

Summary
3’ untranslated region (3’UTR) variants are strongly associated with human traits and diseases, yet few have been causally identified. We developed the Massively Parallel Reporter Assay for 3’UTRs (MPRAu) to sensitively assay 12,173 3’UTR variants. We applied MPRAu to six human cell lines, focusing on genetic variants associated with genome-wide association studies (GWAS) and human evolutionary adaptation. MPRAu expands our understanding of 3’UTR function, suggesting that simple sequences predominately explain 3’UTR regulatory activity. We adapt MPRAu to uncover diverse molecular mechanisms at base-pair resolution, including an AU-rich element of LEPR linked to potential metabolic evolutionary adaptations in East Asians. We nominate hundreds of 3’UTR causal variants with genetically fine-mapped phenotype associations. Using endogenous allelic replacements, we characterize one variant that disrupts a miRNA site regulating the viral defense gene TRIM14, and one that alters PILRB abundance, nominating a causal variant underlying transcriptional changes in age-related macular degeneration.
Graphical Abstract
In Brief
Massively Parallel Reporter Assay for 3′UTRs measures individual regulatory effects of over 12,000 3′UTR variants associated with human disease and evolutionary selection in many cell types, nominating functional genetic variation.
Introduction
Over the past two decades, thousands of variant-trait associations have been identified by genome-wide association studies (GWAS) (Buniello et al., 2019). However, GWAS have been hindered in elucidating the mechanisms of complex disease by two limitations: (1) linkage disequilibrium (LD), the association between alleles at different loci due to strong genetic linkage, causes neighboring neutral polymorphisms to display similarly strong associations as causal loci, greatly increasing the experimental burden for functional validation; (2) over 90% of associations reside in non-coding regions of the genome (Gusev et al., 2014; Maurano et al., 2012), where functional interpretation is much more difficult than in coding regions.
3′ untranslated regions (3’UTRs) contain a particularly important class of noncoding variants that can impact post-transcriptional and translational processes. Causal peripheral blood cis-expression Quantitative Trait Loci (eQTL) variants are 4-fold enriched to be in 3’UTRs, a level matching that of promoter elements (Wang et al., 2020b). Across all tissues in the Genotype-Tissue Expression project (GTEx), eQTLs in 3’UTRs are found to be 2-fold enriched, the largest enrichment amongst all non-coding regions (The GTEx Consortium, 2020). Untranslated regions harbor the largest enrichment of GWAS heritability (5-fold) of all non-coding categories except for transcription start sites, emphasizing a substantial role for post-transcriptional activities in human regulatory variation (Finucane et al., 2015).
Although 3’UTR variants are crucial to understanding human phenotypic variation, only a handful of causal 3’UTR variants have been described. They include BAFF-var in TNFSF13B associated with lupus and multiple sclerosis (Steri et al., 2017), rs13702 in LPL associated with HDL-cholesterol levels (Richardson et al., 2013), and rs12190287 in TCF21 associated with coronary artery disease (Miller et al., 2014). Each case required meta-analysis across multiple trait or population datasets and annotation with well-known regulatory factors, before being pursued with low-throughput luciferase confirmations. These factors demonstrate how current 3’UTR causal variant discovery is burdensome and highlight the need for high-throughput tools to characterize the functional impact of 3’UTR variants on gene expression.
The development of Massively Parallel Reporter Assays (MPRAs) has enabled simultaneous testing of thousands of variants for cis-regulatory activity to nominate causal variants in non-coding regions (van Arensbergen et al., 2019; Choi et al., 2020; Kircher et al., 2019; Klein et al., 2019; Liu et al., 2017; Sample et al., 2019; Tewhey et al., 2016; Ulirsch et al., 2016), Historically MPRA has been primarily applied to understand transcriptional regulation. Several studies have adapted MPRA to test 3’UTR sequences, but genetic variation in 3’UTRs still needs further characterization (Bogard et al., 2019; Litterman et al., 2019; Oikonomou et al., 2014; Siegel et al., 2020; Vainberg Slutskin et al., 2018, 2019; Zhao et al., 2014).
Here, we developed the Massively Parallel Reporter Assay for 3’UTRs (MPRAu) to quantify allelic expression differences for thousands of 3’UTR variants simultaneously in a high-throughput, accurate, and reproducible manner. MPRAu detects distinct aspects of 3’UTR regulation, allowing us to understand general sequence features governing transcript abundance via computational modeling, pinpoint exact sequence architectures underlying variant functionality including RNA structure and RNA-binding protein (RBP) occupancy, and nominate causal variants. We utilize MPRAu to comprehensively test disease-associated, as well as evolutionarily adaptive, 3’UTR genetic variation in six human cell lines. From our functionally nominated causal variants, we also more deeply characterize two variants using CRISPR-induced allelic replacement.
Results
MPRAu reproducibly characterizes the functions of thousands of 3’UTR elements
We applied MPRAu to systematically evaluate the functional effects of genetic variation from 3’UTRs. To do so, we designed and synthesized 100 base pair (bp) oligonucleotides derived from human 3’UTRs, centered on, and differing only with respect to the variant’s ‘reference’ (ref) or ‘alternate’ (alt) alleles (Fig. 1a), for testing using MPRAu. We cloned the oligo pool into the 3’UTR of a plasmid reporter gene controlled by a moderately strong promoter. By transfecting our pool into cell lines of interest, and sequencing both the plasmid pool and mRNA from cells, we could compare steady-state RNA expression effects of each 3’UTR oligonucleotide (from either differential mRNA decay or transcription). We refer to 3’UTR oligo backgrounds that increase mRNA levels as having ‘augmenting’ effects and those that decrease transcript levels as having ‘attenuating’ effects. We also quantify differences between sequences bearing the ref versus alt allele and refer to alleles with a statistically significant ‘allelic skew’ as transcript abundance-modulating variants (tamVars). In addition, MPRAu employs several quality controls to minimize bias, including using random barcodes to ensure adequate library complexity (Methods).
Figure 1: MPRAu reproducibly recapitulates known 3’UTR activity.
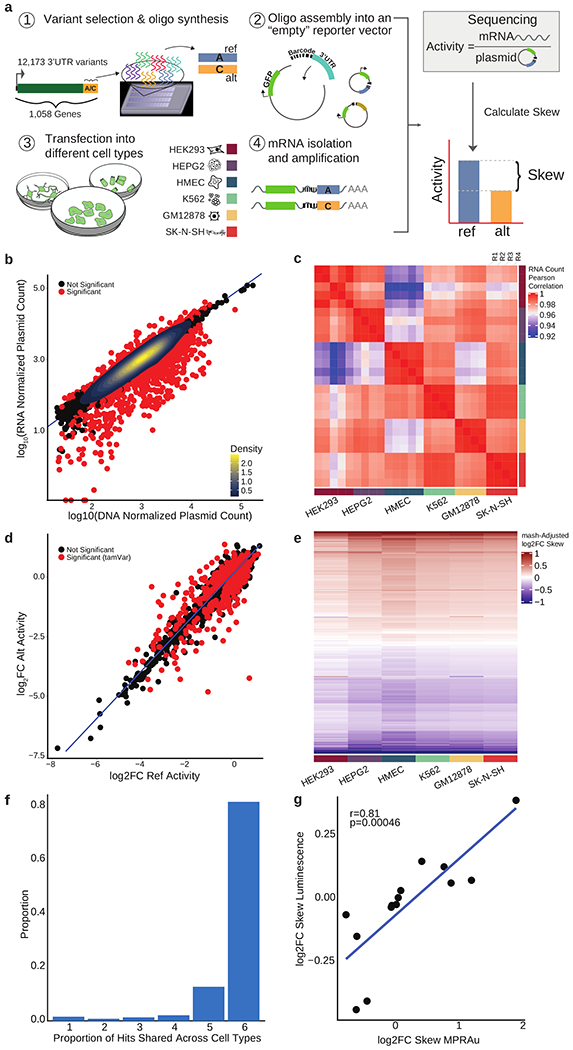
a, Overview of MPRAu: (1) Synthesis of oligonucleotide 3’UTR elements with genomic variants. (2) Oligos are PCR-amplified and cloned into a vector 3’ of GFP and adjacent to a random hexamer barcode. (3) The vector pool is transfected into cells, (4) GFP mRNA is extracted and sequenced. mRNA sequencing counts (4) are compared to plasmid counts (2) to determine the relative expression of ref and alt alleles. b, Scatterplot of normalized RNA versus DNA counts in HEK293 cells. Most oligos with significant activity are observed having attenuating effects. c, Heatmap of the pairwise correlation of RNA counts across all replicates. d, Identification of tamVars (red), variants with significantly different alt versus ref activity (data for HEK293 plotted). e, mash adjusted log2FC allelic skews for all variants with significant effects in at least one cell type (rows) across all tested cell types. f, Barplot depicting tamVar sharing across one to all six cell types. g, tamVar allelic skews concordance with low-throughput luciferase assays. Pearson’s r and its statistical significance are displayed.
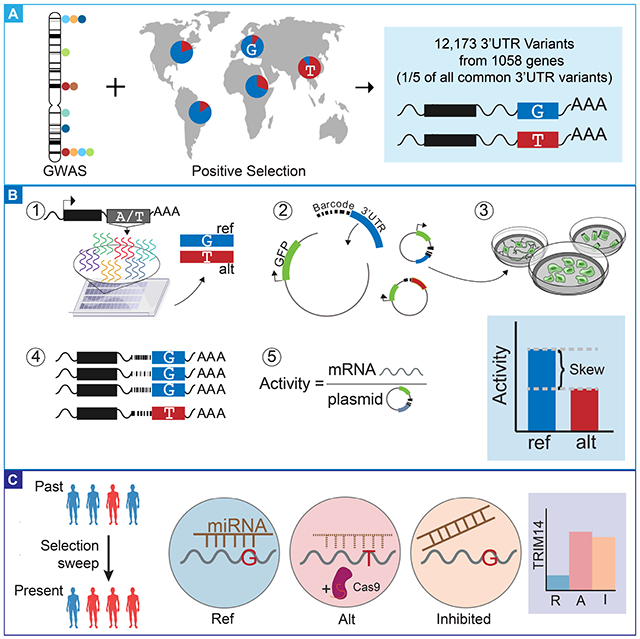
We applied MPRAu to identify functional 3’UTR variants associated with human disease and evolutionary selection, testing 12,173 3’UTR variants. As the causal variant(s) underlying human traits and diseases can be amongst many variants associated with GWAS tagging (tag) SNPs, we tested 3’UTR SNPs and insertion/deletions (indels) (minor allele frequency (MAF)≧5%) in strong genetic linkage, LD, with tag SNPs (LD threshold: minimum r2=0.8) from the NHGRI-EBI GWAS catalog (Welter et al., 2014), totaling 2,153 putative disease-associated variants from 1,556 independent association loci (Supplementary Table 1). We also incorporated a set of 9,325 3’UTR SNPs and indels overlapping regions identified as being under positive selection in humans (Grossman et al., 2013) (Supplementary Table 1). We also included a set of 46 rare 3’UTR variants (minor allele frequency (MAF) ≤0.01 in Europeans) that are in genes with outlier expression signatures across tissues in the Genotype-Tissue Expression (GTEx) project, which are known to have potential deleterious consequences (Li et al., 2017) (Supplementary Table 1). Lastly, across all tested variants, 2,955 were also tested under alternative allelic backgrounds to account for the potential effect of surrounding sequence variants. As genetic variants impacting traits can have tissue-specific effects (GTEx Consortium, 2017; Marbach et al., 2016; Parker et al., 2013), we characterized these variants across six diverse human cellular lines: HEK293 (embryonic kidney), HepG2 (hepatocellular carcinoma), GM12878 (lymphoblastoid), SK-N-SH (neuroblastoma), K562 (leukemia), and a primary cell line (HMEC, mammary epithelial).
We first sought to ensure that our assay was reproducibly capturing expected 3’UTR biological effects. Consistent with the dominant regulatory function of 3’UTRs to attenuate transcript expression, the predominant effect across all MPRAu-tested 3’UTRs was to decrease mRNA abundance (Fig. 1b). This effect is reproducibly observed in the strong correlation of normalized RNA read counts between experimental replicates across all cell types (average Pearson correlation (corr)=0.99) (Fig. 1c).
Confident in our assay’s ability to assess oligos with regulatory activity, we then identified tamVars altering 3’UTR functionality by comparing expression changes between alleles of the same 3’UTR (using as a threshold a Benjamini-Hochberg adjusted p-value (BH p-adj)<0.1) (Fig. 1d). We found 2,368 tamVars in total across all cell types (Supplementary Table 1). To assess cell-specificity of tamVars, we applied mash (Urbut et al., 2019), finding tamVars were largely shared across all six cell types (81.2%) vs specific to one cell type (1.6%) (Fig. 1e,f, Supplementary Fig. 2a,b). Out of the 2,955 variants tested with alternative allelic backgrounds, only 10 tamVars displayed function dependent on its allelic background. To confirm that 3’UTR tamVar effects were reflected in protein levels, we utilized orthogonal methods to measure allelic effects. We performed polysome profiling on HEK293 cells transfected with a subset of our tested library and found polysomal RNA expression highly correlated with steady-state RNA expression (Pearson corr=0.94, p=2.68x10−11,546) (Supplementary Fig. 1a). In addition, variant effects between polysomal and steady-state RNA were concordant (Pearson corr=0.97, p=1.4x10−106) (Supplementary Fig. 1b), recapitulating previous findings that steady-state RNA levels are a good proxy for protein levels (Oikonomou et al., 2014; Zhao et al., 2014). To demonstrate that variant effects are directly translatable to the protein level, we also tested a subset of tamVars and non-tamVar controls via luciferase assays (Supplementary Table 2). We observed a strong correlation between luciferase luminescence assays and the MPRAu-measured allelic skew (Pearson corr=0.81, p=4.6x10−4) (Fig. 1g) across a wide range effect sizes (between 0 and 2 log2FC Skew), which also highlights MPRAu’s ability to triage 3’UTR variant effects over low-throughput luciferase approaches. Together, polysome profiling and luciferase concordance with MPRAu suggests that the assay’s RNA abundance measurements are meaningful at the phenotypic level.
MPRAu sensitively detects 3’UTR regulators and functional sequence variants
To confirm that MPRAu effects are consistent with molecular mechanisms underlying 3’UTR biology, we analyzed features across the entire oligo sequence. We found that GC content and secondary structure, as measured by the predicted minimal free energy, positively correlated with the level of attenuation (percent GC: Pearson corr=−0.24, p=6.59x10−384, mfe: Pearson corr=0.27, p=8.87x10−485) (Fig. 2a). This finding may be explained by the role of high GC content and structuredness in RBP occupancy and therefore functionality (Dominguez et al., 2018; Litterman et al., 2019).
Figure 2: Functional 3’UTR elements overlap known 3’UTR annotations.
