

Abstract
The knowledge of the human microbiome is rapidly expanding and likely a critical factor in the initiation, progression and prognosis of multiple forms of cancer. In this review, we focus on recent investigations to discern putative causative microbial species and/or the microbiome composition and structure currently associated with pro-carcinogenesis and tumorigenesis at select body sites. We specifically highlight forms of cancer, gastrointestinal and non-gastrointestinal, that have significant bacterial associations and well-defined experimental evidence with the aim to generate directions for future experimental and translational investigations to develop a clearer understanding of the multifaceted mechanisms by which microbiota impact cancer formation.
Introduction
The community of bacteria, archaea, fungi, and viruses on and in various sites of the body constitutes the human microbiome. A flourishing literature now links microbiome composition to an assortment of diseases from inflammatory bowel disease to schizophrenia (1,2). Bacteria are an abundant component of the microbiome, particularly in the gut where bacteria are estimated to number ~3.8 × 1013 in total (3). Recent advances in our understanding of the mechanisms by which bacterial members of the microbiome may initiate and/or promote tumorigenesis throughout not just the gastrointestinal tract but also other organs will be the focus of review. Experimental models suggest that bacteria can foster the induction and/or development of tumor formation through multiple mechanisms: (i) direct DNA-damaging effects of bacterial toxins, (ii) bacterial metabolites (such as products of Western diet metabolism), (iii) inflammation driven by direct physical interaction with host cells, chronic infections, and invasive biofilm formation, and (iv) inhibition of anti-tumoral immune responses (Figure 1). Multiple theories have arisen regarding bacterial involvement in tumorigenesis, including the driver-passenger model including extension of the keystone hypothesis, the hit-and-run model, chronic dysbiosis, and alterations in the spatial distribution of the microbiome and/or barrier integrity in epithelial tissues (Figure 2). For the first, an individual or collection of driver pro-carcinogenic bacteria collaborate with microbiome passengers to promote tumorigenesis. In the subtly different keystone hypothesis, the key driver pathogen, even at minute abundances, enables subsequent colonization of putative collaborative, co-occurring passengers. For the hit-and run model, transient colonization and damage by a pro-carcinogenic bacterium is necessary and sufficient to drive tumorigenesis (4). An important note is, with the exception of H. pylori, no additional bacterial species to date has been identified as the causative agent of human tumorigenesis in the absence of a prior host cell genetic mutation. The common link between each model is the disruption of a healthy microbiome, termed dysbiosis. However, a well-defined healthy microbiome membership remains elusive due to significant person-to-person variation. Methods to analyze the composition of the microbiome continue to evolve rapidly with uneven application and ‘best practices’ uncertain. Culturing bacteria from stool samples and other patient tissues is valuable but also inconsistent amongst laboratories with a subset of bacteria remaining unculturable to this day. An early breakthrough in this field was the recognition of the utility of sequencing the 16S rRNA gene to study the human microbiota. Next generation sequencing now allows for increased sequence depth, which when combined with improved computational analyses, allow for in-depth phylogenetic and taxonomic analyses of the microbiome, even to the species level. Sequence detection of all microbial genomes (metagenomics) or transcripts (metatranscriptomics) within a sample have further advanced microbiome analyses. Together, identification of what microbes are present, what genes they contain, and their transcriptional profiles is now feasible in some instances, allowing characterization of both microbiome membership and function. Nonetheless, in tissues or samples with a low abundance of microbes, both accurate taxonomic and functional analyses are challenging. Recent large-scale studies encompassing multiple tumor types have sought: 1) to improve database and computational approaches (particularly to limit sample contaminant reads), thus, facilitating microbiome analyses across cancer cohorts; 2) to develop a deeper understanding of host variables confounding microbiome analyses; and 3) to struggle with the nuances and definition of comparator ‘healthy host’ microbiomes (5–9) (Table 1). Emerging metabolomics and proteomics methods may provide additional insights into the human microbiome. While each approach provides its own unique set of benefits and limitations, a more complete picture of microbiota complex communities and their impact on tumorigenesis will be realized by use of combined methods and incorporation of host cell responses (e.g., RNA-seq).
Figure 1. Examples of bacterial mechanisms of tumorigenesis.
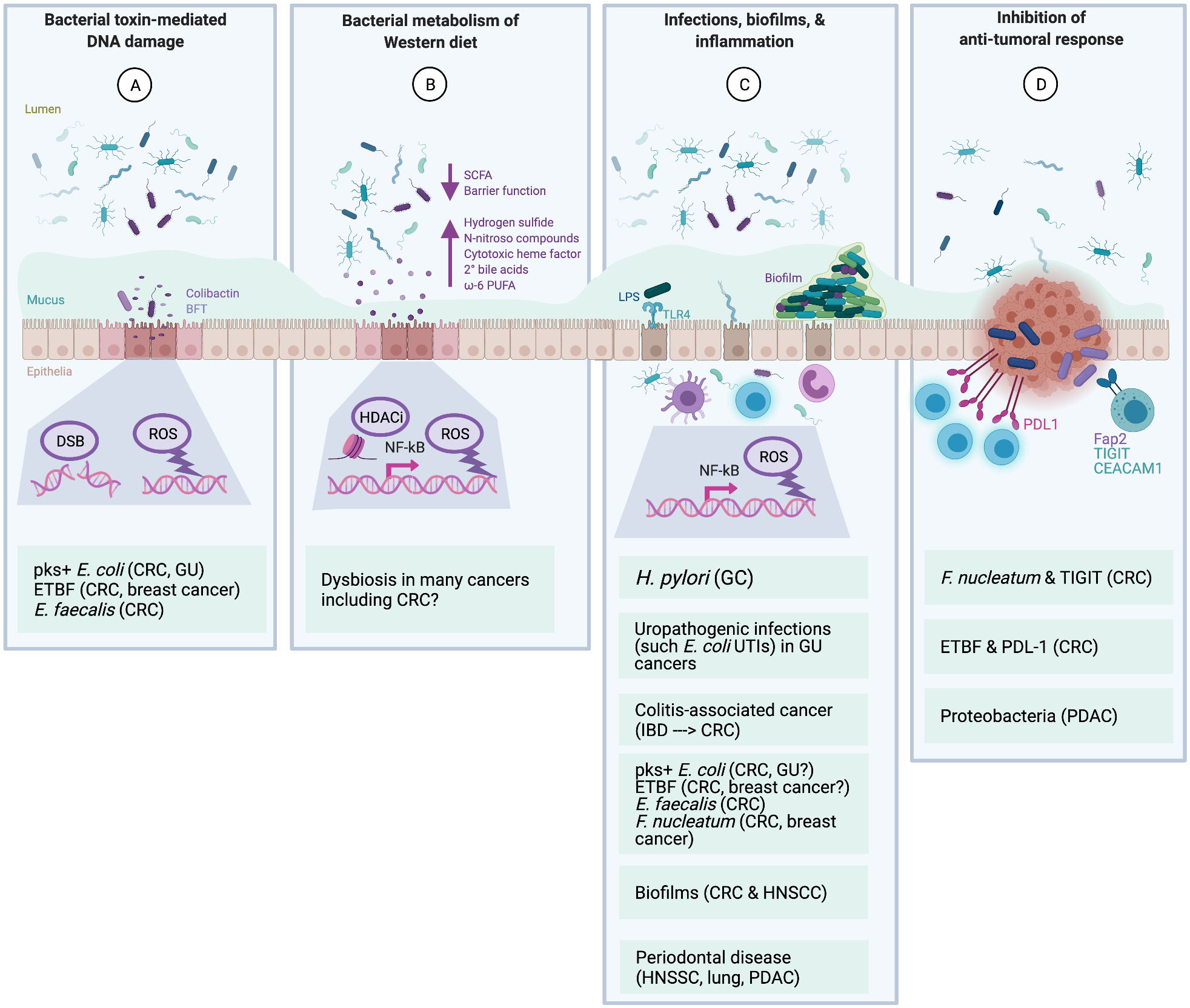
Bacteria have been proposed to contribute to the initiation and/or progression of tumorigenesis via both direct and indirect mechanisms. Many bacteria have overlapping mechanisms that also intersect with lifestyle and host genetic factors. A, Direct DNA-damaging effects of bacterial toxins. The colibactin genotoxin produced by pks+ Escherichia coli induces double-strand DNA breaks (DSB) and other forms of DNA damage that result in a unique mutational signature that is found both in vitro and in vivo in CRC. Certain Enterococcus faecalis strains produce superoxide that can damage host DNA. The BFT toxin from enterotoxigenic Bacteroides fragilis (ETBF) induces ROS via induction of host spermine oxidase in epithelial cells. B, Bacterial metabolism of a Western diet. The microbiota play a critical role in the metabolism of host dietary components. Consumption of a Western diet is associated with increased risk of numerous cancers, likely due to decreased bacterial production of beneficial SCFAs (due to lower fiber levels in diet), decreased barrier function (such as thinner or more permeable mucus in the colon), and increases in numerous pro-carcinogenic metabolites produced by bacteria including hydrogen sulfide, N-nitroso compounds, cytotoxic heme factor, secondary bile acids, and ω−6 PUFAs. These factors lead to ROS production (DNA damage) and HDAC inhibition (altered pro-inflammatory signaling). C, Infections, biofilms, and inflammation. Chronic/recurring bacterial infections and/or invasive bacterial biofilms may promote tumorigenesis via broad, indirect mechanisms related to inflammation and immune response activation through NF-κB transcription, including DNA-damaging ROS production from innate immune cells (e.g., neutrophils and macrophages) and mutations in the pre-cancer cells that accrue due to excessive proliferation. Many cytokines are involved in these pro-tumorigenic inflammatory processes, including IL-6 and IL-17. Helicobacter pylori, the most well-documented case of a bacterium causing cancer, is thought to cause GC not through any direct toxigenic mechanisms, but rather through the induction of such chronic inflammatory responses. Similarly, UTI’s are associated with subsequent increased risk of bladder and kidney cancers. IBD, which has a strong microbial component, is the poster child of colitis-associated CRC. Many of the genotoxic bacteria (from panel A) as well as oral bacteria (such as Fusobacterium nucleatum) may promote inflammation as well. At least some polymicrobial biofilms are also highly pro-inflammatory and may play a role in both HNSCC, where these oral biofilms are known as dental plaque, and CRC, in which mucus-invasive biofilms in the colon are highly prevalent on both hereditary and sporadic CRCs compared to healthy controls. Finally, periodontal disease, which is often linked to oral biofilms, has been associated at the epidemiological level with a number of cancers, including HNSCC, lung cancer, and PDAC. D, Inhibition of anti-tumoral immune responses. Relatively new data suggest that bacteria can inhibit local NK- and T cell-mediated killing of tumor cells. Notable examples include F. nucleatum, which can bind NK and T cell TIGIT receptors via the F. nucleatum adherence factor, Fap2, and ETBF induction of PD-L1 on tumor and/or immune cells in animal models of CRC. Other mechanisms have not yet been fully delineated, yet strongly suggest additional immune modulatory roles, such as Proteobacteria in PDAC. Abbreviations - BFT: B. fragilis toxin; CRC: colorectal cancer; DSB: double strand breaks; ETBF: enterotoxigenic B. fragilis; Fap2: Fusobacterium adherence protein 2; GC: gastric cancer; GU: genitourinary; HDACi: histone deacetylase inhibitor; HNSCC: head and neck squamous cell carcinoma; IBD: inflammatory bowel disease; LPS: lipopolysaccharide; NFkB: Nuclear factor kappa B; NK: Natural killer cells; PDAC: pancreatic ductal adenocarcinoma; PD-L1: Programmed death ligand 1; PUFA: polyunsaturated fatty acids; ROS: reactive oxygen species; SCFA: short chain fatty acids; TIGIT: T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains; TLR4: Toll-like receptor 4; UTI: urinary tract infection
Figure 2. Theories of bacterial involvement in tumorigenesis.
