically, several PRC2-associated proteins have been shown to regulate the recruitment of PRC2 to CGIs. For instance, PCL1 and MTF2 have been shown to bind to the unmethylated CpG motif through an N-terminal winged-helix structure, which is essential for efficient recruitment of PRC2 to CGI-containing promoters [77–79]. In contrast, PRC2.1 accessory protein EPOP has been shown to repress PRC2 binding to CGIs [63]. In mouse ESCs, EPOP occupies most CGIs and depletion of EPOP at PRC2-rich CGIs increases chromatin binding of SUZ12, consequently elevating H3K27me3, and repressing target genes. Ten-eleven translocation 1 (TET1) is another PRC2-binding protein frequently found in CGIs [80, 81]. TET1 mediates DNA demethylation by converting 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC). TET1 and PRC2 have been shown to recruit each other to CGIs through direct interaction. Contrary to PRC2, TET1 reduces DNA methylation levels at CGIs, leading to increased transcription of nearby genes [80]. Therefore, there is a strong coordinated regulation of DNA methylation and H3K27me3 mediated by specific properties of CGIs through interacting with and recruiting PRC2.
II. NON-HISTONE SUBSTRATES OF EZH2
Scientists have known for more than a half a century that proteins can be methylated [82]; however, it was not until the discoveries of histone methyltransferase and demethylase that biological functions of protein methylation started to emerge [83, 84]. Subsequent characterizations of the regulation of protein lysine methylation have suggested that it is a reversible process that controls multiple aspects of biological processes [85]. In the last decade, evidence of methylation on non-histone protein substrates is rapidly accumulating, along with their potential roles in human cancers [86–88]. Here, we summarize EZH2-mediated methylation of non-histone protein substrates, mostly through PRC2-dependent but sometimes PRC2-independent mechanisms, with varying consequences (Table 1 & Fig. 2).
Table 1. EZH2 methylation sites on its histone and non-histone protein substrates.
Shown on each row are the reported substrate, starting aa position, the aa sequence flanking the methylation sites (0 position, highlighted in gray), ending aa position, the functional consequences of the methylation, and the references. AR: a potential substrate, methylation site not mapped.
| Substrate | aa | −8 | −7 | −6 | −5 | −4 | −3 | −2 | −1 | 0 | 1 | 2 | 3 | 4 | 5 | 6 | aa | Functions | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Histone H3 | 19 | Q | L | A | T | K | A | A | R | K | S | A | P | A | T | G | 33 | Transcriptional repression | 6, 7 |
| FOXA1 | 287 | A | K | G | G | P | E | S | R | K | D | P | S | G | A | S | 351 | Protein stabilization | 97 |
| RORa | 30 | P | L | N | Q | E | S | A | R | K | S | E | P | P | A | P | 44 | Protein degradation | 96 |
| PLZF | 422 | H | R | K | L | H | S | G | M | K | T | Y | G | C | E | L | 436 | Protein Degradation | 98 |
| JARID2 | 107 | R | P | R | L | Q | A | Q | R | K | F | A | Q | S | Q | P | 129 | Increase PRC2 enzymatic activity | 38 |
| STAT3 | 132 | F | D | F | N | Y | K | T | L | K | S | Q | G | D | M | Q | 146 | Increase transcriptional activity | 92 |
| GATA4 | 291 | C | N | A | C | G | L | Y | M | K | L | H | G | V | P | R | Decrease transcriptional activity | 94 | |
| EloA | 746 | D | P | R | K | P | A | V | K | K | I | A | P | M | M | A | 760 | Transcriptional repression | 89 |
| Talin1 | 2446 | A | D | Q | D | S | E | A | M | K | R | L | Q | A | A | G | 2460 | Proteolytic cleavage | 101 |
| AR | Transcriptional activation | 90 |
Figure 2. PRC2 directly methylates transcription factors to regulate their functions.
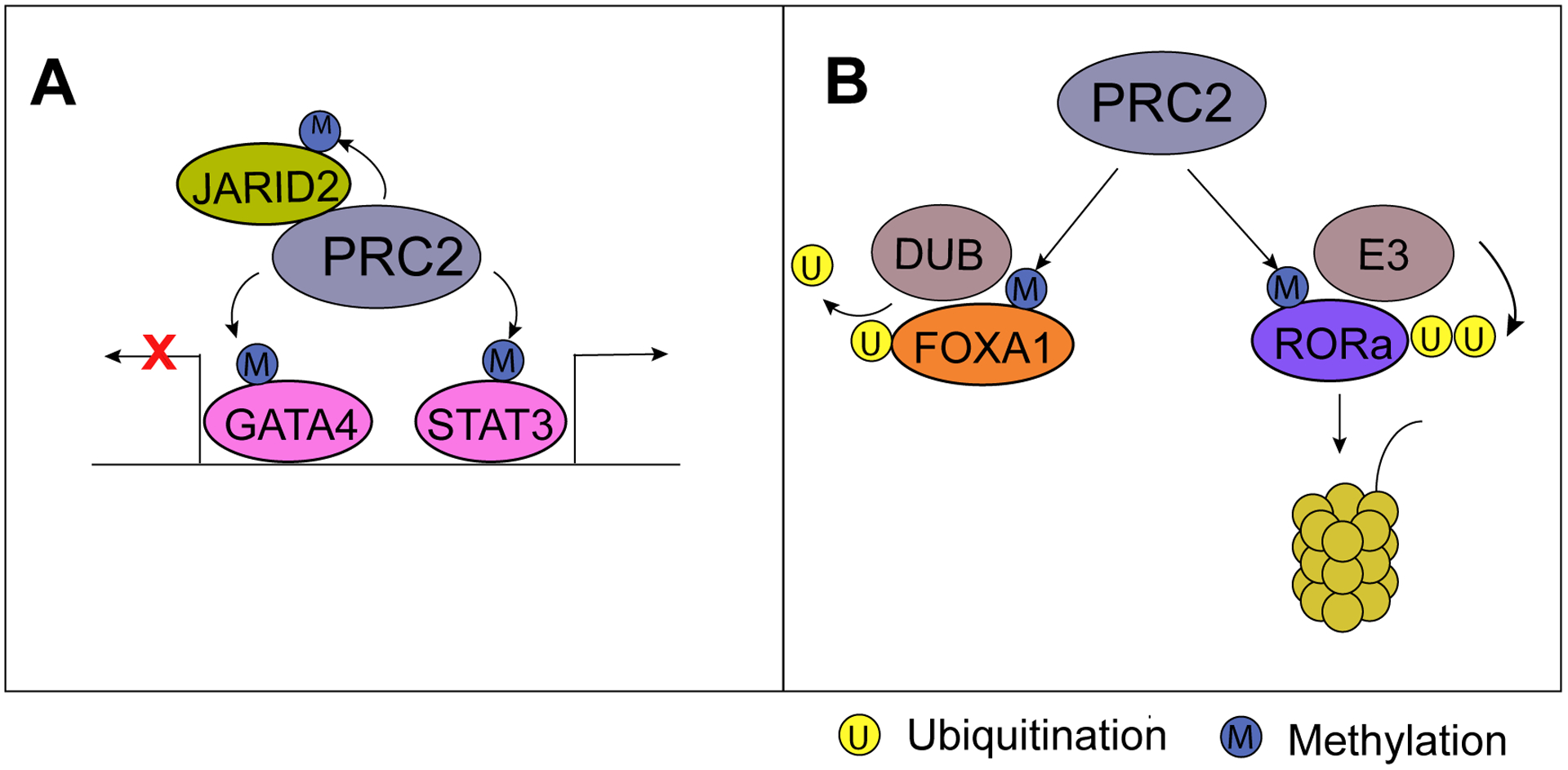
(A) PRC2 methylates and represses GATA4 activity, whereas PRC2 methylation of STAT3 enhances its transcriptional activity. (B) PRC2 methylates FOXA1 protein to stabilize its protein stability by recruiting a deubiquitinase. PRC2 methylation of RORa, on the other hand, recruits an E3 ligase for proteasomal degradation.
