

Abstract
Bacteria resist to the turgor pressure of the cytoplasm through a net‐like macromolecule, the peptidoglycan, made of glycan strands connected via peptides cross‐linked by penicillin‐binding proteins (PBPs). We recently reported the emergence of β‐lactam resistance resulting from a bypass of PBPs by the YcbB L,D‐transpeptidase (LdtD), which form chemically distinct 3→3 cross‐links compared to 4→3 formed by PBPs. Here we show that peptidoglycan expansion requires controlled hydrolysis of cross‐links and identify among eight endopeptidase paralogues the minimum enzyme complements essential for bacterial growth with 4→3 (MepM) and 3→3 (MepM and MepK) cross‐links. Purified Mep endopeptidases unexpectedly displayed a 4→3 and 3→3 dual specificity implying recognition of a common motif in the two cross‐link types. Uncoupling of the polymerization of glycan chains from the 4→3 cross‐linking reaction was found to facilitate the bypass of PBPs by YcbB. These results illustrate the plasticity of the peptidoglycan polymerization machinery in response to the selective pressure of β‐lactams.
Keywords: endopeptidase; Escherichia coli; L,D‐transpeptidase; peptidoglycan; β‐lactam
Subject Categories: Membranes & Trafficking; Microbiology, Virology & Host Pathogen Interaction
Peptidoglycan expansion requires a minimum set of dual specificity endopeptidases that hydrolyse both the most abundant D,D‐transpeptidase crosslinks, but also alternative crosslinks formed by L,D‐transpeptidases.
Introduction
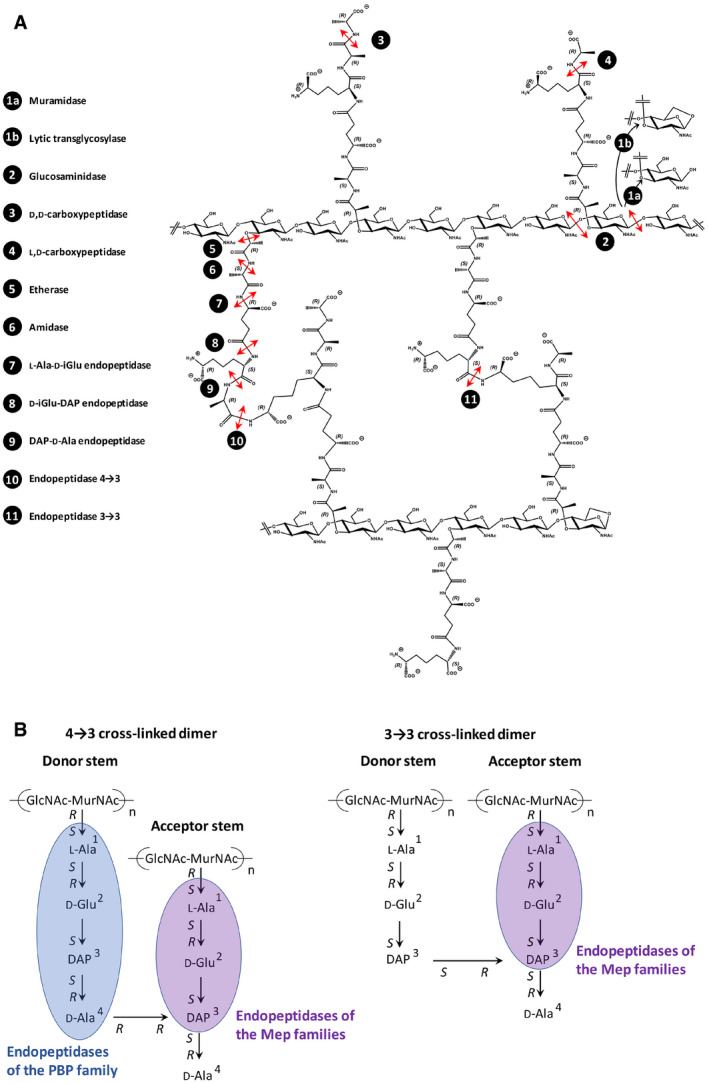
Peptidoglycan (PG) is an essential macromolecule that surrounds the bacterial cell providing resistance to the osmotic pressure of the cytoplasm and determining cell shape (Turner et al, 2014). PG is assembled from a disaccharide peptide subunit consisting of N‐acetylglucosamine (GlcNAc), N‐acetylmuramic acid (MurNAc), and a stem pentapeptide linked to MurNAc (L‐Ala1‐γ‐D‐Glu2‐DAP3‐D‐Ala4‐D‐Ala5 in which DAP is diaminopimelic acid) (Fig 1A). The subunit is assembled by glycosyltransferases that polymerize glycan strands and transpeptidases that form amide bonds between stem peptides linked to adjacent glycan strands. Escherichia coli relies on two types of transpeptidases for the latter reaction (Magnet et al, 2008). The D,D‐transpeptidases, also referred to as penicillin‐binding proteins (PBPs), form the most abundant cross‐links, which connect the fourth residue (D‐Ala4) of an acyl donor to the third residue (DAP3) of an acyl acceptor (4→3 cross‐link) (Fig 1A). The L,D‐transpeptidases (LDTs) form 3→3 cross‐links that connect two DAP residues (Fig 1B). The D‐Ala at the 5th and 4th positions of stem peptides that do not participate in cross‐link formation as donors are fully and partially trimmed by carboxypeptidases of the D,D and L,D specificities, respectively (Fig 1C). PBPs and LDTs are structurally unrelated, rely on different catalytic nucleophiles (Ser versus Cys, respectively), and use different acyl donor stems (pentapeptide versus tetrapeptide, respectively) (Mainardi et al, 2008; Sauvage et al, 2008). PBPs and LDTs also differ by their inhibition profiles since PBPs are potentially inhibited by all classes of β‐lactams (including penams, cephems, monobactams, and carbapenems), whereas LDTs are effectively inhibited only by carbapenems (Mainardi et al, 2005). LDTs are fully dispensable for growth of E. coli, at least in laboratory conditions, and form a minority of the cross‐links during exponential growth (6% of the total cross‐links) (Glauner et al, 1988; Sanders & Pavelka, 2013). The proportion of 3→3 cross‐links is higher in the stationary phase (Pisabarro et al, 1985) and in cells experiencing outer membrane assembly stress (Morè et al, 2019). By‐pass of PBPs by LDTs leads to high‐level resistance to β‐lactams of the penam (such as ampicillin), cephem (ceftriaxone), and monobactam (aztreonam) classes in engineered E. coli strains that overproduce the YcbB L,D‐transpeptidase, also referred to as LdtD, and the guanosine penta‐ and tetra‐phosphate [(p)ppGpp] alarmones (Hugonnet et al, 2016). PG of such strains grown in the presence of β‐lactams exclusively contains 3→3 cross‐links, indicating that the D,D‐transpeptidase activity of PBPs is fully replaced by the L,D‐transpeptidase activity of LDTs.
