

Abstract
Long noncoding RNAs (lncRNAs) are emerging as key players in cancer as parts of poorly understood molecular mechanisms. Here, we investigated lncRNAs that play a role in hepatocellular carcinoma (HCC) and identified NIHCOLE, a novel lncRNA induced in HCC with oncogenic potential and a role in the ligation efficiency of DNA double-stranded breaks (DSB). NIHCOLE expression was associated with poor prognosis and survival of HCC patients. Depletion of NIHCOLE from HCC cells led to impaired proliferation and increased apoptosis. NIHCOLE deficiency led to accumulation of DNA damage due to a specific decrease in the activity of the non-homologous end-joining (NHEJ) pathway of DSB repair. DNA damage induction in NIHCOLE-depleted cells further decreased HCC cell growth. NIHCOLE was associated with DSB markers and recruited several molecules of the Ku70/Ku80 heterodimer. Further, NIHCOLE putative structural domains supported stable multimeric complexes formed by several NHEJ factors including Ku70/80, APLF, XRCC4, and DNA Ligase IV. NHEJ reconstitution assays showed that NIHCOLE promoted the ligation efficiency of blunt-ended DSBs. Collectively, these data show that NIHCOLE serves as a scaffold and facilitator of NHEJ machinery and confers an advantage to HCC cells, which could be exploited as a targetable vulnerability.
INTRODUCTION
Long noncoding RNAs (lncRNAs) have emerged as essential regulators of cell physiology and are involved in the onset and progression of several diseases, including hepatocellular carcinoma (HCC) (1,2). Although most lncRNAs lack protein-coding potential, they are biochemically indistinguishable from messenger RNAs (mRNAs) (3). However, compared to mRNAs, lncRNAs accumulate more in the cell nucleus, are less abundant, and much more tissue- and tumor-specific (4,5).
Recently, a significant focus has been placed on the role of lncRNAs in the DNA damage response (DDR). The accumulation of DNA damage due to defects in the DDR is the main contributor to the genomic instability that characterizes most cancer cells (6). DNA damage can arise from endogenous sources such as transcriptional and replicative stress or from genotoxic insults caused by ionizing radiation (IR) and many chemotherapeutic agents (7). The DDR is divided into subpathways in charge of repairing distinct types of damage, with double-stranded breaks (DSBs) among the most toxic type of DNA lesions. The cell has evolved two main DSB repair mechanisms: homologous recombination (HR) and non-homologous end-joining (NHEJ). HR is error-free as it uses a sister chromatid as a template; therefore, it is only available in the S and G2 phases of the cell cycle and is significantly slower than NHEJ. NHEJ is the preferred pathway to repair DSBs in eukaryotic cells; it is faster-acting and available throughout the cell cycle. However, it is error-prone and usually generates small indels at the site of repair (8).
NHEJ occurs in a stepwise manner and requires the coordinated recruitment and assembly of a repertoire of core and accessory proteins (9). The Ku70/Ku80 heterodimer (Ku) binds to DNA ends at DSBs and recruits the DNA-dependent Protein Kinase catalytic subunit (DNA-PKcs) to assemble the DNA-PK holoenzyme in a flexible long-range synaptic complex. The DNA-PK complex facilitates the processing of incompatible DNA ends while setting the stage for later events (10,11). Through molecular interactions that remain to be fully understood, factors such as Ku, APLF, XLF and XRCC4, fine-tune the alignment of compatible DNA ends in a more compact short-range synaptic complex that is competent for ligation by DNA ligase IV (LIG4), restoring the integrity of the phosphodiester backbone of DNA (11–14). Depending on the complexity of the DNA ends, additional factors may be required for successful ligation (15), and new NHEJ factors continue to be described to promote the assembly and activity of the core NHEJ machinery (12,16). Interestingly, some of these NHEJ factors can bind RNA. In fact, lncRNAs LINP1, SNHG12, and LRIK can interact with components of the NHEJ machinery and contribute to the DDR (17–19).
Here we describe a novel lncRNA that we named NIHCOLE (Noncoding RNA Induced in Hepatocellular Carcinoma with an Oncogenic role in Ligation Efficiency). NIHCOLE binds Ku, supports the formation of multimeric NHEJ complexes, increases ligation efficiency, and is required for effective DNA repair in HCC cells. NIHCOLE depletion increases radiosensitivity of HCC cells, suggesting that NIHCOLE upregulation may be an advantageous malignant adaptation that could be targeted with NIHCOLE inhibitors as a novel therapy for HCC.
MATERIALS AND METHODS
Cell lines and cell transfection
JHH6 cells were kindly provided by Dr. Jessica Zucman-Rossi (INSERM, Paris, France). Huh7 cells were kindly provided by Dr. Chisari (Scripps Research Institute, California, USA). JHH6 and Huh7 cells were not authenticated in our lab; however, the main results with these cell lines were corroborated in recently purchased stocks (2021) from JCRB, Japan (Tebu-bio). All other cell lines, HCC-derived: HEP3B, HepG2 and PLC, SK-HEP-1 (liver adenocarcinoma), 293T (embryonic kidney), A549 (lung cancer) and BJ (foreskin fibroblasts), were obtained from ATCC. JHH6 cells were cultured in Williams Medium E (ThermoFischer, 22551-022). All other cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, 41965-039). All media were supplemented with 2 mM L-Glutamine (Gibco, 25030024), 1% Penicillin/Streptomycin (Gibco, 15140122) and enriched with 10% fetal bovine serum (Gibco, 10270106). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. Cell lines were routinely tested for Mycoplasma sp. contamination using the MycoAlert kit (Lonza, LT07-318) following manufacturer’s instructions. Cells were transfected using Lipofectamine 3000 (Invitrogen) following manufacturer instructions. A final concentration of 50 nM of LNA-gapmers (Qiagen) was transfected in all knock-down experiments. For all plasmid transfections 250 ng of each plasmid were used.
Cell proliferation
Cell proliferation was measure by MTT assay or cell number. For the MTT assay, cells were either transfected directly onto 24-well plates or transfected, trypsinized, counted and re-plated onto 96-well plates at 5000 cells per well. After three hours of incubation with 0.5 mg/ml MTT (Sigma, M5655), formazan crystals were solubilized with 100μl of 1:1 DMSO:methanol and quantified at 570 nm using the SPECTROstar Nano 96-well plate reader (BMG Labtech). Alternatively, cell proliferation was assessed by cell number using automated dual-fluorescence imaging with acridine orange and propidium iodide in a Cellometer K2 Cell Counter (Nexcelom Bioscience) following manufacturer’s instructions.
Human samples
Three different sources of human samples were used. First, public clinical and histological data of patients with HCC from the TCGA (Supplementary Table S1A, TCGA cohort). Second, human HCC and peritumoral samples from patients who underwent hepatic resection or liver transplantation from January 2011 to December 2017 in two hospitals, Clínica Universidad de Navarra in Pamplona and Hospital Clinic in Barcelona (Supplementary Table S1A, BCL-CUN cohort). The study was approved by the Institutional Ethics Committee of each hospital (reference number 121/2015). Third, data from an external cohort of 198 patients from INSERM, Paris (20) (Supplementary Table S1B). All studies were conducted in accordance with the Declaration of Helsinki ethical guidelines. Informed written consent was obtained from all subjects. Clinical and histological features, as well as relevant outcomes, were obtained from medical records. Missing data were not replaced for analysis.
RNA immunoprecipitation
Precipitation of RNA bound to target proteins was performed as previously described with modifications (21). Cell extracts were prepared from sub-confluent 15 cm plates, rinsed twice with cold 1x PBS and crosslinked with either 0.5% formaldehyde for 10min (FA, ThermoScientific, 28908) or UV-light to with 1500 mJ at 254 nm. RIP was performed with whole-cell extracts obtained by shearing 1.0×107 cells in 1 ml of ice-cold RIP Buffer [150 mM KCl, 25 mM Tris-HCl pH 7.4, 5 mM EDTA, 0.5% NP40, 0.5 mM DTT, 100 U/ml RNasin (Promega, N2115) and cOmplete Protease Inhibitor Cocktail (Roche, 11697498001)] with 20 strokes using a Dounce homogenizer. For the RIP using Ku80 and γH2AX antibodies, extracts were sonicated after IR 20 times for 10 seconds in 30-second intervals. All extracts were cleared of debris by centrifugation at 16000x g for 10 min and divided into 200 μl aliquots for different conditions. A 10% volume aliquot was set aside as input. An isotypic IgG and antibodies against DNA-PKcs (Bethyl, A300-516A, Abcam, ab70250), Ku80 (Abcam, ab232381), U1A (Abcam, ab166890) and γH2AX (Abcam, ab81299) were added into the aliquoted extracts (5 μg/200 μl extract) and incubated overnight with gentle rotation. The next day, 40 μl of pre-washed protein G magnetic beads (Invitrogen, 10003D) were added to each tube and further incubated for 1 h. After that, beads were washed 3 times with 500 μl of ice-cold RIP Buffer. Next, 200 μl of Reverse-crosslinking Buffer (2.5 mg/ml Proteinase K (Roche, 3115828001), 0.6% SDS, 60 mM Tris-HCl pH 7.4) was added to the beads of FA- or UV-crosslinked samples and incubated for 45 min at 65°C. Finally, beads were resuspended in 0.5 ml of TRIzol reagent for extraction of total coprecipitated RNA. RNA was reverse transcribed and the enrichment of NIHCOLE and control RNAs was measured with specific primers (Supplementary Table S1C) by qRT-PCR (Supplementary methods). All centrifugations and incubations were carried out at 4°C.
RNA pulldown and mass spectrometry
Proteins bound to NIHCOLE were identified by mass spectrometry, based on a previously described protocol with modifications (22). In summary, around 1×107 cells were resuspended in 2 ml of ice-cold PBS plus 2 ml of ice-cold Nuclear Isolation Buffer (1.28 M sucrose, 40 mM Tris-HCl pH 7.4, 20 mM MgCl2 and 4 % Triton X-100 in DEPC water) and 6 ml of ice-cold DEPC water, mixed well and incubated 20 min with intermittent mixing. Nuclei were spun down by centrifugation at 2500 g for 15min and resuspended in 1 ml of freshly prepared RIP Buffer. Nuclei were sheared on ice with 20 strokes of a Dounce homogenizer and cleared by centrifugation at 16000 g for 10 min. Protein concentration of the supernatant (nuclear extract) was measured by Bradford assay and the nuclear extract was diluted with RIP Buffer to ~ 2.5 μg/μl. The nuclear extract was pre-cleared with Streptavidin T1 magnetic beads (Invitrogen, 65601) in RIP buffer for 1.5 h with rotation. After that, a 10 % volume aliquot was set aside as input. Then, 30 μg of in vitro transcribed biotinylated RNA was incubated with 650 μl of 0.1 μg/μl tRNA-supplemented RIP Buffer and 650 μl of the pre-cleared nuclear extract for 1 h in rotation. Next, 100 μl of pre-washed Streptavidin beads were added to the mix and incubated for one extra hour. Finally, the beads were then washed 3 times with 450 μl of tRNA-supplemented RIP Buffer and 2 more times with RIP Buffer. In the last wash, all the supernatant was carefully removed, and the beads were either sent for mass spectrometry analysis directly or resuspended in protein sample buffer, boiled at 95°C for 10 min, loaded and ran in precast 4–12 % Bis-Tris gel (Invitrogen, NP0321PK2) and stained with SilverQuest silver staining kit (ThermoFischer, LC6070). Alternatively, proteins were transferred to a nitrocellulose membrane and blotted for specific proteins as described in the methods for immunoblotting. All centrifugations and incubations were carried out at 4°C.
Immunofluorescence analysis
Cells were seeded onto 12 mm glass coverslips (Zeiss, 474030-9000-000). After treatment, cells were fixed with cold 3.7 % methanol-free FA for 10 min at RT and then washed with cold 1x TBS, permeabilized with 0.5 % Triton X-100 (Sigma, T8787) for 10 min at RT, washed once with 1x TBS and then incubated with 1 % BSA in TBS for 30 min at RT. Cells were washed twice with 1x TBST (0.05 % Tween in 1x TBS) and immunostained for 1 h with anti-γH2AX (Upstate: 05-636, used at 1:1000 dilution in 1 % BSA). After that, coverslips were washed 6 times with 1x TBST, and then incubated with Alexa 488 conjugated goat anti-mouse (Invitrogen, A28175, used at 1:500 dilution in 1 % BSA) for 30 min in the dark. Cells were washed 6 times with 1x TBST and counterstained with DAPI for 10 min at RT and briefly washed once with deionized water. Finally, coverslips were mounted on microscope slides with Fluoromount-G (Invitrogen, 00-4958-02) and analyzed with a Zeiss Axio ObserverZ1 platform microscope, with a Plan Apochromat 40×/1.3 (oil immersion) objective and an AxioCam MRm Rev.3 camera. Images were captured with Zen Pro (Zeiss) software and analyzed with the open-source software ImageJ.