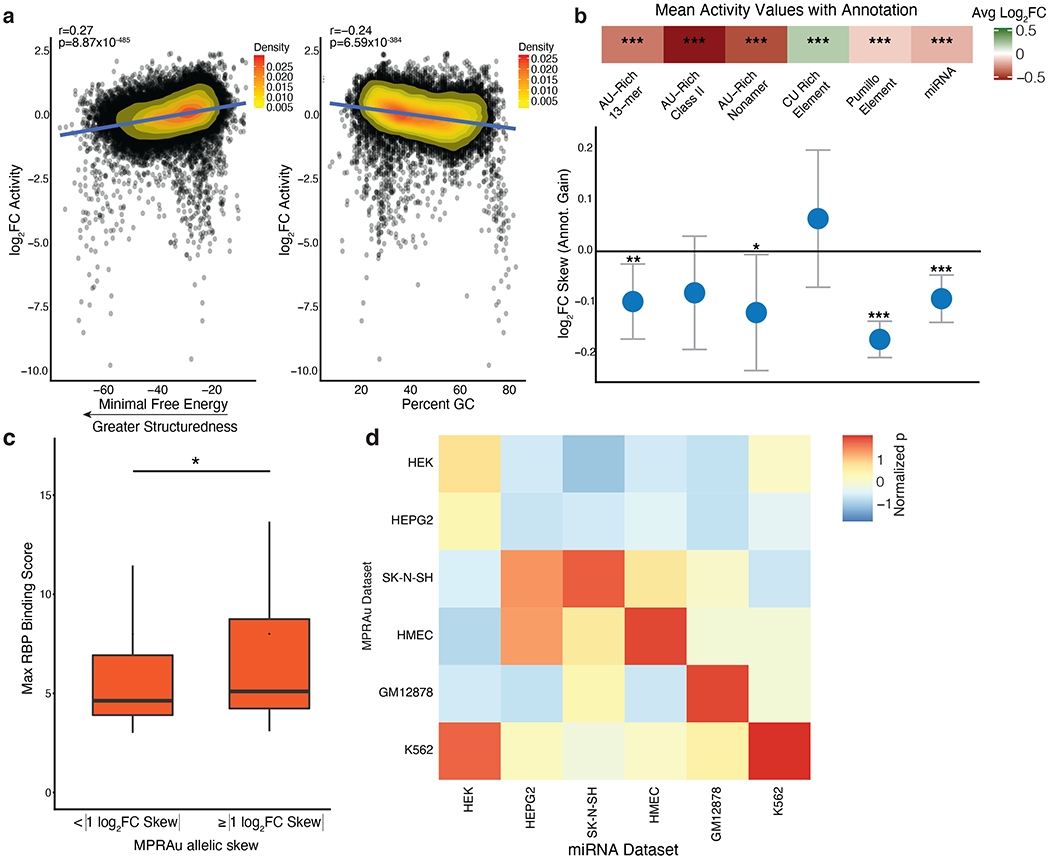
a, Scatterplot comparing correlations between activity effects and 3’UTR structure (more negative minimum free energy, left) and percent GC (right). HMEC log2FC activity data is representative and plotted. Pearson’s r and its statistical significance are displayed. b, (top panel) Enrichment of significantly active 3’UTRs with known 3’UTR attenuating (AU-rich, Pumillo, and miRNA) and augmenting (CU-rich Element) annotations. Average log2FC of 3’UTR activity in MPRAu across all cell types with specified annotation plotted, significance denoted as *** p-value<0.001, ** p-value<0.01, * p-value<0.05, using a two-sided Wilcoxon rank-sum test. (bottom panel) Barplot of the allelic skews for variants that acquire 3’UTR annotations of the class listed above. c, Variants with high allelic skew (| log2FC Skew |≥1 ) have greater in-vivo eCLIP RBP binding scores than variants with a lesser allelic skew (* p-value<0.05, using a one-sided Wilcoxon rank-sum test). d, Each box in the heatmap measures the significance of attenuation (t-test) when cell-type-specific MPRAu-measured 3’UTR activity (y-axis) is subsetted on the top 10 most abundant miRNAs across the cell types tested (x-axis). Across 4 cell types (K562, HMEC, GM12878, SK-N-SH), 3’UTR activity was most significantly attenuated when subsetting on cell-type matched miRNAs.
At the finer sequence level, MPRAu captured the expected attenuation and augmentation effects from the presence of empirical RBP motifs (Bakheet et al., 2001; Holcik and Liebhaber, 1997; White et al., 2001) and predicted miRNA motifs (Friedländer et al., 2012; Friedman et al., 2009). Regulatory signatures such as AU-rich elements, the canonical Pumilio motif, and miRNAs motifs exhibited expected attenuating effects on expression (avg log2 fold change (log2FC) range −0.55 to −0.12, two-sided t-test p<1x10−30 for all attenuating factors) (Fig. 2b). Conversely, CU-rich elements demonstrated their expected augmenting effects on expression (avg log2FC=0.18, two-sided t-test p=1.58x10−14) (Fig. 2b). Variants perturbing these predicted elements abrogate the functional effect (two-sided t-test p<0.05 for all factors except for CU Rich Element and AU-Rich Class II, but skew directionality is consistent for the latter two) (Fig. 2b). Highlighting MPRAu’s ability to capture endogenous perturbation effects, oligos with high background activities (| log 2 FC Activity |≥1) and variants with high allelic skews (| log 2 FC Skew |≥1) also are predominantly in regions with elevated in vivo RBP occupancy signatures (Van Nostrand et al., 2016) (one-sided Wilcoxon rank-sum test p=0.026 for activity, p=0.01 for allelic skew, HepG2 and K562 combined) (Fig. 2c). While 3’UTR effects in MPRAu were found to be strongly correlated across cell types (Fig. 1c), differential expression of trans-acting factors between cell types would be expected to contribute to any cell-type-specific effects that may be present. miRNAs are one such trans-acting factor, and the top 10 expressed miRNAs in each cell line had sequence motifs across 1-3% of our tested sequences. We found attenuation of transcript levels and allelic skew in a given cell line were generally best explained by the miRNAs abundantly expressed within that cell line, rather than miRNAs abundantly expressed in a different cell line, demonstrating the capacity of MPRAu to capture even relatively rare cell-type-specific effects (Fig. 2d, Supplementary Fig. 3a). Lastly, an orthogonal analysis of the barcodes in our MPRAu design also validated the effects of known 3’UTR RBP motifs and provides a valuable resource of potential functional hexamers (Methods, Supplementary Fig. 3b–d, Supplementary Table 3). Together these data demonstrate the ability of MPRAu to detect the effects of known 3’UTR regulators and cell-type-specific regulatory mechanisms.
Computational modeling uncovers features of 3’UTR regulation
Having identified key 3’UTR features underlying MPRAu transcript levels, we trained predictive models for our tested sequences and compared the sensitivity and specificity of several classification models to predict 3’UTR elements with attenuated activity (< −0.5 3’UTR log2FC) (Methods). We used 34 total simple, sequence-specific annotations in our initial models, including features derived from nucleotide and dinucleotide composition, homopolymer content, and secondary structure (Methods). Our best model performed well in all cell types, with an average precision of 0.23-0.48 and an area under the receiver operating characteristic curve of 0.67-0.79 (Fig. 3a,b). Notably, our best model (trained via xgboost) performed better than several other classification models tested, including a one-variable control model using percent U (Supplementary Fig. 4a–d). We also used the same features to predict augmenting expression (>0.5 log2FC) and found comparable performance (Supplementary Fig. 4e).
Figure 3: Computational modeling of 3’UTR activity uncovers features important for accurate prediction.
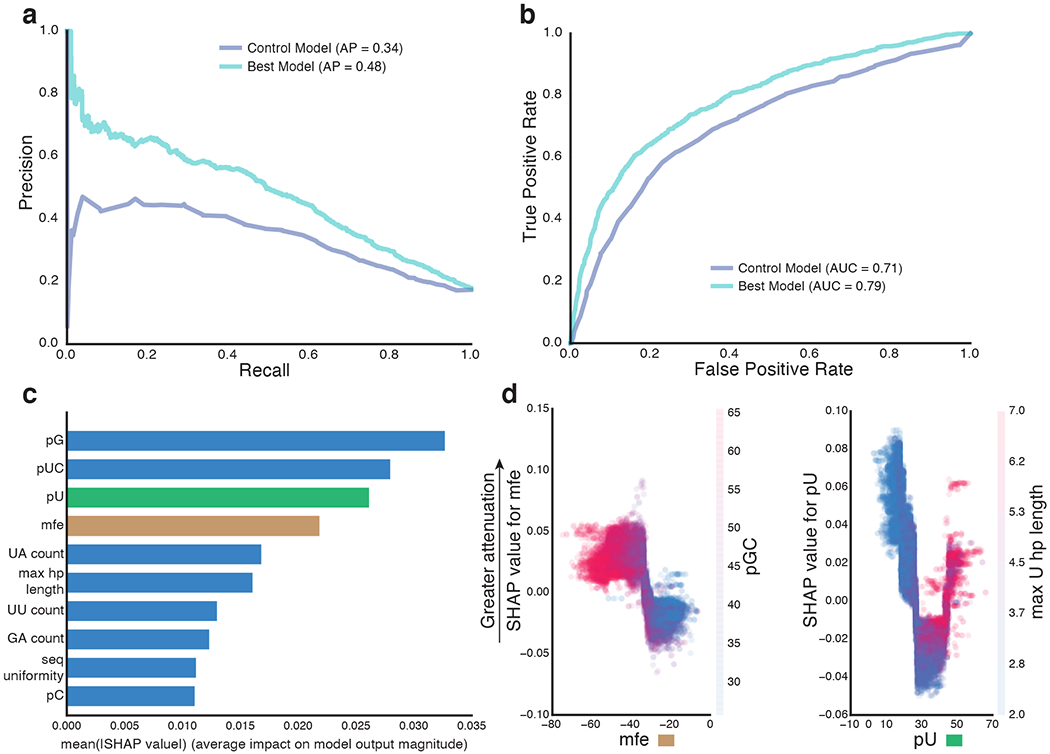
Precision-recall (a) and receiver operating characteristic curve (b) for the best model (xgboost) trained on predicting 3’UTR activity attenuation (data displayed is for HMEC). The results from a control model (one-variable decision tree model using percent U) are also plotted for comparison. c, Plot of the top 10 most important predictor variables of model performance, ranked by mean (| SHAP value |). d, SHAP values of minimal free energy (mfe, left) and percent U (pU, right) compared to that variable’s magnitude. Greater SHAP values indicate higher impact on model prediction towards attenuation. The magnitude of related variables, percent GC (pGC, left) and max U homopolymer (hp) length (right), are depicted by the color scale. mfe displays a monotonic effect on attenuation (left), with high pGC associated with attenuation and low mfe (blue). pU displays a nonlinear effect on attenuation (right), with attenuation especially observed in sequences with long homopolymer Us (red).
Because our initial model included only cell-type agnostic simple sequence features, we expanded our model to incorporate additional features related to cell-type-specific miRNA and RBP occupancy sites. Our expanded feature set includes individual motifs from the top 100 expressed miRNAs for each cell type, as well as aggregated statistics across these highly expressed miRNAs. Furthermore, we also included RNAcontext-k-mer scores from expressed RBPs for each cell type (Methods). However, these features did not improve model accuracy (Supplementary Fig. 4f), suggesting that simple, cell type-agnostic sequence features explain the predominant proportion of regulatory activity in 3’UTRs in the 100 bp context of our assay. This finding is consistent with recent studies showing that RBPs tend to recognize relatively simple sequence motifs (Dominguez et al., 2018; Litterman et al., 2019). Our observation that 84.3% of the tested 3’UTR elements with significant activity in MPRAu are shared across multiple cell types is concordant with this result. A model trained only on miRNA/RBP features achieved similar performance compared to our model with simple sequence variables (Supplementary Fig. 4g), suggesting these simple sequence features are sufficient for prediction.
From our models that utilized only sequence-specific annotations, several features were found to be important for prediction using SHapley Additive exPlanations (SHAP) (Lundberg and Lee, 2017). These features included homopolymer length, sequence diversity, and various features related to uracil content such as percent U/UC and UA/UU dinucleotide count (Fig. 3c, Supplementary Fig. 4h). Investigating the individual effects of some of these important features, we found minimal free energy (mfe) monotonically negatively correlated with attenuation prediction. We also found percent GC to be anticorrelated with mfe and therefore positively correlated with attenuation (Fig. 3d). Surprisingly, we discovered the proportion of uracil content to have a nonlinear effect on attenuation with both low and high uracil content displaying attenuating effects. Specifically, longer uracil homopolymers demonstrate the most attenuating activity (Fig. 3d), concordant with their function as binding motifs for many RBPs (Dominguez et al., 2018; Mukherjee et al., 2019). The identification of attenuation features may be useful for constructing synthetic 3’UTRs with precise expression levels.
MPRAu allelic effects are reflected in gene expression and human phenotype changes
After demonstrating MPRAu’s ability to detect the activity of 3’UTR elements, we asked whether our tamVar allelic effects are supported by causal alleles altering transcriptional outputs and/or phenotypic traits captured by the UK Biobank. We compared tamVars in GM12878 to cell-type matched allele-specific expression (ASE) data from heterozygous individuals in the Geuvadis RNA-seq dataset (Lappalainen et al., 2013) and used this comparison to estimate the positive predictive value (PPV) for tamVars in the assay. We observed moderately strong concordance between our tamVars and endogenously observed ASE (66.1% directionality agreement, binomial p=0.011) (Fig. 4a), which corresponds to a PPV of 32%. With higher stringency ASE calls (two-sided t-test p<0.001, Methods), agreement in directionality increased to 77.5% (binomial p=6.8x10−4, PPV of 55%). We obtained a weaker concordance when overlapping with Geuvadis expression quantitative trait loci (eQTL) data (60.5% agreement in directionality, binomial p=0.22, PPV of 20.9%) (Supplementary Fig. 5a), potentially due to varying regulatory factors (i.e. RBP/miRNA concentrations) muting true allelic effects when aggregating across individuals.
Figure 4: tamVars are responsible for gene expression and phenotype changes.
a, GM12878 tamVar allelic skew correlated with cell-type matched Geuvadis allelic skew. b, Correlation between all-tissue median eQTL allelic skew (posterior beta) in GTEx that have a high probability of being causal via genetic fine-mapping (PIP>0.2) and strong tamVar (BH p-adj<0.05) median skew across all cell types. PIP is an estimate for the probability of a variant to casually affect a gene expression change (measured from GTEx) or a phenotypic trait collected from the UK Biobank. c, GWAS variants from the UK Biobank with increasing PIP cutoffs display increasing enrichment (* p-value<0.05, Fisher’s exact test) for variants significant via MPRAu in at least one tested cell type. d, tamVar allelic skew correlation with RIVER score, a functionality estimate for rare variants (data for HMEC is representative and shown). Pearson’s r and its statistical significance are displayed.
Next, we expanded our analysis to compare tamVars with tissue eQTLs from GTEx (GTEx Consortium, 2013), where we obtained putative causal alleles from genetic fine-mapping (Benner et al., 2016; Ulirsch; Wang et al., 2020a) (Supplementary Table 4). Aggregating allelic effects across cell types and tissues, we observed variants with a high inferred probability of causality (posterior inclusion probability (PIP)>0.2) displayed significant agreement in directionality between aggregated MPRAu and GTEx median effect sizes (concordance=67%, binomial p=0.012) (Fig. 4b). Subsetting on known 3’UTR annotations improved the concordance (82%, binomial p=9.1x10−4). Even at a relaxed significance threshold, we still observed significant concordance (61%, binomial p=0.04), suggesting our findings are robust (Supplementary Fig. 5b). We demonstrate that MPRAu is highly specific to causal variants, as the agreement in directionality is lost at lower PIPs (Supplementary Fig. 5c). PPV estimates based on GTEx tissue-aggregated median effect sizes (34%-64%) are similar to those based on Geuvadis ASE (32%-55%).
We next looked for enrichment of MPRAu tamVars in genetically fine-mapped causal variants associated with 94 traits in the UK Biobank (Benner et al., 2016; Ulirsch; Wang et al., 2020a). We observed greater enrichment in MPRAu functionality with increasing causality (PIP) thresholds (two-sided t-test p=3.64x10−4) (Fig. 4c, Supplementary Table 4). This suggests that in addition to causing in vivo gene expression changes, the tamVars identified in our study have phenotypic consequences and that MPRAu is a powerful approach for dissecting association studies.
As an orthogonal approach to confirm the tamVars identified by MPRAu were correlated with in vivo expression changes, we also assayed a set of rare variants associated with large transcriptional effects (Li et al., 2017). When we compared MPRAu allelic skews with a rare variant functionality metric (RIVER score), we observed a significant positive correlation (Pearson corr=0.42, p=7.2x10−3) (Fig. 4d). This finding suggests MPRAu can identify functionality in common as well as rare 3’UTR variants, which modern association studies have a lower power to detect.
MPRAu SNV and deletion tiling dissects functional sequence motifs
To characterize the molecular mechanisms driving the regulatory effects of several highly significant tamVars with base-pair resolution, we created a MPRAu array that tiled single-nucleotide and deletion variants surrounding the variant (3’UTR tiling). Specifically, we extended MPRAu in two ways: we assayed 1) 5 bp non-overlapping deletions over the entire tested 3’UTR sequence, and 2) all single-nucleotide changes within +/− 10 bp of the tamVar. We carried out this deep analysis on 80 tamVars (Supplementary Table 1), of which three are described.
