

Abstract
Dietary flaxseed (FS) and its components including FS oil (FSO), secoisolariciresinol diglucoside (SDG) and fiber, are processed by the gut microbiota. These data are in support of the article entitled “Discriminatory and cooperative effects within the mouse gut microbiota in response to flaxseed and its oil and lignan components”, Journal of Nutritional Biochemistry [1]. Here we describe data generated by 16S rRNA sequencing of DNA obtained from cecum contents and feces of C57BL/6 female mice fed either a basal diet (BD, AIN93G), or isocaloric diets containing 10% FS, or 10% FS-equivalent amounts of FSO or SDG for 21 days. These include bacterial community composition and inferred KEGG pathways; the raw data are publicly available at the NCBI SRA database (BioProject ID PRJNA683934). Furthermore, this work includes detailed experimentation procedures, total bacterial counts (qPCR) in the cecum content and feces, and correlation analysis between a selected bacterial genus, Bacteroides and a predicted metabolic pathway. FS is utilized worldwide, especially for the prevention and/or treatment of diseases including cardiovascular diseases, diabetes and cancer. These data will be valuable as a reference to study different FS cultivars and SDG- or FSO- enriched products on the gut microbiota, to study gut microbial responses to FS and its components in different mouse strains and mammalian hosts to elucidate individualized effects, and to understand the importance of the gut microbiota for FS benefits.
Keywords: Flaxseed, Flaxseed oil, SDG, Cecal microbiota, Female mice, Fecal microbiota, KEGG pathways
Specifications Table
| Subject | Microbiology: Microbiome |
| Specific subject area | Nutrition, Gut Ecology |
| Type of data | Table Graph Figure FASTQ sequence files from 16S rRNA gene sequencing using primers for variable regions V3-V4. |
| How data were acquired | High-throughput 16S rRNA gene sequencing (pair-end; Illumina MiSeq), quantitative PCR Instruments: Illumina MiSeq™ 500 sequencing system, Applied Biosystems 7900HT Fast Real-Time PCR system |
| Data format | Raw Analyzed Filtered |
| Parameters for data collection | C57BL/6 female mice (4-5 weeks of age) were randomized to either Basal Diet (BD, AIN93G), 10% FS, 3.67% FSO, or 0.15% SDG isocaloric diets for 21 days. After the intervention, the mice were sacrificed and feces and cecum contents were collected for DNA extraction; bacterial quantification, library preparation; sequencing; data processing, and functional predictions |
| Description of data collection | Total DNA was extracted from cecum content and feces collected from the four intervention groups. The V3-V4 16S rRNA library preparation and sequencing were performed following Illumina protocol using the Illumina® MiSeq platform. Data processing was performed using the QIIME V1.8.9 pipeline. Metagenomic predictions were completed using PICRUSt. Quantitative PCR (qPCR) was used to determine total bacterial counts. |
| Data source location | Institution: University of Toronto City/Town/Region: Toronto, Ontario Country: Canada Latitude and longitude (and GPS coordinates, if possible) for collected samples/data: 43.6629° N, 79.3957° W |
| Data accessibility | The data are with the article (qPCR data) and in a public repository (raw 16S rRNA gene amplicon sequences). Repository name: [National center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database] Data identification number: [PRJNA683934] Direct URL to data: [https://www.ncbi.nlm.nih.gov/sra/PRJNA683934] |
| Related research article | co-submission: Amel Taibi, Michelle Ku, Zhen Lin, Giorgio Gargari, Alla Kubant, Dion Lepp, Krista A. Power, Simone Guglielmetti, Lilian U. Thompson and Elena M. Comelli. Discriminatory and cooperative effects within the mouse gut microbiota in response to flaxseed and its oil and lignan components. J Nutr Biochem. 2021;98:108818. |
Value of the Data
-
•
These data provide the first analysis of the gut microbiota in response to flaxseed as a whole food or to its isolated oil and SDG. This is of importance because the microbiota is responsible for at least partially processing FS and its components and the generation of beneficial metabolites. Understanding the effects of FS and its components on the microbiota will help deciphering the mechanisms behind its role in preventative and therapeutic interventions.
-
•
These data will serve as an important reference to study different cultivars of FS and different products containing varying amounts of FSO and SDG. The data may also help scientists and clinical practitioners in developing a gut microbiota-targeted dietary intervention for personalized treatment and specific health outcomes.
