

Abstract
Purpose
In this study, we aimed to characterize the clinical phenotype of a SHANK1-related disorder and define the functional consequences of SHANK1 truncating variants.
Methods
Exome sequencing (ES) was performed for six individuals who presented with neurodevelopmental disorders. Individuals were ascertained with the use of GeneMatcher and Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER). We evaluated potential nonsense mediated decay (NMD) of two variants by making knock-in cell lines of endogenous truncated SHANK1, and expressed the truncated SHANK1 cDNA in HEK293 cells and cultured hippocampal neurons to examine the proteins.
Results
ES detected de novo truncating variants in SHANK1 in six individuals. Evaluation of NMD resulted in stable transcripts, and the truncated SHANK1 completely lost binding with Homer1, a linker protein that binds to the C-terminus of SHANK1. These variants may disrupt protein-protein networks in dendritic spines. Dispersed localization of the truncated SHANK1 variants within the spine and dendritic shaft was also observed when expressed in neurons, indicating impaired synaptic localization of truncated SHANK1.
Conclusion
This report expands the clinical spectrum of individuals with truncating SHANK1 variants and describes the impact these variants may have on the pathophysiology of neurodevelopmental disorders.
INTRODUCTION
The SHANK1 gene (MIM: 604999) is a part of the SHANK gene family, which consists of SHANK1, SHANK2 (MIM: 603290) and SHANK3 (MIM: 606230). SHANK genes encode scaffold proteins located at the postsynaptic density (PSD) of glutamatergic synapses that interact with a variety of membrane and cytoplasmic proteins1,2. SHANK proteins have ankyrin repeats, a SH3 (Src homology) domain, a PDZ (postsynaptic density 95/discs large/zona occludens-1 homology) domain, a proline-rich region and a SAM (sterile alpha motif) domain, which are involved in a variety of protein-protein interactions for organizing receptors, scaffolding proteins and cytosolic components in dendritic spines1,3,4. Mouse models assessing SHANK1 protein function have found that proper regulation of SHANK1 is crucial for appropriate synaptic structure and cognition2,5. Shank1 −/− knockout mice with a deletion of exons 14 and 15 had significantly smaller spine size, reduced PSD thickness and a distinct loss of the largest PSDs compared with wild type (WT) mice5.
SHANK1 expression studies further support the hypothesis that SHANK1 is critical for the development and maintenance of the largest subset of PSDs and synapses. SHANK1 is co-expressed with hippocampal dendritic markers, astrocytes, and microglia in the developing brain, and expression is increased during periods of learning-induced dendritic spine plasticity in the neocortex6,7. SHANK-associated CpG islands are differentially methylated in chronic lymphocytic leukemia and colon cancers, and SHANK is abnormally expressed in cancer tissues8–10. Analysis of CpG island methylation has found epigenetic dysregulation in the SHANK1 gene in individuals with autism spectrum disorder (ASD) when compared to controls11.
The mouse model generated by Hung et al. has been used in several studies to assess the behavioral effects of Shank1 dysregulation5,12–15. In an investigation of the behavioral phenotype of Shank1 −/− mutant mice by a variety of assays, Shank1 −/− mice were found to be less active and displayed increased anxiety-like behavior when compared to WT mice5. Behavioral assays to evaluate ASD-relevant behavioral phenotypes (including social behavior, social cognition memory, communication, repetitive and stereotyped patterns of behavior, and cognitive functioning) have not provided strong evidence for ASD-related deficits in social behavior, but were suggestive of ASD-related deficits in social cognition5,12–16. Studies on communication via ultrasonic vocalizations (USV) in Shank1 −/− mouse models concluded that there were ASD-related communication deficits15,16. Combined, these studies reveal that Shank1 −/− mutant mice displayed high levels of anxiety and ASD-related deficits in social cognition.
There are several reports of microdeletions, microduplications, and truncating variants in coding regions of SHANK1, SHANK2, and SHANK3 in individuals with ASD17–23. Although SHANK3 haploinsufficiency has been implicated as a cause of 22q13 deletion syndrome, also known as Phelan–McDermid syndrome, the effects of SHANK1 and SHANK2 loss-of-function (LOF) variants in humans have not been extensively characterized21,24. Sato et al. first detailed case reports of germline SHANK1 variants17. The group described seven individuals with partial deletions of SHANK1 who all had ASD and/or generalized anxiety, with reduced penetrance in females. Six were from a multi-generational family that carried an inherited 63.8 kb gene deletion in chromosomal region 19q13.33, spanning exons 1–20 of SHANK1 and the entire neighboring CLEC11A (MIM: 604713) gene (hg18), which codes for a growth factor for primitive hematopoietic progenitor cells. Three of the male patients in this family were diagnosed with ASD, a fourth male was diagnosed with a broader autism phenotype, and the two females had severe anxiety. The other, unrelated individual, carried a de novo 63.4 kb deletion spanning the last three exons of SHANK1 and the entire SYT3 (MIM: 600327) gene (hg18), which plays a role in Ca2þ -dependent exocytosis of secretory vesicles17. Shortly thereafter, a novel germline de novo LOF variant was reported in an individual with ASD, intellectual disability (ID), abnormal gait, and regression18. Another novel germline de novo LOF variant was reported in an individual with schizophrenia25. No definitive genotype–phenotype correlation has been established based on the nine individuals with novel LOF variants in SHANK1 previously described in the literature.
We describe six additional individuals with de novo truncating variants in SHANK1, bringing the total number of reported individuals with LOF variants to fifteen. We further explore the clinical phenotype of a SHANK1-related disorder and use molecular studies to help define the functional consequences, phenotype, and pathology of SHANK1 truncating variants.
MATERIALS AND METHODS
Exome sequencing.
Exome sequencing (ES) was performed for six families to identify candidate variants using standard methods. Trio ES was performed for families of individuals 1, 2, 3, 4, and 5, and proband-only ES was performed for individual 6, with subsequent segregation analysis in parents. Trio ES for individual 1 was performed at Columbia University Irving Medical Center after written informed consent was obtained through an institutional review board-approved research study at the Institute for Genomic Medicine at Columbia University (protocol AAAO8410). Variants were analyzed using the methods described in Zhu et al.26. Individuals 2, 3, 4, and 6 were ascertained via contact made by clinicians with the use of GeneMatcher27, a web-based tool for researchers and clinicians working on identical genes. Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER) was also utilized to contact physicians of individual 528. Informed consent was obtained from each family prior to testing. Detailed description of materials and methods used by each laboratory is available in the Supporting Information.
CRISPR KI cell line generation.
CRISPR design was aided by open web resources provided by Synthego and Dharmacon companies. Primers for gRNA were annealed and cloned into the pSpCas9-P2A-puro plasmid (PX458) (Addgene, 48139). Transfected cells were selected with 1.5 μg/ml puromycin and serially diluted for single cell colonies. The CRISPR target regions were also sequenced to confirm genome editions.
