

Abstract
The CD200-CD200R immunoregulatory signaling axis plays an etiological role in the survival and spread of numerous cancers primarily through suppression of anti-tumor immune surveillance. Our previous work outlined a pro-metastatic role for the CD200-CD200R axis in cutaneous squamous cell carcinoma (cSCC) that is independent of direct T cell suppression but modulates the function of infiltrating myeloid cells. To identify effectors of the CD200-CD200R axis important for cSCC metastasis, we conducted RNA-Seq profiling of infiltrating CD11B+Cd200R+ cells isolated from CD200+ versus CD200-null cSCCs and identified the cysteine protease cathepsin K (Ctsk) to be highly upregulated in CD200+ cSCCs. CD11B+Cd200R+ cells expressed phenotypic markers associated with myeloid-derived suppressor cell-like cells and tumor-associated macrophages and were the primary source of Ctsk expression in cSCC. A Cd200R+ myeloid cell-cSCC co-culture system showed that induction of Ctsk was dependent on engagement of the CD200-CD200R axis, indicating that Ctsk is a target gene of this pathway in the cSCC tumor microenvironment. Inhibition of Ctsk, but not matrix metalloproteinases (MMP), significantly blocked cSCC cell migration in vitro. Finally, targeted CD200 disruption in tumor cells and Ctsk pharmacological inhibition significantly reduced cSCC metastasis in vivo. Collectively, these findings support the conclusion that CD200 stimulates cSCC invasion and metastasis via induction of Ctsk in CD200R+ infiltrating myeloid cells.
Keywords: skin carcinogenesis, epithelial metastasis, extracellular matrix, keratinocyte, tumor microenvironment
Introduction
CD200 is a type I membrane-associated glycoprotein and a member of the immunoglobin superfamily. CD200 is expressed in a variety of cells of hematopoietic origin, such as macrophages, B cells and activated T cells, and non-hematopoietic origin, such as neurons, endothelial cells, trophoblasts, and epithelial keratinocytes [1 – 5]. Mice lacking Cd200 exhibit elevated numbers of activated CD11b+ macrophages and granulocytes [6], but exhibit normal myelopoiesis [7]. Phenotypically, Cd200 disruption and concomitant increased activated macrophage levels manifests in chronic nervous system inflammation, early onset of experimental autoimmune encephalomyelitis and susceptibility to experimental autoimmune uveoretinitis [6,8]. These observations underscore an essential role of Cd200 in maintaining tissue homeostasis by tempering the level of activated myeloid cells. Under pathological conditions, Cd200 also plays a key role in directly suppressing Th1-mediated inflammation [9 – 10], thereby orchestrating a balance between effective pathogen clearance and preventing immunopathology.
CD200 expression is reported across most cancer types including hematologic malignancies such as acute myeloid leukemia [11], multiple myeloma [12], and chronic lymphocytic leukemia [13]; solid tumors such as rectal [14], breast [15], colon [16], lung [17], ovarian [18], head and neck [19], glioma [20], pancreatic ductal adenocarcinoma [21], and bladder [22]; and a variety of skin cancers including squamous cell carcinoma [23], basal cell carcinoma [24], Merkel cell carcinoma [25] and melanoma [26]. Collectively, these studies correlate increasing levels of CD200 expression with tumor progression and reduced patient survival, thereby suggesting a broad impact for a functional, pro-tumorigenic role of CD200-mediated immune suppression in human cancer. As such, these studies underscore the importance of a comprehensive understanding of the mechanisms mediated by CD200 to enhance tumor progression and metastasis.
Similar to other immune checkpoint proteins, such as cytotoxic T lymphocyte antigen 4 and program death-1, CD200 pro-tumorigenic function is primarily thought to be mediated via dampening anti-tumor cytotoxic T cell (CTL) responses [27]. Consistent with these data, in syngeneic and xenograft murine models, treatment with anti-CD200 antibodies restored lymphocyte-mediated anti-tumor responses in vivo [15,28]. In normal tissue, the immunosuppressive capacity of CD200 is facilitated via engagement with CD200 receptor (CD200R). CD200R is a transmembrane protein, which upon ligation to CD200 initiates a signaling cascade that ultimately suppresses the activation of the CD200R+ cell [29]. CD200R is primarily expressed by myeloid lineage cells but is also detected on lymphoid lineage cells, such as natural killer (NK) and T cells [30 – 31]. As such, in addition to CTLs, myeloid derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and NK cells are targeted by the CD200-CD200R axis in human cancer [32 – 33]. Adding to this complexity, alternative mechanisms of membrane-bound CD200 in tumorigenesis have recently emerged. CD200 is a target for intracellular cleavage by γ-secretase, resulting in release of the CD200 cytoplasmic tail and subsequent nuclear translocation and DNA binding [34]. DNA binding of the CD200 cytoplasmic tail leads to increased expression of transcription factors associated with leukemic cell growth [34]. ADAM28-mediated CD200 ectodomain shedding leads to increased serum levels of soluble CD200 ectodomain in B-cell CLL patients [35]. Our laboratory previously identified a stimulatory role for membrane-bound Cd200 targeting Cd200r+ MDSC activation in cSCC [23]. Interestingly, we found that cSCC-derived Cd200 was essential for modulation of pro-tumorigenic function of tumor-infiltrating myeloid lineages cells and cSCC metastasis [23]. Collectively, these studies highlight the multiple cell lineages targeted by CD200 in the tumor microenvironment and pleiotropic mechanisms employed by CD200 to facilitate tumor progression and metastasis.
In this study, we set out to understand the mechanism by which the CD200-CD200R axis mediates metastasis in cSCC. We identified the collagen proteinase Cathepsin K (Ctsk) as a downstream target of the Cd200-Cd200r axis in cSCC. We observed that markers for multiple tumor-infiltrating myeloid lineages, including MDSC-like cells (MDSC-LC) and TAMs, were expressed by Cd200r+ infiltrating cells and these two infiltrating phenotypes were critical sources of Ctsk protein in cSCC. Finally, conditional deletion of Cd200 or pharmacological inhibition of Ctsk blocked cSCC cell invasion and metastasis. Collectively, our findings show that Cd200 stimulates cSCC metastasis via regulating the expression of Ctsk from Cd200r+ tumor-infiltrating myeloid lineages and suggest that targeting Ctsk may be a viable option to block cSCC metastasis.
Materials and Methods
Mice
Mice containing a loxP-flanked Cd200 conditional allele (Cd200fl) were re-derived from frozen sperm (B6NTac;B6N-Cd200tm1a(KOMP)Wtsi/H) (EUCOMM MRC Harwell Repository). Mutant mice exhibiting germline Cd200fl transmission were backcrossed ten generations onto a FVB/N genetic background (Taconic Biosciences). Epidermal-targeted Cd200 conditional null mice were generated by crossing Cd200fl/fl mice with Krt14Cre transgenic mice [36] (B6N.Cg-Tg(KRT14-cre)1Amc/J; The Jackson Laboratory) to generate Krt14Cre;Cd200fl/fl (EpiKO) mice (Supplemental Figure 1A). Wild-type animals used were FVB/N (Taconic Biosciences) and C57Bl/6 (Jackson Laboratories). Nu/J immunocompromised mice (The Jackson Laboratory) were chosen for orthotopic studies as they harbor the same MHC H2 haplotype, H-2Kq, as the FVB/N mouse strain. All animals were housed under pathogen-free conditions and received standard rodent chow and water ad libitum according the Institute of Comparative Medicine guidelines. All experiments involving mice were conducted under Columbia University IACUC-approved protocols.
Human and murine cell culture
Normal human epidermal keratinocytes (hKC-1 and hKC-2) were provided by the Stem Cell Imaging and Manipulation Core Facility within the Department of Dermatology Skin Disease Resource Based Center (epiCURE). Human cSCC cell cultures established in our laboratory, SCC-13 and SCC-39, were previously described [37]. LungMet2 SCC cells were previously established in our laboratory from a metastatic SCC lesion in the lung of a DMBA/TPA-treated cSCC-bearing FVB mouse [23]. EpiKO and Wt cSCC cultures were established from enzymatically-dissociated murine cSCCs excised from DMBA/TPA-treated EpiKO and Wt mice (FVB background) as previously described [23]. B16-F10 mouse melanoma cells were purchased from ATCC and were propagated in DMEM (Invitrogen) supplemented with 10% FBS (HyClone). Normal human keratinocytes, used at low passages P0 – P2, cSCC-13 and cSCC-39, murine LungMet2 cSCC and Wt and EpiKO cSCC cultures were propagated in complete FAD medium [38] or CnT-07 as per Manufacturer instruction (CELLnTEC). None of the above cell lines have been tested for mycoplasma.
Co-cultures containing cSCC and cSCC-infiltrating CD11b+CD200r+ cells were generated as previously described [23]. Briefly, CD11b+CD200r+ cells were FACS-sorted from orthotopic EpiKO cSCCs and plated into wells (300,000 cells per well) containing a previously-seeded monolayer of EpiKO or LungMet2 cSCC keratinocytes for 24 hours in complete RPMI medium (Fisher Scientific). After 24 hours, CD11b+CD200r+ and cSCC cells were isolated by flow cytometric analysis for biochemical endpoints described below.
Tumor Studies
Murine cSCCs were induced using a two-stage chemical carcinogenesis protocol as previously described using 100 nmol 7,12-Dimethylbenz(a)anthracene (DMBA) (Sigma-Aldrich) and 5 nmol 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (LC Laboratories) [23] in female EpiKO, Cd200fl/fl and Wt mice on a homogenous FVB background. Primary cSCC development was compared between DMBA/TPA-treated EpiKO and Cd200fl/fl mice by quantifying cSCC burden by visual inspection on a weekly basis [23]. In some cases, newly-emerged cSCC lesions in Wt mice were treated topically with either 6 μg/mL Ctsk inhibitor L-006235 (Santa Cruz Biotechnology) or DMSO (vehicle) once per day for three to four weeks. Orthotopic cSCCs were induced in female Nu/J (The Jackson Laboratory) and FVB mice by sub-cutaneous injection of 1 X 106 EpiKO or LungMet2 [23] cSCC cells on each flank. Similarly, orthotopic melanomas were induced in C57Bl/6 mice by sub-cutaneous injection of 1 X 105 B16 melanoma cells per flank.