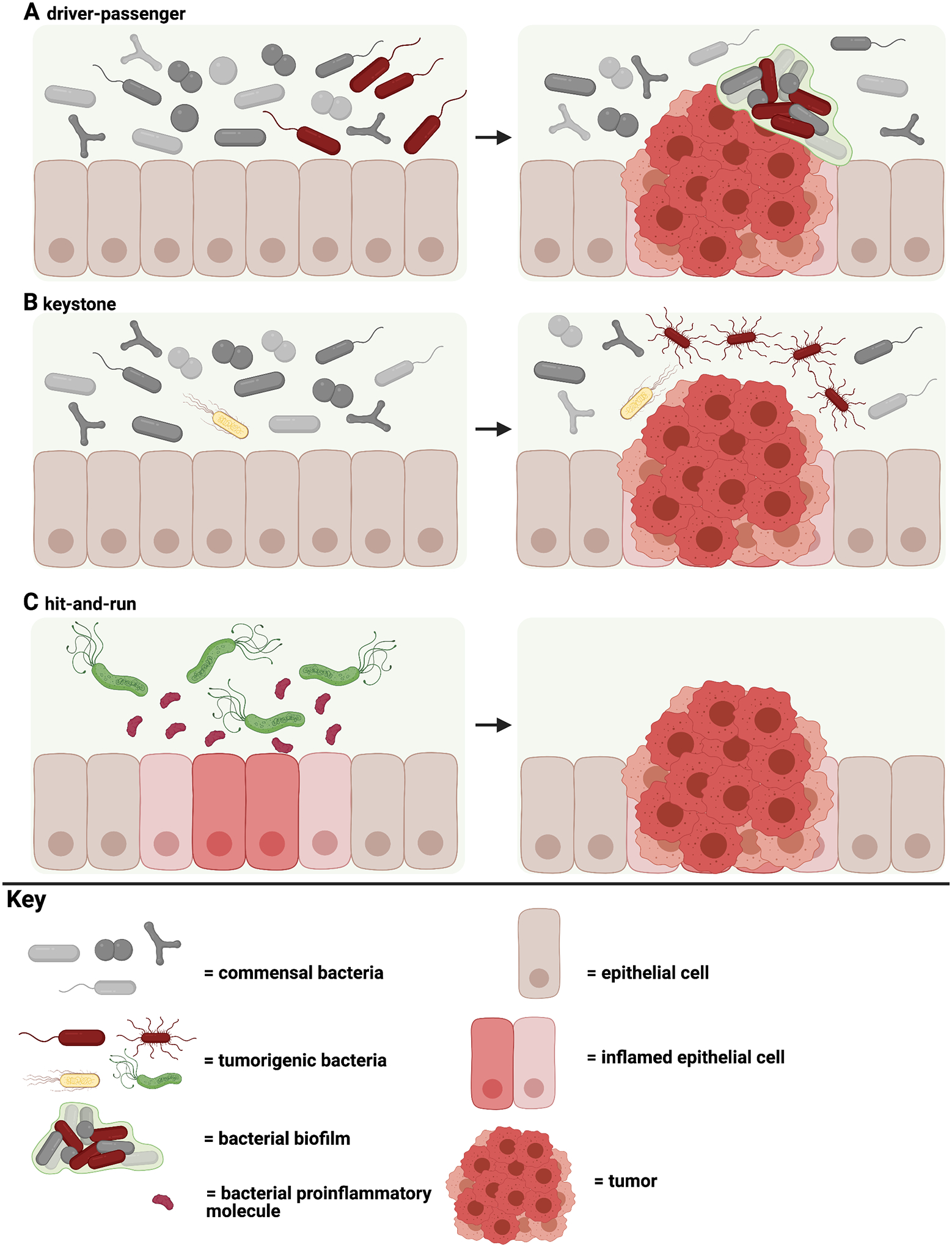
A, The driver-passenger model where a single or group of tumorigenic bacteria recruit or coordinate with members of the microbiome to promote tumorigenesis. This model embraces the concept of the impact of a community of microbes on tumorigenesis. B, The keystone hypothesis states that the presence of a single tumorigenic bacterium even at minimal abundance enables the colonization of additional collaborative pro-carcinogenic bacteria. C, The hit-and-run model is described as temporary colonization and damage by a tumorigenic bacterium that results in tumorigenesis. Additional potential pro-tumorigenic models include chronic dysbiosis and biofilm-mediated changes in tissue function, both extensions of the ‘driver-passenger’ model.
Table 1:
Summary of recent large-scale microbiome papers relevant to cancer.
| Reference | Study Type | Study Goal | Population Studied (N) & Specimen Type(s) | Data & Tumor Types | Major Findings | Comment |
|---|---|---|---|---|---|---|
| Poore GD et al (8) | Cross-sectional | To define unique blood and/or tissue microbial signatures within & between major cancers | The Cancer Genome Atlas (TCGA), 33 cancers from treatment-naïve patients (N=10,183 patients, 17,625 samples) and blood samples analyzed. Details on TCGA sample acquisition & processing in (90).Study included an independent validation cohort (UCSD, N=169) analyzing plasma samples. | Data: Whole genome & whole transcriptome sequencing using polyA-selected RNAseq data. Tumors*: adrenocortical, AML, bladder, brain/GBM, breast, cervical, cholangiocarcinoma, colon, esophageal, gastric, head and neck, kidney, liver, lymphoid, lung, melanoma, mesothelioma, ovarian, pancreas, pheochromocytoma/paraganglioma, prostate, rectum, sarcoma, testicular, thyroid, thymoma, uterine, |
Microbial communities defined at the genus level, carefully normalized & subjected to machine learning pipelines appeared to distinguish between cancer types. | Employed stringent computational removal of predicted contaminating sequences from published data, discarding up to 92.3% of total sequence data in some analyses. Serial analyses raised concern that microbial diagnostics may lack sufficient sensitivity to detect early stage cancers. |
| Nejman D et al (9) | Cross-sectional | To define the bacterial microbiome of select cancers | 7 cancers from 4 countries (Israel, USA, Italy, Netherlands) (N=1010 tumors & 516 mostly normal adjacent tissues; 811 negative controls). See Table S1 in Nejman et al (9). | Data: Real-time qPCR using universal bacterial primers complemented by immunohistochemistry & RNA fluorescence in situ hybridization using snap frozen & formalin-fixed paraffin-embedded samples. Tumors: breast, lung, melanoma, pancreas, ovary, bone, glioblastoma multiforme |
Each tumor type (breast, lung, ovary, pancreas, melanoma, bone, brain) has a distinct microbiome composition. | Breast cancer displayed the most rich & diverse microbiome. Intratumoral bacteria were mostly intracellular in cancer & immune cells. |
| Byrd AL et al (6) |
Prospective recruitment of 1000 healthy men & women, longitudinal sampling of ~50% of cohort. | To define the microbiome associations with host factors & lifestyle parameters in healthy individuals and then begin to test associations with non-GI cancers. | Milieu Intérieur cohort (N=946 healthy French individuals, 1359 gut microbiome samples). Non-GI cancer published cohorts (N=5 cohorts; 283 cancer patients, 375 samples) | Data: Shotgun metagenomic sequencing In the analyses, comparative literature-derived data from non-GI tumors (melanoma, lung, kidney) was used but no primary tumor data included. |
In healthy individuals, identified sex & age as key variables in microbiome composition; in non-GI cancers suggested global microbiome shifts vs healthy individuals. | This paper provides data to define variables important to consider in microbiota studies. Presents the Genome Taxonomy Database (GTDB), a resource for microbiome research aligned with rich clinical metadata. |
| Vujkovic-Cvijin I et al. (5) | Cross-sectional | To determine key exposures determining human gut microbiome heterogeneity | American Gut cohort, the largest publically available human gut bacterial microbiota dataset (N variable by subgroup & analysis, see Table 1 in paper; N=4038–5878, negative controls) | Data: V4 hypervariability region of the 16S rRNA gene Tumors: A subset of enrollees reported ‘cancer’ on the study questionnaire but, in this study, the diagnosis of ‘cancer’ had limited to no impact on the presented analyses. Tumors were not directly analyzed. |
Alcohol consumption & bowel movement quality unexpectedly strong sources of gut microbiome variance | This paper is not cancer-specific but presents important information to consider in future work. The authors propose rigorous matching of exposures between controls vs disease to address contribution of microbiota to human disease. |
| Dohlman AB (7) | Cross-sectional | To define the prevalence of cancer tissue-resident microbiota in GI cancers within the TCGA | TCGA, GI cancer samples (N=3689) as well as TCGA paired normal and blood samples; Duke Hospital healthy plasma samples. | Data: Whole genome sequencing; validation using original TCGA tissue (N=5 CRC samples) Tumors: oropharyngeal, esophageal, GI and colorectal. |
Removed sequencing contaminant reads equi-prevalent across sample types to provide a public database of curated, decontaminated microbiomes from GI cancers termed The Cancer Microbiome Atlas (TCMA). | A potentially useful new resource for GI cancer-associated microbiome research utilizing computational removal of predicted contamination from the published data. |
Abbreviations: AML, acute myeloid leukiemia; GBM, glioblastoma multiforme; GI, gastrointestinal; CRC, colorectal cancer; N/A, not applicable; UCSD, University of California, San Diego
Tumor types may include more than one histopathology subtype.
Thus, the microbiome—microbes and their genomes—presents a tantalizing research frontier for understanding cancer pathogenesis and to devise previously unimagined approaches to cancer prevention, diagnosis and therapy. Herein, we provide a perspective on recent investigations (a collection of literature highlights is found in Table 2) of putative microbiome, primarily bacterial, contributions to the pathogenesis of select gastrointestinal (GI) and non-GI cancers.