-
•
The data can be used for multi-omics analyses for studying microbiome relationships at different intestinal sites and to identify microbial biomarkers to predict the effectiveness of dietary interventions or to develop a specific therapeutic target.
1. Data Description
The data presented in this paper were obtained from female mice receiving either a control basal diet (BD, AIN93G) or a daily dose of 10% FS or 10%-FS equivalent amounts of FSO and SDG for 21 days. Fig. 1 provides a visual representation of these experimental diets.
Fig. 1.

Visual representation of the four experimental diets.
The dietary components are represented as a percentage. Calculation of caloric contents and origin of the dietary components are provided in [1,2].
BD: Basal Diet; FS: Flaxseed; FSO: Flaxseed Oil; SDG: Secoisolariciresinol diglucoside.
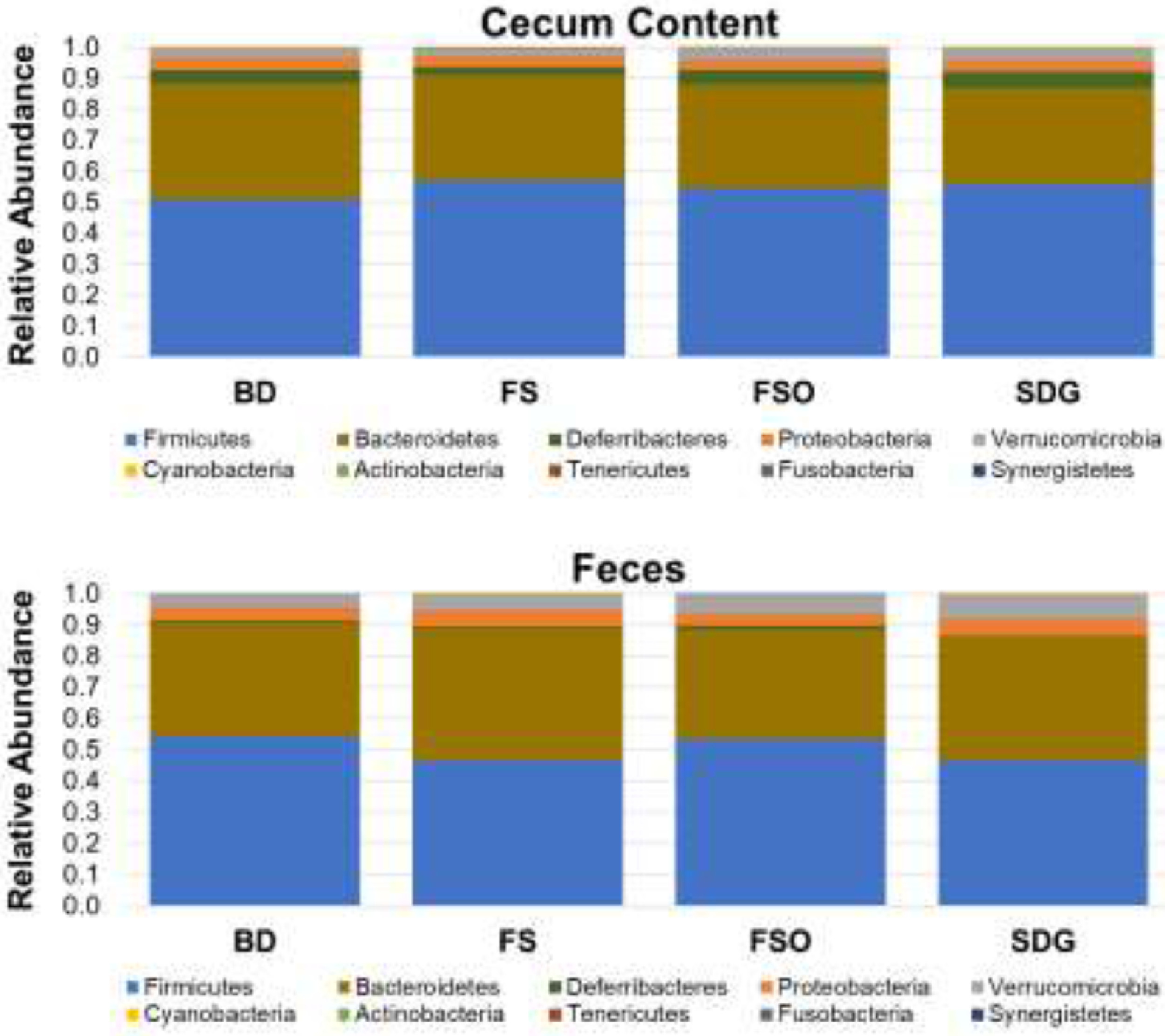
Microbiota data presented here include total bacterial counts in the cecal content and feces of mice at the end of the dietary intervention (Fig. 2) and Illumina sequencing data from cecum content and feces. The raw reads have been deposited at the NCBI SRA (https://www.ncbi.nlm.nih.gov/sra/PRJNA683934). After filtration and quality controls, a total of 5,241,470 reads (Mean ± SD = 107,439 ± 36,329; minimum = 27,172, maximum = 195,448) were obtained (Supplementary Table 1) and were clustered into a total of 2291 and 2397 Operational Taxonomic Units (OTUs) for cecum and feces, respectively (Supplementary Tables 2 and 3). The taxonomic classification of OTUs at the phylum level showed that the microbial communities were dominated by members of Firmicutes (average of 55% and 50% in cecum and feces, respectively) and Bacteroidetes (average of 34% and 39% in cecum and feces, respectively). Conversely, members of Verrucomicrobia, Proteobacteria, and Deferribacteres were present at less than 6% in the cecum and feces (Supplementary Fig. 1). At the genus level, Bacteroides, and a genus from the Clostridiales order were the most dominant in both cecum and feces (Supplementary Fig. 2).
Fig. 2.

Total bacterial counts in cecum content and feces after 21 days intervention.
Data are presented as mean ± SEM of log10 copies of total bacteria/g of cecum content (Top, n=8/group) and feces (Bottom, n=8-14/group). Statistical differences were assessed using Kruskal-Wallis test followed by Dunn's post-hoc multiple comparisons test (p < 0.05).
BD: Basal Diet; FS: Flaxseed; FSO: Flaxseed Oil; SDG: Secoisolariciresinol diglucoside.