Comet assay
The neutral comet assay was performed as previously described (23) with modifications. Briefly, control and treated cells were harvested and resuspended in 150 μl of 1x PBS at 50 000 cells/condition and mixed with 150 μl of 1.2 % low-melting-point agarose (Invitrogen, 16520050) prepared in 1x PBS and prewarmed to 40°C. The agarose cell suspension was quickly layered on top of the previously set 0.8% agarose coat and covered with a coverslip in duplicates and left at 4°C to set for 30 min. From this point and until after electrophoresis, slides were kept in the dark. During this time, the lysis buffer was supplemented with 1 % N-lauryl sarcosine, 0.5 % Triton X-100, and 10% DMSO. Then, slides were submerged in supplemented lysis buffer overnight in a dark container. After lysis, slides were washed 3 times with electrophoresis buffer with freshly added 1% DMSO and placed in a dark electrophoresis chamber covered with supplemented electrophoresis buffer for 30 min. Electrophoresis was carried out at 25 V for 25 min. Slides were briefly washed with water and stained with a 1:10000 dilution of SYBR gold (Invitrogen, S11494) with 4 μg/μl of antifade for 5 min at RT. Excess staining solution was removed and slides were scored on a Zeiss Axioskop microscope using the Comet Assay IV software (Perspective Instruments). At least 50 cells were scored per condition. Tail moment was used as the measurement of the DNA damage extent.
DSB repair reporter assays
The activity of DSB pathways was measured using GFP reporter assays for homologous recombination (DR-GFP), single-strand annealing (SSA-GFP), alternative end-joining (EJ2-GFP), total NHEJ (EJ5-GFP), and distal NHEJ events without indels (EJ7-GFP). The reporters DR-GFP, SSA-GFP, EJ2-GFPand EJ5-GFP were co-transfected with a plasmid encoding the I-SceI endonuclease to introduce a DSB at I-SceI sites in the reporter constructs. The EJ7-GFP reporter was co-transfected with a CRISPR-Cas9 plasmid to induce blunt DSBs at the recognition site of guide RNAs (sgRNA 7a and 7b) in the construct. All reporters were co-transfected with control or NIHCOLE-targetting gapmers. After 48 hours post-transfection GFP was measured in 1×105 cells by FACS. A GFP expression vector was co-transfected in parallel to measure transfection efficiency. DR-GFP (Addgene: 26475), SSA-GFP (Addgene: 41594) and I-SceI (Addgene: 26477) plasmids were a gift from Maria Jasin. EJ2-GFP (Addgene: 44025), EJ5-GFP (Addgene: 44026) and EJ7-GFP reporter, 7a and 7b sgRNA vectors (Addgene: 113617, 113620 and 113624, respectively) were a gift from Jeremy Stark. S. pyogenes CRISPR-Cas9 plasmid (Addgene: 52961) was a gift from Feng Zhang.
Atomic force microscopy
AFM measurements were performed by depositing a 20 μL of sample solution on a mica pre-treated with 50 μL of 30 mM spermidine for 10 minutes, as described previously (24). NIHCOLE samples contained 0.5 nM RNA in 25 mM TrisAc pH 7.5, 12.5 mM KCl, and 5 nM Ku70/80 when incubated with the protein. SM3 samples contained 0.25 nM RNA in 25 mM TrisAc pH 7.5, 12.5 mM KCl, and 10 nM Ku70/80 when incubated with the protein. In samples containing Ku70/80, both RNA and protein were incubated for 5 min at room temperature, prior deposition. After ~60 s of deposition on the mica, the sample was washed with 2 ml of Milli-Q water and dried using nitrogen. Images were taken in tapping mode in air, using an AFM from Nanotech Electronica S.L. with PointProbePlus tips (PPPNCH Nanosensors). Images were processed using the WSxM software (25).
Ku pulldown for electron microscopy
For the pulldown experiments full-length Ku70 containing a twin-strep-tag at the N-teminus and full-length Ku80 including a 10xHis-tag at the N-terminus were co-expressed in baculovirus as previously described (13) in order to produce the Ku heterodimer. Then 10 pmol of purified Ku was incubated with 20 μl Strep-Tactin XT 4Flow (IBA) pre-equilibrated in equilibration buffer EB (25 mM HEPES, pH 7.7, 50 mM NaCl, 1 mM EDTA and 10 % (w/v) glycerol). The mixture was incubated in a thermomixer (Eppendorf) for 20 min at 25°C and 800 rpm and then transferred to a spin column (SigmaPrep spin column, SIGMA) and centrifuged for 1 min at 6,000 rpm. The resin was firstly washed with 50 μl of EB buffer and spun for 1 min at 6,000 rpm. A second wash with 200 μl of EB buffer was performed followed by 1 min centrifugation at 6,000 rpm. The resin with bound Ku was next divided in two tubes: A, B and was further washed with 100 μl of EB buffer and spun for 1 min at 6,000 rpm. Resin in tube B was incubated with 4.25 pmol of NIHCOLE and 100 units of RNasin ribonuclease inhibitor (Promega) for 5 min at RT followed by 1 min centrifugation at 6,000 rpm. Next resins from both tubes were washed with 100 μl of EB buffer and spun for 1 min at 6,000 rpm. Finally bound sample was eluted after incubating resin from tubes A and B with EB buffer supplemented with 50mM biotin (IBA) for 5 min at 25 °C and 800 rpm in a thermomixer (Eppendorf) followed by 1 min centrifugation at 6,000 rpm.
Electrophoretic Mobility Shift Assay
Human Ku70/His-Ku80 was purified from baculovirus-infected insect cells (26) and human APLF and XRCC4 were expressed in bacteria as previously described (12,13). Unlabeled and 5’ FAM labeled RNA and DNA probes were RNase-free synthesized by Integrated DNA Technologies (IDT) (Supplementary Table S1D). Recombinant proteins were incubated with the labeled probes in 1x EMSA binding buffer (25 mM HEPES-KOH pH 7.5, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 10 % glycerol) supplemented with 0.1 μM ultrapure BSA (Invitrogen, AM2616) in a final volume of 20 μl. Samples were incubated at room temperature for 25–30 min in the dark. Then samples were loaded in non-denaturing 5 % acrylamide gels and run in 1x EMSA running buffer (50 mM Tris, 380 mM glycine, 2 mM EDTA) at 100V for 45 min in the dark. After that, gels were imaged using the ImageQuant LAS-4000 (Fujitsu Life Sciences) or a Chemidoc MP Imaging System (Bio-Rad).
NHEJ reconstitution assay
The ligation reactions were conducted in reaction buffer pH 7.5 (20 mM Tris-acetate, 75 mM KAc, and 10 mM MgCl2). In all reactions 40 nM of substrate were first incubated with 0.1 mg/ml neutravidin (Invitrogen, 22832) in reaction buffer for 5 min. Then 0.5 mM ATP, 1 mM DTT, 5% PEG-8000, RNAs, 50 nM Ku70/80 and 100 nM X4L4 were added into the DNA mixture to a final volume of 20 μl. The reaction solution was mixed well and incubated at 37 °C for 90min. After that, reactions were diluted to 48 μl with TE buffer and treated with RNase A (2μg/μl) for 30min RT. The mixture then went through phenol-chloroform (Sigma, 77617) extraction and ethanol precipitation. Then, the ligation products were resolved using a 15% native PAGE in 1x TBE, then gels were stained in a 1/10000 solution of SYBR safe (Invitrogen, S33102) in 1x TBE for 15min in agitation and were later imaged using a Chemidoc MP instrument. Quantitation of ligation product was performed using the ImageLab software (BioRad).
Data availability
RNA-seq data that support the findings of this study, have been deposited in the European Genome Archive (EGA) repository under the study ID accession number EGAS00001002879 (INSERM cohort) or can be accessed from http://cancergenome.nih.gov/ (TCGA cohort).
Statistical analyses
The GBA study was performed using the giTools software (27). Fisher’s exact test using higher/lower than median groups was used to find significant associations. Expression data are shown as means ± standard deviation (SD) or standard error of the mean (SEM) and statistical analyses were performed using Prism 9 (GraphPad Software). A descriptive analysis was carried out to analyze the distribution of the samples with D’Agostino and Pearson normality test. Non-parametric tests were used after normality failure. Differences between two groups were analyzed using two-tailed Student’s t-test or U-Mann Whitney, whereas differences between three groups were analyzed using the Kruskal–Wallis ANOVA-test followed by Dunn’s multiple comparisons test. TCGA paired samples were evaluated with paired t-tests while paired samples from the BCL-CUN cohort were analyzed with Wilcoxon matched-pairs signed-rank tests. Pearson correlation analysis was used to compare functions and associations between lncRNAs. The logrank test was used to compare survival curves between groups of patients. Differences were deemed significant for a real alpha of 0.05 (p < 0.05). Statistical significance is indicated by **** (p < 0.0001), *** (p < 0.001), ** (p < 0.01) or * (p < 0.05). ns indicates non-significant differences.
RESULTS
NIHCOLE is a lncRNA highly expressed in HCC patient samples that correlates with poor prognosis
To identify recurrently deregulated lncRNAs in HCC, we previously analyzed the TCGA and GTEx data sets in a pan-cancer and pan-tissue study (5). From this analysis, we selected LINC02163, which we denote here as NIHCOLE, since further studies showed high expression levels in HCC, good correlation with clinical parameters and positive association with cell cycle and DNA repair (see below). NIHCOLE is mostly uncharacterized, polyadenylated, and shows no significant coding potential, as addressed by PhyloCSF, CPAT, and CPC scores (28) (Fig. S1A). According to current annotations, NIHCOLE is an intergenic lncRNA with chromatin marks consistent with a promoter near its transcription start site (TSS) (Fig. S1B).
NIHCOLE was found significantly upregulated in HCC samples from the TCGA in the differential expression analysis of both peritumor-tumor pairs (Fig. 1A) and all tumors compared to peritumoral samples (Fig. 1B). We and others have found upregulation of NIHCOLE in other tumors (colorectal, gastric, head and neck, lung and breast cancers) (29–32). NIHCOLE expression was not observed in healthy tissues (median TPM ≤ 0.1) according to the GTEx dataset (https://gtexportal.org/home/). To validate TCGA findings, we measured the levels of NIHCOLE in an independent cohort (BCL-CUN) of HCC patient samples (Supplementary Table S1A). In agreement with TCGA data, NIHCOLE’s levels in tumor samples are significantly higher than in the paired peritumor samples or healthy liver biopsies by qRT-PCR (Fig. 1C, D) or RNA-seq in a subset of the paired samples (Fig. 1E).
Figure 1. Validation of NIHCOLE upregulation in HCC and clinical associations.

A, B NIHCOLE levels from RNA-Seq data were evaluated in paired peritumor (PT) and tumor (T) samples (n = 50 pairs) (A) or in all HCC samples (PT n = 50, T n = 374) deposited to TCGA (B). C-E Validation by qRT-PCR in the BCL-CUN cohort in all patient samples including healthy (H) liver biopsies (H n = 10, PT n = 17 and T n = 19) (C), in the paired PT/T pairs (n = 15 pairs) (D) or by RNA-seq (n = 10 pairs) (E). F, G Clinical associations with data from the TCGA (F, G) and INSERM cohort (n = 198) (F). See text for details. Scale, from lowest (red) to <0.05 (black) or >0.05 (blue), indicates p-value. H Survival probability of HCC patients categorized according to median NIHCOLE expression (n is indicated at each time point). I GBA analysis of NIHCOLE related coding-gene expression. Scale, from positive >10 (red) to negative <−10 indicates z-score. The significance of the statistical analysis (two-tailed Wilcoxon matched-pairs signed rank test in A, C, D, two-tailed Mann-Whitney test in B, Kruskal–Wallis ANOVA-test in E, two-tailed Student’s t test in F, G, and the logrank test in H) is indicated in each graph and summarized as: not significant (ns); * <0.05; **<0.01; ***<0.001; ****<0.0001. See algo Figures S1 and S2.