1). EZH2 substrates that regulate global transcription
As described earlier, PRC2 accessory protein JARID2 itself is a non-histone substrate of EZH2 [38]. JARID2 methylation by EZH2 elevates PRC2 HMTase activity especially at genomic loci devoid of H3K27me3 (Fig. 2A). Methylation of JARID2 increases its interaction with the aromatic cage of EED, a domain that is originally shown to promote PRC2 enzymatic activity through its interaction with H3K27me3. Studies show that methylated JARID2 and H3K27me3 compete for the aromatic cage of EED to stimulate PRC2 HMTase activity. Through this feedback loop, JARID2 methylation by PRC2 fine-tunes PRC2 activity depending on the chromatin context. Aside from epigenetic silencing of target genes, PRC2 has also been shown to impose global transcriptional repression through a new non-histone substrate, Elongin A [89]. Elongin A is a general transcription elongation factor that augments the catalytic rate of transcription elongation by Pol II [89]. EZH2 methylation of Elongin A is suggested to impede transcriptional regulation of many target genes, whereas methyl-defective Elongin A mutation allows higher expression of those target genes [89].
2). EZH2 methylation of specific transcription factors
Besides general epigenetic and transcriptional regulation, EZH2 also controls several specific transcription factors. In a study comparing CRPC cell line abl with its androgen-dependent counterpart LNCaP, EZH2 has been shown to activate transcription of a subset of genes that may be key for androgen-independent growth [90]. The study initially identified EZH2-stimulated genes that are highly expressed in CRPC cells. ChIP-sequencing (ChIP-seq) analysis revealed that a portion of the EZH2 binding sites in the abl cell line surprisingly lacks H3K27me3, but rather is significantly enriched with AR binding motifs. Furthermore, the authors found that EZH2 interacts with AR protein to activate AR transcriptional activities. Mechanistically, this is dependent on EZH2 protein phosphorylation at serine 21 (pS21) and requires an intact EZH2 methyltransferase domain that appears to modulate AR-associated lysine methylation. Surprisingly, this function of EZH2 does not require other PRC2 subunits, thus being polycomb-independent. How EZH2 methylates AR independently of PRC2 and why EZH2 is able to act as a co-activator of methylated AR needs further clarification.
EZH2 is highly expressed in glioblastoma multiforme and has been shown to mediate glioblastoma stem-like cell maintenance [91]. One study indicated that this is mediated by EZH2 binding to and methylating STAT3, which leads to STAT3 activation and increased transcription of STAT3-target genes (Fig. 2A) [92]. As in the case of AR, this regulation also requires EZH2 phosphorylation at S21. However, unlike AR, STAT3 also interacts with the PRC2 core subunit SUZ12, suggesting a polycomb-dependent mechanism. Considering that STAT3 has also been linked to glioblastoma stem-like cell survival [93], targeting EZH2 and STAT3 signaling pathways may offer an attractive therapeutic option in glioblastoma. Indeed, 3-Deazaneplanocin A (DZNep), a global histone methylation inhibitor that also induces EZH2 protein depletion, decreases STAT3 methylation and suppresses STAT3 signaling [92]. This suggests that pharmacological disruption of EZH2 may also suppress some of its non-histone substrates, albeit more selective EZH2 inhibitors are yet to be tested.
In another case, EZH2 methylation was shown to repress the activity of a transcription factor. EZH2 interacts with the cardiac transcription factor GATA4 to directly methylate GATA4 at lysine 299 [94]. Furthermore, GATA4 interacts with SUZ12 and EED and their expression is required for EZH2-mediated methylation of GATA4, suggesting a polycomb-dependent mechanism. Earlier studies have reported that the histone acetyltransferase p300 elevates GATA4 transcriptional activity by acetylating lysine residues [95]. Specifically, the studies found that EZH2-mediated methylation of GATA4 reduced its interaction with and acetylation by p300, thereby inhibiting GATA4 transcriptional activity (Fig. 2A). This finding is further bolstered by upregulation of GATA4 target gene, Myh6, upon DZNep-mediated inhibition of EZH2 function. Therefore, this study reveals a crosstalk between PRC2-mediated protein methylation and other post-translational modifications on the same non-histone substrate, indirectly regulating downstream signaling, and it presents important lines of evidence for future investigation of cancers including PCa.
3). EZH2 regulation of protein stability
In addition to regulating transcriptional activities of substrate proteins, EZH2-catalyzed methylation has also been shown to play a crucial role in substrate ubiquitination and protein degradation. EZH2 methylation of orphan nuclear receptor RORα has been shown to promote its ubiquitin-dependent degradation [96]. The study first reported that EZH2 knockout in mouse embryonic fibroblasts resulted in higher RORα protein levels without altering RORα mRNA levels. Moreover, DZNep blocks the EZH2-mediated degradation of RORα in the same cells. Mechanistically, the authors found that EZH2 methylates RORα at an “R-K-S” sequence located within its N-terminal domain, which is highly similar to the region in H3K27 that is methylated by EZH2. Methylated RORα is recognized by a methyl-binding protein DCAF1, which subsequently recruits DDB1/CUL4B E3 ubiquitin ligase complex for RORα ubiquitination and degradation (Fig. 2B). More recently, we reported that EZH2 methylates FOXA1, a transcription factor essential for prostate epithelial differentiation and androgen-dependent PCa growth [97]. Methylation on FOXA1 induces interaction with a WD40 repeat protein BUB3, which recruits USP7, a deubiquitinase, to remove FOXA1 ubiquitination and enhance FOXA1 protein stability.
The Natural Killer T (NKT) cell lineage-defining transcription factor promyelocytic leukemia zin finger (PLZF) is another substrate whose methylation by EZH2 leads to its ubiquitination and protein degradation [98]. Since PLZF promotes NKT cell growth and differentiation [99], T-cell-specific depletion of Ezh2 induces a pronounced expansion of NKT cells without altering H3K27me3. Interestingly, depletion of Suz12 or Eed, on the other hand, ablates NKT cell development, likely through de-stabilizing canonical PRC2 function. This suggests that EZH2-mediated PLZF methylation is independent of PRC2. Similar to the role of EZH2 in T-cell function and immune responses, cytosolic EZH2-containing PRC2 complex has been reported to regulate actin polymerization and control antigen receptor signaling in T cells [100]. Further studies showed that cytosolic EZH2 methylates Talin, which plays a pivotal role in integrin-mediated cell migration [101]. EZH2 methylation of Talin promotes its calpain-mediated cleavage by interfering with the binding of Talin to F-actin. Thus, leukocyte migration is compromised upon EZH2 loss, restricting some disease progression. Further examination of the precise roles of cytoplasmic EZH2 will be important priority for future studies.