Fig. 3 shows the effect of the 4 diets on the alpha–diversity of the microbial communities using the Simpson index.
Fig. 3.

Alpha-diversity in response to dietary interventions. Box-plots illustrating Simpson index in cecum content (n=7-8/group) and feces (n=7-14/group). Each dot represents an individual mouse. Statistical differences were determined using the Kruskal-Wallis test and Dunn's post hoc tests. The same letter (a) denotes no significant difference (P > 0.05).
BD: Basal Diet; FS: Flaxseed; FSO: Flaxseed Oil; SDG: Secoisolariciresinol diglucoside.
The OTU tables were used to predict the functional microbial metagenomes across the dietary groups, the lists of inferred metabolic functions and their relative abundance in cecum contents and feces are provided in Supplementary Tables 4 and 5, respectively.
Fig. 4 shows the relationship between cecal content and fecal Bacteroides relative abundance and the Sphingolipid Metabolism Pathway (map00600).
Fig. 4.

Correlation between Bacteroides spp. and sphingolipid metabolism pathway in the cecum content and feces. Significant correlations were determined by Spearman's test (p < 0.05) between Bacteroides spp. relative abundance and sphingolipid metabolism pathway relative abundance in a) cecum (total n=31); b) feces (total n=41).
2. Experimental Design, Materials and Methods
2.1. Animal study and experimental diets
Information on the study design was provided in [1,2]. Here, we include the basic information that is necessary for understanding the origin of the data and additional details integrating the previous description. Briefly, 56 female C57BL/6 mice were randomized into four groups to receive (1) Basal Diet (BD, AIN93G), (2) 10% FS, (3) 3.67% FSO, or (4) 0.15% SDG isocaloric diets (n=14/group) for 21 days. The full description of diet design and preparation is provided in [1,2] and summarized here in Fig. 1. To reiterate, the amount of FS oil present in 10% flaxseed is equivalent to the amount contained in 3.67 % FSO fed to mice in the FSO group, and the amount of SDG present in 10% flaxseed is equivalent to the amount contained in 0.15% SDG fed to mice in the SDG group. Females were studied because FS and its components hold benefits in the context of menopausal symptoms and breast cancer prevention and treatment [1], [2], [3], [4]). On day 21 (D21), freshly passed feces and cecum content were collected and stored at −80°C for DNA extraction as described in [1].