Primers and ssODN sequences for CRISPR.
gRNA sequences for SHANK1 c.3314del (p.Gly1105fs): 5’-CACCGCCTTGCGGCCGCGGCCCGAG and 5’- AAACCTCGGGCCGCGGCCGCAAGGC. ssODN for SHANK1 c.3314del (p.Gly1105fs): 5’- CCATGTACG TGCCCGCCCGCTCGGGCCGCGCCGCAAGGGCCCGCTGGTCAAGCAGACCAA (60 nt). gRNA sequences for SHANK1 c.3355G>T (p.Glu1119*): 5’- CACCGGTGGAAGGCGAGCCCCAGA and 5’- AAACTCTGG GGCTCGCCTTCCACC. ssODN for SHANK1 c.3355G>T (p.Glu1119*): 5’- CCGCTGGTCAAGCAGACCAAGGTG GAAGGCTAGCCCCAGAAGGGCGGCGGCCTCCCGCCCG (61 nt). Primers for genotyping: 5’- CGC GAGAAGAGCCTGTACCA and 5’- CGGCGCGCCTCATTCTG.
Real-time PCR for relative gene expression.
RNA was prepared from the cell line using a mini spin column (Qiagen, 74104). cDNA was synthesized from 1 μg RNA with random hexamer primers per manufacturer’s instruction (Thermo, 4368814). 50 ng cDNA was used for a single reaction mixture of SYBR Green PCR mix (Thermo, 4368577). CT values are acquired using StepOne™ software (Applied Biosystems) and ΔΔCT values are calculated by manual. Primers for quantitative PCR: 5’-GGCAGAGACTCTCCGACGAC and 5’-CTGTAGGGGAGACCCCTGTT for SHANK1. 5’-AATGGGCAGCCGTTAGGAAA and 5’-GCCCAATACGACCAAATCAGAG for GAPDH.
Plasmids and antibodies.
Human SHANK1 was cloned into the pcDNA3.1(+)-N-HA plasmid and mutagenesis for p.Gly1105fs and p.Glu1119* was performed using GenScript (Piscataway, NJ). Homer1b, and pClneo-GKAP-myc plasmids were used for biochemical and imaging experiments. The commercial antibodies used in this study were mouse rabbit anti-Homer1/2/3 (Synaptic Systems, 160103), mouse anti-PSD-95 (Neuromab, clone K28/43), guinea pig anti-VGLUT1 (Millipore, AB5905), mouse anti-myc (Cell Signaling, clone 9B11), rabbit anti-HA (Abcam, ab9110), rat anti-HA (Roche), rabbit anti-β tubulin (Sigma, T2200), and mouse anti-β actin (ABM, G043).
Transfection and immunoblot.
HEK293 cells were transfected with Lipofectamine2000 and 2 days after transfection the cells were lysed in a TBS buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% Triton X-100. Lysates were centrifuged at 16,000g for 15 min at 4°C and supernatants were used for the analysis. For coimmunoprecipitation, lysates were incubated with an appropriate antibody at 4°C overnight and protein-A-Sepharose beads (GE Healthcare) at 4°C for 1 hour. All chemiluminescence blots were captured with a ChemiDoc™ Imaging System (Bio-Rad).
Neuronal cultures.
Briefly, embryonic hippocampal or cortical tissues were dissociated at 37°C for 30 min by 0.05% trypsin in 10 mM HBSS containing 1.37 mg/ml DNase I. Neurons were plated and maintained in serum-free Neurobasal Medium supplemented with 2% (vol/vol) B-27 and 2 mM L-Glutamine. We adhered to the guidelines of the National Institutes of Health’s Animal Care and Use Committee regarding the care and use of animals for this study (protocol #1171).
Immunocytochemistry.
Cultured hippocampal neurons were grown on glass coverslips precoated with poly-D-lysine (Sigma). To label HA-SHANK1, the transfected neurons were fixed with 4% paraformaldehyde and 4% sucrose in PBS for 10 min. After fixation, the neurons were permeabilized with 0.25% Triton X-100 in PBS and blocked with 10% goat serum. The neurons were labeled with rat anti-HA antibody and Alexa 555-conjugated anti-rat secondary antibody (Molecular Probes). For endogenous PSD-95 staining, neurons were prepared as above, then labeled with anti−PSD-95 (NeuroMab, clone K28/43).
Statistical analysis.
All images were captured with a 63× objective on a Zeiss LSM 510 confocal microscope and analyzed with the Image J and MetaMorph Version 7. Data analysis was carried out in Image LabTM Software (Bio-Rad) or GraphPad Prism (GraphPad Software).
RESULTS
In this study, we present six individuals with novel de novo truncating variants in SHANK1. The individuals present with a spectrum of neurodevelopmental disorders including ASD, neuropsychiatric disturbances, cognitive delays/ID and/or learning disabilities, motor delays, speech and language delays and brain abnormalities. The de novo variants identified include (GenBank: NM_016148.5) c.3488dup (p.Ser1164fs) in individual 1, c.3355G>T (p.Glu1119*) in individual 2, c.3314del (p.Gly1105fs) in individual 3, c.1198C>T (p.Arg400*) in individual 4, c.4496_4499del (p.Gln1499fs) in individual 5, and c.650del (p.Leu217*) in individual 6. All SHANK1 variants were validated by Sanger sequencing, and the de novo origins were confirmed by trio ES or parental-segregation studies (Fig S1). None of the variants are present in variant databases such as the Genome Aggregation Database (gnomAD). Detailed variant information is included in Table 1 and in the Supporting Information. The distribution of variants observed in our cohort is shown in Figure 1.
Table 1.
