

Abstract
Complement plays a key role in immunosurveillance and homeostasis. When dysregulated or overactivated, complement can become a pathological effector, as seen in several inflammatory disorders, including periodontal disease. Recently, clinical correlative studies and preclinical mechanistic investigations have collectively demonstrated that complement is hyperactivated during periodontitis and that targeting its central component (C3), provides therapeutic benefit in non-human primates. The preclinical efficacy of a C3-targeted drug candidate combined with excellent safety and pharmacokinetic profiles supported its use in a recent phase 2a clinical study, in which C3 inhibition resolved gingival inflammation in patients with periodontal disease. We posit that C3-targeted intervention might represent a novel and transformative host-modulation therapy meriting further investigation in phase 3 clinical trials for the treatment of periodontitis.
Rationale for host modulation in periodontal disease
Periodontitis is a microbiome-driven chronic inflammatory disease of the tissues that surround and support the dentition in mammals (Box 1) [1]. A milder form of periodontal disease, gingivitis, represents inflammation contained within the gingival epithelium and the underlying connective tissue, which commonly precedes the onset of periodontitis. Although gingivitis is reversible, it often persists as chronic inflammation despite daily self-performed control of the dental plaque biofilm and periodic professional care and, therefore, presents a constant risk for periodontitis, which is irreversible in susceptible individuals. Periodontal disease (ranging from mild to severe) affects about 50% of human adults, whereas the severe form of the disease afflicts approximately 10% of the adult population [2, 3]. According to the Global Burden of Disease Study, severe periodontitis is the 6th most prevalent condition worldwide [4]. Untreated periodontitis may result in tooth loss, compromised mastication and esthetics, as well as poorer quality of life [5–9]. Moreover, periodontitis increases the risk of other inflammatory disorders, including cardiovascular disease, rheumatoid arthritis, and Alzheimer’s disease [10, 11].
Box 1. Inflammation in periodontal diseases.
The dental plaque biofilm-induced forms of periodontal disease, gingivitis, and periodontitis, are prevalent chronic inflammatory conditions that affect distinct and overlapping components of the periodontium, i.e., the tissues that support the dentition, namely the gingiva, cementum, periodontal ligament and alveolar bone [1]. The clinical manifestations of gingivitis, which is a reversible condition, include degradation of the epithelial and connective tissue attachment of the gingiva to the teeth and the progressive deepening of the gingival crevice. In contrast to gingivitis, the inflammatory cell infiltrate in periodontitis is not restricted within the gingival epithelium and the underlying connective tissue, but also extends into the deeper compartments of the periodontium. This extensive inflammatory attack leads to the degradation of the cementum, periodontal ligament, and alveolar bone, resulting in tooth mobility and ultimately, tooth loss. In gingivitis and periodontitis, inflammation is initiated by dysbiosis of the local tooth-associated microbiota [50]. Specifically, the transition of complex microbial communities from a commensal to a pathogenic entity can compromise tissue homeostasis and lead to destructive periodontal tissue inflammation [50]. The destructive inflammatory response involves the participation and cross-talk of cells and molecules of innate and adaptive immunity such as complement, neutrophils, and IL-17-producing CD4+ T cells (Th17); the inflammatory events arising from these interactions can lead to pathologic activation of osteoclasts, which resorb the alveolar bone supporting the teeth [35, 37, 55]. Periodontal inflammation reinforces and sustains microbial dysbiosis by generating a nutritionally conducive environment, specifically through the accumulation of inflammatory tissue breakdown products (e.g., degraded collagen, a source of amino acids, and heme-containing compounds, a source of iron) [50]. This generates a feed-forward vicious cycle in which destructive inflammation and dysbiosis are reciprocally reinforced and lead to the chronification of periodontal disease.
The aim of the standard-of-care periodontal therapy is to eliminate the pathogenic biofilm by mechanical debridement, often with adjunctive anti-microbial approaches. Current therapy, however, is at times ineffective, especially in highly susceptible patients, or does not prevent episodic recurrences; hence, periodontitis remains a significant public health challenge that results in considerable health care expenditure, which is expected to rise given the increase in population longevity [7, 9, 12–14]. In 2018, the total cost (both direct and indirect due to productivity losses) of periodontal disease in the USA and Europe (32 countries) were estimated at $154.06 billion and €158.64 billion, respectively [15]. Since tissue destruction in periodontitis is mediated primarily by an exaggerated inflammatory response, intentional alteration of the host response (host modulation) as an adjunctive therapy may contribute to its management and reduce the risk of associated comorbidities [16]. In contrast, ineffective control of the host’s immune response in patients with periodontitis results in long-term episodic progression of the disease over a lifetime.
The concept of host-modulation therapy in periodontal disease, however, has remained underdeveloped. Therefore, the potential of innovative host modulatory strategies to enhance clinical outcomes beyond those achieved via conventional periodontal treatment warrants their development from benchtop to the clinic. Here, we review basic immunological, preclinical, and clinical studies that have collectively provided the foundation for our hypothesis suggesting that complement-targeted intervention is a novel host-modulation therapy that might revolutionize the treatment of human periodontal disease.
The host inflammatory response and the role of complement
Although historically perceived as a closed system charged with tagging and killing microbes, complement is now appreciated as a key immunological hub for the induction and regulation of diverse immune and inflammatory functions [17]. Complement can modulate the overall mammalian host immune response by engaging in crosstalk interactions with other immune and physiological systems. For instance, complement cleavage (activation) products cross-talk with pattern-recognition receptors, such as Toll-like receptors (TLRs) (Figure 1); components of the coagulation system, such as thrombin; and adaptive immune cells, such as T helper 1 (Th1) and Th17 cells [18–20]. Although these properties endow complement with key roles in host immunosurveillance and homeostasis, its dysregulation or excessive activation can drive pathological inflammation in multiple disorders, such as paroxysmal nocturnal hemoglobinuria, systemic lupus erythematosus, and age-related macular degeneration [21, 22].