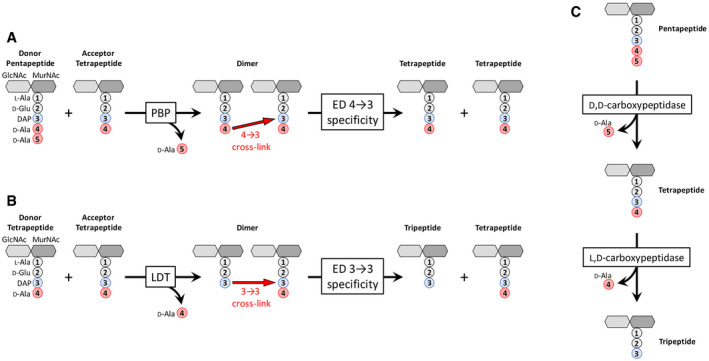
Figure 1. Metabolism of PG cross‐links and maturation of free stem peptides.
-
A, BFormation and hydrolysis of (A) 4→3 and (B) 3→3 cross‐links. The disaccharide pentapeptide unit is assembled from N‐acetylglucosamine (GlcNAc), N‐acetylmuramic acid (MurNAc), and five amino acids including meso‐diaminopimelic acid (DAP), which is linked via its L (S) center to the γ‐carboxyl group of D‐Glu.
-
CHydrolysis of the D‐Ala4‐D‐Ala5 and DAP3‐D‐Ala4 peptide bonds by carboxypeptidases of D,D and L,D specificities, respectively.
Expansion of PG is thought to require highly regulated hydrolytic activities that spatially control the insertion of new subunits into the growing network of cross‐linked glycan strands (Singh et al, 2012; Vollmer, 2012). Due to the covalent net‐like structure of PG, endopeptidases are predicted to be required for insertion of new subunits leading to the expansion of the PG layer (Fig EV1) (De Jonge et al, 1989; Höltje & Heidrich, 2001). The E. coli genome encodes eight endopeptidase paralogues that belong to five enzyme families (Singh et al, 2012; Chodisetti & Reddy, 2019; Pazos & Peters, 2019). PBP4, PBP7, and AmpH belong to the acyl‐serine transferase superfamily, which also comprises D,D‐transpeptidases and D,D‐carboxypeptidases. Members of this superfamily are inhibited by β‐lactam antibiotics. The NIpC/P60 cysteine peptidase family comprises two paralogues (MepH and MepS). Metallo‐enzymes are represented by three enzyme families, LAS metallopeptidases, lysostaphin/M23 peptidases, and M15 peptidases, each contributing one paralogue (MepA, MepM, and MepK, respectively). The specificity of these eight paralogues as endopeptidases or carboxypeptidases (Fig 1) has been explored by using sacculi or purified PG fragments as substrates (Keck & Schwarz, 1979; Korat et al, 1991; Engel et al, 1992; Romeis & Holtje, 1994; Gonzalez‐Leiza et al, 2011; Singh et al, 2012; Chodisetti & Reddy, 2019). PBP4, PBP7, AmpH, MepH, and MepS hydrolyze 4→3 cross‐links but PG dimers containing 3→3 cross‐links were not tested. In contrast, MepA, MepM, and MepK were fully characterized revealing that MepA hydrolyzes both 4→3 and 3→3 cross‐links, MepM is specific to 4→3 cross‐links, and MepK displays a marked preference for 3→3 cross‐links. The endopeptidases of E. coli are redundant, and their essential roles can only be revealed by introducing multiple chromosomal deletions. One study unambiguously showed that hydrolysis of 4→3 cross‐links by endopeptidases is essential as the triple deletion of genes encoding MepH, MepM, and MepS was not compatible with growth of E. coli in laboratory conditions (Singh et al, 2012; comment by Vollmer, 2012). Several endopeptidases interact genetically and physically with the outer membrane‐anchored adaptor protein NlpI supporting overlapping functions during the cell cycle (Banzhaf et al, 2020). Various roles have been assigned to endopeptidases in bacteria other than E. coli such as the determination of cell shape in Helicobacter pylori (Bonis et al, 2010), release of NOD agonist in Neisseria gonorrhoeae (Lenz et al, 2017), or persistence in Mycobacterium tuberculosis (Healy et al, 2020).