Review of systems in our cohort of six patients
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | |
|---|---|---|---|---|---|---|
| Variant (Hg19; NM_016148.5) | 19-51171728-C-CG | 19-51171862-C-A | 19-51171902-GC-G | 19-51207401-G-A | 19-51170717-GGGCT-G | 19-51217196-CA-C |
| c.3488dup | c.3355G>T | c.3314del | c.1198C>T | c.4496_4499del | c.650del | |
| p.Ser1164ValfsTer4 | p.Glu1119* | p.Gly1105AlafsTer238 | p.Arg400* | p.Gln1499fs | p.Leu217* | |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo |
| ACMG/AMP Classificationa | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic |
| Gender | Male | Female | Male | Male | Male | Female |
| Age at last examination | 11y | 6y | 7y | 9y | 10y9m | 11y3m |
| Height at last examination | 143cm (−0.07 SD) | 109cm (+1.60 SD) | 115cm (+1.34 SD) | 134.5cm (+0.16 SD) | 144.5cm (+0.88 SD) | 136cm (−19.98 SD) |
| Weight at last examination | 36kg (+0.01 SD) | 18.1kg (+0.76 SD) | 21.5kg (+0.99 SD) | 28.7kg (+0.02 SD) | unknown | 24kg (−2.36 SD) |
| Head circumference at last examination | unknown | 50cm (−0.32 SD) | 50.5cm (−0.36 SD) | 55.5cm (+2.17 SD) | 58.5cm (+3.97 SD) | 49.8cm (−2.29 SD) |
| Ethnicity | Italian | Egyptian | European, Mexican, Pakistani | European | European | Brazilian |
| Motor development/ delays | Walked at 16mo, overall mild gross motor delay | Walked at 18–19mo | Walked at 16mo | Walked at 2yo, mild motor delay | Walked at 20mo, poor balance | Never walked |
| Speech & language development/ delays | First words at 2yo, still concerns with speech at 3yo; significant expressive language deficits and articulation disorder, low facial tone, compromised oral motor skills | First words at 2yo; speech delay, mixed receptive expressive language disorder | First words at 12mo; mixed receptive expressive language disorder | First words at 2yo; low facial tone; gross language delay | Babbled at about 10mo, then no further words until about 5yo; at 10yo only says occasional single words, understands simple commands | At 11y3mo nonverbal |
| Cognitive delays/ID/ learning disabilities | Auditory processing disorder, difficulty with executive functioning, mathematic concepts, verbal reasoning and problem solving | Intellectual disability | Intellectual disability | Severe learning difficulties | Intellectual disability | Severe intellectual disability |
| ASD | No | Yes | No | Yes | Yes | No |
| Other neuropsychiatric and behavioral issues | Anxiety, ADHD | Hyperactivity and hair pulling when upset | No | Anxiety | Anxiety, tic (head jerk), anger outbursts, self-biting | Puts hands inside the mouth, but won’t bite hard |
| Seizures (seizure type, age of seizure onset) | No | No, abnormal EEG consistent with a seizure disorder of multifocal origin; has not had a clinical seizure | Yes, one generalized tonic-clonic seizure, one seizure consisting of general convulsions lasting 3–4 minutes, both onset at 7yo; abnormal EEG | No | No | Yes, myoclonic seizures onset at 11mo; evolved into focal to bilateral tonic-clonic seizures at 14mo; abnormal EEG |
| Hypotonia | Yes (as a newborn) | Yes (mild) | Yes (mild) | No | Yes (mild) | No |
| Headaches/migraines | Yes | No | No | No | No | No |
| Macrocephaly (≥ 2SD) | No | No | No | Yes (+2.17 SD) | Yes (+3.97 SD) | No |
| Microcephaly (≤ 2SD) | No | No | No | No | No | Yes (−2.29 SD) |
| Brain imaging abnormalities | N/A | Normal MRI | Normal MRI | N/A | N/A | Abnormal MRI: absent cerebellar vermis, possible dysgenesis with partial fusion of the cerebral hemispheres and thalami |
| Dysmorphic features | No | Prominent forehead, midface hypoplasia | Clinodactyly, left epicanthal fold | Dolichocephaly, high implantation of the hair with bilateral frontal upsweep, posteriorly rotated ears, short philtrum, thick lower lip, diastema, tapering fingers, long toes | Pronounced cupid’s bow, pes planus, soft skin | No |
| Other | Feeding difficulties | Exotropia, constipation | Unilateral clubfoot | Joint laxity; soft, doughy skin, GERD | Partial holoprocencephaly |
SI conversion factors: To convert height and head circumference to inches, divide values by 2.54. To convert weight to pounds, divide values by .45.
ACMG/AMP: American College of Medical Genetics and Genomics and Association of Molecular Pathology, ASD: autism spectrum disorder, ADHD: attention deficit hyperactivity disorder, EEG: electroencephalogram, GERD: gastroesophageal reflux disease, MRI: magnetic resonance image, N/A: not assessed.
ACMG/AMP classification according to the guidelines of Richards et al43.
Figure 1. De novo SHANK1 variants identified in patients with developmental delay and autism.
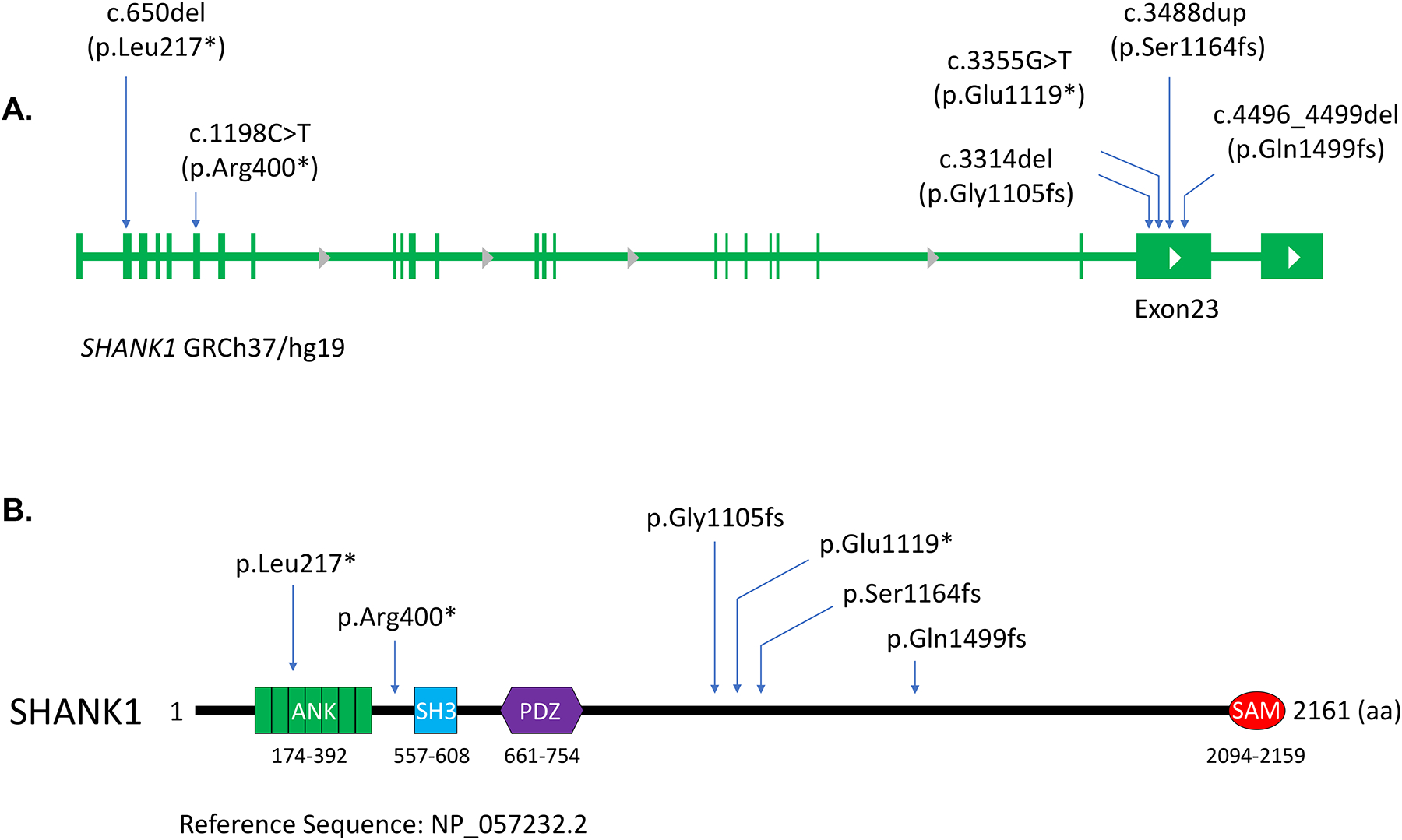
(A) Schematic figures show the distribution of six de novo variants observed in our cohort in the SHANK1 genome. (B) Amino acid changes caused by the SHANK1 variants are indicated in the diagram of the SHANK1 protein. Protein domains and amino acid numbers are indicated.