Figure 1: Complement activation and its effects in periodontitis.
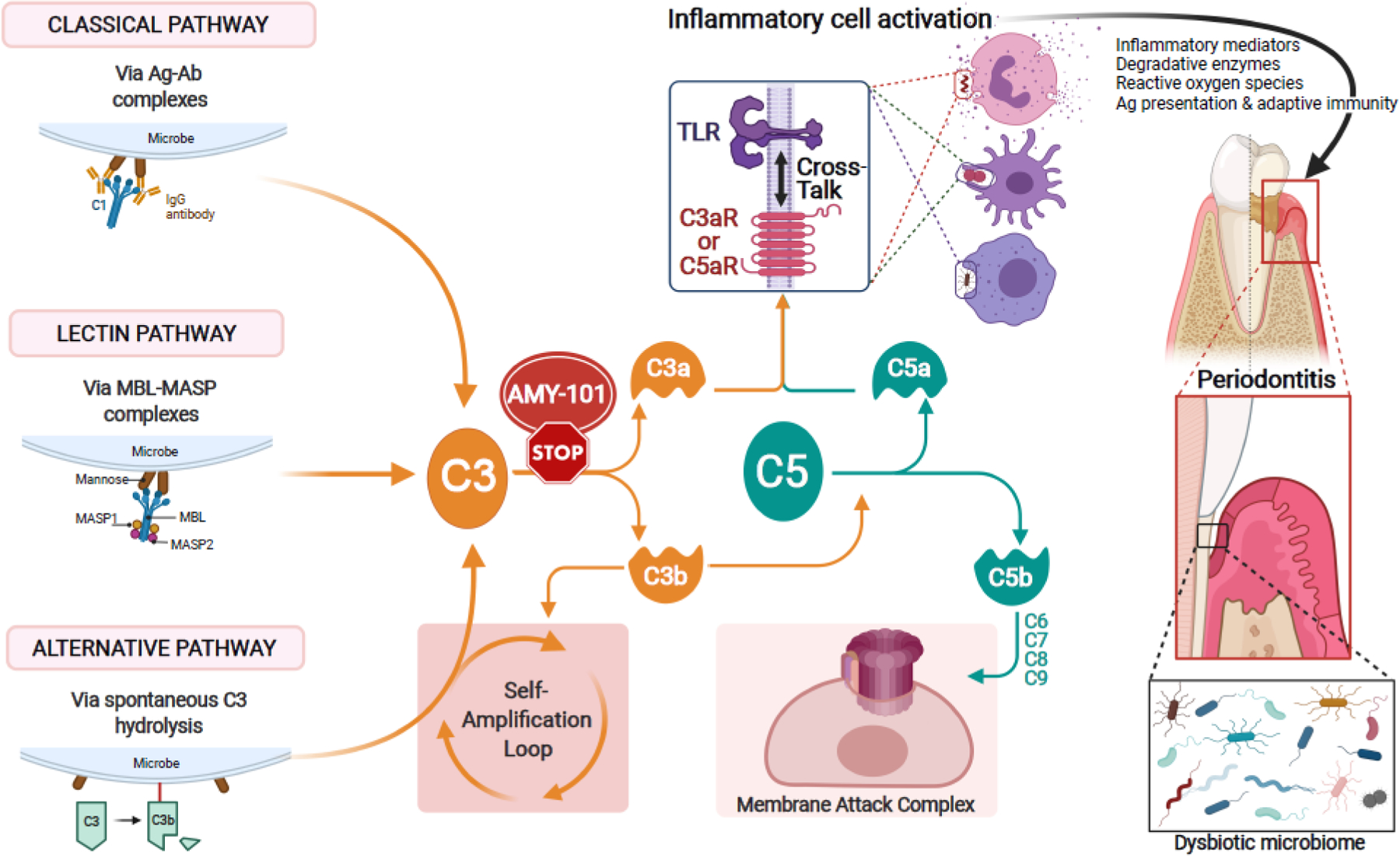
The complement cascade can be triggered by distinct initiation mechanisms: The classical pathway is initiated by antigen (Ag) – antibody (Ab) complex–mediated activation of the C1 complex. The lectin pathway is activated when complexes of mannose-binding lectin (MBL) and MBL-associated serine proteases (MASPs) bind to microbial surfaces. The alternative pathway can be triggered by a ‘tick-over’ mechanism involving spontaneous hydrolysis of C3, if a regulatory mechanism is absent (as is typically the case with microbial surfaces). The so-called ‘alternative pathway (AP)-amplification loop’ (in which more C3 is cleaved into more C3b which further fuels the loop) amplifies complement activation independently of the initiating mechanism, thereby contributing most of the downstream terminal pathway-mediated effector responses (i.e., C5a, membrane attack complex [MAC]). All three mechanisms of complement activation and amplification converge at C3, which is thus an attractive target of pharmacological interception, e.g., by the cyclic inhibitory peptide AMY-101. If C3 is not blocked, downstream effects include the generation of effectors (C3a and C5a) that promote inflammation and the generation the C5b-C9 MAC with potential antimicrobial but also tissue destructive capacity. Whereas the role of MAC in periodontitis is uncertain, C3a and C5a activate their cognate G-protein-coupled receptors (C3aR and C5aR1), which cross-talk with Toll-like receptors. This cross-talk synergistically activates inflammatory leukocytes, which can directly or indirectly mediate destructive periodontal tissue inflammation and bone loss in periodontitis [46, 56]. Complement-mediated inflammation also promotes the dysbiosis of the periodontal microbial community [32, 33]. C3 blockade can prevent these downstream effects and thereby offer broader protection against periodontitis [33, 37]. TLR, toll-like receptor.