Table 2:
Recent1 Literature Highlights in Cancer Microbiome Research
| Reference | Study Type | Study Goal | Human Population Studied (N) | Major Findings |
|---|---|---|---|---|
| GI Cancers | ||||
| Colorectal Cancer | ||||
| Dejea CM et al (39) | Mouse model & human samples | To study the role of biofilm formation in the progression of hereditary colon cancer. | 5 FAP patients, 1 juvenile polyposis syndrome patient | Biofilms containing co-colonization with ETBF and pks+ E. coli promotes carcinogenesis through mucus degradation enabling pks+ E. coli adherence and subsequent DNA damage as well as IL-17 induction by both bacteria. |
| Kitamoto S et al (35) | Mouse model | To investigate how periodontal inflammation exacerbates gut inflammation | N/A | Oral pathobionts and oral pathobiont-reactive Th17 translocate to the gut and cause development of colitis. |
| Pleguezlos-Manzano C et al (14) | Organoid & human samples | To identity mutagenic characteristics of pks+ E. coli | 5786 cancer genomes | Revealed a distinct mutational signal in organoids injected with pks+ E. coli that was detected in a subset of predominantly CRC human cancer genomes. |
| Wilson MR et al (22) | Cell lines & mouse model | To determine the molecular mechanism of the genotoxic effects of colibactin | N/A | Colibactin alkylates DNA in vitro and the metabolite was identified in mice colonized with pks+ E. coli. |
| Pancreas | ||||
| Geller LT et al (55) | Mouse model & human samples | To study impact of microbes on PDAC chemotherapy | 113 human PDAC samples | Mouse model: Bacteria, likely Gammaproteobacteria, metabolize the chemotherapeutic drug gemcitabine via long isoform of cytidine deaminase conferring gemcitabine resistance; Human samples: PDACs contain Gammaproteobacteria populations. |
| Pushalkar S et al (56) | Mouse model & human samples | To define PDAC microbiome-mediated immune mechanisms of oncogenesis | Fecal samples (N=32 patients with PDAC; N=31 healthy volunteers); Pancreas tissue samples (N=5 healthy or PDAC patients each) | Mouse model: The PDAC microbiome promotes disease progression through innate immune & T-cell intratumoral immunosuppressive mechanisms that can enable response to checkpoint-based immunotherapy. Human samples: Proteobacteria are prominent in PDAC tissues. Comparison of patients with both gut & PDAC microbiome analysis suggest increase translocation of Proteobacteria to the pancreas. |
| Riquelme E et al (57) | Mouse model & human samples | To identify microbiome mechanisms contributing to long-term survival in PDAC patients. | PDAC tissues from short-term survivors (STS) (N=22 primary cohort, 10 validation cohort) & long-term survivors (LTS) (N=21 primary cohort, 15 validation cohort); stools from PDAC STS, LTS-no disease & healthy controls (N=8–17/group) | Mouse model: Human-to-mouse FMT from STS, LTS or controls differentially modulated the tumor microbiome, TME and tumor progression, mirroring patient outcomes. Human samples: STS and LTS PDAC patients display distinct tumor microbiomes with LTS PDAC enriched in Proteobacteria, Actinobacteria and Bacillus clausii. |
| Gastric | ||||
| Choi IJ et al (47) | Human samples | To determine whether antibiotic clearance of H. pylori can prevent development of metachronous gastric cancer | Prospective clinical trial of 470 patients who had prior endoscopic resection of early gastric cancer or high-grade adenoma and received either antibiotics (to clear H. pylori) or placebo | H. pylori antibiotic clearance reduced the incidence of metachronous gastric cancer by nearly 50% (13.4% vs. 7.2% treatment vs. placebo) and improved gastric corpus atrophy. |
| Non-GI Cancers | ||||
| Lung | ||||
| Greathouse KL et al (66) | Human samples | To define the microbiome associations of lung cancer vs patient-matched normal lung tissues. | Retrospective analysis of prospective National Cancer Institute-Maryland study; N=106 matched pairs of lung tumor and non-tumor tissues. Includes a TCGA-derived validation cohort. | Identified microbiome-gene and microbiome-exposure interactions in squamous cell carcinoma lung cancer tissues. Specifically, enrichment of Acidovorax spp. in smoking-associated squamous cell carcinoma lung cancers with TP53 mutations. |
| Jin C et al (67) | Mouse model | To identify the contribution of the local lung microbiota to lung cancer development | N/A | Local lung microbiota promotes lung cancer development in KP mice. Local lung dysbiosis induces tumor-promoting inflammation attributable to ɣδ –T17 cells and myeloid cells. |
| Tsay J-C J et al (68) | Mouse model & human samples | To define human microbial signatures associated with lung cancer prognosis & disease mechanisms. | N=83 prospectively enrolled lung cancer patients | Human samples: A lower airway microbiota signature enriched with oral commensals associated with worse lung cancer prognosis. Human samples & mouse model: Lung cancer dysbiosis was associated with upregulation of IL-17, PI3K-AKT, MAPK and ERK pathways as well as IL-6/IL-8. Veillonella parvula was the most abundant taxon driving the association. |
| Breast | ||||
| Parhi L et al (70) | Mouse model & human samples | To investigate the contribution of Fusobacterium nucleatum to breast cancer development. | N=50 FFPE breast cancer samples with N=30 matched adjacent non-tumor tissues | Human samples: Gal-GalNAc levels are increased in breast cancer samples. Using 16S rRNA amplicon sequencing, ~30% of breast cancer samples displayed increased F. nucleatum reads. Mouse model: IV inoculation of F. nucleatum into an orthotropic breast cancer model resulted in Fap2-mediated F. nucleatum tumor colonization and enhanced tumor growth inhibited by antibiotics. |
| Parida S et al (71) | Mouse model & human samples | To investigate the breast microbiome | Utilized available human datasets comparing benign & malignant breast tumors as well as nipple aspirate fluids of breast cancer survivors & healthy volunteers | Human data: Meta-analysis of breast cancer microbiome studies identified Bacteroides fragilis in breast tumor tissues. Mouse model: Gut or breast intraductal colonization with a toxin-producing molecular subset of B. fragilis (ETBF) induced growth and metastasis of breast cancer cells potentially mediated by β–catenin and Notch1 signaling. |
| Head & Neck | ||||
| Hayes RB et al (64) | Human samples | To define whether changes in the oral microbiome precede HNSCC | Oral rinse samples from 383 patients from the CPS-II and PLCO studies, including 129 incident cases of HNSCC and 254 controls | The strongest microbial associations identified were protective effects of Kingella and Corynebacterium genera in larynx cancer and smokers, a biologically plausible mechanism due to the cigarette toxin-neutralizing capabilities of these taxa. |
| Genitourinary | ||||
| Shrestha E et al (83) | Human samples | To define whether the urinary microbiome is associated with prostate cancer | Urine samples from 135 men with or without prostate cancer | Total prostate cancer cases did not cluster differently from controls; however, a cluster of cases harbored a striking flora containing 6 pro-inflammatory bacteria suggesting possible subsets of prostate cancer that may be driven by the urinary microbiome. |
Abbreviations: CRC, colorectal cancer; ETBF, enterotoxigenic Bacteroides fragilis; FAP, familial adenomatous polyposis; Fap2, Fusobacterium adherence protein 2; FFPE, formalin-fixed paraffin-embedded; FMT, fecal microbiota transplantation; GI, gastrointestinal; KP, mice bearing Kras mutation and p53 loss; N/A, not applicable; PDAC, pancreatic ductal adenocarcinoma; TCGA, The Cancer Genome Atlas; TME, tumor immune microenvironment.
Recent defined as 2016 or later
Gastrointestinal Cancers
Colorectal cancer
Colorectal cancer (CRC) is the second highest diagnosed cancer and ranks second in cancer-related deaths worldwide (10). The induction of oncogenes and suppression of tumor-suppressor genes in the large bowel results from a series of mutations and epigenetic changes over time, leading to the onset of tumor formation (11,12). Inherited genetic predisposition syndromes, such as familial adenomatous polyposis (FAP), Lynch syndrome, and Peutz-Jeghers syndrome, constitute a minority of CRC cases (11), with the heritability of CRC estimated to be between 12–35% (13). Therefore sporadic CRC developing from environmental stimuli constitutes the majority of cases (11). Lifestyle factors including obesity, diabetes, a Western diet, alcohol consumption, and smoking are recognized as CRC risk factors (11). Each of the aforementioned CRC risk factors alter the gut microbiome. These data, combined with the knowledge that individual strains of bacteria harbor the ability to contribute to tumorigenesis, provided strong support for investigations into the role of the gut microbiome in the initiation and progression of colorectal carcinogenesis (11,13).
Individual bacterial species or strains associated with CRC
Multiple models of bacteria-induced tumorigenesis have been theorized (see Introduction and Figure 1, 2), yet a defined sequence of bacteria-driven events in CRC and the fraction of CRC most clearly linked to bacterial species remain poorly defined (14). This lack of clarity suggests that multiple microbial and non-microbial mechanisms may contribute to CRC development. Despite this, specific bacterial strains have been linked to human CRC using a combination of human epidemiological studies that demonstrate plausible associations with tumorigenesis and animal models that demonstrate potential mechanisms (15,16).
Bacteroides fragilis is a commensal member of the human microbiota composing 0.1 – 0.5% of total gut bacteria (17). The majority of B. fragilis strains remain benign, however a subset of strains produces the zinc-dependent metalloprotease toxin, Bacteroides fragilis toxin (BFT) (17). Toxin-producing strains (enterotoxigenic B. fragilis [ETBF]), induce colon inflammation, which has been associated with diarrhea, inflammatory bowel disease, and CRC (17). ETBF stimulates carcinogenesis by activating host colonic epithelial cell (CEC) NF-κB and STAT3 pathways while additionally recruiting procarcinogenic myeloid inflammation and inducing mucosal IL-17 production (17). BFT binds to CECs through an unknown receptor triggering E-cadherin cleavage resulting in increased barrier permeability, activation of CEC Wnt signaling, induction of c-Myc expression, and amplified CEC proliferation (17). ETBF modulates host gene expression through BFT, increasing chromatin accessibility. Genes impacted by BFT include upregulation of CEACAM6, that functions as a receptor for adherent-invasive E. coli, and downregulation of MUC2, the primary glycoprotein of colonic mucus (18). Combined with its cytoskeletal impact and ability of all B. fragilis to digest mucin, ETBF modifies colon barrier function and the CEC apical membrane. Additional alterations to the host genome stem from ETBF-induced genomic hypo- and hypermethylation of human CRC cell line genomes and tumors (18–20). While some results support an association between methylation changes and, for example, reduced expression of genes with known tumor-suppressive functions, direct, consistent linkage of methylation changes to transcriptional changes remained uncertain and vary based on tumor type (18,20,21). Furthermore, bft has been identified more frequently in CRC patients versus controls in both the colon mucosa and in the stool (15). Together, these data lend support for ETBF to be a potent contributor to colorectal tumorigenesis.
Escherichia coli is another common gut commensal bacterium that is mainly considered benign. Harmful strains produce the genotoxin colibactin through a 50-kb hybrid polyketide-nonribosomal peptide synthase operon (pks+ E. coli) (14). Detailed studies have demonstrated colibactin causes DNA inter-strand crosslinks, DNA double-strand breaks, chromosomal aberrances, and cell cycle arrest in human cells in vitro (12). Despite the unstable nature of colibactin, specific DNA adducts are formed leading to cytotoxicity and mutations (22). The damage to DNA in combination with intestinal inflammation fosters tumorigenesis in mice (12). In clonal organoids injected with pks+ E. coli, a mutational signal was identified that was absent in organoids injected with an isogenic pks− E. coli (14). These data provide evidence that colibactin can modify CEC DNA, which is required for CRC development (14). Despite this mutational signature being present in up to 16% of human cancer genomes and associated with APC mutation, predominantly in CRC, whether certain pks+ E. coli strains initiate CRC remains undefined (14). Ultimately, these results suggest pks+ E. coli modify host genetics to favor the initiation and propagation of tumorigenesis in the colon.