To evaluate the relevance of NIHCOLE overexpression in HCC, we studied its clinical associations. Using TCGA data, we found that NIHCOLE associates with previously published molecular classifications (Hoshida’s and iCluster) (33,34) and clinical parameters such as the presence of the macrotrabecular massive (MTM) histopathological finding, a feature of the G3 molecular subgroup in Bouyault’s classification that is strongly associated with bad prognosis (Fig. 1F) (35). Similarly, significant associations were found with expression levels of TERT-related genes and mutations in the TERT promoter, TP53, and CTNNB1, which are well-known HCC drivers. In these analyses, we observed that the expression levels of NIHCOLE could segregate patients according to molecular classifications and were significantly higher in samples from patients with more aggressive phenotypes (i.e., high expression levels of TERT-regulated genes and mutated TP53) (Fig. 1G).
Clinical annotations of HCC samples from the TCGA are limited. Therefore, we analyzed NIHCOLE’s associations in an additional thoroughly annotated cohort of 198 patients (INSERM) (20). In this cohort, around 90% of the patients present underlying liver cirrhosis with diverse etiologies that better recapitulate the natural history of HCC (Supplementary Table S1B). Analysis of this cohort showed a significant DNA copy number gain in NIHCOLE’s genomic location (Fig. S1C, D) which can partially explain (in about 20% of the samples) the significant upregulation of NIHCOLE (p = 3.0 × 10−10) (Fig. S2A) and correlates with Boyault’s transcriptomic groups G3 and G6 (36), and with TP53 and CTNNB1 gene mutations as in the TCGA data (Fig. 1F and Fig. S2B–D). Remarkably, when patients were stratified according to NIHCOLE expression levels, there was a significant correlation between decreased patient survival and high expression of NIHCOLE (Fig. 1H). NIHCOLE upregulation was also associated with poor prognosis as per the 5-gene score (37) (Fig. S2E) and the presence of vascular invasion (Fig. S2F); and with advanced disease stage (according to the Barcelona Clinic Liver Cancer (BCLC) staging) and less differentiated tumors (following the World Health Organization (WHO) differentiation score) (Fig. 1F and Fig. S2G, H). Interestingly, since NIHCOLE expression associates with aggressive tumors and CTNNB1 mutations, which are usually less lethal tumors, we evaluated whether NIHCOLE levels can segregate the CTNNB1-mutated tumors with a more aggressive phenotype. Indeed, CTNNB1-mutated tumors with higher levels of NIHCOLE are significantly associated with a worse prognosis using the 5-gene score (Fig. S2I).
To predict the function of NIHCOLE, we performed a guilt-by-association (GBA) analysis. The results indicate that expression of NIHCOLE correlates positively (z-score >10) with cell cycle, DNA repair, and gene expression, and negatively (z score <10) with cell adhesion and motility, signal transduction, and the immune response, all of them key cancer hallmarks (Fig. 1I).
NIHCOLE is required for HCC cell proliferation
We measured NIHCOLE’s expression levels in cell lines derived from HCC (JHH6, HEP3B, PLC/PRF/5, Huh7 and HepG2) or, as controls, liver adenocarcinoma (SK-HEP-1), embryonic kidney (293T), and lung tumor (A549). NIHCOLE is expressed to higher levels in the HCC cancer cell lines JHH6, Huh7, HepG2 and PLC but not in HEP3B or the other non-HCC cell lines (Fig. 2A). Absolute copy number quantification of NIHCOLE revealed that it is indeed highly expressed to an average of 293 ± 33 copies per JHH6 cell, whereas Huh7 cells express around 385 ± 77 copies per cell. Expression analysis from RNA-seq data in a collection of 33 HCC-derived cell lines categorized in an epithelial-mesenchymal transition (EMT) differentiation gradient (38), showed similar expression levels of NIHCOLE in all three clusters without significant differences between groups (Fig. 2B), suggesting NIHCOLE’s levels are unlikely related to EMT.
Figure 2. Characterization of NIHCOLE expression and depletion in HCC cells.
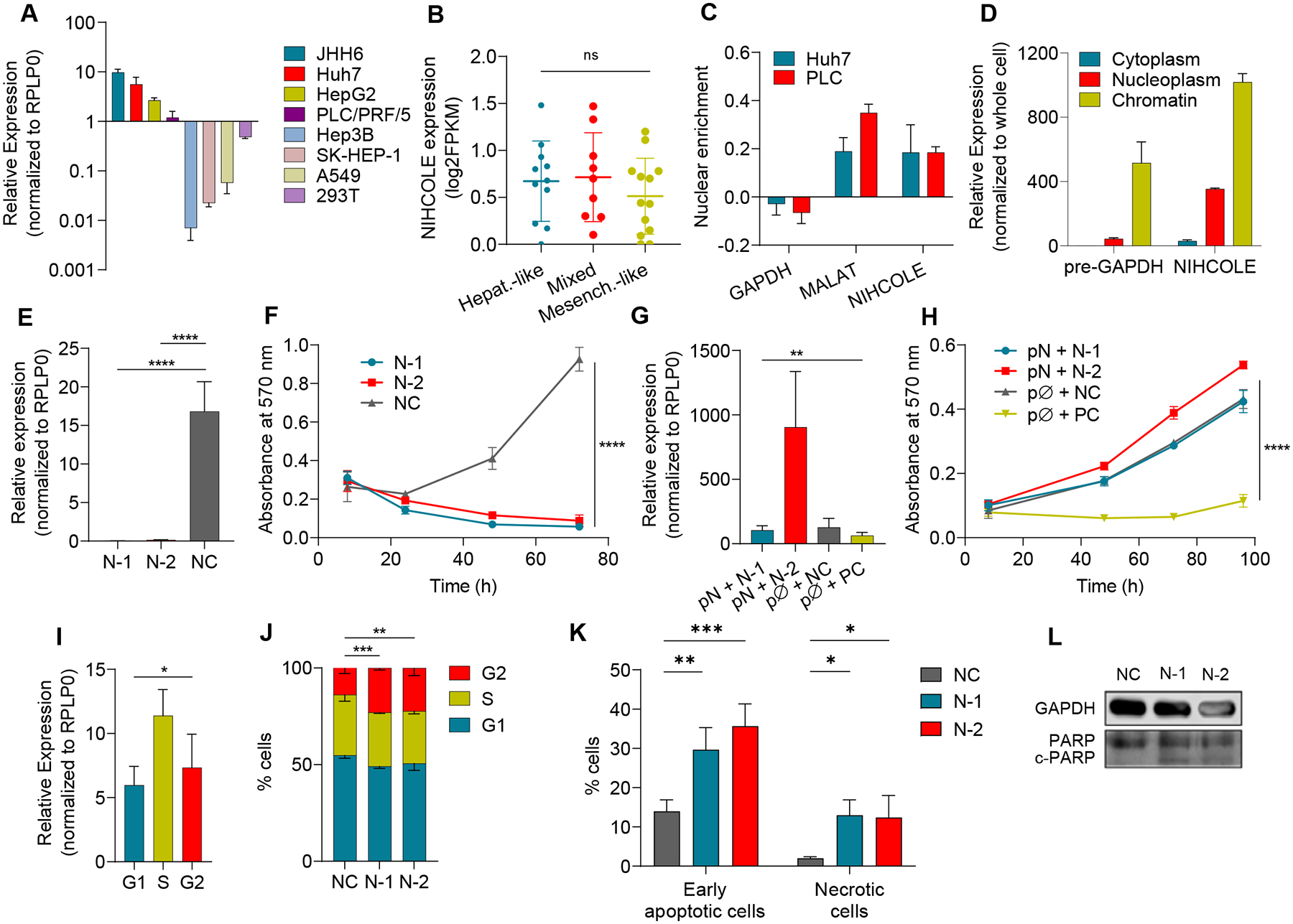
A NIHCOLE levels in HCC and non-HCC cell lines (in lighter shades) by qRT-PCR. Mean ± SD (n = 3–6) B Expression of NIHCOLE from RNA-Seq data of 33 HCC cell lines in a differentiation gradient from hepatoblast-like (Hepat.-like, n = 11), mixed epithelial-mesenchymal (Mixed, n = 9) to mesenchymal-like (Mesench.-like, n = 13). C, D Enrichment of NIHCOLE in the nucleus versus cytoplasm (C) or nucleoplasm versus chromatin (D). Mean ± SD (n = 3). GAPDH mRNA or pre-mRNA and MALAT1 levels were used as controls. E, F Huh7 cells were transfected with two specific gapmers (N-1 and N-2) and a non-targeting negative control (NC). NIHCOLE expression 48 hours post-transfection (E; mean ± SD (n = 5)) and proliferation by MTT assays (F; mean ± SEM (n = 3)). G, H Adding back experiments. NIHCOLE expressing plasmid (pN) was co-transfected with NIHCOLE’s gapmers. Empty plasmid (pØ), NC gapmer or an ACTN1-specific gapmer (PC) were used as controls. NIHCOLE levels were measured 48h post-transfection (n = 3) (G) and proliferation was measured by MTT assays (H). Mean ± SEM (n = 3). I NIHCOLE expression by qRT-PCR in synchronized Huh7 cells. Mean ± SD (n = 3). J Cell cycle distribution of NIHCOLE-depleted cells. Mean ± SD (n = 3) K, L Apoptosis was measured by Annexin V staining and FACs analysis (K) and by immunoblotting of PARP cleavage (c-PARP) (L) in control and NIHCOLE-depleted cells. GAPDH was used as loading control (n = 3). The significance of the statistical analysis (two-tailed Student’s t test in B, E, F, G, H, I and two-way ANOVA in J) is indicated and summarized as: not significant (ns); * <0.05; **<0.01; ***<0.001; ****<0.0001. See also Figure S3.
Subcellular fractionation shows that NIHCOLE is highly enriched in the nucleus of HCC cells (Fig. 2C). This enrichment is mostly due to its retention in the chromatin fraction, as happens with GAPDH pre-mRNA used as control (Fig. 2D). Nuclear enrichment disfavors efficient targeting by siRNAs. Therefore, we designed two antisense LNA-gapmers against NIHCOLE that target sequences in exon three (N-1) and intron three (N-2) (Fig. S1B). NIHCOLE levels were efficiently decreased upon gapmer transfection compared to a non-targeting negative control (NC) (Fig. 2E). Analysis of the expression of neighboring genes sharing the same TAD as NIHCOLE showed no alteration after NIHCOLE depletion. More importantly, NIHCOLE depletion dramatically decreased cell numbers in NIHCOLE-expressing cells (Fig. 2F and Fig. S3A) while cells without NIHCOLE expression remained unaffected (Fig. S3B). To validate the specificity of this effect, we cloned the cDNA of NIHCOLE in a mammalian expression vector and performed adding-back experiments by co-transfecting the NIHCOLE-expressing vector (pN) or an empty vector (pØ) with control or NIHCOLE-targeting gapmers. Of note, much higher levels of NIHCOLE were observed for N-2 as this gapmer targets an intronic sequence that is not found in the cloned transcript (Fig. 2G). Re-expression of NIHCOLE restored the proliferation of NIHCOLE-depleted HCC cells (Fig. 2H). As expected, this effect was not observed in cells co-transfected with a positive control gapmer (PC) that affects cell number by targeting the essential structural protein actinin alpha-1 (ACTN1) (Fig. 2H). Of note, NIHCOLE overexpression does not significantly increase cell proliferation beyond basal levels (Fig. 2H and Fig. S3C, D). Overall, these results support specific targeting of NIHCOLE and strongly suggest NIHCOLE is required for the growth of HCC cells, likely through a trans-acting mechanism. Further characterization of NIHCOLE expression showed that NIHCOLE is more highly expressed in S-phase cells (Fig. 2I). Interestingly, depletion of NIHCOLE leads to significant G2/M transition arrest (Fig. 2J), apoptosis and necrosis (Fig. 2K, L), supporting the involvement of NIHCOLE in cell cycle-regulated events such as cell cycle progression and DNA damage repair as suggested by the GBA analysis.