The complement system comprises some 50 proteins, including the classic serum proteins (C1-9), pattern-recognition molecules, convertases and other proteases, fluid-phase or cell-associated regulatory proteins, and receptors that interact with a variety of immune mediators [23]. The complement cascade can be initiated by distinct mechanisms (i.e the classical, lectin, or alternative pathways), all of which converge at the third complement component, C3 [24]. C3 is thus a central target for therapeutic regulation of the downstream immune and inflammatory pathways (Figure 1).
Complement involvement in periodontitis
Pioneering clinical studies by independent groups in the 1970s and 1980s associated periodontitis with increased abundance of complement activation products in gingival tissue biopsies and the gingival crevicular fluid (GCF) obtained from patients relative to healthy control samples [25–29]. An experimental human gingivitis study demonstrated progressive increase of complement activation products that correlated with elevated clinical periodontal inflammation [29]. Consistently, successful periodontal therapy that resolved periodontal inflammation resulted in reduced C3 activation in the GCF [30]. A recent study in human patients with periodontitis and periodontally healthy controls suggested that the complement activation product C3c might be a potential salivary biomarker for periodontitis [31].
These observational human studies indicated that complement may be involved in the pathogenesis of periodontitis, a notion that was conclusively demonstrated by cause-and-effect studies in preclinical models of periodontitis. Indeed, mice globally genetically deficient in C3 (C57BL/6 C3−/−), or in the receptor for the complement anaphylatoxin C3a (C57BL/6 C3ar1−/−), were protected from developing gingival inflammation and alveolar bone loss, as compared to their wild-type littermate controls [32, 33] (Figure 2). Notably, the protective effect seen in C3−/− mice was confirmed in three distinct disease models, namely, ligature-induced periodontitis, Porphyromonas gingivalis-induced periodontitis, and naturally-occurring periodontitis [33]. Specifically, in all three models, C3−/− mice developed significantly less alveolar bone loss than wild-type littermate controls (C57BL/6 C3+/+ mice). In the same study [33], analysis of the gingival tissue by quantitative real-time PCR showed that compared to wild-type mice, C3−/− mice exhibited reduced mRNA amounts of cytokines known to drive inflammatory bone loss in periodontitis; namely, interleukin (IL)-23 and IL-17 -- derived mainly from antigen-presenting cells [34] and Th17 cells [35], respectively. This finding is not only consistent with the ability of complement to cross-talk with and regulate innate and adaptive immune responses [23], but also implies that complement inhibition alone might have a broader therapeutic effect than previously thought by blocking downstream inflammation mediated by diverse effectors.
Figure 2: Milestones to the development of complement C3-targeted intervention in periodontitis.
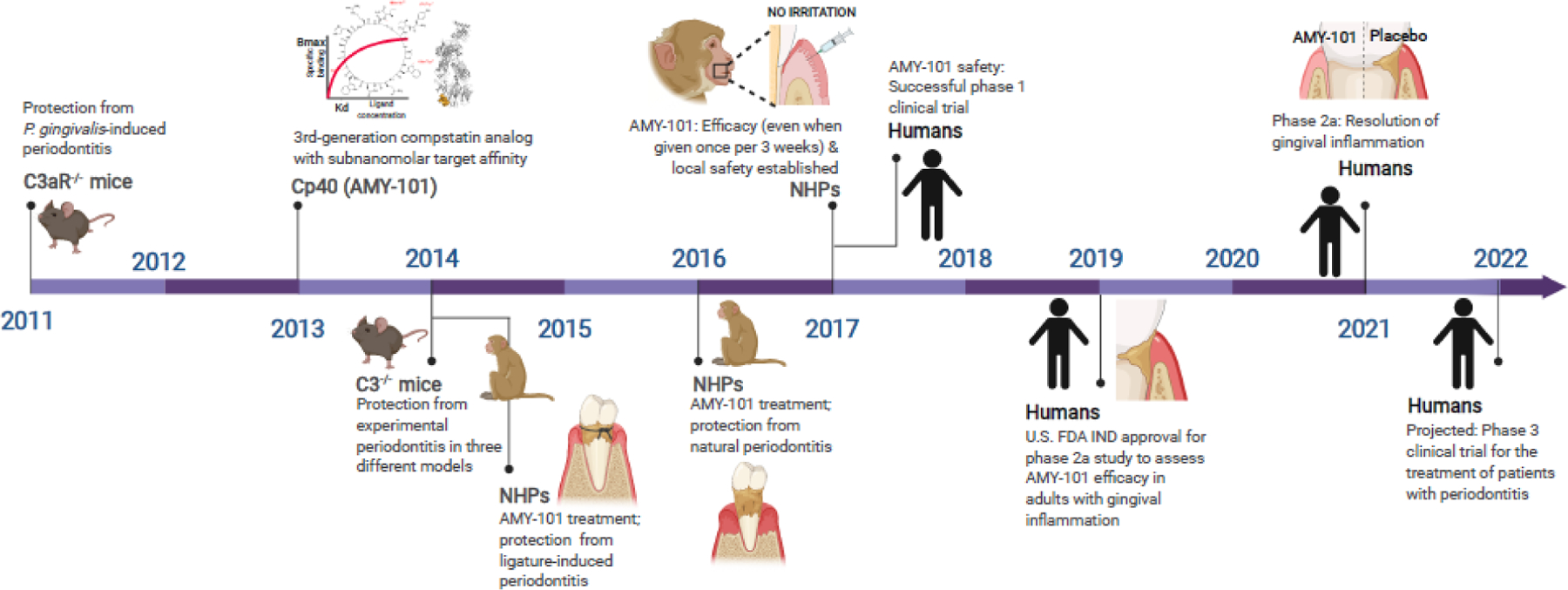
A timeline is shown of key preclinical and clinical studies leading to the development of complement C3-targeted host-modulation using the peptide-based C3 therapeutic AMY-101 as an adjunctive therapy in mammalian periodontal disease (see text for details) [24, 32, 33, 37, 41, 51]. NHP, non-human primates.