Phenotypic details for the six individuals in our cohort are provided in Table 1, and complete clinical summaries for these individuals are available in the Supporting Information. Ages of the six individuals described in this report were 6 to 11 years. All individuals share overlapping clinical features of a neurodevelopmental disorder including cognitive delays/ID and/or learning disabilities, speech and language delays, and behavioral problems. All individuals (6 of 6) had delayed speech and language development and either intellectual disability or learning disabilities. Three individuals (50%) have been diagnosed with mixed receptive-expressive language disorder. Three individuals (50%) have motor delays. Three individuals (50%) have a diagnosis of ASD. Two individuals (33%) have had seizures. Additionally, five (83%) exhibit neuropsychiatric symptoms including anxiety and attention deficit hyperactivity disorder (ADHD). Behavioral problems observed include, but are not limited to, anger outbursts and self-biting.
Two individuals have had seizures. Individual 3 had two separate incidents of seizures at 7 years of age. The first was a generalized tonic-clonic seizure with a subsequent abnormal EEG. The second looked like 3–4 minutes of generalized convulsions and vomiting and consisted of approximately 10 minutes of very agitated post-ictal state with coughing, then a more placid post-ictal time. He was started on levetiracetam to control his seizures. Levetiracetam caused behavioral problems, so he was switched to topiramate, which has been tolerated. Individual 6 presented with myoclonic seizures at 11 months of age, which evolved into focal to bilateral tonic-clonic seizures at 14 months of age, associated with abnormal EEG. She is taking a combination of sodium valproate, lamotrigine and clobazam to control her seizures. This individual also has abnormalities on brain MRI including absent cerebellar vermis, and possible dysgenesis with partial fusion of the cerebral hemispheres and the thalami, suggestive of a tubulinopathy. Individual 2 had an abnormal EEG showing sharp waves in the occipital head regions bilaterally and also independently on the left and right, but has had no clinical seizures. The presence of dysmorphic features was documented in four individuals, but taken together these features are inconsistent.
Molecular studies were performed on two of the variants, c.3314del (p.Gly1105fs) and c.3355G>T (p.Glu1119*). Both variants occur in penultimate exon 23 of SHANK1 (Fig. 1A), which encodes a large proline-rich region including a Homer binding site. The c.3314del variant generates a premature stop codon, which results in a truncation of the WT sequence coupled with the addition of 237 incorrect amino acids (Fig. 1B and 3A). The c.3355G>T nonsense variant also leads to a premature stop codon (p.Glu1119*) and the removal of the C-terminal domain of the resulting protein (Fig. 1B and 3A). To examine transcription levels of these de novo SHANK1 variants, we established cell lines in which these two SHANK1 variants were introduced into the genome using a CRISPR knockin (KI) protocol (Fig. 2A and 2B). We observed a significant increase in the SHANK1 transcript containing c.3355G>T (p.Glu1119*) compared to WT and to the transcript containing the c.3314del (p.Gly1105fs) variant (Fig. 2C), indicating that mRNAs containing these two variants have the capacity to escape nonsense-mediated decay (NMD) and translate into a mutant protein.
Figure 3. Truncated SHANK1 proteins abolished Homer1 binding.
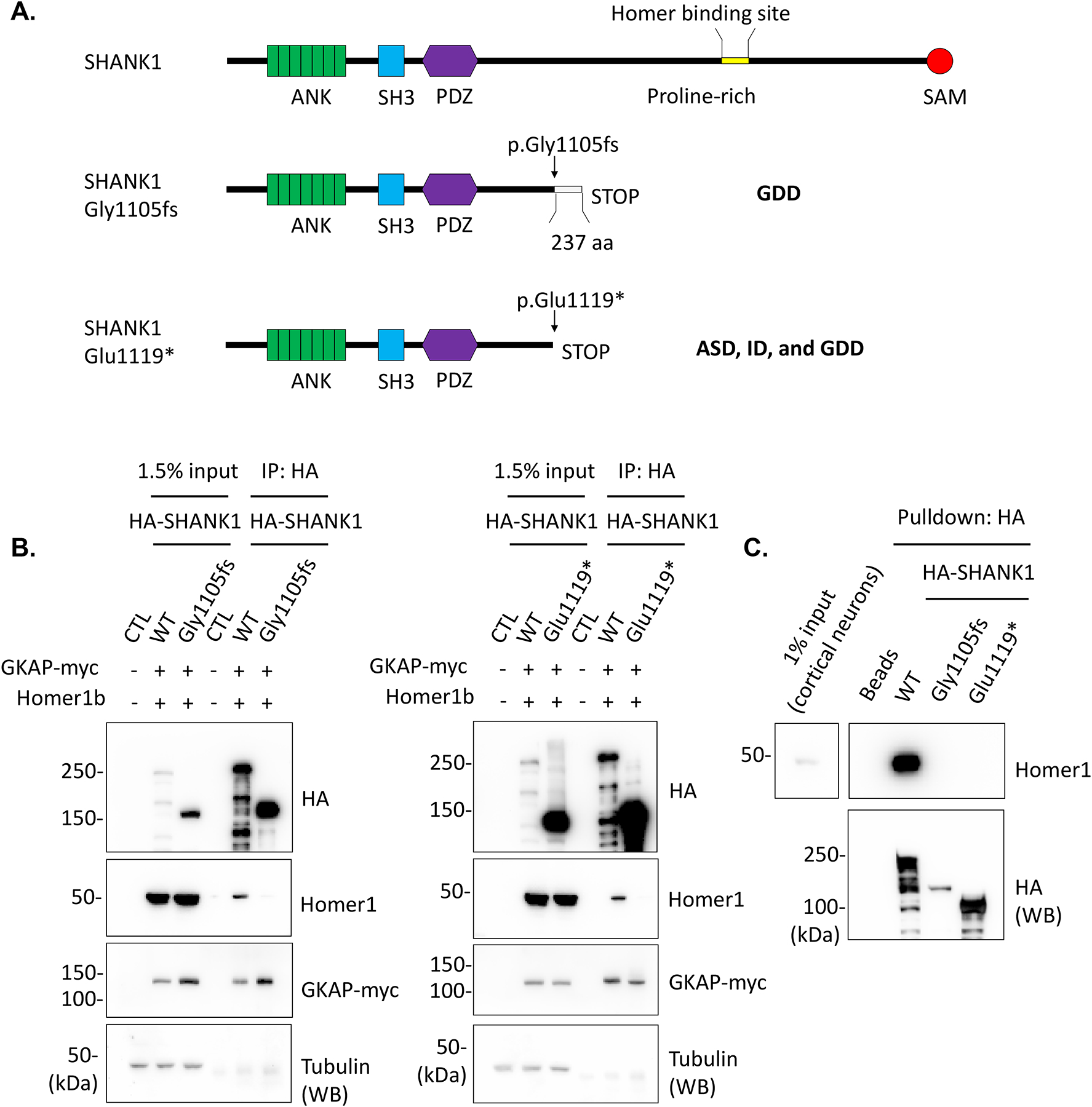
(A) Schematic figures denote SHANK1 c.3314del (p.Gly1105fs) and c.3355G>T (p.Glu1119*) variants and resulting SHANK1 protein truncations. (B) HA-SHANK1 (WT, Gly1105fs, or Glu1119*), GKAP-myc and Homer1b were co-transfected in HEK293 cells as indicated in the figures. GKAP-myc and Homer1b in the immunoprecipitates with HA antibody were analyzed by immunoblotting. (C) The immunoprecipitated HA-SHANK1 (WT, Gly1105fs, or Glu1119*) beads from HEK293 cells were incubated with cultured rat cortical neuron lysates for pulldown assays. Endogenous Homer1 binding with the immunoprecipitated HA-SHANK1 was analyzed by immunoblotting.
