

Abstract
Systemic inhibition of Notch with γ-secretase inhibitors (GSI) decreases multiple myeloma (MM) tumor growth, but the clinical use of GSI is limited due to its severe gastrointestinal toxicity. In this study, we generated a GSI Notch inhibitor specifically directed to the bone (BT-GSI). BT-GSI administration decreased Notch target gene expression in the bone marrow, but it did not alter Notch signaling in intestinal tissue or induce gut toxicity. In mice with established human or murine MM, treatment with BT-GSI decreased tumor burden and prevented the progression of MM-induced osteolytic disease by inhibiting bone resorption more effectively than unconjugated GSI at equimolar doses. These findings show that BT-GSI has dual anti-myeloma and anti-restorative properties, supporting the therapeutic approach of bone-targeted Notch inhibition for the treatment of MM and associated bone disease.
Keywords: Notch, myeloma, tumor microenvironment, cancer, bone, resorption
Introduction
Multiple myeloma (MM) is a plasma cell malignancy characterized by the growth of clonal plasma cells in the bone marrow (BM) and is currently incurable (1). The bone/BM niche plays a critical role in MM onset and progression (2). MM cells reside in specialized niches in the bone/BM where they interact with multiple cell types. These interactions transform the bone/BM compartment into an ideal microenvironment for the migration, proliferation, and survival of MM cells (2, 3). In addition, MM cells suppress osteoblast differentiation and bone formation and increase osteoclast activity and bone resorption, leading to bone destruction and the development of bone lesions that rarely heal, which can result in fractures, negatively affect quality of life, and are a leading cause of morbidity and mortality in MM patients (4, 5). The development of drugs targeting the tumor and its microenvironment has significantly improved patient outcomes (6, 7). Thus, current research efforts focus on identifying signaling pathways that mediate communication between the MM cells and cells of the host niche as novel molecular targets.
Notch is a highly conserved signaling pathway that mediates cell to cell communication by transducing short-range signals via interactions between cell-surface receptors (Notch 1-4) with transmembrane ligands (Jagged1-2, Delta 1-4) (8). Several components of the Notch signaling pathway are dysregulated in MM cells, facilitating the delivery of MM signals to both adjacent MM cells (homotypic) and cells in the bone/BM niche (heterotypic) (9–11). Notch communication favors MM progression by increasing MM proliferation (12, 13), promotes chemotherapy resistance (14), and stimulates angiogenesis (15). In addition, Notch communication increases osteoclast differentiation and function and induces apoptosis of osteocytes to further promote bone resorption and destruction (10, 13).
The multifunctional role of Notch communication in MM provides a strong rationale to block Notch signaling in the MM tumor niche. However, the specific role of Notch components in MM is poorly defined, and Notch receptors can have distinct functions. Thus, signaling inhibition downstream of the four Notch receptors with γ-secretase inhibitors (GSIs) has been used to inhibit Notch in MM (16). GSIs decrease MM growth and mitigate the bone disease in animal models of MM (17–19). Despite these promising results, the use of GSIs in the clinic has been prevented because of undesirable, dose-limiting severe toxicities, particularly gastrointestinal toxicity, derived from the systemic inhibition of Notch in different tissues (16, 20–22). To circumvent the unwanted side-effects of GSI while retaining its ability to block Notch communication in the MM niche, we generated a bone-targeted Notch inhibitor (BT-GSI). We used a modified bisphosphonate with a high affinity for bone as a bone-targeting moiety and linked it to GSI-XII using a hydrazine acid-sensitive linker designed to be cleaved in areas of the bone/BM with low pH (Figure 1a). We report here that BT-GSI selectively inhibits Notch signaling in bone/BM but not in other tissues. Further, mice with established MM treated with BT-GSI exhibit decreased MM tumor growth and reduced MM-induced bone destruction at doses in which unconjugated GSI is ineffective, and are free of gastrointestinal toxicity.
Figure 1. Generation of a bone-targeted gamma-secretase Notch inhibitor (BT-GSI).
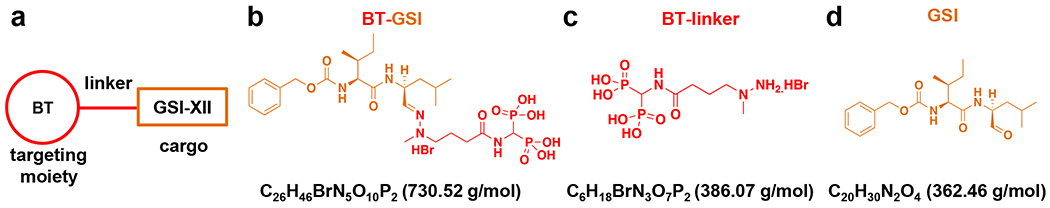
(a) Diagram showing the principle of the bone targeting strategy, including a modified bisphosphonate with high bone affinity, a pH-sensitive linker, and gamma-secretase inhibitor (GSI) XII. Molecular weight and chemical formulation of BT-linker BT-GSI (b, shown in orange and red), (c, shown in red), and GSI (d, shown in orange).
Methods
Reagents.
Commercial γ-secretase inhibitor (GSI-XII) was purchased from Calbiochem (San Diego, CA, US); Minimum Essential Media (MEM) α, RPMI-1640 media, fetal bovine and calf serum, penicillin/streptomycin, and normocin were obtained from Invitrogen Life Technologies (Grand Island, NY, US). PCR primer-probe sets were from Applied Biosystems (Foster City, CA, US) and Roche Applied Science (Indianapolis, IN, US).
Design and synthesis of a bone-targeted GSI inhibitor.
Detailed chemical steps for generating BT-GSI are described in the supplementary methods. Briefly, following previously published approaches (23–25), we generated the bone-targeted GSI (BT-GSI; Figure 1a–b), a conjugate formed by 1) a modified, less active bisphosphonate moiety with high bone affinity designed to direct the conjugate to the skeleton (BT; Figure 1c), 2) a pH-sensitive labile linker that binds the BT to the cargo, and 3) the cargo, the small molecule GSI-XII that inhibits Notch signaling by preventing the cleavage of the Notch intracellular domain of Notch receptors (Figure 1d). The molecular weight of the BT containing the cleaved linker structure is 386.07 g/mol, and GSI-XII has a molecular weight of 362.46 g/mol. Because the molecular weight of BT-GSI is 730.52 g/mol (Figure 1b), for the in vivo studies, we used half the mg/kg doses of BT and GSI to achieve equimolar concentrations. BT, GSI, and BT-GSI were dissolved in DMSO, whereas zoledronic acid was dissolved in PBS. All drugs were diluted in PBS for animal dosing. Control animals received injections of 5% DMSO v/v (vehicle) diluted in PBS.
Cells and culture conditions.
Human JJN3 (RRID: CVCL_2078) and murine 5TGM1 (RRID: CVCL_VI66) MM cells were provided by N. Giuliani (University of Parma, Parma, Italy) and B. Oyajobi (University of Texas at San Antonio, USA), respectively. Cell lines were routinely tested for mycoplasma and authenticated for morphology, gene expression profile, and tumorigenic capacity. JJN3/5TGM1 MM cells were cultured in RPMI-1640 medium and treated with 15μM of BT or unconjugated GSI for 4h-24 hours. To test if acidic conditions activate BT-GSI (inactive under physiological pH), BT, BT-GSI, and GSI were pre-incubated in PBS at pH 7.5, 6.5, and 5.5 overnight. pH was adjusted using a hydrochloric acid solution. Then, pH-incubated compounds were added to the media at a 15μM concentration. Control cells for these experiments were treated with DMSO (0.5% v/v). Then treatments were performed in MM cells cultured in media at physiological pH, which remained unchanged by adding the treatments.
Animal studies.
For these studies, we used immunocompetent (C57BL/KaLwRijHsd) (26–28) and immunodeficient (C57BL/6 Prkdcscid; Jackson’s Lab, Bar Harbor, ME, US; RRID: IMSR) murine models of established MM disease that closely resemble the human disease, as shown before (13, 29). Briefly, six to eight-week-old female and male mice were injected intratibially with 105 murine 5TGM1 or human JJN3 MM cells, and control mice were injected with saline. Three weeks after MM cell inoculation, mice were randomized by tumor levels and bone lytic disease and assigned to subgroups receiving i.p. injections of either 1) vehicle (DMSO), 2) GSI (0.1mol/L (5 mg/kg), 3 times a week), 3) zoledronic acid (0.1 mg/kg, 2 times a week), 4) BT (0.1mol/L (5mg/kg), 0.05mol/L (2.5mg/kg) or 0.02mol/L (1mg/kg), 3 times a week) or 5) BT-GSI (0.1mol/L (10mg/kg), 0.05mol/L (5mg/kg) or 0.02mol/L (2mg/kg), 3 times a week). Treatments were administered in random order and given for a total of three weeks. For the gut toxicity studies, six to eight-week-old naïve C57BL/6 female mice were assigned to subgroups receiving i.p. injections of either 1) vehicle (DMSO), 2) GSI LY3039478 (Selleck Chemical LLC, Houston, TX 77014; 0.4mol/L (20 mg/kg), 2 times a day), 3) GSI (0.4mol/L (20/mg/kg), 2 times a day) or 4) BT-GSI (0.4mol/L (40 mg/kg), 2 times a day) for one week. Mice were fed a regular diet (Harlan, Indianapolis, IN, US), received water ad libitum, and maintained a 12-hour light/dark cycle. Animal studies were approved by the Institutional Animal Care and Use Committee of the Indiana University School of Medicine and the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences. Animal studies and data reporting follow ARRIVE guidelines.