C3-targeted intervention in non-human primate periodontitis
The aforementioned studies in mice suggested that inhibition of complement C3 might provide a therapeutic benefit in the management of periodontitis. The potential efficacy of C3 blockade was addressed using Cp40, a third-generation analog of the compstatin family of cyclic peptidic inhibitors of human and non-human primate (NHP) C3 [24, 36]. In human plasma, Cp40 exhibits a half-life of 48 h that exceeds the standards for most peptidic drugs and has a sub-nanomolar affinity for C3 [36]. The binding of Cp40 to C3 prevents its cleavage by C3 convertases, thereby blocking the release of the anaphylatoxin C3a and the generation of C3b [24]. The latter inhibitory effect suppresses the amplification of the complement response via the alternative pathway of complement triggering (Figure 1) [24]. Moreover, by blocking the activation of C3 and the assembly of C3b-containing convertases, Cp40 can additionally prevent downstream effector responses, such as the generation of the anaphylatoxin C5a and the assembly of the membrane attack complex [17] (Figure 1). Cp40 was clinically developed for human use as ‘AMY-101’ [24] and is hereafter referred to as such.
The suitability of C3 as a therapeutic target in periodontitis was first tested in NHPs (Figure 2) [33]. Specifically, 4 young adult cynomolgus monkeys (3 to 7 years of age) were subjected to ligature-induced periodontitis under a split-mouth experimental design, i.e., having test and control tooth sites in the same animal [33]. Sites that were locally treated with intragingival injections of AMY-101 (0.1 mg/interdental papilla) showed significantly less alveolar bone loss, as evidenced radiographically, relative to sites treated with a sequence-scrambled control peptide [33]. Moreover, AMY-101 significantly prevented gingival inflammation and clinical attachment loss, as measured by clinical indices. These clinical effects were associated with lower GCF concentrations of pro-inflammatory and osteoclastogenic cytokines (e.g., IL-17 and RANKL), as well as diminished osteoclastogenesis in bone biopsies from AMY-101-treated sites relative to control peptide-treated sites [33].
A follow-up study examined the effect of AMY-101 in a therapeutic, rather than preventive, setting. Specifically, the drug was tested for its ability to reverse naturally-occurring periodontitis in 10 aged NHPs (7 to 15 years of age) [37]. This study involved a 6-week treatment period with injections of AMY-101 in the interdental papillae (0.1 mg/site) and a 6-week follow-up period in the absence of treatment. Regardless of administration frequency (once or three times weekly), AMY-101 significantly reduced clinical indices related to inflammation (Gingival Index and Bleeding on Probing), periodontal pocket formation and tissue destruction (Probing Pocket Depth and Clinical Attachment Loss), as well as tooth mobility, as a result of severe periodontitis [37]. Consistent with the clinical findings, AMY-101 administration was associated with a significant reduction of pro-inflammatory mediators (such as, C3a, IL-1β, IL-6, and IL-17) in the GCF and osteoclast numbers in bone biopsies relative to baseline values (i.e., just prior to treatment). The therapeutic effects of AMY-101 persisted for at least six weeks after treatment completion. GCF samples from the animals of the aforementioned study [37] were subjected to hypothesis-free proteomics characterization [38]. Gene Ontology analysis using Protein Analysis Through Evolutionary Relationships suggested the involvement of the alternative and classical complement pathways as well as ‘leukocyte degranulation’ in NHP periodontitis [38]. The latter implied that AMY-101 might also suppress neutrophil exocytosis -- a major mechanism of inflammatory tissue destruction [39]. Consistent with this, a later study showed that C3 inhibition by AMY-101 blocked neutrophil extracellular trap release in the plasma of patients with COVID-19, as assessed by myeloperoxidase/DNA complex ELISA [40].
In another study of NHPs with naturally occurring periodontitis, the efficacy of AMY-101 (given at 0.1 mg/site) was tested with decreased frequency of administration, specifically once every two weeks (in 5 monkeys; 7 to 10 years of age) or once every three weeks (in a different set of 5 monkeys; 7 to 10 years of age) for a total duration of six weeks [41]. Irrespective of regimen, AMY-101 was associated with significant reduction in clinical indices measuring periodontal inflammation (Gingival Index and Bleeding on Probing) as well as periodontal pocket formation and tissue destruction (Probing Pocket Depth and Clinical Attachment Loss) relative to baseline values. Most of these therapeutic effects remained statistically significant for at least 6 weeks after treatment completion, indicating substantial sustainability of its pharmacological effects [41]. Taken together, these studies established that AMY-101 mediated clinically relevant anti-inflammatory effects in naturally occurring periodontitis in NHPs.
