

Abstract
Myocardial infarction ranks first for the mortality worldwide. Because the adult heart is unable to regenerate, fibrosis develops to compensate for the loss of contractile tissue after infarction, leading to cardiac remodeling and heart failure. Adult mesenchymal stem cells (MSC) regenerative properties, as well as their safety and efficacy, have been demonstrated in preclinical models. However, in clinical trials, their beneficial effects are controversial. In an experimental model of arthritis, we have previously shown that PPARβ/δ deficiency enhanced the therapeutic effect of MSC. The aim of the present study was to compare the therapeutic effects of wild-type MSC (MSC) and MSC deficient for PPARβ/δ (KO MSC) perfused in an ex vivo mouse model of ischemia-reperfusion (IR) injury. For this purpose, hearts from C57BL/6J mice were subjected ex vivo to 30 min ischemia followed by 1-h reperfusion. MSC and KO MSC were injected into the Langendorff system during reperfusion. After 1 h of reperfusion, the TTC method was used to assess infarct size. Coronary effluents collected in basal condition (before ischemia) and after ischemia at 1 h of reperfusion were analyzed for their cytokine profiles. The dose-response curve for the cardioprotection was established ex vivo using different doses of MSC (3.105, 6.105, and 24.105 cells/heart) and the dose of 6.105 MSC was found to be the optimal concentration. We showed that the cardioprotective effect of MSC was PPARβ/δ-dependent since it was lost using KO MSC. Moreover, cytokine profiling of the coronary effluents collected in the eluates after 60 min of reperfusion revealed that MSC treatment decreases CXCL1 chemokine and interleukin-6 release compared with untreated hearts. This anti-inflammatory effect of MSC was also observed when hearts were treated with PPARβ/δ-deficient MSC. In conclusion, our study revealed that the acute cardioprotective properties of MSC in an ex vivo model of IR injury, assessed by a decreased infarct size at 1 h of reperfusion, are PPARβ/δ-dependent but not related to their anti-inflammatory effects.
Keywords: myocardial infarction, reperfusion injury, inflammation, PPAR, mesenchymal stem cells, cardioprotection
Introduction
Acute myocardial infarction (AMI) is the leading cause of cardiovascular mortality worldwide and a provider of heart failure (1). Prompt revascularization of the culprit artery with primary coronary angioplasty or thrombolysis is associated with deleterious side effects called ischemia-reperfusion (IR) injury due to abrupt restoration of blood flow and oxygen.
The release of DAMPS (Damage-associated molecular patterns) from dead cells, in concert with the activation of the complement cascade and reactive oxygen species (ROS), triggers an acute pro-inflammatory response at the onset of AMI that activates the resident immune cells of the heart. Reperfusion exacerbates this inflammatory response to eliminate necrotic cells and repair the infarcted myocardium (2). Interleukin 1 (IL-1), and interleukin 6 (IL-6) are the major cytokines that mediate this short but strong inflammatory burst, which contributes to cell death and irreversible IR injury (2). At the site of the injury, many cell types, including cardiomyocytes, vascular cells, fibroblasts, and immune cells, are involved in this inflammatory response. Necrotic cardiomyocytes in the infarcted area provide the main stimulus for the post-infarction inflammatory response through the release of DAMPs. In the border zone, surviving cardiomyocytes, once activated by IL-1, Toll Like Receptor (TLR) ligands, and ROS, will produce and secrete cytokines such as IL-6 (3), TNFα (4), and chemokines such as CXCL1 (KC/GRO) and MIP-2 (5) to trigger inflammatory activation. Endothelial cells, the most abundant non-cardiomyocytes in the heart, when activated by TNFα, produce CXCL1 (6). In addition to a local inflammatory response, myocardial cells sense tissue necrosis and trigger the post-infarction inflammatory response, stimulating the release and recruitment of bone marrow (BM)-derived leukocytes. However, the relative contribution of cardiomyocyte-derived inflammatory mediators in the progression and extension of post-infarction inflammation remains unknown.
Previous studies have shown that the application of ischemic postconditioning (PostC) i.e., repeated brief episodes of IR in the myocardial tissue applied at the onset of reperfusion, was able to specifically inhibit IR injury (7, 8). PostC, considered a gold standard strategy for cardioprotection in animal models of AMI, was reported to be mediated by multiple intracellular cascades leading to anti-apoptotic and anti-inflammatory effects resulting in cardioprotection (7, 9, 10). Various targets have been identified in PostC signaling pathways, however, no product of potential clinical utility, including anti-inflammatory drugs, has emerged from all candidates identified as cardioprotective in preclinical studies (11, 12). This suggests that other strategies with pleiotropic mechanisms of action are clearly needed (13).
Preclinical studies have shown that MSC-based therapy improves myocardial functional recovery after AMI by promoting endogenous cell survival, proliferation, and angiogenesis. In addition, MSC exert pleiotropic effects, including reduction of inflammation and apoptosis through their ability to release bioactive molecules (14, 15). Based on these promising results, MSC were then tested in clinical trials that demonstrated their safety and promising efficacy in phase I and II, but yielded inconclusive results in phase III trials (16). Indeed, no significant long-term beneficial effects in AMI patients has been reported based on recent meta-analyses (17, 18).This failure to translate preclinical results into human clinical trials could be attributed to, in part, trial design differences, the source and dose of MSC used, and the route and timing of MSC injection (18). To bridge the gap between preclinical and clinical studies, the development of “preconditioning” methods to improve MSC therapeutic potential has been widely investigated (19) mainly focusing on the enhancement of their anti-inflammatory properties. For example, MSC treated with IGF-1 before transplantation into the ischemic heart reduce the production and expression of proinflammatory cytokines, including TNFα, IL-1β, and IL-6 and improve cardiac functions (20). Although promising, this approach of enhancing the anti-inflammatory properties of MSC to improve their therapeutic potential in AMI has been poorly investigated.
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors expressed in three different isoforms, PPARα, PPARβ/δ, and PPARγ, which heterodimerize with the retinoid X receptor (RXR) and act as transcriptional regulators after ligand binding. Peroxisome proliferator-activated receptor isoforms exert multiple functions depending on tissue ligands and cofactors (21). PPARβ/δ, a proangiogenic member of the PPAR family, is ubiquitously expressed (22–24) in contrast to PPARα, which is mainly detected in brown adipose tissue, intestine, heart, liver, kidney, and PPARγ, which is expressed in immune cells, intestine, white, and brown adipose tissue. The potent anti-inflammatory actions of PPARβ/δ on several immune cells including macrophages have been previously reported. Indeed, the capacity of IL-4 and IL-13 to direct macrophages to an M2-like anti-inflammatory phenotype in mouse adipose tissue and liver depends on PPARβ/δ expression (25–27). Recently, in an experimental model of the auto-immune and inflammatory disorder in mice, “collagen-induced arthritis (CIA),” we demonstrated that PPARβ/δ expression level could predict the immunoregulatory potential of MSC and that its inhibition increased their immunoregulatory and therapeutic activities (28).
Given that reperfusion injury is associated with acute inflammation, we hypothesized that inactivation of PPARβ/δ might impact the cardioprotective properties of MSC during IR injury associated to local inflammation (26). In mouse models, in order to get closer to the classical clinical conditions of MSC administration (29–37), the local injection of MSC at the acute phase, avoiding the systemic route is prefered. Thus, in the present study, we explored the contribution of PPARβ/δ in the acute local cardioprotective effect mediated by MSC during reperfusion in an ex vivo mouse model of isolated heart subjected to IR injury.
Materials and Methods
Ethics
Studies involving animals were reviewed and approved by the Institute's SBEA (Structure Bien-être Animal) commitee in accordance with the European directive 2010/63/EU and the French Ministerial Order of February 01, 2013.