Serum biochemistry
Human Kappa light chain, produced by JJN3 myeloma cells, and IgG2b, produced by 5TGM1 cells, are well-established biomarkers of MM tumor growth (27, 28, 30, 31). Serum levels of human Kappa Light chain (Bethyl Laboratories Inc., Montgomery, TX, USA) and mouse IgG2b (Thermo Fisher, Waltham, MA, USA) paraproteins were measured using enzyme-linked immunosorbent assays (ELISA) kits and following manufacturer protocols, as previously described (13, 29). Serum levels of the marker of bone formation N-terminal propeptide of type I procollagen (P1NP) (32) and the maker of bone resorption C-terminal telopeptides of type I collagen (CTX) (33) were quantified as previously published (13, 29).
Analysis of the skeletal phenotype.
X-ray radiography and micro-computed tomography (microCT) analyses were performed as previously described (13, 29). Lytic lesions were evaluated in X-ray radiographs taken before and after 3 weeks of treatment. BioQuant software (Nashville, TN, USA, RRID: SCR_016423) was used to determine osteolytic lesion area/number (13, 29).
Histological analysis.
Tartrate-resistant acid phosphatase (TRAP) staining and static histomorphometric analyses were performed using the OsteoMeasure High-Resolution Digital Video System (OsteoMetrics, Decatur, GA, USA) as previously described (34–37). Immunostaining for Proliferating Cell Nuclear Antigen (PCNA) was performed in de-paraffinized bone tissue sections. Antigen retrieval was performed using Antigen Unmasking Solution (Vector Labs). Sections were blocked with 5% goat serum (Vector S-1000, Vector Laboratories, CA, USA, RRID: AB_2336615) for 1h at RT and then incubated with anti-PCNA (1:100, ab18197, Abcam, RRID: AB_444313) overnight at 4°C. After washing with PBS, slides were incubated with Cyanine5 conjugated goat anti-rabbit secondary antibody (Invitrogen, A-10523, RRID: AB_2534032) for 30 min, then mounted with 4′,6-diamidino-2-phenylindole (DAPI) mounting medium (Vector Laboratories). Tissue sections were photographed using a Leica digital image system at 20x magnification. From each photograph, localized to areas in the bone marrow showing tumor cells were selected. The number of PCNA+ve pixels/total number of DAPI +ve pixels was calculated as shown before (38). Formalin-fixed paraffin-embedded sections (5 μm) of intestinal tissue were stained with Alcian Blue (Newcomer Supply, Middleton, WI, USA) to assess mucin-containing goblet cells as previously described (39). Goblet cells were counted in 5 individual villi per section.
Ex vivo MM-bone organ cultures.
Ex vivo MM-bone organ cultures were established with 5TGM1 MM cells and calvarial bones from C57BL/KaLwRijHsd mice as previously shown (13, 29, 40). Briefly, 50,000 MM cells were plated on calvarial disks and incubated for 24h. Then, calvarial disks containing MM cells were transferred to a 96-well plate and treated with 15μM of BT, GSI, BT-GSI, or DMSO (0.5% v/v) as control. Media and treatments were refreshed every 3 days.
Analysis of mRNA gene expression.
RNA was isolated from cells, bones, or intestinal tissue, and mRNA gene expression was performed as previously described (13, 29). Relative mRNA gene expression levels were normalized to the housekeeping gene ribosomal protein S2 and/or GAPDH, and absolute expression was calculated using the ΔCt method (41). Fold changes were calculated by dividing the treatment values by the control/vehicle values.
Analysis of protein expression.
Protein lysates were prepared from MM cells or bones as previously described (13, 42). Cell lysates (50-100 μg) were boiled in the presence of sodium dodecyl sulfate (SDS) sample buffer (NuPAGE LDS sample buffer, Invitrogen) for 10 minutes and subjected to electrophoresis on 10% SDS-PAGE (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were transferred to PVDF membranes using a semidry blotter (Bio-Rad) and incubated in blocking solution (5% nonfat dry milk in TBS containing 0.1% Tween-20) for 1 hour to reduce nonspecific binding. Immunoblots were performed using anti-GAPDH (1:1000, RRID: AB_561053), HES1 (1:1000, RRID: AB_1209570), and beta-ACTIN (1:1000, RRID: AB_306371), antibodies followed by goat anti-rabbit (1:5000, Invitrogen, RRID: AB_2534782) or goat anti-mouse antibodies (1:2000, Santa Cruz Biotechnology, RRID: AB_631736), conjugated to horseradish peroxidase in 5% milk. Western blots were developed using an enhanced chemiluminescence detection assay following the manufacturer’s directions (Bio-Rad).
Statistical analysis.
Data were analyzed using SigmaPlot 12.0 (Systat Software, Inc., San Jose, CA, RRID: SCR_003210). Data were analyzed by one-way ANOVA-Dunn’s test using MM (vehicle) as the control group or by student’s t-test. Means ± standard deviation (S.D.) are reported. P values ≤0.05 were considered significant. Samples sizes were estimated based on previous experience and results obtained with the in vivo models and in vitro systems employed in this manuscript. Data exceeding two standard deviations from the mean were considered as outliers and removed from the analysis. All these analyses were performed by two independent investigators in a blinded manner.
Data and Materials availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request. There are restrictions to the availability of BT-GSI due to the limited amount of this compound available and ongoing efforts to commercialize the invention through patent or licensing. We may require a completed Materials Transfer Agreement.
Results
BT-GSI is activated under acidic conditions.
We first determined in vitro the pH sensitivity of BT-GSI and its ability to inhibit Notch signaling in MM cells and bone. Before treatment, compounds were pre-incubated in PBS at various pH levels resembling pH changes induced by osteoclasts and the growth of MM cells in the marrow (43–48). In vitro treatment of 5TGM1 murine MM cells with unconjugated GSI pre-incubated at pH 7.5 decreased the expression of the Notch target gene Hes1 in MM cells by ~30%, whereas no effects were seen with BT-GSI pre-incubated at the same pH (Figure 2a). In contrast, pre-incubation at pH ≤ 6.5 activated BT-GSI and decreased Hes1 mRNA expression in MM cells to a similar extent as GSI (Figure 2a). BT had no effects on Hes1 mRNA expression at any of the pH levels tested. To test the effects of BT-GSI on bone under physiological conditions, we used ex vivo bone organ cultures, which reproduce the in vivo acidic conditions induced by osteoclastic resorption in the bone microenvironment (13, 40, 42). Ex vivo treatment with GSI and BT-GSI similarly decreased in bone tissue the mRNA expression of the Notch target genes Hes1 and Hey1 by ~75% (Figure 2b), whereas treatment with BT did not affect the expression of Notch target genes. Further, we established ex vivo MM-bone organ cultures with 5TGM1 murine MM cells to examine the effects of BT-GSI on the growth of MM cells. GSI and BT-GSI decreased the levels of 5TGM1-derived IgG2b, a well-established biomarker of MM tumor growth, in conditioned media by ~50% after 3 days of treatment (Figure 2b). In contrast, treatment with BT induced a non-significant ~10% decrease in IgG2b levels. Together, these findings demonstrate that BT-GSI is activated under acidic conditions in bone, reduces the mRNA expression of Notch target genes, and decreases MM cell growth.
Figure 2. BT-GSI is activated at low pH, and Notch inhibition decreases MM cell growth and osteoclast formation.