Tissue Harvesting
All mouse skin tumors and enlarged skin-draining lymph nodes and lung and liver tissues were surgically excised and portions were either flash frozen in liquid nitrogen or embedded in optimal cutting temperature (O.C.T.) compound (Sakura) and cryopreserved. De-identified human cSCC and normal skin specimens were obtained from the clinical services of the Dermatology Department at Columbia University Irving Medical Center (IRB AAAA4588).
Immunofluorescence labeling
Thin, 6 – 8 μm, histological frozen sections of normal skin and cSCC were fixed in acetone and probed with primary antibodies overnight at 4°C followed by detection with Alexa Fluor-conjugated species-specific secondary antibodies (Molecular Probes). Please see Supplemental Figure 2A-H for secondary antibody control tissue section staining. In some cases, a M.O.M. Immunodetection Kit was used with mouse primary antibodies as per Manufacturer instruction (Vector Laboratories). Please see Supplemental Table 1 for antibody details. Immunofluorescence was captured using a Zeiss LSM 5 Exciter confocal microscope and renderings of confocal image stacks were acquired using NIH ImageJ software.
Flow Cytometry
Single cell suspensions were generated from enzymatically-dissociated EpiKO and Cd200fl/fl mouse epidermis [39], dermis [40] or EpiKO and LungMet2 orthotopic cSCCs [23] as previously described. Epidermal and dermal cell suspensions from normal skin were labelled with directly-conjugated primary antibodies (Supplemental Table 1) and analyzed using a BD LSRFortessa flow cytometer (BD Biosciences). Cell suspensions from cSCCs were labeled with antibodies for CD45-PE/Cy7 (Biolegend), Cd200r1 (R&D Systems), and CD11b-PE (Biolegend), Ly6C-Alexa Fluor 647 (Biolegend), Ly6G-PerCP/Cy5.5 (Biolegend). Cd200r1 labeling was detected with species-specific Alexa-Fluor secondary antibodies (Invitrogen). CD11b+Cd200r1+ cells were sorted using a BD Influx flow cytometer (BD Biosciences). For quantification, all flow cytometry data were analyzed using the FlowJo software (BD Biosciences).
RNA-sequencing
Total RNA was isolated from 1 – 2 x 106 FACS-sorted CD11b+Cd200r1+ cells from orthotopic cSCCs (n = 2 replicates per group). Poly-A pull-down enriched mRNAs from total RNA samples (200 ng – 1 μg per sample, RIN > 8) were used for library preparation (Illumina TruSeq RNA prep kit). Libraries were sequenced using Illumina HiSeq2000 at the Columbia Sulzberger Genome Center (RNA TRUSEQ 30M reads, single ends). Relative abundance (expression level) of genes was rendered using cufflinks [41] with default settings. DeSeq analysis [42] of normalized reads using a Benjamini-Hochberg statistical correction was conducted to identify differentially regulated genes in FACS-sorted CD11b+Cd200r1+ cells harvested from LungMet2 versus EpiKO cSCCs. Sequencing data deposition number: GSE178496.
Quantitative PCR
Total RNA extracted from LungMet2 and EpiKO cSCC co-cultures was subjected to first strand cDNA synthesis as per Manufacturer instruction using the SuperScript III First-Strand Synthesis SuperMix kit (ThermoFisher). Quantitative PCR (QPCR) analyses for Ctsk, Cd200r1 and Gapdh mRNA levels were performed with SYBR® Green Master Mix (Bio-Rad) in a MyiQ Real-Time PCR Detection System (Bio-Rad). Each sample was run in a triplicate. The above protocol was repeated for sorted CD45+CDllb+ and CD45+CDllb- cells from orthotopic cSCCs for analysis of Ctsk and Cd200r1 mRNA levels. Oligonucleotide primer sequences are listed in Supplemental Table 2.
Invasion assays
Human and murine cSCC cells were seeded onto cell culture inserts (Fisher Scientific) at 25,000 cells per insert containing either 0.3 μg/mL bovine Type I collagen solution (Advanced Biomatrix) or 0.3 μg/mL bovine Type II collagen solution (Southern Biotech). Type I and Type II collagen gel matrices were prepared as per Manufacturer protocol. Invasion chambers were incubated for 24 hours following cell seeding. After which, membranes were removed, fixed in 4% PFA, and stained with DAPI. Cells were visualized by DAPI fluorescence using a Zeiss LSM 5 Exciter confocal microscope and the number of invasive cells on five randomly chosen fields of view per insert were counted. The average number of cells per membrane was statistically compared using the Student’s t-test between groups (N = 3 inserts/group). In some cases, culture media was supplemented with 10 μM of the broad spectrum MMP inhibitor GM6001 (Selleckchem) or 10 μM of the CTSK inhibitor MK-0822 (Selleckchem).
T cell activation assays
Untouched CD8 T cells were purified from FVB or C57Bl/6 mouse spleens (non-tumor bearing) using the EasySep Mouse CD8+ T Cell Isolation Kit as per Manufacturer instruction (Stem Cell Technologies). CD8 T cell purity was routinely confirmed between 92 – 94% by flow cytometry (Supplemental Figure 3A-H). For T cell receptor stimulation, CD8 T cells were plated at 2 X 105 cells per well on uncoated or 5 μg/mL CD3 antibody-coated (BioLegend) wells in complete RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated FBS (HyClone) and 2 μg/mL CD28 antibody (BioLegend) for 24 (activation) or 72 (proliferation) hours. For proliferation endpoints, CD8 T cells were loaded with CFSE as per Manufacturer instruction (BD Biosciences) immediately prior to seeding. To assess MDSC immunosuppressive function, in some cases, T cells were co-cultured with CD11b+Ly6G-Ly6Chi (M-MDSC) or CD11b+Ly6G+Ly6Clo (PMN-MDSC) cells [43 – 44] isolated by FACS from dissociated LungMet2 cSCC or spleens of cSCC- or B16 melanoma-bearing mice. For co-cultures, the ratio of MDSC cell subsets to CD8 T cells was either 1:10, 1:3 or 1:2. Following stimulation, T cells were harvested and labeled with antibodies against CD3, CD8, Tnf-α and Ifn-γ (BioLegend) and analyzed by flow cytometry.
Results
Characterization of Cd200 conditional null mice
The B6NTac;B6N-Cd200tm1a(KOMP)Wtsi/H Cd200 allele with exons 3 and 4 flanked by loxP sites (Supplemental Figure 1A), was used to generate Cd200 conditional mice. The Cd200fl allele transmits at the expected frequency as detected by PCR analysis of genomic DNA (Supplemental Figure 1B). To target Cd200 disruption to the skin epidermis, we crossed Cd200fl mice with Krt14Cre transgenic mice [36] to generate Krt14Cre;Cd200fl/fl conditional null (EpiKO) mice. Krt14Cre mice express Cre recombinase in the entire basal epidermal layer in the skin including the stem cell compartment [36], which results in ablation of Cd200 in the entire skin epidermis. Epidermal ablation of Cd200 was confirmed at mRNA (Supplemental Figure 1C) and protein (Supplemental Figure 1D-E) levels in EpiKO mice. No differences in the numbers of hair follicle α6+CD34+ bulge stem cells were observed between EpiKO and control Cd200fl/fl mice (Supplemental Figure 1F). EpiKO mice are viable and show a normal gross appearance (Supplemental Figure 4A) and no pathological manifestations were observed in histological sections of EpiKO adult skin (Supplemental Figure 4B-C). Finally, we quantified the levels of lymphoid and myeloid lineages in the skin dermis and observed comparatively similar levels of total CD45+ and CD3+ and CD11b+ subsets in the skin of adult EpiKO compared to Cd200fl/fl mice (Supplemental Figure 4D-E). Consistent with the characterization of Cd200 germline null mouse skin [6], the phenotype of EpiKO skin is unremarkable and highly similar to Wt skin (Supplemental Figures 1 & 4).
Ctsk is a CD200-dependent transcript in stromal MDSCs
Our laboratory previously identified CD200 as a signature molecule of human and murine poorly differentiated (high-risk) cSCCs and metastatic SCCs targeting a number of secondary organs [23]. We also demonstrated functional relevance for Cd200 where shRNA-silencing of Cd200 expression abrogated the ability of cSCC-derived metastatic cells to initiate secondary tumors [23]. We previously detected dense populations of infiltrating Cd200r1+ cells localized to the stroma of cSCCs, the vast majority of which expressed markers consistent with a MDSC (CD11b+Gr-1+) phenotype [23]. Importantly, using a co-culture system, we demonstrated that the activity of cSCC-infiltrating CD11b+Gr-1+ cells was dependent on engagement of the Cd200-Cd200r axis [23], an observation that may hold significance for the metastatic spread of cSCC and potentially other CD200-expressing tumors. However, the mechanistic basis for CD200-dependent genes expressed by CD11b+Gr-1+CD200R+ infiltrating cells in cSCC metastasis remains poorly understood.