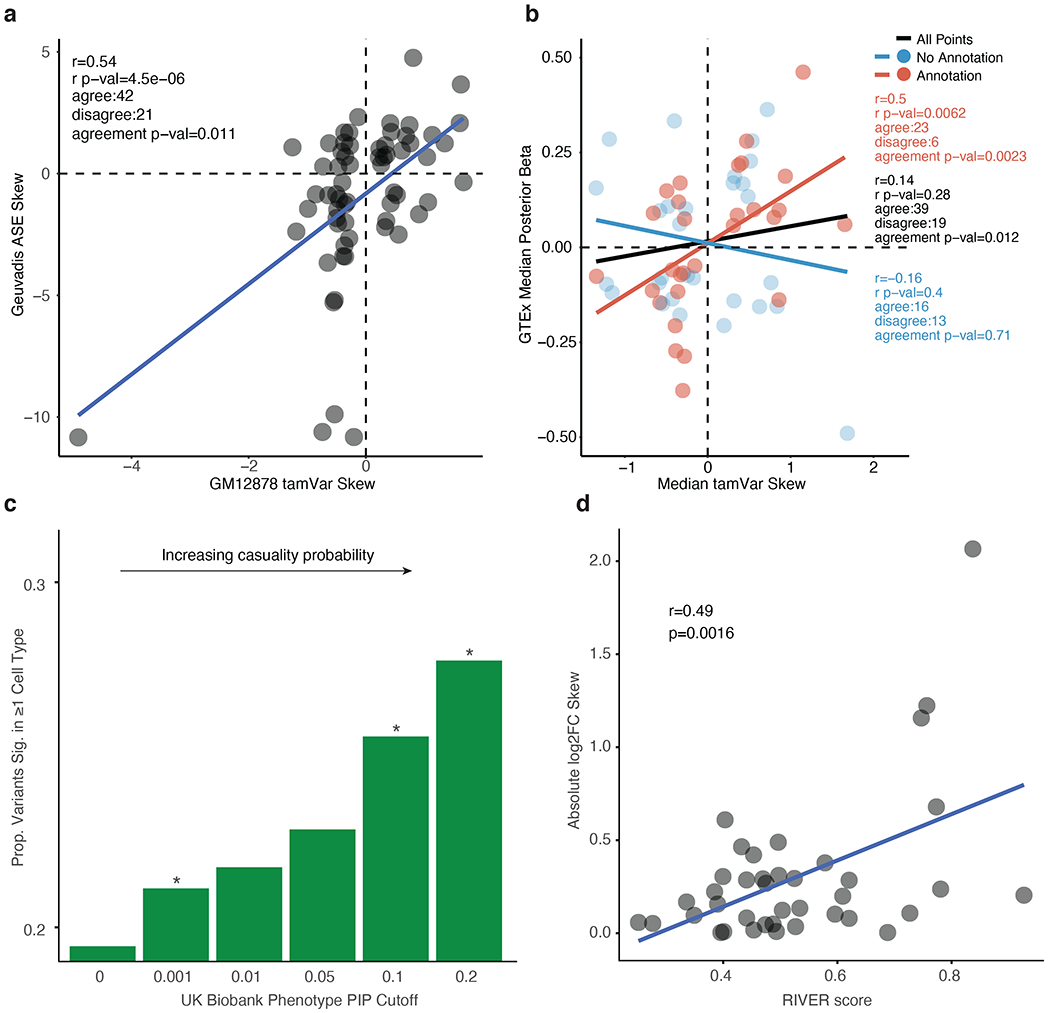
We found 3’UTR tiling useful to identify RBP sequence motifs. At rs16975240 (CIBAR2), which in our screen displayed a strong allelic skew (log2FC=2.06, BH p-adj=1.33x10−60), the ref allele demonstrated strong expression attenuation (log2FC=−2.29). However, the alt allele exhibited a muted effect (log2FC=−0.24), which suggests the perturbation of an attenuating element. Deletions between the variant position and 10 bps upstream on the ref background identified such an element that when removed, alleviated this attenuation (+3.55-4.46 FC increase). As expected in the alt background, deletions of the same sequence yielded minimal changes (+1.13-1.16 FC increase). Saturation mutagenesis identified a 10 bp motif in the ref region perfectly matching the U1 snRNP binding sequence consensus (Fig. 5a). The alt allele disrupts this motif potentially explaining the allelic skew, but functional interrogation at the endogenous locus is needed to confirm.
Figure 5: MPRAu uncovers functional sequence architectures via SNV and deletion tiling.
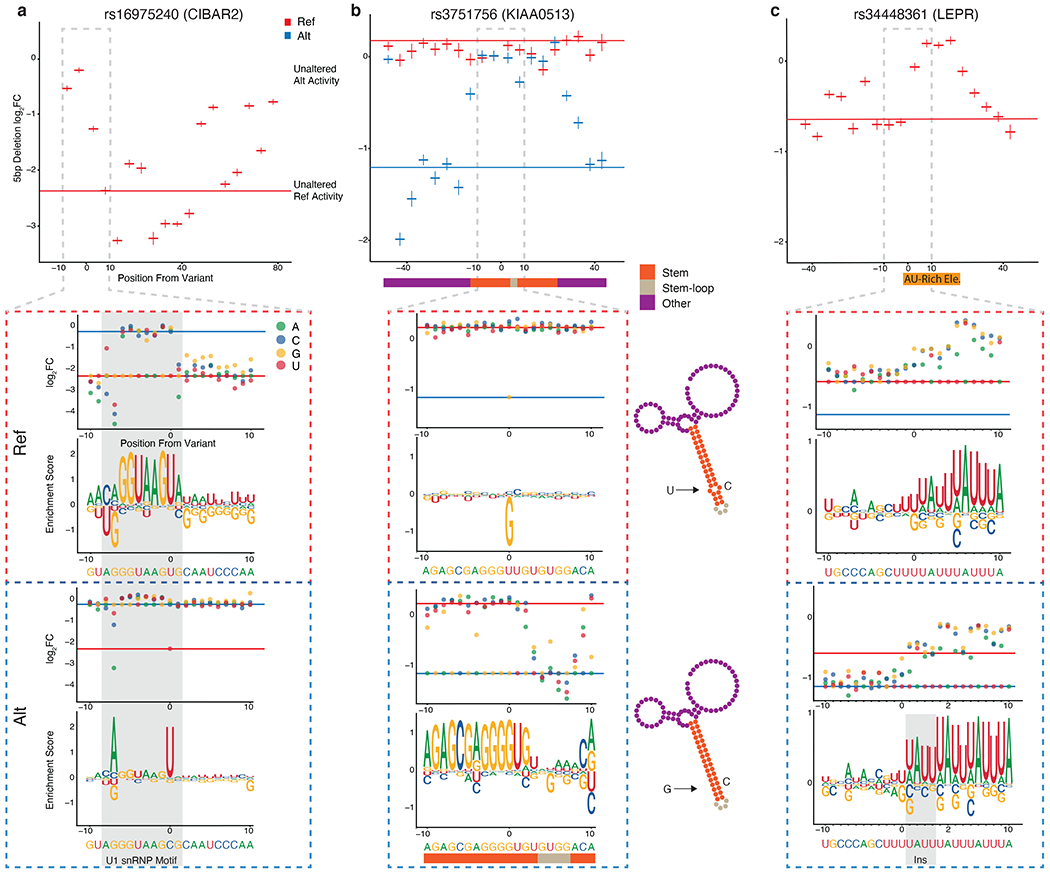
For a-c, Top, 5 bp deletion tiling of the targeted variant using the ref (red) or alt (blue) sequence context across 100 bp. Bottom, SNV tiling +/− 10 bp around each variant showing log2 fold change and motif enrichment score in both variant contexts. HEK293 3’UTR activity data is plotted for all panels. a, Plots for tiling at rs16975420 (CIBAR2), including the sequence motif for U1 snRNP (gray shading). b, Plots for tiling at rs3751756 (KIAA0513), overlaid with a predicted structure of the RNA element (bottom middle). Large magnitude changes observed in SNV tiling are predicted to disrupt bases in the stem structure. c, Plots for tiling at rs34448361 (LEPR) with an AU-rich element denoted.
3’UTR tiling also can be used to detect three-dimensional structures with the potential to influence RNA stability. rs3751756 (KIAA0513), displayed a large allelic skew (log2FC=−1.38, BH p-adj=2.65x10−40), with the alt allele demonstrating a significant attenuating effect that is unobserved with the ref allele. On the alt background, deletions within a 45 bp window around the tamVar restored expression to near ref levels, which suggests an overlying functional element. RNAfold (Gruber et al., 2008) predicts a stem-loop structure in the region, potentially mediating this attenuation (Fig. 5b). While the non-functional ref allele (U) creates a bulge in the stem, the alt allele (G) is predicted to create a stable stem, potentially amenable to RBP occupancy and subsequent functional attenuation. Moreover, nearly every base alteration disrupting pairing of the stem in our saturation mutagenesis alleviated repression (average Z-test p=6.13x10−4), in contrast to tolerated changes in the loop that yielded attenuation. Highlighting the sensitivity of MPRAu to small effect sizes, we recapitulated the expected effects of wobble base pairing, observing A to G mutations along the stem have the smallest effect sizes.
We similarly investigated the indel tamVar rs34448361 (LEPR) due to its large allelic skew (log2FC=−0.55, BH p-adj=2.19x10−11), and its previous identification as being within a genomic region with evidence of recent natural selection (Grossman et al., 2010). Both 3’ UTR alleles displayed attenuation, but the alt allele exhibited a significantly stronger effect (ref log2FC=−0.56, alt log2FC=−1.12), which was validated in a luciferase assay (two-sided t-test p=4.4x10−4) (Supplementary Fig. 6a). Deletions mapped a 25 bp AU-rich element consisting of AUUUA pentamer repeats which are known to impose attenuating effects (Siegel et al., 2020) (Fig. 5c). The ref 3’UTR contains four pentamers while the alt allele is a 4 bp insert that creates a fifth pentamer which is in agreement with the alt allele showing an increased effect. SNV tiling recapitulated this finding, with only disruption of AUUUA pentamers abrogating attenuation in both allelic contexts (Fig. 5c). The alt allele is highly represented in East Asian populations (allele frequency=0.79) versus other populations (African allele frequency=0.15) (1000 Genomes Project Consortium et al., 2015). rs34448361 is in the 3’UTR of a LEPR isoform (LepRb) (Supplementary Fig. 6b,c), and matches the direction of effect for an eQTL for LepRb across diverse tissues contexts including adipose (eQTL β=−0.26, p=2.35x10−7) and IFN stimulated macrophage (eQTL β=−0.50, p=6.61x10−5) (Kerimov et al., 2020). This isoform has been implicated in traits highly essential for survival, including satiety and obesity (Münzberg and Morrison, 2015), as well as activation of various immune cells (Abella et al., 2017). However, the region surrounding LEPR has also been implicated in cold adaptation in modern and archaic humans (Sazzini et al., 2014). While the exact phenotype under selection is still unknown, MPRAu provides strong functional evidence for rs34448361 as a causal variant driving the evolutionary signature in the genomic region and provides evidence towards the variant’s regulatory mechanism.
We further highlight three additional tamVars where SNV/deletion tiling elucidated functional 3’UTR elements, including rs632255 (CNTLN), which overlaps a functionally identified AU-rich element (Supplementary Fig. 7a). Two tamVars, (rs482356 (STX3) and rs1049508 (GATM)), perturb unknown 3’UTR regulatory motifs (Supplementary Fig. 7b,c). STX3 and GATM are associated with microvillus inclusion disease and cerebral creatine deficiency syndrome respectively (Nouioua et al., 2013; Wiegerinck et al., 2014). This finding highlights MPRAu’s ability to both discover individual variants with allelic effects as well as to characterize the underlying motifs potentially responsible for regulating human disease-associated genes.
MPRAu identifies causal 3’UTR variants related to human evolution and disease
A key attribute of MPRAu is the ability to parse genetic associations based on functional evidence to nominate causal variants for a variety of traits and diseases. Amongst all 2,153 GWAS-associated 3’UTR variants tested, we found 677 GWAS loci with a significant tamVar in at least one cell type. From this tamVar set, several previously nominated causal variants were identified, including rs13702, which disrupts a miRNA binding site in LPL and has been suggested to underlie associations for triglyceride levels and type 2 diabetes (Ban et al., 2010; Richardson et al., 2013; Tang et al., 2010). Our set also provides functional evidence for 10 variants convincingly nominated for causality from genetics alone, such as rs5891007 (LSM1, Schizophrenia risk) (Shi et al., 2011) and rs1140711 (LIN7C, bone mineral density) (Kemp et al., 2014), among others (Table 1). While MPRAu nominates hundreds of phenotypically-relevant 3’UTR variants, a potential limitation is the episomal-based assay not capturing endogenous gene expression effects. To more directly confirm the effects of tamVars, we performed endogenous allelic replacements via CRISPR-mediated homology-directed repair (HDR) on two tamVars linked to specific human disease processes: rs705866 and rs1059273.
Table 1:
GWAS variants nominated for functional causality by MPRAu.
| MPRAu Tested SNP | tag SNP | Gene | Trait | PMID | OR/Beta | tamVar log2FC | tamVar P value |
|---|---|---|---|---|---|---|---|
| rs1056801# | rs3771570 | SEPT2 | Prostate cancer | 23535732 | 1.12 | −1.2 | 7.7x10-12 |
| rs1140711 | rs10835187 | LIN7C | Bone mineral density | 24945404 | 0.127 | −0.43 | 0.061 |
| rs12190287# | rs12190287 | TCF21 | Coronary heart disease | 21378990 | 1.08 | 0.47 | 0.0021 |
| rs13702# | rs10105606 | LPL | Triglycerides | 20864672 | 0.07 | 0.31 | 1.00E-08 |
| rs13702# | rs331 | LPL | HDLC | 19936222 | 1.459 | 0.31 | 1.00E-08 |
| rs5891007 | rs16887244 | LSM1 | Schizophrenia | 22037555 | 1.19 | 0.36 | 0.004 |
| rs705866# | rs7803454 | PILRB | Age-related macular degeneration | 26691988 | 1.13 | 0.3 | 0.083 |
| rs708723 | rs823118 | RAB7L1 | Parkinson’s disease | 25064009 | 1.122 | 0.37 | 0.067 |
| rs71396950 | rs1805007 | SPATA33 | Freckles | 17952075 | 4.37 | −0.43 | 0.095 |
| rs71396950 | rs1805007 | SPATA33 | Red hair color | 17952075 | 12.47 | −0.43 | 0.095 |
| rs71396950 | rs35063026 | SPATA33 | Squamous cell carcinoma | 26829030 | 1.33 | −0.43 | 0.095 |
| rs8066731 | rs9902453 | SLC6A4 | Coffee consumption | 25288136 | 0.03 | −0.65 | 0.0039 |
| rs35274349‡ | rs10948363 | CD2AP | Alzheimer’s disease | 24162737 | 1.1 | 0.48 | 6.30E-10 |
(Most significant adjusted p-value, and its corresponding log2FC, from the MPRAu screen is shown)
denotes variant perturbation of a top 100 expressed miRNA,
denotes variant perturbation of a potential RBP motif within an eCLIP-seq peak region.
rs705866 (PILRB) is associated with age-related macular degeneration (odds ratio (OR)=1.13, p=5x10−9), residing amongst 151 SNPs in strong LD to a tag-SNP (r2=0.808 with rs7803454) (Fritsche et al., 2016) (Fig. 6a). MPRAu identified a suggestive allelic effect (log2FC Skew=0.3, BH p-adj=0.08) with the ref allele showing an attenuating effect on expression (Fig. 6b). Other variants in this credible set (the smallest set of variants that contain the true causal variant with 95% probability) include multiple missense SNPs, although their impacts are predicted to be benign (Supplementary Fig. 6d,e). Recent work identified rs705866 as an eQTL for PILRB specifically within the focal disease tissue, the retinal macular region (eQTL β=1.25, p=5.72x10−29) (Orozco et al., 2020).
Figure 6: rs705866 and rs1059273 are endogenously validated tamVars impacting human disease and adaptation phenotypes.
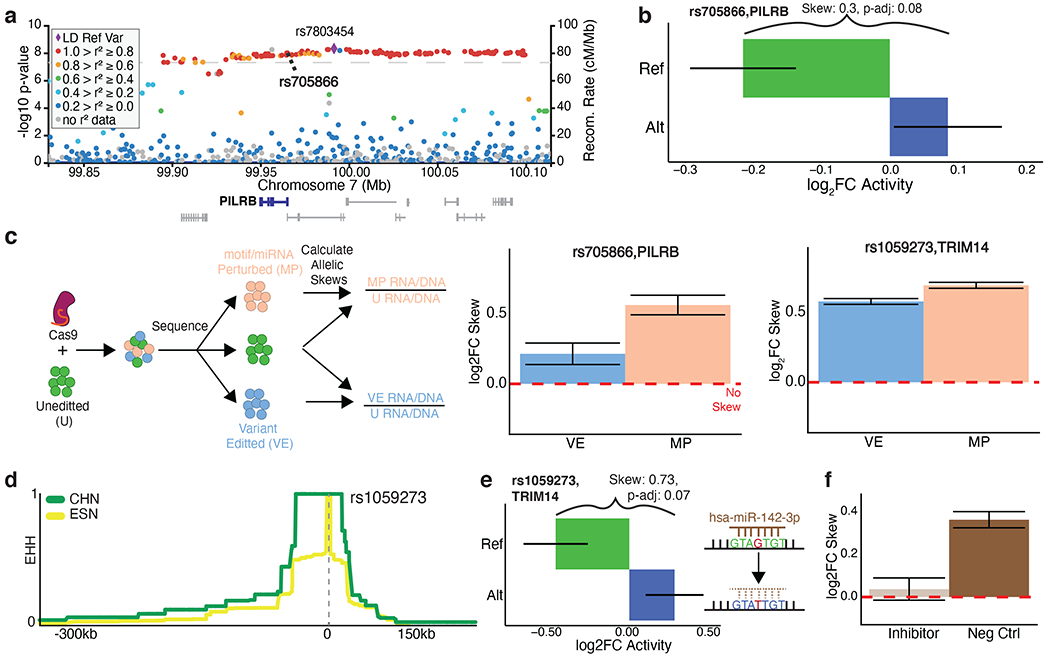
a, Age-related macular degeneration GWAS association plot surrounding the tag SNP rs7803454 with rs705866 (MPRAu tested SNP) in bold. b, MPRAu allelic results for rs705866. c, Schematic of the HDR experimental pipeline (left). Allelic skews estimated from HDR for rs705866 (center), and rs1059273 (right) exhibit the same directionality as the MPRAu results. d, EHH score surrounding rs1059273 suggests evolutionary selection in Han Chinese (CHN). For comparison, EHH scores for Esan in Nigeria (ESN) are also shown. e, MPRAu shows significant attenuating activity in the allelic background (ref) with the unperturbed miRNA binding site for rs1059273. f, Allelic skew result after transfection of a miRNA inhibitor for hsa-miR-142-3p versus a negative control miRNA inhibitor.