The Gram-negative anaerobic oral commensal Fusobacterium nucleatum has been identified as a potential CRC biomarker in stool and is predominantly found in the tumor microenvironment (TME) (23). However, as with the previously described tumorigenic bacteria, the causality of F. nucleatum in CRC is uncertain. Surprisingly, F. nucleatum encodes limited canonical virulence factors and no currently described toxins (23). F. nucleatum includes four major subspecies: F. nucleatum animalis, F. nucleatum nucleatum, F. nucleatum polymorphum, F. nucleatum vincentii. To date, using repetitive inoculation, only select strains of F. nucleatum impact colon carcinogenesis in SPF ApcMin/+ mice (e.g., EAVG_002; 7_1 (24)). No strain has been found to induce colon tumors in germ-free mice (25). F. nucleatum utilizes the virulence factor FadA to bind to the extracellular domain of E-cadherin, which induces colon cancer cell proliferation through activation of host CEC Wnt/β-catenin signaling in addition to TLR4-activated signaling to NF-κB (23). In support of this mechanism, FadA gene expression in human CRC tissue is significantly upregulated compared to healthy tissue controls (26). F. nucleatum further induces a proinflammatory tumor-promoting microenvironment by expanding myeloid derived-immune cells. Interestingly, F. nucleatum also inhibits anti-tumor responses by Fap2 binding of the human TIGIT receptor, subsequently inhibiting the cytotoxic function of Natural Killer (NK) cells and other tumor-infiltrating lymphocytes (TILs) (23). Importantly, this interaction only occurs in human cells, as F. nucleatum does not bind to mouse TIGIT (27). It remains unclear as to whether this lack of murine TIGIT binding may partly explain the disconnect between the strong association studies with CRC in human cross-sectional cohorts vs. the relatively weak and variable tumorigenesis seen in mouse models. Finally, F. nucleatum was found to persist with its associated microbiome in distant liver metastases of CRC tumors (28), suggesting that F. nucleatum and its associated metastatic microbiota affects on anti-tumoral responses and could play a role in metastatic lesions (29). Taken together, F. nucleatum appears to colonize and expand in the TME to promote CRC through impacts on the host immune response; well-defined F. nucleatum procarcinogenic virulence factors deserve more study.
Collectively, the list of putative tumorigenic bacteria and their connection to CRC continues to expand. Similar to F. nucleatum, Streptococcus gallolyticus has been shown to modify the TME. S. gallolyticus induces a proinflammatory state marked by high NF-κB and IL-8 messenger RNA tissue expression while simultaneously recruiting TILs and myeloid cells that cause an immune-suppressive microenvironment promoting neoplasia (30). Peptostreptococcus stomatis and P. anaerobius are other candidate bacteria that have been shown to modulate the TME. P. stomatis contributes to acidity and hypoxia while P. anaerobius leads to ROS accumulation promoting bacterial colonization and cellular proliferation respectively (31,32).
Recently, a meta-analysis of CRC 16S rRNA amplicon sequence data revealed a limited consortium of bacteria, B. fragilis, F. nucleatum, P. stomatis, Parvimonas micra and Gemella morbillorum, was reliably associated with human CRC (15). Of this consortia all are members of the oral microbiome and were detected both in the oral cavity and tumors of CRC patients presenting a strong trend between CRC pathogenesis and the oral microbiome (15,33,34). Notably, periodontitis was found to generate oral microbiome reactive Th17 cells imprinted with gut tropism, which subsequently migrate to the inflamed gut. These translocated (mouth to gut) Th17 cells led to the development of colitis (35). The precise mechanism of translocation of oral bacteria to cancerous lesions, whether by descent through the digestive tract or by hematogenous routes as a result of chewing or dental hygiene and procedures, remains undefined. Furthermore, recent evidence suggests that at least some gut strains of F. nucleatum are identical to those found in the oral cavity in patients, and that hematogenous (tail vein injection) administration of F. nucleatum led to better gut colonization in mice compared to oral gavage (36). Whether the route of administration alters tumorigenesis in mouse models – let alone in patients - remains to be understood but provides another important area of consideration for future preventative measures. A continued exploration into the gut microbiome of CRC patients followed by experimental analyses is imperative to divulge key bacteria and their interactions that function to initiate and/or promote CRC.
The impact of the collective microbiome community on CRC
The collective microbiome community likely demonstrates, at a minimum, an equal and critical contribution vs. individual bacteria in influencing CRC. Immune activation through TLR and NOD-like receptor (NLR) signaling during dysbiosis results in low-level colon inflammation, a known common factor of all CRC (2). Furthermore, the diverse metabolites produced by the microbiome in response to host diet directly interface with CECs (Figure 1). Consumption of a Western diet, rich in red meats, processed foods, and low in dietary fiber is associated with an increased risk of CRC, potentiated by lower levels of beneficial short chain fatty acids (SCFAs) and increases in potentially deleterious metabolites such as secondary bile acids, hydrogen sulfide, and others. Fermenting bacteria produce SCFAs such as butyrate following the consumption of non-digestible carbohydrates (13). Butyrate elicits anti-inflammatory properties by downregulating pro-inflammatory cytokines, modulating colonic Treg cells, regulating gene expression through inhibition of histone deacetylases, and inducing apoptosis in CRC cell lines (2). In contrast to these anti-carcinogenic properties, butyrate induced proliferation of harvested MSH2-deficient CECs in vitro (37). These data suggest butyrate may require tight regulation to benefit the host, potentially depending on host cell genotype. The conversion of primary bile acids to secondary structures is dependent on the gut microbiome (13). A high abundance of secondary bile acids have the potential to induce oxidative DNA damage and stimulate colon tumor formation (13), despite these molecules providing numerous benefits. While the data describing the influence and subtle balance of the metabolites produced by the gut microbiome are far from comprehensive, they lend to the significance of the community composition in CRC.
Highly concentrated communities of bacteria that invade the inner, dense, typically sterile mucus layer of the colon, colonic biofilms, are assemblies that may provide continual microbial interaction with the host CECs (3). Mucus-invasive biofilms were present on over 50% of sporadic CRCs in both a US and a Malaysian cohort compared to <15% in healthy colonoscopy controls (15,38). Interestingly, colonic biofilms on histologically normal colonic tissues displayed increased STAT3 activation and a loss of E-cadherin in CECs (38). Three major microbiologically divergent colonic biofilms types were described in sporadic CRC patient samples: polymicrobial (Bacteroidetes, Lachnospiraceae), polymicrobial with Fusobacteria blooms, and Proteobacteria-dominant (15). Human colonic biofilms from both CRC patients and otherwise healthy controls were found to be directly tumorigenic in murine models (25), whereas biofilm-negative normal mucosal tissues were not tumorigenic; biofilm-positive colonic tissues may enable pro-carcinogenic bacteria adherent to the colonic epithelium to deliver virulence factors to CECs. Patients with FAP also harbored nearly ubiquitous mucus-invasive biofilms, although they were primarily composed of ETBF and pks+ E. coli (39). The compounding effects of a dual-pathogen ecosystem were tested in mouse and in vitro models, in which ETBF promoted mucin degradation that facilitated mucosal colonization of pks+ E. coli and led to a coordinated increase in tumorigenesis compared to either bacteria alone (39). In subsequent work, human colon biofilms were found to be directly tumorigenic in murine models (25). Together, these data suggest the structure and physical position of the bacterial community in the colon have a critical role in CRC tumorigenesis.
Gastric cancer
The recognition of Helicobacter pylori as causal to gastric cancer provides some of the most compelling evidence for the microbiome as a cancer promoter and a roadmap for demonstrating microbial causality in other malignancies (Figure 3, (40)). Gastric cancers of the intestinal type follow a pro-inflammatory trajectory (termed the Correa cascade), advancing from inflamed mucosa/gastritis to gastric atrophy, intestinal metaplasia, intraepithelial neoplasia, and, finally, gastric cancer. As reviewed by Engstrand and Graham, the earliest studies (late 1800’s) that examined gastric cancer microflora identified Lactobacilli overgrowth initially presumed to contribute to cancer initiation itself; however, later studies demonstrated that the Lactobacilli and fungal overgrowth was, in fact, likely a bystander effect due to increase in pH (hypo- or achlorhydria), making these bystander organisms, thriving due to the more hospitable environment (41). In the 1980’s, however, H. pylori emerged as a potential cause of gastric cancer and is now recognized as a type I human carcinogen, with ~90% of gastric cancer cases worldwide considered attributable to H. pylori infection (42) and ~10% to Epstein-Barr virus infection. Additional microbial players are now being investigated subsequent to the advent of next-generation sequencing (see below).
Figure 3: Application of Koch’s postulates to tumorigenic bacteria.
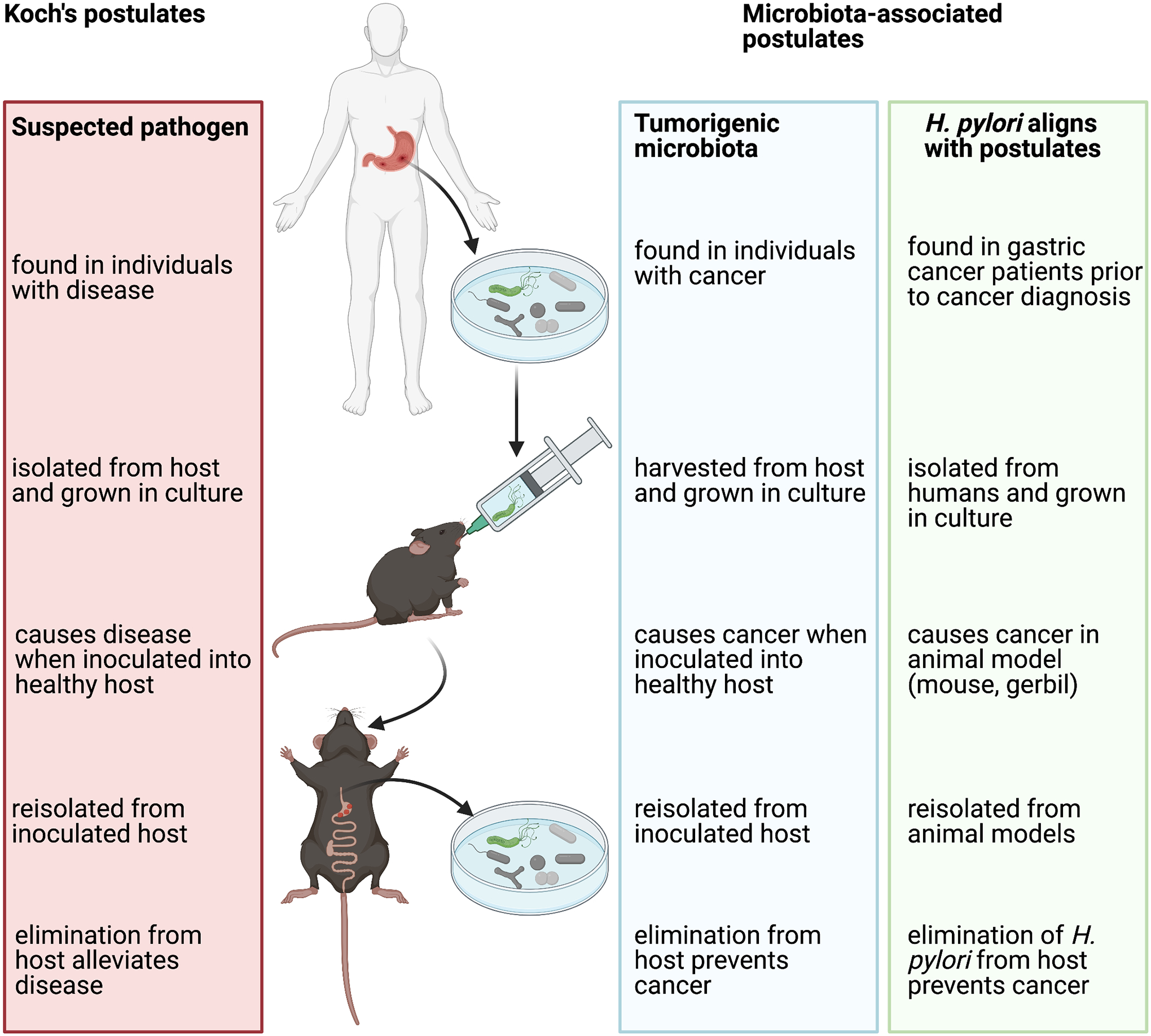
The identification of Helicobacter pylori in humans and then gastric cancer patients and the ability of this bacterium to be harvested, transferred, replicate disease in a healthy host and be reisolated from the inoculated host aligns directly with Koch’s postulates. A critical criterion for causality requires that elimination or control of the microbe or community then reduces disease onset, a criterion also met by H. pylori. The application of Koch’s postulates to H. pylori as a cancer promoter provides a roadmap for microbiota-associated postulates of tumorigenesis. However, in cancer to date, most microbiota association studies are cross-sectional, rather than prospective and longitudinal, and, thus, causality is unable to be defined as this requires demonstration that the microbe or community is present antecedent to tumor onset. Furthermore, the majority of animal models utilized to investigate causality are genetically modified and thus do not fulfill the requirement as a “healthy host.” An additional consideration for investigators in studies of the microbiota and cancer is the use of germ-free vs specific pathogen-free models which, in limited direct comparisons to date, can yield differing results (24,25,91,92). Collectively, available studies more aptly suggest that the cancer microbiome contributes to cancer progression, not initiation. Future studies should carefully analyze whether the results suggest cancer initiation and/or progression, two distinct frameworks in which the microbiome may well contribute to cancer pathogenesis. Adapted from Finlay et al (40).