Figure EV1. Insertion of PG subunits into the growing PG network.
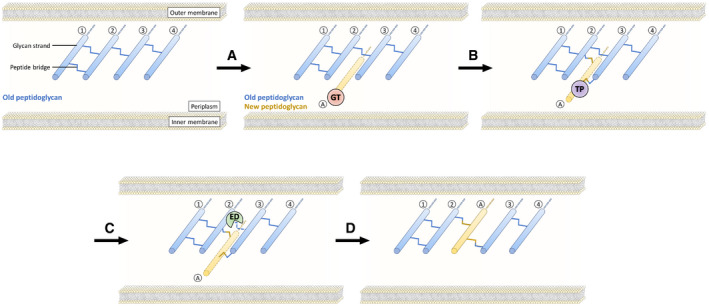
According to this model, one glycan strand (circled A) is polymerized by glycosyltransferases (GTs; step A) and attached to the pre‐existing polymer (strands 2 and 3) by transpeptidases (TPs; step B). Hydrolysis of the cross‐links connecting strands 2 and 3 by endopeptidases (EDs; step C) results in the expansion of the PG layer (step D). Of note, this model, which applies to the synthesis of the lateral wall, accounts for incorporation of new subunits sheltered from the cytoplasm osmotic pressure (De Jonge et al, 1989). There are other models in which whole glycan strands are removed (Höltje & Heidrich, 2001). All models predict that amide bonds should be cleaved for extension of the peptidoglycan network.
The dual capacity of E. coli to use transpeptidases of the D,D and L,D specificities raises the possibility that polymerization of PG containing 4→3 or 3→3 cross‐links involves two overlapping sets of endopeptidases. To address this question, we used an E. coli strain that conditionally and exclusively relies on the formation of 3→3 cross‐links for growth in the presence of ampicillin or ceftriaxone (Hugonnet et al, 2016). By introducing serial deletions of endopeptidase genes, we showed that the 4→3 and 3→3 modes of PG polymerization both require hydrolysis of cross‐links. We identified distinct sets of endopeptidases that are essential for growth involving the two modes of PG cross‐linking. Strikingly, impaired digestion of nascent glycan strands by a lytic transglycosylase was found to favor PG cross‐linking mediated by LDTs. These results highlight the functional plasticity of PG polymerization complexes to accommodate various PG cross‐linking enzymes and hydrolases.
Results
MepM is essential for β‐lactam resistance mediated by the YcbB L,D‐transpeptidase
The role of endopeptidases was assessed in E. coli BW25113 ΔrelA pKT2(ycbB) pKT8(relA′), BW25113(ycbB, relA′) in short, which enables controlling the relative contribution of formation of 4→3 and 3→3 cross‐links to PG polymerization (Hugonnet et al, 2016). In this strain, the ycbB L,D‐transpeptidase gene carried by plasmid pKT2 is expressed under the control of an IPTG‐inducible promoter. Plasmid pKT8 carries an L‐arabinose‐inducible copy of the relA′ gene encoding a truncated version of RelA (residues 1–455), which synthesizes the (p)ppGpp alarmone in an unregulated manner due to the absence of the C‐terminal ribosome binding module (Schreiber et al, 1991). In the presence of both inducers, production of YcbB and RelA′ is sufficient for full bypass of the D,D‐transpeptidase activity of PBPs by the L,D‐transpeptidase activity of YcbB (Fig 2A). This enables bacterial growth in the presence of ampicillin or ceftriaxone since these drugs do not inhibit the YcbB L,D‐transpeptidase. Testing for the inducible expression of β‐lactam resistance in BW25113(ycbB, relA′) therefore provides a means to identify genes that are essential for growth when PG cross‐linking is exclusively mediated by the YcbB L,D‐transpeptidase. We used this phenotypic assay to assess the individual role of seven of the eight endopeptidases of E. coli following single‐gene deletions in the BW25113(ycbB, relA′) strain (Fig 2B). The remaining endopeptidase MepK could not be tested by this approach since deletion of the corresponding gene was not compatible with the presence of plasmid pKT2(ycbB) (see below). Mutants with deletion of mepA, mepH, dacB, pbpG, or ampH were resistant to ceftriaxone. Deletion of mepS decreased plating efficiency in the presence of the drug. Deletion of mepM abolished the expression of β‐lactam resistance. These results are surprising since MepM and MepS were not reported to hydrolyze 3→3 cross‐links (Singh et al, 2012; Chodisetti & Reddy, 2019).
Figure 2. MepM is essential for YcbB‐mediated β‐lactam resistance.