To confirm the MPRAu allelic effect in the endogenous genomic context, we used CRISPR HDR to perform allelic replacement of rs705866 (T (ref) to C (alt)) in neuronal SK-N-SH cells. Allelic ratios of RNA and DNA from cells with the desired edit were compared with unperturbed cells, and an allelic skew was found (log2FC=0.21, Fisher p=3.39x10−8) to be concordant with the effects measured by MPRAu (Fig. 6c). Due to imperfect editing from CRISPR HDR, additional SNVs and indels perturbing the surrounding sequence containing the allele were also generated. Aggregating effects of these imprecise edits led to an even larger functional disruption (log2FC=0.56, Fisher p=1.53x10−57) (Fig. 6c). Investigating the sequence context surrounding the variant suggests it may be bound by several RBPs (BCLAF1, PPIG, and RBFOX2 (Van Nostrand et al., 2016)) or a miRNA (hsa-miR-374a-5p).
rs1059273 (TRIM14) lies within a genomic region that experienced positive selection in East Asian populations (Grossman et al., 2010). Han Chinese genomes display an extended haplotype heterozygosity score surrounding rs1059273, but identifying a causal allele has remained difficult (Fig. 6d). MPRAu identified attenuating effects on the ref background, but not the alt (log2FC Skew=0.73, BH p-adj=0.07) (Fig. 6e). rs1059273 disrupts a miRNA binding site (hsa-miR-142-3p), potentially explaining higher expression from the alt allele (Fig. 6e). TRIM14 has many functions, including antiviral and antimicrobial activity as part of the Type I Interferon pathway (Chen et al., 2016; Tan et al., 2017; Zhou et al., 2014). Knockouts of TRIM14 in macrophages more effectively control Mycobacterium tuberculosis replication (Hoffpauir et al., 2020), and TRIM14 has also been found to suppress Influenza A replication (Wu et al., 2019). rs1059273 is a significant eQTL for TRIM14 in T cells and NK cells (T cell, CD4, Th1/17, eQTL β=0.91, p=7.2x10−9; NK cell, CD56dim CD16+, eQTL β=0.90, p=9.7x10−9) (Schmiedel et al., 2018). Fine-mapping also assigns a high likelihood of the variant causally affecting TRIM14 expression (PIP=0.53, 0.55 in Th1, Th17 cells), and finds no other potentially causal (PIP≥0.1) variants with functional annotations within the credible set containing rs1059273 (Kerimov et al., 2020). These associations further link this variant’s potential evolutionary impact to an immunological role.
We performed allelic replacement of rs1059273 (G (ref) to T (alt)) in lymphoblastoid cells and confirmed the variant’s effects on TRIM14 (allele replacement: log2FC=0.58, Fisher p=6.79x10−699). Similar to rs708566, we aggregated the effects from cells with unspecific edits over the miRNA binding site mutations and observed an effect size larger than by rs1059273 alone (log2FC=0.69, Fisher p=3.32x10−953) (Fig. 6c). Transfecting an inhibitor for the miRNA abrogated the allelic skew (log2FC=0.04, Fisher p=0.16), which was not observed using a negative control inhibitor (log2FC=0.36, Fisher p=4.44x10−78) (Fig. 6f), providing additional evidence that hsa-miR-142-3p is mechanistically responsible for the allelic skew.
Discussion
We developed MPRAu, a high-throughput tool to functionally characterize 3’UTR variants and used it to identify 2,368 3’UTR variants that modulate transcript abundance across six cell lines. We built powerful predictive models of 3’UTR function and identified modes of 3’UTR regulation. This resource characterizing GWAS, selection signals, and common variation in 3’UTRs will be useful to ongoing future studies of human adaptation and disease. We expect MPRAu will be a common experimental paradigm to test variants of unknown significance and rare variants going forward. In the future, MPRAu may be further modified to specifically detect variants impacting a particular regulatory mechanism of interest, such as transcription termination (Shalem et al., 2015) or mRNA localization (Andreassi and Riccio, 2009; Berkovits and Mayr, 2015; Tushev et al., 2018), as well as investigate the proposed miRNA/RBP mechanisms proposed for many of the tamVars we identifed. From a recent exhaustive alternative polyadenylation eQTL analysis across tissues in GTEx (Li et al., 2021), we found APA eQTLs overlapping 217 of our tamVars. While our current assay is not optimally designed to derive 3’UTR variant effects on APA, future modifications can allow us to more comprehensively detect these variant functions as well.
In addition to our reporter assay, we provide additional evidence using 3’UTR tiling and endogenous allelic replacement for three variants (rs1059273, rs705866, and rs34448361) with potentially important consequences to understanding human disease and evolution. Further experiments are needed to assess each candidate variant’s contribution in the relevant cellular context. For example, while TRIM14 expression is suppressed in multiple viral infections, including the SARS-CoV-2 virus responsible for the COVID-19 pandemic (Blanco-Melo et al., 2020), the exact extent of rs1059273 in modulating viral infectivity is unknown. While we tested our assay across six different cellular contexts, additional phenotypic-relevant variants may be found when applying our assay in disease-specific tissues.
Currently, potentially causal variants in 3’UTR elements underlying complex human diseases are largely overlooked because of the lack of tools to characterize them. The vast addition of 3’UTR measurements, especially in the context of phenotype-relevant genetic variation, may additionally inform future models of genome function. In total, MPRAu provides a framework for prioritizing regulatory variation in 3’UTRs based on functionality. Our study helps further a more comprehensive understanding of the regulatory processes important for non-coding variant function.
Limitations of the Study
While MPRAu bridges a tremendous gap linking 3’UTR genomic variation, functional effects, and ultimately phenotypes, the assay has important limitations. The 100 bp tested sequence may prevent comprehensive interrogation of all human 3’UTRs, which have an average length of 800-1000 bp (Sood et al., 2006). Additionally, while the assay’s episomal-based nature may not fully recapitulate endogenous effects, it does enable assaying variant functions in 3’UTRs from lowly and/or tissue-specific expressed genes regulated by common RBPs/miRNAs. Furthermore, RNA steady-state levels may not be perfectly linked with protein levels (Battle et al., 2015; Chick et al., 2016); however, flow-cytometry assays have shown overwhelmingly strong agreement of RNA expression from episomal reporter assays with protein abundance (Oikonomou et al., 2014; Zhao et al., 2014). Although tamVars allelic effects were found to be strongly shared across different cellular contexts, the diversity of cell types tested, and cell-type-specific experimental factors may affect these estimates.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to James Xue (jxue@broadinstitute.org).
Materials Availability
Oligo libraries used in this study are available upon request. CRISPR-modified GM12878s for rs1059273 and CRISPR-modified SK-N-SH for rs705866 are available upon request. All additional unique/stable reagents generated in this study are available from the Lead Contact without restriction, or with a Materials Transfer Agreement.
Data and Code Availability
Raw sequencing reads have been deposited to GEO, the ENCODE portal (https://www.encodeproject.org/awards/UM1HG009435/), and SRA. Processed MPRAu screen data are available in Supplementary Table 1 and will also be available on the ENCODE portal. Read counts per oligo are included in Supplementary Table 1.
Analyses was performed with standard analysis packages cited in the text, and plotted using custom R scripts that are available upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293 cells were grown in DMEM (ThermoFisher, 10564) supplemented with 10% FBS (ThermoFisher, A3160401), 1X NEAA (ThermoFisher, 11140050), and 1 mM Sodium Pyruvate (ThermoFisher, 11360070) at 37C.
GM12878s (Coriell) were grown in RPMI (ThermoFisher, 61870036) supplemented with 15% FBS and 1% 10X Penicillin-Streptomycin (Pen-Strep; ThermoFisher, 15140122; Corning, 30-002-CI) at 37C.
K562s (ATCC, CCL-243) were grown in RPMI supplemented with 10% FBS and 1% 10X Pen-Strep at 37C.
HepG2s (ATCC, HB-8065) were grown in 25 mL MEM Alpha (ThermoFisher, 32561037) + 10% FBS + 1% Pen-Strep on 15 cm plates at 37C.
SK-N-SH (ATCC, HTB-11) were grown in 90 mL EMEM (ATCC, 30-2003) supplemented with 10% FBS and 1% Pen-Strep on Nunc Triple Flasks (VWR, 89498-706) at 37C.
HMECs (ThermoFisher, A10565) were grown in 60 mL MEGM (Lonza, CC-3150) in T225 flasks at 37C.
METHOD DETAILS
Variant selection and oligo design
We designed our entire tested 3’UTR sequence/variant set in two separate oligonucleotide libraries. One library contained predominantly variants in regions associated with recent human evolutionary adaptation derived from a previous scan of natural selection (Grossman et al., 2013). As this scan for natural selection relied on a method called the “Composite of Multiple Signals” (CMS), we refer to this UTR library and associated experiments as the “CMS array” throughout the paper. The other library contained variants derived from GWAS, and referred to as the “GWAS array.” A full table of tested oligos/variants is listed in Supplementary Table 1, and a more thorough description of each of the library contents is described immediately following.
For the CMS array, we selected 9,325 common 3’UTR variants (SNPs and indels up to 10 bp in length, from Phase 1 of the Thousand Genomes Project with a global MAF≥5% amongst individuals from the pilot-phase Thousand Genomes populations) that fell within positively selected regions identified by the Composite of Multiple Signals test (Grossman et al., 2013). We additionally selected 415 common (MAF≥5%) 3’UTR variants at random from the genome.
For the GWAS array, we selected 2,153 common 3’UTR variants (SNPs and indels up to 10 bp from Phase 1 of the Thousand Genomes Project with a MAF≥5% amongst individuals from European populations) which were linked (r2>0.8 using PLINK v1.9) (Purcell et al., 2007) with a variant that met genome-wide significance (p-value<5x10−8) in the 2/13/2017 freeze of the NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas). 95 indels (4.2% of the initial set of 2,248 variants) were excluded from subsequent analysis due to a reference allele coding error. We additionally included all variants from an initial pilot CMS array performed in HEK293 cells (see MPRAu transfection experiments) which significantly impacted expression (134 variants) to measure the reproducibility of variant effects across different libraries, and noticed concordant effects between the GWAS and CMS array (r=0.79, p=2.5x10−28). Furthermore, for our 3’UTR “tiling” experiment, we incorporated SNV and deletion mutations of both the reference and alternate backgrounds of 80 variants with magnitude skew 1.5-fold or more in the CMS array (Supplementary Table 1). For SNV tiling, we included all SNVs from 10 bp upstream of the variant site to 10 bp downstream of the variant site. For deletion tiling, we performed 5 bp non-overlapping deletions of the entire tested 100 bp sequence surrounding the variant. Finally, we included 281 rare 3’UTR variants, 46 of which were associated with outlier expression in GTEx (Li et al., 2017) and did not have rare structural variants within 200 kb (Supplementary Table 1). The remaining 235 control variants were selected by first identifying non-outlier genes ((gene, individual) tuples with |Median Z-score| < 1), then extracting rare variants within 10 kb of the gene, (gene, individual) tuples with at least one rare SV within 200 kb of the gene, including the gene body, were excluded in selecting control variants. Control variants were also required to have the same gene and variant type (indel, SNP) as the outlier variants - each control variant was matched to at least one outlier variant at the same gene with the same variant type. Both rare and control variants are also annotated by the Ensembl Variant Effect Predictor (McLaren et al., 2016) as 3_prime_UTR_variant.
Both CMS and GWAS libraries were synthesized as 133 bp sequences containing a maximum of 101 bp of 3’UTR context and sequence adapters on either end (5’CGAGCTCGCTAGCCT [maximum of 101 bp of 3’UTR sequence] AGATCGGAAGAGCGTCG3’) (Fig. 1a). To select 3’UTRs, we searched the Gencode v19 database for 3’UTR-annotated entries downstream of CDS-annotated entries, filtering out entries annotated for nonsense-mediated decay and non-stop decay. Ideally, 50 bp of 3’UTR sequence context on each side of the variant was obtained; if the variant was within 50 bp of the 5’ (stop codon) or 3’ (termination site) end of the 3’UTR, its sequence context ended prematurely, and if possible, additional sequence was taken from the other end to obtain 100 bp total. If a variant’s oligo sequence context overlapped one or more of the other variants tested in our set, we designed a reference and alternate oligo in which all other variants were assigned the reference allele (as in the reference human genome, “reference background”), as well as a reference and alternate oligo in which all other variants were assigned the alternate allele (“alternate background”). For example, a variant Y flanked by one or more variants, such as variants X and Z, would be referenced in our variant datasets within our supplementary tables as follows: Y would refer to a comparison between X_ref/Y_ref/Z_ref and X_ref/Y_alt/Z_ref, whereas Y_2 would refer to a comparison between X_alt/Y_ref/Z_alt and X_alt/Y_alt/Z_alt. We have flagged 117 variants from the CMS library with an indexing error in Y_2 alternate background sequences in Supplementary Table 1. This indexing error does not affect the Y reference background sequences for each of these variants, and thus their allelic skew can be independently assessed.
Oligo synthesis and amplification
The CMS array oligos were synthesized by CustomArray and the GWAS array oligos were synthesized by Twist Biosciences. Post-synthesis, for the CMS array, 6 bp random hexamer barcodes and additional adapter sequences were added by performing 20 PCR reactions, each 50 μL in volume, containing 5.7 ng of oligo, 25 μL of Q5 NEBNext MasterMix (NEB, M0541S), 1 unit Q5 HotStart polymerase (NEB, M0493S), 0.5 μM oligo_BAR_Bmt_F and oligo_pmir_R_min primers, and 20 μg BSA (NEB, B9000) (Supplementary Table 5). PCR cycling conditions used are as follows: 98°C for 30 seconds, 22 cycles (98°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min), 72°C for 2 min. For the GWAS array oligos, PCR was performed using the same primers and cycling conditions with the following exceptions: 1) 24 50 μL PCR reactions instead of 20, 2) 1 ng of oligo in each reaction instead of 5.7 ng, 3) use of the NEBNext Ultra II Q5 Master Mix (NEB, M0544L) instead of the Q5 HotStart polymerase, 4) and 12 cycles for amplification instead of 22 cycles. PCR reactions from both arrays were purified by performing a 2.5X SPRI purification using Agencourt AMPure XP SPRI (Beckman Coulter, A63881) beads according to manufacturer instructions.
MPRAu vector assembly
To create our MPRAu plasmid pool for the CMS array, barcoded oligos were inserted into a modified pmirGLO (Promega, E1330) vector, pmirGLO:Δluc::gfp ΔAmpR::KanR, which contains the GFP gene driven by the pgk promoter. Oligos were inserted by Gibson Assembly (NEB, E2611) using 1 μg of BmtI/XbaI (NEB, R0658S; NEB, R0145S) digested vector and 415 ng of amplified oligos (10:1 plasmid to oligo molar ratio) in a 40 μL reaction incubated for 60 min at 50°C followed by 2X SPRI purification and eluted in 25 μL EB. The elution was then concentrated to 10 μL by vacuum centrifugation. The ligated vector was then split into 5 μL aliquots, each of which was transformed into 100 μL of 10-beta e.coli (NEB, C3020K) by electroporation (2kV, 200 ohm, 25 μF). Electroporated bacteria were immediately split into five 1 mL aliquots of SOC (NEB, B9020S) and recovered for 1 hour at 37°C then independently expanded in 1L of LB supplemented with 50 μg/mL of kanamycin (Teknova, K2125) on a floor shaker at 37°C for 12 hours. After outgrowth, aliquots were pooled before plasmid purification (Qiagen, 12991). For each of the aliquots, we plated serial dilutions after SOC recovery and estimated a library size of >108CFUs. For the GWAS array, a similar vector assembly protocol was followed except the Gibson Assembly reaction time and size was doubled. Each electroporation also was split into four 1 mL aliquots of SOC, subsequently pooled, and grown into four final 1L LB supplemental with 50 μg/mL of kanamycin in a floor shaker.