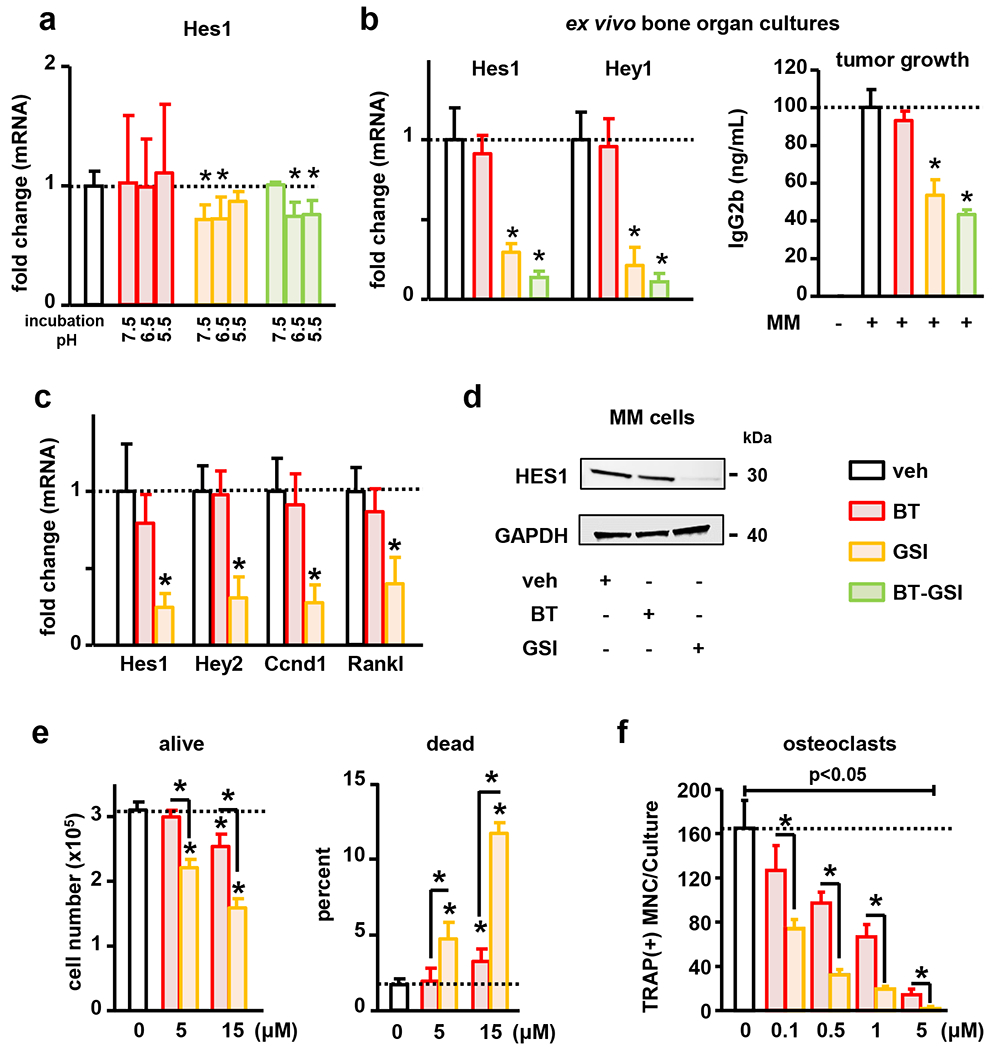
(a) Compounds were incubated at various pH in PBS before treatments. 4h treatment of JJN3 MM cells with BT-GSI pre-incubated at pH≤ 6.5 decreased Hes1 expression in MM cells (n=4/group). (b) 3-day treatment with GSI and BT-GSI, but not BT, decreased Hes1 and Hey1 mRNA expression (n=3-6/group) and reduced the levels of the MM tumor biomarker IgG2b in ex vivo whole bone organ cultures bearing 5TGM1 MM cells that reproduce acidic conditions in the bone microenvironment (n=5-6/group). (c-e) Treatment of 5TGM1 MM cells with BT and GSI decreased proliferation and induced cell death, but only GSI downregulated the expression of Hey2, Hes1, Cyclin D1 (Ccnd1), and Rankl (n=4/group). (f) Treatment with BT and GSI inhibited RANKL-induced pre-osteoclast differentiation in a dose-dependent manner (n=6/group). Bars represent means ± SD. Representative experiments out of two are shown. Horizontal dotted lines indicate the mean value for vehicle conditions. Fold changes in mRNA expression were calculated by dividing the treatment values by the control/vehicle values. *p<0.05 vs. veh.
Notch inhibition decreases MM cell proliferation and osteoclast number.
To examine the effects of Notch inhibition on MM growth and osteoclast differentiation, we cultured 5TGM1 MM cells and osteoclast precursors in the presence/absence of BT and GSI. GSI inhibited Notch signaling, decreased MM cell proliferation, and increased the number of MM dead cells (Figures 2c–f). Consistent with the reduced proliferation, GSI downregulated the expression of Cyclin D1 by 87% (Figure 2c). In addition, GSI decreased Rankl expression in MM cells (Figure 2c) and inhibited Rankl-induced osteoclast formation in a dose-dependent manner (Figure 2f). BT did not affect the expression of Notch target genes or Rankl in MM cells (Figure 2c–d). Higher doses of BT decreased MM cell number (Figure 2e) but did not affect the expression of the proliferating marker Cyclin D1 (Figure 2c), suggesting that this effect is due to MM cell death (Figure 2e). BT also decreased osteoclast differentiation but with lower efficacy than GSI (Figure 2f). These in vitro findings suggest that the local release of GSI to the bone/bone marrow niche and the consequent Notch inhibition could decrease MM growth and block resorption by inhibiting Rankl production and osteoclast differentiation. Further, these results suggest that BT could also reduce MM tumor burden and mitigate osteoclast-mediated bone resorption at higher doses.
BT-GSI inhibits tumor growth and osteolytic disease progression more efficiently than GSI.
We next examined the in vivo effects of BT-GSI (0.1 mol/L) on tumor growth and MM-induced bone destruction and compared its effects to those resulting from equimolar administration of unconjugated GSI and the bone targeting moiety BT without cargo, and to a well-established regime of zoledronic acid, the gold-standard for the clinical management of MM bone disease (Figure 3a) (4). Three weeks after cell injection, mice injected with 5TGM1 MM cells exhibited a 2.5 fold increase in serum levels of the tumor biomarker IgG2b compared to saline-injected mice, and evident osteolytic lesions in the injected tibias (Figure 3b and 4a–b), indicative of effective engraftment and active tumor growth and established MM bone disease. After 3 weeks of treatment, mice bearing MM and treated with vehicle exhibited a 6-fold increase in tumor growth. Similar tumor progression was observed in mice receiving zoledronic acid or unconjugated GSI. In contrast, MM tumor burden did not significantly increase since treatment initiation in mice treated with BT-GSI or BT, and both treatments decreased MM tumor growth by 50% compared to vehicle-treated mice (Figure 3b). Consistent with these results, tumors in mice treated with vehicle, zoledronic acid, or GSI exhibited abundant PCNA + MM cells (0.15±0.03 vs. 0.20±0.05 vs. 0.18±0.03 PCNA+ pixels/DAPI+ pixels, respectively). BT-GSI decreased the number of PCNA+ MM cells (0.07±0.02 PCNA+ pixels/DAPI+ pixels), whereas tumors in mice treated with BT still showed PCNA+ cells (0.18±0.03 PCNA+ pixels/DAPI+ pixels) (Figure 3c). These results, together with our in vitro data, suggest that BT-GSI decreases tumor growth by inhibiting MM cell proliferation, while BT might reduce tumor burden by promoting MM cell death.
Figure 3. BT-GSI decreases tumor growth and the number of proliferating MM cells.
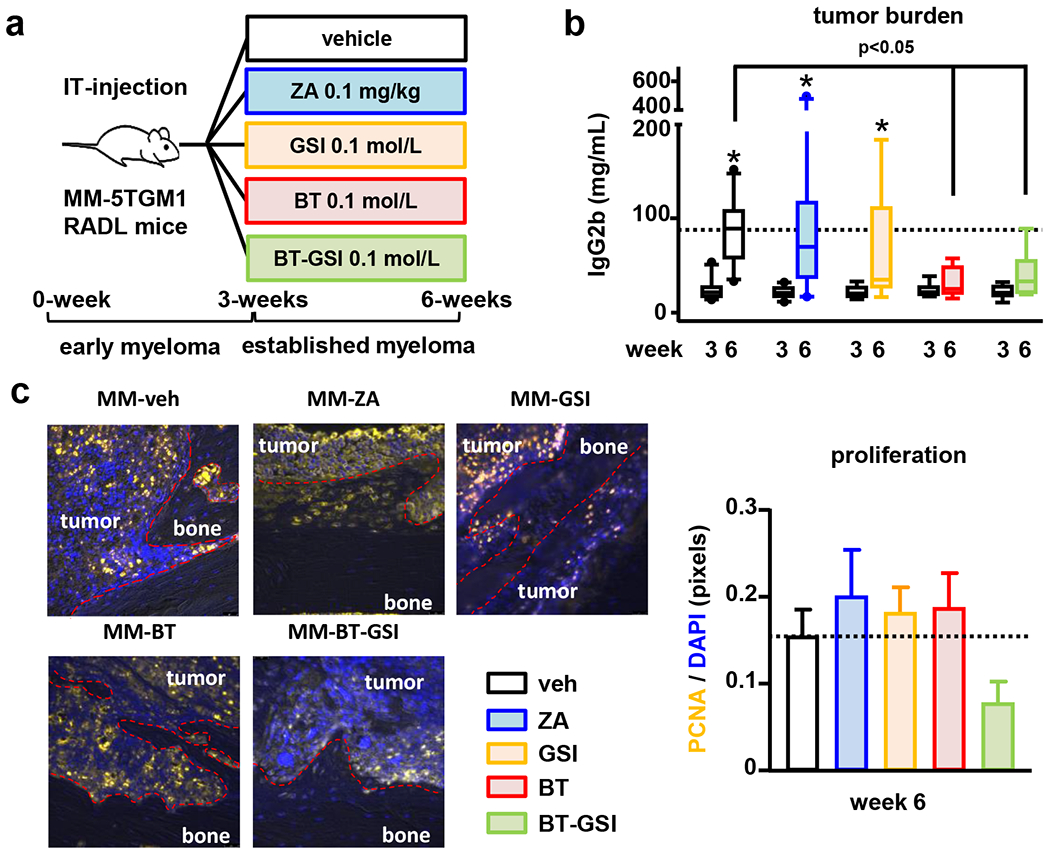
(a) Experimental design (105 5TGM1 MM cells, IT-intratibial injection). Effects of zoledronic acid (ZA, 0.1 mg/kg, two times a week, for 3 weeks) and equimolar dosing (0.1mol/L, three times a week, 3 weeks) of GSI, BT, and BT-GSI on (b) serum levels of the 5TGM1 tumor biomarker IgG2b, and (c) on bone tumors (week 6) stained for Proliferating Cell Nuclear Antigen (PCNA). Bars represent means ± SD. n=6-11/group. *p<0.05 vs. week 3 (c). Representative immunostaining images per group are shown. Red dotted lines delimit bone tissue. The horizontal dotted line indicates the mean value for vehicle-treated mice bearing MM tumors.