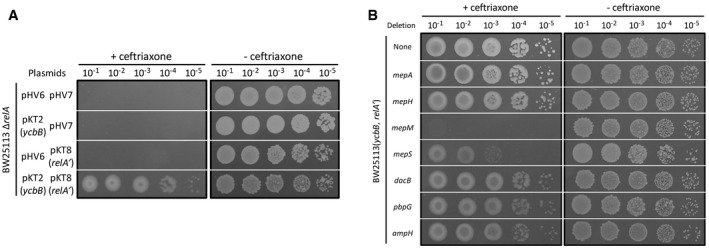
- BW25113 ΔrelA derivatives harboring plasmids pKT2(ycbB), pKT8(relA′) and the vectors pHV6 and pHV7 used to construct these plasmids, respectively. Expression of β‐lactam resistance requires induction of both ycbB and relA′.
- BW25113(ycbB, relA′) and its derivatives obtained by individual deletion of endopeptidase genes. BW25113(ycbB, relA′) is an abbreviated name for BW25113 ΔrelA pKT2(ycbB) pKT8(relA′).
Production of YcbB is lethal in the absence of MepK
The gene encoding MepK, an endopeptidase with the dual 4→3 and 3→3 specificities, was readily deleted from the chromosome of BW25113 ΔrelA. The resulting strain, BW25113 ΔrelA ΔmepK, was transformed with pKT2(ycbB), pKT8(relA′), or both plasmids in combination (co‐transformation). Tetracycline and chloramphenicol were used to select transformants that acquired pKT2(ycbB) and pKT8(relA′), respectively. Plasmid pKT8(relA′) was readily introduced into BW25113 ΔrelA ΔmepK by transformation (108 transformants per µg of DNA). Plasmid pKT2(ycbB) alone or in combination with pKT8(relA′) could not be introduced into BW25113 ΔrelA ΔmepK (< 5 transformants per µg of DNA). The same plating efficacies were observed in selective media containing IPTG, L‐arabinose, or both inducers, in addition to tetracycline and chloramphenicol. These results show that production of the YcbB L,D‐transpeptidase is lethal in the absence of MepK, in agreement with a recent report (Chodisetti & Reddy, 2019). Thus, cleavage of 3→3 cross‐links by MepK is essential for bacterial growth when the proportion of 3→3 cross‐links is increased in the presence of a plasmid copy of ycbB. Quantitatively, the basal level of ycbB expression in the absence of IPTG was sufficient for the lethal phenotype associated with the mepK deletion. Under non‐inducing conditions, the relative proportion of 4→3 and 3→3 cross‐links in the PG extracted from exponential phase cultures of BW25113(ycbB, relA′) was in the order of 60 and 40%, respectively (data not shown). Thus, the cleavage of 3→3 cross‐links by MepK was essential even if these cross‐links co‐existed with 4→3 cross‐links formed by the PBPs.
The hydrolytic activity of MepM is essential for β‐lactam resistance
Deletion of the mepM gene abolished YcbB‐mediated β‐lactam resistance (above, Fig 2B) even though this endopeptidase was not reported to cleave 3→3 cross‐links (Singh et al, 2012; Chodisetti & Reddy, 2019). We therefore considered the possibility that the essential role of mepM in resistance could involve an as yet unknown function in addition to its 4→3‐endopeptidase activity. MepM (440 residues) comprises a LytM (lysostaphin/M23 peptidase) domain and a LysM PG‐binding domain (Pfam: P0AFS9). Complementation analysis of the mepM deletion in BW25113(ycbB, relA′) was performed with plasmids encoding (i) MepM, (ii) MepM H393A harboring an Ala residue at position 393 in place of an essential catalytic His residue conserved in members of the M23 peptidase family, and (iii) MepM ΔLytM lacking the C‐terminal endopeptidase catalytic domain (Fig 3). Expression of β‐lactam resistance by BW25113(ycbB, relA′) ΔmepM was only restored by the plasmid harboring an intact copy of the mepM gene. Thus, the complementation analysis led to the conclusion that the endopeptidase activity of MepM is essential for YcbB‐mediated resistance to β‐lactams.
Figure 3. MepM endopeptidase activity is required for YcbB‐mediated β‐lactam resistance.
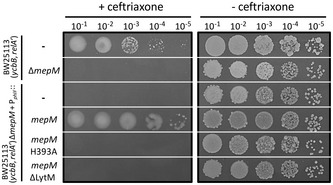
Growth of BW25113(ycbB, relA′), BW25113(ycbB, relA′) ΔmepM, and its derivatives harboring plasmids encoding MepM, MepM H393A, and MepM ΔLytM were tested in the presence of ceftriaxone at 8 µg/ml (+ ceftriaxone) or in the absence of the drug (− ceftriaxone) in BHI agar plates supplemented with 40 µM IPTG and 1% L‐arabinose for induction of ycbB and relA′, respectively. The genes encoding MepM, MepM H393A, and MepM ΔLytM were inserted into the vector pHV9 under the control of the PphlF promoter, which is inducible by 2,4‐diacetylphloroglucinol (DAPG). Basal level of expression of mepM under the control of PphlF was sufficient to restore ceftriaxone resistance in the absence of the inducer.