MPRAu transfection experiments
We performed our initial pilot experiment using the CMS array into HEK293 cells (specifically the HEK293FT cell line (Thermofisher, R70007)). HEK293 cells were grown in DMEM (ThermoFisher, 10564) supplemented with 10% FBS (ThermoFisher, A3160401), 1X NEAA (ThermoFisher, 11140050), and 1 mM Sodium Pyruvate (ThermoFisher, 11360070). For all 5 replicate transfections (Figure 1C), cells were plated in a 15 cm plate and grown to a density of ~80-90%. Cells were then transfected with 80 μL Lipofectamine 2000 (ThermoFisher, 11668027) and 20 μg DNA, and incubated with the transfection reagents for 24 hours, monitored by fluorescent microscopy. Afterward, transfected cells were then split 1:3 into 2 new 15 cm plates. After an additional 24 hours (48 hours post-transfection), media from each plate was replaced with 30 mL DMEM + 100 μg/mL cycloheximide (CHX) (Sigma-Aldrich, C4859) and incubated for 5 minutes at 37°C. Both plates were then washed with 10 mL cold PBS (ThermoFisher, 14040) + 100 μg/mL CHX, scraped in 1 mL cold PBS + 100 μg/mL CHX, pooled and centrifuged for 5 minutes at 500 x g, and finally resuspended in 350 μL lysis buffer composed of 5 mM Tris pH 7.5 (ThermoFisher, 15567), 2.5 mM Magnesium Chloride (ThermoFisher, AM9530G), 1.5 mM Potassium Chloride (ThermoFisher, AM9640G), 2 μM DTT (VWR, 97061-340), 100 μg/mL CHX, 5 mg/mL Triton-X (Sigma-Aldrich, T8787), and 5 mg/mL sodium deoxycholate (Sigma-Aldrich, 30970). The cell lysate was then centrifuged for 5 minutes at 12,000 x g and the supernatant was flash-frozen in vapor phase nitrogen. For polysome profiling, approximately 200 μL lysate was loaded onto 10%-50% sucrose gradients in 15 mM Tris pH 7.5, 15 mM Magnesium Chloride, 150mM Sodium Chloride (ThermoFisher, AM9760G), and 100 μg/mL CHX. Gradients were centrifuged in SW41Ti rotor at 35,000 rpm for 2.5 hours at 4°C and 0.5 mL fractions were collected.
We then transfected the GWAS array into HEK293 cells, following the same protocol above, except using two 15 cm plates per replicate, and excluding the polysome profiling step. The CMS & GWAS array were then pooled together into one library (CMS+GWAS) for subsequent transfections across other cell types, with protocols described below.
GM12878s (Coriell) were grown in RPMI (ThermoFisher, 61870036) supplemented with 15% FBS and 1% 10X Penicillin-Streptomycin (Pen-Strep; ThermoFisher, 15140122; Corning, 30-002-CI). Each replicate (4 total, grown on different days) was grown to ~ 1x106 cells/mL, 150 million cells were then collected via centrifugation at 300 x g and suspended in 1.2 mL RPMI with 150 μg library. Subsequently, cells were electroporated with the Neon transfection system with the 100 μL kit (ThermoFisher, MPK10096) using 3 pulses of 1200V, 20 ms each. Following transfection, each replicate was grown in 150mL RPMI + 15% FBS without Pen-Strep, to recover for 48 hours. Cells were split 1:2 after the first 24 hour recovery time to prevent overgrowth. The cells were then spun down, washed once with PBS, flash-frozen via liquid nitrogen, and subsequently stored at −80°C.
K562s (ATCC, CCL-243) were grown in RPMI supplemented with 10% FBS and 1% 10X Pen-Strep. Each replicate (4 total, grown on different days) was grown to ~ 1x106 cells/mL, 150 million cells were then collected via centrifugation at 300 x g and suspended in 1.2 mL RPMI with 150 μg library. Subsequently, cells were electroporated with the Neon transfection system with the 100 μL kit using 3 pulses of 1450 V, 10 ms each. Following transfection, each replicate was grown in 150 mL RPMI + 15% FBS without Pen-Strep to recover for 48 hours. Cells were split 1:2 after the first 24 hour recovery time to prevent overgrowth. The cells were then spun down, washed once with PBS, flash-frozen via liquid nitrogen, and subsequently stored at −80°C.
HepG2s (ATCC, HB-8065) were grown in 25 mL MEM Alpha (ThermoFisher, 32561037) + 10% FBS + 1% Pen-Strep on 15 cm plates. Cells were grown to 60%-80% confluency. For all 4 replicates, grown on different days, two 15 cm plates per replicate were transfected with 87.5 μL Lipofectamine 3000 (ThermoFisher, L3000015) and 35 μg library. Following transfection, each replicate was grown in 25 mL MEM Alpha + 10% FBS without Pen-Strep to recover for 48 hours. The cells were then trypsinized, spun down at 300 x g at 4°C, washed once with PBS, flash-frozen via liquid nitrogen, and subsequently stored at −80°C.
SK-N-SH (ATCC, HTB-11) were grown in 90 mL EMEM (ATCC, 30-2003) supplemented with 10% FBS and 1% Pen-Strep on Nunc Triple Flasks (VWR, 89498-706). Each replicate (4 total, grown on different days) was grown to 80%-100% confluency. Cells were trypsinized, and 40 million cells were collected and resuspended in 400 μL Buffer R with 25 μg library. Subsequently, cells were electroporated with the Neon transfection system with the 100 μL kit using 3 pulses of 950 V, 30 ms each. Following transfection, each replicate was grown in 45 mL EMEM + 10% FBS without Pen-Strep to recover for 48 hours. The cells were then trypsinized, spun down at 300 x g at 4°C, washed once with PBS, flash-frozen via liquid nitrogen, and subsequently stored at −80°C.
HMECs (ThermoFisher, A10565) were grown in 60 mL MEGM (Lonza, CC-3150) in T225 flasks. For each replicate (5 total, grown on different days), 6 confluent flasks were grown to 80%-100%. Cells were then resuspended in buffer R and DNA to get a final concentration of 10 million cells/mL and 25 μg DNA/mL. Subsequently, cells were electroporated with the Neon transfection system with the 100 μL kit using 3 pulses of 950 V, 30 ms each. Following transfection, each replicate was grown in 4 T225 flasks (each with 60 mL MEGM) to recover for 48 hours. The cells were then trypsinized, spun down at 250 x g at 4°C, washed once with PBS, flash-frozen via liquid nitrogen, and subsequently stored at −80°C.
Transfection efficiency was monitored across all cell types by assessing GFP fluorescence. Across all cell lines, greater than 50% of live cells fluoresced after transfection, signifying acquisition of the MPRA construct. K562, HepG2, and HEK293 had the highest transfection efficiency (>80%), while HMEC had the worst (50%).
RNA extraction and cDNA synthesis
For all cell type replicates, RNA was extracted with TRIzol LS (ThermoFisher, 10296) according to manufacturer instructions. Total RNA was purified from the cell lysate. Polysomal RNA was purified from sucrose fractions, pooling fractions corresponding to three or more ribosomes. 7.5 μg GlycoBlue (ThermoFisher, AM9515) was added to each sample to visualize the pellet. mRNA was purified from total RNA using oligo d (T)25 magnetic beads (NEB, S1419S) according to the manufacturer’s instructions, and eluted at 80°C. Purified total and polysomal mRNA were then subjected to Turbo DNase treatment (ThermoFisher, AM2239). The reaction was terminated in 2 mg/mL SDS (ThermoFisher, AM9822) and purified by performing a 2X SPRI purification using Agencourt RNAClean XP SPRI (Beckman Coulter, A63987) beads according to manufacturer instructions. DNase-treated mRNA for each of the tested replicates was diluted to the concentration of the lowest concentration sample, and first-strand cDNA was synthesized from concentration-normalized mRNA with Superscript III (ThermoFisher, 18080) and a gene-specific primer 162 bp downstream of the oligo (oligo_RT_R, Supplementary Table 5). For the Superscript III reaction, we used the manufacturer’s recommended protocol, except by increasing the total reaction volume to 40 μL and performing the elongation step at 55°C for 80 minutes. Single-stranded cDNA was purified by performing a 2X SPRI purification using Agencourt RNAClean XP SPRI beads.
qPCR and library construction
cDNA concentrations from the cell type replicates were estimated via qPCR using 1 μL of cDNA sample in a 10 μL reaction that contained 5 μL Q5 NEBNext master mix, 1.7 μL SYBR Green I diluted 1:10,000 (Life Technologies, S-7567), and 0.5 μM of PE_PCR_P1 and PE_PCR_P2_BMT primers (Supplementary Table 5), and under the following conditions: 98°C for 30 seconds, 40 cycles (98°C for 10 sec, 65°C for 30 sec, and 72°C for 30 sec), 72°C for 5 minutes. The cDNA samples across all cell types had a cycle threshold (CT) between 7 and 15 cycles.
Samples across all tested replicates from all cellular cDNA samples were then aliquoted to achieve the same input going into the next amplification step based on the CT values derived from the previous step. Specifically, we matched all input amounts to achieve a CT of 11. The plasmid pool was also amplified in five independent PCR reactions (technical replicates), also adjusting the input amount per reaction to achieve an expected CT of 11. Samples were amplified in a 50 μL PCR reaction containing 25 μL NEBNext Ultra II Q5 Master Mix and 0.5 μM of PE_PCR_P1 and PE_PCR_P2_Bmt (Supplementary Table 5), and under the following conditions: 98°C for 30 seconds, 9 cycles (98°C for 10 sec, 65°C for 30 sec, 72°C for 30 sec), 72°C for 5 minutes. 2.5X AMPure SPRI purification was then performed, and another round of PCR (as above, except with 5 cycles) was performed using a set of Illumina P5 index primers and a set of Illumina P7 index primers in a 100 μL reaction. Another 2.5X AMPure SPRI purification was performed afterward. Samples were then pooled according to molar estimates from the Agilent 2200 TapeStation (using the D1000 screentape reagents (Agilent, 5067-5585)) and then subsequently sequenced using a S4 flowcell (2 x 150 bp) on a NovaSeq using the Broad Institute’s walk-up sequencing service.
Sample preparation for the initial pilot CMS array pilot library in HEK293 cells was processed separately in an analogous manner just described, with the main difference being the use of the SYBR Green Master Mix (ThermoFisher, 4367659) to quantify CT. The pilot library samples were sequenced using 2 x 150 bp chemistry on an Illumina HiSeq through the Broad Institute’s walk-up sequencing service.
Read alignment to 3’UTR sequences
Paired-end 150 bp reads were merged into single amplicons using Flash v1.2.11 (flags: -M 150, -O) (Magoč and Salzberg, 2011). For data from the NovaSeq, only Read 1 sequences were used due to lower quality sequences from Read 2. Amplicon sequences were retained for quantification if the sequence surrounding the barcode met the following conditions: (1) a perfect match was found to the 10 bp sequence on either the left or right side of the barcode, (2) the 10 bp sequences on both the left and right sides of the barcode matched with a Levenshtein distance of 3 or less, and (3) the 2 bp immediately surrounding each side of the barcode matched perfectly. Oligo sequences from the passing reads were then mapped back to the expected oligo sequences using BWA mem version 0.7.12 (flags: -M) (Li, 2013). We calculated our own alignment scores to assess the quality of the alignments. These scores were calculated as the number of matching bases divided by the expected oligo size. Reads with alignment scores of less than 0.95 were discarded. Oligo libraries were extremely complex, with an average of 70-330 unique hexamer barcodes per oligo per replicate sample, which would minimize effects from any outlier barcodes that would have functional effects. As a result, oligo reads were pooled across barcodes for oligo analyses and barcode reads were pooled across oligos for barcode analyses. On average, each oligo contained 1100-3300 reads across all tested cell type/plasmid replicates (Supplementary Table 1). Due to the manner that BWA calls multi-mapped reads and our strict filtering of reads that multi-mapped, we noticed some oligos received inadequate reads to call any effects. This issue prevented ~1% of tested variants from being assessed for allelic skew, as well as 5 bp deletion sequences at the 3’ end from the deletion tilling set from be assessed for activity. These sequences have close to zero counts in the count table in Supplementary Table 1. All sequences were still retained for subsequent DESeq2 analysis, which inherently accounts for low counts in its modeling.
Functional 3’UTR element and tamVar calling
Oligo counts from all samples were passed into DESeq2 and a median-of-ratios method was used to normalize samples for varying sequencing depths (Love et al., 2014). Normalized read counts of each oligo were then modeled by DESeq2 as a negative binomial distribution. DESeq2 estimates variance for each NB by pooling all oligo counts across samples and fitting a trend line to model the relationship between oligo counts and observed dispersion. It then applies an empirical Bayes shrinkage by taking the observed relationship as a prior and performing a maximum a posteriori estimate of the dispersion for each oligo. The overall result is that DESeq2 can obtain an estimate for dispersion of each oligo with greatly reduced bias by pooling information from all oligos.
We first used DESeq2 to estimate oligo fold changes between our sample types (plasmid pool, total RNA, polysomal RNA) for our initial pilot HEK293 dataset with just the CMS array. To calculate total RNA expression and polysomal RNA expression, we normalized total RNA counts in the lysate and polysomal RNA counts respectively by the baseline counts in the plasmid pool (design = ~ Replicate+Sample_Type). We estimated fold changes between the reference and alternate alleles (RNAskew and POLYskew) by adding an interaction term (design = ~ Replicate+Sample_Type+Variant+Sample_Type:Variant) and using a Wald test with the Bonferroni multiple test correction. In all models, a replicate term was added to pair samples from the same transfection. We used this initial model to look at the concordance between polysomal and total RNA data (Supplementary Fig. 1).
Upon expanding the assay to the 5 other cell types other than HEK293, we ran DESeq2 separately for each cell type to derive functional 3’UTR elements and tamVars using a revised model. The DESeq2 model was revised as follows: design = ~ Variant+Type+Variant:Type, where Type corresponds to total RNA or the plasmid pool. Wald tests were used with contrasts to derive reference and alternate specific activity (fold changes of RNA over plasmid) and the difference between alternate activity and reference activity (allelic skew). The Benjamini-Hochberg test correction was performed via DESeq2 to correct for multiple hypothesis testing. Variants with significant skew (tamVars) were designated if the adjusted p-value from any of the tested allelic backgrounds was less than 0.1. The output from this DESeq2 analysis is the one reported in Supplementary Table 1.
tamVar cell-specificity analysis
mash was used to estimate tamVar effect sharing across cell types (Urbut et al., 2019). mash requires an input of user-specified data-driven covariance matrices. Using variants with strong MPRAu-measured allelic effects (BH p-adj<10−3), we derived the following data-driven covariance matrices: 1) the empirical covariance matrix, 2) flash matrix factorization of the empirical covariance matrix (Wang and Stephens, 2021), and 3) SVD rank 4 approximation of the empirical covariance matrix. In addition, rank 1 covariance matrices derived from flash factors with large components (defined as containing at least two rows with values greater than 1/sqrt(6)) were added to the data-driven covariance matrix set. Extreme deconvolution (ED) was also applied to the full set of data-driven covariance matrix set mentioned (Bovy et al., 2011), and the subsequent ED output matrices was used as the final matrix set for analysis. The exchangeable effects model was also used over the exchangeable Z due to its better performance (measured by likelihood) from cross-validation testing. Variants were shared between cell types X and Y if the local false sign rate was less than 0.05 for both X and Y.
Luciferase assays
To validate the expression values obtained by MPRAu, we selected 18 oligos consisting of nine ref/alt pairs. Five of the oligos were selected as no-skew controls for having uncorrected p-values of greater than 0.01. We designed the same 101 bp sequence that was tested by MPRAu as a gBlock (IDT) and cloned each into the pmirGLO dual-luciferase reporter vector (Promega, E1330). Cells were plated in a 96 well plate and grown to a density of 80%-90%, then transfected with a mixture of 0.2 μL Lipofectamine 2000, 500 pg of the cloned dual-luciferase vector, and 49.5 ng of pGL4.23, a promoterless control vector (Promega, E8411). We performed six transfection replicates per oligo (all on the same 96-well plate). Cells were incubated with transfection reagents for 24 hours, monitored by fluorescent microscopy, and then split 1:3 into a new 96-well plate. After 24 hours (48 hours post-transfection), firefly and Renilla luminescence were read from each well using the Dual-Glo Luciferase Assay (Promega, E2920). Firefly luciferase luminescence for each well was normalized to the Renilla luciferase luminescence for the same well, and each experiment was normalized as a log-ratio value relative to the mean of a control oligo with an MPRAu RNA/DNA ratio of −0.2 (Supplementary Table 2).