To identify key downstream targets of the CD200-CD200R axis in cSCC-infiltrating myeloid cells that are critical for metastasis, we conducted RNA-Seq profiling of CD11b+Cd200r1+ cells purified by flow cytometry from orthotopic cSCCs propagated from Cd200+ (LungMet2) and Cd200- (EpiKO) cells (See Figure 1A for experimental design). No difference in the total numbers of infiltrating CD11b+Cd200r1+ cells were observed in Cd200+ versus Cd200- cSCC (Figure 1B-D) indicating that Cd200 does not primarily influence myeloid cell recruitment. However, principal component and MA scatter plot analyses indicated highly significant gene expression differences in CD11b+Cd200r1+ cells isolated from each cSCC genotype (Figure 1E-F; Padj < 0.05). DeSEq analysis of normalized mRNA read counts identified a total of 203 downregulated and 312 upregulated genes in CD11b+Cd200r1+ cells isolated from EpiKO cSCCs (Figure 1G-H; Padj < 0.05). Next, we conducted functional enrichment analysis [45] to identify key pathways that may be downstream of the Cd200-Cd200r axis in CD11b+Cd200r1+ cells. Interestingly, GO analysis identified osteoclast differentiation as the top downregulated pathway in CD11b+Cd200r1+ cells from EpiKO cSCC (Figure 1J-K). The CD200-CD200R signaling axis plays an important role in osteoclast differentiation [46]. Cd200 germline null mice exhibit decreased osteoclast numbers but also display increased bone density compared to Wt mice [46]. These observations suggest that, in addition to regulating osteoclast differentiation, CD200 may be required for induction of key mediators of bone resorption. Cathepsin K (Ctsk), which ranked as the second highest downregulated transcript in CD11b+Cd200r1+ infiltrating cells from EpiKO cSCC (Supplemental Table 3), is a collagenolytic peptidase and the primary osteoclast factor implicated in bone remodeling [47 – 49]. Collectively, these observations suggest that the Cd200-Cd200r axis ectopically induces an expression signature in Cd200r+ cSCC-infiltrating cells that mimics osteoclast differentiation and stimulates collagen degradation. However, a direct link between Cd200-Cd200r signaling and induction of Ctsk expression has not been reported.
Figure 1. Profiling Cd200-dependent genes in Cd200r+ cSCC-infiltrating myeloid cells.
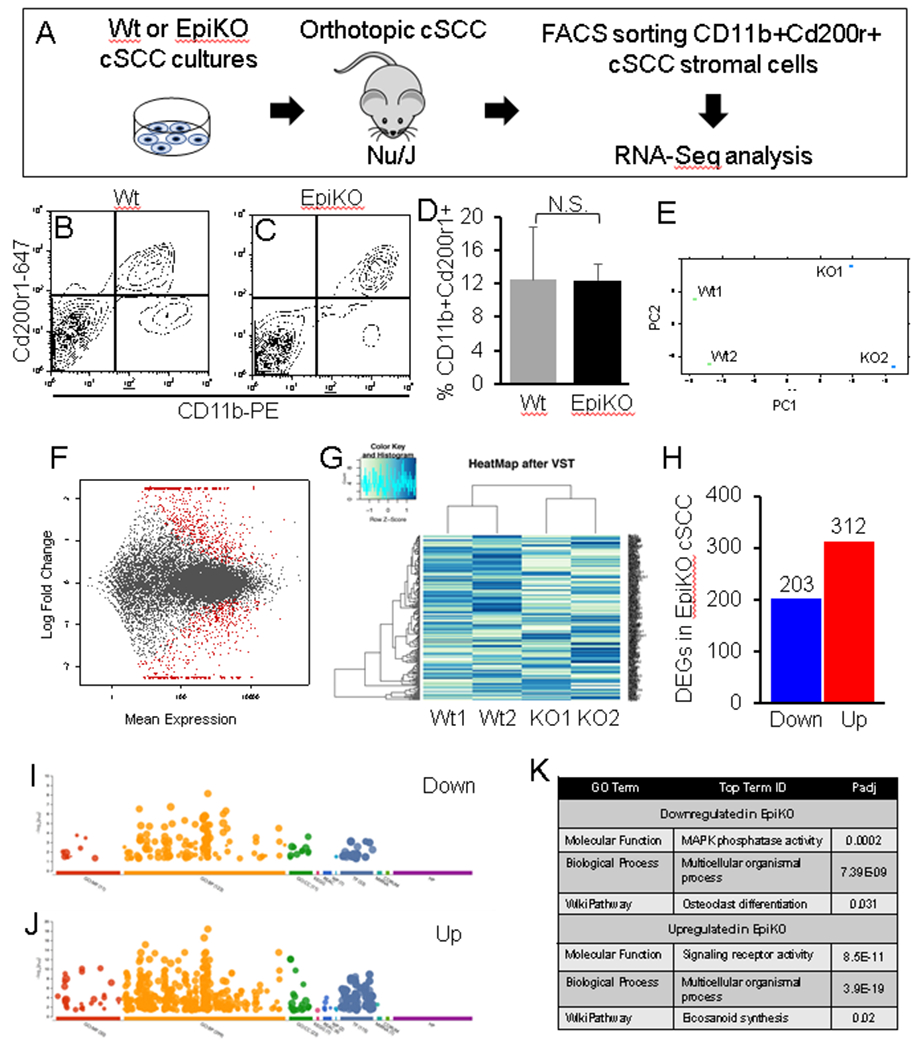
(A) Schematic illustration of experimental design to identify CD200-dependent target genes in Cd200r+ myeloid cells purified from orthotopic Wt (CD200+) or EpiKO (CD200-) cSCCs. (B-C) Representative FACS plots showing gating for CD11b+Cd200r1+ cSCC myeloid cells. (D) Bar graph showing quantification of the percentage of CD11b+Cd200r1+ cSCC cells from tumors propagated by Wt or EpiKO cSCC cells (n = 4 tumors per group). (E-F) Principal component analysis (E) and MA scatter plot (F) showing log fold change versus mean expression of mRNA read counts in CD11b+Cd200r1+ cells from each cSCC genotype. Significant differentially expressed genes (P < 0.05 cutoff, Student’s t-test with Benjamini-Hochberg correction; N = 2 cSCCs per group) highlighted in red (F). (G) Heat Map of CD11b+Cd200r1+ cell expression profiles in Wt versus EpiKO cSCCs. (H) Bar graph showing total numbers of differentially expressed genes in CD11b+Cd200r1+ cells from EpiKO versus Wt cSCCs (P < 0.05 cutoff, Student’s t-test with Benjamini-Hochberg correction). (I-J) Functional enrichment analysis plots showing GO terms (x-axis) and statistical significance (y-axis) from downregulated (I) and upregulated (J) gene sets. (K) Top upregulated and downregulated GO Terms from I-J, enriched in CD11b+Cd200r1+ myeloid cells isolated from EpiKO cSCCs. All error bars are represented as Standard Deviation.
cSCC-infiltrating myeloid cells expressing Cd200r are the primary source of Ctsk
While our RNA-Seq profiling identified Ctsk to be highly enriched in CD11b+Cd200r+ infiltrating cells isolated from Wt (Cd200+) cSCC, whether other immune lineages may serve as a significant source of Ctsk independent of Cd200-Cd200r signaling is unknown. To address this, we purified CD11b+ and CD11b- cell subsets from the total pool of CD45+ cSCC-infiltrating cells by flow cytometry (Figure 2A-C). Next, QPCR analysis of Cd200r and Ctsk mRNA expression levels in these two CD45+ subsets demonstrated that CD11b+ cSCC stromal cells are the primary source of both Cd200r and Ctsk (Figure 2D-E).
Figure 2. CD11b+Cd200r+ infiltrating cells are the primary source of Ctsk.