H. pylori is a common gastric mucosa inhabitant, colonizing approximately 50% of individuals worldwide, and is typically acquired in childhood then persists for life unless treated. H. pylori colonization always induces chronic inflammation, and although most individuals are asymptomatic, this inflammation can cause numerous sequelae, including peptic ulcers (10% of infected individuals), gastric adenocarcinomas (1–3%), and mucosa-associated lymphoma (<0.1%) (43). In seminal studies in the early 1990’s, prospective case-control studies revealed an association between anti-H. pylori antibodies in banked serum samples with increased risk of gastric cancer several years later (44,45). A recent meta-analysis established that clearance of H. pylori with antibiotics reduced the risk of incident gastric cancer from 3% to 1.6% in healthy, asymptomatic H. pylori-positive individuals over a range of 4–22 years follow-up (46). In a separate study, antibiotic clearance of H. pylori similarly reduced the risk of metachronous gastric cancer nearly in half, from 13.4% to 7.2% in H. pylori-positive patients with prior endoscopic resection of early gastric cancer or high-grade adenoma (47). As gastric tumors emerge, the gastric environment and TME are less hospitable to ongoing H. pylori colonization. Consistent with these observations, a recent study did not observe an enrichment of H. pylori in gastric tumors compared to adjacent normal tissue, providing support for the ‘hit-and-run’ model of bacterial-initiated tumorigenesis, albeit after likely protracted, chronic colonization as occurs in H. pylori gastritis in humans (8). Thus, absence of a bacterium at the time of tumor diagnosis does not exclude that a bacterium contributed to tumor pathogenesis.
Mechanistically, inflammation is hypothesized to drive H. pylori-associated cancer, as H. pylori does not contain any directly genotoxic virulence factors. Rather, H. pylori induces the recruitment of neutrophils and macrophages, which, in turn, produce ROS and nitrogen species (RNS). As recently reviewed by Kidane, inflammation-mediated ROS/RNS can directly trigger single-strand DNA breaks (SSB) and/or induce the NF-κB pro-inflammatory pathway that can trigger double-strand DNA breaks (DSB) (48). H. pylori virulence factors (CagA, VacA) may influence the degree of stomach inflammation, and thus strain differences as well as polymorphisms in host inflammatory cascades influence risk of H. pylori-associated gastric cancer (43). For example, H. pylori peptidoglycans delivered to host cells via the cag pathogenicity island type IV secretion system are recognized by host Nod1, activating NF-κB signaling; mice deficient in Nod1 are more susceptible to H. pylori colonization compared to wild-type mice (43). Co-infection with Epstein-Barr Virus (EBV) may also exacerbate H. pylori-associated gastric cancer in ~10% of cases (41). Finally, the pro-inflammatory responses induced by H. pylori, including IL-1β and TNF-α, inhibit stomach parietal cell acid secretion; the resulting hypochlorhydria may promote secondary bacterial overgrowth that may modify chronic gastric inflammation, consistent with the earliest associations between benign Lactobacilli overgrowth and gastric cancer (43).
Although H. pylori is the strongest risk factor for gastric cancer, recent next-generation sequencing studies identified oral microbes - similar to those seen in CRC and HNSCC – with gastric cancer (49). Support for a role of oral microbes in gastric cancer also comes from a meta-analysis demonstrating a potential link between tooth loss (a marker for periodontal disease) and risk of gastric cancer (50), although this linkage remains controversial and needs validation. Other studies highlight additional, but inconsistent, microbes putatively contributory to gastric cancer; thus, a meta-analysis of the taxa and functional predictions from published 16S rRNA sequencing data may be of value to assess associations with gastric cancer.
In summary, gastric cancer and H. pylori currently represent the strongest link between a single bacterium and cancer causality (Figure 3, (40)), with prospective data demonstrating that H. pylori precedes tumorigenesis, while clearance of the organism with antibiotics reduces rates of gastric cancer. While other microbes may influence this tumorigenesis process, the dozens of other microbes associated with gastric cancer from 16S rRNA gene sequencing studies – including the enrichment of oral microbes – are inconsistent, lack clear mechanisms and prospective studies to demonstrate causality. Indeed, the lessons from the early observations of enrichment of Lactobacillus in gastric cancer represent an important cautionary tale regarding tumorigenesis associations vs. causality.
Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) cancer is lethal, with few patients surviving 5 years after diagnosis, in part, because diagnosis most often occurs at an advanced stage of disease. Thus, novel approaches to early diagnosis and treatment are being sought, including exploration of gut and tumor microbiota. The normal human pancreas tissue may harbor a microbiota, both bacterial and possibly fungal, as well as produce antimicrobial peptides (10% of the proteins in exocrine pancreatic fluid) suggesting the pancreas modulates both its intrinsic microbiota and that in the duodenal lumen and gut (51). The mouth, duodenum and gut microbiota all likely seed the pancreatic microbiota. Exactly how the human pancreatic microbiota modulates pancreatic function, immunologic homeostasis and/or susceptibility to pancreatic disease remains unknown.
As with CRC, oral microbiota have been associated with PDAC in 16S rRNA microarray and sequencing studies as well as by detection of plasma antibodies to oral bacteria (51–53). These results are consistent with the epidemiologic association of PDAC with periodontitis. Further, in intraductal papillary mucinous neoplasms (IPMNs), increased intracystic bacterial copy numbers enriched in oral bacteria taxa and inflammatory signals (IL-1β) associate with IPMNs with high-grade dysplasia or cancer compared to non-IPMN pancreatic cystic neoplasms suggesting that oral bacteria, likely through chronic inflammation, contribute to PDAC pathogenesis. Putative contributing oral microbiota members include bacteria with positive (e.g., Porphyromonas gingivalis), negative (e.g., Neisseria elongata, Streptococcus mitis) and variable (e.g., Fusobacterium nucleatum) associations with PDAC, as well as associations with more complex consortia of oral bacterial species (51). Studies of fecal microbiota in patients with PDAC display lower α–diversity (within sample diversity) and phyla shifts vs healthy individuals, a result consistent with a wide range of microbiota comparisons in disease vs health (51,54). Nonetheless, inter-individual variability of the fecal microbiome among PDAC patients, similar to healthy individuals, is high and preliminary observations found little concordance between PDAC-associated microbial signals and the microbiota in pre-cancerous lesions, suggesting limited utility for use in early detection of pancreatic neoplasia (54).
Informative translational studies of the microbiota in the PDAC tumor bed combined with pre-clinical studies in mouse models have emerged. Overall, in short-term survivors of PDAC (the vast majority of patients), the microbiome assessment of tumor resection samples supports dominance of the phyla Proteobacteria, Firmicutes and Bacteroidetes. Among the Proteobacteria, Enterobacteriaceae (family), Pseudomonas (genus) and Elizabethkingia (genus) appear notable and, in part, likely represent translocation from the gut into the pancreas where Proteobacteria may flourish (55–57). In one murine study, a pro-tumorigenic role for the species Bifidobacterium pseudolongum (phylum Actinobacteria) is highlighted (56), contrasting with work in non-pancreas tumors suggesting that B. pseudolongum or B. longum promote anti-tumorigenic mechanisms (58,59). Using preclinical mouse models, both germ-free mice and antibiotic-treated mice display limited PDAC growth, strongly supporting that the intratumoral and/or fecal microbiota can promote PDAC progression. Mechanistically, murine models support that the intratumoral microbiota promote PDAC progression through innate- and adaptive-immunosuppression mechanisms whereas microbial ablation with antibiotics fosters T-cell proliferation and immune activation including tumor responsiveness to checkpoint inhibitor therapy (56). The intratumoral bacteria (likely Proteobacteria) may foster PDAC therapeutic resistance by metabolizing gemcitabine, a common PDAC chemotherapeutic, to an inactive form (55). Consistent with these observations, retrospective clinical data suggest that co-incident antibiotic therapy may improve outcomes in PDAC patients treated with gemcitabine (55–57,60–62). In marked contrast, in a study focused on examining the infrequent long term PDAC survivors (LTS; survival >5 years), Riquelme et al (57) presented, using multiple approaches, compelling data that distinct intratumoral microbiome features, increased α-diversity and a consortium of the genera Saccharopolyspora (phylum Actinobacteria), Pseudoxanthomona (phylum Proteobacteria), Streptomyces (phylum Actinobacteria) and species Bacillus clausii (phylum Firmicutes) are highly predictive of LTS and, again, likely act through modulation of the tumor microenvironment. Further, PDAC LTS has been linked to identical circulating and intratumoral T-cell clones reactive to both high-quality tumor neoantigens and infectious disease-derived sequences consistent with neoantigen molecular mimicry, suggesting the hypothesis that an individual’s exposure to particular microbes over time may serve, in part, to prime an effective neoantigen-specific immune response as PDAC outgrowth emerges (63).
Collectively, the assessment of the PDAC microbiome at the time of PDAC therapy initiation may offer insight into prognosis and help design experimental studies to improve patient survival through modulation of the pancreatic and/or gut microbiome. Longitudinal and familystudies of microbiota characteristics and onset of PDAC might further enhance the understanding of microbiota contributions to PDAC derived, to date, only from cross-sectional studies.
Non-Gastrointestinal Cancers
Lung cancer
The lung, with the largest mucosal surface area in the body, displays a complex microbiota molded by both intrinsic (e.g., upper vs lower lobe) and extrinsic environmental factors (e.g., air-borne microbes, smoking, oral microbiome). Lung cancer is a highly heterogeneous tumor and largely cross-sectional studies provide evidence of lung cancer-associated microbiome dysbiosis but with highly variable results (64,65). One study employing both primary lung tissue samples and a validation cohort from The Cancer Genome Atlas (TCGA) suggested Proteobacteria overall increase in the lung cancer microbiome whereas increased Acidovorax (phylum Proteobacteria) abundance was specifically found in squamous cell carcinoma with TP53 mutations in smokers, suggesting microbiome-gene and microbiome-exposure interactions (66). Two recent studies provide pivotal data on the local lung microbial and immune mechanisms contributing to lung cancer whereas data supporting gut-lung axis mechanisms are lacking. Jin and colleagues (67) used the KP murine model of lung adenocarcinoma (LUAD), driven by an activating point mutation of Kras and loss of p53, to clearly demonstrate that an increased lung bacterial load, and even a limited bacterial consortium or bacterial molecules, accelerate LUAD by activating a myeloid cell-lung-resident γδ-T cell amplification loop that drives pro-carcinogenic inflammation and tumor cell proliferation through IL-17 and polymorphonuclear cell-mediated mechanisms. In a prospective study, Tsay and colleagues (68) tackled the human lung microbiota of Stages I-IIIA vs Stages IIIB-IV lung cancer providing evidence that, independent of disease stage, a lower airway microbiota enriched for oral commensals (e.g., Streptococcus, Prevotella, Veillonella, termed SPT, supraglottic predominant taxa, pneumotype) associated with a worse prognosis as well as upregulation of inflammatory cancer-related pathways (e.g., ERK/MAPK, PI3K/AKT). As proof-of-concept, using the KP mouse model, addition of V. parvula alone to the lung microbiota accelerated LUAD progression. It is hoped these results will provide translational approaches to improve the lethality of lung cancer.