The essential role of the endopeptidase activity of MepM in β‐lactam resistance is not restricted to the transition from 4→3 to 3→3 cross‐links triggered by the induction of ycbB and relA′
The experiment reported above did not rule out the possibility that hydrolysis of 4→3 cross‐links by MepM might be transiently essential to enable bypass of PBPs by YcbB, i.e., cleavage of 4→3 cross‐links by MepM could be initially essential to enable insertion of new PG subunits into the PG network by YcbB. Ultimately, this would lead to replacement of 4→3 by 3→3 cross‐links and could then suppress the essential role of 4→3 cross‐link cleavage by MepM. According to this hypothesis, MepM would only be essential during the transition between the two modes of PG cross‐linking. To test this possibility, we sought a plasmid construct enabling tight regulation of the mepM gene. In a first attempt, mepM was cloned under the control of the L‐rhamnose‐inducible promoter (PrhaBAD) of the pHV30 vector. Complementation of the mepM deletion of BW25113(ycbB, relA′) was obtained both in the presence or absence of L‐rhamnose indicating that the un‐induced level of ycbB afforded by this plasmid construct was too high (Fig 4A). To address this issue, the level of mepM expression was reduced by replacing the sequence containing the translation initiation signal (TIS1) of mepM by a weaker translation initiation signal (TIS2). TIS1 (aAAGAGGAGAAAtgacataATG) combined an ATG initiation codon to a “strong” ribosome binding site (RBS) with extensive complementarity (underlined) to the 3′ OH extremity of 16S rRNA (5′‐ AUCACCUCCUUA‐3′OH) (Elowitz & Leibler, 2000). TIS2 (acacAGGAcacttaTTG) combined a TTG initiation codon to an RBS with limited complementarity to 16S rRNA (5′‐ AUCACCUCCUUA‐3′OH) (Ringquist et al, 1992; Vellanoweth & Rabinowitz, 1992; Hecht et al, 2017). In contrast to the results obtained with TIS1, the presence of L‐rhamnose was required for β‐lactam resistance if mepM was expressed under the control of TIS2 (Fig 4A). L‐rhamnose requirement for growth in the presence of ceftriaxone was not abolished by pre‐exposure to the inducer (Fig 4B). These results indicate that the essential role of MepM in β‐lactam resistance is not limited to the transition between the two modes of PG cross‐linking, i.e., from 4→3 to 3→3. Since all D,D‐transpeptidases are inhibited by ceftriaxone, these results also indicate that the hydrolytic activity of MepM is essential in conditions in which 4→3 cross‐links are not detectable (Kocaoglu & Carlson, 2015; Hugonnet et al, 2016).
Figure 4. MepM is essential for β‐lactam resistance beyond the transition from 4→3 to 3→3 PG cross‐linking.
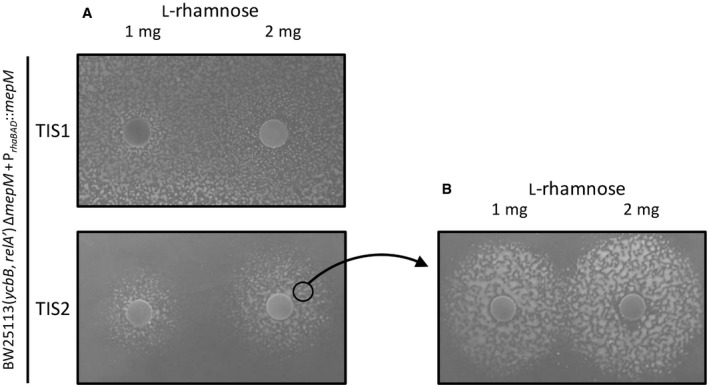
- For TIS2, growth around paper disks containing 1 or 2 mg of L‐rhamnose indicated that induction of the expression of mepM was required for ceftriaxone resistance. In contrast, a higher level of translation from TIS1 was sufficient for ceftriaxone resistance in the absence of the inducer.
- The experiment was repeated with bacteria pre‐exposed to L‐rhamnose that were harvested at the vicinity of the disk containing 2 mg of L‐rhamnose. Expression of ceftriaxone resistance remained dependent on the presence of L‐rhamnose indicating that the requirement for MepM is not transient. The diameter of the growth zones is larger in panel (B) than in panel (A) as expected from the fact that bacteria in the inoculum used in (B) had been grown in the presence of the inducer and already contained MepM. In (A) growth is only possible after diffusion of L‐rhamnose in the medium prior to the action of ceftriaxone. At a distance from the disk, diffusion was not sufficiently rapid to observe resistance.