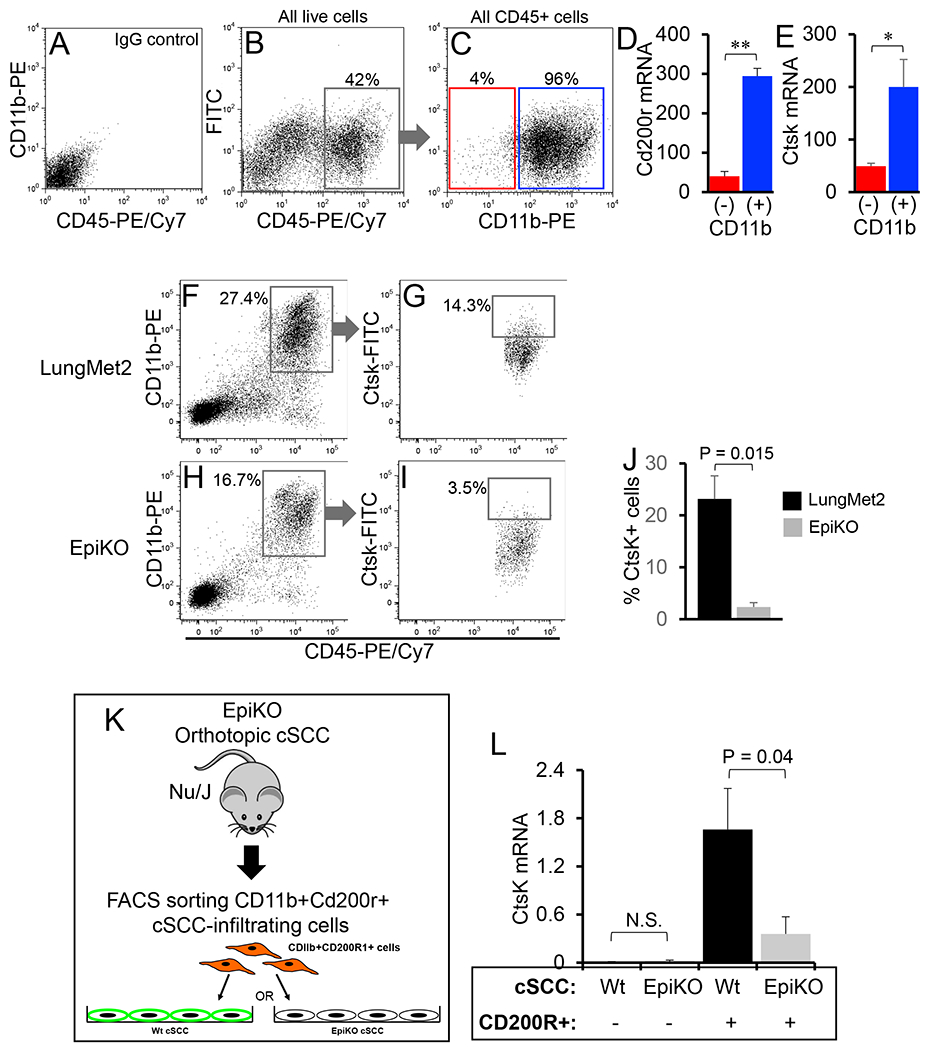
(A-C) Representative FACS plots showing gating of CD11b+ and CD11b- tumor-infiltrating immune (CD45+) lineages. IgG-stained control cells are shown in panel A. The total population of CD45+ cells were gated (B) and subsequently partitioned into CD11b- and CD11b+ populations (C). (D-E) Bar graphs showing relative Cd200r (D) and Ctsk (E) mRNA levels quantified in CD11b+ and CD11b- cSCC cells by QPCR (P values generated by unpaired Student’s t-test (*, P = 0.008; **, P = 0.004). (F-J) Representative FACS plots showing gating of CD45+CD11b+ (F,H) and the percentage of Ctsk+ cells (G,I) in LungMet2 (F-G) and EpiKO (H-I) cSCCs. (J) Bar graph showing quantification of the percentage of Ctsk+ cells from the total pool of CD11b+Cd200r+ cells in each group (n = 4 replicates per group; P value generated from unpaired Student’s t-test). All error bars are represented as Standard Deviation. (K) Schematic of experimental design for propagation of co-cultures containing Wt or EpiKO cSCC cells with FACS-sorted CD11b+Cd200r1+ cSCC stromal cells used for QPCR analysis in D. (L) Bar graph showing relative Ctsk mRNA levels determined by QPCR analysis of cultures of Wt or EpiKO cSCC cells plated alone or co-cultures consisting of Wt or EpiKO cSCC cells plated with CD11b+Cd200r1+ cSCC stromal cells. Ctsk transcript levels were normalized to Gapdh mRNA levels (N = 3 replicates per group, P values generated from unpaired Student’s t-test). All error bars are represented as SEM.
Ctsk expression is dependent on Cd200-Cd200r signaling in cSCC
Next, we investigated whether Ctsk expression in Cd200r+ cSCC-infiltrating cells may be dependent on engagement of the Cd200-Cd200r axis. First, we compared Ctsk protein levels in CD11b+Cd200r+ cells isolated from LungMet2 (CD200+) versus EpiKO (CD200-) cSCCs by FACS analysis (Figure 2F-I). We observed an approximate ten-fold increase in the average percentage of Ctsk+CD11b+Cd200r+ cells in LungMet2 versus EpiKO cSCCs (Figure 2J; n = 4 cSCCs per group). Second, we employed our co-culture model to quantify Ctsk expression induction in the presence or absence of engagement of the Cd200-Cd200r axis. CD11b+Cd200r+ infiltrating cells were purified from EpiKO cSCC and plated over monolayers of Wt (Cd200+) or EpiKO (Cd200-) cSCC cells (Figure 2K). CD200 expression levels were confirmed by flow cytometric analysis in Wt and EpiKO cSCCs (Supplemental Figure 5A-C). CD11b+Cd200r+ cells were isolated from EpiKO cSCC so that the baseline levels of Cd200-Cd200r signaling prior to co-culture seeding would be largely negligent. No Ctsk mRNA expression was observed in Wt or EpiKO cSCC cells plated in the absence of stromal cells (Figure 2L). An approximate 4-fold increase in Ctsk mRNA levels were observed in CD11b+Cd200r+ cells purified from co-cultures containing Wt versus EpiKO cSCC cells (Figure 2L) confirming that Ctsk expression in cSCC-infiltrating CD11b+Cd200r+ cells is dependent on engagement of the Cd200-Cd200r axis.
Phenotyping of Cd200r+ cSCC-infiltrating cells
Next, we examined tissue sections of cSCC, induced by DMBA/TPA, by immunofluorescence labeling to i) confirm co-expression of Cd200r and Ctsk in naturally occurring cSCC in vivo and to determine the proximity of Cd200r+ cells to Cd200+ cSCC cells. Therefore, we focused on the leading edge of the cSCC invasive front (Figure 3A), as leading-edge cytokeratin Krt14+ cSCC cells are highly enriched for Cd200 expression compared cSCC areas distally located to the leading edge (Figure 3B-D). As expected, no Cd200 immunodetection was observed in invasive front Krt14+ cells from EpiKO cSCCs (Figure 3E-G). In the stromal areas of cSCC proximal to leading edge Krt14+ cells (Figure 3H), we observed co-immunofluorescence labeling of Cd200r1 and Arginase 1 suggesting a MDSC-like phenotype in Cd200r+ cSCC-infiltrating cells (Figure 3I-K). We found that virtually all Ctsk+ cells co-localized with Cd200r1+ cells (Figure 3L-O). Little to no Ctsk immunolabeling was observed in cytokeratin+ cSCC cells (Figure 3O). In normal skin, Cd200r mRNA expression is reported in Langerhans cells and dendritic epidermal T cells [50]. We also examined normal skin by immunofluorescence labeling for co-localization of Ctsk in Cd200r+ dermal cells. Cd200r immunopositive cells were identified throughout murine skin dermis; however, no Ctsk co-labeling was observed (Supplemental Figure 6A-D). Collectively, these observations confirm that Cd200r+ cSCC-infiltrating cells in close proximity to the tumor invasive front are the primary source of Ctsk and that Ctsk expression is a unique feature of the skin tumor microenvironment.
Figure 3. Ctsk expression is localized to stromal Cd200r1+ MDSCs in mouse cSCCs.
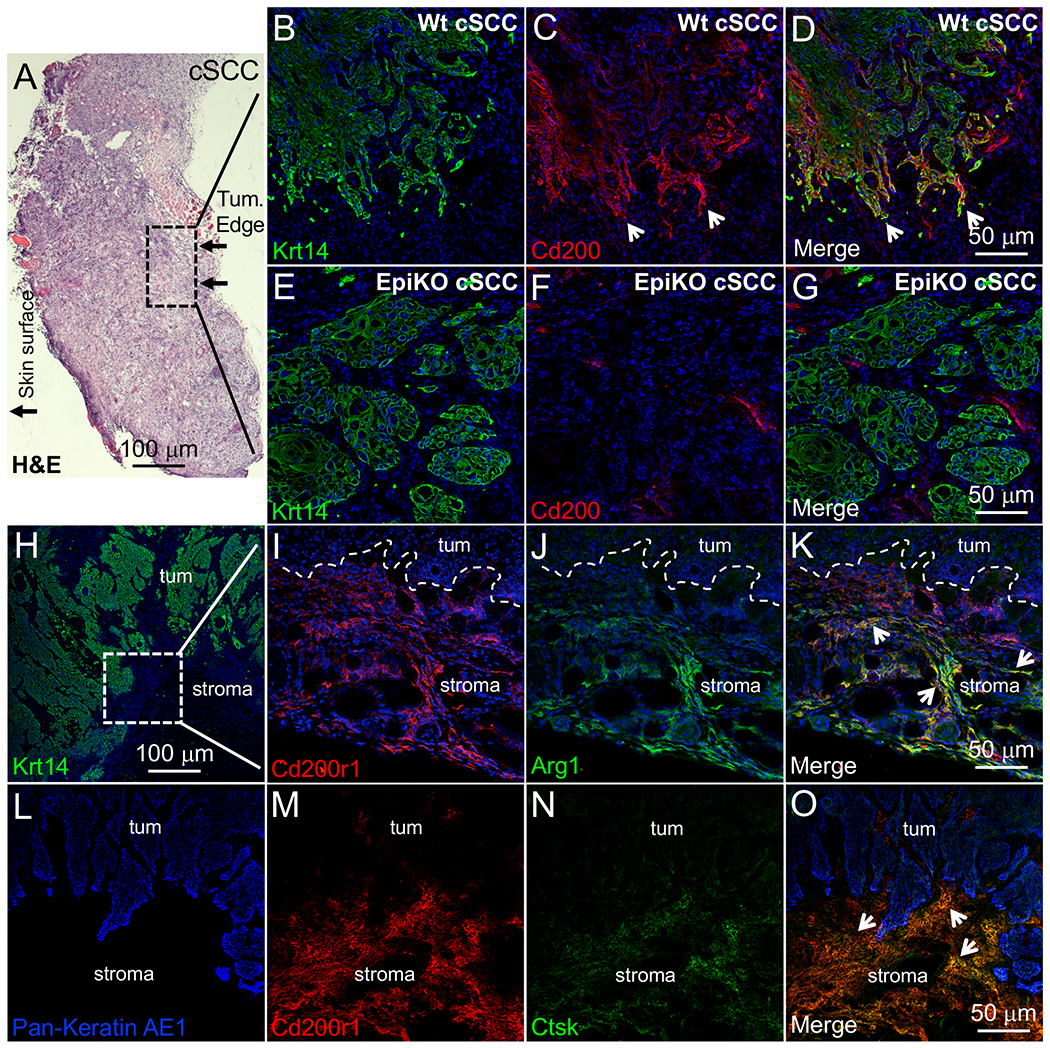
(A) Low magnification field of H&E-stained murine cSCC. Hashed box designates invasive tumor front penetrating the underlying striated muscle. (B-G) Krt14 and Cd200 co-immunofluorescence labeling in the invasive front of Wt (B-D) and EpiKO (E-G) cSCCs. Arrowheads designate enriched Cd200 immunolabeling in leading edge cSCC cells (C-D). Panels B-D and E-G represent the same field of view. (H) Low magnification of Krt14 immunolabeling in Wt cSCC. Hashed-line box designates an area of tumor stroma just below the tumor invasive front that was used for further immunolabeling in panels I-O. (I-K) Cd200r1 and Arg1 co-immunofluorescence labeling in the stroma below the invasive front of Wt cSCCs. In the merged panel (K), arrows designate stromal cells co-labeled for Cd200r1 and Arg1. (L-O) Pan Keratin, Cd200r1 and Ctsk co-immunofluorescence labeling in the stroma below the invasive front of Wt cSCCs. Panels L-O represent the same field of view. In the merged panel (O), arrows designate stromal cells co-labeled for Cd200r1 and Ctsk. Panels B-K were counterstained with DAPI (blue) to visualize nuclei.