III. POST-TRANSLATIONAL MODIFICATIONS AS MOLECULAR SWITCHES OF EZH2 FUNCTIONS
Post-translational modifications including ubiquitination, acetylation, O-GlcNAclation, and sumoylation have been shown to dynamically alter EZH2 protein functions (reviewed in [102]). Here, we highlight EZH2 phosphorylation, methylation, and PARylation in their regulation of EZH2 functions.
1). EZH2 phosphorylation
The phosphoinositide 3-kinase (PI3K)-AKT signaling pathway is one of the most frequently altered pathways in primary and metastatic PCa, with alteration frequencies of 42% and 100%, respectively [103]. It was first discovered in breast cancer that AKT1 directly interacts with and phosphorylates EZH2 at S21 to reduce its affinity for canonical substrate histone H3 without changing either EZH2 subcellular localization or its interaction with SUZ12 and EED [104]. Subsequent studies have shown that pS21 changes EZH2 substrate preference from histone to non-histone substrates. Xu et al. specifically identified some EZH2-associated genomic sites that lack H3K27me3 but contain active epigenetic marks, such as H3K4me2/3 and Pol II in CRPC cells [90]. Further investigation showed a molecular switch of EZH2 methyltransferase (MTase) preference from histone H3 toward a non-histone protein such as AR, which is dependent on EZH2 pS21 executed by hyper-activated PI3K-AKT signaling in CRPC cells. Immunohistochemistry analysis of tissue microarrays comparing neoadjuvant-treated prostate tumors versus CRPC showed significantly increased levels of EZH2 and pS21 in CRPC, whereas H3K27me3 levels were surprisingly decreased, supporting a disconnect between EZH2 and H3K27me3 in CRPC. Similar observations were made in glioblastoma multiforme and non-small cell lung carcinoma where EZH2 S21 phosphorylation changes its MTase preference from histone H3 to STAT3 [105, 106]. Hence, pS21 enhances STAT3 methylation and activates STAT3 signaling for subsequent tumorigenesis independently of H3K27me3. Interestingly, a positive feedback loop has been described in which STAT3 induces AKT1 activation and EZH2 S21 phosphorylation [107]. More precise crystallographic analysis and mass spectrometry-based assays are necessary to elucidate how post-translational modification of EZH2 shifts its specificity between histone substrate and non-histone protein substrates. Further, a recent study indicates that AKT-dependent pS21 may induce cytosolic localization of EZH2 [107]. Although the underlying mechanism remains unclear, phosphorylation-dependent translocation of EZH2 could be one of the potential ways to promote oncogenic roles of EZH2 outside of the nucleus.
Besides AKT1, mitogen-activated protein kinase 14 (MAPK14, also called p38α), when activated by TNF, has been shown to phosphorylate EZH2 at threonine 372 (T372), which promotes PRC2 interaction with YY1, leading to the formation of repressive chromatin on the Pax7 promoter. Thus, PRC2 represses the expression of Pax7 and expansion of muscle stem cells [108]. Moreover, cyclin-dependent kinase 1 (CDK1) and 2 (CDK2) have also been shown to phosphorylate EZH2 at different regions [109, 110]. CDK-mediated phosphorylation at T350 of EZH2 showed no effect on its histone methyltransferase activity, PRC2 complex formation, and the half-life of the EZH2 protein. However, it increased EZH2 binding with ncRNAs HOTAIR and Xist, and this binding is important for the recruitment of EZH2 and maintenance of H3K27me3 levels at EZH2-target genomic loci [109, 110]. In contrast, phosphorylation of EZH2 at T487 does not affect its binding to HOTAIR but disrupts EZH2 interaction with other PRC2 core subunits SUZ12 and EED, thereby inhibiting EZH2 MTase activity [110, 111]. Furthermore, phosphorylation at T492 has been shown to promote dissociation of EZH2 from SUZ12 and EED and suppress EZH2 HMTase activity. It would be interesting to identify EZH2 phosphorylation in PCa cells and investigate its roles in regulating PCa progression.
2). PRC2 automethylation
Many post-translational modification enzymes self-regulate their own activities. Protein kinases are particularly prominent in self-regulating their activity by auto-phosphorylating a specific site in the domain called the activation loop [112]. Similarly, methyltransferases including PRC2 have also been shown to have auto-methylation activities. As described earlier, PRC2 accessory protein JARID2 is methylated by PRC2 and this modification is recognized by EED, a core subunit of PRC2, leading to allosteric activation of PRC2’s enzymatic activity, forming a feedforward loop [38]. Further, PRC2 auto-methylates EZH2 and SUZ12 in the presence or absence of nucleosomal substrates [113]. Methylation on both EZH2 and SUZ12 can be enhanced by the addition of H3K27me3-peptide, an allosteric activator of PRC2 activity, and tightly regulated by PRC2 accessory proteins such as AEBP2 and JARID2. Auto-methylation of EZH2 occurs on three lysine residues (K510, K514, K515) on a disordered but highly conserved loop between its SANT2L and CXC domains [113, 114]. Such modifications were shown to increase HTMase activity of PRC2, promote PRC2 accessibility to the histone tail, and are required for attaining proper H3K27me3 levels. These studies demonstrate a self-regulatory loop of PRC2 in modulating its own HMTase activity through auto-methylation. EZH2 auto-methylation is drastically reduced in diffuse intrinsic pontine glioma with H3K27M mutation. PRC2 auto-methylation and its aberrant regulation in other cancer types have not been reported and will be an interesting area for future investigation.
3). EZH2 PARylation
Whole exome and transcriptome sequencing have identified relatively higher frequency of aberrations in the DNA repair pathway (22.7%) in CRPC patient samples compare to primary prostate cancer [115]. Alterations in DNA repair genes such as BRCA2, BRCA1, and ATM suggest the potential value for targeting DNA repair pathways in PCa. Notably, a recent Phase II clinical trial tested efficacy of the poly (ADP-ribose) polymerase (PARP) inhibitor Olaparib in a subset (33%) of CRPC patients with DNA-repair gene mutations and concluded that 88% of patients responded to Olaparib, including all 7 patients with BRCA2 loss [116]. Of note, the use of PARP inhibitors has been shown to induce the expression of EZH2 and increase global levels of H3K27me3 [117]. Further, EZH2 is a direct target of PARP1-mediated PARylation. PARylation of EZH2 leads to PRC2 complex dissociation, inhibits PRC2 HMTase activity, and reduces H3K27me3 at EZH2-target genes [118, 119]. PARP1 inhibition, on the other hand, increases EZH2 expression and HMTase activity, and promotes cancer stem cell properties [119]. Accordingly, EZH2 inhibitors were shown to further sensitize BRCA-mutant breast and ovarian cancer cells to PARP inhibitors. It is unclear whether such synergy between these two inhibitors will occur in PCa with DNA-repair gene mutations. Moreover, there is at least one study arguing against combinatorial use of these inhibitors, as low EZH2 expression was shown to predict chemoresistance and poor survival in patients with BRAC2-mutated tumors [120]. Using murine Brca2−/− breast tumor models, the authors further demonstrated that inhibition of Ezh2 is associated with stabilization of the replication fork and acquired resistance to PARP inhibitors. Therefore, these studies suggest novel roles of PRC2 in DNA damage response and repair, which may provide novel opportunities for cancer treatment, warranting further investigation.