Breast Cancer
In 2014, demonstration of a breast microbiome, potentially sourced from the skin, mouth and/or gut, emerged. Highly variable studies suggest that, while breast cancer patients display dysbiosis at the genus and species level, the phyla Proteobacteria, Firmicutes and Bacteroidetes are prominent in both healthy breast and breast cancer tissues; however, Lactobacilli (Phylum Firmicutes) abundance may be lower in breast cancer tissues. To date, limited and inconsistent taxa differences appear to distinguish tumor and adjacent normal tissue microbiota and benign vs malignant tumors although breast cancer subtypes may possess distinct microbial signatures (69). Bacterial regulation of estrogen bioavailability or induction of DNA damage in the breast are proposed to mediate carcinogenesis. In contrast, increased Fusobacterium nucleatum gDNA in breast cancer tissues has been associated with breast tissue Gal-GalNAc levels that increase with breast cancer progression; results consistent with the known binding of Fap2, a F. nucleatum adherence protein, to Gal-GalNAc. In a murine model, Fap2-sufficient, but not -deficient, F. nucleatum colonized orthotopic breast cancer and suppressed tumor-infiltrating T cells to promote tumor growth and metastasis (70). Whether the gut microbiome impacts breast cancer biology is unknown. However, Parida and colleagues (71) bioinformatically identified increased Bacteroides fragilis, a common colon anaerobe, in both benign and malignant breast tumor tissue. Using murine models, both breast intraductal or colon colonization with toxin-producing (ETBF, see CRC section), but not non-toxin-producing, B. fragilis promoted breast tumor growth and metastatic progression involving β-catenin and Notch1 pathways. Notably, breast tissue cells exposed to the B. fragilis toxin retained pro-carcinogenic memory, potentially increasing disease risk. The breast and/or gut microbiome in breast cancer patients may help stratify breast cancer risk and/or response to therapy.
Head and neck squamous cell carcinoma
The majority of head and neck cancers arise from mucosal membrane squamous epithelial cells [termed head and neck squamous cell carcinomas (HNSCC)]. HNSCC encompasses a broad number of malignancies, including cancers of the oral cavity, pharynx (including nasopharyngeal, oropharyngeal, and hypopharyngeal), larynx, paranasal sinuses and nasal cavity, and salivary glands. Multiple general and tumor type-specific risk factors for HNSCC exist including Human papillomavirus (HPV) type 16 (primarily in oropharyngeal cancers), Epstein-Barr virus (EBV) (nasopharyngeal and salivary gland cancers), preserved or salted foods (nasopharyngeal cancers), radiation (salivary gland cancers), poor oral hygiene (i.e., periodontitis in oral cavity cancers) along with tobacco and alcohol products (linked to >75% of all HNSCC cases), occupational exposures (e.g., asbestos, pesticides, and industrial solvents in multiple HNSCC types), and host genetics (www.cancer.gov). Bacteria may play a direct (e.g., periodontal disease) or indirect (e.g., alcohol metabolism) role in the tumorigenesis pathways of many of these risk factors. Approximately 15% of HNSCC lack known risk factors, spurring increased interest in the role for novel bacteria or groups of bacteria in the etiology of HNSCC.
The oral microbiome is a diverse community of over 750 species (Human Oral Microbiome Database, homd.org), many of them anaerobes, that form complex multi-species biofilms on both tooth surfaces and on oral cavity mucosal membranes. Within a healthy individual, the oral microbiome is largely stable. Left unchecked through poor dental hygiene, however, periodontal biofilms can trigger disease resulting in tooth decay, tooth loss, and potentially oral cavity cancers. Distinct oral microbial niches (>25) and sampling tools exist, from buccal swabs to oral rinses to tumor biopsies, which markedly complicates microbiome-based analyses.
Most studies analyzing the microbiome and HNSCC have focused on oral cavity squamous cell carcinomas (OSCC). Using both sequencing- and culture-based methods, OSCC biopsies appear to harbor dozens of both intratumoral and surface biofilm-associated bacterial species not found in healthy oral biopsy samples from the same patients [see review by Minarovits (72)], and include several species also found at higher abundances in CRC tissues compared to normal colon tissues: Fusobacterium nucleatum, Fusobacterium periodonticium, Gemella morbillorum, and Peptostreptococcus stomatis (72). Bacterial biofilms found on OSCCs also harbored a higher overall abundance of total anaerobic and aerobic bacteria by colony forming units (CFU), which parallels data from the colon where CRC-associated mucus-invasive biofilms are thicker and harbor more bacteria than paired normal tissues (38). However, inconsistencies between studies exist and each studied cohort was relatively small. In a larger study of oral rinse samples from 197 OSCC patients compared to 52 healthy individuals, the Fusobacteria phylum again strongly associated with OSCC, increasing from a relative abundance (RA) of 2.98% in healthy controls to 7.92% RA in stage 4 OSCC alongside decreases in Bacteroidetes and Actinobacteria phyla. At the species level, this trend was largely driven by Fusobacterium periodonticium (~1% RA in healthy tissues to 2% RA, stage 1; 3% RA, stage 2 and 3; and 5% RA, stage 4 OSCC) (73). Four other bacteria also increased alongside tumor stage in the oral rinse samples: Parvimonas micra (also increased in CRC-associated tissues), Streptococcus constellatus, Haemophilus influenzae, and Filifactor alocis (73). A meta-analysis of Fusobacterium in swab and/or tissue samples from 13 HNSCC studies found the Fusobacterium genus consistently enriched in tumor sites compared to non-tumor controls with F. nucleatum the most abundant Fusobacterium sp. detected, followed by F. naviforme, F. periodonticum, and others (74).
Only a single large prospective study exists to date for HNSCC, which did not validate the above organisms, although all HNSCC were included (not solely OSCC) (75). In this nested case-control study of >100,000 patients within the American Cancer Society Prevention Study II Nutrition Cohort (CPS-II) and the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO), 129 new cases of HNSCC were identified over an average of 3.9 years of follow-up. Prediagnostic, baseline oral mouthwash samples were examined by 454 pyrosequencing of the 16S rRNA V3-V4 region. HNSCC cases associated with more tobacco usage, alcohol consumption and HPV-16 carriage compared to controls, consistent with prior data. The oral microbiome β-diversity was not different between the total HNSCC cases or specific cancer types compared to controls (75). Surprisingly, the strongest trends observed were for protective bacteria against HNSCC. The genera Corynebacterium (phylum Actinobacteria) and Kingella (phylum Proteobacteria) were independently associated with reduced HNSCC risk. The protective effects of Corynebacterium and Kingella remained even after excluding current smokers or HPV-16-positive patients. Results remained similar by age, sex, and alcohol usage. Divided by cancer sub-type, the genera Corynebacterium, Kingella, Neisseria, Abiotrophia, Capnocytophaga, and species Kingella dentificans and Streptococcus sanguinins were associated with reduced risk of larynx cancer; Actinomyces oris and Veillonella denticariosi with reduced risk of pharynx cancer; and Parvimonas micra and Neisseria siccas with reduced risk of oral cavity cancer. The only bacteria positively associated with HNSCC were phylum Actinobacteria (HNSCC overall) and Actinomyces (oral cavity cancer). The microbial associations in this prospective study were strongest in patients with a history of tobacco usage and in those who developed larynx cancer, considered biologically plausible because larynx cancer is the HNSCC most strongly associated with tobacco usage, and Corynebacterium and Kingella may neutralize several toxicants found in cigarette smoke. Thus, amongst tobacco users, having Corynebacterium and/or Kingella spp. may be particularly protective against larynx cancer. However, the average time from sample collection to HNSCC detection in this cohort (3.9 years) may be insufficient to truly demonstrate potential microbial modulators of cancer. Replication studies are needed.
Despite the absence of a clear ‘smoking gun’ microbial trigger for HNSCC, at a mechanistic level, oral bacteria may facilitate tumorigenesis via both specific metabolic activities and broad inflammatory properties. For example, oral bacteria facilitate the metabolism of alcohol into its carcinogenic by-product, acetaldehyde, at levels that can induce DNA damage and enhance epithelial cell proliferation (76). The genus Neisseria is particularly adept at alcohol dehydrogenase activity, whereas Lactobacillus metabolizes acetaldehyde to relatively non-toxic forms. Interestingly, alcohol consumption associated with increased Neisseria RA and decreased Lactobacillus RA in oral wash samples from a large >1,000-person cohort (76). However, Neisseria have not been elevated in OSCC patient samples, either in comparison to paired normal control biopsies from the same patient or in oral wash samples from cancer-free individuals (72). This disconnect between the alcohol-associated microbiome and the OSCC-associated microbiome suggests that early microbial drivers or potential contributors to OSCC (i.e., mediators of alcohol-induced damage) may be lost by the time the tumor has overtly formed (hit-and-run model).
Alternatively, oral bacteria may promote tumorigenesis via inflammation induction. Using the functional prediction tool PICRUSt, Al-Hebshi et al proposed that OSCC tissues harbor an “inflammatory bacteriome” characterized by enrichment of genes for bacterial mobility, flagellar assembly, bacterial chemotaxis, and LPS synthesis, while control samples were enriched for more homeostatic genes such as amino acid biosynthesis, DNA repair, purine metabolism, ribosome biogenesis, and glycolysis/gluconeogenesis (77). Similarly, the link between periodontal disease and HNSCC revolves around pro-inflammatory mechanisms. Although links between periodontal disease and OSCC are inconsistent, in studies using clinical measurements of periodontal disease, a 4–10-fold higher risk of HNSCC associated with severe periodontitis (78). Periodontal disease has also variably been linked to total cancer risk, as well as specifically lung, PDAC, and CRC (78). Mechanistically, the link between periodontal disease and cancer has primarily focused on the pro-inflammatory bacteria F. nucleatum and Porphyromonas gingivalis. P. gingivalis is a keystone periodontal disease pathogen that may inhibit apoptosis and enhance proliferation pathways via increased JAK/STAT signaling, suppression of Bcl2, altering p53 pathway cyclins, and/or alteration of DC-SIGN (78).
In summary, HNSCC are associated with numerous oral microbiome changes that vary from pre-cancer to the cancer stage. Risk factors (e.g., alcohol consumption and periodontal disease) induce early microbial changes, although data are lacking on whether these bacteria, a mixture of oral microbes led by the Fusobacterium genus, directly impact human cancer initiation. However, these associations may be merely bystander associations. The only prospective cohort study yielded no species identified as HNSCC risk factors, rather Corynebacterium and Kingella appeared protective. Prospective cohorts in other study populations would be invaluable. A meta-analysis of all 16S rRNA or metagenomic studies in HNSCC examining all potential bacteria (not solely Fusobacterium) would be beneficial.