CRISPR allelic replacement
All crRNA and ssODN were designed and ordered via IDT (Supplementary Table 5). Cas9/Cpf1 reagents were also ordered from IDT. Two replicate experiments were performed for each target. rs1059273_GuideRNA_Cas9 (Cas9 crRNA) and rs1059273_ssODN (ssODN) were used for both replicates of rs1059273. For rs705866, rs705866_GuideRNA_Cpf1_1 (Cpf1 crRNA) and rs705866_ssODN_1 (ssODN) were used for the first replicate, and rs705866_GuideRNA_Cpf1_2 (Cpf1 crRNA) and rs705866_ssODN_2 (ssODN) were used for the second replicate. rs1059273 experiments were performed in GM12878 and rs705866 experiments were performed in SK-N-SH. Consistent effects were observed across both replicate experiments for both targets (Fig. 6c, Supplementary Fig. 6f,g). Cells were grown in the following media conditions: RPMI, supplemented with 15% FBS for GM12878s, and EMEM supplemented with 10% FBS for SK-N-SH. The HDR protocol used was adapted from IDT’s provided one: http://sfvideo.blob.core.windows.net/sitefinity/docs/default-source/protocol/homology-directed-repair-alt-r-crispr-cas9-ultramer-oligos.pdf?sfvrsn=9750707_8
For the Cas9 HDR experiments, the following protocol was used per target. First, 0.9 μL of 200 μM Alt-R CRISPR-Cas9 target-specific crRNA, 0.9 μL of 200 μM Alt-R CRISPR-Cas9 tracrRNA (IDT, 1072533), and 1.5 μL Nuclease-Free Duplex Buffer (IDT, 1072570) were combined and heated at 95°C for 5 minutes. The crRNA:tracrRNA solution was then cooled at room temperature. 3 μL of the crRNA:tracrRNA solution was then combined with 2 μL Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT, 1081059) and incubated at room temperature for 10-20 minutes to form the RNP complex. 100K cells per electroporation were washed with PBS, then resuspended in 7.69 μL of Neon Resuspension Buffer R. Next, 1.61 μL of the RNP complex, 7.69 μL of 100K cells in Neon Resuspension Buffer R, 0.3 μL of 100 μM ssODN, and 0.4 μL of Alt-R Cas9 Electroporation Enhancer (IDT, 1075916) were combined for one electroporation using the Neon transfection system with the 10 μL kit (ThermoFisher, MPK1025). Each target underwent two electroporations using set electroporation conditions (3 pulses of 1200 V, 30 ms each for GM12878s). Both electroporations were transferred to a well containing 0.4 mL of recovery media (regular media supplemented with 30 μM HDR enhancer (IDT, 1081072)) in a 24-well plate and grown for 12-24 hours. The recovery media was then changed to regular media afterward. Cells were grown and expanded until we achieved a population of at least 6-8 million cells, then ~ 6-8 million cells were extracted, washed with PBS, and flash-frozen afterward.
For the Cpf1 HDR experiments, the following protocol was used per target. First, 2.5 μL of Alt-R CRISPR-Cpf1 target-specific crRNA was combined with 2.5 μL Alt-R A.s. Cas12a (Cpf1) Ultra (IDT, 10001273) and incubated at room temperature for 10-20 minutes to form the RNP complex. Following the formation of the RNP complex, the protocol follows exactly as the Cas9 HDR protocol, except with the use of 0.3 μL of Alt-R Cpf1 Electroporation Enhancer (IDT, 1076300) (cells were also resuspended in 7.79 μL Neon Resuspension Buffer R instead of 7.69 μL to account for the 0.1 μL decrease in volume), and the use of a different electroporation setting for SK-N-SH (3 pulses of 950 V, 30 ms each ).
DNA/RNA was extracted from the frozen samples using the AllPrep DNA/RNA Mini Kit (Qiagen, 80204). Extracted RNA was DNase treated and purified via 2X SPRI using Agencourt RNAClean XP SPRI beads. DNase was inactivated via 2 mg/mL SDS (ThermoFisher, AM9822). DNase-treated RNA was then used to generate target-specific cDNA using Superscript III and a gene-specific primer (rs1059273_R) for target rs1059273. For target rs705866, we used the same gene-specific primer but utilized Superscript IV VILO instead (we switched enzymes due to the lower expression levels of the gene). 17 20 μL reactions with 500 ng RNA in each reaction were performed for target rs1059273 and 12 20 μL reactions with 500 ng RNA in each reaction for target rs705866. The entire 20 μL from each reaction was then directly used to amplify the target amplicon via PCR using the NEBNext Ultra II Q5 Master Mix with 0.5 μM rs1059273_F and rs1059273_R primers and the following cycling conditions: 95°C for 20 seconds, 15 cycles (95°C for 20 sec, 68°C for 20 sec, 72°C for 30 sec), 72°C for 2 minutes (Supplementary Table 5). rs705866 had the same cycling conditions except with using primers rs705866_F and rs705866_R and 12 instead of 15 cycles for amplification (Supplementary Table 5). Purified DNA was also amplified via PCR using the same target primers and subject to the same cycling conditions for each target, except with 50 individual PCR reactions for target rs1059273 and 12 individual PCR reactions for target rs 1059273. For each target DNA/RNA, the individual post-PCR reactions were then pooled together, subject to a 1X AMPure SPRI purification, and concentrated via vacuum centrifugation. Another round of PCR was then performed (same cycling conditions as above, except with 8 cycles and 64°C for the annealing temperature) to attach p7 and p5 Illumina adapters with unique sample indices. The PCR products were then subject to another 2X SPRI and eluted in 30 μL. The resulting purified PCR products across all targets were then molar pooled from Agilent 2200 TapeStation quantifications (using D1000 screentape reagents) and subsequently sequenced using 2 x 150 bp chemistry on an Illumina MiSeq.
CRISPR HDR was found to be efficient across all replicates. For rs705866, 3.5% of alleles from the first replicate and 14.5% of the alleles from the second replicate obtained perfect edits. For rs1059273, 31.4% of the alleles from the first replicate and 34.3% of the alleles from the second replicate acquired perfect edits. Furthermore, for rs705866, 3.8% of alleles from the first replicate and 3.8% from the second replicate had additional sequence perturbations (+/− 5 bp from the variant position) surrounding the variant on the ref background, which allowed us to quantify the effects of other SNVs/indels overlying the potential functional element (Fig. 6c). Similarly, for rs1059273, 22.5% of alleles from the first replicate and 24.1% from the second replicate had additional perturbations over the expected miRNA motif on the ref background. CRISPResso was used to derive the allele proportions from the sequencing data (Clement et al., 2019).
miRNA Inhibitor Experiments
miRNA inhibitor for rs1059273 (hsa-miR-142-3p, HSTUD0219) and a negative control (ath-miR416, NCSTUD001) were designed by Sigma-Aldrich. GM12878 (for target rs1059273) were grown in the same conditions as the CRISPR allelic replacement experiments.
For introducing the hsa-miR-142-3p/negative control inhibitor into GM12878s, cells were grown to greater than 6M cells. Then, cells were collected via centrifugation, washed with PBS, and resuspended in Neon Resuspension Buffer R. 50 nM of inhibitor was transfected with 2M cells using the Neon transfection system with the 100 μL kit across 3 separate electroporations (6M cells total, using 3 pulses of 1200V, 20 ms each).
DNA/RNA was also processed and sequenced the same way as the CRISPR allelic replacement experiments, with the following alterations: 1) for RNA processing, 6 different Superscript IV VILO reactions with 1 μg of RNA in each reaction were performed, 2) for DNA processing, 6 different PCR reactions were used, and 3) the number of cycles for attaching the first set of primers (rs1059273_F/ rs1059273_R) was 14 instead of 12.
3’UTR element annotation derivation
We identified AU-rich elements in multiple ways, including using the AUUUA pentamer that often occurs in multiples, the UUAUUUAWW nonamer that has been associated with rapid mRNA decay, and finally two 13 bp motifs from Bakheet et al. 2001 (Bakheet et al., 2001) (WWWUAUUUAUWWW and WWAUUUAUUUAWW, with one mismatch allowed in the flanking sequence outside of AUUUA and UUUAUUU respectively). We identified CU-rich elements using the (C/U)CCANxCCC (U/A) (C/U)yUC (C/U)CC consensus sequence that has been shown to increase mRNA stability (Holcik and Liebhaber, 1997). We identified GU-rich elements using the UGUUUGUUUGU consensus sequence that has been associated with short-lived transcripts (Vlasova et al., 2008). We identified Pumilio binding sites using the UGUANAUA motif identified through human Pumilio immunoprecipitation experiments (Galgano et al., 2008; Hafner et al., 2010; Morris et al., 2008).
To predict microRNA binding sites, we used TargetScan6 (Friedman et al., 2009) to identify 7mer and 8mer seed matches to the top 10 and top 100 most abundant microRNAs in each of the cell lines tested. To obtain the top 10 and top 100 most abundant microRNAs, we used miRDeep2 (Friedländer et al., 2012) to quantify microRNA abundance in short RNA sequencing experiments from ENCODE and the Sequence Read Archive (see below). For SK-N-SH we were unable to obtain sequencing data and instead used short RNA sequencing of the SH-SY5Y line, a neuroblast-like subline of SK-N-SH. To generate the heatmaps in Fig. 2d and Supplementary Fig. 3a, for each cell type with multiple miRNA datasets, we used the miRNA dataset that demonstrated the most significant effect when overlapping with the 8mer annotations from the dataset’s top 10 most abundant miRNAs. For Fig. 2d, significance of effect was measured by the p-value derived from a t-test on the expression values of the oligos that contained an 8mer motif from the dataset’s top 10 most abundant miRNAs (comparing against a null of zero, with an alternative hypothesis of the mean expression values being less than zero). For Supplementary Fig. 3a, significance of effect was measured by the p-value derived from a t-test on the allelic skew results for variant perturbations that led to a gain an 8mer motif from the dataset’s top 10 most abundant miRNAs (comparing against a null of zero, with an alternative hypothesis of the mean expression values being less than zero). We used the same best miRNA dataset per cell type to extract features for our functional 3’UTR element computational modeling work.
| Cell Line | Archive | Dataset ID |
|---|---|---|
| HEK293 | SRA | • SRR1240816 • SRR1240817 |
| SH-SY5Y | SRA | • SRR1304311 |
| HepG2 | SRA | • ERR738415 • ERR738403 • ERR738417 |
| HepG2 | ENCODE | • ENCFF175ZOB |
| HMEC | SRA | • DRR041459 • DRR041581 • DRR041472 • SRR5127224 |
| GM12878 | ENCODE | • ENCFF878BGO • ENCFF440XMV • ENCFF322QWE |
| K562 | ENCODE | • ENCFF119JCH • ENCFF756CSN • ENCFF691LGG |
The RBP dataset used in generating Supplementary Fig. 3b was derived from Table S3 from Dominguez et al. 2018. Motifs with Stepwise_R-1 greater than 0.1 were classified as having a “strong RBP motif”, and motifs with Stepwise_R-1 between 0 and 0.1 were classified as having a “weak RBP motif.”
RBP expression data derivation
RNA expression data (for deriving RBP expression, and used for our computational prediction work) were downloaded as tsv files from the following sources:
| Cell Line | Source | Link/ENCODE file accession |
|---|---|---|
| HEK293 | Protein Atlas | https://www.proteinatlas.org/download/rna_celline.tsv.zip |
| HepG2 | ENCODE | ENCFF004HYK |
| HMEC | ENCODE | ENCFF380GBC |
| GM12878 | ENCODE | ENCFF599JTV |
| K562 | ENCODE | ENCFF172GIN |
| SK-N-SH | ENCODE | ENCFF389TFR |
Hexamer barcode analysis
We used the barcodes included in our MPRAu design to directly measure the activity of all possible 4,096 hexamer sequences across every tested cell type (Fig. 1a, Supplementary Table 3). For each cell type/plasmid replicate, counts were derived for each of the 4,096 hexamers by summing up the counts of all CMS array oligos that were tagged with the corresponding hexamer. For assembling the hexamer count table, only oligos that belonged to the CMS array were used to ensure that hexamers were derived from a diverse oligo pool. The GWAS oligo set was overrepresented by 3’UTR tiling oligos, comprising 69% of the GWAS oligo set. Since these 3’UTR tiling oligos were designed based on oligos significant effects in the CMS array, hexamers obtained from these oligos were inherently biased to have significant effects. To not bias hexamer activity with interactions with these oligos, the GWAS oligos were excluded from calculating hexamer activity. DESeq2 was also utilized to calculate the fold change of RNA over DNA independently for each cell type. The DESeq2 model used was design = ~ Type, where Type corresponds to RNA or the plasmid backbone. The Wald test was used to derive p-values, and the Benjamini-Hochberg test correction was performed via DESeq2 to correct for multiple hypothesis testing. Using the DESeq2 derived adjusted p-values, we were able to derive the hexamers having the most significant effects per cell type.
Examining the hexamers with the most significant effects (| log2FC Skew |≥0.5, BH p-adj<0.1), we identified up to 28 hexamers per cell type with large attenuating or augmenting effects. Our tested hexamers confirmed effects expected at known RBP motifs (Dominguez et al., 2018) (Supplementary Fig. 3b) and variants perturbing functional hexamers abrogated the intact element’s effect (two-sided Wilcoxon rank-sum test p<0.05 across most tested cell types) (Supplementary Fig. 3c,d). As each 3’UTR was linked to many unique barcodes (Supplementary Table 3), the few functional barcodes are not expected to impact our measurements, as evidenced by the high correlation observed between cell type replicates in Fig. 1c (Methods).
3’UTR element computational modeling
The original MPRAu dataset was first filtered to remove variants that had an average plasmid count of less than 20 (average calculated across all plasmid replicates). For each cell type, for predicting attenuation, oligos were classified as one if their log2FC was less than −0.5, and zero otherwise. For predicting augmentation, oligos were classified as one if their log2FC was greater than 0.5, and zero otherwise. We performed predictions independently for each cell type. For prediction, two sets of variables were utilized: the minimal model, and the full model.
For the minimal model, the following variables were calculated across each oligo and used for prediction: nucleotide percentage (across four bases, four variables), dinucleotide percentages (six variables), exact dinucleotide counts (16 variables), minimal free energy (as measured by RNAFold), maximum homopolymer length (for each base type A, U, C, G, and across all base types, five variables), maximum dinucleotide length across all bases, and a measure of sequence uniformity (let “seq” be the tested sequence, then it is calculated as the following: for i in range (1,len (seq),1): if seq[i]==seq[ (i-1)]: seq_uniformity=seq_uniformity+1)
For the full model, all of the variables in the minimal model were used, and the following variables, calculated across the entire oligo, were added per cell type: the number of RBP motifs for AU-rich, GU-rich, CU-rich, GU-rich, constitutive decay, and Pumilio elements (6 variables), RNAcontext-k-mer scores (Orenstein et al., 2016) for expressed RBPs (79-86 variables total, dependent on which RBPs were expressed in specific cell types), the number of binding sites for each of the top 100 expressed miRNAs within the cell type (100 variables total), the number of hexamers for each of the top 300 most significant activity hexamers within the cell type (ranked by adjusted p-value, 300 variables total), the total number of overlapped miRNA motifs from the top 10/25/100 expressed miRNAs subsetting on 8mer/7mer-1A/7mer-m8/8mer+7mer-1A+7mer-m8/8mer+7mer-m8 annotations (15 variables), and the number of overlapped hexamers derived from the top 10/20/50/100/200/300 most significant positive/negative/all hexamers (ranked by adjusted p-value, 18 variables). We note that all the hexamer and miRNA-associated variables are derived from cell-type-specific datasets, thus are different in each cell type. For the miRNA annotations, if a cell type contained multiple miRNA expression datasets, we used the dataset that had the most significant attenuating effect for oligos that contained motifs from the top 10 most highly expressed miRNAs (subsetting on the 8mer+7mer-m8 annotation, significance was calculated via a t-test, with the null mean centered at zero).
For each cell type, under both the minimal and full model tested, the following procedure was performed for training. Approximately 20% of the dataset was used for validating the final model, and ~80% of the dataset was used to train a predictive model using xgboost. The initial ~20% testing was selected by iteratively selecting all the oligos for a specific gene, weighted by the number of oligos corresponding to the gene, until the size of the validation set reached at least 20%. The rest of the oligos that were not selected in the testing set were used in the training set. Some genes had overlapping 3’UTRs, and the oligos that they overlapped were grouped together as being derived from a single gene in this selection strategy. This splitting ensured that oligos in the same gene did not overlap in the validation/training sets.
The xgboost implementation in python was used then used to train models with the following range of parameters (in the following format: [ parameter_name: start, stop, step_size ]): [ colsample_bytree: 0.75, 1, 0.25 ], [ gamma: 1, 7, 2 ], [ min_child_weight: 1, 7, 2 ], [ max_depth: 2, 10, 2 ], and [ n_estimators: 50, 150, 50 ] across all cell types. This range of parameters yielded a total of 480 combinations. We also used SVM and logistic regression, and the implementations in the python sklearn package were used. For parameter tuning, the following parameters were used - for SVM: [ C: 0.0001, 0.001, 0.01, 0.1, 1, 10, 100, 1000 ], [ gamma: 0.001, 0.002, 0.01, 0.02, 0.03, 0.05, 0.1, 1, 2], [ kernel: string, rbf ], and for logistic regression: [ C: 0.0001, 0.001, 0.01, 0.1, 1, 10, 100, 1000 ], [penalty: I1, I2 ]. Lastly, a one-variable decision tree model trained using percent U.was used as a control.