NIHCOLE binds to effector proteins of the NHEJ pathway of DSB repair
To gain mechanistic insights into NIHCOLE’s function, we studied the NIHCOLE-binding proteome. Since NIHCOLE is highly enriched in the nucleus, we incubated Huh7 nuclear extracts with in vitro transcribed (ivt) and biotinylated full-length NIHCOLE (Fig. 3A). After precipitation with streptavidin beads, electrophoresis, and silver staining, we observed a differential high molecular weight band coprecipitating with NIHCOLE and not with the control RNA (Fig. 3B, blue arrow). We excised the region of the differential band and a similar region in the control lane and analyzed them by mass-spectrometry (MS). This analysis identified a significant enrichment for DNA-PKcs (Fig. 3C, Supplementary Table S1E). DNA-PKcs binds to the Ku70/80 heterodimer through the C-terminal domain of Ku80 to form the DNA-PK complex, part of the NHEJ machinery of DSB repair. Therefore, we analyzed the NIHCOLE-bound material by immunoblotting for the presence of DNA-PKcs, Ku80, and the abundant RNA-binding protein (RBP) U1A used as a control. We could detect DNA-PKcs and Ku80 but not U1A, while no interaction was observed in the absence of RNA (Fig. 3D). Similarly, when we used crosslinked nuclear extracts and specific DNA-PKcs and Ku80 antibodies or control IgG to capture RNAs by immunoprecipitation (Fig. 3E), we found, by qRT-PCR, a significant enrichment for NIHCOLE but not for control RNA or ACTB mRNA (Fig. 3F, G).
Figure 3. Proteomic analyses of NIHCOLE-binding proteins.
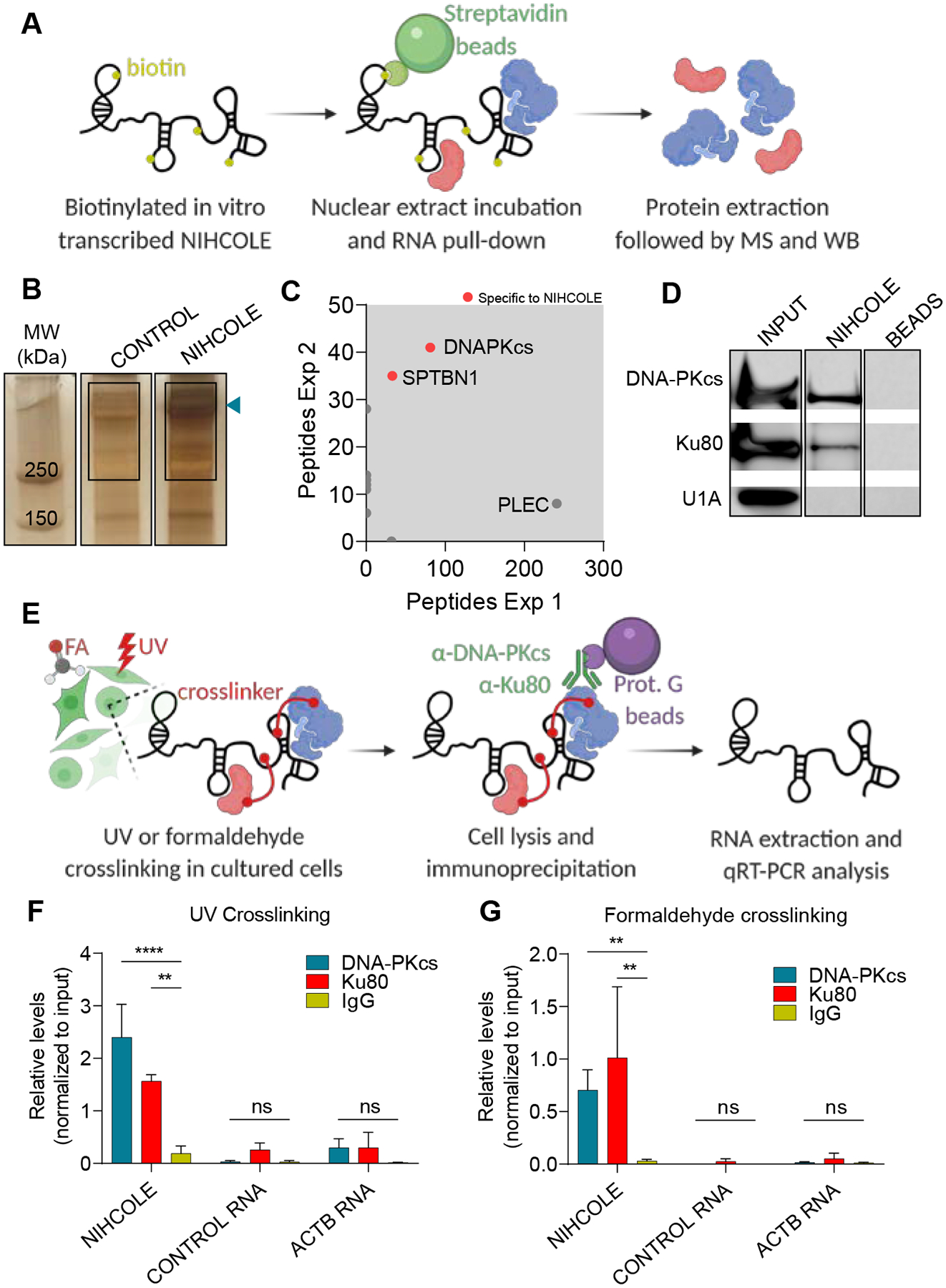
A Schematic representation of the RNA pull-down and mass spectrometry analysis. B Silver staining of protein complexes bound to NIHCOLE or control RNAs. Blue arrow, differential band. Rectangles, gel fragment sent for MS. C NIHCOLE-bound proteins identified in two independent experiments are plotted according to peptide enrichment. Specific NIHCOLE interactors are highlighted with red dots while unspecific interactors are depicted as gray dots. Proteins identified above 250 kDa are shown. D Immunoblotting for DNA-PKcs, Ku80 and U1A in the protein complexes obtained from the pull-down of NIHCOLE or beads alone. INPUT, 10% of the nuclear extract. E Schematic overview of the RNA immunoprecipitation assays. F, G Relative levels of NIHCOLE, control RNA, and actin-beta mRNA (ACTB) from UV (F) or formaldehyde (G) crosslinked co-precipitates, using Ku80 and DNA-PKcs specific antibodies and mouse IgG as non-specific control. Graph shows mean ± SD (n = 2, Ku80 antibody; n = 3 DNA-PKcs antibody). Significance of the statistical analysis (two-way ANOVA in F and G) is indicated and summarized as: not significant (ns); **<0.01; ***<0.001; ****<0.0001.
NIHCOLE-depleted cells accumulate DNA damage and show decreased NHEJ activity
Given that NIHCOLE interacts with DNA-PKcs and Ku80, we hypothesized that their binding could mediate a potential role of NIHCOLE in the DDR. To address this, we performed a single cell electrophoresis of damaged DNA also known as the “comet assay” (Fig. 4A). Quantitation of tail moment in control and NIHCOLE-depleted cells, untreated (24h after gapmer transfection) and at different time points after ionizing radiation, shows a drastic increase in tail moment at one-hour post-IR. The tail moment gradually returns to background levels after 24h in control treated cells. Surprisingly, NIHCOLE-depleted cells show a similar pattern than control cells except at 24h post-IR, when a highly significant accumulation of damage is observed (Fig. 4B). To corroborate this result, we evaluated the number of DNA damage foci by immunostaining of histone H2AX serine 139 phosphorylation, also known as γH2AX, one of the most widely used read-outs of DNA damage. We measured γH2AX foci of control and NIHCOLE-depleted in a similar scheme as in the comet assay (Fig. 4C). Basal levels of γH2AX foci were observed in cells at 24 hours after transfection (No IR). This is likely due to the high transcriptional stress of these cells, which can be a considerable source of DSBs. After IR, as with the comet assay, NIHCOLE-depleted cells show a highly significant accumulation of damage at 24h post radiation, contrary to control cells (Fig. 4D). A similar increase in the number of foci is observed when cells are treated with the DNA-PKcs inhibitor NU7441 instead of IR. Remarkably, both γH2AX and Ku80 antibodies are able to immunoprecipitate NIHCOLE at early times after radiation (Fig. 4E and F), suggesting that NIHCOLE accumulates in the vicinity of DNA damage sites, especially since NIHCOLE levels do not change significantly after damage (Fig. S3E). These results support that NIHCOLE depletion could lead to a significant defect in DSB repair. To evaluate if defective NHEJ mediates this defect, we used reporter assays in which NHEJ-mediated repair reconstitutes a GFP gene in a substrate plasmid cleaved by the I-SceI endonuclease to measure total NHEJ activity (39), or cleaved by CRISPR-Cas9 to measure distal NHEJ without indels (40). Both reporter assays show significantly decreased NHEJ repair efficiency in NIHCOLE-depleted Huh7 (Fig. 4G) and JHH6 (Fig. 4H) cells compared to the same number of control cells. Importantly, NHEJ activity was unaltered upon NIHCOLE gapmer transfection in cells that do not express NIHCOLE (Fig. S3F). Further, the defect observed upon NIHCOLE depletion is specific for NHEJ-mediated repair, since reporter assays to measure alternative DSB repair pathways including homologous recombination (HR), single-strand annealing (SSA), and alternative end-joining (Alt-EJ), did not show differences between NIHCOLE-depleted and control Huh7 or JHH6 cells (Fig. S3G–I).
Figure 4. Evaluation of DNA damage defects in NIHCOLE-depleted HCC cells.