Safety considerations
In one of the NHP studies, the potential of a therapeutic dose of AMY-101 (0.1 mg/site) to cause local irritation after injection was assessed daily on the healthy gingiva of NHPs without signs of irritation throughout the monitoring period [41]. Although the possibility exists that intragingival AMY-101 injections may result in systemic exposure, the amount of systemically absorbed AMY-101 appears to be negligible and cannot affect complement functions in circulation or extra-oral tissues, as explained below. In particular, even if the entire amount of locally administered AMY-101 (1.5 mg needed for treating 15 gingival sites [37]) were injected directly into the circulation, this might result in only 0.2–0.3 mg AMY-101/kg bodyweight in NHPs (or, in case of humans, 0.02–0.03 mg/kg bodyweight, given that an average human adult has an approximately 10-fold higher bodyweight than an average cynomolgus monkey adult). However, a considerably higher systemic AMY-101 dose (1 to 2 mg/kg bodyweight) is required to achieve target-exceeding drug concentrations [42]. Thus, any amounts of locally injected AMY-101 leaking into the blood circulation should presumably be readily bound by excess C3 (1.0 to 1.5 mg/ml) in the blood. No off-target effects have been observed in animal or human studies with AMY-101, which has also been recently used for the treatment of severe COVID-19 immunopathology with favorable clinical outcomes and without evidence of any systemic toxicity [40]. Moreover, the up to 3 months-monitoring of NHPs under systemic exposure of inhibitory AMY-101 (injected subcutaneously at 2 mg/kg bodyweight every 12h for a total of 15 injections) showed no significant changes in biochemical, hematological, or immunological parameters (such as, hemoglobin, hematocrit, white and red blood cell counts, and expression of C3 and proinflammatory cytokines, including IL-1β, IL-6, IL-10, and IL-12) in blood or tissues, as compared to vehicle alone-treated NHP controls [43]. Moreover, the recent US FDA approval of the compstatin-based C3 therapeutic pegcetacoplan for the treatment of paroxysmal nocturnal hemoglobinuria adds support to the clinical safety and efficacy of performing compstatin-mediated C3 inhibition [44].
A potential concern is whether complement blockade could impair the competency of antimicrobial defenses in the periodontal tissue. However, we argue that local inhibition of complement is unlikely to lead to uncontrolled microbial growth in periodontitis. In part, this notion is based on findings that complement is exploited by periodontal pathogens to subvert the host immune response and promote the persistence of a dysbiotic microbial community [32, 45, 46]. For instance, P. gingivalis manipulates the crosstalk of complement with TLR signaling in neutrophils and macrophages to uncouple bactericidal mechanisms from inflammation and thereby promote microbial dysbiosis in mice [32, 45, 46]. Thus, complement activation might favor rather than restrain periodontal pathogens, many of which have developed mechanisms to protect themselves from the antimicrobial action of complement [47, 48]. Accordingly, C57BL/6 C3−/− mice subjected to experimental periodontitis exhibit decreased periodontal bacterial burden compared to wild-type littermate controls, assessed in the periodontium using 16S rRNA gene quantitative real-time PCR [33]. The diminished periodontal inflammation seen in C3 deficiency might also contribute to the observed reduced microbial burden. This notion is supported by the fact that inflammation represents a major ecological factor driving the selective expansion of pathogenic species in periodontitis, presumably by generating a nutritionally favorable environment through accumulation of tissue breakdown products (nutrients for bacteria) [49, 50]; however, further robust studies are warranted to fully evaluate these relationships.
Pharmacological inhibition of C3 in human periodontal disease
In 2017, a phase 1 safety trial of AMY-101 was successfully completed in human volunteers (NCT03316521I) [24]. This was a prospective, single-center, open-label, first-in-human clinical study in healthy male volunteers, which evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of a single ascending dose and a multiple dose of AMY-101. The primary outcome was ‘treatment-related adverse events’ and AMY-101 was shown to be safe and well tolerated, as there were no serious or other significant adverse events during the study [24]. Two years later, AMY-101 received Investigational New Drug approval by the U.S. FDA for the first clinical study to evaluate its efficacy in adults with periodontal inflammation (NCT03694444II) (Figure 2). A randomized, placebo-controlled, double-blind phase 2a clinical trial was conducted to evaluate the safety and efficacy of AMY-101 in 40 patients with existing periodontal inflammation [51]. The primary efficacy outcome was the change in gingival inflammation, measured by the modified gingival index (MGI), whereas bleeding-on-probing (BOP) was a secondary outcome. AMY-101 was shown to be safe and well-tolerated in all study participants, as evidenced by clinical and laboratory assessments of safety performed for each visit, including vital signs (blood pressure, heart rate, and respiratory rate), examination of oral soft and hard tissues and injection sites for ulceration, and determination of liver enzyme concentrations (alkaline phosphatase, aspartate transaminase, and alanine transaminase). Corroborating the NHP proof-of-concept studies of C3 inhibition [33, 37, 41], a once-per-week intragingival injection of AMY-101 for 3 weeks resulted in pronounced and sustainable resolution of gingival inflammation in human subjects. In particular, the clinical efficacy of AMY-101 was reflected by statistically significant reductions in MGI and BOP, two key periodontal indices measuring gingival inflammation [51]. Consistent with preclinical studies of C3 inhibition in NHP periodontitis [33, 37, 41], AMY-101 treatment exerted a prolonged anti-inflammatory effect in patients with periodontal inflammation including at 3 months after the initiation of treatment. In light of these promising results, AMY-101 will be further tested in a pivotal Phase 3 study as an adjunctive therapeutic in patients with periodontitis [51].