IV. DIVERSE FUNCTIONAL ROLES OF EZH2 IN PROSTATE CANCER
EZH2 is highly upregulated in aggressive PCa and plays important roles in promoting PCa progression. EZH2 depletion has been shown to induce G2/M cell cycle arrest, abolish cell proliferation and invasion in vitro, and quench tumor growth in vivo, while EZH2 overexpression has been shown to promote oncogenic behaviors [121–125]. Much of these activities have been associated with EZH2-mediated epigenetic silencing of tumor suppressor genes [6, 126] (Fig. 3A). In addition to its epigenetic targets, our recent studies revealed FOXA1 as a non-histone substrate of EZH2 [97]. EZH2 methylates FOXA1, which recruits deubiquitinase to block FOXA1 degradation, thereby increasing its protein levels (Fig. 3B). EZH2 and FOXA1 co-regulated genes are largely involved in cell cycle processes and their elevated expression levels were associated with poor prognoses. Further, PCa cells with high FOXA1 expression are more sensitive to inhibitors of EZH2 MTase activities that lead to FOXA1 protein degradation. Re-expression of FOXA1 can rescue the growth-inhibitory effects of these enzymatic EZH2 inhibitors. Nevertheless, the efficacy of these enzymatic EZH2 inhibitors in PCa is in general much lower than that in hematologic cancers, suggesting the existence of crucial EZH2 target genes that are insensitive to PRC2 MTase activity.
Figure 3. Diverse roles of EZH2 in prostate cancer.
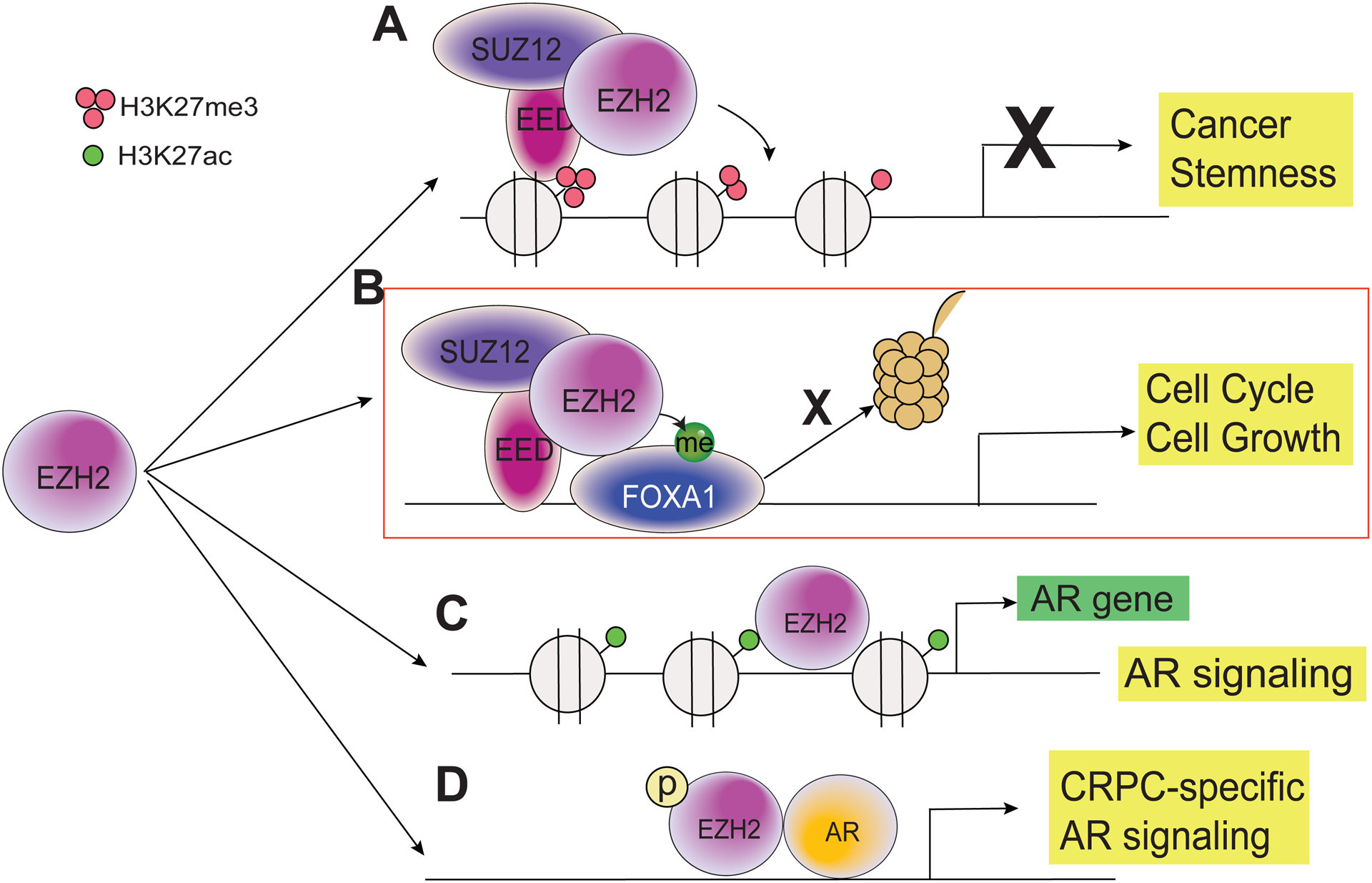
(A) The canonical function of EZH2 is to form the PRC2 complex with other core subunits SUZ12 and EED to catalyze H3K27me3, which leads to epigenetic silencing of tumor suppressor genes, rendering cancer cell stemness. (B) FOXA1 is a non-histone substrate of EZH2 in prostate cancer. EZH2 methylates FOXA1 and protect it from protein degradation. EZH2 and FOXA1 co-regulates cell cycle progression and prostate cancer growth. (C) EZH2 directly binds at the AR gene promoter to induce AR gene transcription. This function is independent of PRC2 and EZH2 MTase activities. (D) In CRPC cell line LNCaP-abl, phosphorylated EZH2 has been shown to act as an AR co-activator to drive AR signaling and CRPC progression.