Animal Housing and Care
Experiments were performed in C57BL/6J mice (Charles River laboratory) in accordance with the European Communities Council directive of November 1986 and in accordance with the Guide for the Care and Use of Laboratory Animals" published by the US National Institutes of Health (NIH publication 8th Edition, 2011). All mice were maintained under controlled environmental conditions (22 ± 2°C, 12 h light /12 h dark cycle) in the Institute's animal facility.
Ex vivo Experiments
Male mice were anesthetized with an intraperitoneal injection (IP) of ketamine (14 mg/kg, Imalgène® Merial), xylazine (14 mg/kg, Rompun® Bayer) followed by an injection of pentobarbital (IP; 76.6 mg/kg; Sanofi-Aventis). The anesthetized mice received 250 U heparin (IP) in order to prevent blood clot formation. After sternotomy, the heart was excised, cannulated through the ascending aorta, and quickly mounted on the Langendorff perfusion system. Prewarmed Tyrode's solution (NaCl 140 mM, KCl 5.4 mM, MgCl 1 mM, Hepes 5 mM, glucose 5.5 mM, CaCl2 1.8 mM, pH 7.4) was perfused at constant pressure (70 mmHg) and temperature (37°C).
Ischemia-Reperfusion Protocol
On the Langendorff perfusion system, the heart was perfused with prewarmed Tyrode solution for 15 min (stabilization). Global ischemia was obtained by stopping the perfusion flow (no-flow) for 30 min. A reperfusion step (60 min) was achieved by restoring the flow. Mesenchymal stem cells treatment (prepared in Tyrode solution) was applied during reperfusion as a non-recirculating perfusate. The control condition (IR) was obtained using only Tyrode. A positive control of cardioprotection was obtained by applying an ischemic postconditioning stimulus, comprising three cycles of 1 min ischemia-1 min reperfusion at the onset of reperfusion (PostC group). Perfusates containing coronary effluents were collected at the apex of the heart at both 10 min of the stabilization (basal) phase and at 15, 30, and 60 min after the onset of reperfusion and were stored at −80°C for further experiments.
Infarct Size Measurement
At the end of the IR protocol, the heart was harvested from the apparatus. The left ventricle (LV) was embedded in agar (4% w/v), and transversely sliced (1 mm) with a vibratome. To reveal tissue viability, slices were incubated in a 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich) for 15 min at 37°C. After a fixation step (4% paraformaldehyde, 48 h), each slice was photographed from each side. The infarct area was quantified by planimetric measurements with ImageJ software.
MSC Culture
Isolation, amplification, and characterization of murine MSC were performed as previously described (33). Briefly, BM was flushed out from the long bones of Ppardfl/flsox2cretg PPARβ/δ-deficient mice and their wild-type littermates (Ppardfl/+) kindly provided by Gerhard Krönke laboratory (Institute of Rheumatology and Immunology, Erlangen, Germany) (38) to isolate KO MSC and MSC, respectively. Cells were cultured in minimal essential medium (MEM)-α containing 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 ng/ml human basic fibroblast growth factor (bFGF) at a density of 0.5 × 106 cells/cm2. Phenotypic and functional characterization of MSC has been performed previously (28). To confirm the effects of PPARb/d inactivation, mesenchymal stem cells were pre-incubated 24 h with 5 μM of PPARβ/δ selective antagonist GSK0660.
MSC Labeling With CM-DiI
The stock solution of the fluorescent cell-tracer CM-DiI (Molecular Probes) was reconstituted in dimethyl sulfoxide (DMSO) at a concentration of 1 μg/μl. Mesenchymal stem cells were collected and suspended at the concentration of 1 × 107 cells/10 μg CM-DiI in 5 ml PBS. Cells were incubated at 37°C for 5 min followed by 15 min at 4°C, in the dark. Unincorporated fluorescent dye was then removed by centrifugation at 300 g for 5 min and two washes with PBS were performed. Labeled cells were resuspended in Tyrode's solution and maintained at 4°C prior being injected into the myocardium.
Cytokine Level Quantification
For quantification of cytokine levels, coronary effluents from perfused hearts were collected under basal conditions (t0), after 10 min of stabilization, and after IR injury at 15, 30, and 60 min after the onset of reperfusion. The different perfusates were stored at −80°C until the assay was performed using the Meso Scale Discovery (MSD) V-Plex Plus Proinflammatory Panel 1 (mouse) kit at the “Plateforme de Protéomique Clinique de Montpellier” according to the manufacturer's protocol. As compared to other methods, MSD is the most suitable assay for samples with low endogenous levels of the cytokines although cytokine's levels can be below the limits of detection of this immunoassays. The V-Plex Plus Proinflammatory Panel 1 includes IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-6, KC/GRO (CXCL1), IL-10, IL-12p70, and TNFα. When the concentration of a cytokine was undistinguishable from background (i.e., below the limit of detection), the sample was considered negative for that cytokine.
Immunochemistry
At the end of ex vivo experiments, LV were fixated in 4%-PFA and embedded in paraffin. Each LV was cut from apex to base (sections of 4 μM each 150 μM). The paraffin-embedded sections were deparaffinized then rehydrated through an alcohol gradient. Left ventricle sections were incubated with a primary anti α-actinin antibody (1:100, mouse monoclonal; Sigma-Aldrich). Cell nuclei were stained with Hoechst (Life technologies SAS) and endothelial cells with Isolectin B4 (FITC Conjugate; Sigma-Aldrich). After incubation with primary antibodies, sections were washed in PBS, and then incubated (3 h) with secondary antibodies (1:2,000, Jackson ImmunoRes Laboratories, Inc.). Primary and secondary antibodies were diluted in PBS containing 3% BSA and 0.1% Triton X100. Stained sections were mounted in Mowiol (Biovalley). Images were obtained with a Zeiss Axioimager Z3 fluorescent microscope after observation of six different sections of the LV harvested on n = 2 hearts treated by MSC labeled with CM-DiI and analyzed using ImageJ and Adobe Photoshop to prepare the final figures.
Statistical Analysis
Data expressed as mean ± SD values were compared among groups using non-parametric Mann-Whitney (two groups) and Kruskal-Wallis (multiple comparison) methods. P-values < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001(****) were considered statistically significant. Analysis and graphical representation were performed using Graph-Pad Prism™ software (GraphPad).
Results
Induction of a Pro-inflammatory Response in Isolated Perfused Heart Subjected to Ischemia-Reperfusion Injury Ex vivo
For this study, we have developed an ex vivo model of global ischemia followed by reperfusion (IR protocol) to evaluate the short-term therapeutic effects of MSC. Isolated hearts were mounted and perfused on a Langendorff system and subjected to 30 min of global ischemia (no-flow) followed by 60 min of reperfusion (see protocol in Figure 1A). The first part of the study was devoted to the characterization of our model. Hearts after myocardial IR injury were characterized by an infarct size with a mean value of 56.4% ± 10.3 expressed as percentage of the LV (Figure 1B).
Figure 1.

C57BL/6J mouse hearts were mounted on a Langendorff system and subjected to IR injury. (A) The ex vivo protocol included a 15 min-stabilization period, followed by 30 min of global ischemia achieved by stopping the flow in the aorta (no-flow). Reperfusion was achieved by restoring the Tyrode infusion during 60 min. For the SHAM condition, the heart was perfused throughout the protocol without any ischemic induction. At the end of the protocol, infarct size was measured using the TTC-staining method. Coronary effluents were collected at two time points during the ex vivo protocol: during stabilization to evaluate the “basal” level of cytokine release and at the end of the reperfusion phase to evaluate the “IR60” cytokine production after IR injury. (B) Scatter plots and bars (mean ± SD) were represented for infarct size (in % of LV) in IR (n = 12) and SHAM (n = 3) hearts. Representative pictures of TTC-stained LV slices were shown for each group. (C) Scatter plots with bars (mean ± SD) are presented for quantification of cytokines within coronary effluents collected before ischemia (Basal) and after 60 min of reperfusion after the IR protocol (IR60) using the Meso Scale Discovery (MSD) V-Plex Plus Proinflammatory Panel 1 (mouse) kit. Statistical analysis was performed using the Mann-Whitney test. For CXCL1 (pg/ml), **** was noted for p < 0.0001, for IL-6 (pg/ml), **** was noted for p< 0.0001 and for TNFα (pg/ml), *** was noted for p = 0.0008.