Concluding Remarks
Novel functions attributed to complement in the past two decades have revolutionized our perception of this system from a blood-based antimicrobial effector to a global modulator of immune and inflammatory responses [17]. The multifaceted interactions of complement with other immune cells and physiological systems are reflected in the diversity of inflammatory disorders driven, or exacerbated by complement dysregulation or overactivation [21, 22]. Compelling evidence accumulated over the years indicates that complement is causally linked to periodontitis by inducing destructive inflammation and promoting microbial dysbiosis (Figure 1). Accordingly, inhibition of the central complement component C3 by AMY-101 provides a targeted therapeutic benefit without adverse toxicities [51, 52]. In the studies conducted thus far, AMY-101 was successfully applied as a stand-alone treatment. However, this C3-targeted drug is intended for use as an adjunctive therapy to the standard periodontal treatment, which by itself, is not effective for all patients and thus, periodontitis persists as a serious public health and economic burden [7, 12–14]. Conceivably, C3-targeted interventions might also be useful in a preventive personalized setting in the case of high-risk individuals (e.g., patients with diabetes or cigarette smokers). Moreover, since gingivitis is a major risk factor and a necessary precursor of periodontitis [53, 54], we argue that AMY-101 might also be applicable in the treatment of gingivitis after development of convenient modes of delivery, or perhaps as a consumer product.
Whereas basic and translational research in the complement and periodontal disease fields has come a long way, many questions remain to be addressed (see Outstanding Questions Box). The documented safety and efficacy of AMY-101 in early-phase clinical trials merits further investigation in future phase 3 clinical trials for the treatment of human periodontitis (Figure 2). The phase 3 trial will expand AMY-101 testing to diverse populations examining its therapeutic effect on both surrogate (i.e., clinical or biological measures of periodontitis) and true periodontal endpoints, such as maintenance of periodontal health or tooth loss. Ultimately, the goal will be to develop a sustainable intervention, which might overcome the existing limitations of the current standard-of-care treatment of periodontitis.
Outstanding Questions Box.
How long can the host-modulatory effects of C3-targeted inhibition be sustained after periodontitis treatment in patients? What are the mechanisms underlying a sustained therapeutic effect? The answers to these questions may not only provide insights into the homeostatic mechanisms of the periodontium but can also help develop and optimize clinical treatment regimens.
Besides regulating the host inflammatory response, does complement blockade in human periodontitis also modulate the dysbiotic microbiome toward a health-compatible configuration? The notion that inflammation is a key ecological factor for dysbiosis is supported by in vitro and in vivo preclinical studies, but clinical evidence is largely lacking.
Can C3-targeted treatment of patients with periodontitis attenuate systemic inflammatory markers and improve surrogate markers of inflammatory comorbidities? If this can be proven in future studies, it may strengthen the notion that periodontitis is a modifiable risk factor for systemic comorbidities.
Could local C3 blockade be used as a preventive tool against disease recurrence in patients who have already been treated for periodontitis and are under standard maintenance therapy?
Could C3 inhibitors be locally delivered into periodontal pockets in a sustained-release formulation?
Is complement also involved in the pathogenesis of other oral inflammatory conditions such as peri-implant mucositis and peri-implantitis? If so, could complement-targeted intervention be applied to protect against these hitherto intractable conditions?
Highlights Box.
Complement is a key contributor to immunosurveillance and homeostasis; however, when dysregulated or overactivated, complement can mediate pathological inflammation.
Periodontitis is a prevalent chronic inflammatory disease of mammalian tissues that surround and support the teeth. It constitutes a significant healthcare and socioeconomic burden.
Clinical studies have shown that complement is overactivated in periodontitis and that there is a correlation between periodontal inflammation and complement activation.
Complement involvement in the pathogenesis of periodontitis was demonstrated in preclinical mouse and non-human primate studies, identifying C3 as a potential target of therapeutic intervention.
A randomized, placebo-controlled, double-blind phase 2a clinical trial showed that C3-targeted inhibition blocks gingival inflammation in patients with periodontal disease.
Acknowledgments
The cited preclinical complement studies were supported by grants from the U.S. National Institutes of Health (AI068730, AI030040, DE015254 and DE021685) and the European Commission (FP7-DIREKT 602699). The figures were generated using Biorender.com.
Glossary
- Compstatins
Family of disulfide-bridged peptides (13–17 aa); potent, selective inhibitors of the central complement protein C3. The original compstatin was a cyclic tridecapeptide isolated from a phage-display peptide library. A second-generation compstatin-based C3 therapeutic (pegcetacoplan/APL-2) was recently approved for the treatment of paroxysmal nocturnal haemoglobinuria. AMY-101 represents a third-generation compstatin-based C3 therapeutic.
- Dysbiosis
Imbalanced, disease-provoking interaction among microbial constituents of a community and/or between the microbial community and the host immune system; results from alterations in abundance and/or influence of individual microbial species in disease relative to their abundance or influence in health. Dysbiotic inflammatory disorders include periodontitis and inflammatory bowel disease.
- Gingival crevicular fluid (GCF)
Serum exudate originating in the gingival capillaries and flowing into the gingival crevice (periodontal pocket). The quantity of this fluid increases with increasing inflammation and contains an abundance of neutrophils as well as locally produced immune and inflammatory mediators (e.g. complement activation fragments, antimicrobial peptides, cytokines).