We previously observed greater than 90% of Cd200r+ cSCC-infiltrating cells exhibited a MDSC phenotype, albeit with a limited antibody marker panel (CD11b+Gr-1+), by flow cytometry and in cSCC tissue sections [23]. To further investigate the phenotype of Cd200r+ cSCC-infiltrating cells, we labeled single cell suspensions from dissociated LungMet2 cSCCs with antibody panels specific for MDSC subsets M-MDSC or PMN-MDSC [43 – 44] or TAM, F4/80 and MHCII [43 – 44]. From the total pool of CD45+CD11b+ cSCC-infiltrating cells, we detected Ly6G-Ly6Chi (M-MDSC) and Ly6G+Ly6Clo (PMN-MDSC) populations, both of which contained Cd200r+ cells (Figure 4A-D). We also detected F4/80+MHCII+ TAM within the CD45+Cd11b+ pool (Figure 4A, E-F). The average percentage of Cd200r+ cells was higher in Ly6G-Ly6Chi cells (approximately 50%) compared to Ly6G+Ly6Clo cells (approximately 10%), and approximately 40% of the F4/80+ population were Cd200r+ (Fig. 4G). Collectively, these observations depict a heterogeneous pool of Cd200r+ cSCC-infiltrating cells. In support of this concept, we further interrogated our RNA-Seq gene list of Cd11b+Cd200r+ cSCC-infiltrating cells (Figure 1) and observed high mean read values for multiple lineage markers consistent with MDSCs and TAMs but not T or NK cells (Supplemental Table 4). To further assess a functional immunosuppressive phenotype, we performed T cell immunosuppression assays using FACS-sorted Ly6G-Ly6Chi or Ly6G+Ly6Clo cells isolated from cSCC or the spleens of cSCC- or B16 melanoma-bearing mice. Purified CD8 T cells were subjected to CD3/CD28 antibody stimulation for either 24 hours (activation) or 72 hours (proliferation) in the presence of different T cell-to-MDSC subset cell ratios. Rather than immunosuppression, for both tumor types we observed myeloid cell number-dependent increases in CD8 T cell production of pro-inflammatory cytokines Tnf-α and Ifn-γ (Supplemental Figure 3). However, CD8 T cell proliferation was suppressed by both M-MDSC (35% reduction) and PMN-MDSC (22% reduction) (Supplemental Figure 3). The observed stimulation of CD8 T cell cytokine production at 24 hours and suppression of proliferation at 72 hours by MDSC subsets following CD3/CD28 stimulation is consistent with other published studies [43-44] and suggests that cSCC-infiltrating Ly6Chi and Ly6G+Ly6Clo cells that express Cd200r possess a MDSC-LC phenotype.
Figure 4. Phenotyping Cd200r+ cSCC-infiltrating cells.
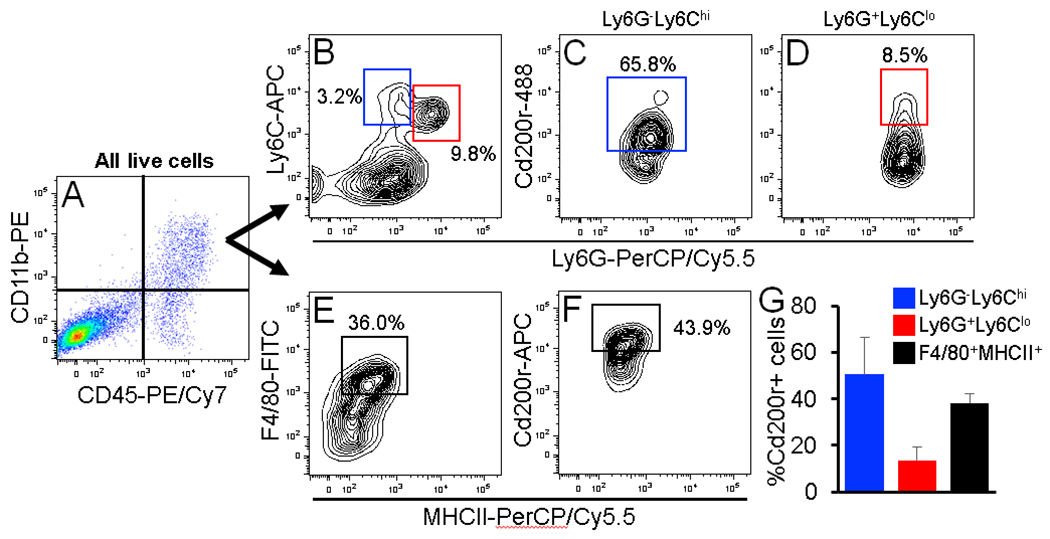
(A-F) Representative FACS dot and contour plots showing the gating strategy for identification of myeloid cell subsets expressing Cd200r. From the total pool of CD45+CD11b+ cSCC-infiltrating cells (A), cells were analyzed for M-MDSC and PMN-MDSC marker panels Ly6G-Ly6Chi and Ly6G+Ly6Clo, respectively (B) or a TAM marker panel F4/80/MHCII (E). Each subset was individually analyzed for Cd200r (C-D,F). (G) Bar graph showing the average percentage of Cd200r+ cells in each infiltrating cell subset (n = 3 cSCCs). All error bars are represented as Standard Deviation.
Overall, the variety of immune lineage markers expressed by Cd200r+ tumor-infiltrating cells (Supplemental Table 4) indicates that Cd200r is a common marker for a heterogeneous pool of tumor-infiltrating cells comprised of MDSC-LC, TAMs and granulocytes, while Cd200r+ cells exhibiting markers consistent with MDSC-LC and TAM phenotypes represent the critical sources of Ctsk (Figures 3 & 4).
Ctsk facilitates cSCC invasion
To determine the functional significance of Ctsk induction in Cd200r+ cSCC-infiltrating cells, we investigated human and murine cSCCs for the presence of stromal-localized collagen substrates and the impact of Ctsk inhibition on invasion through collagen gels. Ctsk has demonstrated proteolytic activity against Type I and Type II collagens [51 – 52]. Type I collagen is selectively expressed in normal skin dermis (Supplemental Figure 7A-B); whereas, Type II collagen expression is absent in skin (Supplemental Figure 7C-D) and restricted to cartilage [52]. However, whether Type II collagen may be ectopically expressed in cSCC is poorly understood. We observed positive immunofluorescence labeling for Type 1 and Type II collagens in murine and human cSCC histological sections (Figure 5A-D). However, ectopic stromal Type II collagen expression was detected in far fewer (48%) of cSCCs analyzed compared to Type I collagen (100%) (Figure 5E). Next, for each human cSCC specimen, we quantified the percentage of the dermal surface area (290 μm2) immunopositive for Type I and Type II collagens as follows: dense immunostaining, ≥ 75% surface area; sparse immunostaining, ≤ 15%. Within the Type II collagen immunopositive cSCC subset, labeling was sparsely detected across the cSCC stromal surface area in the majority (70%) of cSCCs. Whereas, Type I collagen expression was densely distributed throughout 100% of cSCCs examined (Figure 5E).
Figure 5. Impact of Ctsk inhibition on human and murine cSCC invasion through collagen.