We therefore asked whether induction of IR injury was associated with excessive release of pro-inflammatory cytokines in coronary effluents collected 60 min after the onset of reperfusion (time point at which infarct size was evaluated) compared with the basal level before ischemia (collected during stabilization). Of the 10 cytokines quantified using the MSD VPlex Plus Proinflammatory, seven including IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-10, IL-12p70 were not considered because of their undetectable levels. However, the concentration of three cytokines, KC/GRO (CXCL1), IL-6, and TNFα, were showed to be significantly increased in the coronary effluents of the hearts after IR injury (after ischemia and 60 min of reperfusion) compared with the basal conditions (collected at 10 min stabilization, basal) (Figure 1C). Overall, these results reveal that myocardial IR injury of C57BL/6 mouse hearts subjected ex vivo to 30 min of ischemia followed by 1 h of reperfusion is associated with the release of pro-inflammatory cytokines.
MSC Exerted a Potent Cardioprotective Effect When Administered During Reperfusion in an Ex vivo Model of Global Ischemia
To reduce IR injury-related inflammation and thus infarct size, we used two well-known cardioprotective strategies, ischemic PostC and MSC-based therapy, to compare for the first time their effects in an ex vivo mouse model. Mesenchymal stem cells were administered during reperfusion in isolated hearts subjected to 30 min of global ischemia as described in the protocol shown in Figure 2A. Different concentrations of MSC in the perfusion solution were tested (Figure 2B). The dose-response curve was established using different concentrations of MSC and showed that 5,000 cells/mL (6.105 cells/heart) and 20,000 cells/ml (24.105 cells/heart) induced cardioprotective effects by decreasing infarct size (24.01% ± 11.8, n = 9 for MSC 5,000 vs. 56.4% ± 10.3, n = 12 for IR; p = 0.0009, and 17.0% ± 4.1; n = 6 for MSC 20,000 vs. 56.4% ± 10.3, n = 12; p = 0.0003) as opposed to 2,500 cells/ml (3.105 cells/heart), which had no significant beneficial effect (34.1% ± 13.9, n = 6 for MSC 2,500 vs. 56.4 % ± 10.3, n = 12 for IR; p = 0.1571). The concentration of 5,000 cells/ml was chosen for all experiments in our study because it was the minimum concentration giving maximum cardioprotection in the ex vivo mouse model. Furthermore, we demonstrated that reperfusion with MSC at 5,000 cells/ml (6.105 cells/heart) reduced infarct size to the same extent as ischemic PostC, taken as a positive control in our experiments (18.3 % ± 9.7, n = 10 for PostC vs. 24.1% ± 11.8, n = 9 for MSC 5,000; p > 0.99). Immunohistological analysis revealed that ex vivo perfused MSC labeled with CM-DiI were 100% co-localized with Isolectin B4-positive endothelial cells in coronary microvessels after 1 h of reperfusion (Figure 2D).
Figure 2.
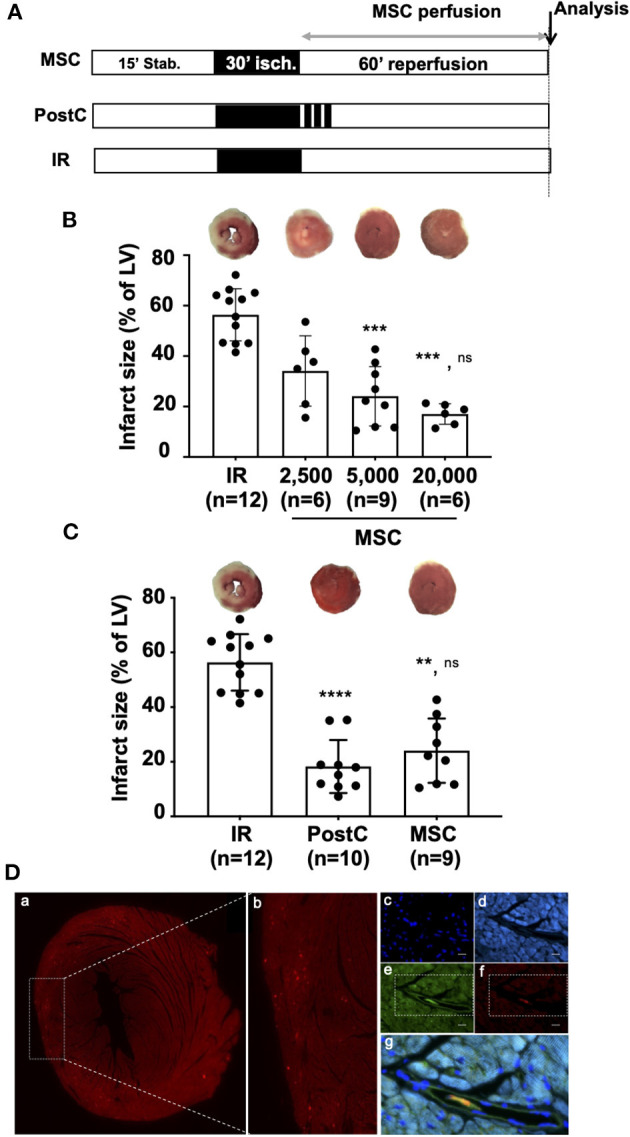
(A) Isolated hearts perfused ex vivo on the Langendorff system were submitted to perfusion protocol similar to that described in Figure 1A. In the MSC group, reperfusion was achieved with a solution of MSC cells prepared in a Tyrode buffer at various concentrations (2,500; 5,000; or 20,000 cells/mL). For the PostC group, a postconditioning stimulus comprising three cycles of 1 min ischemia-1 min reperfusion was applied at the onset of reperfusion. In the control condition (IR), hearts were reperfused with Tyrode solution alone (control condition). Histological analysis was performed at the end of the protocols for infarct size measurement and immunochemistry. (B) Scatter plots and bars (mean ± SD) were represented for infarct size (in % of LV) in IR (n = 12), MSC 2,500 cells/ml (n = 6), MSC 5,000 cells/ml (n = 9), and MSC 20,000 (n = 6). Representative pictures of TTC-stained LV slices were shown for each group. Statistical analysis was performed using Kruskal-Wallis with the Dunn's post hoc test for multiple comparison. Statistical significance is noted *** for p = 0.0009 (MSC 5,000 vs. IR), *** for p = 0.0003 (MSC 20,000 vs. IR) and ns for p > 0.99 (MSC 20,000 vs. MSC 5,000). (C) Scatter plots and bars (mean ± SD) were represented for infarct size (in % of LV) in IR (n = 12), PostC (n = 10), and MSC (5,000 cells/ml, n = 9). Representative pictures of TTC-stained LV slices were shown for each group. Statistical analysis was performed using Kruskal-Wallis with the Dunn's post hoc test for multiple comparison. Statistical significance is noted **** for p > 0.0001 (PostC vs. IR), ** for p = 0.0018 (MSC vs. IR) and ns for p > 0.99 (PostC vs. MSC). (D) Representative pictures of microscopic observations among 12 (data not shown) for an MSC-treated heart section (a) and (b) corresponding enlarged immunostaining images (Original magnification: x40 oil immersion) showing (c) cell nuclei (DAPI), (d) alpha-actinin, (e) microvessels (isolectin B4), (f) DI-I labeled MSC, and (g) merge allowing to show that MSC are located in the microvessels after 60 min of reperfusion (same time point of infarct size evaluation).