MepM hydrolyzes both 4→3 and 3→3 cross‐links in vitro
The essential role of the endopeptidase activity of MepM in the context of LDT‐mediated PG cross‐linking (above) led us to reconsider the specificity of the enzyme. Previous analyses were based on incubation of MepM and lysozyme with an E. coli PG preparation containing minute amounts of 3→3 cross‐links (Singh et al, 2012; Chodisetti & Reddy, 2019). Analyses of rpHPLC profiles revealed that the major dimers containing 4→3 cross‐links were digested by MepM but minor peaks corresponding to dimers containing 3→3 cross‐links remained unchanged in the presence of the enzyme leading to the conclusion that MepM was specific to 4→3 cross‐links (Singh et al, 2012; Chodisetti & Reddy, 2019). To improve the sensitivity of the assay, we purified MepM and reproduced this analysis with a PG preparation of E. coli BW25113 grown to stationary phase in minimal medium, which contained a higher proportion of 3→3 cross‐links (Fig 5A, upper panel). The muropeptides corresponding to the indicated peaks are shown in Fig 5B (see Fig EV2 for determination of the structure of muropeptides by mass spectrometry). Full digestion of all dimers was observed upon incubation of this PG preparation with MepM (5 µM) indicating that the endopeptidase hydrolyzes both 4→3 and 3→3 cross‐links to completion (Fig 5A, lower panel). Incubation of the PG preparation with lower concentrations of MepM led to partial hydrolysis of the dimers (Fig 5C). Comparison of the relative abundance of muropeptides based on the integration of peak areas in the chromatograms showed the expected increase in monomers upon digestion of dimers (Fig 5D). The concentrations of MepM required for hydrolysis of half of the muropeptides containing 4→3 and 3→3 cross‐links were 0.4 and 0.7 µM, respectively, revealing similar apparent hydrolysis efficacies for the two types of cross‐links under the assay conditions (Fig 5D). These results indicate that hydrolysis of 3→3 cross‐links may account for the essential role of MepM in conditions in which the L,D‐transpeptidase activity of YcbB fully replaces the D,D‐transpeptidase activity of the PBPs, as inferred from expression of ceftriaxone resistance by BW25113(ycbB, relA′) but not by its ΔmepM derivative (Fig 2B).
Figure 5. Hydrolysis of 4→3 and 3→3 cross‐links by purified MepM.
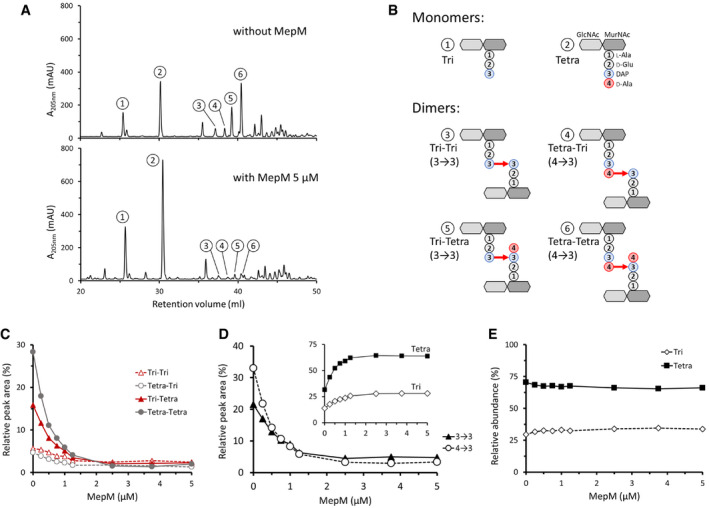
- rpHPLC chromatograms of sacculi isolated from BW25113 grown in minimal medium to stationary phase and digested by lysozyme (upper panel) or by lysozyme and 5 µM MepM (lower panel). Absorbance was monitored at 205 nm (mAU, milli‐absorbance unit).
- Structure of the muropeptides as determined by mass spectrometry (Fig EV2).
- Hydrolysis of the four types of dimers by MepM. Sacculi were incubated with lysozyme and MepM at various concentrations. The relative abundance of the muropeptides was estimated by calculating the relative peak areas.
- Hydrolysis of dimers containing 4→3 and 3→3 cross‐links by MepM. The relative peak areas of Tri‐Tri and Tri‐Tetra containing 3→3 cross‐links and that of Tetra‐Tri and Tetra‐Tetra containing 4→3 cross‐links were combined. The inset shows variations in the relative peak areas of the Tri and Tetra monomers.
- Relative abundance of Tri and Tetra stems in all muropeptides.
Figure EV2. Analysis of endopeptidases activity by mass spectrometry.
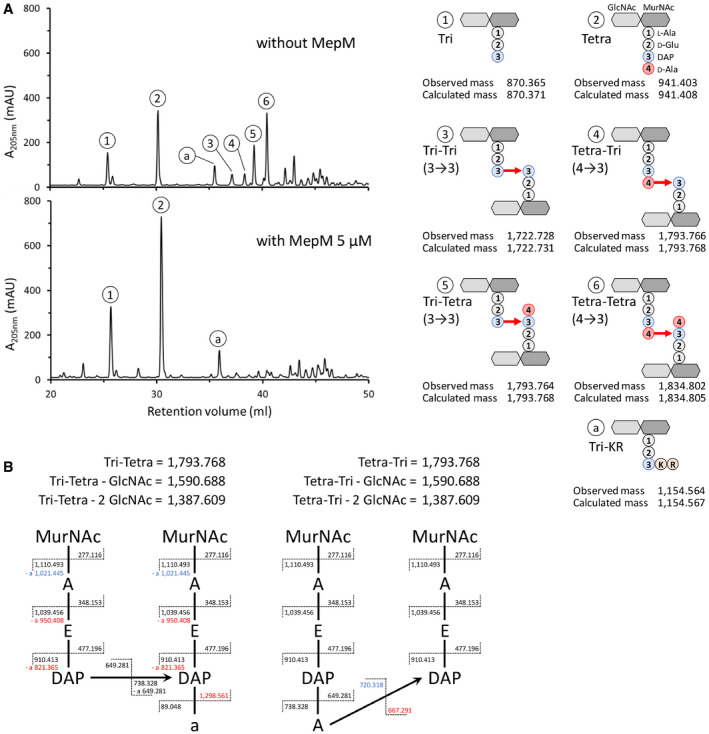
- Mass spectrometry analysis of muropeptides obtained by digestion of sacculi from BW25113 with lysozyme (upper panel) or lysozyme plus MepM (lower panel). The observed and calculated monoisotopic mass is indicated in Dalton. Peak a corresponds to a disaccharide‐tripeptide substituted by a Lys‐Arg (KR) dipeptide originating from digestion of the covalently‐bond Braun lipoprotein by trypsin (Magnet et al, 2008).