Figure 2. SHANK1 truncating variants were transcribed in CRISPR KI HEK293 cell lines.
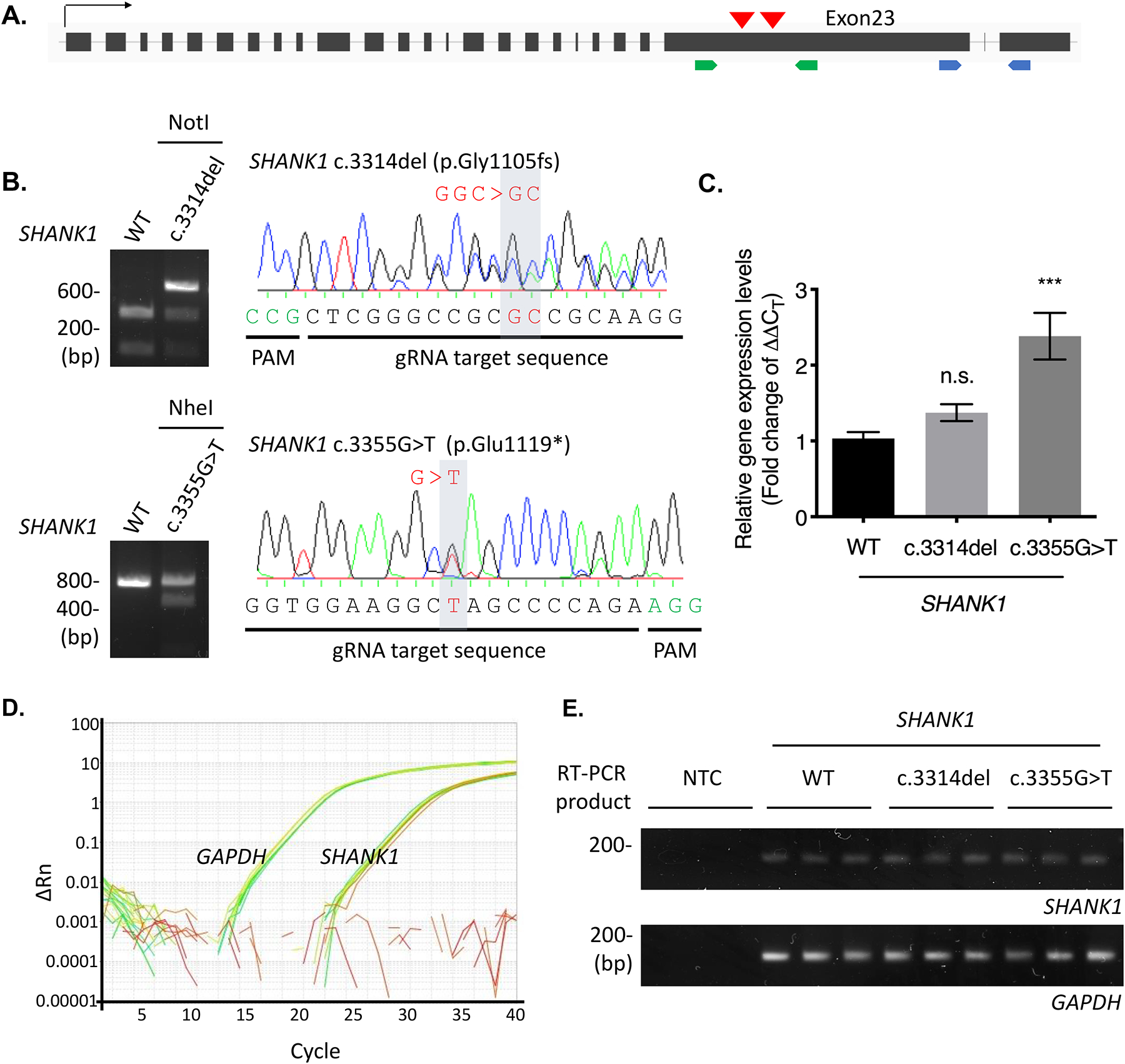
(A) Two de novo SHANK1 variants, c.3314del (p.Gly1105fs) and c.3355G>T (p.Glu1119*), in the penultimate exon of SHANK1 gene are selected for generation of CRISPR KI cell lines. Red arrow heads indicate brief location of the variants in the SHANK1 genome. Green arrows indicate binding sites of the primer set for genotyping. Blue arrows represent exons spanning primers binding sites for real-time PCR. (B) gDNA prepared from the SHANK1 c.3314del CRISPR and c.3355G>T CRISPR cell lines were amplified for the KI region by PCR. The PCR products were digested by NotI restriction enzyme and sequenced to check SHANK1 c.3314delG (top). The PCR products were digested with NheI restriction enzyme and sequenced to confirm SHANK1 c.3355G>T (bottom). (C) Total transcripts were prepared from the CRISPR cell lines and real-time PCR was performed. Fold change of the SHANK1 transcript levels (2^-(ΔΔCT)) was calculated. Graph indicates mean ± SEM (n = 9). The statistical significance between the mean of WT and the mean of each SHANK1 variant was calculated using one-way ANOVA with Dunnett’s multiple comparison test. ***P = 0.0001. n.s., not significant. (D) Representative SYBR green amplification graphs of real-time PCR analysis for SHANK1 and GAPDH. (E) Final product of the real-time PCR were run onto 2% agarose gel to check specific amplification.
To examine molecular aspects of the SHANK1 variants, we introduced the c.3314del (p.Gly1105fs) and c.3355G>T (p.Glu1119*) variants into the HA-tagged human SHANK1 cDNA plasmid. HA-SHANK1 (WT, Gly1105fs, or Glu1119*), GKAP-myc and Homer1b plasmids were co-transfected in HEK293 cells. The resulting SHANK1 Gly1105fs and Glu1119* proteins displayed reduced apparent molecular weights and a robust increase in protein expression levels when analyzed by SDS PAGE followed by immunoblotting (Fig. 3B). We also observed that the expressed SHANK1 Gly1105fs and Glu1119* proteins completely lose Homer1b binding, whereas GKAP-myc still binds to both mutant SHANK1 proteins (Fig. 3B). To examine potential protein interactions between mutant SHANK1 protein and endogenous Homer protein, we expressed SHANK1 (WT, Gly1105fs, or Glu1119*) in HEK293 cells, and SHANK1 immunoprecipitates were incubated with rat cultured cortical neuron lysates for a pulldown assay. As with the coimmunoprecipitation results, SHANK1 Gly1105fs and Glu1119* did not bind to endogenous Homer (Fig. 3C).
We investigated the neuronal localization of SHANK1 WT and SHANK1 Gly1105fs and Glu1119* in cultured rat hippocampal neurons with immunofluorescence confocal microscopy. Intriguingly, whereas SHANK1 WT displayed highly enriched localization in spines, both SHANK1 Gly1105fs and Glu1119* displayed a dispersed localization throughout the spine and dendritic shaft (Fig. 4A).