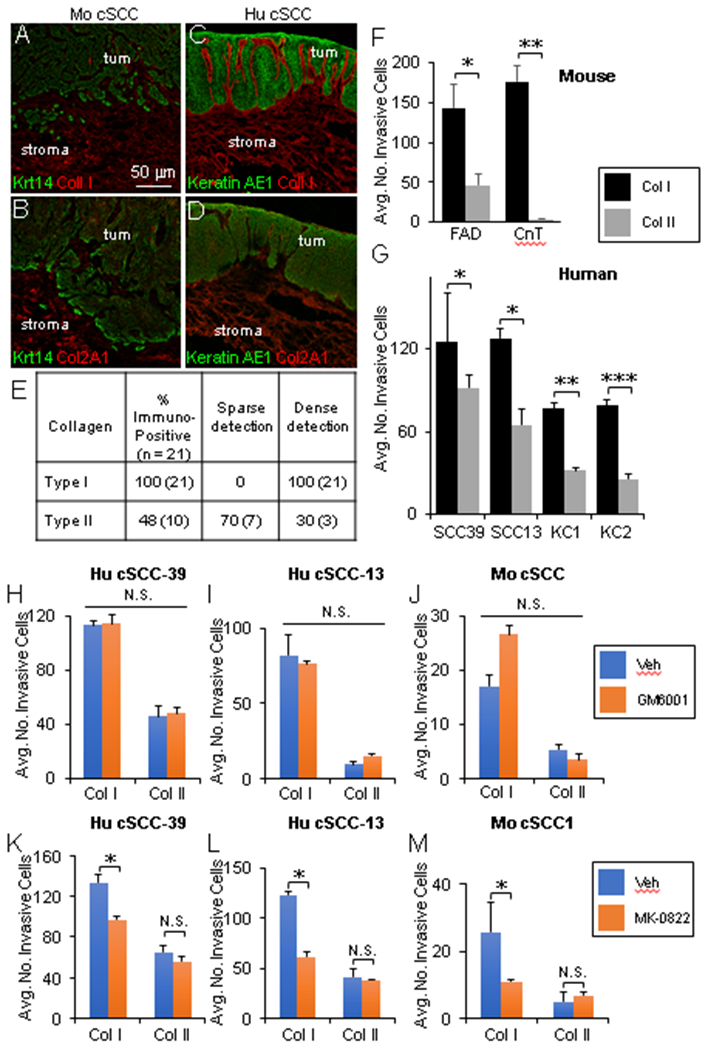
(A-D) Representative co-immunofluorescence labeling in murine (A-B) and human (C-D) cSCC histological sections with antibodies against Krt14 and Type I Collagen (A), Krt14 and Type II Collagen (B), Pan cytokeratin AE1 and Type I Collagen (C), or Pan cytokeratin AE1 and Type II Collagen. (E) Quantitative summary of Type I and II Collagen expression status in 21 human cSCC samples determined by immunofluorescence labeling; dense immunostaining, ≥ 75% surface area; sparse immunostaining, ≤ 15%. (F) Bar graph showing the average number of invasive murine Wt cSCC cells in Type I vs. Type II collagen under serum-containing (FAD) and serum-free (CnT) media conditions (N = 5 fields of view per insert and 3 inserts per group, P values from unpaired Student’s t-test: (*)- P < 0.05; (**)- P < 0.01). (G) Bar graph showing the average number of invasive human cSCC (cSCC39, cSCC13) and normal keratinocytes (KC1, KC2) in Type I versus Type II collagen (N = 5 fields of view per insert and 3 inserts per group, P values from unpaired Student’s t-test: (*)- P < 0.05; (**)- P < 0.01; (***)- P < 0.001). (H-M) Quantification of invasive human (H-I, K-L) and murine (J, M) cSCC cells in Type I and Type II collagen either untreated or treated with inhibitors targeting MMPs (10 μm GM6001) (H-J) or Ctsk (10 μm MK-0822) (K-M) (N = 5 fields of view per insert and 3 inserts per group, P values from unpaired Student’s t-test: (*)- P < 0.05). All error bars are represented as SEM.
To determine whether CTSK plays a role in cSCC invasion, we first quantified cSCC cell invasion through three-dimensional gel matrices constructed with Type I or II collagen. Both murine and human cSCC cells demonstrated increased invasion through Type I compared to Type II collagen (Figure 5F-G) and the levels of invasion through Type I collagen were not impacted by serum-containing or serum-free media (Figure 5F). Since collagen triple helices are also substrates for matrix metallopeptidase (MMP) family enzymes [53], we quantified invasive cells in Type I and II collagen gels in media supplemented with the broad spectrum MMP inhibitor GM6001 or vehicle (Figure 5H-J). No significant difference in human or murine cSCC invasion through Type I or II collagens was observed in response to MMP inhibition (Figure 5H-J). However, treatment with the Ctsk inhibitor MK-0822 led to significant decreases in human and murine cSCC invasion in Type I collagen but not Type II (Figure 5K-M). Given the high density of Type I collagen in human and murine cSCC tissue relative to Type II (Figure 5E), our observations support the concept that CTSK may play a role in CD200-mediated cSCC metastasis by facilitating invasion through Type I collagen in the stroma.
Ctsk inhibition blocks cSCC metastasis in vivo
To determine the in vivo relevance of our above observations of CTSK inhibition on cSCC invasion through collagen gel matrices, we assessed the impact of Ctsk inhibition on the metastatic spread of chemically-induced cSCCs in Wt mice. FVB mice represent a robust strain to investigate cSCC metastasis where the ratio of metastatic-to-primary cSCC is typically 1 [23,54]. Mice bearing benign papillomas were observed daily by gross inspection to detect malignant conversion to cSCC [23,54]. Upon emergence in the skin, cSCCs were treated topically with 6 μg/ml of the Ctsk inhibitor L-006235 or vehicle once per day for a period up to 4 weeks. Systemic administration of 10 – 500 mg/kg L-006235 in rodents for periods ranging from two days to six weeks were effective at inhibiting Ctsk activity while averting signs of toxicity [55-58]. In vitro administration of 180 μM L-006235 for 72 hours leads to 50% death of neuroblastoma cells [59]. Therefore, we opted for a 13 μM (6 μg/ml) L-006235 for topical cSCC treatment, as it was greater than ten-fold lower than the cytotoxic dose observed in culture but still in the efficacious range observed in vivo. L-006235 topical treatment did not elicit overt signs of toxicity as it did not impact cSCC growth in the skin as tumors were harvested at 2 cm diameter size endpoints in both groups, which typically occurred in 3 – 4 weeks. All enlarged skin draining lymph nodes were harvested along with primary cSCCs from mice from each cohort. Liver and lungs were also harvested and examined for metastatic cSCC as previously described [23]; however, no metastatic lesions in the liver or lungs were observed in either cohort. Histological sections from enlarged nodes were examined by Krt14 (cSCC cell-specific marker) and CD45 (immune cell-specific marker) co-immunofluorescence labeling (Figure 6A-L) to confirm the presence or absence of metastatic disease. Krt14 marks virtually all cSCC keratinocytes in primary skin lesions (Figure 6A-C) but is absent in lymph nodes harvested from normal mice (Figure 6D-F). Only enlarged lymph nodes exhibiting positive Krt14 immunoreactivity (Figure 6G-L) were scored as a cSCC metastasis to the lymph node (MetLN). The average number of MetLN per cSCC were scored and statistically compared between L-006235- and vehicle-treated cSCC cohorts. Treatment with L-006235 did not impact the persistence of cSCC lesions (Figure 6M) or the total number of enlarged lymph nodes per cSCC bearing mouse compared to vehicle-treated mice (Figure 6N). However, a dramatic 16-fold reduction in the average MetLN per cSCC was observed in L-006235-treated mice (Figure 6O).
Figure 6. Impact of Ctsk inhibitor L-006235 or Cd200 conditional disruption on cSCC metastasis.
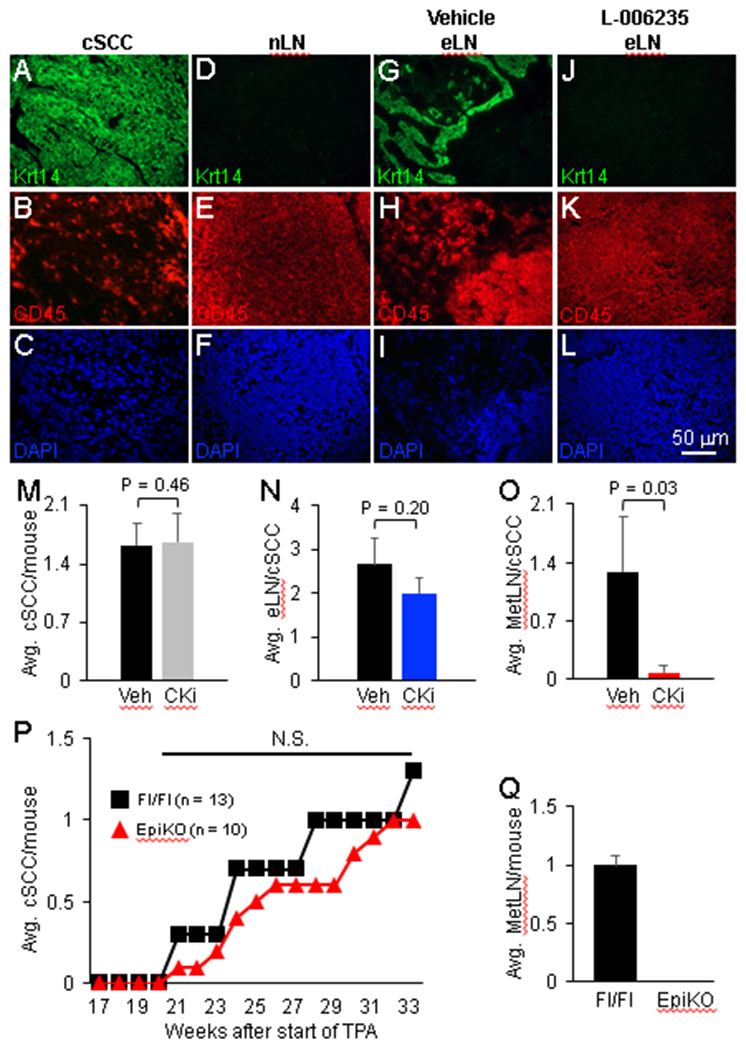
(A-L) Representative micrographs showing Krt14 and CD45 co-immunofluorescence labeling in primary cSCC (A-C), normal lymph node (nLN) (D-F), and enlarged lymph node (eLN) (G-L). DAPI counterstain (blue) was conducted to visualize nuclei (C,F,I,L). Panels A-C, D-F, G-I and J-L represent the same field of view. Primary cSCC and nLN specimens were utilized to validate immunodetection of Krt14+ keratinocytes and CD45+ immune cells. Krt14 immunolabeling in eLNs was used to confirm the presence (G) or absence (J) of metastatic disease. (M-O) Bar graphs showing the average number of cSCCs per mouse (M), enlarged lymph nodes (eLN) per cSCC (N) and lymph node metastases per cSCC (O) in vehicle- and 6 μg/ml L-006235-treated groups (N = 8 vehicle-treated cSCCs and 7 L-006235-treated cSCCs). (P) Line graph showing the average number of cSCC malignancies per mouse observed in Cd200fl/fl (control) and EpiKO mouse skin subjected to a DMBA/TPA two-stage carcinogenesis protocol (N = 13 Cd200fl/fl and 11 EpiKO mice). (O) Bar graph showing the average number of metastatic lymph node lesions per mouse in Cd200fl/fl (Fl/Fl) and EpiKO mice. All error bars are represented as Standard Deviation. Abbreviation: CKi, Cathepsin K inhibitor; N.S., not significant.