Figure 4. BT-GSI decreases MM-induced osteolytic lesions and osteoclasts in immunocompetent mice with established MM.
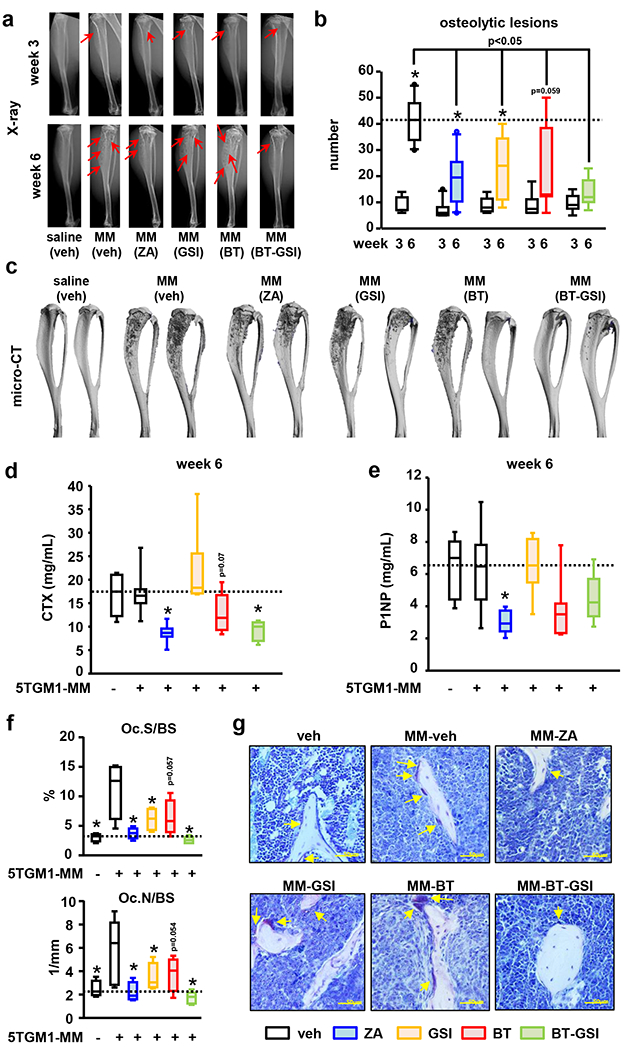
Effects of zoledronic acid (ZA, 0.1 mg/kg, two times a week , for 3 weeks) and equimolar dosing (0.1mol/L, three times a week, 3 weeks) of GSI, BT, and BT-GSI on (a-b) osteolytic lesion number per bone, (c) bone destruction (microCT 3D reconstruction), (d) serum levels of the bone resorption marker CTX (week 6), (e) the bone formation marker P1NP (week 6), and on (f) bone surface covered by osteoclasts (Oc.S/BS) and osteoclast number (Oc.N/BS). (g) Representative images of TRAP stained bone histological sections. Yellow arrows indicate TRAP+ osteoclasts. Bars represent means ± SD. n=6-11/group. *p<0.05 vs week 3 (b), vs MM (veh; d-e), or vs naïve mice (f). Red arrows indicate osteolytic lesions. Horizontal dotted lines indicate the mean value for vehicle-treated mice bearing MM tumors (b, d, e) or naïve mice (f).
During the treatment period, the number of osteolytic lesions increased by 5-fold in vehicle-treated mice bearing MM (Figure 4a–c). Although the osteolytic disease also progressed in MM mice receiving zoledronic acid, GSI, and BT, these treatments reduced the number of osteolytic lesions by 45% compared to mice receiving vehicle. Mice treated with BT-GSI showed no significant increases in osteolytic lesion number compared to the number at the start of treatment and exhibited a 70% reduction in osteolytic lesion number compared to vehicle-treated mice (Figure 4a–c). BT-GSI and zoledronic acid similarly decreased the systemic levels of the bone resorption marker CTX by 50% compared to vehicle-treated mice (Figure 4d). BT treatment resulted in a modest 25% reduction of CTX, while GSI had no effects on systemic bone resorption. No significant changes were found in the serum levels of the bone formation biomarker P1NP in mice treated with BT, GSI, or BT-GSI. In contrast, treatment with zoledronic acid led to a significant reduction in P1NP (Figure 4e). Moreover, BT-GSI and zoledronic acid decreased to control levels the elevated number of osteoclasts and bone surface covered by osteoclasts induced by inoculation of MM cells (Figure 4f–g). GSI also decreased osteoclasts in bones bearing MM tumors, whereas BT showed a trend to reduce osteoclast indexes that did not reach statistical significance. Collectively, these results demonstrate that at equimolar doses, BT-GSI inhibits MM tumor growth at doses of which GSI is ineffective and that BT-GSI decreases bone resorption and stops MM-induced bone destruction more efficiently than GSI.
BT-GSI selectively inhibits Notch signaling in bone and does not induce gut toxicity.
We investigated the effects of intermittent administration of BT-GSI and GSI (0.1mol/L, 3 times a week) on intestinal tissue. BT-GSI decreased further the expression of Notch target genes in bone tissue compared to GSI (Figure 5a–b). However, BT-GSI did not alter Notch signaling expression in intestinal tissue or increase the expression of the gut toxicity biomarker Adipsin (Figure 5c) (20, 49). Further, BT-GSI did not alter the morphology of intestinal tissue or increase goblet cell numbers or mucous cell hyperplasia, common features of gut toxicity induced by continuous GSI treatment (Figure 5c–d) (20, 49). Treatment with GSI decreased the mRNA expression of Hes5 and Hes7 in the gut and induced a 3.5-fold increase in the expression of Adipsin (Figure 5c), suggesting that GSI initiated the signaling cascade leading to the development of gut toxicity. Yet, with this suboptimal intermittent regimen of GSI, we did not find the development of goblet hyperplasia in intestinal tissue (Figure 5d). Treatment with BT or zoledronic acid did not affect Notch signaling in either tissue or induced gut toxicity (Supplementary figure 1a–c). We also examined the effects of all these treatments on complete blood counts (Supplementary Table 1). Treatment with BT-GSI or any of the other treatments had no significant effects on blood cell counts.
Figure 5. BT-GSI decreases Notch signaling specifically in bone and does not induce gut toxicity.