Genitourinary cancers
Cancers of the genitourinary (GU) tract (i.e., adrenal, bladder, kidney, penile, prostate, and testicular cancer) have limited – but growing - data suggesting a role of the microbiome in disease etiology. Urine, previously assumed to be a sterile fluid, is now known to contain a limited but variable microbiome that may impact GU cancers. The urinary microbiome is proposed as a source of pro-inflammatory bacteria that reflux to, for example, the prostate or kidney. The urinary microbiome varies by sex, potentially contributing to the higher rates of several GU cancers in men (bladder, renal, and prostate) compared to women; as men harbor more Actinobacteria including Corynebacterium and Propionibacterium in healthy urine samples, whereas women tend to harbor more Lactobacillales (79).
A microbial role in bladder cancer has been evident for decades, as infection with the parasitic flatworm Schistosoma haematobium is associated with very high rates of bladder cancer in endemic areas, including the Middle East and Africa, particularly before effective treatments for schistosomiasis were developed (80). Inflammation triggered by the embedding of parasitic eggs into the bladder wall may be the primary disease mechanism. The resident bacterial microbiome may still contribute, however, as even in Schistosoma-positive cases, multiple genera including Fusobacterium, Bacteroides, Veillonella, Aerococcus and Facklamia were enriched in patients with bladder cancer compared to those with only Schistosoma infection or no infection (81). Additionally, in the US population where Schistosoma is not endemic, a history of three or more urinary tract infections (UTIs) is an established risk factor for bladder cancer (82). The vast majority (>70%) of UTIs are E. coli, with preliminary data suggesting ~20% of UTI patients harbor DNA-damaging pks+ E. coli (see CRC section). While diverse genera are reported as enriched in bladder cancers, taxa overlap between studies is limited suggesting further study is needed (reviewed in (80).
Studies in men with prostate cancer similarly report differentially abundant microbes encompassing broad genera, with little overlap between studies with the exception of Bacteroides and Streptococcus [reviewed in Table 3 in Nicolar et al (80)]. However, these studies were performed on diverse samples (urine, rectal swabs, or stool samples). In one of the largest studies to date, urine obtained from men with and without prostate cancer did not reveal broad clustering of cancer vs. non-cancer patients, although, 6 largely uropathogenic bacteria (capable of inducing inflammation) were enriched in a subset of prostate cancer cases (Streptococcus anginosus, Anaerococcus lactolyticus, Anaerococcus obesiensis, Actinobaculum schaalii, Varibaculum cambriense, Propionimicrobium lymphophilum) (83). In mouse models, uropathogens (e.g., E. coli) lead to long-lasting inflammation even once the pathogen subsides to lower levels, suggesting a plausible mechanism for infection-associated cancers (79). Epithelium damage in bacterial-induced prostatitis may lead to impaired antimicrobial defenses, potentiating a feed-forward cycle of recurrent bacterial infections and epithelial damage resulting in chronic inflammation (84).
The microbiome of kidney (renal cell) cancer (RCC) is the least well-studied of the GU cancers. However, a history of UTIs of the bladder or kidney in a US population associated with increased risk for RCC, particularly in men who smoked. Complex interactions between bacteria and other epidemiological risk factors in RCC may exist (85).
Use of the microbiome in prevention and therapy of cancer
The growing associations between the microbiome and various cancers offer an opportunity to develop screening modalities that may target cancer prevention and treatment. Current cancer diagnosis usually requires invasive techniques (e.g., colonoscopy for CRC, biopsies of potential tumorigenic tissue) (11). Other less invasive tests including, blood tests, urine tests, imaging, fecal immuno-chemical tests, and/or multi-target stool DNA tests, provide alternative but less accurate diagnosis (4,11). Early cancer detection is key to patient outcomes. Exploiting defined microbial signatures specific to individual cancer types may enhance accurate early and less invasive methods of diagnosis. Currently, no microbial screening tools exist outside of H. pylori for gastric cancer, but the ever-increasing investigations provide promise for future development.
Microbial therapeutics
The concept of targeting microbes in cancer originates from the removal of H. pylori as a treatment for gastric cancer (Figure 3, (13)). Therefore, targeting the microbiome for other cancers may influence therapeutic outcomes as well as provide an unparalleled opportunity to develop microbial-targeted treatments. Microbiota manipulation is an intense research area (86). Dietary intervention, prebiotics, synbiotics, and probiotics that enrich or provide beneficial bacteria in the gut are currently under investigation to prevent or improve therapy (2). Naturally-occurring or engineered probiotic bacteria may outcompete detrimental species through colonization displacement and niche exclusion or by producing therapeutic molecules in situ (86). The complete replacement or restoration of a patient’s microbiome through fecal microbiota transfer (FMT) is another area of focused research. Nonetheless, human FMT has resulted in patient infections with enteric as well as multiple drug-resistant bacteria leading to patient deaths due to lack of quality control and/or use of well-defined microbial products (87). Removal of pro-carcinogenic members of the microbiota through narrow-spectrum antibiotics, monoclonal antibody therapy, or species-specific bacteriophages, provides putative direct therapeutic approaches to microbial modulation (13). To date, antibiotics have been primarily utilized as a tool to discern the contribution of the microbiota to tumorigenesis in various murine disease models (88). In human studies, only associations, not causal links, between antibiotic exposure and onset of cancers have been reported. Further, the potential impact of antibiotic exposure on cancer pathogenesis likely varies by route of administration, the timing of exposure and antibiotic class with even cancer-specific impacts (89). Vaccination against the microbial virulence factors involved in tumorigenesis, such as toxins or adhesion factors, or the specific bacteria themselves has the potential to elicit an immuno-modulatory benefit. Lastly, designing therapeutic molecules to scavenge or negate tumorigenic molecules produced by the microbiome (e.g., preventing DNA modifications) may emerge. Despite numerous theoretical approaches for microbiota therapy, the application of these ideas remains in their infancy. However, clinical trials are ongoing for numerous types of cancers and diseases suggesting live microbial therapeutics might become available as a treatment option (86).
Conclusions
Survivability of every type of cancer described in this review can be attributed to the magnitude of disease progression when first detected. While many factors propel cancer progression, the state of the microbiome is now entertained as a harbinger, cause and/or promoter of disease. Microbial signals in cancer are likely highly confounded by the heterogeneity of cancer combined with observations that single bacterial species-oncogene interactions modify cancer biology (20,66), making well-defined cancer microbiome ‘signatures’ difficult to classify or detect. Herein, we focused on how specific bacterial microbiome members as well as the microbiome community structure impact cancer progression. We delineated mechanisms, when described, by which the production of harmful microbial molecules, microbe-driven host immune responses and/or microbe-triggered changes in the host cell function and/or genome may contribute to cancer biology. Beyond viruses, H. pylori, and schistosomiasis, microbiota causality in cancer is not yet established, in part, due to the absence of longitudinal microbiome studies antecedent to human cancer. Further investigations to understand microbiome transitions in cancer emergence and oncogenic mechanisms may direct development of targeted screening modalities and microbial-based treatments with the vital objective of enhancing patient care and outcomes. The ever-increasing importance of the microbiome in a multitude of human cancers heralds boundless research opportunities to inform and direct new standards of cancer clinical practice.
Statement of Significance.
Emerging and, for some cancers, strong experimental and translational data support the contribution of the microbiome to cancer biology and disease progression. Disrupting microbiome features and pathways contributing to cancer may provide new approaches to improving cancer outcomes in patients.
Acknowledgements.
The authors regret the inability to cite additional authors and their primary research within the constraints of this review. The authors thank the many members of the Sears laboratory for the inspiration they provided over time in considering the role of the microbiome in cancer. Figures 1, 2, and 3 are unpublished original works created with a licensed copy of BioRender.com.
Funding statement.
Support for this manuscript was provided by Cancer Research UK CRUK (#C10674/A27140, CLS), Bloomberg Philanthropies (CLS), NCI R01CA196845 (CLS), NCI R00CA230192 (JLD) and support from the Johns Hopkins School of Medicine.
Footnotes
Conflict of Interest Statement
CLS reports research funding from Janssen and Bristol Myers Squibb.
References
- 1.Golofast B, Vales K. The connection between microbiome and schizophrenia. Neurosci Biobehav Rev 2020;108:712–31 doi 10.1016/j.neubiorev.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Tilg H, Adolph TE, Gerner RR, Moschen AR. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018;33(6):954–64 doi 10.1016/j.ccell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Domingue JC, Drewes JL, Merlo CA, Housseau F, Sears CL. Host responses to mucosal biofilms in the lung and gut. Mucosal Immunol 2020;13(3):413–22 doi 10.1038/s41385-020-0270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol 2020;28(5):401–23 doi 10.1016/j.tim.2020.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y. Host variables confound gut microbiota studies of human disease. Nature 2020;587(7834):448–54 doi 10.1038/s41586-020-2881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd AL, Liu M, Fujimura KE, Lyalina S, Nagarkar DR, Charbit B, et al. Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218(1) doi 10.1084/jem.20200606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dohlman AB, Arguijo Mendoza D, Ding S, Gao M, Dressman H, Iliev ID, et al. The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants. Cell Host Microbe 2021;29(2):281–98.e5 doi 10.1016/j.chom.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020;579(7800):567–74 doi 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368(6494):973–80 doi 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71(3):209–49 doi 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 11.Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 2019;16(12):713–32 doi 10.1038/s41575-019-0189-8. [DOI] [PubMed] [Google Scholar]
- 12.Allen J, Sears CL. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Med 2019;11(1):11 doi 10.1186/s13073-019-0621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol 2019;16(11):690–704 doi 10.1038/s41575-019-0209-8. [DOI] [PubMed] [Google Scholar]