The observed decrease in cSCC metastasis frequency in L-006235-treated mice correlates with our previous work, which demonstrated that shRNA silencing of Cd200 expression did not influence orthotopic cSCC formation but did block cSCC metastasis in a tail vein assay [23]. Subsequent work with Cd200 germline null mice showed decreased development of benign papillomas following DMBA/TPA treatment that was attributed to tumor cell-independent Cd200 function [60]. However, this study did not assess malignant development of papillomas to cSCC, where Cd200 expression in tumor cells is first observed [23], or skin tumor metastasis. As such, the impact of targeted genetic disruption of Cd200 in tumor cells on cSCC development and/or metastasis in a natural carcinogenesis setting has not been reported. Here, we employed a DMBA/TPA two-stage chemical carcinogenesis model to induce cSCC in Cd200fl/fl (control) and EpiKO mice. No difference in primary cSCC formation was observed between Cd200fl/fl and EpiKO mice (Figure 6P). Skin tumor-bearing mice from each cohort were also examined for enlarged lymph nodes and lung and liver metastases. No difference in total number of enlarged lymph nodes was observed between each cohort; however, cSCC metastasis, as determined by Krt14 immunopositive lymph nodes, was abolished in cSCC-bearing EpiKO mice (Figure 6Q).
Discussion
Our findings identify Ctsk as a critical Cd200-dependent gene expressed by multiple tumor-infiltrating myeloid lineages in the tumor microenvironment and collectively support the conclusion that Cd200-mediated cSCC metastasis may depend on Ctsk production by Cd200r+ infiltrating cells. As such, our data provide a novel role for tumor-infiltrating myeloid cells in remodeling or degradation of extracellular matrix proteins to facilitate cSCC invasion and metastasis out of the skin. Finally, we identify Ctsk as a potential therapeutic target for CD200-expressing metastatic cancer.
As previously mentioned, the CD200-CD200R signaling axis plays an important role in osteoclast differentiation. Cd200 germline null mice exhibit decreased osteoclast numbers but also display increased bone density compared to Wt mice [46]. These observations suggest that, in addition to regulating osteoclast differentiation, CD200 may be required for induction of key mediators of bone resorption, such as Ctsk. However, this previous study did not explicitly address osteoclast expression of Ctsk in Cd200 germline null mice. Since we do not observe Ctsk expression in Cd200r+ cells in healthy skin, our observations of Cd200-dependent ectopic expression of Ctsk in invasive cSCC suggest that Cd200r regulation of Ctsk in infiltrating myeloid lineages during tumor metastasis may mimic a mechanism similar to what is observed for Cd200r in osteoclasts during bone remodeling.
It is well established that matrix degradation, both basement membrane and connective tissue, is a featured component of cancer cell invasion and metastasis [61]. CTSK has been implicated in bone metastasis, whereby the conventional role for osteoclast-derived CTSK in bone remodeling supports conditioning of the bone microenvironment for seeding from migratory prostate and breast cancer cells [62 – 63]. However, to our knowledge a direct role for CTSK outside of the bone in promoting the exit of tumor cells from the primary lesion has not been demonstrated. Given the prevalence of CD200 expression on a variety of aggressive cancer types and the ubiquitous presence of infiltrating myeloid lineages in the tumor microenvironment, our findings suggest that CD200-dependent induction of CTSK may have broad impact on the metastatic spread of a variety of cancer types.
Synthetic MMP inhibitors have been the focal point of most clinical trials designed to prevent connective tissue destruction and tumor metastasis [64]. However, multiple MMP inhibitors have failed at clinical trial level to prevent metastasis [65] suggesting that other protease families may play a pivotal role in connective tissue destruction in the tumor stroma. In the skin, bundles of Type I collagen constitute the vast majority of connective tissue that comprises the dermal scaffold supporting the upper layers of the epidermis. It is these collagen bundles that represent the greatest physical impediment to cSCC cell invasion downward through the skin and subsequent metastasis. CTSK is well characterized as the major osteoclastic proteolytic enzyme responsible for bulk collagen degradation during bone remodeling [47 – 49]. Our identification of Ctsk as a Cd200-target gene in cSCC-infiltrating Cd11b+Cd200r+ cells raises the key question: why would targeting Ctsk be efficacious when targeting other collagenases in the MMP family has done little to block tumor metastasis? One possible explanation lies in the unique ability of Ctsk to cleave the triple helix of collagen bundles at multiple sites [51]. To date this ability has not been observed in any other human collagenases, which typically recognize a single cleavage site in collagen bundles [51]. Previously reported biochemical assessments of Ctsk structure and function suggest that in vivo collagen degradation by Ctsk may be substantially more severe compared to MMP collagenases [51]. We posit that the increase in collagen cleavage sites by Ctsk may be required for sufficient connective tissue degradation to enable cSCC metastasis. Our findings specifically that Ctsk, but not Mmp, inhibition blocks cSCC cell invasion through collagen gel matrices support this concept.
Supplementary Material
Statement of Significance.
Findings highlight the relationship between CD200-CD200R and cathepsin K in cutaneous squamous cell carcinoma metastasis and suggest that either of these components may serve as a viable therapeutic target in this disease.
Acknowledgements
Normal human epidermal keratinocytes were provided by the Stem Cell Imaging and Manipulation (SCIM) Core Facility within the Department of Dermatology Skin Disease Resource Based Center (NIAMS P30AR069632). We are grateful to Olga Yarygina in the SCIM for processing and cutting tissue sections and performing H&E stains. The re-derivation of Cd200 conditional mice from frozen sperm was performed by Charles River Laboratories. Computational analysis of RNA-Seq data sets was conducted by Eve Byington in the Columbia Sulzberger Genome Center through the Genomics and High Throughput Screening Shared Resource. We are grateful to Wei Wang in the HICCC Flow Cytometry Shared Resource for assistance with sorting orthotopic cSCC stromal cells. The Genetically Modified Mouse Models, Genomics and High Throughput Screening and Flow Cytometry Shared Resources within the HICCC are supported by NCI 5P30CA013696-44. I. Khan was supported by the Barnard College SURF/SRI fellowship and NIH P30AR069632. J. Cho was supported by a Cancer Research Institute CLIP Award. R. Du was supported by NIH P30AR069632. A. Shao and A. Cohen are supported by NYDOH DOH01-C32582GG. D. Owens was supported by a Cancer Research Institute CLIP Award and NIH P30AR069632.
Abbreviations:
- BM
bone marrow
- Ctsk
Cathepsin K
- cSCC
cutaneous squamous cell carcinoma
- EpiKO
K14Cre;CD200fl/fl epidermal-targeted conditional CD200 knock out
Footnotes
Conflicts of Interest: The Authors have no conflicts of interest to report.