In CRPC cells, EZH2 has been shown to exhibit an oncogenic function that is polycomb-independent and instead involves the ability of EZH2 to act as a coactivator of AR [90] (Fig. 3C). This activating function of EZH2 is dependent on EZH2 protein phosphorylation and also requires its intact methyltransferase domain. Further, we recently found that EZH2 induces the expression of AR by direct binding to AR promoter for transcriptional activation (Fig. 3D). EZH2 activates AR transcription in both primary PCa and CRPC cell lines, enhancing AR signaling, and rendering resistance to enzymatic EZH2 inhibitors. This function of EZH2 is independent of both its methyltransferase activities and the PRC2 complex [127]. Structural analysis of EZH2 revealed a partially disordered domain that resembled transcriptional activation domain of p53, which might mediate potential gene activation function by EZH2 [128].
In addition to PRC2-independent EZH2 in activating AR, we also noted a small fraction of PRC2 binding at the AR promoter, which exerts its canonical function in epigenetic silencing. This is consistent with the conventional role of EZH2 in suppressing developmental regulators and promoting cell stemness [6, 126] (Fig. 3A). Consequently, enzymatic EZH2 inhibitors have been shown to slightly increase AR levels in PCa cells [127]. However, upon EZH2 knockdown, we observed a decrease of total AR protein, suggesting that EZH2-mediated transcriptional activation of AR dominates over its epigenetic role at the AR promoter. These results warrant the development of EZH2-degrading compounds that will not only abolish the epigenetic function of EZH2 but also its PRC2-independent role in activating AR.
Due to the increased use of high-affinity AR pathway inhibitors (ARPI), such as enzalutamide, approximately 20% of end-stage PCa patients develop treatment-induced neuroendocrine PCa (NEPC), most of which no longer express AR, and NEPC is essentially incurable [129]. NEPC tumors are resistant to all hormonal treatments and exhibit features of neuroendocrine differentiation (NED), commonly labeled with multiple neuroendocrine markers, including synaptophysin, CD56, neuron-specific enolase (NSE), and chromogranin A [130]. NEPC patients have a median overall survival of less than a year after diagnosis and a 5-year cancer-specific survival rate of only 17.95% [131, 132]. NEPC patients have poor clinical outcomes likely attributed to increased expression of genes involved in cellular proliferation and cell cycle and mitosis [133]. RNA-Seq comparing 30 localized PCa with 7 NEPC tumors showed that EZH2 is among the most highly overexpressed genes in NEPC compared to CRPC [134]. Further analyses of patient-derived xenografts revealed several key genetic differences between parental PCa and relapsed NEPC models. Tumor suppressor retinoblastoma (Rb) is absent in the majority of NEPC patient samples [135]. Loss of Rb1 and p53 induces the pathogenesis of NEPC in transgenic mouse models [136]. MYCN gene encoding the N-Myc protein was amplified in 40% of NEPC tumors, compared to 5% of prostate adenocarcinoma tumors [134]. Accordingly, much higher levels of MYCN were detected in NEPC compared with CRPC in clinical specimens, and N-Myc overexpression in engineered mouse model leads to the development of NEPC tumors [137]. Numerous chromatin regulators, including EZH2, were found differentially expressed in NEPC as compared to CRPC, suggesting the epigenetic nature of NED [138]. Moreover, genes that were silenced in NEPC models were preferentially enriched with PRC2 target genes and associated with poor outcomes, suggesting a functional significance of EZH2 overexpression. In the MYCN-driven NEPC models, epigenetic targets of EZH2/PRC2 are significantly enriched in the N-Myc-downregulated genes. N-Myc was shown to interact with EZH2 and SUZ12 proteins to redirect PRC2 activity for the repression of AR signaling, thereby contributing to NED. Interestingly, upregulation of EZH2 expression has been observed in tumors derived from prostate basal cells overexpressing N-Myc and myrAKT1 [139]. Further, increased EZH2 expression in NEPC tumors was validated in a larger cohort of patient tumors as well as organoids derived from needle biopsies of metastatic lesions from end-stage PCa patients [140].
Multiple studies have attempted to understand the molecular functions of EZH2 in NEPC tumors. Importantly, elevated expression of EZH2 in NEPC tumors was found to be accompanied by concomitant increase in H3K27me3, supporting the epigenetic role of EZH2 in NEPC [140]. One of the direct targets of EZH2-mediated epigenetic silencing during NEPC is Thrombospondin 1 (TSP1), an inhibitor of angiogenesis [141]. TSP1 expression is negatively correlated with EZH2 and NED markers in clinical samples. Enzymatic EZH2 inhibitors have demonstrated much better efficacy in NEPC models than CRPC tumors [140]. However, treatment still requires high doses of the inhibitors (approximately 5μM) in order to achieve 50% growth inhibition, suggesting a role for methylation-independent PRC2 functions. Interestingly, several studies have found that AR is also a direct target of EZH2-mediated epigenetic silencing in AR-negative NEPC cells and that EZH2 inhibitors restore AR expression [136, 142]. Both EZH2 depletion and enzymatic EZH2 inhibitors were found to increase AR levels in NEPC cells, in contrast to their opposing effects in AR-positive cells [127]. It is likely that the AR-activating role of EZH2 is lost as PCa progresses to NEPC, which may contribute to the AR-negative nature of most NEPC cells, expressing high levels of EZH2. Nevertheless, such results suggest that EZH2 inhibitors might re-sensitize NEPC cells to ARPI, supporting their combinatorial use. Indeed, combination of GSK503 and Enzalutamide showed synergistic effects in suppressing the growth of post-castration DKO (with Pten and Rb1 loss) tumors. As the field continues to see an increased incidence of NEPC tumors, there is an urgent need for improved therapeutics, and with EZH2 being overexpressed in these NEPC tumors, these improved therapeutics can be expeditiously translated into the clinic. Whereas most studies have suggested a primary role of the epigenetic function of EZH2 in NEPC, at least one study has demonstrated that EZH2 function can be switched from H3K27 to non-histone substrate STAT3, wherein its methylation promotes NED in enzalutamide-induced NEPC [143]. These divergent mechanistic findings warrant further characterization of EZH2 functions, specifically defining non-histone substrates and interacting partners, in NEPC, which will further guide the use of existing EZH2 inhibitors in combination therapies and further support the development of novel PRC2 inhibitors to fully block all EZH2 functions.
V. THERAPEUTIC TARGETING OF EZH2 IN PROSTATE CANCER
There has been intense interest in the development of new small molecule EZH2 inhibitors along with an emphasis on new therapeutic modalities such as EZH2 degradation [144]. The first orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity to receive FDA approval is Tazemetostat, also known as EPZ-6438 [145–147]. This drug was approved in January 2020 for the treatment of epithelioid sarcoma and follicular lymphoma, where gain-of-function mutations of EZH2 are prevalent [148]. It is also currently being tested in clinical trials for other types of lymphomas and solid tumors. Valemetostat, known as DS-3201 [149] by Daiichi Sankyo, is a highly specific orally bioavailable EZH1/2 dual inhibitor in Phase 1/2 clinical trials against a variety of hematologic and solid tumors. Jiangsu Hengrui Medicine Co., Ltd and Pfizer currently have EZH2 inhibitors SHR554 and PF-6821497 [150], respectively, in Phase 1 testing for safety and efficacy in lymphomas. Constellation Pharmaceuticals [151, 152] currently has its second-generation EZH2 inhibitor CPI-0209 in Phase 1 clinical trials as a potential treatment for solid and hematologic tumors. Constellation’s original EZH2 inhibitor (CPI-1205), which was in Phase 2 studies for metastatic castration-resistant prostate cancer (mCRPC), was discontinued in 2020 due to lack of efficacy. In addition to the EZH2 inhibitors in clinical development listed above, a significant number of other EZH2 inhibitor programs have shown promising pre-clinical activity and have been reviewed elsewhere [144]. Finally, Novartis is currently developing a small molecule EED inhibitor that blocks binding of EED to H3K27, and consequently prevents EZH2 activation. This compound, MAK683, is in Phase 1/2 trials for Diffuse Large B-cell Lymphoma (DLBCL).