- Discrimination of isomers containing 3→3 (Tri‐Tetra) and 4→3 (Tetra‐Tri) cross‐links by tandem mass spectrometry. All fragments lost both GlcNAc molecules. Fragments that are specific of each isomer are shown in red. Fragments specific of an isomer but which can also be found in the other isomer following loss of a water molecule are shown in blue. Mass of fragments is shown in Dalton. A, L‐Ala or D‐Ala; a, C‐terminal D‐Ala; E, D‐Glu; DAP, diaminopimelic acid.
The sum of the relative proportion of tripeptide and tetrapeptide stems in all monomers and dimers did not vary upon addition of MepM (Fig 5E). This observation indicates that MepM did not hydrolyze D‐Ala4 from tetrapeptide monomers or from tetrapeptide stems located in the acceptor position of dimers. Thus, MepM did not display any L,D‐carboxypeptidase activity.
Design of an assay to investigate the redundancy of endopeptidases required for YcbB‐mediated β‐lactam resistance
Single deletion of endopeptidase genes revealed four phenotypes (above, Fig 2B). (i) Deletion of mepK was not compatible with production of YcbB. (ii) Deletion of mepM abolished YcbB‐mediated ceftriaxone resistance. (iii) Deletion of mepS impaired growth in the presence of ceftriaxone. (iv) Deletion of mepA, mepH, dacB, pbpG, or ampH had no impact on growth in the presence of ceftriaxone. The absence of any phenotypic alteration associated with the individual deletion of the latter genes does not necessarily imply that the corresponding endopeptidases are unable to participate in the hydrolysis of 3→3 cross‐links. Indeed, the function of these enzymes may be redundant. Alternatively, their level of production may be insufficient under the tested growth conditions. To investigate these possibilities, each of the eight endopeptidase genes was independently cloned under the control of the “strong” TIS1 translation initiation signal downstream from the PrhaBAD promoter of the vector pHV30 in order to modulate the level of endopeptidase production based on induction by L‐rhamnose. The plasmids were introduced into BW25113(ycbB, relA′) ΔmepM, and growth of the resulting strains was tested in the presence or absence of L‐rhamnose and in the presence or absence of ceftriaxone in all combinations (Fig 6).
Figure 6. Complementation of the mepM deletion by plasmids encoding L‐rhamnose‐inducible copies of the eight endopeptidase genes.
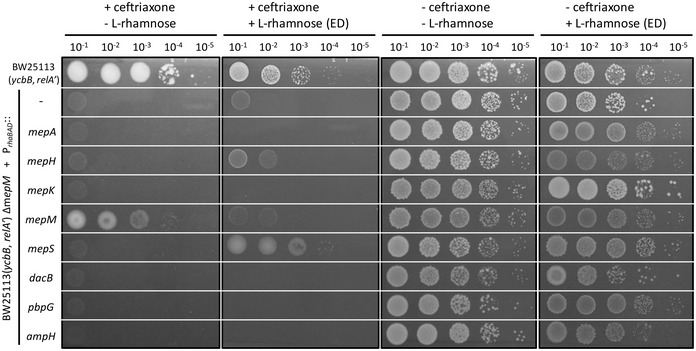
Functional complementation of the mepM deletion in BW25113(ycbB, relA′) ΔmepM was performed with the pHV30 vector or recombinant plasmids encoding each of the eight endopeptidases under the control of the PrhaBAD promoter and of the TIS1 translation initiation signal. Induction of endopeptidase (ED) genes was performed with 0.2% L‐rhamnose in the presence or absence of 8 µg/ml ceftriaxone. BHI agar plates contained 40 µM IPTG and 1% L‐arabinose for induction of ycbB and relA′, respectively.
Overexpression of mepM is toxic in the presence of ceftriaxone
The assay described above revealed that the basal level of expression of the plasmid copy of mepM in the absence of the inducer was sufficient to restore growth of BW25113(ycbB, relA′) ΔmepM in the presence of ceftriaxone (Fig 6). Induction of the mepM gene by L‐rhamnose prevented growth in the presence of ceftriaxone but not in the absence of the drug. Importantly, MepM toxicity was only observed when its production was under the control of the effective TIS1 translation initiation signal, but not of the weaker TIS2 signal used for the experiment described in Fig 4. These results suggest that overproduction of MepM inhibits growth by cleavage of 3→3 cross‐links if 4→3 cross‐links are absent due to the inactivation of the PBPs by ceftriaxone.