Altogether, these results demonstrate that 6.105 MSC/heart provide a similar cardioprotection to that of PostC, considered as a positive control in our study.
The Potent Cardioprotective Effect of Both Ischemic PostC and MSC Is Associated With a Decrease in Pro-inflammatory Cytokines in the Injured Myocardium
To determine whether the beneficial effect of PostC or MSC administration on IR injury was associated with the regulation of the immune response, we quantified pro-inflammatory cytokines in coronary effluents collected at 15, 30, and 60 min after the onset of reperfusion (Figure 3A). Among the 10 cytokines quantified by the MSD approach, CXCL1 (KC/GRO), IL-6, and TNFα, were detected with significant levels only at 60 min of reperfusion in the samples from the untreated IR hearts. IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-10, and IL-12p70 were not detected. None of the 10 cytokines were detectable at 15 and 30 min post-reperfusion (data not shown). In addition, the levels of CXCL1 (KC/GRO) (Figure 3B) were significantly lower in coronary effluents from hearts treated by PostC or MSC (16.61 pg/ml ± 10.53, n = 10 for IR vs. 3.10 pg/ml ± 5.9, n = 10 for PostC; p*** = 0.0006, and vs. 2.56 pg/ml ± 3.79, n = 10 for MSC; p* = 0.018). Similar results were obtained for IL-6 (21.83 pg/ml ± 11.08, n = 10 for IR vs. 6.16 pg/ml ± 8.95, n = 10; p** = 0.021, and vs. 5.43 pg/ml ± 3.02, n = 10 for PostC; p* = 0.029 for MSC; Figure 3C). For TNFα, there was a slight reduction of the effluent levels upon PostC treatment (without reaching significativity), which was not observed in the MSC group (pns > 0.99; Figure 3D).
Figure 3.
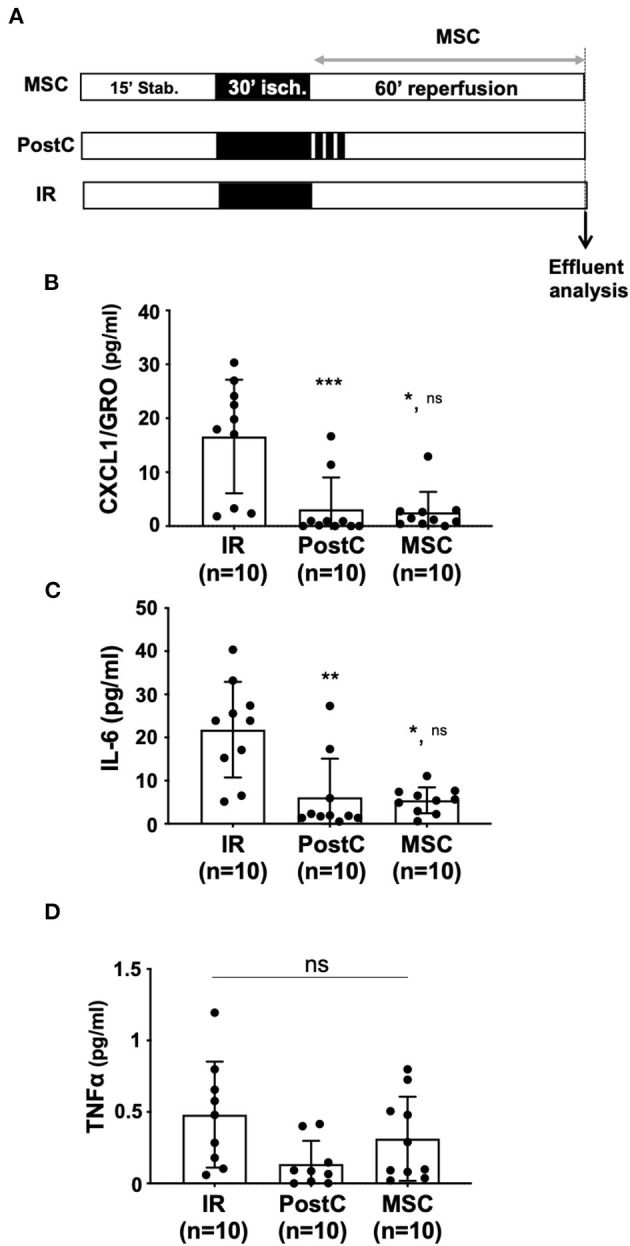
(A) Isolated hearts perfused ex vivo on the Langendorff system were submitted to the perfusion protocol similar to that described in Figure 1A. In the MSC group, reperfusion was achieved with a MSC Tyrode solution (5,000 cells/ml). For the PostC group, a postconditioning stimulus comprising three cycles of 1 min ischemia-1 min reperfusion was applied at the onset of reperfusion. In the control condition (IR), hearts were reperfused with Tyrode solution alone (control condition). Coronary effluents were collected at the end of the reperfusion phase to evaluate cytokine production after IR, PostC or MSC protocols. (B–D) Scatter plots with bars (mean ± SD) are presented for quantification of cytokines within coronary effluents collected after 60 min of reperfusion using the Meso Scale Discovery (MSD) V-Plex Plus Proinflammatory Panel 1 (mouse) kit. Statistical analysis was performed using the Kruskal-Wallis test followed by the Dunn's post test. (B) For CXCL1 (pg/mL), *** was noted for p = 0.0006 (PostC vs. IR), ** for p = 0.018 (MSC vs. IR) and ns for p > 0.999 (MSC vs. PostC). (C) For IL-6 (pg/ml), ** was noted for p = 0.0021 (PostC vs. IR), * for p = 0.0287 (MSC vs. IR) and ns for p > 0.999 (MSC vs. PostC) and (D): for TNFα (pg/ml), ns was noted for p = 0.059.
This result indicates that the protective effects of both PostC and MSC were associated with a potent anti-inflammatory effect assessed by quantification of pro-inflammatory cytokines in coronary effluents.
PPARβ/δ Is Involved in the Cardioprotective Effects Mediated by MSC Against IR Injury
Recently, we showed that PPARβ/δ is pivotal for the MSC immunoregulatory and therapeutic functions in an experimental model of arthritis (28). However, the role of PPARβ/δ on the cardioprotective activity of MSC and the relevance of PPARβ/δ to the anti-inflammatory properties of MSC in the inflamed myocardium have never been addressed. To determine whether PPARβ/δ is essential for the cardioprotective properties of MSC, we compared the effect of MSC isolated from PPARβ/δ−/− deficient mice (KO MSC) and those obtained from their PPARβ/δ+/+ control littermates (MSC). Isolated hearts were perfused during reperfusion with solutions containing MSC at the optimal dose of 6.105cells/heart (see protocol Figure 4A). Under these conditions, the drastic decrease in infarct size induced by MSC (24.1% ± 11.8, n = 9 for MSC vs. 56.4% ± 10.3, n = 12 for IR; p** = 0.001) was abolished when KO MSC were infused into isolated hearts after the ischemic insult (48.4% ± 25.4, n = 13 for KO MSC vs. 56.4% ± 10.3, n = 12 for IR; pns = 0.75 and 48.4% ± 25.4, n = 13 for KO MSC vs. 24.1% ± 11.8, n = 9 for MSC; p* = 0.029) (Figure 4B). A similar absence of cardioprotective effet was observed after the infusion of MSC pharmacologically inactivated for PPARβ/δ using GSK0660, a selective antagonist of PPARβ/δ (data not shown). To determine whether the loss of therapeutic effect of MSC in response to PPARβ/δ knockdown was associated with a loss of their ability to reduce inflammation in infarcted myocardium, we quantified pro-inflammatory cytokines within coronary effluents collected 60 min after the onset of reperfusion. Quantification of cytokines by MSD was performed in coronary effluents from hearts treated with either MSC or KO MSC and compared with those of untreated hearts. We demonstrated that PPARβ/δ knockdown in MSC did not alter their anti-inflammatory potential as revealed by the measured levels of CXCL1 (2.56 pg/ml ± 3.79, n = 10 for MSC vs. 3.73 pg/ml ± 1.65, n = 8 for KO MSC, pns = 0.28; Figure 4C) and IL-6 (5.43 pg/ml ± 3.02, n = 10 for MSC vs. 7.65 pg/ml ± 5.04, n = 8 for KO MSC, pns > 0.99; Figure 4D) in coronary effluents collected during reperfusion. There was no difference in the TNFα levels among groups (pns = 0.60; Figure 4E).