Protein degradation has recently become a promising new avenue for therapeutics development [153–155]. There are currently at least two different proteolysis, targeting chimeras (PROTACs) in clinical development, both from Arvinas, for the treatment of prostate and breast cancer. As discussed above, EZH2 possesses histone and non-histone methylation activity, as well as methylation-independent functions that promote carcinogenesis and proliferation. Whereas inhibiting EZH2 will inhibit methylation activity (e.g., H3K27me3), non-canonical methylation-independent functions of EZH2 would be unaffected by enzymatic inhibitors. Thus, degradation of EZH2 could be a viable approach to target the non-canonical methylation-independent functions of EZH2 functions. To this end, the first targeted degrader of EZH2 was recently reported [156]. This compound, MS1943, was reported in 2020 and was shown to induce EZH2 degradation in various cancer cell lines, including the non-malignant prostate cancer cell line PNT2. It was found that MS1943 induced apoptosis partly by causing ER stress and strongly activating the unfolded protein response (UPR) pathway. Considering the lack of enzymatic EZH2 inhibitors in PCa, EZH2-degrading compounds will be of great interest for further exploration.
VI. CONCLUSIONS/FUTURE DIRECTIONS
In the present review, we focus on the methyltransferase activities of EZH2, its histone vs non-histone substrates, and regulators of its activities. We also reviewed some studies on the non-catalytic activities of EZH2 that are either independent of its catalytic domain or the entire PRC2 complex, which have been more widely reported in other cancer types. In estrogen receptor (ER)-negative breast cancer, EZH2 has been shown to form a complex with RelA and RelB to activate canonical and non-canonical Nuclear Factor-kappaB (NF-κB) signaling, independent of the MTase activity of EZH2 [157]. EZH2 has also been shown to act as a scaffold protein, bridging the estrogen and Wnt signaling pathways in breast cancer, specifically mediating an interaction between ERα and B-catenin at the c-Myc and cyclin D1 promoters [158]. It was speculated that EZH2-interacting mediator complex might enhance the transactivation activity at specific loci via regulation of RNA polymerase II. EZH2 has been shown to interact with TRIM28 and the chromatin-remodeling SWI/SNF (SWItch/Sucrose Non-Fermentable) complex [159]. This novel EZH2/TRIM28/SWI/SNF complex coordinately activates target genes involved in breast cancer. This finding is particularly intriguing as growing evidence has indicated an epigenetic antagonism between PRC2 and SWI/SNF complexes [160]. Moreover, cancer cell lines with mutations in SWI/SNF complex were more dependent on EZH2, primarily on its non-catalytic roles in the stabilization of the PRC2 complex, further supporting that EZH2 enzymatic inhibitors may not fully suppress the oncogenic functions of EZH2 [161]. Of relevance, SWI/SNF complex was recently reported to be deregulated in NEPC and to play important roles in lineage plasticity [162].
In summary, EZH2 is an epigenetic regulator that is strongly and persistently upregulated in clinical samples and mouse models of aggressive PCa, such as CRPC and NEPC. EZH2 was thought essential to epigenetic reprogramming and lineage plasticity of NEPC cells, and many PRC2-target genes are indeed deregulated in NEPC. However, enzymatic inhibitors of EZH2 remain ineffective in NEPC, despite NEPC being AR-negative, suggesting potential roles of other non-epigenetic targets of EZH2. All of these findings provide the impetus for future investigations to focus on identifying non-histone substrates of EZH2 and delineating non-epigenetic roles of EZH2 in PCa, especially NEPC, where it is most highly upregulated. Such understanding will be critical to guide the use of catalytic inhibitors of EZH2 in combination therapeutic regimens and the development of novel approaches to effectively target EZH2 in late-stage PCa.
ACKNOWLEDGEMENT
This work was supported in part by the NIH/NCI R01CA227918 (to JY), training grant T32CA09560 (to SP), prostate SPORE P50CA180995 (to JY), and Department of Defense grants W81XWH-17-1-0405 and W81XWH-17-1-0578 (to JY).
Footnotes
Conflict of Interest: All authors declare that they have no competing interests.
REFERENCES
- 1.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature 1978; 276: 565–570. [DOI] [PubMed] [Google Scholar]
- 2.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011; 469: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis 2011; 2: e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv 2015; 1: e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nature Medicine 2016; 22: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013; 4: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 2004; 14: 155–164. [DOI] [PubMed] [Google Scholar]
- 8.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 9.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002; 111: 185–196. [DOI] [PubMed] [Google Scholar]
- 10.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 2002; 16: 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002; 111: 197–208. [DOI] [PubMed] [Google Scholar]
- 12.Zee BM, Britton LM, Wolle D, Haberman DM, Garcia BA. Origins and formation of histone methylation across the human cell cycle. Mol Cell Biol 2012; 32: 2503–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 2014; 53: 49–62. [DOI] [PubMed] [Google Scholar]
- 14.Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 2003; 12: 1577–1589. [DOI] [PubMed] [Google Scholar]
- 15.Hojfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A et al. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nature structural & molecular biology 2018; 25: 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015; 350: aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 2016; 7: 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009; 461: 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CH, Holder M, Grau D, Saldana-Meyer R, Yu JR, Ganai RA et al. Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol Cell 2018; 70: 435–448 e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CH, Yu JR, Kumar S, Jin Y, LeRoy G, Bhanu N et al. Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin. Mol Cell 2018; 70: 422–434 e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R et al. Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol Cell 2018; 70: 1149–1162 e1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 2004; 23: 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Jiao L, Shubbar M, Yang X, Liu X. Unique Structural Platforms of Suz12 Dictate Distinct Classes of PRC2 for Chromatin Binding. Mol Cell 2018; 69: 840–852 e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nekrasov M, Wild B, Muller J. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep 2005; 6: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holoch D, Margueron R. Mechanisms Regulating PRC2 Recruitment and Enzymatic Activity. Trends Biochem Sci 2017; 42: 531–542. [DOI] [PubMed] [Google Scholar]
- 26.Laugesen A, Hojfeldt JW, Helin K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol Cell 2019; 74: 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao R, Wang H, He J, Erdjument-Bromage H, Tempst P, Zhang Y. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol 2008; 28: 1862–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 2008; 28: 2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 2010; 24: 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY et al. PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem Cells 2011; 29: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conway E, Jerman E, Healy E, Ito S, Holoch D, Oliviero G et al. A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Mol Cell 2018; 70: 408–421 e408. [DOI] [PubMed] [Google Scholar]
- 32.Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol Cell 2011; 42: 330–341. [DOI] [PubMed] [Google Scholar]
- 33.Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde ME, Hong Z et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nature structural & molecular biology 2012; 19: 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nature structural & molecular biology 2012; 19: 1273–1281. [DOI] [PubMed] [Google Scholar]
- 35.Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizan P et al. EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol Cell 2016; 64: 645–658. [DOI] [PubMed] [Google Scholar]