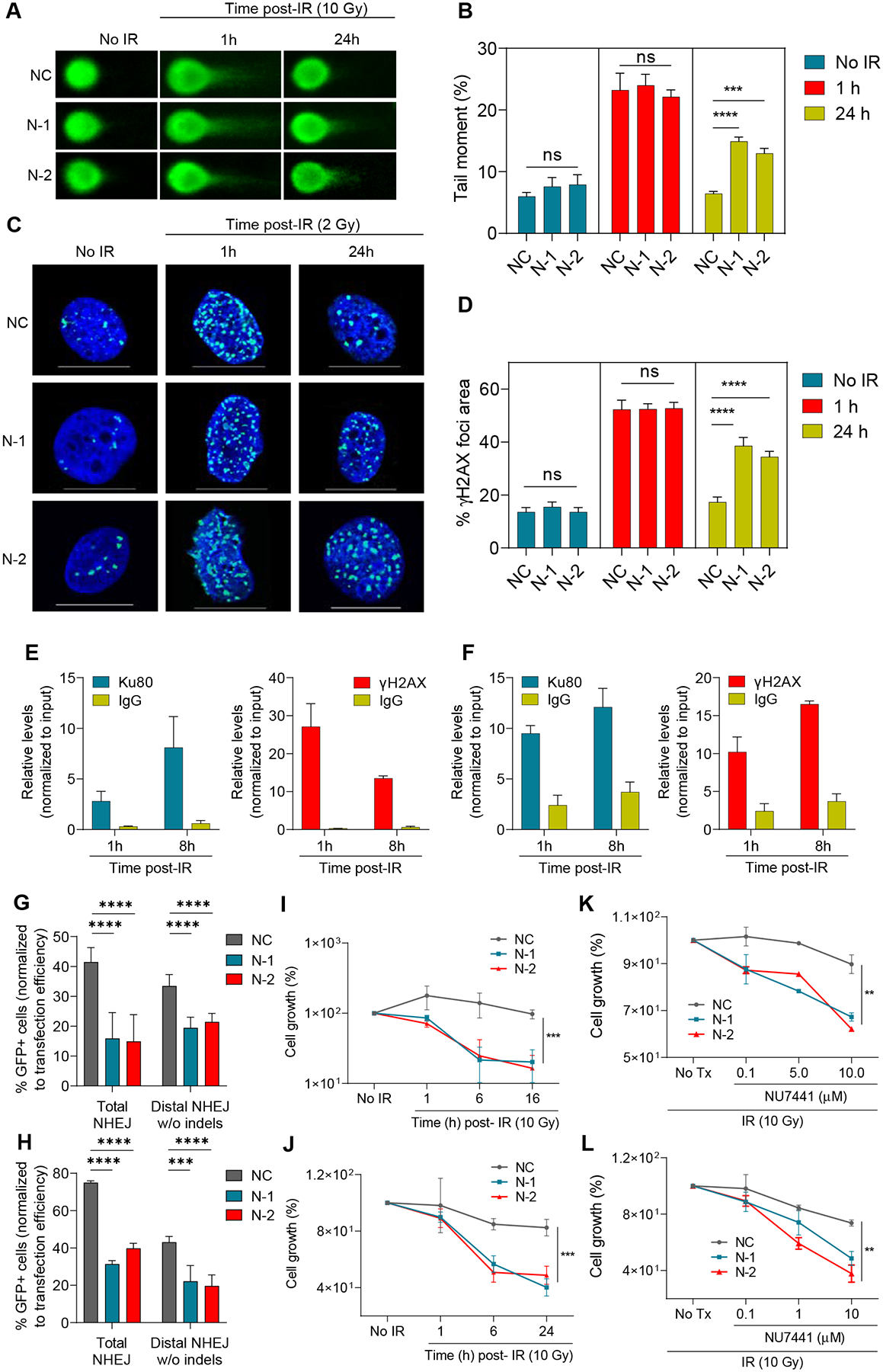
A-D At 24h post-transfection NIHCOLE-depleted, and control cells were either fixed (no IR) or irradiated and fixed at one or 24 hours after irradiation. Cells were then submitted to single-cell electrophoresis for comet scoring. Representative images are shown (A). DNA damage was measured as comet tail-moment (B). Mean ± SEM is shown (n > 90 cells) from two independent experiments. Similarly treated cells were immunostained for γH2AX to detect DNA damage foci (C) Representative images of γH2AX-positive foci (green) and nuclear DNA with DAPI (blue) are shown. Scale bars, 20 μm (D) Percentage of γH2AX foci area over DAPI-stained nuclei. Graph shows mean ± SEM (n > 45 cells) from two independent experiments. E, F RNA immunoprecipitation of NIHCOLE by Ku80 and yH2AX antibodies after radiation. Ku80 and yH2AX antibodies and IgG were used to immunoprecipitate RNA in formaldehyde crosslinked Huh7 (E) and JHH6 (B) cells at early time-points after ionizing irradiation with 2Gy. NIHCOLE was quantified by qRT-PCR. Graph shows mean ± SEM (n = 3 in E, n = 2 in F). G, H Two GFP reporter assays were used to assess NHEJ repair of NIHCOLE-depleted Huh7 (G) and JHH6 (H) cells. The percentage of GFP positive cells was normalized to transfection efficiency. Graph shows mean ± SEM (n = 3 in G, n = 2 in H). I-L Cell growth was measured 48h post-gapmer transfection in control (No IR) or at different times after damage induction by IR in Huh7 (I) and JHH6 (J) cells, or after IR, in mock treated (No Tx) or cells treated with increasing concentrations of DNA-PKcs inhibitor NU7441 in Huh7 (K) and JHH6 (L) cells. Cell number in No IR and No Tx cells was adjusted to 100% to discard the single effect of NIHCOLE depletion on cell growth. Graph shows mean ± SEM (n = 3). The significance of the statistical analysis (one-way ANOVA B, D, G and H; and two-tailed Student’s t test in I-L) is indicated and summarized as: not significant (ns); **<0.01; ***<0.001; ****<0.0001. See also Figure S3.
Our results are compatible with a role of NIHCOLE in DNA repair that may be relevant for liver cancer cell growth. To address this, we followed the viability of NIHCOLE-depleted HCC cells after DNA damage induction with ionizing radiation. Under these conditions, NIHCOLE depletion caused an additional impact on the growth of Huh7 (Fig. 4I) and JHH6 (Fig. 4J) cells. Further cell death was observed when the same cells were treated with 10Gy IR and increasing concentrations of DNA-PKcs inhibitor NU7441 (Fig. 4K and L). Conversely, similarly treated HEP3B cells that do not express NIHCOLE showed no differences in cell growth (Fig. S3J). Overall, these results suggest that NIHCOLE depletion, together with DNA damage induction, have an additive antiproliferative effect on NIHCOLE-expressing HCC cells.
NIHCOLE and predicted structural motifs bind to recombinant Ku
To directly address the ability of NIHCOLE to bind Ku70/80, we used single-molecule atomic force microscopy imaging (AFM) of NIHCOLE alone or mixed with recombinant Ku. We observed that NIHCOLE folds into what appear to be complex dsRNA secondary structures (Fig. 5A, NIHCOLE alone, and Fig. S4A). In the presence of Ku, most NIHCOLE molecules colocalize with several molecules of Ku, suggestive of cooperative binding (Fig. 5A, NIHCOLE + Ku and Fig. S4A). The images also support the possibility of the recruitment of Ku molecules around one or several molecules of NIHCOLE, as Ku alone remains monodispersed (Fig. S4A, Ku alone). In fact, Ku molecules can be distinguished from RNA molecules by image analysis of AFM experiments. This allowed us to calculate that the average NIHCOLE-Ku cluster includes around three NIHCOLE molecules that can bind an average of 14 Ku molecules (Fig. S4B, C). Such hyper stoichiometric relationship would support the potential of NIHCOLE to promote phase separation as a way to favor repair kinetics, especially given the apparent enrichment of NIHCOLE at DNA damage sites. As expected, since Ku has been shown to be an RNA-binding protein, the RNA messenger of luciferase (LUC) used as a control for these experiments also showed cooperative binding of Ku into clusters of overall similar sizes (Fig. S4D) although through differential stoichiometries (Fig. S4B, C).
Figure 5. Characterization of Ku binding by NIHCOLE and NIHCOLE’s putative structural motifs.
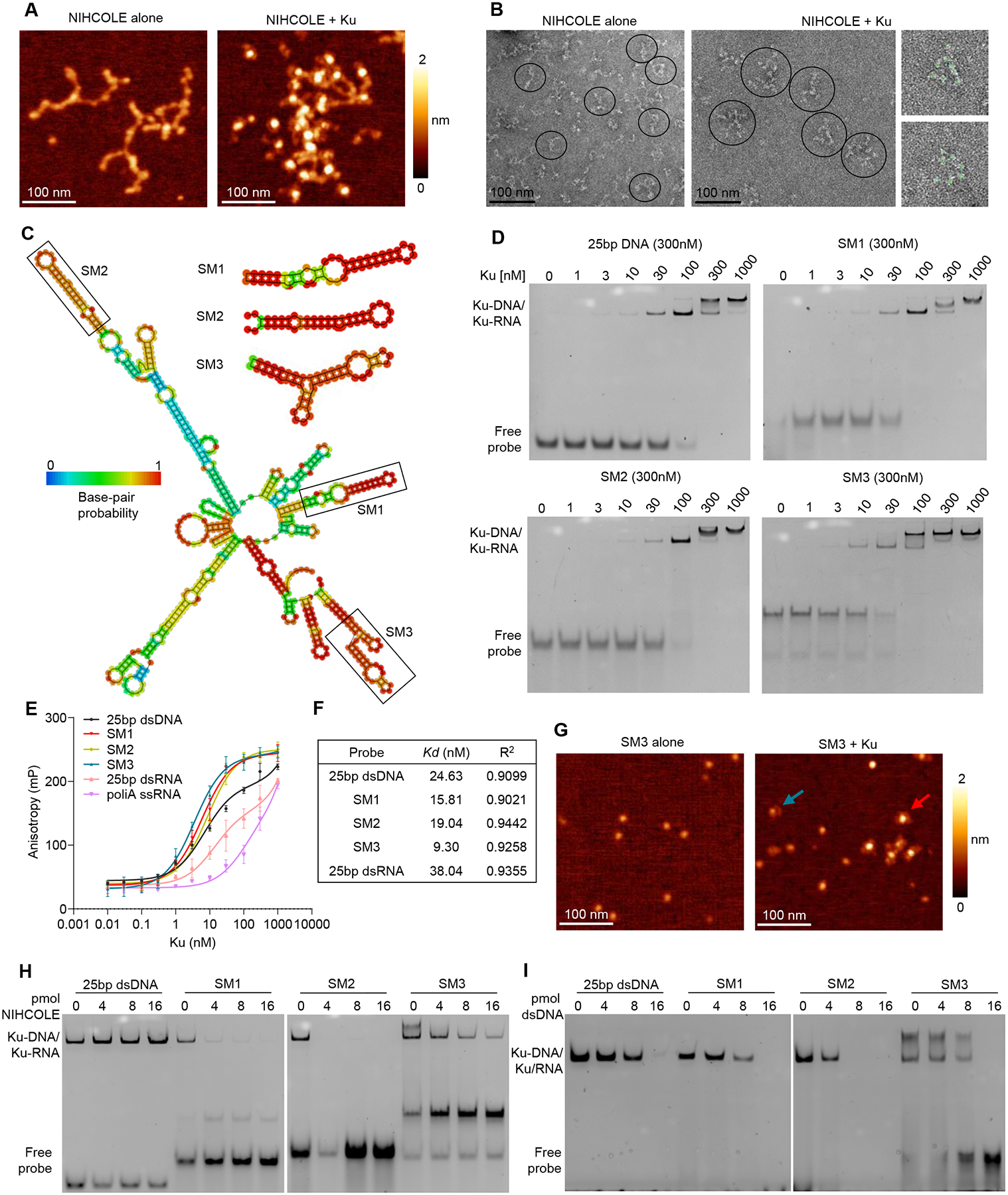
(A) Representative AFM images of full-length ivt NIHCOLE alone or bound to Ku. z-scale from dark to bright is 0–2 nm. Scale bar, 100 nm (B) Representative negative staining-EM images of NIHCOLE alone or bound to Ku. Representative molecules are indicated within circles. Scale bar, 100 nm. Right panels show two clusters of NIHCOLE-Ku complexes, showing Ku as globular densities identified with light green circles). (C) Representative output from RNAfold predicted minimum free energy structure for the full-length NIHCOLE and the three putative structural motifs (SMs). Location of SM1, SM2 and SM3 is shown. Color scale shows base-pair probability. (D-F) Increasing concentrations of recombinant Ku were assessed for binding to FAM-labeled RNA probes by native electrophoresis (D) or fluorescence polarization anisotropy (E) Graph shows mean ± SD (n = 3). Duplex 25 bp DNA was used as control. Representative images from three independent experiments are shown. Nonlinear regression based on FPA data was used to determine Kd, and R2 (F). (G) Representative AFM images of ivt SM3 alone or SM3 in the presence of Ku. Red arrow, SM3-Ku complex; blue arrow, individual Ku molecule. Z-scale from dark to bright is 0–2 nm. (H, I) Recombinant Ku was assessed for binding to RNA by native electrophoresis in the presence of increasing molar ratios of ivt full-length NIHCOLE (H) or unlabeled duplex 25bp DNA (I). A representative image of two independent experiments is shown. See algo Figure S4 and S5.
Since there are no reports in the literature of the interaction of RNAs and RBPs by AFM, to substantiate this result, we performed a similar experiment using negative stain electron microscopy (EM) of NIHCOLE alone (Fig. 5B, NIHCOLE alone and Fig. S4E) or incubated with Twin-Strep-tagged Ku. After pulldown of Ku, we could observe Ku as globular densities decorating NIHCOLE clusters (Fig. 5B, NIHCOLE + Ku, and Fig. S4E) while Ku alone remained monodispersed (Fig. S4E, Ku alone) as previously observed in AFM. Negative stain EM does not allow the visualization of RNA and Ku at the same time; however, Ku globular densities can be isolated by image analysis. Estimations from hundreds of clusters suggest a mean of 8.2 ± 2.9 Ku molecules per NIHCOLE-Ku cluster (Fig. S4F) which is within the error range of our AFM-based estimations.
We next sought to characterize the interaction of NIHCOLE with Ku using electrophoretic mobility shift assays (EMSA). To do this, the structure of NIHCOLE was analyzed using the RNAFold (41) and UNAFold (42) tools. We selected three structural motifs (SMs): SM1, SM2, and SM3 as they appeared in most NIHCOLE predicted structures with the highest base-pair probability and were small enough to be assayed by EMSA (Fig. 5C).