- Host modulation
Aims to alter the status or function of the host to treat a disease. In an inflammatory disease, host modulation entails efforts to manipulate the immune response to prevent or mitigate tissue damage. In periodontitis, the purpose is to disrupt a chronic, self-sustained, vicious cycle linking and reinforcing microbial dysbiosis and destructive inflammation.
- Ligature-induced periodontitis
Experimental model (small and large animals) involving the placement of a silk ligature around teeth. Ligature placement generates a subgingival biofilm-retentive milieu leading to microbial dysbiosis, gingival inflammation and bone loss, simulating periodontitis.
- Microbiome
Collective term for a diverse microbial community (microbiota) and its combined genetic material and functions within a defined anatomical niche (e.g., the subgingival tooth surface).
- Neutrophil extracellular traps
Web-like chromatin scaffolds that concentrate released antimicrobial molecules and proteins; can potentially trap and kill extracellular microorganisms.
- Membrane attack complex
Assembly of fluid-phase complement proteins into a complex that can form a pore across the cell membrane, leading to lysis of susceptible microbial pathogens.
- Pattern-recognition receptors
Germline-encoded receptors typically found in innate immune cells; recognize molecular motifs from microbes (‘microbe-associated molecular patterns’) or molecules released by damaged host cells (‘damage-associated molecular patterns’).
- T helper 1 (Th1) and Th17 cells
Specific subsets of pro-inflammatory CD4+ T helper cells defined by their production of interferon-γ (Th1) or interleukin 17 (Th17).
- Toll-like receptors
Family of pattern-recognition receptors; can collectively respond to broad classes of microbes; each member recognizes distinct molecular motifs conserved within specific microbial classes.
- Periodontal pocket
Physiological narrow gap between the root of the teeth and the free gingiva, known as subgingival crevice; this crevice deepens during periodontitis progression: ‘periodontal pocket’.
- Periodontal therapy
Involves mechanical debridement (‘scaling and root planing’) to remove the microbial biofilm (dental plaque) and calculus (tartar) from tooth surfaces, including beneath the gingiva. This facilitates inflammation resolution, and smoothens dental root surfaces to deter further buildup of dental plaque and calculus.
- Porphyromonas gingivalis
Gram-negative anaerobic and inflammophilic bacterium implicated in human periodontal disease. From mouse model mechanistic studies, this bacterium is thought to act as a keystone pathogen, i.e., a species whose influence on its community is disproportionate relative to its abundance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs such as AMY-101). J.D.L. is inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. J.D.L. and G.H. have a joint patent that describes the use of complement inhibitors for therapeutic purposes in periodontitis. J.D.L. is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives such as APL-2/pegcetacoplan/Empaveli). The other authors declare no competing interest.
Resources
This trial is listed in https://clinicaltrials.gov/ct2/show/NCT03316521
This trial is listed in https://clinicaltrials.gov/ct2/show/NCT03694444
References
- 1.Kinane DF et al. (2017) Periodontal diseases. Nat Rev Dis Primers 3, article No.17038. [DOI] [PubMed] [Google Scholar]
- 2.Eke PI et al. (2015) Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J Periodontol 86 (5), 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frencken JE et al. (2017) Global epidemiology of dental caries and severe periodontitis - a comprehensive review. J Clin Periodontol 44 (S18), S94–S105. [DOI] [PubMed] [Google Scholar]
- 4.Kassebaum NJ et al. (2014) Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res 93 (11), 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brauchle F et al. (2013) Impact of periodontal disease and periodontal therapy on oral health-related quality of life. Int Dent J 63 (6), 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapple IL (2014) Time to take periodontitis seriously. BMJ 348, g2645. [DOI] [PubMed] [Google Scholar]
- 7.Peres MA et al. (2019) Oral diseases: a global public health challenge. The Lancet 394 (10194), 249–260. [DOI] [PubMed] [Google Scholar]
- 8.Saito A et al. (2010) Effect of initial periodontal therapy on oral health-related quality of life in patients with periodontitis in Japan. J Periodontol 81 (7), 1001–1009. [DOI] [PubMed] [Google Scholar]
- 9.GBD 2017 Disease and Injury Incidence and Prevalence Collaborators (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392 (10159), 1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajishengallis G and Chavakis T (2021) Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol 21 (7), 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schenkein HA et al. (2020) Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000 83 (1), 90–106. [DOI] [PubMed] [Google Scholar]
- 12.Tonetti MS et al. (2011) Biological approaches to the development of novel periodontal therapies--consensus of the Seventh European Workshop on Periodontology. J Clin Periodontol 38 Suppl 11, 114–118. [DOI] [PubMed] [Google Scholar]
- 13.Tonetti MS et al. (2017) Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J Clin Periodontol 44 (5), 456–462. [DOI] [PubMed] [Google Scholar]
- 14.Righolt AJ et al. (2018) Global-, Regional-, and Country-Level Economic Impacts of Dental Diseases in 2015. J Dent Res 97 (5), 501–507. [DOI] [PubMed] [Google Scholar]
- 15.Botelho J et al. (2021) Economic burden of periodontitis in the United States of America and Europe – an updated estimation. J Periodontol Online ahead of print: doi: 10.1111/jper.10813. [DOI] [PubMed] [Google Scholar]
- 16.Balta MG et al. (2021) Host Modulation and Treatment of Periodontal Disease. J Dent Res 100 Online ahead of print: doi: 10.1177/0022034521995157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hajishengallis G et al. (2017) Novel mechanisms and functions of complement. Nat Immunol 18 (12), 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekdahl KN et al. (2016) Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev 274 (1), 245–269. [DOI] [PubMed] [Google Scholar]