- 36.Liefke R, Karwacki-Neisius V, Shi Y. EPOP Interacts with Elongin BC and USP7 to Modulate the Chromatin Landscape. Mol Cell 2016; 64: 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hauri S, Comoglio F, Seimiya M, Gerstung M, Glatter T, Hansen K et al. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep 2016; 17: 583–595. [DOI] [PubMed] [Google Scholar]
- 38.Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol Cell 2015; 57: 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Son J, Shen SS, Margueron R, Reinberg D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev 2013; 27: 2663–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moussa HF, Bsteh D, Yelagandula R, Pribitzer C, Stecher K, Bartalska K et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat Commun 2019; 10: 1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004; 431: 873–878. [DOI] [PubMed] [Google Scholar]
- 42.Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 2012; 45: 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LLP et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014; 157: 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 2014; 7: 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nature structural & molecular biology 2014; 21: 569–571. [DOI] [PubMed] [Google Scholar]
- 46.Blackledge NP, Fursova NA, Kelley JR, Huseyin MK, Feldmann A, Klose RJ. PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol Cell 2020; 77: 857–874 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamburri S, Lavarone E, Fernandez-Perez D, Conway E, Zanotti M, Manganaro D et al. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol Cell 2020; 77: 840–856 e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J 2013; 32: 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhatnagar S, Gazin C, Chamberlain L, Ou J, Zhu X, Tushir JS et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014; 516: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol Cell 2016; 61: 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 2003; 17: 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 2003; 17: 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441: 349–353. [DOI] [PubMed] [Google Scholar]
- 54.Kim TG, Chen J, Sadoshima J, Lee Y. Jumonji represses atrial natriuretic factor gene expression by inhibiting transcriptional activities of cardiac transcription factors. Mol Cell Biol 2004; 24: 10151–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grijzenhout A, Godwin J, Koseki H, Gdula MR, Szumska D, McGouran JF et al. Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development 2016; 143: 2716–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Vire E et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007; 11: 513–525. [DOI] [PubMed] [Google Scholar]
- 57.Atchison L, Ghias A, Wilkinson F, Bonini N, Atchison ML. Transcription factor YY1 functions as a PcG protein in vivo. EMBO J 2003; 22: 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S et al. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol 2008; 28: 6473–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest 2010; 120: 3209–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boulay G, Dubuissez M, Van Rechem C, Forget A, Helin K, Ayrault O et al. Hypermethylated in cancer 1 (HIC1) recruits polycomb repressive complex 2 (PRC2) to a subset of its target genes through interaction with human polycomb-like (hPCL) proteins. J Biol Chem 2012; 287: 10509–10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A 2013; 110: 12331–12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mukhopadhyay NK, Kim J, You S, Morello M, Hager MH, Huang WC et al. Scaffold attachment factor B1 regulates the androgen receptor in concert with the growth inhibitory kinase MST1 and the methyltransferase EZH2. Oncogene 2014; 33: 3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liefke R, Shi Y. The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discov 2015; 1: 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A. A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2004; 2: E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ et al. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget 2015; 6: 41045–41055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long Y, Hwang T, Gooding AR, Goodrich KJ, Rinn JL, Cech TR. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat Genet 2020; 52: 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beltran M, Yates CM, Skalska L, Dawson M, Reis FP, Viiri K et al. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res 2016; 26: 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW et al. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nature structural & molecular biology 2017; 24: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 2008; 4: e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanay A, O’Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci U S A 2007; 104: 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010; 143: 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS et al. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 2010; 6: e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lynch MD, Smith AJ, De Gobbi M, Flenley M, Hughes JR, Vernimmen D et al. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J 2012; 31: 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jermann P, Hoerner L, Burger L, Schubeler D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci U S A 2014; 111: E3415–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 2014; 55: 347–360. [DOI] [PubMed] [Google Scholar]
- 77.Choi J, Bachmann AL, Tauscher K, Benda C, Fierz B, Muller J. DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nature structural & molecular biology 2017; 24: 1039–1047. [DOI] [PubMed] [Google Scholar]
- 78.Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017; 549: 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H et al. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat Genet 2018; 50: 1002–1010. [DOI] [PubMed] [Google Scholar]
- 80.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011; 473: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R et al. Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol 2013; 14: R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paik WK, Paik DC, Kim S. Historical review: the field of protein methylation. Trends Biochem Sci 2007; 32: 146–152. [DOI] [PubMed] [Google Scholar]
- 83.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000; 406: 593–599. [DOI] [PubMed] [Google Scholar]
- 84.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–953. [DOI] [PubMed] [Google Scholar]
- 85.Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 2007; 25: 1–14. [DOI] [PubMed] [Google Scholar]
- 86.Clarke SG. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem Sci 2013; 38: 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biggar KK, Li SS. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol 2015; 16: 5–17. [DOI] [PubMed] [Google Scholar]
- 88.Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer 2015; 15: 110–124. [DOI] [PubMed] [Google Scholar]
- 89.Ardehali MB, Anselmo A, Cochrane JC, Kundu S, Sadreyev RI, Kingston RE. Polycomb Repressive Complex 2 Methylates Elongin A to Regulate Transcription. Mol Cell 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012; 338: 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 2009; 69: 9211–9218. [DOI] [PubMed] [Google Scholar]
- 92.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013; 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009; 27: 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 2012; 26: 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takaya T, Kawamura T, Morimoto T, Ono K, Kita T, Shimatsu A et al. Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J Biol Chem 2008; 283: 9828–9835. [DOI] [PubMed] [Google Scholar]
- 96.Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell 2012; 48: 572–586. [DOI] [PubMed] [Google Scholar]
- 97.Park SH, Fong KW, Kim J, Wang F, Lu X, Lee Y et al. Posttranslational regulation of FOXA1 by Polycomb and BUB3/USP7 deubiquitin complex in prostate cancer. Sci Adv 2021; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep 2017; 18: 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol 2008; 9: 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R et al. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell 2005; 121: 425–436. [DOI] [PubMed] [Google Scholar]
- 101.Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat Immunol 2015; 16: 505–516. [DOI] [PubMed] [Google Scholar]