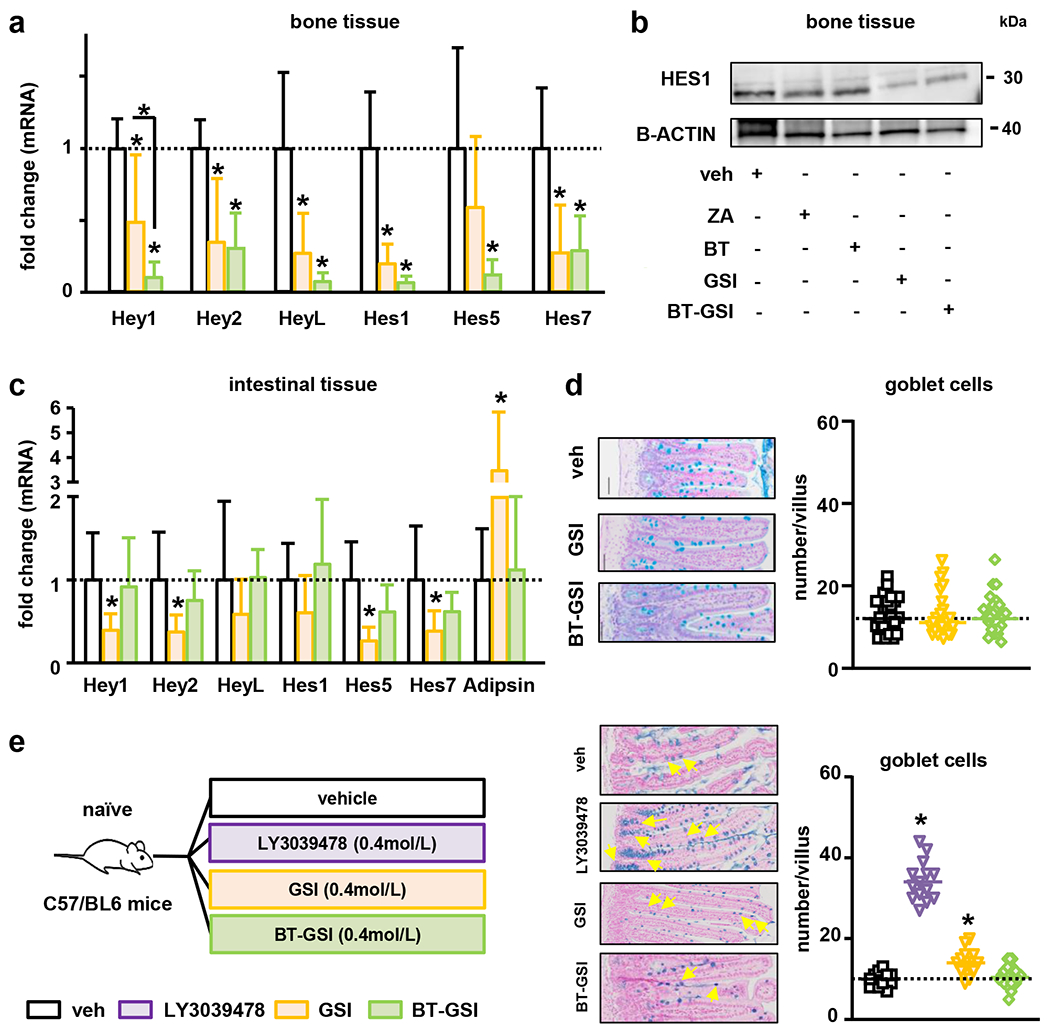
(a-b) Treatment with BT-GSI (0.1mol/L, three times a week, 3 weeks) decreased the expression of Notch target genes in bones not bearing MM but did not alter Notch mRNA expression or increased the expression of the recognized biomarker of gastrointestinal toxicity Adipsin in intestinal tissue (c) (n=6-11/group). Fold changes in mRNA expression were calculated by dividing the treatment values by the control/vehicle values. (d) Alcian blue staining of intestinal tissue revealed no presence of goblet cell metaplasia in BT-GSI treated mice. (e) Experimental design (C57BL/6 naïve mice). Alcian blue staining of intestinal tissues showing an increased number of goblet cells in GSI and LY3039478 (0.4mol/L, two times a day, 1 week) treated mice. Treatment with BT-GSI (0.4 mol/L, twice a day, 1 week) did not change the number of goblet cells per villi (n=3/group). Bars represent means ± SD. Representative images of Alcian blue staining of intestinal tissues are shown. Horizontal dotted lines indicate the mean value for vehicle conditions. *p<0.05 vs. veh.
To further examine the gastrointestinal safety of BT-GSI, we compared its effects versus a daily regime of GSI and LY3039478, a γ-secretase inhibitor known to rapidly induce gut toxicity in rodents and humans and recently tested in mouse models of MM and MM patients (49–51) (Figure 5e). Daily treatment with LY3039478 and GSI for only a week increased the number of goblet cells in intestinal villi, a hallmark of GSI-induced gut toxicity (20, 49). In contrast, no significant changes in goblet cell number were observed in the intestines of mice receiving equimolar doses of BT-GSI (Figure 5e). Together, these results show that bone-targeted Notch inhibition with BT-GSI, even with daily high doses, circumvents the gut toxicity seen with systemic administration of GSIs.
Low doses of BT-GSI retain high anti-MM efficacy and preserve bone compared to BT.
A critical aspect of BT-GSI is that it is designed to accumulate at higher concentrations in the skeleton in vivo, thereby exerting its bioactivity locally in bone. Thus, we hypothesized that lower doses of BT-GSI should still decrease MM growth and prevent bone destruction. To test this hypothesis, we treated mice with active tumor growth and established bone disease with equimolar doses of BT and BT-GSI that were two-times (0.05 mol/L) or five-times (0.02 mol/L) lower than the dose used in studies shown in Figure 3 (Figure 6a). At the end of the treatment period, mice receiving vehicle injections exhibited a 5-fold increase in MM tumor burden (Figure 6b). At these lower doses, BT did not affect tumor growth (Figure 6b), whereas mice receiving BT-GSI 0.02 mol/L or 0.05 mol/L had a 51% and 60% decrease in tumor burden, respectively, compared to control MM mice (Figure 6b). The progression of the osteolytic disease was nearly identical in mice receiving vehicle or either dose of BT. In contrast, BT-GSI doses reduced the number of osteolytic lesions compared to vehicle-treated mice (41% and 52%, respectively; Figure 6c–e) and to a similar extent than the 0.1mol/L BT-GSI dose (Supplementary figure 2). At these doses, both BT and BT-GSI retained their ability to decrease systemic bone resorption, as shown by the decrease in serum CTX levels compared to vehicle-treated mice (Figure 6f). In addition, no significant differences were found in P1NP levels between any of the groups (Figure 6g). These results demonstrate that lower doses of BT-GSI retain potent anti-MM and anti-resorptive activities and can slow tumor progression and MM-induced bone disease.
Figure 6. Lower doses of BT-GSI effectively inhibit tumor growth and prevent bone destruction in immunocompetent mice with established MM.
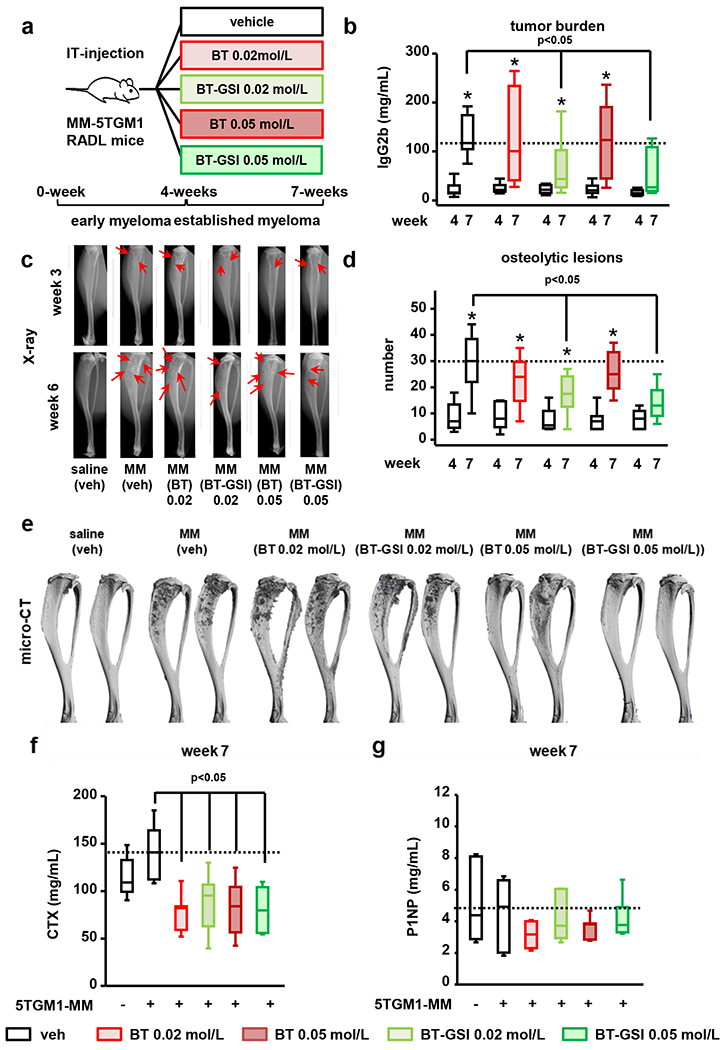
(a) Experimental design (105 5TGM1 MM cells, IT-intratibial injection). Effects of equimolar doses (0.05 mol/L and 0.02 mol/L, three times a week, 3 weeks) of BT-GSI and BT on (b) serum levels of the 5TGM1 tumor biomarker IgG2b, (c-d) osteolytic lesion number per bone, (e) bone destruction (microCT 3D reconstruction), (f) serum levels of the bone resorption marker CTX (week 7) and (g) of the bone formation marker P1NP (week 7). Bars represent means ± SD. n=8-9/group. *p<0.05 vs week 4 (b-d) or vs MM-veh (f-g). Red arrows indicate osteolytic lesions. Horizontal dotted lines indicate the mean value for vehicle-treated mice bearing MM tumors.