For each parameter combination, five-fold cross-validation, grouped by genes (so that oligos will not overlap between the testing and training folds), was performed to obtain the combination’s performance. The mean of the average precision across the five folds was utilized as the scoring metric for model performance under each parameter setting. The model parameter combination with the best mean average precision score was chosen as the best combination. To obtain the validation average precision score, the best parameter combination was used to train a model on the entire training set, and then evaluated by the validation set which was never seen in the training procedures. Across all tested cell types, we consistently found xgboost outperforming all models in all predictions (Supplementary Fig. 4).
Concordance with Geuvadis and GTEx datasets
Both ASE and eQTL data were downloaded from https://www.ebi.ac.uk/arrayexpress/experiments/E-GEUV-1/files/analysis_results/
Allele-specific expression data was derived from the file GD462.ASE.COV8.ANNOT_PTV.txt.gz and eQTL data was derived from combining the files YRI89.gene.cis.FDR5.all.rs137.txt.gz and EUR373.gene.cis.FDR5.all.rs137.txt.gz.
GM12878 MPRAu variant data was overlapped with Geuvadis ASE data using chromosome, position, ref, and alt allele as the identifier to match between datasets. The overlapped set was then further filtered by only retaining effect sizes which have a p-value less than 0.05 for Geuvadis and a BH p-adj less than 0.1 for MPRAu. Variants were further filtered by keeping the ones that had a pooled (across individuals) Geuvadis ASE significant effect (two-sided t-test p <0.01). From the remaining filtered variants, the median Geuvadis ASE value from all individuals per variant was used for plotting in Fig. 4a. If multiple MPRAu variants had the same identifier (for example, due to the same variant lying in different backgrounds), the median was taken as the point for plotting.
eQTL data from both YRI and EUR were concatenated together into a single file. GM12878 data was extracted from our MPRAu data set and overlapped with Geuvadis data using chromosome, position, ref, and alt allele, and Ensembl gene name as the identifier to match between datasets. If multiple Geuvadis eQTL entries overlap with a single MPRAu variant, then the median was taken as the representative point for plotting in Supplementary Fig. 5a. Similarly, if multiple MPRAu variants had the same identifier (for example, due to the same variant lying in different backgrounds), the median was taken as the point for plotting.
GTEx v8 fine-mapped eQTL effect sizes (Ulirsch) were correlated with MPRAu allelic skews in Fig. 4b and Supplementary Fig. 5b. Since there was not a direct mapping of MPRA tested cell type to GTEx tissue type, and variants that have significant allelic skew effects across multiple tissues appear to have concordant directional effects across these same tissues (Fig. 1e), we created aggregated scores to compare our MPRAu experiments with the GTEx fine-mapping dataset. To do this, for each variant, we took the median eQTL effect size across all GTEx tissues with a fine-mapped signal greater or lesser than a certain PIP cutoff (greater than 0.2 for Fig. 4b, and Supplementary Fig. 5b, lesser than 0.01 for Supplementary Fig. 5c) and compared it against the median tamVar skew across significant skew values (BH p-adj<0.05 for Fig. 4b and <0.1 for Supplementary Fig. 5b,c) from all cell types. We further included only variants that are annotated by the Ensembl Variant Effect Predictor (version 85) (McLaren et al., 2016) to have the 3’UTR annotation as the most severe consequence in comparing allelic effect concordance. MPRAu data was overlapped with the GTEx v8 fine-mapping data using chromosome, position, ref, and alt allele, and Ensembl gene name as the identifier to match between datasets.
UK Biobank fine-mapping enrichment
Genetic fine-mapping of 94 traits in up to 361,194 individuals from the UKBB was performed using FINEMAP (Benner et al., 2016) and the Sum of Single Effects (SuSiE) method (Wang et al., 2020a) (https://www.finucanelab.org/data). Fine-mapped variants were overlapped with MPRAu variant data and across all PIP filters, the proportion of tested 3’UTR variants significant in at least one MPRAu tested cell type was used to create Fig. 4c. If a variant was tested across multiple sequence backgrounds, it was considered significant if strong allelic effects (BH p-adj<0.1) were observed in any of the tested contexts. We only included variants that were annotated by the Ensembl Variant Effect Predictor (version 85) (McLaren et al., 2016) to have 3’UTR annotation as the most severe consequence. 3’UTR variants that were part of a 95% SuSiE credible set with another highly likely causal variant (defined as PIP>0.3 and in a coding or promoter region) were also excluded.
3’UTR SNV and deletion tiling analysis
Log2FC of RNA over plasmid was quantified via DESeq2. To generate the sequence logo for SNV tiling, the sign of the log2FC of the unaltered oligo was first multiplied to the log2FC of all oligos that tiled the specific region. Then, per oligo position, all four log2FC values (corresponding to the position with A, U, C, or G) were normalized by the sum of the total log2FC for that position. These steps created a normalized weight matrix per position across the entire oligo. EDLogo (Dey et al., 2018) then used these matrices to generate the motifs for SNV tiling. In contrast to conventional sequence logos, EDLogo allows both enrichment and depletion of the activity of characters to be displayed at each specific position. Formally, enrichment/depletion for a specific character at a specific position is interpreted to be relative to the median enrichment/depletion across all possible characters for the position.
We multiplied the sign of the log2FC of the unaltered oligo first to all the other oligos to ensure that underlying motifs (regardless of the direction of its effect), if perturbed, show positive enrichment scores. For example, at a specific position, if a base is part of a functional element, and all other bases perturb the element, then the log2FC magnitude observed for the other bases would be less in magnitude than the one observed by the unaltered oligo. In this manner, the base of the unaltered oligo would have the highest enrichment score (as seen in the AU-rich element in Fig. 5c and the U1 snRNP motif in Fig. 5a). However in the rare case of a single base creating a novel functional element in the opposite effect of the unaltered oligo log2FC, then we would see the base having a very low enrichment score (as seen in the motif derived in the ref SNV tiling plot in Fig. 5b).
Further analysis of rs705866 and rs1059273
The surrounding LD block around rs705866 contains approximately 150 variants, as estimated by looking at the LD block via Haploreg (r2>0.6) (Ward and Kellis, 2016), or the credible set given in Supplementary Data Set 3 from Fritsche et al. 2016. Most noticeably, amongst these ~150 variants, three are missense variants. Although the three missense SNPs are in LD (rs11771799, rs35986051, and rs11761306) with rs705866, none of them look to be strongly conserved (Supplementary Fig. 6d), and all of them are also annotated as benign according to the Ensembl Variant Effect Predictor (McLaren et al., 2016). Examining the protein structure of PILRB, all three also lie on non-secondary structure elements, far away from the active site for the gene, which further gives support of these variants being non-functional (Supplementary Fig. 6e). While another variant may still have causal regulatory effects, it is still likely that rs705866 is a causal variant due to the concordance in MPRAu/HDR allelic expression directionality with the reported retina eQTL allelic effect (i.e. higher expression in the alt allele).
For rs1059273, FsT was calculated between all pairwise non-admixed populations in the 1000 Genomes Dataset (1000 Genomes Project Consortium et al., 2015) using the StAMPP package (https://www.rdocumentation.org/packages/StAMPP/versions/1.5.1/topics/StAMPP-package) (Pembleton et al., 2013). The highest significantly elevated fixation index was observed between Han Chinese and Esan. These populations were then used to calculate EHH scores on ancestral and derived haplotypes centered at rs1059273. Related individuals were excluded from all analyses.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of exact statistical analyses, packages, tests, and other procedures used can be found in the main text, figure legends, and STAR Methods.
Supplementary Material
Supplementary Fig. 1: Polysomal RNA-derived MPRAu metrics are correlated with Total RNA-derived MPRAu metrics, related to Figure 1.
a, HEK293 Polysomal RNA-derived MPRAu activity correlation with total RNA-derived HEK293 MPRAu activity. b, HEK293 Polysomal RNA-derived MPRAu skew correlation with HEK293 total RNA-derived MPRAu skew for significant hits (BH p-adj<0.1 for either Polysomal RNA or total RNA variants).
Supplementary Fig. 2: mash covariance structure with the largest weight demonstrates extensive allelic effect sharing amongst tamVars, related to Figure 1.
a, mash covariance matrix weights. b, Correlation matrix of the covariance structure with the highest weight (ED SVD) from a.
Supplementary Fig. 3: 3’UTR annotations with known effects provide evidence of MPRAu’s functionality, related to Figure 2.
a, Each box in the heatmap measures the significance of attenuation (t-test) when cell-type-specific MPRAu-measured 3’UTR allelic skews (y-axis) are subsetted on the variants with sequence perturbations that lead to a gain of a top 10 most abundant miRNAs across the cell types tested (x-axis). b, Magnitude of MPRAu activity when subsetting on RBP motifs derived from Dominguez et al., 2018. c, MPRAu allelic skews of 3’UTR variants with sequence perturbations that lead to a gain a top 20 barcode with negative activity (ranked by p-adj) versus MPRAu skews from 3’UTR variants that do not gain such barcodes. d, MPRAu allelic skews of 3’UTR variants with sequence perturbations that lead to a gain a top 200 barcode with positive activity (ranked by p-adj) versus MPRAu skews from 3’UTR variants that do not gain such barcodes. For b-d, reported p-values are derived from two-sided Wilcoxon rank-sum tests.
Supplementary Fig. 4: xgboost modeling additional information, related to Figure 3.
a, xgboost precision-recall curve across all six tested cell types (right). xgboost ROC curve across all six tested cell types (left). b, SVM precision-recall curve across all six tested cell types (right). SVM ROC curve across all six tested cell types (left). c, Logistic regression precision-recall curve across all six tested cell types (right). Logistic regression ROC curve across all six tested cell types (left). d, One-variable percent U decision tree precision-recall curve across all six tested cell types (right). One-variable percent U decision tree ROC curve across all six tested cell types (left). e, xgboost precision-recall curve across all six tested cell types for predicting augmenting activity (right). xgboost ROC curve across all six tested cell types for predicting augmenting activity (left). f, xgboost precision-recall curve across all six tested cell types for the full model, incorporating the top 100 expressed miRNAs for each cell type and RNAcontext-k-mer scores from expressed RBPs for each cell type, for predicting attenuation (right). xgboost ROC curve across all six tested cell types for the full model for predicting attenuation (left). g, xgboost precision-recall curve across all six tested cell types for using only miRNA and RBP-specific features for predicting attenuation (right). xgboost ROC curve across all six tested cell types using only miRNA and RBP-specific features for predicting attenuation (left). h, SHAP summary plot for the top 10 most important features in Fig. 3c. Each individual point plotted shows the effect of each feature from each training data point towards attenuation prediction. The features are ordered in terms of feature importance (top most important), and colored by feature value. Higher SHAP values denote greater contribution of prediction towards attenuation. “↑” next to variable name indicates higher values of the variable contribute to greater attenuation. Similarly, “↓,“ indicates lesser values of the variable contribute to greater attenuation. Some features, such as pU, display subtle non-linear effects which are not captured here.
Supplementary Fig. 5: Geuvadis eQTLs and GTEx fine-mapped eQTL concordance with MPRAu results, related to Figure 4.
a, Geuvadis eQTL effect size correlation with GM12878 3’UTR MPRAu allelic skews. b, Correlation between all-tissue median eQTL allelic skew (posterior beta) in GTEx that have a high probability of being causal via genetic fine-mapping (PIP>0.2) and tamVar (BH p-adj<0.1) median skew across all cell types. c, The same as in b, except with the all-tissue median eQTL allelic skew calculated only on points with PIP<0.01. Pearson’s r and its statistical significance are displayed.
Supplementary Fig. 6: Additional information relevant to 3’UTR variants functionally characterized, related to Figures 5 and 6.
a, Luciferase assay results on rs34448361. b, The alternate allele (insertion that extends the AU-rich element overlying the variant) has high frequency specifically in East Asian populations. c, rs34448361(green star) lies specifically in the 3’UTR of the longest isoform of LEPR ENST00000349533 (LepRb). d, Multiple sequence alignment across 30 mammals show that rs11771799, rs35986051, and rs11761306 (all in LD with rs705866) are in bases without strong conservation. e, Protein structure of PILRB demonstrates that rs11771799, rs35986051, and rs11761306 (green) lie far from the active site and are likely non-functional mutations. Shown in yellow are residues within 5 Å of the sialic acid ligand (purple). f, Allelic skew effects from the second replicate of a HDR allelic replacement of rs705866 (VE: Fisher p=1.43x10−15, MP: Fisher p=5.52x10−122). g, Allelic skew effects from the second replicate of a HDR allelic replacement of rs1059273 in GM12878 (VE: Fisher p=5.40x10−76, MP: Fisher p=1.83x10−287). VE: variant edited, MP: motif perturbed.
Supplementary Fig. 7: Additional sequence architectures via MPRAu SNV and deletion tiling, related to Figure 5.
For a-c, Top, 5 bp deletion tiling of the targeted variant using the ref (red) or alt (blue) sequence context across 100 bp. Bottom, SNV tiling +/− 10 bp around each variant showing log2 fold change and motif enrichment score in both variant contexts. Bottom, SNV tiling showing log2 fold change and motif enrichment score in both sequence contexts. HEK293 3’UTR activity data is plotted for all panels. a, Plots for tiling at rs632255 (CNTLN) with multiple AU-rich elements (AREs) denoted. b, Plots for tiling at rs482356 (STX3). c, Plots for tiling at rs1049508 (GATM).
Supplementary Table 1: MPRAu results, related to STAR Methods
Supplementary Table 2: Luciferase assay results, related to Figure 1
Supplementary Table 3: MPRAu barcode effects, related to STAR Methods
Supplementary Table 4: tamVars with gene expression and phenotype fine-mapping support, related to Figure 4
Supplementary Table 5: Primers, HDR ssODN/guides, related to STAR Methods
Assayed thousands of GWAS and adaptation associated 3’UTR variants in 6 cell lines
Nominated hundreds of causal GWAS variants with functional evidence of activity
Characterized mechanistic regulatory motifs at base-pair resolution
Used allelic replacement on causal variants for macular degeneration and viral defense
Acknowledgments
We thank S. Schaffner, S. Gosai, S. Weingarten-Gabbay, A.E. Lin, D. Kotliar, C. Myhrvold, and C. Tomkins-Tinch for thoughtful conversations and help with editing the manuscript. We thank D. P. Bartel as well as his lab for thoughtful discussions. We thank I. Shlyakhter for providing the CMS regions. This work, D. Griesemer, J.R. Xue, S.K. Reilly and R. Tewhey were supported as an ENCODE Functional Characterization Center (UM1HG009435), a Broad SPARC grant, NSF DEB-1401237, and the Howard Hughes Medical Institute. D. Griesemer was partially supported by F30GM114940. S.K. Reilly was partially supported by K99HG010669 and F32HG00922. J.Luban is supported by R37AI147868. R. Tewhey is supported by R00HG008179.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
P.C.S. is a co-founder of and consultant to Sherlock Biosciences and Board Member of Danaher Corporation. She is a shareholder in both companies.