- 14.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks. Nature 2020;580(7802):269–73 doi 10.1038/s41586-020-2080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017;3:34 doi 10.1038/s41522-017-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med 2019;25(4):679–89 doi 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valguarnera E, Wardenburg JB. Good Gone Bad: One Toxin Away From Disease for Bacteroides fragilis. J Mol Biol 2020;432(4):765–85 doi 10.1016/j.jmb.2019.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Allen J, Hao S, Sears CL, Timp W. Epigenetic Changes Induced by Bacteroides fragilis Toxin. Infect Immun 2019;87(6) doi 10.1128/IAI.00447-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiuri AR, Savant SS, Podicheti R, Rusch DB, O’Hagan HM. DNA methyltransferase inhibition reduces inflammation-induced colon tumorigenesis. Epigenetics 2019;14(12):1209–23 doi 10.1080/15592294.2019.1634986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeStefano Shields CE, White JR, Chung L, Wenzel A, Hicks JL, Tam AJ, et al. Bacterial-driven inflammation and mutant BRAF expression combine to promote murine colon tumorigenesis that is sensitive to immune checkpoint therapy. Cancer Discov 2021. doi 10.1158/2159-8290.CD-20-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maiuri AR, Peng M, Podicheti R, Sriramkumar S, Kamplain CM, Rusch DB, et al. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer Res 2017;77(13):3467–78 doi 10.1158/0008-5472.CAN-17-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carrá A, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019;363(6428) doi 10.1126/science.aar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol 2019;17(3):156–66 doi 10.1038/s41579-018-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013;14(2):207–15 doi 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomkovich S, Dejea CM, Winglee K, Drewes JL, Chung L, Housseau F, et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J Clin Invest 2019;129(4):1699–712 doi 10.1172/JCI124196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013;14(2):195–206 doi 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015;42(2):344–55 doi 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017;358(6369):1443–8 doi 10.1126/science.aal5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertocchi A, Carloni S, Ravenda PS, Bertalot G, Spadoni I, Lo Cascio A, et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell 2021;39:5:708–24 doi 10.1016/j.ccell.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Weng Y, Gan H, Zhao X, Zhi F. Streptococcus gallolyticus conspires myeloid cells to promote tumorigenesis of inflammatory bowel disease. Biochem Biophys Res Commun 2018;506(4):907–11 doi 10.1016/j.bbrc.2018.10.136. [DOI] [PubMed] [Google Scholar]
- 31.Purcell RV, Visnovska M, Biggs PJ, Schmeier S, Frizelle FA. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep 2017;7(1):11590 doi 10.1038/s41598-017-11237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology 2017;152(6):1419–33.e5 doi 10.1053/j.gastro.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Komiya Y, Shimomura Y, Higurashi T, Sugi Y, Arimoto J, Umezawa S, et al. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut 2019;68(7):1335–7 doi 10.1136/gutjnl-2018-316661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flemer B, Warren RD, Barrett MP, Cisek K, Das A, Jeffery IB, et al. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018;67(8):1454–63 doi 10.1136/gutjnl-2017-314814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG, Hayashi A, Imai J, et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020;182(2):447–62.e14 doi 10.1016/j.cell.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, Emgård JEM, et al. Colon Cancer-Associated Fusobacterium nucleatum May Originate From the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front Cell Infect Microbiol 2020;10:400 doi 10.3389/fcimb.2020.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014;158(2):288–99 doi 10.1016/j.cell.2014.04.051. [DOI] [PubMed] [Google Scholar]
- 38.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 2014;111(51):18321–6 doi 10.1073/pnas.1406199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018;359(6375):592–7 doi 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finlay BB, Humans C, Microbiome. Are noncommunicable diseases communicable? Science 2020;367(6475):250–1 doi 10.1126/science.aaz3834. [DOI] [PubMed] [Google Scholar]
- 41.Engstrand L, Graham DY. Microbiome and Gastric Cancer. Dig Dis Sci 2020;65(3):865–73 doi 10.1007/s10620-020-06101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 2012;13(6):607–15 doi 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 43.Peek RM Jr., Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol 2006;208(2):233–48 doi 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 44.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991;325(16):1127–31 doi 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 45.Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991;325(16):1132–6 doi 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- 46.Ford AC, Yuan Y, Forman D, Hunt R, Moayyedi P. Helicobacter pylori eradication for the prevention of gastric neoplasia. Cochrane Database Syst Rev 2020;7:CD005583 doi 10.1002/14651858.CD005583.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY, Kim CG, et al. Helicobacter pylori Therapy for the Prevention of Metachronous Gastric Cancer. N Engl J Med 2018;378(12):1085–95 doi 10.1056/NEJMoa1708423. [DOI] [PubMed] [Google Scholar]
- 48.Kidane D Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. Int J Mol Sci 2018;19(10) doi 10.3390/ijms19102891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018;67(6):1024–32 doi 10.1136/gutjnl-2017-314281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin XH, Wang YD, Luo H, Zhao K, Huang GL, Luo SY, et al. Association between Tooth Loss and Gastric Cancer: A Meta-Analysis of Observational Studies. PLoS One 2016;11(3):e0149653 doi 10.1371/journal.pone.0149653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas RM, Jobin C. Microbiota in pancreatic health and disease: the next frontier in microbiome research. Nat Rev Gastroenterol Hepatol 2020;17(1):53–64 doi 10.1038/s41575-019-0242-7. [DOI] [PubMed] [Google Scholar]
- 52.Farrell JJ, Zhang L, Zhou H, Chia D, Elashoff D, Akin D, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012;61(4):582–8 doi 10.1136/gutjnl-2011-300784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjønneland A, et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013;62(12):1764–70 doi 10.1136/gutjnl-2012-303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Half E, Keren N, Reshef L, Dorfman T, Lachter I, Kluger Y, et al. Fecal microbiome signatures of pancreatic cancer patients. Sci Rep 2019;9(1):16801 doi 10.1038/s41598-019-53041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357(6356):1156–60 doi 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov 2018;8(4):403–16 doi 10.1158/2159-8290.CD-17-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019;178(4):795–806.e12 doi 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350(6264):1084–9 doi 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369(6510):1481–9 doi 10.1126/science.abc3421. [DOI] [PubMed] [Google Scholar]
- 60.Imai H, Saijo K, Komine K, Otsuki Y, Ohuchi K, Sato Y, et al. Antibiotic therapy augments the efficacy of gemcitabine-containing regimens for advanced cancer: a retrospective study. Cancer Manag Res 2019;11:7953–65 doi 10.2147/CMAR.S215697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakano S, Komatsu Y, Kawamoto Y, Saito R, Ito K, Nakatsumi H, et al. Association between the use of antibiotics and efficacy of gemcitabine plus nab-paclitaxel in advanced pancreatic cancer. Medicine (Baltimore) 2020;99(39):e22250 doi 10.1097/MD.0000000000022250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weniger M, Hank T, Qadan M, Ciprani D, Michelakos T, Niess H, et al. Influence of Klebsiella pneumoniae and quinolone treatment on prognosis in patients with pancreatic cancer. Br J Surg 2020. doi 10.1002/bjs.12003. [DOI] [PubMed] [Google Scholar]
- 63.Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551(7681):512–6 doi 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goto T Airway Microbiota as a Modulator of Lung Cancer. Int J Mol Sci 2020;21(9) doi 10.3390/ijms21093044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martins D, Mendes F, Schmitt F. Microbiome: A Supportive or a Leading Actor in Lung Cancer? Pathobiology 2020:1–10 doi 10.1159/000511556. [DOI] [PubMed] [Google Scholar]
- 66.Greathouse KL, White JR, Vargas AJ, Bliskovsky VV, Beck JA, von Muhlinen N, et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol 2018;19(1):123 doi 10.1186/s13059-018-1501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019;176(5):998–1013.e16 doi 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsay JJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov 2021;11(2):293–307 doi 10.1158/2159-8290.CD-20-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parida S, Sharma D. The power of small changes: Comprehensive analyses of microbial dysbiosis in breast cancer. Biochim Biophys Acta Rev Cancer 2019;1871(2):392–405 doi 10.1016/j.bbcan.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun 2020;11(1):3259 doi 10.1038/s41467-020-16967-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parida S, Wu S, Siddharth S, Wang G, Muniraj N, Nagalingam A, et al. A pro-carcinogenic colon microbe promotes breast tumorigenesis and metastatic progression and concomitantly activates Notch and βcatenin axes. Cancer Discov 2021;11(5):1138–57 doi 10.1158/2159-8290.CD-20-0537. [DOI] [PubMed] [Google Scholar]
- 72.Minarovits J Anaerobic bacterial communities associated with oral carcinoma: Intratumoral, surface-biofilm and salivary microbiota. Anaerobe 2020:102300 doi 10.1016/j.anaerobe.2020.102300. [DOI] [PubMed] [Google Scholar]
- 73.Yang CY, Yeh YM, Yu HY, Chin CY, Hsu CW, Liu H, et al. Oral Microbiota Community Dynamics Associated With Oral Squamous Cell Carcinoma Staging. Front Microbiol 2018;9:862 doi 10.3389/fmicb.2018.00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bronzato JD, Bomfim RA, Edwards DH, Crouch D, Hector MP, Gomes B. Detection of Fusobacterium in oral and head and neck cancer samples: A systematic review and meta-analysis. Arch Oral Biol 2020;112:104669 doi 10.1016/j.archoralbio.2020.104669. [DOI] [PubMed] [Google Scholar]
- 75.Hayes RB, Ahn J, Fan X, Peters BA, Ma Y, Yang L, et al. Association of Oral Microbiome With Risk for Incident Head and Neck Squamous Cell Cancer. JAMA Oncol 2018;4(3):358–65 doi 10.1001/jamaoncol.2017.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan X, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Freedman ND, et al. Drinking alcohol is associated with variation in the human oral microbiome in a large study of American adults. Microbiome 2018;6(1):59 doi 10.1186/s40168-018-0448-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Hebshi NN, Nasher AT, Maryoud MY, Homeida HE, Chen T, Idris AM, et al. Inflammatory bacteriome featuring Fusobacterium nucleatum and Pseudomonas aeruginosa identified in association with oral squamous cell carcinoma. Sci Rep 2017;7(1):1834 doi 10.1038/s41598-017-02079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung M, York BR, Michaud DS. Oral Health and Cancer. Curr Oral Health Rep 2019;6(2):130–7 doi 10.1007/s40496-019-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sfanos KS, Yegnasubramanian S, Nelson WG, De Marzo AM. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol 2018;15(1):11–24 doi 10.1038/nrurol.2017.167. [DOI] [PubMed] [Google Scholar]
- 80.Nicolaro M, Portal DE, Shinder B, Patel HV, Singer EA. The human microbiome and genitourinary malignancies. Ann Transl Med 2020;8(19):1245 doi 10.21037/atm-20-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adebayo AS, Suryavanshi MV, Bhute S, Agunloye AM, Isokpehi RD, Anumudu CI, et al. The microbiome in urogenital schistosomiasis and induced bladder pathologies. PLoS Negl Trop Dis 2017;11(8):e0005826 doi 10.1371/journal.pntd.0005826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kantor AF, Hartge P, Hoover RN, Narayana AS, Sullivan JW, Fraumeni JF. Urinary tract infection and risk of bladder cancer. Am J Epidemiol 1984;119(4):510–5 doi 10.1093/oxfordjournals.aje.a113768. [DOI] [PubMed] [Google Scholar]
- 83.Shrestha E, White JR, Yu SH, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the Urinary Microbiome in Men with Positive versus Negative Biopsies for Prostate Cancer. J Urol 2018;199(1):161–71 doi 10.1016/j.juro.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Bono JS, Guo C, Gurel B, De Marzo AM, Sfanos KS, Mani RS, et al. Prostate carcinogenesis: inflammatory storms. Nat Rev Cancer 2020;20(8):455–69 doi 10.1038/s41568-020-0267-9. [DOI] [PubMed] [Google Scholar]
- 85.Parker AS, Cerhan JR, Lynch CF, Leibovich BC, Cantor KP. History of urinary tract infection and risk of renal cell carcinoma. Am J Epidemiol 2004;159(1):42–8 doi 10.1093/aje/kwh014. [DOI] [PubMed] [Google Scholar]
- 86.Jimenez M, Langer R, Traverso G. Microbial therapeutics: New opportunities for drug delivery. J Exp Med 2019;216(5):1005–9 doi 10.1084/jem.20190609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, et al. Drug-Resistant. N Engl J Med 2019;381(21):2043–50 doi 10.1056/NEJMoa1910437. [DOI] [PubMed] [Google Scholar]
- 88.DeStefano Shields CE, Van Meerbeke SW, Housseau F, Wang H, Huso DL, Casero RA, et al. Reduction of Murine Colon Tumorigenesis Driven by Enterotoxigenic Bacteroides fragilis Using Cefoxitin Treatment. J Infect Dis 2016;214(1):122–9 doi 10.1093/infdis/jiw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang J, Haines C, Watson AJM, Hart AR, Platt MJ, Pardoll DM, et al. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989–2012: a matched case-control study. Gut 2019;68(11):1971–8 doi 10.1136/gutjnl-2019-318593. [DOI] [PubMed] [Google Scholar]
- 90.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173(2):291–304.e6 doi 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009;15(9):1016–22 doi 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun 2009;77(4):1708–18 doi 10.1128/IAI.00814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]