2.2. Bacterial quantification using quantitative PCR (qPCR)
Total bacterial counts in the cecum contents and feces were assessed using the 7900HT Fast Real-Time PCR machine (Applied Biosystems™, Thermo Fisher Scientific). The qPCR was carried out in triplicate in 384 wells optical plates using TaqMan™ Gene Expression Master Mix (Applied Biosystems™, Thermo Fisher Scientific, Foster City, CA, USA), a custom TaqMan® assay targeting the 16S rRNA gene [5], and 10 ng of DNA in a final volume of 10 µl per reaction. Bacterial counts were calculated using a standard curve generated from 10-fold serial dilutions of the constructed pGEM T-Easy-16S rRNA plasmid DNA, as described by Dumonceaux et al. [6]. Data are represented as log10 copies/g of cecum content or feces.
2.3. 16S rRNA gene library preparation, sequencing and data analysis
The extracted DNA was used for library preparation of the 16S rRNA hypervariable region V3-V4 following the Illumina® 16S Metagenomic Sequencing Library Preparation guide (Rev. B.), as described in [7]. Briefly, primers Bakt_341F (5′-CCTACGGGNGGCWGCAG-3′) and Bakt_805R (5′-GACTACHVGGGTATCTAATCC-3′) were used to amplify a 550 bp fragment. The PCR amplification was performed in 25 µl for 25 cycles as described in [7]. The PCR products were purified and ligated with the sequencing adaptors, then loaded on an Illumina® MiSeq and sequenced using the MiSeq 600-cycle V3 kit (Illumina®, San Diego, CA, USA). This work was performed at the Guelph Food Research Centre, Agriculture and Agri-Food Canada, Guelph, ON, Canada.
The resulting FASTQ files, containing 300-bp dual-indexed paired-end reads, were processed using QIIME V1.8.9. The reads were assembled by aligning and overlapping sequences with fastq-join, then quality-filtered. The retained reads were clustered at 97% similarity with uclust [8] and the operational taxonomy units were picked using a closed-reference approach and the GreenGenes database as a reference (gg_otus_13_8) [9]. In addition to alpha-diversity metrics used in [1], we calculated here the Simpson index.
2.4. Prediction of microbial metabolic functions
The microbial functional potential was predicted using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) [10], as described in [1].
2.5. Correlation analysis
Spearman's correlations were performed between Bacteroides genus (relative abundance) and the predicted sphingolipid metabolism pathway (relative abundance) in the cecum content (all groups combined) and feces (all groups combined). The correlations were run using GraphPad Prism 8.4.0 (GraphPad Software, Inc., La Jolla, CA, USA). Significance was set at p < 0.05.
Ethics Statement
All the animal procedures followed the ARRIVE guidelines, the Regulations of the Animals for Research Act in Ontario, and the Guidelines of the Canadian Council on Animal Care. The study was approved by the animal ethics committee of the University of Toronto (Protocol #: 20011734).
CRediT authorship contribution statement
Amel Taibi: Supervision, Data curation, Formal analysis, Writing – original draft. Michelle Ku: Data curation, Formal analysis, Writing – review & editing. Zhen Lin: Visualization, Writing – review & editing. Giorgio Gargari: Data curation, Formal analysis, Writing – review & editing. Alla Kubant: Data curation, Formal analysis, Writing – review & editing. Dion Lepp: Visualization. Krista A. Power: Writing – review & editing. Simone Guglielmetti: Writing – review & editing. Lilian U. Thompson: Conceptualization, Writing – review & editing. Elena M. Comelli: Conceptualization, Writing – original draft.
Declaration of Competing Interest
All funding agencies have been listed in the Acknowledgments section above.
EMC has received research support from Lallemand Health Solutions and Ocean Spray and has received consultant fees or speaker or travel support from Danone, Nestlé, and Lallemand Health Solutions (All are outside of this work). The authors declare that they have no known competing financial interests or personal relationships which have or could be perceived to have influenced the work reported in this article.
Acknowledgments
This project was funded by the Natural Sciences and Engineering Research Council of Canada (Discovery grants- L.U.T, E.M.C, K.A.P). E.M.C holds the Lawson Family Chair in Microbiome Nutrition Research at the University of Toronto. Z.L was the recipient of an NSERC Canada Graduate Scholarship for Master's (CGSM). A.K was the recipient of an Undergraduate Research Opportunity Program (UROP) Award at the University of Toronto.