BT-GSI decreases tumor growth and prevents bone destruction in a human xenograft mouse model of established MM disease.
To determine the effects of BT-GSI on human MM disease, we treated immunodeficient mice bearing JJN3 human MM cells with BT-GSI (Figure 7a). BT-GSI decreased the expression of Notch related genes in bone by ~50%. In contrast, BT-GSI treatment did not change the expression of Notch genes or Adipsin in intestinal tissue (Figure 7b–c). Three weeks after cell inoculation, mice injected with JJN3 MM cells exhibited detectable levels of the tumor biomarker human Kappa light chain in the serum and osteolytic lesions (Figure 7d–f). Mice bearing MM treated with vehicle showed a 6-fold increase in MM tumor burden 3 weeks after treatment initiation. BT-GSI decreased MM tumor growth by 40% compared to vehicle-treated mice (Figure 7d). Vehicle-treated mice bearing MM had a ~10-fold increase in the osteolytic area since treatment initiation. In contrast, MM-injected mice receiving BT-GSI had no significant increases in the lytic area compared to before treatment and a 50% reduction in osteolytic lesion area compared to vehicle-treated mice at the end of the study (Figure 7e–g). This bone protective effect of BT-GSI was accompanied by a 35% decrease in the systemic CTX levels (Figure 7h), while no significant differences were found in P1NP serum levels (Figure 7i). We also tested the effects of equimolar dosing of the BT moiety, without its cargo, in this human xenograft mouse model with established MM disease. After three weeks of treatment with BT, tumor burden levels remained indistinguishable from those detected in vehicle-treated MM mice (Figure 7j). Further, mice bearing MM tumors and receiving either vehicle or BT injections exhibited a similar loss of cancellous bone volume in the tibia (Figure 7k).
Figure 7. BT-GSI decreases tumor growth and reduces the progression of the osteolytic disease in immunodeficient mice with established human MM.
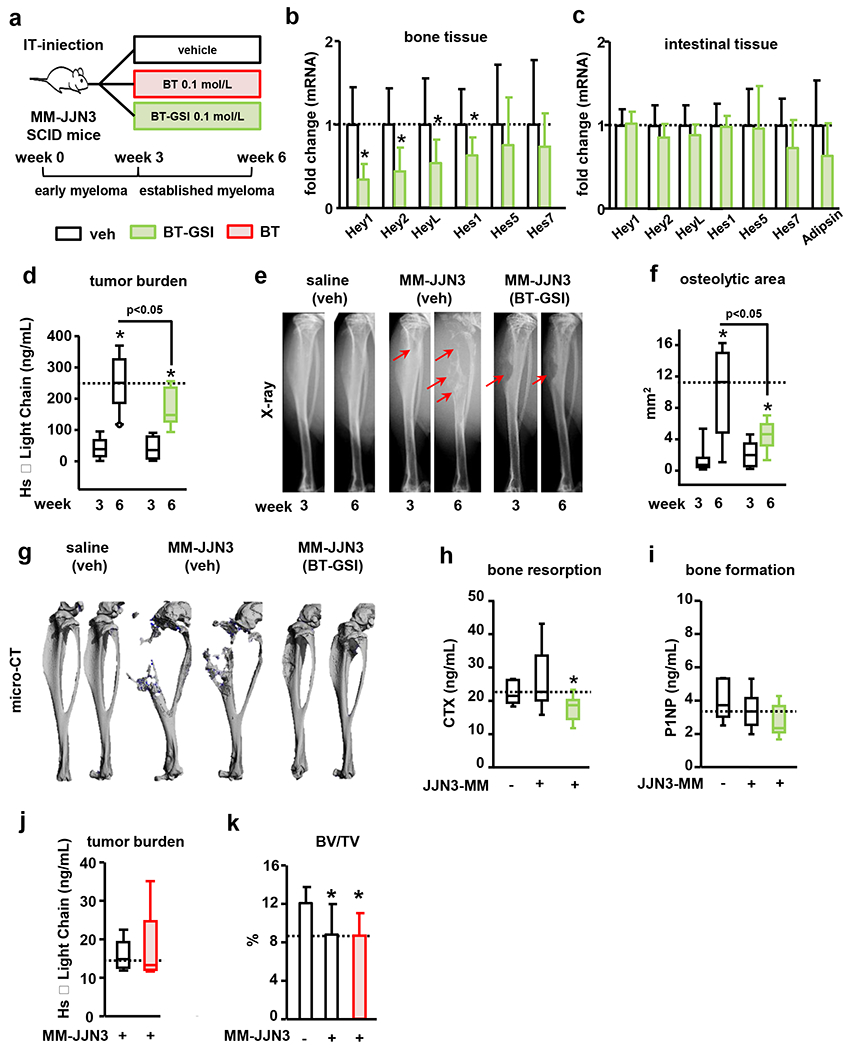
(a) Experimental design for BT-GSI and BT (105 JJN3 cells, IT-intratibial injection). Effects of BT-GSI (0.1mol/L, three times a week, 3 weeks) on (b-c) mRNA gene expression of Notch targets in bone and intestinal tissue, (d) serum human Kappa light chain, (e) tibia X-rays, (f) area of osteolytic lesions, (g) 3D microCT reconstruction images of PBS/MM cells injected tibias, (h) serum levels of CTX and (i) P1NP. Bars represent means ± SD. n=7-11/group. Effects of BT on serum human Kappa light chain 6 weeks after MM cell inoculation (j), and bone volume over tissue volume (BV/TV) in the cancellous bone of tibias inoculated with JJN3 MM cells (k). Bars represent means ± SD. n=9-10/group. *p<0.05 vs 3 weeks (d-f) or vs MM-veh (b, c, l). Red arrows indicate osteolytic lesions. Horizontal dotted lines indicate the mean value for vehicle-treated mice bearing MM tumors. Fold changes in mRNA expression were calculated by dividing the treatment values by the control/vehicle values.
Discussion
Notch signaling mediates MM cell communication with adjacent cancer and host cells in the tumor niche and has been recognized as a major contributor to MM progression and osteolysis (10, 11). To safely inhibit this signaling pathway in the MM niche and avoid toxic effects in other tissues, we generated a bisphosphonate-linked GSI (BT-GSI) analog designed to target the bone and release GSI in the acidic microenvironment of the bone/tumor niche. BT-GSI selectively inhibited Notch signaling in bone but not in other organs, decreased MM growth, and reduced osteolytic disease progression by inhibiting bone resorption in both immunocompetent and immunodeficient models of established MM. Remarkably, BT-GSI inhibits MM growth and bone destruction at doses in which systemic administration of GSI is ineffective. BT-GSI did not induce gut toxicity or alter blood cell counts, which are important toxicities seen with systemic administration of GSI (16, 20). These results show that BT-GSI is a Notch inhibitor targeted to the bone/BM niche and suggest that BT-GSI inhibition of Notch signaling in the MM niche is a promising and less toxic approach to simultaneously inhibit the growth of MM cells and prevent bone loss in MM patients.
The high bone affinity of bisphosphonates has shown in vivo utility to transport other pharmacologically active drugs to the bone, particularly areas of the skeleton with higher turnover (52, 53). In the current studies, we used a novel hydrazone acid-sensitive linker designed for use with the aldehyde functionality of GSI-XII. Our in vivo and in vitro findings support that BT can direct its cargo GSI to the bone and that acidic conditions (pH ≤6.5) are needed to cleave the linker and activate the conjugate. We hypothesized that extracellular acidification by osteoclasts (pH range 4-6) (43–45) and/or MM-induced acidification of the bone/BM (pH range 6-7) (46–48) is sufficient to stimulate the release of GSI from the BT moiety. The Notch inhibition seen in bone tissue, together with the anti-MM and anti-resorptive efficacy displayed by BT-GSI in our in vivo studies, support the functionality of our “target and release” approach and suggest that the acidic conditions of the MM-bone niche lead to an adequate cleavage of the hydrazine linker and the local release of GSI in the bone/MM tumor niche.
BT-GSI decreased MM tumor burden in vivo in a preclinical model of established MM disease, whereas the unconjugated GSI counterpart or zoledronic acid did not. The decrease in MM growth seen with BT-GSI could be due to the inhibition of homotypic and/or heterotypic Notch communication in the MM niche. Notch inhibition decreases Cyclin D1 expression and MM proliferation, and induces caspase-3-dependent apoptosis in MM cells (14, 54). Further, Notch blockade reduces the proliferation of MM cells supported by direct contact with stromal cells and osteocytes (13, 55, 56). Intriguingly, treatment with high doses of the bone targeting molecule BT alone without the GSI cargo increased MM cell death in vitro in MM cell cultures and reduced tumor growth in vivo in immunodeficient mice bearing 5TGM1 tumors. Thus, we cannot exclude the possibility that once hydrolyzed, the modified bisphosphonate BT contributes to the decrease in MM growth by increasing MM cell death, as seen with other bisphosphonates in vitro or administered for longer periods (57–61). Yet, this effect of BT on MM cells is independent of Notch regulation, as BT did not affect the expression of Notch target genes in bone or MM cells. Taken together, these findings show that targeted inhibition of Notch signaling in the MM niche with BT-GSI is a potent approach to inhibit MM growth.