- 102.Lu H, Li G, Zhou C, Jin W, Qian X, Wang Z et al. Regulation and role of post-translational modifications of enhancer of zeste homologue 2 in cancer development. Am J Cancer Res 2016; 6: 2737–2754. [PMC free article] [PubMed] [Google Scholar]
- 103.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005; 310: 306–310. [DOI] [PubMed] [Google Scholar]
- 105.Chen X, Hao A, Li X, Du Z, Li H, Wang H et al. Melatonin inhibits tumorigenicity of glioblastoma stem-like cells via the AKT-EZH2-STAT3 signaling axis. J Pineal Res 2016; 61: 208–217. [DOI] [PubMed] [Google Scholar]
- 106.Riquelme E, Behrens C, Lin HY, Simon G, Papadimitrakopoulou V, Izzo J et al. Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Res 2016; 76: 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen B, Liu J, Chang Q, Beezhold K, Lu Y, Chen F. JNK and STAT3 signaling pathways converge on Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells induced by arsenic. Cell Cycle 2013; 12: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010; 7: 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 2010; 12: 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 2010; 24: 2615–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 2011; 13: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang ZX, Wu JW. Autophosphorylation kinetics of protein kinases. Biochem J 2002; 368: 947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee CH, Yu JR, Granat J, Saldana-Meyer R, Andrade J, LeRoy G et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev 2019; 33: 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X, Long Y, Paucek RD, Gooding AR, Lee T, Burdorf RM et al. Regulation of histone methylation by automethylation of PRC2. Genes Dev 2019; 33: 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161: 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015; 373: 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martin KA, Cesaroni M, Denny MF, Lupey LN, Tempera I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Mol Cell Biol 2015; 35: 3934–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caruso LB, Martin KA, Lauretti E, Hulse M, Siciliano M, Lupey-Green LN et al. Poly(ADP-ribose) Polymerase 1, PARP1, modifies EZH2 and inhibits EZH2 histone methyltransferase activity after DNA damage. Oncotarget 2018; 9: 10585–10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018; 37: 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 2017; 19: 1371–1378. [DOI] [PubMed] [Google Scholar]
- 121.Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004; 23: 5759–5769. [DOI] [PubMed] [Google Scholar]
- 122.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bryant RJ, Cross NA, Eaton CL, Hamdy FC, Cunliffe VT. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate 2007; 67: 547–556. [DOI] [PubMed] [Google Scholar]
- 124.Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007; 12: 419–431. [DOI] [PubMed] [Google Scholar]
- 125.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med 2010; 16: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jain P, Di Croce L. Mutations and deletions of PRC2 in prostate cancer. BioEssays : news and reviews in molecular, cellular and developmental biology 2016; 38: 446–454. [DOI] [PubMed] [Google Scholar]
- 127.Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep 2018; 25: 2808–2820 e2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiao L, Shubbar M, Yang X, Zhang Q, Chen S, Wu Q et al. A partially disordered region connects gene repression and activation functions of EZH2. Proc Natl Acad Sci U S A 2020; 117: 16992–17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018; 36: 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol 2008; 32: 65–71. [DOI] [PubMed] [Google Scholar]
- 131.Alanee S, Moore A, Nutt M, Holland B, Dynda D, El-Zawahry A et al. Contemporary Incidence and Mortality Rates of Neuroendocrine Prostate Cancer. Anticancer Res 2015; 35: 4145–4150. [PubMed] [Google Scholar]
- 132.Zaffuto E, Pompe R, Zanaty M, Bondarenko HD, Leyh-Bannurah SR, Moschini M et al. Contemporary Incidence and Cancer Control Outcomes of Primary Neuroendocrine Prostate Cancer: A SEER Database Analysis. Clin Genitourin Cancer 2017; 15: e793–e800. [DOI] [PubMed] [Google Scholar]
- 133.Parimi V, Goyal R, Poropatich K, Yang XJ. Neuroendocrine differentiation of prostate cancer: a review. Am J Clin Exp Urol 2014; 2: 273–285. [PMC free article] [PubMed] [Google Scholar]
- 134.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer discovery 2011; 1: 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2014; 20: 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017; 355: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016; 30: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin Epigenetics 2015; 7: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016; 29: 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun 2018; 9: 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun 2018; 9: 4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kleb B, Estecio MR, Zhang J, Tzelepi V, Chung W, Jelinek J et al. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics 2016; 11: 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luo J, Wang K, Yeh S, Sun Y, Liang L, Xiao Y et al. LncRNA-p21 alters the antiandrogen enzalutamide-induced prostate cancer neuroendocrine differentiation via modulating the EZH2/STAT3 signaling. Nat Commun 2019; 10: 2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martin MC, Zeng G, Yu J, Schiltz GE. Small Molecule Approaches for Targeting the Polycomb Repressive Complex 2 (PRC2) in Cancer. J Med Chem 2020; 63: 15344–15370. [DOI] [PubMed] [Google Scholar]
- 145.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013; 110: 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kuntz KW, Campbell JE, Keilhack H, Pollock RM, Knutson SK, Porter-Scott M et al. The Importance of Being Me: Magic Methyls, Methyltransferase Inhibitors, and the Discovery of Tazemetostat. J Med Chem 2016; 59: 1556–1564. [DOI] [PubMed] [Google Scholar]
- 147.Kurmasheva RT, Sammons M, Favours E, Wu J, Kurmashev D, Cosmopoulos K et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2017; 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fan W, Morinaga H, Kim JJ, Bae E, Spann NJ, Heinz S et al. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J 2010; 29: 4223–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morishima S, Ishitsuka K, Izutsu K, Kusumoto S, Makiyama J, utsunomiya A et al. First-in-Human Study of the EZH1/2 Dual Inhibitor Valemetostat in Relapsed or Refractory Non-Hodgkin Lymphoma (NHL) - Updated Results Focusing on Adult T-Cell Leukemia-Lymphoma (ATL). Blood 2019; 134 [Google Scholar]
- 150.Kung PP, Bingham P, Brooun A, Collins M, Deng YL, Dinh D et al. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2- dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J Med Chem 2018; 61: 650–665. [DOI] [PubMed] [Google Scholar]
- 151.Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J Med Chem 2016; 59: 9928–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Taplin M-EH A; Shah S; Shore ND; Edenfield WJ; Sartor OA; Nordquist LT; Agrawal M; Clark W; Wise DR; Oh WK; Fleming MT; Butrynski JE; Chatta GS; Bupathi M; Lebedinsky C; Senderowicz A; Li J; Colak G; Nash D; Trojer P; Bradley WD; Piel J; Antonarakis ES, . Abstract CT094: Phase Ib results of ProSTAR: CPI-1205, EZH2 inhibitor, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res 2019; 79. [Google Scholar]
- 153.Cromm PM, Crews CM. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol 2017; 24: 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov Today Technol 2019; 31: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nalawansha DA, Crews CM. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem Biol 2020; 27: 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol 2020; 16: 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell 2011; 43: 798–810. [DOI] [PubMed] [Google Scholar]
- 158.Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 2007; 27: 5105–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li J, Xi Y, Li W, McCarthy RL, Stratton SA, Zou W et al. TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene 2017; 36: 2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18: 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med 2015; 21: 1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cyrta J, Augspach A, De Filippo MR, Prandi D, Thienger P, Benelli M et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat Commun 2020; 11: 5549. [DOI] [PMC free article] [PubMed] [Google Scholar]