The authors thank Omega Nutrition Inc. (Vancouver, BC, Canada) for kindly providing the flaxseed and flaxseed oil.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.dib.2021.107409.
Contributor Information
Lilian U. Thompson, Email: lilian.thompson@utoronto.ca.
Elena M. Comelli, Email: elena.comelli@utoronto.ca.
Appendix. Supplementary materials
Supplementary Fig. 1: Relative abundance of phyla in cecum content and feces.
The bar charts show the relative abundance of OTUs at the phylum level detected in the cecum content and feces of female mice after 21-days dietary intervention. Data are mean values of 7–14 mice/group.
BD: Basal Diet; FS: Flaxseed; FSO: Flaxseed Oil; SDG: Secoisolariciresinol diglucoside.
{kind=link}
Supplementary Fig. 2: Relative abundance of the top 30 genera in cecum content and feces.
The bar charts show the relative abundance of OTUs at the genus level detected in the cecum content and feces of female mice after 21-days dietary intervention. Data are mean values of 7-14 mice/group.
BD = Basal Diet; FS = Flaxseed; FSO = Flaxseed Oil; SDG = Secoisolariciresinol diglucoside.
{kind=link}
References
- 1.Taibi A., Ku M., Lin Z., Gargari G., Kubant A., Lepp D. Discriminatory and cooperative effects within the mouse gut microbiota in response to flaxseed and its oil and lignan components. J. Nutr. Biochem. 2021;98 doi: 10.1016/j.jnutbio.2021.108818. [DOI] [PubMed] [Google Scholar]
- 2.Taibi A., Lin Z., Tsao R., Thompson L.U., Comelli E.M. Effects of flaxseed and its components on mammary gland MiRNome: Identification of potential biomarkers to prevent breast cancer development. Nutrients. 2019;11:2656. doi: 10.3390/nu11112656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mason J.K., Thompson L.U. Flaxseed and its lignan and oil components: can they play a role in reducing the risk of and improving the treatment of breast cancer? Appl. Physiol. Nutr. Metab. 2014;39:663–678. doi: 10.1139/apnm-2013-0420. [DOI] [PubMed] [Google Scholar]
- 4.Parikh M., Maddaford T.G., Austria J.A., Aliani M., Netticadan T., Pierce G.N. Dietary flaxseed as a strategy for improving human health. Nutrients. 2019;11:1171. doi: 10.3390/nu11051171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furet J.P., Firmesse O., Gourmelon M., Bridonneau C., Tap J., Mondot S. Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol. Ecol. 2009;68:351–362. doi: 10.1111/j.1574-6941.2009.00671.x. [DOI] [PubMed] [Google Scholar]
- 6.Dumonceaux T.J., Hill J.E., Briggs S.A., Amoako K.K., Hemmingsen S.M., Van Kessel A.G. Enumeration of specific bacterial populations in complex intestinal communities using quantitative PCR based on the chaperonin-60 target. J. Microbiol. Methods. 2006;64:46–62. doi: 10.1016/j.mimet.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Power K.A., Lepp D., Zarepoor L., Monk J.M., Wu W., Tsao R. Dietary flaxseed modulates the colonic microenvironment in healthy C57Bl/6 male mice which may alter susceptibility to gut-associated diseases. J. Nutr. Biochem. 2016;28:61–69. doi: 10.1016/j.jnutbio.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 8.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 9.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langille M.G., Zaneveld J., Caporaso J.G., McDonald D., Knights D., Reyes J.A. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1: Relative abundance of phyla in cecum content and feces.
The bar charts show the relative abundance of OTUs at the phylum level detected in the cecum content and feces of female mice after 21-days dietary intervention. Data are mean values of 7–14 mice/group.
BD: Basal Diet; FS: Flaxseed; FSO: Flaxseed Oil; SDG: Secoisolariciresinol diglucoside.
Supplementary Fig. 2: Relative abundance of the top 30 genera in cecum content and feces.
The bar charts show the relative abundance of OTUs at the genus level detected in the cecum content and feces of female mice after 21-days dietary intervention. Data are mean values of 7-14 mice/group.
BD = Basal Diet; FS = Flaxseed; FSO = Flaxseed Oil; SDG = Secoisolariciresinol diglucoside.