Figure 4.

(A) Isolated hearts perfused ex vivo on the Langendorff system were submitted to perfusion protocol similar to that described in Figure 1A. Reperfusion was achieved with a Tyrode solution alone in the IR group or with a Tyrode solution prepared at a concentration of 5,000 cells/ml with MSC (MSC group) and with KO MSC (KO MSC group). At the end of the protocol, infarct size analysis was performed on the isolated heart (B) and cytokine production was analyzed (C–E) using the Meso Scale Discovery (MSD) V-Plex Plus Proinflammatory Panel 1 (mouse) kit. Statistical analysis was performed using the Kruskal-Wallis test with the Dunn's post test. (B) Scatter plots and bars (mean ± SD) were represented for infarct size (in % of LV) in IR (n = 12), MSC (5,000 cells/ml, n = 9), and KO MSC (5,000 cells/ml, n = 13). Statistical significance is noted ** for p = 0.001 (MSC vs IR), * for p = 0.029 (KO MSC vs. IR), and ns for p = 0.075 (KO MSC vs. MSC). (C–E) Scatter plots with bars (mean ± SD) are presented for quantification of TNFα, CXCL1, and IL-6 within coronary effluents collected at 60 min after the onset of reperfusion from untreated hearts (IR), hearts treated with wild-type MSC (MSC) and MSC deficient for PPARβ/δ (KO MSC) using the Meso Scale Discovery kit. Statistical analysis was performed using the Kruskal-Wallis test. (C) For CXCL1 (pg/ml), ** was noted for p = 0.0017 (MSC vs. IR), ns was noted for p = 0.34 (KO MSC vs. IR) and for p = 0.28 for (KO MSC vs. MSC); (D) For IL-6 (pg/ml), ** was noted for p = 0.0021 (MSC vs. IR), * for p = 0.028 (KO MSC vs. IR), and ns for p > 0.999 (KO MSC vs. MSC); (E) for TNFα (pg/ml), ns was noted for p = 0.60.
Discussion
Our study evaluated, for the first time, the role of PPARβ/δ in MSC-induced cardioprotective effects in an ex vivo mouse model of myocardial IR injury. The rationale comes from data recently reported from our laboratory showing a pivotal role of these receptors for the immunoregulatory and therapeutic functions of MSC in an experimental model of arthritis (28). We first demonstrated that the ex vivo protocol of global ischemia (30 min) followed by 60 min-reperfusion used in that study induced a proinflammatory response assessed by an increase in the level of TNFα, IL-6, and CXCL1 in coronary effluents collected at 60 min of reperfusion. We then identified the optimal dose of MSC, 6.105 cells/heart, to provide significant cardioprotection in the ex vivo mouse model with minimal cell concentration. Of note, at this selected dose of MSC, we observed similar cardioprotection to that of ischemic postconditioning (PostC) taken as a positive control in our study. Indeed, both treatments reduced the pro-inflammatory response of IR injury and decreased infarct size with the same efficacy. Moreover, this study revealed that the acute cardioprotective properties of MSC during the first hour of reperfusion are PPARβ/δ-dependent but not related to their anti-inflammatory effects on the release of CXCL1 and IL-6 in coronary effluents.
Numerous studies have demonstrated in isolated perfused hearts (ex vivo) the cardioprotective effect of PostC or various pharmacologic drugs applied at the onset of reperfusion. Considering most in vivo studies, the protocol is quite similar since PostC or protective drugs are applied at the onset of reperfusion and infarct size measured often at 1 h reperfusion, an effect maintained at 24 h or at several months of reperfusion (39). This means that major local events occur very early during reperfusion. These rapid local events, which are triggered during reperfusion, include inflammatory processes and/or paracrine effects of secreted molecules such as cytokines by endogenous or exogenous cells. The ex vivo model is well-suited to identify these pathways, as short-term protection is critical for long term protection.
Despite its benefical effects in AMI patients, reperfusion therapy induces local and systemic inflammation, termed sterile inflammation, which will enhance the initial inflammatory response triggered to clear necrotic cells after AMI and to repair the infarcted myocardium. To develop and evaluate innovative therapeutic approaches, we designed an ex vivo model that provides strong IR injury assessed by 56% infarct size (expressed as percentage of the LV) and the release of pro-inflammatory cytokines at significant levels in coronary effluents, as reported in vivo in patients with acute coronary syndrome (40). This inflammatory response is a potential therapeutic target to improve the post-AMI clinical state because it plays a crucial role in determining infarct size and subsequent left ventricular remodeling. To protect the myocardium, many therapeutic strategies targeting indirectly or directly the proinflammatory response after AMI have been tested. In this context, approaches to decrease the levels of inflammatory cytokines and chemokines in animal models have been developed and used with promising results regarding the control of inflammation (2). However, most clinical trials based on the use of broad-spectrum immunosuppressive drugs such as corticosteroids or immunosuppressants in AMI patients have shown no benefit on infarct size or clinical outcomes (2). Since rapid activation of this inflammatory response is a consequence of an inappropriate activation of the innate immune system triggered by DAMPs release, targeting key components of the innate immune system appeared as a promising approach for limiting IR injury (41). Thus, more refined strategies have been proposed instead of fully suppressing the immune system. In this context, promising results have been obtained although they have been mitigated by negative results as reported for inhibitors of IL-1 (42, 43). Targeting inflammation in AMI patients is quite challenging because innate immunity has been reported to contribute to myocardial repair and remodeling (44). This makes it difficult to determine the right dose and timing of administration of therapeutic agents to avoid compromising innate immunity-induced cardiac repair. Mesenchymal stem cells-based therapy in this context appears as an interesting strategy in creating local inflammation permissive to regeneration.
Ischemic postconditioning, considered the gold standard of cardioprotection against IR injury, has been reported to activate a myriad of intracellular cascades leading to inhibition of regulated cell death and also an anti-inflammatory response to induce a strong cardioprotective effect at 1 h of reperfusion (9). In the ex vivo model of IR injury used in the present study, the PostC cardioprotective strategy reduced both infarct size by 75.1% (PostC vs. IR, p*) and IL-6 and CXCL1 levels by 71.8 and 91.4% compared with IR, respectively, in coronary effluents from isolated hearts. Taken together, these results suggest that regulation of the inflammatory response is associated with cardioprotection against IR injury mediated by PostC.
Therefore, global therapies such as PostC, have emerged as promising approaches to treat multifaceted ischemic heart disease and restore cardiac function. Indeed, considering the pleiotropic effects of MSC that include their immunoregulatory, antifibrotic, and anti-apoptotic capabilities, MSC-based therapy could counteract the three main pathogenic axes of AMI and thus have been considered a breakthrough in this incurable disease with unmet medical needs. Safety and efficacy were evaluated by assessment of adverse events and the improvement of the left ventricular ejection fraction (LVEF) and mortality rate, respectively. Although the results obtained were marked by significant heterogeneity, MSC injection did not appear to be associated with acute adverse events but induced an improvement in LVEF in patients. No significant differences in mortality were reported. Other outcomes of interest were rarely studied, which limits the conclusions. In our study, MSC exerted a cardioprotective effect similar to that provided by PostC, considered a positive control in our study. Indeed, infarct size was decreased by 69.3% using MSC-based cell therapy (PostC vs. MSC, p = ns). In addition, MSC treatment (6.105 cells/heart) had a strong anti-inflammatory effect, as assessed by MSD quantification of IL-6 and CXCL1, which were decreased by 75.1 and 84.6%, respectively.