Treatment with GSIs results in unacceptable side-effects, limiting the administration of effective doses and its clinical efficacy, ultimately impeding FDA clinical approval (20, 49–51). The bone selectivity of BT-GSI enables the inhibition of Notch signaling only in bone, without affecting this pathway in intestinal tissues, a major target of GSIs (20). We found that lower doses of GSI increased Adipsin in intestinal tissue but did not induce the typical GSI-induced goblet hyperplasia in histological sections (20, 39). This observation is not entirely unexpected and could be due to the suboptimal regime of GSI used in this particular study. In contrast, daily administration of higher doses of GSI increased the number of goblet cells per villi, a hallmark of gut toxicity (20, 49–51). In contrast, equimolar administration of high or low doses of BT-GSI did not result in gut toxicity and preserved the intestinal morphology as seen in histological sections. Further, BT-GSI administration did not decrease body weight (Supplementary figure 3a–c). Of note, the gut safety displayed by BT-GSI contrasts with the marked increase in goblet cell hyperplasia seen in mice administered LY3039478, a GSI currently being tested in clinical trials as an anti-cancer agent. Altogether, these findings demonstrate that BT-GSI exhibits selective inhibition of Notch in bone and shows a safer profile than systemic GSIs by avoiding side effects of inhibition of Notch in the intestines and other organs.
BT-GSI inhibited systemic bone resorption (even at low doses), decreased osteoclast number/surface, and prevented the progression of the osteolytic disease to a greater extent than GSI or zoledronic acid, the mainstay therapy for MM-induced bone disease (4). The effects of BT-GSI on osteoclasts are further supported by our mechanistic in vitro studies showing that GSI reduces Rankl expression in MM cells and inhibits Rankl-mediated osteoclast differentiation; and by recent reports indicating that Notch signaling mediates the increased osteoclastic resorption induced by MM cells (10, 17, 19). Our results show that the bone-targeting moiety BT has some residual anti-resorptive activity, as it was able to inhibit osteoclast differentiation in vitro and decrease systemic bone resorption in vivo. It is important to note that the BT dose used for these studies was ~50 times higher than the dose used for zoledronic acid. However, despite decreasing systemic CTX, BT only had modest effects in preventing the osteolytic disease or reducing osteoclast number, suggesting BT itself is not potent enough to inhibit osteoclast activity locally in bones with active MM growth. Together, our results indicate that, in addition to its anti-MM efficacy and safer profile, BT-GSI has potent anti-resorptive effects. Of note, BT-GSI did not halt bone remodeling or decrease bone formation, unlike zoledronic acid. These results suggest that BT-GSI preserves physiological bone formation and open the possibility to combine it with bone anabolic agents (i.e., Sclerostin neutralizing antibodies) (29, 62, 63).
There are several aspects of BT-GSI that will require further investigation. For instance, Notch inhibition has been shown to enhance the efficacy of anti-MM therapies, including bortezomib and glucocorticoids (14, 56, 64). Hence, further studies are needed to evaluate in vivo BT-GSI’s anti-MM efficacy when co-administered with other therapeutics. Although unlikely due to the high availability of bone surfaces in bones colonized by MM cells, whether administration of bisphosphonates, commonly used in the clinic for MM-related skeletal events (4), affects BT-GSI’s anti-MM and anti-resorptive activities remains to be determined. Future studies are also needed to evaluate the effects of prolonged administration and withdrawal of BT-GSI.
In summary, we show here that targeted inhibition of Notch signaling to the MM niche with BT-GSI is a promising therapeutic approach with dual anti-MM and anti-resorptive properties, enabling simultaneous inhibition of tumor growth and prevention of bone destruction in MM. Because BT-GSI is tissue-specific and selectively blocks Notch signaling in bone, it lacks gut toxicity and circumvents the deleterious side effects that limit the clinical use of GSIs for MM and potentially other cancers that grow in bone.
Supplementary Material
Statement of significance.
Development of a bone-targeted Notch inhibitor reduces multiple myeloma growth and mitigates cancer-induced bone destruction without inducing gastrointestinal toxicity typically associated with inhibition of Notch.
Acknowledgments
This work was supported by the National Institutes of Health (R37-CA251763 to J.D.C., R01-CA209882 to G.D.R. and T.B., and R01CA241677 to G.D.R.), the Arkansas COBRE program (NIGMS P20GM125503; to J.D.C.), a Scholar Award by the American Society of Hematology Scholar Award (to J.D.C), and a Brian D. Novis Award by the International Myeloma Foundation (to J.D.C.). The authors thank Dr. Gabriel Pagnotti and Stuart Berryhill for their assistance with microCT evaluations.
Footnotes
Conflict of interest: R.L and M.V are employees of Cellestia Biotech AG. F.H.E. is an employee of BioVinc LLC. All authors declare no competing financial interests.
References
- 1.Rajkumar SV. Myeloma today: Disease definitions and treatment advances. Am J Hematol 2016;91:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croucher PI, McDonald MM, and Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer 2016;16:373–86. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi G, and Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015;125:3049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terpos E, Berenson J, Raje N, and Roodman GD. Management of bone disease in multiple myeloma. Expert Rev Hematol 2014;7:113–25. [DOI] [PubMed] [Google Scholar]
- 5.Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004;350:1655–64. [DOI] [PubMed] [Google Scholar]
- 6.Roodman GD. Targeting the bone microenvironment in multiple myeloma. J Bone Miner Metab 2010;28:244–50. [DOI] [PubMed] [Google Scholar]
- 7.Lomas OC, Tahri S, and Ghobrial IM. The microenvironment in myeloma. Curr Opin Oncol 2020;32:170–5. [DOI] [PubMed] [Google Scholar]
- 8.Meurette O, and Mehlen P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018;34:536–48. [DOI] [PubMed] [Google Scholar]
- 9.Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, et al. Notch-directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia 2013;27:1009–18. [DOI] [PubMed] [Google Scholar]
- 10.Colombo M, Thummler K, Mirandola L, Garavelli S, Todoerti K, Apicella L, et al. Notch signaling drives multiple myeloma induced osteoclastogenesis. Oncotarget 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colombo M, Galletti S, Garavelli S, Platonova N, Paoli A, Basile A, et al. Notch signaling deregulation in multiple myeloma: A rational molecular target. Oncotarget 2015;6:26826–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colombo M, Galletti S, Bulfamante G, Falleni M, Tosi D, Todoerti K, et al. Multiple myeloma-derived Jagged ligands increases autocrine and paracrine interleukin-6 expression in bone marrow niche. Oncotarget 2016;7:56013–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res 2016;76:1089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, and Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood 2008;111:2220–9. [DOI] [PubMed] [Google Scholar]
- 15.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002;2:584–93. [DOI] [PubMed] [Google Scholar]
- 16.Imbimbo BP. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr Top Med Chem 2008;8:54–61. [DOI] [PubMed] [Google Scholar]
- 17.Ramakrishnan V, Ansell S, Haug J, Grote D, Kimlinger T, Stenson M, et al. MRK003, a gamma-secretase inhibitor exhibits promising in vitro preclinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia 2012;26:340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Chen F, Clifton N, Sullivan DM, Dalton WS, Gabrilovich DI, et al. Combined inhibition of Notch signaling and Bcl-2/Bcl-xL results in synergistic anti-myeloma effect. Mol Cancer Ther 2010;9:3200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarzer R, Nickel N, Godau J, Willie BM, Duda GN, Schwarzer R, et al. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J 2014;4:e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem 2003;278:46107–16. [DOI] [PubMed] [Google Scholar]
- 21.Fabbro D, Bauer M, Murone M, and Lehal R. Notch Inhibition in Cancer: Challenges and Opportunities. Chimia (Aarau) 2020;74:779–83. [DOI] [PubMed] [Google Scholar]
- 22.Kreft AF, Martone R, and Porte A. Recent advances in the identification of gamma-secretase inhibitors to clinically test the Abeta oligomer hypothesis of Alzheimer’s disease. J Med Chem 2009;52:6169–88. [DOI] [PubMed] [Google Scholar]