To study the binding affinities of SMs to recombinant Ku, we synthesized 5’ FAM-labeled SM1, SM2, SM3, and a 25bp dsDNA was used as a positive control (Supplementary Table S1D). Then, equal concentrations of labeled probes were incubated with increasing concentrations of purified Ku and visualized after native gel electrophoresis (Fig. 5D) or by measuring fluorescence polarization anisotropy (FPA) (Fig. 5E). As expected, dsDNA binds Ku efficiently (Fig. 5D, 25bp dsDNA); instead, a linear poly(A) RNA and a control 25 bp dsRNA did not bind Ku (Fig. 5E). In contrast, SM1, SM2, and SM3 bound Ku with similar affinity to dsDNA (Fig. 5D). In addition, the highest concentrations of Ku led to a supershifted complex of lower mobility in all cases, which is consistent with published data that suggests the binding of two heterodimers of Ku (43). The concentration of Ku required for the supershift is lowest for SM3 compared to dsDNA, SM1, and SM2 (Fig. 5D). These results were corroborated by calculating the dissociation constants (Kds) of the Ku-DNA/RNA complexes by FPA (Fig. 5F). Interestingly, by non-linear regression estimations, the Kd of dsDNA corresponds with that reported in the literature to be around 10−9 M (44). Similarly, SM1, SM2, and SM3 have calculated Kds in the same order as DNA, suggesting a high affinity of Ku for NIHCOLE structures, especially for SM3 (Fig. 5F). In fact, when we analyzed SM3 by itself or in the presence of Ku by AFM, we observed a direct interaction between SM3 and Ku (Fig. 5G).
Full-length NIHCOLE is too large to enter native gels. Nonetheless, we were able to use it in competition experiments to help determine whether Ku releases dsDNA, SM1, SM2, or SM3, in the presence of NIHCOLE. For these experiments, complexes between labeled dsDNA, SM1, SM2 or SM3, and Ku were formed and evaluated by native electrophoresis in the absence or presence of increasing concentrations of ivt NIHCOLE. Full-length NIHCOLE did not compete with DNA but effectively competed with the SM RNAs for binding, as evidenced by the clear displacement of the labeled probe with increasing concentration of NIHCOLE (Fig. 5H). Notably, the best competition was observed for SM2, followed by SM1 and SM3, with the latter remaining in complex with Ku even in the presence of a four-fold molar excess of NIHCOLE. To determine whether the sequence or the structure of NIHCOLE was essential for outcompeting the SMs, we repeated this experiment using NIHCOLE antisense (NIHCOLEas) (Fig. S5A) or LUC (Fig. S5B), and we observed similar results to NIHCOLE, suggesting that large structured RNAs, can bind to Ku and compete with the binding of smaller RNA structures depending on their affinity for Ku. When a similar competition was performed using unlabeled 25 bp dsDNA (Fig. 5I), as expected, we observed an equilibrium between labeled and unlabeled DNA bound to Ku (Fig. 5I, 25bp dsDNA). Instead, Ku-RNA complexes are outcompeted by DNA binding, suggesting that Ku cannot bind DNA and RNA at the same time at molar excess of DNA. However, SM1 and SM3 showed higher strength of binding to Ku, as they were more difficult to displace than SM2. Also, SM3 supershift was retained until it was released from the complex (Fig. 5I, SM3 panel). It should be noted that the unlabeled 25 bp dsDNA used for competition is likely able to bind only one Ku molecule, since structural analysis have shown that each Ku covers about 20 bp, making 20 bp dsDNA approximately the unit site for Ku binding (43). Instead, competition with full length NIHCOLE, with numerous SMs, should bind several molecules of Ku, resulting in a stronger competitor than 25 bp dsDNA (compare Fig 5H and I). Competitions with DNA were corroborated by FPA analysis (Fig. S5C). Using the same dsDNA as a competitor for the Ku-RNA complex, binding curves and calculated constants of inhibition (Ki) showed that higher DNA concentrations are required to inhibit SM1- or SM3-Ku binding compared to SM2 (Fig. S5C). Overall, these results are in accordance with recent literature that describes Ku as an RNA binding protein (45) and supports NIHCOLE, and particularly SM3, as a relevant structural motif for Ku binding.
NIHCOLE supports multimeric complexes with NHEJ factors and promotes the ligation efficiency of DSBs
Ku is known to interact with several NHEJ factors besides DNA-PKcs (46). To test for binding to additional NHEJ factors, we incubated the labeled SMs and control dsDNA with a battery of NHEJ proteins available to us including XRCC4 alone, XRCC4 and LIG4 complex (X4L4), APLF or XLF and evaluated complex formation by EMSA. Neither of these proteins alone could bind RNA or DNA (Fig. S6A). However, APLF, a recently described accessory factor in the NHEJ response (13), was able to interact with Ku and dsDNA or SM3 to form higher-order complexes (Fig. 6A). In fact, APLF can bridge the interaction between Ku and XRCC4 (12), likely stabilizing the binding between Ku and the ligation complex X4L4 required for DNA repair (47). Therefore, to determine whether SM3 can form a complex with Ku, APLF, and X4L4, we used purified proteins in band shift experiments. Under these experimental conditions, Ku and X4L4 do not form a stable complex with SM3 or DNA (Fig. 6A, lanes 4 and 8). However, in the presence of APLF, there is a supershift of the Ku-APLF complex bound to SM3 or DNA after incubation with X4L4 (Fig. 6A, lanes 2 and 3 for SM3 and lanes 6 and 7 for DNA). This has been reported previously for DNA (13) but not for any RNA structure. Interestingly, in the presence of equimolar concentrations, both SM3 and DNA supercomplexes can coexist (Fig. 6A, lane 11). Of note, as opposed to SM3, a similar SM2 complex was outcompeted by equimolar concentrations of DNA (Fig. 6A, lanes 9), and even in the absence of competing DNA, multimeric complexes were much less stable with SM2 (Fig. S6B) following our previous results.
Figure 6. Analysis of NIHCOLE and SM3 binding to other NHEJ factors and their effect on ligation efficiency.
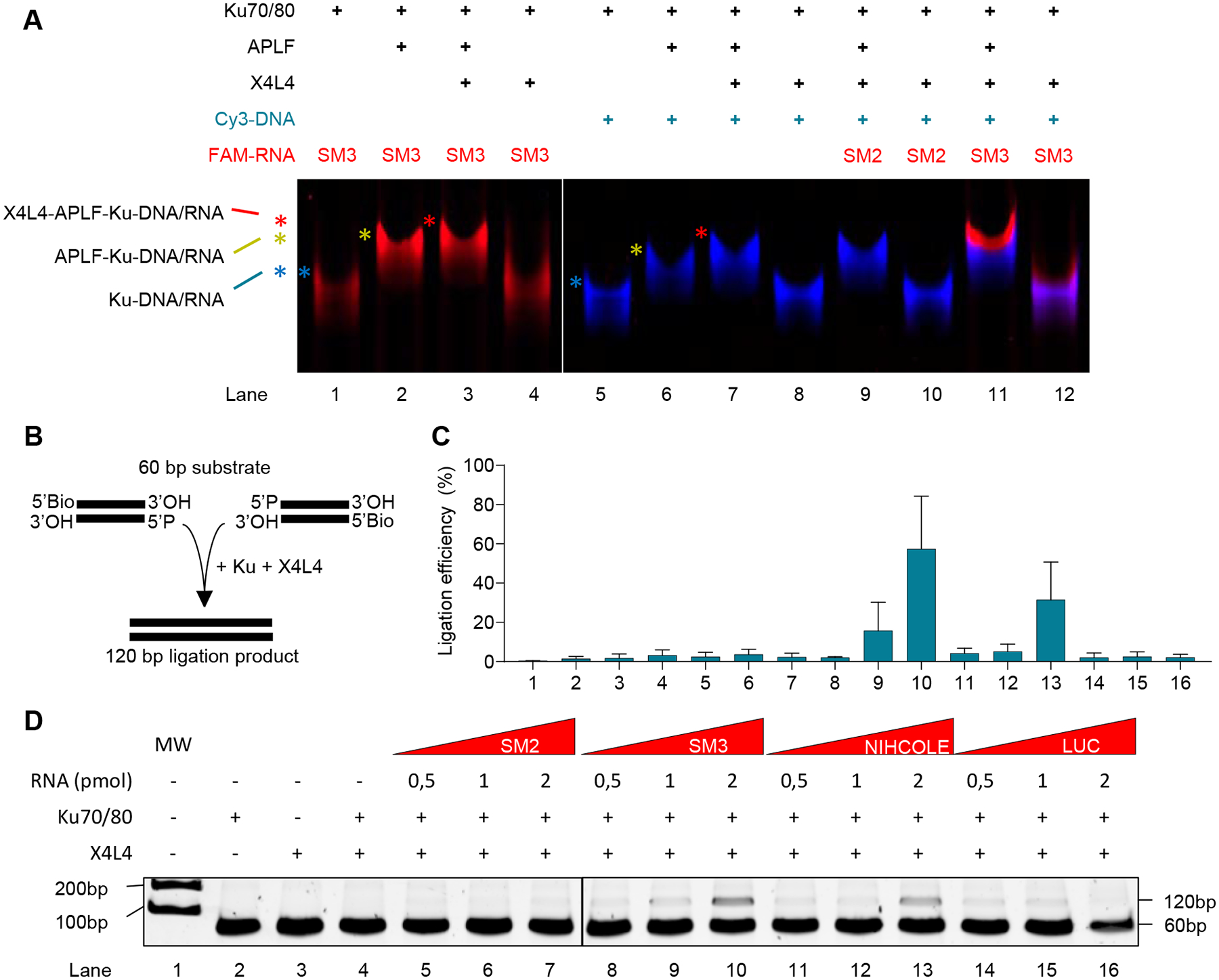
A Recombinant Ku, APLF, and X4L4 were assessed for binding to FAM-labeled RNA and Cy3-labeled DNA by native electrophoresis. A representative image from three independent experiments is shown. B-D A NHEJ reconstitution assay with recombinant Ku and X4L4 (B) was used to evaluate the effect of in vitro transcribed NIHCOLE, SM3 and control RNAs on the ligation of 60bp dsDNA oligos. DNA was visualized by neutral PAGE in 15% acrylamide gel and imaged after SYBRsafe staining (C). Ligation efficiency of the 120bp ligation product was calculated against unligated substrate (D). Results show mean ± SEM (n = 4). See also Figure S6.
To gain insights into the functional implications of NIHCOLE-mediated scaffolding, we performed NHEJ reconstitution assays. To evaluate the effect of NIHCOLE on ligation efficiency, we reconstituted the minimal NHEJ machinery required for the ligation of compatible unoxidized DNA ends (Fig. 6B). As previously described, Ku and X4L4 alone are capable of ligating blunt-ended DSBs. In fact, additional NHEJ factors such as DNA-PKcs and/or XLF actually reduce the ligation efficiency in this setting (15,47). Recent structural insights into the ligation complex provide a mechanism in which DNA-PKcs prevents the alignment of the DNA ends for ligation, likely to make room for end-processing enzymes, and instead, release of DNA-PKcs from the ligation complex is required to allow DNA end-ligation. Therefore, we combined human recombinant Ku and X4L4 proteins (expressed in baculovirus-infected insect cells (Fig. S7A, B)) with a 60bp dsDNA substrate (Supplementary Table S1D) in a ligation-competent buffer without or with increasing concentrations of RNAs. After ligation, we treated the reaction with RNase, precipitated the DNA, and resolved the samples by gel electrophoresis (Fig. 6C, D). Although highly inefficient, Ku and X4L4 alone are able to generate a ligation product of 120bp (Fig. 6C, D lane 4). Strikingly, results show that the presence of SM3 and NIHCOLE (Fig. 6D, lanes 8–13) but not control RNAs SM2 and LUC (Fig. 6D, lanes 5–7 and 14–16 respectively) increase the ligation efficiency in an RNA concentration-dependent manner as corroborated after quantitation of the ligation product by gel electrophoresis (Fig. 6C).
DISCUSSION
The number of identified and characterized lncRNAs continues to expand, challenging our understanding of the multiple levels at which genomes are regulated. Since it became evident that lncRNAs constitute a significant layer of genome regulation, many have been reported to act in the loss of genomic integrity leading to disease, especially in cancer. To date, only a handful of lncRNAs have been described to be involved in the repair of DSBs, the most toxic type of DNA lesion. The NHEJ branch of DSB repair is a double-edged sword. It has been depicted as a genome guardian (48), as it allows fast repair kinetics throughout the cell cycle, thereby protecting cells from agents that induce DSBs. However, in cancer cells, DNA repair pathways, especially NHEJ, can be repurposed to sustain malignant proliferation despite cumulating DNA damage arising from high replicative stress or even from anticancer treatments, causing therapy resistance (7,49).