In this context, we tested the therapeutic role of MSC knockdown for PPARβ/δ previously described by our laboratory to exhibit an enhanced ability to inhibit both T-cell proliferation, in vitro, and arthritic development and progression in CIA in vivo compared with naive MSC (28). When primed with TNFα and IFNγ, MSC deficient for PPARβ/δ express increased levels of mediators of MSC immunosuppression, including VCAM-1, ICAM-1, and nitric oxide (NO), compared with their wild-type counterparts (28).
In the present study, we observed that knockdown of PPARβ/δ in MSC did not alter their anti-inflammatory properties when injected into the infarcted myocardium. Indeed, both wild-type and PPARβ/δ KO MSC significantly decreased pro-inflammatory cytokine levels within coronary effluents after 60 min of reperfusion after an ischemic insult. Therefore, the loss of MSC therapeutic effect in the myocardium subjected to IR injury reported here for PPARβ/δ KO MSC could be attributed to an impairment of other functions of MSC pivotal for their beneficial effect in AMI or to a reduction of their survival in perfused hearts. We have recently shown that PPARβ/δ modulation impacts on MSC metabolism (45). Indeed, inactivation of PPARβ/δ promoted the metabolic switch of MSC from oxidative phosphorylation to glycolysis (45). This is in agreement with the role of PPARβ/δ previously described in energy-demanding cells and tissues in which PPARβ/δ promotes fatty acid oxidation that leads to increased ATP production contributing, not only to cell survival but also to cell protection and maintenance of its function (46).
Therefore, it is tempting to anticipate that PPARβ/δ deficiency in MSC could affect their survival rate and reduce their engraftment once injected in vivo since PPARβ/δ has been described to promote survival of several cell types, including cancer cells and cardiomyocytes (47, 48) [for review see (49)]. In endothelial cells, anti-apoptotic role of PPARβ/δ has been reported and the underlying mechanisms were related to endothelial 14-3-3 upregulation (50, 51). Similarly, in the cardiomyoblast cell line H9c2, activation of PPARβ/δ by the selective agonist, GW501516, was described to protect the heart from H2O2-induced cell death (52). PPARβ/δ is highly expressed by MSC (28) but its role on MSC anti-apoptotic and cardioprotective properties has never been investigated. Moreover, further studies are required to identify key mediators regulated by PPARβ/δ and involved in the acute cardioprotective effect of MSC.
In conclusion, our study shows for the first time that PPARβ/δ plays a key role in the acute local cardioprotective effect of MSC against myocardial IR injury ex vivo. Moreover, PPARβ/δ knockdown does not affect the anti-inflammatory properties of MSC assessed in isolated hearts after 1 h of reperfusion. Altogether, these results highlithing the crucial role of PPARβ/δ in MSC cardioprotective properties pave the way toward the development of novel strategies for MSC-based therapy for AMI patients. Further in vivo studies will be required to evaluate the contribution of the peripheral immune system to the inflammatory response of IR injury and the subsequent cardiac remodeling.
Study Limitation
This study had some limitations because the coronary flow was not assessed in our ex vivo experiments performed on a conventional Langendorff system (glass coils and tubes) in contrast to previous studies in our laboratory performed with a fully integrated system (discarded here because cells adhered to the long polyethylene tubes) (53, 54). This technical limitation could introduce biases in the quantification of cytokines in the perfusates because the concentration of circulating molecules depends on the coronary flow.
To interpret results on Figure 1C, we can refer to data from the literature obtained with the same ex vivo model of 30-min global ischemia followed by 60 min of reperfusion showing that coronary flow at 60 min of reperfusion is about 80% of baseline (55). In this context, the decrease in coronary flow at 60 min of reperfusion could contribute to the increase in the measured levels of cytokines in the perfusates compared with basal conditions. Our interpretation was toward an increased release of proinflammatory cytokines in our IR model, consistent with the widely reported pro-inflammatory effects of IR injury (2) even in vivo in patients with acute coronary syndrome (40). Considering the results presented in Figure 3C, we observed decreased amounts of cytokines (CXCL1 and IL-6) in the PostC condition vs. IR, whereas Maruyama et al. have reported similar values of coronary flow values at 60 min of reperfusion in both IR and PostC conditions (55). On the basis of these results, we can therefore suggest that the decrease in cytokine levels at IR60 observed after a PostC (see Figure 3C) may result mainly from a decrease in the release of cytokines vs. IR, in accordance with the anti-inflammatory effect of PostC described in vivo (10, 56). However, we cannot exclude an improvement in the coronary flow upon PostC, as described for many other cardioprotective strategies evaluated in our laboratory (53, 54) or others (57), which could also contribute to the reduction in measured amounts of cytokines.
Considering MSC effects (Figures 3, 4), the authors are confident in their interpretation of the results showing a decrease in cytokine release vs. IR, consistent with the anti-inflammatory effect of MSC widely described in the literature (14, 15). Furthermore, these results are corroborated by those showing reduced levels of IL-6 in the serum of pigs subjected to myocardial IR injury in vivo (58) and, more importantly, by other data showing decreased protein production and gene expression of inflammatory cytokines (including IL-6) that are not dependent on the coronary flow (20). In addition, it can be assumed that the coronary flow might be decreased when MSC are administered during reperfusion because of their relatively large size in small-diameter coronary vessels, as suggested by in vivo data showing microvascular obstruction after intracoronary injection of MSC (59–61), especially with high doses of MSC (44.106 cells) leading to an increase in intravascular resistance, a subsequent decrease in coronary flow, and also to microinfarction (62, 63). However, this deleterious effect does not seem to occur here in our experiments because (i) the mean values of cytokine levels assessed in the MSC group were close to those obtained for PostC (Figures 3B,C), (ii) the dose of MSC was relatively low (6.105 cells/heart), and (iii) a drastic decrease in infarct size was observed upon MSC treatment (Figures 2B,C, 4B).
Overall, despite the absence of coronary flow assessment, the results on decreased cytokine concentration presented here seem to be related to the anti-inflammatory effects of PostC and MSC treatment against IR injury.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Studies involving animals were reviewed and approved by the Institute's SBEA (Structure Bien-être Animal) committee in accordance with the European directive 2010/63/EU and the French Ministerial Order of February 01, 2013.
Author Contributions
FD and SB-L designed the all project and the experiments. CJ, CP, JN, and AV contributed to design the experimental protocols and interpret the data for the work. CS, CB, GT, RC, PL-C, and NN performed the experiments and analyzed the results. FD and SB-L wrote the manuscript with the input of CJ, JN, AV, SK, and CP. All authors revised and gave final approval of the manuscript.
Funding
This work was supported by Inserm, the University of Montpellier (FD, SB-L, and CJ) and by the CNRS (SB-L). We thank the Fonds Marion et Elisabeth Brancher for the financial support of this project (CS) and also the PHC program of Campus France (project number 33858WM; SK and SB-L) as well as the Agence Nationale de Recherche for the LabEx ICST ANR (ANR-11-LABX-0015; SB-L, JN, AV, CB, and CP) and for the PPAROA ANR (ANR-18-CE18-0010-02; FD, CJ, and RC) grants, SATT AxLR n°19/0150 contract, the ECOS-Sud (action ECOS n°C18S03) and La Fondation Arthritis. We also thank the University of Naresuan for the staff development travel grant (NN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Biocampus facilities and in particular, Julien Cau and Amélie Sarrazin for assistance with microscopy (MRI), Laura Gallot for her expertise in ex vivo experiments, Nelly Pirot for histological preparations (RHEM) as well as Denis Greuet, Luc Forichon and Karim Mesbah for animal care (iExplore/RAM).