Figure 4. Truncated SHANK1 proteins disrupted synaptic localization.
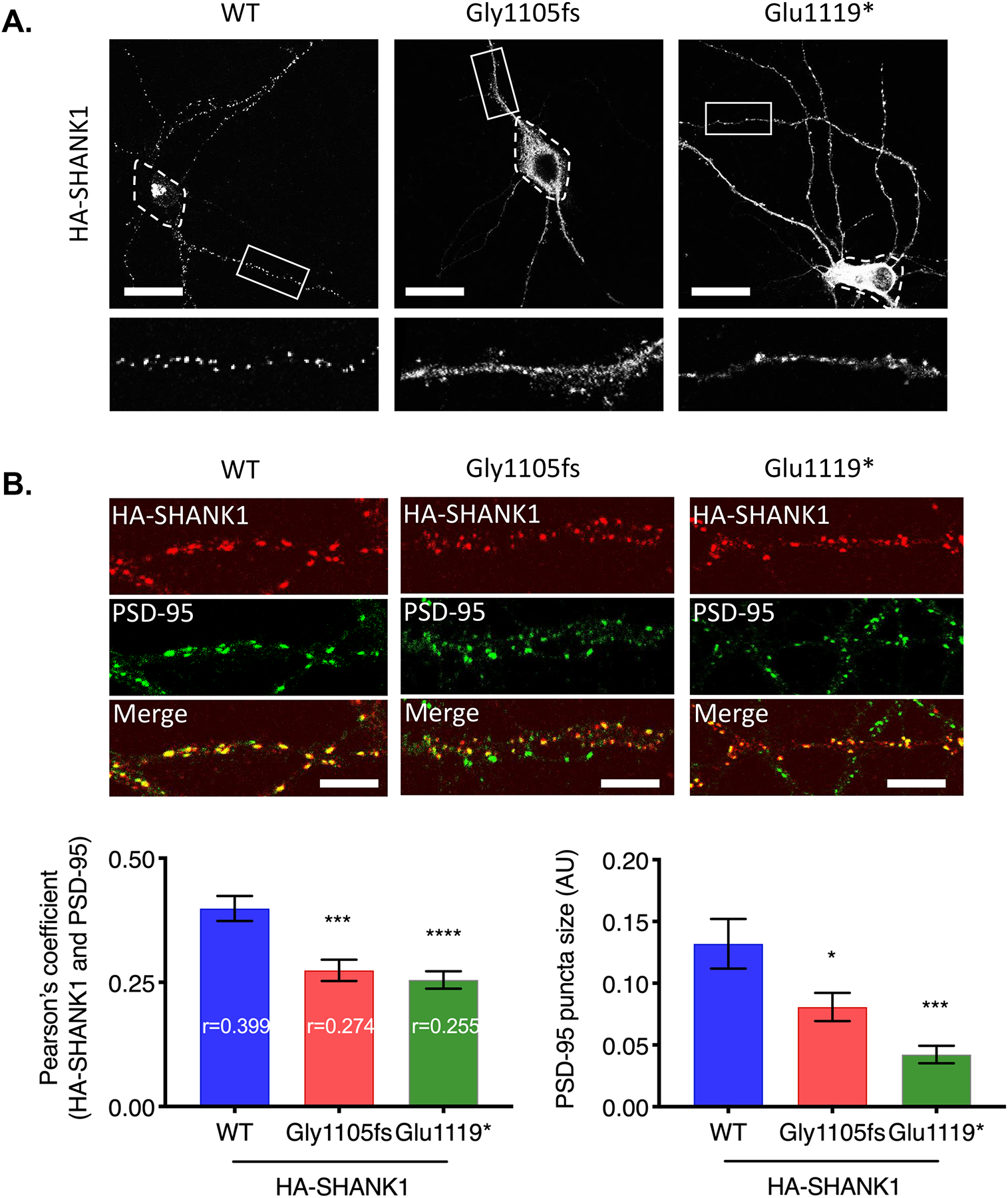
(A) HA-SHANK1 (WT, Gly1105fs, or Glu1119*) was transfected in cultured rat hippocampal neurons. HA-SHANK1 was labeled with anti-HA and Alexa 555-conjugated secondary antibody (white). Enlarged images of the boxed regions are shown below each panel. Dashed regions indicate soma. (Scale bar, 25 μm.) (B) HA-SHANK1 (WT, Gly1105fs, or Glu1119*) was expressed in cultured rat hippocampal neurons. HA-SHANK1 was labeled with anti-HA and Alexa 555-conjugated secondary antibody (red). Endogenous PSD-95 was labeled with anti-PSD-95 and Alexa 488-conjugated secondary antibody (green). Regions from three dendrites per each neuron were analyzed for Pearson’s coefficient. Graph indicates mean ± SEM (n = 15~19). *P < 0.05, ***P < 0.0005, ****P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (Scale bar, 5 μm.)
Next, we examined the colocalization of SHANK1 proteins with PSD-95, a postsynaptic protein often used to visualize the PSD. Hippocampal neurons were transfected with HA-SHANK1 (WT, Gly1105fs, or Glu1119*), and double labeled for HA and endogenous PSD-95 to evaluate synaptic localization. Unexpectedly, SHANK1 Gly1105fs and Glu1119* still colocalized with PSD-95 but to a lesser extent compared to WT (Fig. 4B), which indicates impaired synaptic localization of SHANK1 Gly1105fs and Glu1119*. Moreover, PSD-95 puncta size was decreased in the neurons expressing SHANK1 Gly1105fs and Glu1119* (Fig. 4B), suggesting reduced functional synaptic maturation. We further examined colocalization of SHANK1 proteins with Homer and VGLUT1, an excitatory presynaptic marker. Hippocampal neurons were transfected as above, and double labeled for HA and endogenous Homer or VGLUT1. Intriguingly, SHANK1 WT displayed strong colocalization with endogenous Homer, whereas SHANK1 Gly1105fs and Glu1119* depleted endogenous Homer from synapses (Fig. S3A). Both SHANK1 Gly1105fs and Glu1119* displayed comparable association with VGLUT1 to SHANK1 WT (Fig. S3B), indicating robust effect of Gly1105fs and Glu1119* variants on SHANK1 protein binding in the PSD. All these results indicate that truncating variants impair synaptic localization and postsynaptic protein interaction of SHANK1.