References
- 1.Webb M, Barclay AN. Localisation of the MRC OX-2 glycoprotein on the surfaces of neurones. J Neurochem 1984;43:1061–7. [DOI] [PubMed] [Google Scholar]
- 2.Bukovsky A, Presl J, Zidovsky J. Association of some cell surface antigens of lymphoid cells and cell surface differentiation antigens with early rat pregnancy. Immunology 1984;52:631–40. [PMC free article] [PubMed] [Google Scholar]
- 3.Dick AD, Broderick C, Forrester JV, Wright GJ. Distribution of OX2 antigen and OX2 receptor within retina. Invest. Ophthalmol Vis Sci 2001;42:170–6. [PubMed] [Google Scholar]
- 4.Wright GJ, Jones M, Puklavec MJ, Brown MH, Barclay AN. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology 2001;102:173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenblum MD, Olasz EB, Yancey KB, Woodliff JE, Lazarova Z, Gerber KA, et al. Expression of CD200 on epithelial cells of the murine hair follicle: a role in tissue-specific immune tolerance? J Invest Dermatol 2004;123:880–7. [DOI] [PubMed] [Google Scholar]
- 6.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000;290:1768–71. [DOI] [PubMed] [Google Scholar]
- 7.Rikjers ES, de Ruiter T, Buitenhuis M, Veninga H, Hoek RM, Meyaard L. Ligation of CD200R by CD200 is not required for normal murine myelopoiesis. Eur J Haematol 2007;79:410–6. [DOI] [PubMed] [Google Scholar]
- 8.Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol 2002;161:1669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorczynski RM, Chen Z, Lee L, Yu K, Hu J. Anti-CD200R ameliorates collagen-induced arthritis in mice. Clin Immunol 2002;104:256–64. [DOI] [PubMed] [Google Scholar]
- 10.Rygiel TP, Rijkers ESK, de Ruiter T, Stolte EH, van der Valk M, Rimmelzwaan GF, et al. Lack of CD200 enhances pathological T cell responses during influenza infection. J Immunol 2009;183:1990–6. [DOI] [PubMed] [Google Scholar]
- 11.Tonks A, Hills R, White P, Rosie B, Mills KI, Burnett AK, et al. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia 2007;21:566–8. [DOI] [PubMed] [Google Scholar]
- 12.Moreaux J, Hose D, Reme T, Jourdan E, Hundemer M, Legouffe E, et al. CD200 is a new prognostic factor in multiple myeloma. Blood 2006;108:4194–7. [DOI] [PubMed] [Google Scholar]
- 13.McWhirter JR, Kretz-Rommel A, Saven A, Maruyama T, Potter KN, Mockridge CI, et al. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc Natl Acad Sci USA 2006;103:1041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisgin A, Meng w-J, Adell G, Sun X-F. Interaction of CD200 overexpression on tumor cells with CD200R1 overexpression on stromal cells: an escape from the host immune response in rectal cancer patients. J Oncol 2019;5689464:doi: 10.1155/2019/5689464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorczynski RM, Chen Z, Diao J, Khatri I, Wong K, Yu K, et al. Breast cancer cell CD200 expression regulates immune response to EMT6 tumor cells in mice. Breast Cancer Res Treat 2010;123:405–15. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun 2007;364:778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohling SD, Davis E, Thompson K, Kussick SJ, Love J. Flow cytometric analysis of CD200 expression by pulmonary small cell carcinoma. Cytometry B Clin Ctyom 2016;90:493–8. [DOI] [PubMed] [Google Scholar]
- 18.Siva A, Xin H, Qin F, Oltean D, Bowdish KS, Kretz-Rommel A. Immune modulation by melanoma and ovarian tumor cells through expression of the immunosuppressive molecule CD200. Cancer Immunol Immunother 2008;57:987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung YS, Vermeer PD, Vermeer DW, Lee SJ, Goh AR, Ahn HJ, et al. CD200: association with cancer stem cell features and response to chemoradiation in head and neck squamous cell carcinoma. Head Neck 2015;37:327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moertel CL, Xia J, LaRue R, Waldron NN, Andersen BM, Prins RM, et al. CD200 in CNS tumor-induced immunosuppression: the role for CD200 pathway blockade in targeted immunotherapy. J Immunother Cancer 2014;2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawlor RT, Dapra V, Girolami I, Pea A, Pilati C, Nottegar A, et al. CD200 expression is a feature of solid pseudopapillary neoplasms of the pancreas. Virchows Arch 2019;474:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rexin P, Tauchert A, Hanze J, Heers H, Schmidt A, Hofmann R, et al. The immune checkpoint molecule CD200 is associated with tumor grading and metastasis in bladder cancer. Anticancer Res 2018;38:2749–54. [DOI] [PubMed] [Google Scholar]
- 23.Stumpfova M, Ratner D, Desciak EB, Eliezri YD, Owens DM. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res 2010;70:2962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colmont CS, Benketah A, Reed SH, Hawk NV, Telford WG, Ohyama M, et al. CD200-expressing human basal cell carcinoma cells initiate tumor growth. Proc Natl Acad Sci USA 2013;110:1434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaiser MR, Weiss CA, Gaiser T, Jiang H, Buder-Bakhaya K, Herpel E, et al. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory T cells. Oncoimmunology 2018;7:e1426517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petermann KB, Rozenberg GI, Zedek D, Groben P, McKinnon K, Buehler C, et al. CD200 is induced by ERK and is a potential therapeutic target in melanoma. J Clin Invest 2007;117:3922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol 2007;178:5595–605. [DOI] [PubMed] [Google Scholar]
- 28.Gorczynski RM, Chen Z, Hu J, Kai Y, Lei J. Evidence of a role for CD200 in regulation of immune rejection of leukaemic tumor cells in C57Bl/6 mice. Clin Exp Immunol 2001;126:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenmalm MC, Cherwinski H, Bowman EP, Phillips JH, Sedgwick JD. Regulation of myeloid cell function through the CD200 receptor. J Immunol 2006;176:191–9. [DOI] [PubMed] [Google Scholar]
- 30.Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol 2002;23:285–90. [DOI] [PubMed] [Google Scholar]
- 31.Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol 2003;171:3034–46. [DOI] [PubMed] [Google Scholar]
- 32.Coles S, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011;25:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulations of protumoral macrophages. Oncotarget 2017;8:8633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z, Kapus A, Khatri I, Kos O, Zhu F, Gorczynski RM. Cell membrane-bound CD200 signals both via an extracellular domain and following nuclear translocation of a cytoplasmic fragment. Leuk Res 2018;69:72–80. [DOI] [PubMed] [Google Scholar]
- 35.Twito T, Chen Z, Khatri I, Wong K, Spaner D, Gorczynski R. Ectodomain shedding of CD200 from the B-CLL cell surface is regulated by ADAM28 expression. Leuk Res 2013;37:816–21. [DOI] [PubMed] [Google Scholar]
- 36.Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates tooth growth and morphogenesis. Development 2000;127:4775–85. [DOI] [PubMed] [Google Scholar]
- 37.Bachelor MA, Lu Y, Owens DM. L-3-Phosphoserine phosphatase (PSPH) regulates cutaneous squamous cell carcinoma proliferation independent of L-serine biosynthesis. J Dermatol Sci 2011;63:164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 1975;6:331–43. [DOI] [PubMed] [Google Scholar]
- 39.Jensen UB, Xan X, Triel C, Woo SH, Christensen R, Owens DM. A distinct population of clonogenic and multipotent murine follicular keratinocytes residing in the upper isthmus. J Cell Sci 2008;121:609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai Z, Xing L, Cerise J, Wang EH, Jabbari A, de Jong A, et al. CXCR3 blockade inhibits T cell migration into the skin and prevents development of alopecia areata. J Immunol 2016;197:1089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Comm 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor bearing mice. J Immunol 2008;181:5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler — a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucl Acids Res 2007;35:doi. 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui W, Cuartas E, Ke J, Zhang Q, Einarsson HB, Sedgwick JD, et al. CD200 and its receptor, CD200R, modulate bone mass via the differentiation of osteoclasts. Proc Natl Acad Sci USA 2007;104:14436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996;273:1236–8. [DOI] [PubMed] [Google Scholar]
- 48.Inui T, Ishibashi O, Inaoka T, Origane Y, Kumegawa M, Kokubo T, et al. Cathepsin K antisense oligodeoxynucleotide inhibits osteoclastic bone resorption. J Biol Chem 1997;272:8109–12. [DOI] [PubMed] [Google Scholar]
- 49.Votta BJ, Levy MA, Badger A, Bradbeer J, Dodds RA, James IE, et al. Peptide aldehyde inhibitors of cathepsin K inhibit bone resorption both in vitro and in vivo. J Bone Miner Res 1997;12:1396–406. [DOI] [PubMed] [Google Scholar]
- 50.Rosenblum MD, Woodliff JE, Madsen NA, McOlash LJ, Keller MR, Truitt RL. Characterization of CD200-receptor expression in the murine epidermis. J Invest Dermatol 2005;125:1130–8. [DOI] [PubMed] [Google Scholar]
- 51.Novinec M, Lenarcic B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem 2013;394:1163–79. [DOI] [PubMed] [Google Scholar]
- 52.Dejica VM, Mort JS, Laverty S, Percival MD, Antoniou J, Zukor DJ, et al. Cleavage of type II collagen by cathepsin K in human osteoarthritic cartilage. Am J Pathol 2008;173:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Overall CM. Molecular determinants of the metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol Biotechnol 2002;22:51–86. [DOI] [PubMed] [Google Scholar]
- 54.Owens DM, Romero MR, Gardner C, Watt FM. Suprabasal α6 4 integrin expression in epidermis results in enhanced tumourigenesis and disruption of TGFβ signaling. J Cell Sci 2003;116:3783–91. [DOI] [PubMed] [Google Scholar]
- 55.Desmarias S, Black WC, Oballa R, Lamontagne S, Riendeau D, Tawa P, et al. Effect of Cathepsin K inhibitor basicity on in vivo off-target activities. Mol Pharmacol 2008;73:147–56. [DOI] [PubMed] [Google Scholar]
- 56.Soung DY, Gentile MA, Duong LT, Drissi H. Effects of pharmacological inhibition of cathepsin K on fracture repair in mice. Bone 2013;55:248–55. [DOI] [PubMed] [Google Scholar]
- 57.Bonnet N, Brun J, Rousseau JC, Duong LT, Ferrari SL. Cathepsin K controls cortical bone formation by degrading periostin. J Bone Min Res 2017;32:1432–41. [DOI] [PubMed] [Google Scholar]
- 58.Svelander L, Erlandsson-Harris H, Astner L, Grabowska U, Klareskog L, Lindstrom E, et al. Inhibition of cathepsin K reduces bone erosion, cartilage degradation and inflammation evoked by collagen induced arthritis in mice. Eur J Pharmacol 2009;613:155–62. [DOI] [PubMed] [Google Scholar]
- 59.Cartledge DM, Colella R, Glazewski L, Lu G, Mason RW. Inhibitors of cathepsins B and L induce autophagy and cell death in neuroblastoma cells. Invest New Drugs 2013;31:20–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rygiel TP, Karnam G, Goverse G, van der Marel APJ, Greuter MJ, van Schaarenburg RA, et al. CD200-CD200R signaling suppresses anti-tumor responses independently of CD200 expression on the tumor. Oncogene 2012;31:2979–88. [DOI] [PubMed] [Google Scholar]
- 61.Quintero-Fabian S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argaez V, Lara-Riegos J, et al. Role of matrix metalloproteinases in angiogenesis and cancer. Front Oncol 2019;9:doi: 10.3389/fonc.2019.01370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Podgorski I, Linebaugh BE, Sloane BF. Cathepsin K in the bone microenvironment: link between obesity and prostate cancer? Biochem Soc Trans 2007;35:701–3. [DOI] [PubMed] [Google Scholar]
- 63.Le Gall C, Bonnelye E, Clezardin P. Cathepsin K inhibitors as treatment of bone metastasis. Curr Opin Support Palliat Care 2008;2:218–22. [DOI] [PubMed] [Google Scholar]
- 64.Young D, Das N, Anowai A, Dufour A. Matrix metalloproteases as influencers of the cells’ social media. Int. J Mol Sci 2019;20:doi: 10.3390/ijms20163847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 2002;295:2387–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.