References
- 1.Roth G, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study (2017). Lancet. (2018) 392:1736–88. 10.1016/S0140-6736(18)32203-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: M33ultiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. 10.1016/j.pharmthera.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. (1999) 99:546–51. 10.1161/01.CIR.99.4.546 [DOI] [PubMed] [Google Scholar]
- 4.Sharma HS, Das DK. Role of cytokines in myocardial ischemia and reperfusion. Mediators Inflamm. (1997) 6:175–83. 10.1080/09629359791668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K, Wen S, Jiao J, Tang T, Zhao X, Zhang M, et al. IL-21 promotes myocardial ischaemia/reperfusion injury through the modulation of neutrophil infiltration. Br J Pharmacol. (2018) 175:1329–43. 10.1111/bph.13781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo HM, Lai TH, Li CH, Wu WB. TNF-α induces CXCL1 chemokine expression and release in human vascular endothelial cells in vitro via two distinct signaling pathways. Acta Pharmacol Sin. (2014) 35:339–50. 10.1038/aps.2013.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heusch G. Treatment of myocardial ischemia/reperfusion injury by ischemic and pharmacological postconditioning. Compr Physiol. (2015) 5:1123–45. 10.1002/cphy.c140075 [DOI] [PubMed] [Google Scholar]
- 8.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. (2003) 285:H579–88. 10.1152/ajpheart.01064.2002 [DOI] [PubMed] [Google Scholar]
- 9.Roubille F, Franck-Miclo A, Covinhes A, Lafont C, Cransac F, Combes S, et al. Delayed postconditioning in the mouse heart in vivo. Circulation. (2011) 124:1330–6. 10.1161/CIRCULATIONAHA.111.031864 [DOI] [PubMed] [Google Scholar]
- 10.Zhao ZQ, Vinten-Johansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res. (2006) 70:200–11. 10.1016/j.cardiores.2006.01.024 [DOI] [PubMed] [Google Scholar]
- 11.Lefer DJ, Marban E. Is cardioprotection dead? Circulation. (2017) 136:98–109. 10.1161/CIRCULATIONAHA.116.027039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossello X, Yellon DM. Cardioprotection: the disconnect between bench and bedside. Circulation. (2016) 134:574–5. 10.1161/CIRCULATIONAHA.116.022829 [DOI] [PubMed] [Google Scholar]
- 13.Hausenloy DJ, Garcia-Dorado D, Botker HE, Davidson SM, Downey J, Engel FB, et al. Novel targets and future strategies for acute cardioprotection: position paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res. (2017) 113:564–85. 10.1093/cvr/cvx049 [DOI] [PubMed] [Google Scholar]
- 14.Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. (2015) 116:1413–30. 10.1161/CIRCRESAHA.116.303614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanina C, Hare JM. Mesenchymal stem cells as a biological drug for heart disease: where are we with cardiac cell-based therapy? Circ Res. (2015) 117:229–33. 10.1161/CIRCRESAHA.117.306306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong H, Yim HW, Park HJ, Cho Y, Hong H, Kim NJ, et al. Mesenchymal stem cell therapy for ischemic heart disease: systematic review and meta-analysis. Int J Stem Cells. (2018) 11:1–12. 10.15283/ijsc17061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodayari S, Khodayari H, Amiri AZ, Eslami M, Farhud D, Hescheler J, et al. Inflammatory microenvironment of acute myocardial infarction prevents regeneration of heart with stem cells therapy. Cell Physiol Biochem. (2019) 53:887–909. 10.33594/000000180 [DOI] [PubMed] [Google Scholar]
- 18.Hénon P. Key Success factors for regenerative medicine in acquired heart diseases. Stem Cell Rev Rep. (2020) 16:441–58. 10.1007/s12015-020-09961-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Chen L, Zeng C, Wang WE. Functionally improved mesenchymal stem cells to better treat myocardial infarction. Stem Cells Int. (2018) 2018:7045245. 10.1155/2018/7045245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J, Zheng D, Li WF, Li HR, Zhang AD, Li ZC. Insulin-like growth factor 1 treatment of MSCs attenuates inflammation and cardiac dysfunction following MI. Inflammation. (2014) 37:2156–63. 10.1007/s10753-014-9949-3 [DOI] [PubMed] [Google Scholar]
- 21.Wagner KD, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol Ther. (2010) 125:423–35. 10.1016/j.pharmthera.2009.12.001 [DOI] [PubMed] [Google Scholar]
- 22.Xin X, Yang S, Kowalski J, Gerritsen ME. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem. (1999) 274:9116–21. 10.1074/jbc.274.13.9116 [DOI] [PubMed] [Google Scholar]
- 23.Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol. (2000) 129:823–34. 10.1038/sj.bjp.0703149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J Biol Chem. (1999) 274:17042–8. 10.1074/jbc.274.24.17042 [DOI] [PubMed] [Google Scholar]
- 25.Adhikary T, Wortmann A, Schumann T, Finkernagel F, Lieber S, Roth K, et al. The transcriptional PPARβ/δ network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res. (2015) 43:5033–51. 10.1093/nar/gkv331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. 10.1016/j.cmet.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7:496–507. 10.1016/j.cmet.2008.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luz-Crawford P, Ipseiz N, Espinosa-Carrasco G, Caicedo A, Tejedor G, Toupet K, et al. PPARbeta/delta directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann Rheum Dis. (2016) 75:2166–74. 10.1136/annrheumdis-2015-208696 [DOI] [PubMed] [Google Scholar]
- 29.Chen SL, Fang WW, Qian J, Ye F, Liu YH, Shan SJ, et al. Improvement of cardiac function after transplantation of autologous bone marrow mesenchymal stem cells in patients with acute myocardial infarction. Chin Med J (Engl). (2004) 117:1443–8. [PubMed] [Google Scholar]
- 30.Gao LR, Chen Y, Zhang NK, Yang XL, Liu HL, Wang ZG, et al. Intracoronary infusion of Wharton's jelly-derived mesenchymal stem cells in acute myocardial infarction: double-blind, randomized controlled trial. BMC Med. (2015) 13:162. 10.1186/s12916-015-0399-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao LR, Pei XT, Ding QA, Chen Y, Zhang NK, Chen HY, et al. A critical challenge: dosage-related efficacy and acute complication intracoronary injection of autologous bone marrow mesenchymal stem cells in acute myocardial infarction. Int J Cardiol. (2013) 168:3191–9. 10.1016/j.ijcard.2013.04.112 [DOI] [PubMed] [Google Scholar]
- 32.Houtgraaf JH, den Dekker WK, van Dalen BM, Springeling T, de Jong R, van Geuns RJ, et al. First experience in humans using adipose tissue-derived regenerative cells in the treatment of patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. (2012) 59:539–40. 10.1016/j.jacc.2011.09.065 [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Cho JH, Lee YH, Lee JH, Kim SS, Kim MY, et al. Improvement in left ventricular function with intracoronary mesenchymal stem cell therapy in a patient with anterior wall ST-segment elevation myocardial infarction. Cardiovasc Drugs Ther. (2018) 32:329–38. 10.1007/s10557-018-6804-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, Lee SH, Youn YJ, Ahn MS, Kim JY, Yoo BS, et al. A randomized, open-label, multicenter trial for the safety and efficacy of adult mesenchymal stem cells after acute myocardial infarction. J Korean Med Sci. (2014) 29:23–31. 10.3346/jkms.2014.29.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musialek P, Mazurek A, Jarocha D, Tekieli L, Szot W, Kostkiewicz M, et al. Myocardial regeneration strategy using Wharton's jelly mesenchymal stem cells as an off-the-shelf 'unlimited' therapeutic agent: results from the acute myocardial infarction first-in-man study. Postepy Kardiol Interwencyjnej. (2015) 11:100–7. 10.5114/pwki.2015.52282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Xi WC, Wang F. The beneficial effects of intracoronary autologous bone marrow stem cell transfer as an adjunct to percutaneous coronary intervention in patients with acute myocardial infarction. Biotechnol Lett. (2014) 36:2163–8. 10.1007/s10529-014-1589-z [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, Zhang F, Ma W, Chen B, Zhou F, Xu Z, et al. A novel approach to transplanting bone marrow stem cells to repair human myocardial infarction: delivery via a noninfarct-relative artery. Cardiovasc Ther. (2010) 28:380–5. 10.1111/j.1755-5922.2009.00116.x [DOI] [PubMed] [Google Scholar]