Using our previous pan-cancer and pan-tissue lncRNA expression profiling study (32), we identified the novel lncRNA NIHCOLE, and we now show that it is upregulated in HCC and expressed to higher levels in patients with decreased survival, worse prognosis, and mutations in key HCC drivers (Fig. 1). In addition, we show that NIHCOLE is essential for the proliferation and NHEJ-mediated DNA damage repair of HCC cells (Fig. 2F, H; Fig. S3A and Fig. 4G, H). Importantly, we provide evidence to support that NIHCOLE binds to Ku (Fig. 5A, B, and H) and enhances the ligation efficiency of DSBs (Fig. 6C, D). Since all healthy tissues deposited to GTEx, healthy liver biopsies (Fig. 1C), and non-HCC cell lines (Fig. 2A) lack significant NIHCOLE expression, it is unlikely that NIHCOLE is a core factor of the NHEJ machinery but rather a specific adaptation of HCC and, possibly, other cancer cells.
We hypothesize that NIHCOLE is working as a scaffold for the NHEJ machinery, sustaining the assembly of multimeric NHEJ repair complexes and thus promoting the efficiency of end-ligation (Fig. 7). Scaffolding and multimerization of repair factors at DNA breaks are crucial preconditions for repair. Previous findings show that the interaction among APLF, Ku, DNA-PKcs, XRCC4, and LIG4, can effectively bridge DNA ends and facilitate ligation (12,47). More recent studies suggest that such complexes could be helped by DNA-PKcs dimers (10) and Ku dimer-of-dimers (17), facilitating alignment and tethering of DNA ends prior to ligation (50). Interestingly, it has been described that factors that transiently stabilize the interactions formed at DNA ends can promote short-range synapsis. When the ligation complex is well-positioned, they could also contribute to enhancing ligation efficiency (8). We propose that this is the case of NIHCOLE (Fig. 7). NIHCOLE can bind Ku, and the addition of X4L4 and blunt DNA ends to the Ku/NIHCOLE mixture results in enhanced ligation, consistent with a role of NIHCOLE in the short-range synaptic complex. Similar results are specifically observed when the small dsRNA SM3 is used, allowing us to suggest that SM3 is a natural domain within NIHCOLE that may contribute to NIHCOLE function in NHEJ. Interestingly, using SM3, but not other putative small dsRNAs within NIHCOLE, we are able to detect a stable complex containing SM3-Ku-APLF-X4L4 (Fig. 6A). Further experiments are required to understand whether this complex releases the RNA to bind DNA ends and whether this transfer of the pre-assembled machinery results in a more efficient reaction. Nonetheless, we cannot rule out the possibility of NIHCOLE working by promoting macromolecular crowding through lncRNA-induced liquid-liquid phase separation (LLPS) that could modulate the physicochemical properties of the repair factors. LncRNAs have been described to form RNA condensates through RNA-RNA interactions, as suggested for LINP1 (17) and our in vitro results (Fig. 5A, B and Fig. S4A–C, E, F). In our case, binding to an intrinsically disordered protein such as APLF could promote the formation of phase-separated repair hubs where NHEJ is promoted (Fig. 7). In addition, APLF has been shown to favor NHEJ activity in vivo (13) and to have histone chaperone functions working as an anchor (51) potentially favoring multimerization and scaffolding in the repair condensates together with NIHCOLE in HCC cells. Nevertheless, the study of LLPS in biologically-relevant models is currently limited (52). Other lncRNAs have been reported to interact with Ku (19,53), DNA-PKcs (18) or both (54); however, only recently, mechanistic insights into LINP1 function in NHEJ reported a Ku-DNA-PKcs-LINP1-mediated synapse stabilization, suggesting that LINP1 works in the long-range synaptic complex as opposed to NIHCOLE (17). Remarkably, our results showing the ability of an RNA to sustain multicomponent assemblies of NHEJ factors including Ku70/80, APLF and X4L4, and to increase ligation efficiency (Fig. 6), raise interesting structural questions that need to be addressed in the future.
Figure 7. Proposed mechanism of action of NIHCOLE.
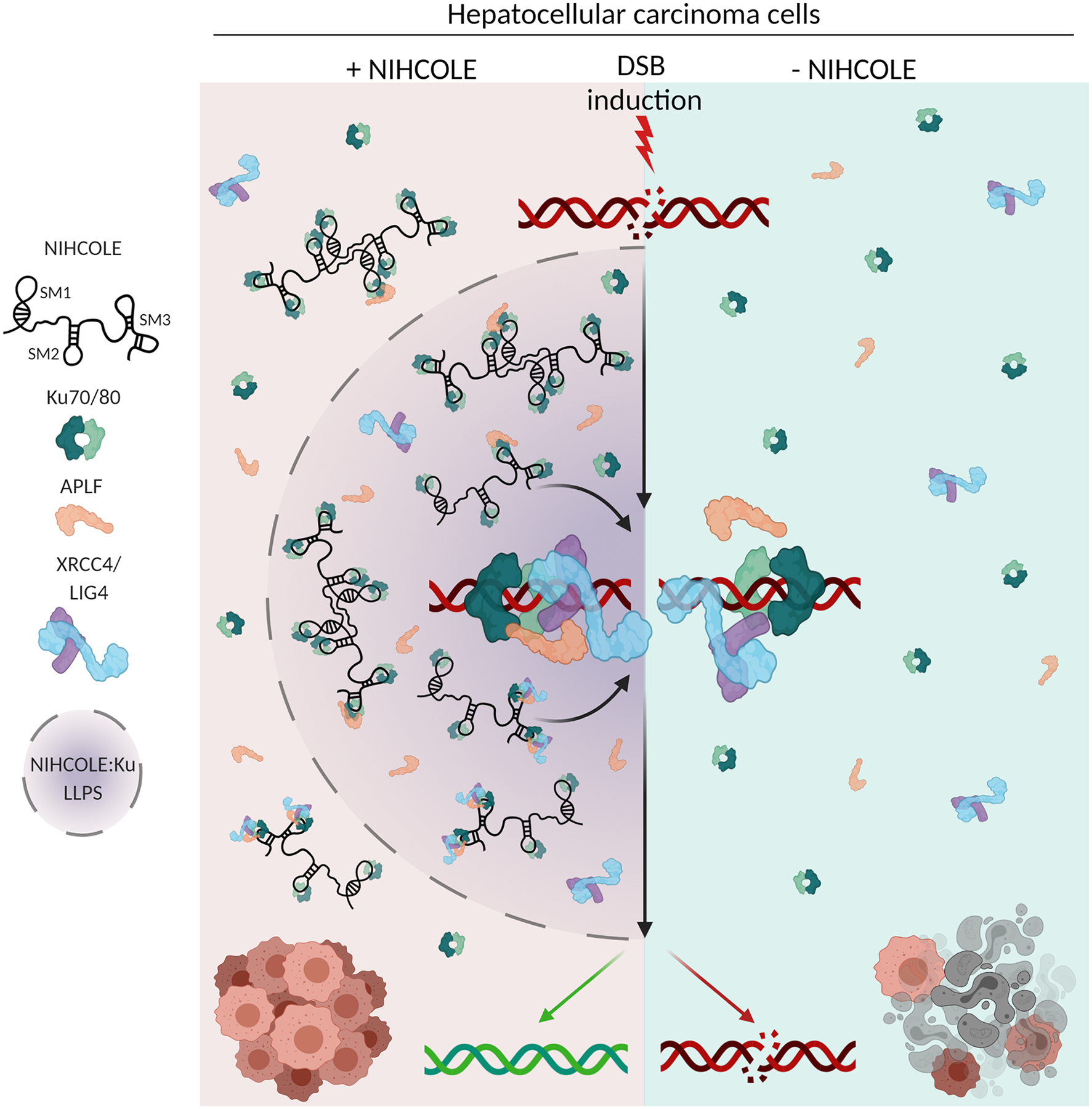
HCC cells expressing NIHCOLE lncRNA, through its structural motifs, especially SM3, are able to cooperatively support the binding of several molecules of Ku and the modular assembly of other repair factors into multimeric complexes to help the scaffolding, multimerization and stabilization required by the short-range synaptic complex to allow DNA-end ligation by the X4L4 complex. In addition, NIHCOLE’s multivalent interactions with Ku and intrinsically disordered APLF could potentially induce phase-separation to form repair hubs where NHEJ activity is promoted. In contrast, cells lacking NIHCOLE expression have less efficient repair machineries that makes them more sensitive to radiation or DNA damaging agents.
Indeed, we also show an observable and cooperative interaction between Ku and lncRNA NIHCOLE (Fig. 5A and Fig. S4A, E) or NIHCOLE’s structural motif SM3 by AFM (Fig. 5G). The use of AFM for the imaging of lncRNA-protein complexes offers a unique opportunity to face the challenges of structural, biochemical, and biophysical studies of lncRNAs. While the basis of Ku-RNA interaction remains to be elucidated, it is likely that it is sequence-independent as it happens for DNA (43). Recent eCLIP data analysis found no specific RNA sequence motifs for Ku-RNA binding (17). Interestingly, yeast Ku has been shown to bind a stem loop structure in TLC1, the RNA component of yeast telomerase (55) and while yeast and human Ku differ significantly, the conservation of the RNA-binding capacity of Ku highlights its importance for the cell. The observation of the interaction between Ku and SM3 allows us to propose that the presence of SM3-like structural features in lncRNAs could outperform other RNAs in their ability to functionally interact with the NHEJ machinery, as suggested by previous studies (17,56).
Overall, our results support that lncRNA NIHCOLE confers an advantageous malignant adaptation to HCC cells by promoting the ligation efficiency of NHEJ-mediated DSB repair. Cancer cells could have evolutionarily favored the upregulation of specific lncRNAs that are especially suited to promote DNA end ligation, likely imparting a fitness advantage to overcome the DNA damage overload arising from higher replicative stress. Consequently, our findings could have significant repercussions for cancer therapy, as increased DNA damage is one of the major outcomes of conventional radio and chemotherapeutics, which could work in combination with lncRNA depletion to increase anti-tumor efficacy. In this context, NIHCOLE appears as a novel and potentially useful therapeutic target for tumors like HCC, where most genetic alterations are non-druggable, and systemic therapies remain inefficient.
Supplementary Material
SIGNIFICANCE.
This study characterizes the role of a lncRNA NIHCOLE in DNA repair and cellular fitness in hepatocellular carcinoma, thus implicating it as a therapeutic target.
ACKNOWLEDGEMENTS
We particularly acknowledge the patients for their participation and the Biobank of the University of Navarra for its collaboration. We thank all members of the Fortes Lab for helpful discussions and Guillermo Serrano and Dr. Victor Segura, from CIMA bioinformatics unit, for excellent expert assistance. We thank R. Ye and Dr. Anne Vaahtokari for technical assistance, S. Fang (University of Calgary) for purification of XRCC4 and APLF; Dr. Laurence H. Pearl (Genome Damage and Stability Centre, School of Life Sciences, University of Sussex) for providing X4L4, and Ku70/80 for some of the experiments in this work. We also thank the flow cytometry facility and the Arnie Charbonneau microscopy facility at Cumming School of Medicine, University of Calgary for expert assistance. The results shown here are in part based upon data generated by the TCGA Research Network, (http://cancergenome.nih.gov/) and by the GTEx Project, (https://gtexportal.org/). Figures 3A, 3E, 7 and S3H were created with BioRender.com.