- 19.Hajishengallis G and Lambris JD (2016) More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev 274 (1), 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeley S et al. (2016) The “ins and outs” of complement-driven immune responses. Immunol Rev 274 (1), 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricklin D et al. (2016) Complement in disease: a defence system turning offensive. Nat Rev Nephrol 12 (7), 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mödinger Y et al. (2018) Complement involvement in bone homeostasis and bone disorders. Sem Immunol 37, 53–65. [DOI] [PubMed] [Google Scholar]
- 23.Ricklin D et al. (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11 (9), 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mastellos DC et al. (2019) Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov 18 (9), 707–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Courts FJ et al. (1977) Detection of functional complement components in gingival crevicular fluid from humans with periodontal diseases. J Dent Res 56 (3), 327–331. [DOI] [PubMed] [Google Scholar]
- 26.Schenkein HA and Genco RJ (1977) Gingival fluid and serum in periodontal diseases. II. Evidence for cleavage of complement components C3, C3 proactivator (factor B) and C4 in gingival fluid. J Periodontol 48 (12), 778–784. [DOI] [PubMed] [Google Scholar]
- 27.Niekrash CE and Patters MR (1986) Assessment of complement cleavage in gingival fluid in humans with and without periodontal disease. J Periodontal Res 21 (3), 233–242. [DOI] [PubMed] [Google Scholar]
- 28.Nikolopoulou-Papaconstantinou AA et al. (1987) Deposits of immunoglobulins, complement, and immune complexes in inflamed human gingiva. Acta Odontol Scand 45 (3), 187–193. [DOI] [PubMed] [Google Scholar]
- 29.Patters MR et al. (1989) Assessment of complement cleavage in gingival fluid during experimental gingivitis in man. J Clin Periodontol 16 (1), 33–37. [DOI] [PubMed] [Google Scholar]
- 30.Niekrash CE and Patters MR (1985) Simultaneous assessment of complement components C3, C4, and B and their cleavage products in human gingival fluid. II. Longitudinal changes during periodontal therapy. J Periodontal Res 20 (3), 268–275. [DOI] [PubMed] [Google Scholar]
- 31.Grande MA et al. (2021) Complement split product C3c in saliva as biomarker for periodontitis and response to periodontal treatment. J Periodontal Res 56 (1), 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hajishengallis G et al. (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10 (5), 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maekawa T et al. (2014) Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J. Immunol 192 (12), 6020–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allam JP et al. (2011) IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol 38 (10), 879–886. [DOI] [PubMed] [Google Scholar]
- 35.Dutzan N et al. (2018) A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med 10 (463), eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu H et al. (2013) New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 218 (4), 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maekawa T et al. (2016) Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol 43, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bostanci N et al. (2018) Gingival Exudatome Dynamics Implicate Inhibition of the Alternative Complement Pathway in the Protective Action of the C3 Inhibitor Cp40 in Nonhuman Primate Periodontitis. J Proteome Res 17 (9), 3153–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley K et al. (2018) Neutrophils: New insights and open questions. Sci Immunol 3 (30), eaat4579. [DOI] [PubMed] [Google Scholar]
- 40.Mastellos DC et al. (2020) Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin Immunol 220, 108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kajikawa T et al. (2017) Safety and Efficacy of the Complement Inhibitor AMY-101 in a Natural Model of Periodontitis in Non-human Primates. Mol Ther Methods Clin Dev 6, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Risitano AM et al. (2014) Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood 123 (13), 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reis ES et al. (2018) Safety profile after prolonged C3 inhibition. Clin Immunol 197, 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullard A (2021) First approval of a complement C3 inhibitor opens up autoimmune and inflammatory opportunities. Nat Rev Drug Discov 20, doi: 10.1038/d41573-41021-00094-41578. [DOI] [PubMed] [Google Scholar]
- 45.Wang M et al. (2010) Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal 3 (109), ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maekawa T et al. (2014) Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 15 (6), 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller DP et al. (2014) Analysis of the complement sensitivity of oral treponemes and the potential influence of FH binding, FH cleavage and dentilisin activity on the pathogenesis of periodontal disease. Mol Oral Microbiol 29 (5), 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slaney JM et al. (2006) Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infect. Immun 74 (9), 5352–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilian M et al. (2016) The oral microbiome - an update for oral healthcare professionals. Br Dent J 221 (10), 657–666. [DOI] [PubMed] [Google Scholar]
- 50.Lamont RJ et al. (2018) The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol 16 (12), 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amyndas (2021) https://www.businesswire.com/news/home/20210319005042/en/Amyndas-Announces-Positive-Top-Line-Phase-2-Results-for-Investigational-AMY-101-in-Adults-With-Periodontal-Inflammation-and-Gingivitis.
- 52.Hajishengallis G et al. (2019) Complement-Dependent Mechanisms and Interventions in Periodontal Disease. Front Immunol 10, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramseier CA et al. (2017) Natural history of periodontitis: Disease progression and tooth loss over 40 years. J Clin Periodontol 44 (12), 1182–1191. [DOI] [PubMed] [Google Scholar]
- 54.Lang NP et al. (1990) Absence of bleeding on probing. An indicator of periodontal stability. J Clin Periodontol 17 (10), 714–721. [DOI] [PubMed] [Google Scholar]
- 55.Tsukasaki M et al. (2018) Host defense against oral microbiota by bone-damaging T cells. Nat Commun 9, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abe T et al. (2012) Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 189 (11), 5442–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]