DISCUSSION
Our study adds six individuals to the existing nine described in the literature with germline truncating variants in SHANK1, bringing the total number to fifteen. As summarized in Table S1, all fifteen individuals share overlapping clinical features of a neurodevelopmental disorder and present with at least one of the following: ASD, neuropsychiatric disturbances, motor delays, speech and language delays, cognitive delays/ID, and/or learning disabilities. The most commonly observed findings amongst all affected individuals are speech and language delays, observed in ten of fourteen assessed individuals (71%). Although this is the most commonly observed finding across all reported individuals, the high frequency in which we observed speech and language delays in our cohort contrasts reported literature, in which Sato et al. reported that four of the five individuals with ASD in their cohort had an IQ in the normal range with no clinically significant language delays17. Unlike Sato et al. who reported reduced penetrance in females, we saw no clear evidence of reduced penetrance in either sex17. The second most frequently observed finding was hypotonia, observed in four of the six assessed individuals (67%). ASD was observed in nine of fourteen individuals for which this finding was documented (64%). Neuropsychiatric and behavioral issues were observed in nine of the fifteen individuals (60%). Individual 6 in our cohort presents with severe brain abnormalities on MRI including an absent cerebellar vermis suggesting dysgenesis with partial fusion of the cerebral hemispheres and basal ganglia. There were no abnormal MRI findings reported in the other two patients in our cohort who underwent imaging. Of the two other previously reported patients with LOF variants in SHANK1 who have had brain imaging, one had a normal brain MRI, and the other had a positron emission tomography (PET) scan that showed mild hyperfusion in the left temporal region. The possibilities that individual 6 in our cohort has a secondary diagnosis that explains these MRI abnormalities or other findings, or that the other five individuals in our cohort have secondary diagnoses to explain their various neurodevelopmental findings, cannot be ruled out. We have also identified an individual with developmental delay, ID, ASD, ADHD, and multiple neuropsychological diagnoses with an in-frame indel c.2265_2267dup (p.Val756dup), who inherited this variant from his mother. These individuals were not included in our cohort as effect of the in-frame indel is unknown. More information regarding these patients is available in the supporting information.
The nonsense-mediated mRNA decay (NMD) pathway is a RNA surveillance mechanism that detects and removes premature termination codon (PTC)-bearing transcripts29. Usually, an intron ~55 nucleotides downstream of the PTC triggers NMD in mammalian cells. However, NMD efficiency varies among genes, PTC positions, and different tissues30. NMD is known as an important factor to consider when trying to determine haploinsufficiency and dominant-negative phenotypes for single nucleotide variants and small deletions in ASD-related genes. However, we have limited information on the molecular regulation for the mutated or truncated SHANK proteins in human patients22,23,31,32. Recently, Zhou et al., generated homozygous Shank3 InsG3680 and Shank3 Arg1117* mutant mice33. The two variants were separated by only 325 nucleotides, but the Shank3 InsG3680 variant was associated with ASD and Shank3 Arg1117* was linked to schizophrenia. In the study, the Shank3 InsG3680 variant resulted in a complete loss of Shank3 mRNA and Shank3 protein, whereas the Shank3 Arg1117* variant generated truncated Shank3 protein in vivo33. These results indicate that the NMD pathway is important, but not the only mechanism to regulate PTC-bearing transcripts and their translation.
To examine NMD regulation on our de novo SHANK1 variants, we established CRISPR KI cell lines for the SHANK1 c.3314del (p.Gly1105fs) and c.3355G>T (p.Glu1119*) variants (Fig. 2). In our study, we did not observe a robust reduction in SHANK1 transcript levels in the KI cell lines, and SHANK1 transcript levels of the SHANK1 c.3355G>T (p.Glu1119*) cell lines were even significantly higher than WT (Fig. 2C). Although there are potential differences between processing in HEK293 cell lines and neuronal tissues, our results show an escape of SHANK1 truncating variants from NMD in some cell types. Truncated forms of SHANK1 proteins may be generated and act as a dominant negative to cause SHANK1 dysfunction. However, the study of the effect of the mutants endogenous SHANK1 proteins was not feasible due to limited resources. Instead, the corresponding truncated SHANK1 protein sequences were analyzed by bioinformatics tools34. Interestingly, the truncated SHANK1 proteins are predicted to have overall enhanced protein stability compared to WT protein. The amino acid compositions in the sequence were changed (% of proline: 14.35% for WT, 12.98% for Gly1105fs, and 10.55% for Glu1119*), and flexible loop secondary structure was reduced in the truncated SHANK1 proteins (% of loop structure: 84.82% for WT, 76.36% for Gly1105fs, 71.91% for Glu1119*) (Fig. S2), which indicate the mutant SHANK1 protein structures become more rigid34. Future studies evaluating human patient tissues will be important.
Homer is one of the most abundant scaffolding proteins in the PSD and binds to several synaptic molecules such as metabotropic glutamate receptors (mGluRs), IP3 receptors (IP3Rs), dynamin3, and SHANKs35. The Homer-SHANK1 complex is known as a critical structural platform for interactions with other signaling molecules, which are required for proper spine localization of SHANK135. Sala et al. demonstrated the importance of SHANK1 and Homer binding for spine maturation and synaptic function4. In SHANK1 knockout mice, Homer protein levels and dendritic spine size were reduced, and basal synaptic transmission was decreased in CA1 hippocampus5. Recent studies using a Shank3 knockout model of ASD displayed aberrant synaptic functional connectivity due to an altered Homer-mGluR5 scaffold36,37. In our current study, we observed complete loss of Homer binding with SHANK1 Gly1105fs and Glu1119* in the biochemical assays (Fig. 3B and 3C). Also, SHANK1 Gly1105fs and Glu1119* depleted endogenous Homer from synapses (Fig. S3A), which is consistent with the notion that SHANK1 binding is required for synaptic location of Homer3,4. In contrast, SHANK1 Gly1105fs and Glu1119* displayed similar colocalization with presynaptic VGLUT1 compared to SHANK1 WT (Fig. S3B). These findings indicate that the truncation variants have the largest effect on the SHANK1 protein interactions with binding proteins in the PSD. As we also observed abnormal neuronal localization and reduced PSD-95 colocalization of SHANK1 Gly1105fs and Glu1119* (Fig. 4), our data strengthen the idea that impaired synaptic signaling by disrupted SHANK1 and Homer interactions is one of the main pathogenic mechanisms of development of ASD.
All six of the variants identified in our cohort are previously unreported de novo LOF variants. Interrogation of gnomAD demonstrates that SHANK1 is significantly depleted of loss-of-function variation (Probability of loss of function intolerance (pLI) = 1.00). Interestingly, truncating variants in gnomAD Browser control individuals have been reported for SHANK3, which also has a pLI of 1. The exact explanation for these gnomAD Browser variants that overlap the disease-associated variants for the three SHANK1 genes remains unclear, but possibilities include somatic mosaicism, reduced penetrance, or variable expressivity. Finally, no evidence of genotype-phenotype correlations can be extracted from our data.
Although SHANK1 has previously been associated with ASD, neuropsychiatric disturbances and cognitive delays/ID, we report an additional six individuals with a spectrum of neurodevelopmental disorders including ASD, neuropsychiatric disturbances, cognitive delays/ID and/or learning disabilities, motor delays, speech and language delays and brain abnormalities. We evaluated patients with multiple de novo SHANK1 variants, but observed no significant correlation between our SHANK1 variant positions in the gene and clinical phenotypes (Table 1). To date, genotype and phenotype comparison studies in the cases of SHANK truncating variants with ASD have not been extensively performed38. Thus far, studies have mostly focused on SHANK3 truncating variants, but the clinical data vary and are still limited and thus have not revealed any meaningful relationship between SHANK3 variants and clinical features22,23,32,38–41. So far, how different variants in the same SHANK gene cause distinct clinical phenotypes is not fully understood42. By studying a larger cohort of patients with SHANK1 truncation and frameshift variants, we hope to understand any genotype-phenotype correlation.