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gómez-Reino JJ, Mera A, Lago F, Gómez R, and Gualillo O (2017). Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol 13, 100–109. [DOI] [PubMed] [Google Scholar]
- Andreassi C, and Riccio A (2009). To localize or not to localize: mRNA fate is in 3’UTR ends. Trends Cell Biol. 19, 465–474. [DOI] [PubMed] [Google Scholar]
- van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, van der Weide RH, Teunissen H, Võsa U, Franke L, et al. (2019). High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet 51, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet T, Frevel M, Williams BR, Greer W, and Khabar KS (2001). ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 29, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H-J, Heo JY, Oh K-S, and Park K-J (2010). Identification of Type 2 Diabetes-associated combination of SNPs using Support Vector Machine. BMC Genet. 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, and Gilad Y (2015). Impact of regulatory variation from RNA to protein. Science 347, 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C, Spencer CCA, Havulinna AS, Salomaa V, Ripatti S, and Pirinen M (2016). FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovits BD, and Mayr C (2015). Alternative 3’ UTRs act as scaffolds to regulate membrane protein localization. Nature 522, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, et al. (2020). Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 181, 1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogard N, Linder J, Rosenberg AB, and Seelig G (2019). A Deep Neural Network for Predicting and Engineering Alternative Polyadenylation. Cell 178, 91–106.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovy J, Hogg DW, and Roweis ST (2011). Extreme deconvolution: Inferring complete distribution functions from noisy, heterogeneous and incomplete observations. Ann. Appl. Stat 5. [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang R-F, et al. (2016). TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 64, 105–119. [DOI] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, and Gygi SP (2016). Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Zhang T, Vu A, Ablain J, Makowski MM, Colli LM, Xu M, Hennessey RC, Yin J, Rothschild H, et al. (2020). Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun 11, 2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, et al. (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey KK, Xie D, and Stephens M (2018). A new sequence logo plot to highlight enrichment and depletion. BMC Bioinformatics 19, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 70, 854–867.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, ReproGen Consortium, Schizophrenia Working Group of the Psychiatric Genomics Consortium, The RACI Consortium, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, and Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK-H, Burge CB, and Bartel DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, et al. (2016). A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, and Gerber AP (2008). Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PloS One 3, e3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Shlyakhter I, Shylakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, et al. (2010). A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327, 883–886. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, Park DJ, Griesemer D, Karlsson EK, Wong SH, et al. (2013). Identifying recent adaptations in large-scale genomic data. Cell 152, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, and Hofacker IL (2008). The Vienna RNA Websuite. Nucleic Acids Res. 36, W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, et al. (2014). Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet 95, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffpauir CT, Bell SL, West KO, Jing T, Wagner AR, Torres-Odio S, Cox JS, West AP, Li P, Patrick KL, et al. (2020). TRIM14 Is a Key Regulator of the Type I IFN Response during Mycobacterium tuberculosis Infection. J. Immunol 205, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik M, and Liebhaber SA (1997). Four highly stable eukaryotic mRNAs assemble 3’ untranslated region RNA-protein complexes sharing cis and trans components. Proc. Natl. Acad. Sci. U. S. A 94, 2410–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JP, Medina-Gomez C, Estrada K, St Pourcain B, Heppe DHM, Warrington NM, Oei L, Ring SM, Kruithof CJ, Timpson NJ, et al. (2014). Phenotypic Dissection of Bone Mineral Density Reveals Skeletal Site Specificity and Facilitates the Identification of Novel Loci in the Genetic Regulation of Bone Mass Attainment. PLoS Genet. 10, e1004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimov N, Hayhurst JD, Manning JR, Walter P, Kolberg L, Peikova K, Samoviča M, Burdett T, Jupp S, Parkinson H, et al. (2020). eQTL Catalogue: a compendium of uniformly processed human gene expression and splicing QTLs (Genomics). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, and Ahituv N (2019). Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun 10, 3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JC, Keith A, Rice SJ, Shepherd C, Agarwal V, Loughlin J, and Shendure J (2019). Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun 10, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv13033997 Q-Bio. [Google Scholar]
- Li L, Huang K-L, Gao Y, Cui Y, Wang G, Elrod ND, Li Y, Chen YE, Ji P, Peng F, et al. (2021). An atlas of alternative polyadenylation quantitative trait loci contributing to complex trait and disease heritability. Nat. Genet [DOI] [PubMed] [Google Scholar]
- Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, Hess GT, Zappala Z, Strober BJ, Scott AJ, et al. (2017). The impact of rare variation on gene expression across tissues. Nature 550, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litterman AJ, Kageyama R, LeTonqueze O, Zhao W, Gagnon JD, Goodarzi H, Erle DJ, and Ansel KM (2019). A massively parallel 3’ UTR reporter assay reveals relationships between nucleotide content, sequence conservation, and mRNA destabilization. Genome Res. 29, 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Zhang Q, Wu J, Liang J, Yu S, Wei G-H, White KP, and Wang X (2017). Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 18, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg S, and Lee S-I (2017). A Unified Approach to Interpreting Model Predictions. ArXiv170507874 Cs Stat. [Google Scholar]
- Magoč T, and Salzberg SL (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinforma. Oxf. Engl 27, 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, and Bergmann S (2016). Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 13, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Haas U, Diaz R, Leeper NJ, Kundu RK, Patlolla B, Assimes TL, Kaiser FJ, Perisic L, Hedin U, et al. (2014). Coronary Heart Disease-Associated Variation in TCF21 Disrupts a miR-224 Binding Site and miRNA-Mediated Regulation. PLoS Genet. 10, e1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, and Keene JD (2008). Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol. Cell. Biol 28, 4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Wessels H-H, Lebedeva S, Sajek M, Ghanbari M, Garzia A, Munteanu A, Yusuf D, Farazi T, Hoell JI, et al. (2019). Deciphering human ribonucleoprotein regulatory networks. Nucleic Acids Res. 47, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzberg H, and Morrison CD (2015). Structure, production and signaling of leptin. Metabolism 64, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouioua S, Cheillan D, Zaouidi S, Salomons GS, Amedjout N, Kessaci F, Boulahdour N, Hamadouche T, and Tazir M (2013). Creatine deficiency syndrome. A treatable myopathy due to arginine–glycine amidinotransferase (AGAT) deficiency. Neuromuscul. Disord 23, 670–674. [DOI] [PubMed] [Google Scholar]
- Oikonomou P, Goodarzi H, and Tavazoie S (2014). Systematic identification of regulatory elements in conserved 3’ UTRs of human transcripts. Cell Rep. 7, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein Y, Wang Y, and Berger B (2016). RCK: accurate and efficient inference of sequence- and structure-based protein–RNA binding models from RNAcompete data. Bioinformatics 32, i351–i359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco LD, Chen H-H, Cox C, Katschke KJ, Arceo R, Espiritu C, Caplazi P, Nghiem SS, Chen Y-J, Modrusan Z, et al. (2020). Integration of eQTL and a Single-Cell Atlas in the Human Eye Identifies Causal Genes for Age-Related Macular Degeneration. Cell Rep. 30, 1246–1259.e6. [DOI] [PubMed] [Google Scholar]
- Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, NISC Comparative Sequencing Program, et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pembleton LW, Cogan NOI, and Forster JW (2013). StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour 13, 946–952. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PUNK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson K, Nettleton JA, Rotllan N, Tanaka T, Smith CE, Lai C-Q, Parnell LD, Lee Y-C, Lahti J, Lemaitre RN, et al. (2013). Gain-of-Function Lipoprotein Lipase Variant rs13702 Modulates Lipid Traits through Disruption of a MicroRNA-410 Seed Site. Am. J. Hum. Genet 92, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, and Seelig G (2019). Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol 37, 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazzini M, Schiavo G, De Fanti S, Martelli PL, Casadio R, and Luiselli D (2014). Searching for signatures of cold adaptations in modern and archaic humans: hints from the brown adipose tissue genes. Heredity 113, 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, et al. (2018). Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 175, 1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sharon E, Lubliner S, Regev I, Lotan-Pompan M, Yakhini Z, and Segal E (2015). Systematic dissection of the sequence determinants of gene 3’ end mediated expression control. PLoS Genet 11, e1005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J, Zhang F, Chen J, Zhou G, Ji W, et al. (2011). Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet 43, 1224–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DA, Tonqueze OL, Biton A, Zaitlen N, and Erle DJ (2020). Massively Parallel Analysis of Human 3’ UTRs Reveals that AU-Rich Element Length and Registration Predict mRNA Destabilization (Genomics). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood P, Krek A, Zavolan M, Macino G, and Rajewsky N (2006). Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci 103, 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, Sidore C, Faà V, Floris M, Deiana M, et al. (2017). Overexpression of the Cytokine BAFF and Autoimmunity Risk. N. Engl. J. Med 376, 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P, He L, Cui J, Qian C, Cao X, Lin M, Zhu Q, Li Y, Xing C, Yu X, et al. (2017). Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Mol. Cell 68, 293–307.e5. [DOI] [PubMed] [Google Scholar]
- Tang W, Apostol G, Schreiner PJ, Jacobs DR, Boerwinkle E, and Fornage M (2010). Associations of lipoprotein lipase gene polymorphisms with longitudinal plasma lipid trends in young adults: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circ. Cardiovasc. Genet 3, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. (2016). Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tushev G, Glock C, Heumüller M, Biever A, Jovanovic M, and Schuman EM (2018). Alternative 3’ UTRs Modify the Localization, Regulatory Potential, Stability, and Plasticity of mRNAs in Neuronal Compartments. Neuron 98, 495–511.e6. [DOI] [PubMed] [Google Scholar]
- Ulirsch J. in prep. [Google Scholar]
- Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, et al. (2016). Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbut SM, Wang G, Carbonetto P, and Stephens M (2019). Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet 51, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainberg Slutskin I, Weingarten-Gabbay S, Nir R, Weinberger A, and Segal E (2018). Unraveling the determinants of microRNA mediated regulation using a massively parallel reporter assay. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainberg Slutskin I, Weinberger A, and Segal E (2019). Sequence determinants of polyadenylation-mediated regulation. Genome Res. 29, 1635–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, et al. (2008). Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol. Cell 29, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and Stephens M (2021). Empirical Bayes Matrix Factorization. ArXiv180206931 Stat. [PMC free article] [PubMed] [Google Scholar]
- Wang G, Sarkar A, Carbonetto P, and Stephens M (2020a). A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B Stat. Methodol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QS, Kelley DR, Ulirsch J, Kanai M, Sadhuka S, Cui R, Albors C, Cheng N, Okada Y, The Biobank Japan Project, et al. (2020b). Leveraging supervised learning for functionally-informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs (Genomics). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, and Kellis M (2016). HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. (2014). The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EK, Moore-Jarrett T, and Ruley HE (2001). PUM2, a novel murine puf protein, and its consensus RNA-binding site. RNA N. Y. N 7, 1855–1866. [PMC free article] [PubMed] [Google Scholar]
- Wiegerinck CL, Janecke AR, Schneeberger K, Vogel GF, van Haaften–Visser DY, Escher JC, Adam R, Thöni CE, Pfaller K, Jordan AJ, et al. (2014). Loss of Syntaxin 3 Causes Variant Microvillus Inclusion Disease. Gastroenterology 147, 65–68.e10. [DOI] [PubMed] [Google Scholar]
- Wu X, Wang J, Wang S, Wu F, Chen Z, Li C, Cheng G, and Qin FX-F (2019). Inhibition of Influenza A Virus Replication by TRIM14 via Its Multifaceted Protein–Protein Interaction With NP. Front. Microbiol 10, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Pollack JL, Blagev DP, Zaitlen N, McManus MT, and Erle DJ (2014). Massively parallel functional annotation of 3’ untranslated regions. Nat. Biotechnol 32, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Jia X, Xue Q, Dou Z, Ma Y, Zhao Z, Jiang Z, He B, Jin Q, and Wang J (2014). TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. U. S. A 111, E245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1: Polysomal RNA-derived MPRAu metrics are correlated with Total RNA-derived MPRAu metrics, related to Figure 1.
a, HEK293 Polysomal RNA-derived MPRAu activity correlation with total RNA-derived HEK293 MPRAu activity. b, HEK293 Polysomal RNA-derived MPRAu skew correlation with HEK293 total RNA-derived MPRAu skew for significant hits (BH p-adj<0.1 for either Polysomal RNA or total RNA variants).
Supplementary Fig. 2: mash covariance structure with the largest weight demonstrates extensive allelic effect sharing amongst tamVars, related to Figure 1.
a, mash covariance matrix weights. b, Correlation matrix of the covariance structure with the highest weight (ED SVD) from a.
Supplementary Fig. 3: 3’UTR annotations with known effects provide evidence of MPRAu’s functionality, related to Figure 2.
a, Each box in the heatmap measures the significance of attenuation (t-test) when cell-type-specific MPRAu-measured 3’UTR allelic skews (y-axis) are subsetted on the variants with sequence perturbations that lead to a gain of a top 10 most abundant miRNAs across the cell types tested (x-axis). b, Magnitude of MPRAu activity when subsetting on RBP motifs derived from Dominguez et al., 2018. c, MPRAu allelic skews of 3’UTR variants with sequence perturbations that lead to a gain a top 20 barcode with negative activity (ranked by p-adj) versus MPRAu skews from 3’UTR variants that do not gain such barcodes. d, MPRAu allelic skews of 3’UTR variants with sequence perturbations that lead to a gain a top 200 barcode with positive activity (ranked by p-adj) versus MPRAu skews from 3’UTR variants that do not gain such barcodes. For b-d, reported p-values are derived from two-sided Wilcoxon rank-sum tests.
Supplementary Fig. 4: xgboost modeling additional information, related to Figure 3.
a, xgboost precision-recall curve across all six tested cell types (right). xgboost ROC curve across all six tested cell types (left). b, SVM precision-recall curve across all six tested cell types (right). SVM ROC curve across all six tested cell types (left). c, Logistic regression precision-recall curve across all six tested cell types (right). Logistic regression ROC curve across all six tested cell types (left). d, One-variable percent U decision tree precision-recall curve across all six tested cell types (right). One-variable percent U decision tree ROC curve across all six tested cell types (left). e, xgboost precision-recall curve across all six tested cell types for predicting augmenting activity (right). xgboost ROC curve across all six tested cell types for predicting augmenting activity (left). f, xgboost precision-recall curve across all six tested cell types for the full model, incorporating the top 100 expressed miRNAs for each cell type and RNAcontext-k-mer scores from expressed RBPs for each cell type, for predicting attenuation (right). xgboost ROC curve across all six tested cell types for the full model for predicting attenuation (left). g, xgboost precision-recall curve across all six tested cell types for using only miRNA and RBP-specific features for predicting attenuation (right). xgboost ROC curve across all six tested cell types using only miRNA and RBP-specific features for predicting attenuation (left). h, SHAP summary plot for the top 10 most important features in Fig. 3c. Each individual point plotted shows the effect of each feature from each training data point towards attenuation prediction. The features are ordered in terms of feature importance (top most important), and colored by feature value. Higher SHAP values denote greater contribution of prediction towards attenuation. “↑” next to variable name indicates higher values of the variable contribute to greater attenuation. Similarly, “↓,“ indicates lesser values of the variable contribute to greater attenuation. Some features, such as pU, display subtle non-linear effects which are not captured here.
Supplementary Fig. 5: Geuvadis eQTLs and GTEx fine-mapped eQTL concordance with MPRAu results, related to Figure 4.
a, Geuvadis eQTL effect size correlation with GM12878 3’UTR MPRAu allelic skews. b, Correlation between all-tissue median eQTL allelic skew (posterior beta) in GTEx that have a high probability of being causal via genetic fine-mapping (PIP>0.2) and tamVar (BH p-adj<0.1) median skew across all cell types. c, The same as in b, except with the all-tissue median eQTL allelic skew calculated only on points with PIP<0.01. Pearson’s r and its statistical significance are displayed.
Supplementary Fig. 6: Additional information relevant to 3’UTR variants functionally characterized, related to Figures 5 and 6.
a, Luciferase assay results on rs34448361. b, The alternate allele (insertion that extends the AU-rich element overlying the variant) has high frequency specifically in East Asian populations. c, rs34448361(green star) lies specifically in the 3’UTR of the longest isoform of LEPR ENST00000349533 (LepRb). d, Multiple sequence alignment across 30 mammals show that rs11771799, rs35986051, and rs11761306 (all in LD with rs705866) are in bases without strong conservation. e, Protein structure of PILRB demonstrates that rs11771799, rs35986051, and rs11761306 (green) lie far from the active site and are likely non-functional mutations. Shown in yellow are residues within 5 Å of the sialic acid ligand (purple). f, Allelic skew effects from the second replicate of a HDR allelic replacement of rs705866 (VE: Fisher p=1.43x10−15, MP: Fisher p=5.52x10−122). g, Allelic skew effects from the second replicate of a HDR allelic replacement of rs1059273 in GM12878 (VE: Fisher p=5.40x10−76, MP: Fisher p=1.83x10−287). VE: variant edited, MP: motif perturbed.
Supplementary Fig. 7: Additional sequence architectures via MPRAu SNV and deletion tiling, related to Figure 5.
For a-c, Top, 5 bp deletion tiling of the targeted variant using the ref (red) or alt (blue) sequence context across 100 bp. Bottom, SNV tiling +/− 10 bp around each variant showing log2 fold change and motif enrichment score in both variant contexts. Bottom, SNV tiling showing log2 fold change and motif enrichment score in both sequence contexts. HEK293 3’UTR activity data is plotted for all panels. a, Plots for tiling at rs632255 (CNTLN) with multiple AU-rich elements (AREs) denoted. b, Plots for tiling at rs482356 (STX3). c, Plots for tiling at rs1049508 (GATM).
Supplementary Table 1: MPRAu results, related to STAR Methods
Supplementary Table 2: Luciferase assay results, related to Figure 1
Supplementary Table 3: MPRAu barcode effects, related to STAR Methods
Supplementary Table 4: tamVars with gene expression and phenotype fine-mapping support, related to Figure 4
Supplementary Table 5: Primers, HDR ssODN/guides, related to STAR Methods
Data Availability Statement
Raw sequencing reads have been deposited to GEO, the ENCODE portal (https://www.encodeproject.org/awards/UM1HG009435/), and SRA. Processed MPRAu screen data are available in Supplementary Table 1 and will also be available on the ENCODE portal. Read counts per oligo are included in Supplementary Table 1.
Analyses was performed with standard analysis packages cited in the text, and plotted using custom R scripts that are available upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.