This work was supported by the European FEDER funding (to the activities of the groups directed by P.F, O.L and F.M-H) and grants from the Ministry of Economy and Competitiveness (SAF2015-70971-R to P.F. and BFU2017-83794-P (AEI/FEDER, UE) to F.M.-H.)); MCIU/AEI/FEDER/UE (RTI2018-101759-B-I00 to P.F.), NIH program (CA92584 to S.P.L-M.), Ligue National Contre le Cancer, Équipe Labellisée and ITMO Cancer: Consortium HETCOLI (to J.Z.-R.), NIH program (P01CA092584 to G.W), NSERC (RGPIN-2018-04327 to G.W), and CFI (RCP-18-023-SEG to G.W), Gobierno de Navarra (33/2015 to P.F.), Scientific Foundation of the Spanish Association Against Cancer (AECC IDEAS20169FORT to P.F.); Fondo de Investigación Sanitaria (PI19/00742 to B.S.), financed by the National Institute of Health Carlos III and FEDER. CNIO and CIBERehd are funded by the National Institute of Health Carlos III. J.P.U was a recipient of a University of Navarra’s Asociación de Amigos fellowship. L.P.-M is a recipient of a PFIS fellowship (FI20/00074) by the National Institute of Health Carlos III and FSE “Investing in Your Future”. This work was also funded by grants from the Autonomous Region of Madrid (Tec4Bio – S2018/NMT-4443 and NanoBioCancer – Y2018/BIO-4747 to O.L. and F.M.-H.) and co-funded by the European Social Fund. F.M.-H. acknowledges support from the European Research Council (ERC) under the European Union Horizon 2020 Research and Innovation Program (grant agreement 681299). The GTEx project was supported by the NIH and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Footnotes
AUTHORS’ DISCLOSURES
The authors declare no competing interests.
REFERENCES
- 1.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet [Internet].2015;47:199–208. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25599403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unfried JP, Sangro P, Prats-Mari L, Sangro B, Fortes P. The Landscape of lncRNAs in Hepatocellular Carcinoma: A Translational Perspective. Cancers (Basel) [Internet].2021;13:2651. Available from: https://www.mdpi.com/2072-6694/13/11/2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet [Internet]. Nature Publishing Group; 2016;17:47–62. Available from: http://www.nature.com/articles/nrg.2015.10 [DOI] [PubMed] [Google Scholar]
- 4.Ulitsky I Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat Rev Genet [Internet]. Nature Publishing Group; 2016;17:601–14. Available from: 10.1038/nrg.2016.85 [DOI] [PubMed] [Google Scholar]
- 5.Unfried JP, Serrano G, Suarez B, Sangro P, Ferretti V, Prior C, et al. Identification of coding and long noncoding RNAs differentially expressed in tumors and preferentially expressed in healthy tissues. Cancer Res. 2019;79. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011. page 646–74. [DOI] [PubMed] [Google Scholar]
- 7.Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell [Internet]. Cell Press; 2017. [cited 2018 Mar 20];168:644–56. Available from: https://www.sciencedirect.com/science/article/pii/S0092867417300053?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The molecular basis and disease relevance of non-homologous DNA end joining. Nat Rev Mol Cell Biol. Springer US; 2020;21:765–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA. Structural insights into NHEJ: Building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst) [Internet].2014;17:110–20. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1568786414000469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaplin AK, Hardwick SW, Liang S, Kefala Stavridi A, Hnizda A, Cooper LR, et al. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat Struct Mol Biol. 2020; [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Lee L, Naila T, Fishbain S, Wang A, Tomkinson AE, et al. Structural basis of long-range to short-range synaptic transition in NHEJ. Nature [Internet].2021;593:294–8. Available from: http://www.nature.com/articles/s41586-021-03458-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammel M, Yu Y, Radhakrishnan SK, Chokshi C, Tsai MS, Matsumoto Y, et al. An intrinsically disordered APLF links Ku, DNA-PKcs, and XRCC4-DNA ligase IV in an extended flexible non-homologous end joining complex. J Biol Chem. 2016;291:26987–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, Manley K, et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J [Internet]. Nature Publishing Group; 2013;32:112–25. Available from: 10.1038/emboj.2012.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, Melo De AJ, et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature. 2016;535:566–9. [DOI] [PubMed] [Google Scholar]
- 15.Chang HHY, Watanabe G, Gerodimos CA, Ochi T, Blundell TL, Jackson SP, et al. Different DNA end configurations dictate which NHEJ components are most important for joining efficiency. J Biol Chem. 2016;291:24377–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thapar R, Wang JL, Hammel M, Ye R, Liang K, Sun C, et al. Mechanism of efficient double-strand break repair by a long non-coding RNA. Nucleic Acids Res. 2020;48:10953–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haemmig S, Yang D, Sun X, Das D, Ghaffari S, Molinaro R, et al. Long noncoding RNA SNHG12 integrates a DNA-PK–mediated DNA damage response and vascular senescence. Sci Transl Med. 2020; [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Zhou Z, Wu E, Ouyang C, Wei G, Wang Y, et al. LRIK interacts with the Ku70–Ku80 heterodimer enhancing the efficiency of NHEJ repair. Cell Death Differ. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirsch TZ, Negulescu A, Gupta B, Caruso S, Noblet B, Couchy G, et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J Hepatol. 2020;72:924–36. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Feng Y, Hu Z, Hu X, Yuan C-X, Fan Y, et al. Characterization of Long Noncoding RNA-Associated Proteins by RNA-Immunoprecipitation. Genomic Elem Heal Dis Evol [Internet].New York, NY: Springer New York; 2016. page 19–26. Available from: http://link.springer.com/10.1007/978-1-4939-3070-8_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marín-Béjar O, Huarte M. RNA Pulldown Protocol for In Vitro Detection and Identification of RNA-Associated Proteins. In: Carmichael GG, editor. New York, NY: Springer New York; 2015. page 87–95. Available from: http://link.springer.com/10.1007/978-1-4939-1369-5 [DOI] [PubMed] [Google Scholar]
- 23.Moore S, Berger ND, Luijsterburg MS, Piett CG, Stanley FKT, Schräder CU, et al. The CHD6 chromatin remodeler is an oxidative DNA damage response factor. Nat Commun [Internet]. Springer US; 2019;10. Available from: 10.1038/s41467-018-08111-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilmore JL, Yoshida A, Takahashi H, Deguchi K, Kobori T, Louvet E, et al. Analyses of nuclear proteins and nucleic acid structures using atomic force microscopy. In: Nakagawa S, Hirose T, editors. Methods Mol Biol. New York, NY: Springer New York; 2015. page 119–53. [DOI] [PubMed] [Google Scholar]
- 25.Horcas I, Fernández R, Gómez-Rodríguez JM, Colchero J, Gómez-Herrero J, Baro AM. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev Sci Instrum. 2007;78:13705. [DOI] [PubMed] [Google Scholar]
- 26.Radhakrishnan SK, Lees-Miller SP. DNA requirements for interaction of the C-terminal region of Ku80 with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). DNA Repair (Amst) [Internet].2017;57:17–28. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1568786416304347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol [Internet].2004;5:R80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15461798%5Cnhttp://www.ncbi.nlm.nih.gov/sites/entrez [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volders P-J, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P, et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res [Internet].2019;47:D135–9. Available from: https://academic.oup.com/nar/article/47/D1/D135/5146198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong L, Hong H, Chen X, Huang Z, Wu W, Wu F. LINC02163 regulates growth and epithelial-to-mesenchymal transition phenotype via miR-593–3p/FOXK1 axis in gastric cancer cells. Artif Cells, Nanomedicine Biotechnol. 2018;46:607–15. [DOI] [PubMed] [Google Scholar]
- 30.Ma J, Zhang L, Shang A, Song H, Huo J, Zhang M, et al. LINC02163 promotes colorectal cancer progression via miR-511–3p/AKT3 axis. Artif Cells, Nanomedicine Biotechnol. 2020;48:961–8. [DOI] [PubMed] [Google Scholar]
- 31.Qin C, Jin L, Li J, Zha W, Ding H, Liu X, et al. Long non-coding RNA LINC02163 accelerates malignant tumor behaviors in breast cancer by regulating the microRNA-511–3p/HMGA2 axis. Oncol Res Featur Preclin Clin Cancer Ther. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Unfried JP, Serrano G, Suárez B, Sangro P, Ferretti V, Prior C, et al. Identification of Coding and Long Noncoding RNAs Differentially Expressed in Tumors and Preferentially Expressed in Healthy Tissues. Cancer Res [Internet].2019;79:5167–80. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-19-0400 [DOI] [PubMed] [Google Scholar]
- 33.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu A, Cancer Genome Atlas Research Network M, Carlsen R, Chuah E, Clarke A, Dhalla N, et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell [Internet]. Elsevier; 2017. [cited 2018 Jul 2];169:1327–1341.e23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28622513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc J-FF, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol [Internet]. European Association for the Study of the Liver; 2017. [cited 2019 Feb 13];67:727–38. Available from: 10.1016/j.jhep.2017.05.014 [DOI] [PubMed] [Google Scholar]
- 36.Boyault S, Rickman DS, De Reyniès A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. [DOI] [PubMed] [Google Scholar]
- 37.Nault JC, De Reyniès A, Villanueva A, Calderaro J, Rebouissou S, Couchy G, et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology. 2013;145:176–87. [DOI] [PubMed] [Google Scholar]
- 38.Caruso S, Calatayud AL, Pilet J, La Bella T, Rekik S, Imbeaud S, et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology [Internet]. Elsevier, Inc; 2019;157:760–76. Available from: 10.1053/j.gastro.2019.05.001 [DOI] [PubMed] [Google Scholar]
- 39.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. Haber JE, editor. PLoS Genet. 2008;4:e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhargava R, Sandhu M, Muk S, Lee G, Vaidehi N, Stark JM. C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat Commun [Internet]. Springer US; 2018;9. Available from: 10.1038/s41467-018-04867-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuker M Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res [Internet].2003;31:3406–15. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arosio D, Cui S, Ortega C, Chovanec M, Di Marco S, Baldini G, et al. Studies on the Mode of Ku Interaction with DNA. J Biol Chem [Internet].2002;277:9741–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925819360946 [DOI] [PubMed] [Google Scholar]
- 44.Fell VL, Schild-Poulter C. The Ku heterodimer: Function in DNA repair and beyond. Mutat Res Mutat Res [Internet]. Elsevier B.V.; 2015;763:15–29. Available from: 10.1016/j.mrrev.2014.06.002 [DOI] [PubMed] [Google Scholar]
- 45.Dalby AB, Goodrich KJ, Pfingsten JS, Cech TR. RNA recognition by the DNA end-binding Ku heterodimer. Rna. 2013;19:841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubin M, Newsome J, Ribes-Zamor A. The Role of Multimerization During Non-Homologous End Joining. New Res Dir DNA Repair [Internet]. InTech; 2013. page 13. Available from: http://www.intechopen.com/books/new-research-directions-in-dna-repair/the-role-of-multimerization-during-non-homologous-end-joining [Google Scholar]
- 47.Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR. The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nat Commun [Internet]. Springer US; 2019;10:3588. Available from: http://www.nature.com/articles/s41467-019-11507-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009. page 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sishc BJ, Davis AJ. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers (Basel). 2017;9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cary RB, Peterson SR, Wang J, Bear DG, Bradbury EM, Chen DJ. DNA looping by Ku and the DNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1997;94:4267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corbeski I, Dolinar K, Wienk H, Boelens R, Van Ingen H. DNA repair factor APLF acts as a H2A-H2B histone chaperone through binding its DNA interaction surface. Nucleic Acids Res. Oxford University Press; 2018;46:7138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell [Internet]. Elsevier Inc.; 2019;176:419–34. Available from: 10.1016/j.cell.2018.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ting NSY, Yu Y, Pohorelic B, Lees-Miller SP, Beattie TL. Human Ku70/80 interacts directly with hTR, the RNA component of human telomerase. Nucleic Acids Res. 2005;33:2090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, He Q, Hu Z, Feng Y, Fan L, Tang Z, et al. Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer. Nat Struct Mol Biol [Internet]. Nature Publishing Group; 2016;23:522–30. Available from: http://www.nature.com/doifinder/10.1038/nsmb.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterson SE, Stellwagen AE, Diede SJ, Singer MS, Haimberger ZW, Johnson CO, et al. The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat Genet. 2001;27:64–7. [DOI] [PubMed] [Google Scholar]
- 56.Shadrina O, Garanina I, Korolev S, Zatsepin T, Van Assche J, Daouad F, et al. Analysis of RNA binding properties of human Ku protein reveals its interactions with 7SK snRNA and protein components of 7SK snRNP complex. Biochimie. Elsevier Ltd; 2020;171–172:110–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data that support the findings of this study, have been deposited in the European Genome Archive (EGA) repository under the study ID accession number EGAS00001002879 (INSERM cohort) or can be accessed from http://cancergenome.nih.gov/ (TCGA cohort).