- 38.Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, et al. PPARβ/δ governs Wnt signaling and bone turnover. Nat Med. (2013) 19:608–13. 10.1038/nm.3146 [DOI] [PubMed] [Google Scholar]
- 39.Covinhes A, Gallot L, Barrère C, Vincent A, Sportouch C, Piot C, et al. Anti-apoptotic peptide for long term cardioprotection in a mouse model of myocardial ischemia-reperfusion injury. Sci Rep. (2020) 10:18116. 10.1038/s41598-020-75154-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kristono GA, Holley AS, Lakshman P, Brunton-O'Sullivan MM, Harding SA, Larsen PD. Association between inflammatory cytokines and long-term adverse outcomes in acute coronary syndromes: a systematic review. Heliyon. (2020) 6:e03704. 10.1016/j.heliyon.2020.e03704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panahi M, Papanikolaou A, Torabi A, Zhang JG, Khan H, Vazir A, et al. Immunomodulatory interventions in myocardial infarction and heart failure: a systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc Res. (2018) 114:1445–61. 10.1093/cvr/cvy145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol. (2010). 105:1371.e1–7.e1. 10.1016/j.amjcard.2009.12.059 [DOI] [PubMed] [Google Scholar]
- 43.Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J. (2015) 36:377–84. 10.1093/eurheartj/ehu272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuurbier CJ, Abbate A, Cabrera-Fuentes HA, Cohen MV, Collino M, De Kleijn DPV, et al. Innate immunity as a target for acute cardioprotection. Cardiovasc Res. (2019) 115:1131–42. 10.1093/cvr/cvy304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Contreras-Lopez RA, Elizondo-Vega R, Torres MJ, Vega-Letter AM, Luque-Campos N, Paredes-Martinez MJ, et al. PPARβ/δ-dependent MSC metabolism determines their immunoregulatory properties. Sci Rep. (2020) 10:11423. 10.1038/s41598-020-68347-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magadum A, Engel FB. PPARbeta/delta: linking metabolism to regeneration. Int J Mol Sci. (2018). 19:2013. 10.3390/ijms19072013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li YJ, Sun L, Shi Y, Wang G, Wang X, Dunn SE, et al. PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia. (2017) 31:1905–14. 10.1038/leu.2016.395 [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Wang G, Shi Y, Sun L, Gorczynski R, Li YJ, et al. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis. (2016) 5:e232. 10.1038/oncsis.2016.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palomer X, Barroso E, Zarei M, Botteri G, Vázquez-Carrera M. PPARβ/δ and lipid metabolism in the heart. Biochim Biophys Acta. (2016) 1861:1569–78. 10.1016/j.bbalip.2016.01.019 [DOI] [PubMed] [Google Scholar]
- 50.Liou JY, Lee S, Ghelani D, Matijevic-Aleksic N, Wu KK. Protection of endothelial survival by peroxisome proliferator-activated receptor-delta mediated 14-3-3 upregulation. Arterioscler Thromb Vasc Biol. (2006) 26:1481–7. 10.1161/01.ATV.0000223875.14120.93 [DOI] [PubMed] [Google Scholar]
- 51.Brunelli L, Cieslik KA, Alcorn JL, Vatta M, Baldini A. Peroxisome proliferator-activated receptor-delta upregulates 14-3-3 epsilon in human endothelial cells via CCAAT/enhancer binding protein-beta. Circ Res. (2007) 100:e59–71. 10.1161/01.RES.0000260805.99076.22 [DOI] [PubMed] [Google Scholar]
- 52.Pesant M, Sueur S, Dutartre P, Tallandier M, Grimaldi PA, Rochette L, et al. Peroxisome proliferator-activated receptor delta (PPARdelta) activation protects H9c2 cardiomyoblasts from oxidative stress-induced apoptosis. Cardiovasc Res. (2006) 69:440–9. 10.1016/j.cardiores.2005.10.019 [DOI] [PubMed] [Google Scholar]
- 53.Boisguerin P, Covinhes A, Gallot L, Barrere C, Vincent A, Busson M, et al. A novel therapeutic peptide targeting myocardial reperfusion injury. Cardiovasc Res. (2020) 116:633–44. 10.1093/cvr/cvz145 [DOI] [PubMed] [Google Scholar]
- 54.Vincent A, Covinhes A, Barrere C, Gallot L, Thoumala S, Piot C, et al. Acute and long-term cardioprotective effects of the Traditional Chinese Medicine MLC901 against myocardial ischemia-reperfusion injury in mice. Sci Rep. (2017) 7:14701. 10.1038/s41598-017-14822-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maruyama Y, Chambers DJ. Ischaemic postconditioning: does cardioplegia influence protection? Eur J Cardiothorac Surg. (2012) 42:530–9. 10.1093/ejcts/ezr305 [DOI] [PubMed] [Google Scholar]
- 56.Zhang XY, Huang Z, Li QJ, Zhong GQ, Meng JJ, Wang DX, et al. Ischemic postconditioning attenuates the inflammatory response in ischemia/reperfusion myocardium by upregulating miR-499 and inhibiting TLR2 activation. Mol Med Rep. (2020) 22:209–18. 10.3892/mmr.2020.11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong LY, Qiu XX, Zhuang Y, Xue S. Y-27632, a Rho-kinase inhibitor, attenuates myocardial ischemia-reperfusion injury in rats. Int J Mol Med. (2019) 43:1911–9. 10.3892/ijmm.2019.4097 [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Chen Z, Dai Q, Zhao J, Wei Z, Hu J, et al. Intravenously delivered mesenchymal stem cells prevent microvascular obstruction formation after myocardial ischemia/reperfusion injury. Basic Res Cardiol. (2020) 115:40. 10.1007/s00395-020-0800-8 [DOI] [PubMed] [Google Scholar]
- 59.Eggenhofer E, Luk F, Dahlke MH, Hoogduijn MJ. The life and fate of mesenchymal stem cells. Front Immunol. (2014) 5:148. 10.3389/fimmu.2014.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grieve SM, Bhindi R, Seow J, Doyle A, Turner AJ, Tomka J, et al. Microvascular obstruction by intracoronary delivery of mesenchymal stem cells and quantification of resulting myocardial infarction by cardiac magnetic resonance. Circ Heart Fail. (2010) 3:e5–6. 10.1161/CIRCHEARTFAILURE.109.931360 [DOI] [PubMed] [Google Scholar]
- 61.Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. (2009) 104:398–402. 10.1161/CIRCRESAHA.108.187724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fiarresga A, Mata MF, Cavaco-Gonçalves S, Selas M, Simões IN, Oliveira E, et al. Intracoronary delivery of human mesenchymal/stromal stem cells: insights from coronary microcirculation invasive assessment in a swine model. PLoS ONE. (2015) 10:e0139870. 10.1371/journal.pone.0139870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong SJ, Hou D, Brinton TJ, Johnstone B, Feng D, Rogers P, et al. Intracoronary and retrograde coronary venous myocardial delivery of adipose-derived stem cells in swine infarction lead to transient myocardial trapping with predominant pulmonary redistribution. Catheter Cardiovasc Interv. (2014) 83:E17–25. 10.1002/ccd.24659 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.