- 23.Arns S, Gibe R, Moreau A, Monzur MM, and Young RN. Design and synthesis of novel bone-targeting dual-action pro-drugs for the treatment and reversal of osteoporosis. Bioorg Med Chem 2012;20:2131–40. [DOI] [PubMed] [Google Scholar]
- 24.Liu CC, Hu S, Chen G, Georgiou J, Arns S, Kumar NS, et al. Novel EP4 receptor agonist-bisphosphonate conjugate drug (C1) promotes bone formation and improves vertebral mechanical properties in the ovariectomized rat model of postmenopausal bone loss. J Bone Miner Res 2015;30:670–80. [DOI] [PubMed] [Google Scholar]
- 25.Morioka M, Kamizono A, Takikawa H, Mori A, Ueno H, Kadowaki S, et al. Design, synthesis, and biological evaluation of novel estradiol-bisphosphonate conjugates as bone-specific estrogens. Bioorg Med Chem 2010;18:1143–8. [DOI] [PubMed] [Google Scholar]
- 26.Asosingh K, Radl J, Van RI, Van CB, and Vanderkerken K. The 5TMM series: a useful in vivo mouse model of human multiple myeloma. Hematol J 2000;1:351–6. [DOI] [PubMed] [Google Scholar]
- 27.Garrett IR, Dallas S, Radl J, and Mundy GR. A murine model of human myeloma bone disease. Bone 1997;20:515–20. [DOI] [PubMed] [Google Scholar]
- 28.Radl J, Croese JW, Zurcher C, MH VdE-V, and de Leeuw AM. Animal model of human disease. Multiple myeloma. Am J Pathol 1988;132:593–7. [PMC free article] [PubMed] [Google Scholar]
- 29.Delgado-Calle J, Anderson J, Cregor MD, Condon KW, Kuhstoss SA, Plotkin LI, et al. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017;31:2686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dallas SL, Garrett IR, Oyajobi BO, Dallas MR, Boyce BF, Bauss F, et al. Ibandronate reduces osteolytic lesions but not tumor burden in a murine model of myeloma bone disease. Blood 1999;93:1697–706. [PubMed] [Google Scholar]
- 31.Tsunenari T, Koishihara Y, Nakamura A, Moriya M, Ohkawa H, Goto H, et al. New xenograft model of multiple myeloma and efficacy of a humanized antibody against human interleukin-6 receptor. Blood 1997;90:2437–44. [PubMed] [Google Scholar]
- 32.Garnero P, Vergnaud P, and Hoyle N. Evaluation of a fully automated serum assay for total N-terminal propeptide of type I collagen in postmenopausal osteoporosis. Clin Chem 2008;54:188–96. [DOI] [PubMed] [Google Scholar]
- 33.Rosen HN, Moses AC, Garber J, Iloputaife ID, Ross DS, Lee SL, et al. Serum CTX: a new marker of bone resorption that shows treatment effect more often than other markers because of low coefficient of variability and large changes with bisphosphonate therapy. Calcif Tissue Int 2000;66:100–3. [DOI] [PubMed] [Google Scholar]
- 34.Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intra-cortical remodeling. J Bone Miner Res 2011;26:1035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012;50:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, et al. Osteocytes mediate the anabolic actions of canonical Wnt/β-catenin signaling in bone. Proc Natl Acad Sci U S A 2015;112:E478–E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hauge E, Mosekilde L, and Melsen F. Missing observations in bone histomorphometry on osteoporosis: implications and suggestions for an approach. Bone 1999;25:389–95. [DOI] [PubMed] [Google Scholar]
- 38.Coyac BR, Salvi G, Leahy B, Li Z, Salmon B, Hoffmann W, et al. A novel system exploits bone debris for implant osseointegration. J Periodontol 2021;92:716–26. [DOI] [PubMed] [Google Scholar]
- 39.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005;435:959–63. [DOI] [PubMed] [Google Scholar]
- 40.Bellido T, and Delgado-Calle J. Ex Vivo Organ Cultures as Models to Study Bone Biology. JBMR Plus 2020;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller PY, Janovjak H, Miserez AR, and Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 2002;32:1372–9. [PubMed] [Google Scholar]
- 42.Delgado-Calle J, Kurihara N, Atkinson EG, Nelson J, Miyagawa K, Galmarini CM, et al. Aplidin (plitidepsin) is a novel anti-myeloma agent with potent anti-resorptive activity mediated by direct effects on osteoclasts. Oncotarget 2019;10:2709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kowada T, Kikuta J, Kubo A, Ishii M, Maeda H, Mizukami S, et al. In vivo fluorescence imaging of bone-resorbing osteoclasts. J Am Chem Soc 2011;133:17772–6. [DOI] [PubMed] [Google Scholar]
- 44.Kikuta J, Wada Y, Kowada T, Wang Z, Sun-Wada GH, Nishiyama I, et al. Dynamic visualization of RANKL and Th17-mediated osteoclast function. J Clin Invest 2013;123:866–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano H, Kikuta J, Furuya M, Kondo N, Endo N, and Ishii M. Intravital bone imaging by two-photon excitation microscopy to identify osteocytic osteolysis in vivo. Bone 2015;74:134–9. [DOI] [PubMed] [Google Scholar]
- 46.Hiasa M, Okui T, Allette YM, Ripsch MS, Sun-Wada GH, Wakabayashi H, et al. Bone Pain Induced by Multiple Myeloma Is Reduced by Targeting V-ATPase and ASIC3. Cancer Res 2017;77:1283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neri D, and Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. [DOI] [PubMed] [Google Scholar]
- 48.Rolvien T, Butscheidt S, Jeschke A, Neu A, Denecke J, Kubisch C, et al. Severe bone loss and multiple fractures in SCN8A-related epileptic encephalopathy. Bone 2017;103:136–43. [DOI] [PubMed] [Google Scholar]
- 49.Lehal R, Zaric J, Vigolo M, Urech C, Frismantas V, Zangger N, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci U S A 2020;117:16292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019;134:1585–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Even C, Lassen U, Merchan J, Le Tourneau C, Soria JC, Ferte C, et al. Safety and clinical activity of the Notch inhibitor, crenigacestat (LY3039478), in an open-label phase I trial expansion cohort of advanced or metastatic adenoid cystic carcinoma. Invest New Drugs 2020;38:402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Xiao L, Tao J, Srinivasan V, Boyce BF, Ebetino FH, et al. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sedghizadeh PP, Sun S, Junka AF, Richard E, Sadrerafi K, Mahabady S, et al. Design, Synthesis, and Antimicrobial Evaluation of a Novel Bone-Targeting Bisphosphonate-Ciprofloxacin Conjugate for the Treatment of Osteomyelitis Biofilms. J Med Chem 2017;60:2326–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu D, Hu J, Xu S, De BE, Menu E, Van CB, et al. Dll1/Notch activation accelerates multiple myeloma disease development by promoting CD138+ MM-cell proliferation. Leukemia 2012;26:1402–5. [DOI] [PubMed] [Google Scholar]
- 55.Jundt F, Probsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004;103:3511–5. [DOI] [PubMed] [Google Scholar]
- 56.Nefedova Y, Cheng P, Alsina M, Dalton WS, and Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood 2004;103:3503–10. [DOI] [PubMed] [Google Scholar]
- 57.Ural AU, Yilmaz MI, Avcu F, Pekel A, Zerman M, Nevruz O, et al. The bisphosphonate zoledronic acid induces cytotoxicity in human myeloma cell lines with enhancing effects of dexamethasone and thalidomide. Int J Hematol 2003;78:443–9. [DOI] [PubMed] [Google Scholar]
- 58.Gordon S, Helfrich MH, Sati HI, Greaves M, Ralston SH, Culligan DJ, et al. Pamidronate causes apoptosis of plasma cells in vivo in patients with multiple myeloma. Br J Haematol 2002;119:475–83. [DOI] [PubMed] [Google Scholar]
- 59.Tassone P, Forciniti S, Galea E, Morrone G, Turco MC, Martinelli V, et al. Growth inhibition and synergistic induction of apoptosis by zoledronate and dexamethasone in human myeloma cell lines. Leukemia 2000;14:841–4. [DOI] [PubMed] [Google Scholar]
- 60.Aparicio A, Gardner A, Tu Y, Savage A, Berenson J, and Lichtenstein A. In vitro cytoreductive effects on multiple myeloma cells induced by bisphosphonates. Leukemia 1998;12:220–9. [DOI] [PubMed] [Google Scholar]
- 61.Croucher PI, De Hendrik R, Perry MJ, Hijzen A, Shipman CM, Lippitt J, et al. Zoledronic acid treatment of 5T2MM-bearing mice inhibits the development of myeloma bone disease: evidence for decreased osteolysis, tumor burden and angiogenesis, and increased survival. J Bone Miner Res 2003;18:482–92. [DOI] [PubMed] [Google Scholar]
- 62.McDonald MM, Reagan MR, Youlten SE, Mohanty ST, Seckinger A, Terry RL, et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017;129:3452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eda H, L Santo, Wein MN, Hu DZ, Cirstea DD, Nemani N, et al. Regulation of Sclerostin Expression in Multiple Myeloma by Dkk-1; A Potential Therapeutic Strategy for Myeloma Bone Disease. J Bone Miner Res 2016;31:1225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, and Nefedova Y. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr) 2011;34:545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.