Supplementary Material
Acknowledgments
We thank all affected individuals and family members for their participation in this work. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email fromcontact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome. Those who carried out the original analysis and collection of the Data bear no responsibility for the further analysis or interpretation of the data. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873; the Japan Agency for Medical Research and Development (AMED) under grant numbers JP20ek0109486, JP20dm0107090, JP20ek0109301, JP20ek0109348, and JP20kk0205012 (to N. Matsumoto); and by JSPS KAKENHI grant numbers JP17H01539 (to N. Matsumoto) and JP19K17865 (to Y. Uchiyama); the NINDS Intramural Research Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflicts of Interest
Aida Telegrafi and Richard Person are employees of GeneDx, Inc. David Goldstein is a founder of and holds equity in Q State Biosciences and Praxis Therapeutics; holds equity in Apostle Inc., and serves as a consultant to AstraZeneca, Gilead Sciences, GoldFinch Bio and Gossamer Bio. No other authors have conflicting interests to disclose.
Data Availability
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author on request.
Ethics Declaration
This study has approval through institutional review board-approved research studies at the Institute for Genomic Medicine at Columbia University (protocol AAAO8410) and UK Research Ethics Committee (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). Written informed consent was obtained for patients 1 and 5 as required by the IRB and REC, and data was de-identified. Written informed consent was obtained for patients 2, 3, 4, and 6 as required by their respective healthcare institutions as well as by the labs that performed their clinical testing. Data was de-identified before being used for the purposes of this study.
Supplementary Information
Document S1: Case Reports, Supplementary Methods, Figure S1–3, Table S1, and Supplementary Resources
References
- 1.Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113 (Pt 11):1851–1856. [DOI] [PubMed] [Google Scholar]
- 2.Mao W, Watanabe T, Cho S, et al. Shank1 regulates excitatory synaptic transmission in mouse hippocampal parvalbumin-expressing inhibitory interneurons. Eur J Neurosci. 2015;41(8):1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romorini S, Piccoli G, Jiang M, et al. A functional role of postsynaptic density-95-guanylate kinase-associated protein complex in regulating Shank assembly and stability to synapses. J Neurosci. 2004;24(42):9391–9404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–130. [DOI] [PubMed] [Google Scholar]
- 5.Hung AY, Futai K, Sala C, et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J Neurosci. 2008;28(7):1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins SM, Belagodu AP, Reed SL, Galvez R. SHANK1 is differentially expressed during development in CA1 hippocampal neurons and astrocytes. Dev Neurobiol. 2018;78(4):363–373. [DOI] [PubMed] [Google Scholar]
- 7.Collins SM, Galvez R. Neocortical SHANK1 regulation of forebrain dependent associative learning. Neurobiol Learn Mem. 2018;155:173–179. [DOI] [PubMed] [Google Scholar]
- 8.Loi E, Moi L, Fadda A, et al. Methylation alteration of SHANK1 as a predictive, diagnostic and prognostic biomarker for chronic lymphocytic leukemia. Oncotarget. 2019;10(48):4987–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadda A, Gentilini D, Moi L, et al. Colorectal cancer early methylation alterations affect the crosstalk between cell and surrounding environment, tracing a biomarker signature specific for this tumor. Int J Cancer. 2018;143(4):907–920. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Lv Y, Liu G. The roles of SHANK1 in the development of colon cancer. Cell Biochem Funct. 2020;38(5):669–675. [DOI] [PubMed] [Google Scholar]
- 11.Bahado-Singh RO, Vishweswaraiah S, Aydas B, et al. Artificial intelligence analysis of newborn leucocyte epigenomic markers for the prediction of autism. Brain Res. 2019;1724:146457. [DOI] [PubMed] [Google Scholar]
- 12.Silverman JL, Turner SM, Barkan CL, et al. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011;1380:120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sungur AO, Vorckel KJ, Schwarting RK, Wohr M. Repetitive behaviors in the Shank1 knockout mouse model for autism spectrum disorder: developmental aspects and effects of social context. J Neurosci Methods. 2014;234:92–100. [DOI] [PubMed] [Google Scholar]
- 14.Wohr M Ultrasonic vocalizations in Shank mouse models for autism spectrum disorders: detailed spectrographic analyses and developmental profiles. Neurosci Biobehav Rev. 2014;43:199–212. [DOI] [PubMed] [Google Scholar]
- 15.Sungur AO, Jochner MCE, Harb H, et al. Aberrant cognitive phenotypes and altered hippocampal BDNF expression related to epigenetic modifications in mice lacking the post-synaptic scaffolding protein SHANK1: Implications for autism spectrum disorder. Hippocampus. 2017;27(8):906–919. [DOI] [PubMed] [Google Scholar]
- 16.Wohr M, Roullet FI, Hung AY, Sheng M, Crawley JN. Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS One. 2011;6(6):e20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato D, Lionel AC, Leblond CS, et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am J Hum Genet. 2012;90(5):879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang T, Guo H, Xiong B, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berkel S, Marshall CR, Weiss B, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42(6):489–491. [DOI] [PubMed] [Google Scholar]
- 20.Pinto D, Pagnamenta AT, Klei L, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leblond CS, Nava C, Polge A, et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10(9):e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moessner R, Marshall CR, Sutcliffe JS, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81(6):1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phelan K, McDermid HE. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol Syndromol. 2012;2(3–5):186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17(10):774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84(4):524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36(8):801–808. [DOI] [PubMed] [Google Scholar]
- 30.Linde L, Boelz S, Nissim-Rafinia M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117(3):683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durand CM, Perroy J, Loll F, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonaglia MC, Giorda R, Beri S, et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 2011;7(7):e1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Y, Kaiser T, Monteiro P, et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron. 2016;89(1):147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32(Web Server issue):W321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiraishi-Yamaguchi Y, Furuichi T. The Homer family proteins. Genome Biol. 2007;8(2):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Bey AL, Katz BM, et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun. 2016;7:11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vicidomini C, Ponzoni L, Lim D, et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol Psychiatry. 2017;22(5):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarasua SM, Dwivedi A, Boccuto L, et al. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J Med Genet. 2011;48(11):761–766. [DOI] [PubMed] [Google Scholar]
- 39.Boccuto L, Lauri M, Sarasua SM, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet. 2013;21(3):310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gauthier J, Spiegelman D, Piton A, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(3):421–424. [DOI] [PubMed] [Google Scholar]
- 41.Waga C, Okamoto N, Ondo Y, et al. Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr Genet. 2011;21(4):208–211. [DOI] [PubMed] [Google Scholar]
- 42.Inoue K, Khajavi M, Ohyama T, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36(4):361–369. [DOI] [PubMed] [Google Scholar]
- 43.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web Resources
- ClinVar browser: http://www.ncbi.nlm.nih.gov/clinvar/
- Consensus Coding Sequence (CCDS), https://www.ncbi.nlm.nih.gov/CCDS/
- Ensembl genome assembly GRCh37: http://grch37.ensembl.org/Homo_sapiens/Info/Index.
- Ensembl Variant Effect Predictor (VEP): http://grch37.ensembl.org/Homo_sapiens/Tools/VEP.
- GenBank, https://www.ncbi.nlm.nih.gov/genbank/
- gnomAD, https://gnomad.broadinstitute.org/
- OMIM, http://www.omim.org/
- Pubmed: https://www.ncbi.nlm.nih.gov/pubmed.
- The Human Gene Mutation Database (HGMD): http://www.hgmd.cf.ac.uk/ac/index.php.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.