Overexpression of mepS complements the mepM deletion for expression of YcbB‐mediated β‐lactam resistance
MepS restored growth of BW25113(ycbB, relA′) ΔmepM only in the presence of the inducer (Fig 6). Thus, MepM and MepS have overlapping functions although overproduction of MepS was required to compensate for the absence of MepM. As mentioned above (Fig 2B), deletion of mepS impaired but did not abolish ceftriaxone resistance in BW25113(ycbB, relA′). Together, these results indicate that expression of mepS in its native chromosomal environment contributes to resistance but the level of its expression is not sufficient to compensate for the absence of MepM.
Partial complementation of the mepM deletion by mepH
The plasmid encoding MepH partially restored growth of BW25113(ycbB, relA′) ΔmepM on ceftriaxone only in the presence of L‐rhamnose (Fig 6). This result indicates that MepH, like MepS, replaces MepM for the expression of ceftriaxone resistance if MepH is overproduced.
Purified MepS and MepH hydrolyze 4→3 and 3→3 cross‐links (endopeptidase activity) and the DAP‐D‐Ala bond of tetrapeptide stems (L,D‐carboxypeptidase activity)
Complementation of ΔmepM by overproduction of MepS and MepH prompted us to evaluate the specificity of these enzymes, as described above for MepM. MepH and MepS both hydrolyzed 4→3 and 3→3 cross‐links (Fig 7A and B). MepS showed no preference for 4→3 or 3→3 cross‐links, while MepH displayed a strong preference for 4→3 cross‐links. The weak hydrolytic activity of MepH on 3→3 cross‐links may account for the fact that the overproduction of MepH can only partially compensate for the absence of MepM (Fig 6, above). Both MepS and MepH displayed L,D‐carboxypeptidase activity leading to rapid conversion of tetrapeptide stems into tripeptide stems (Fig 7A and B).
Figure 7. Hydrolysis of 4→3 and 3→3 cross‐links by purified endopeptidases.
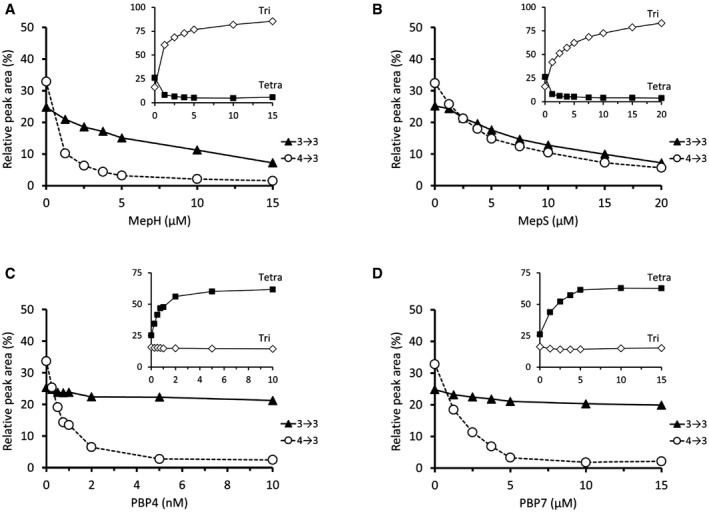
-
A–DSacculi were incubated with lysozyme and purified MepH (A), MepS (B), PBP4 (C), and PBP7 (D). The relative peak areas of Tri‐Tri and Tri‐Tetra containing 3→3 cross‐links and that of Tetra‐Tri and Tetra‐Tetra containing 4→3 cross‐links were combined. MepH preferentially hydrolyzed dimers containing 4→3 cross‐links. MepS hydrolyzed dimers containing 4→3 and 3→3 cross‐links with similar efficacies. PBP4 and PBP7 only hydrolyzed dimers containing 4→3 cross‐links. The insets show variations in the relative peak areas of the Tri and Tetra monomers. MepH and MepS displayed L,D‐carboxypeptidase activity, but not PBP4 and PBP7.
MepA and MepK do not compensate the absence of MepM
In spite of the fact that MepA and MepK were previously shown to cleave 3→3 cross‐links (Engel et al, 1992; Chodisetti & Reddy, 2019), growth of BW25113(ycbB, relA′) ΔmepM on ceftriaxone was not restored by overproduction of these enzymes (Fig 6). Thus, there was not a strict correlation between the ability of the endopeptidases to hydrolyze 3→3 cross‐links in vitro and their ability to restore growth of BW25113(ycbB, relA′) ΔmepM. This absence of correlation was particularly striking for MepK since this endopeptidase, which preferentially hydrolyze 3→3 cross‐links in vitro (Chodisetti & Reddy, 2019), did not complement the mepM deletion although it was required for growth of BW25113(ycbB, relA′) expressing the ycbB L,D‐transpeptidase gene.
These results indicate that functional properties of the endopeptidases, beyond their mere hydrolytic specificity, are relevant to the bypass of the D,D‐transpeptidase activity of PBPs by the L,D‐transpeptidase activity of YcbB. These properties may include the interaction of the endopeptidases with other proteins that regulate their spatiotemporal activity (see Discussion section).
Endopeptidases of the PBP family do not compensate the absence of MepM
Complementation of the mepM deletion was not observed for PBP4, PBP7, or AmpH in the presence or absence of induction of the corresponding genes by L‐rhamnose (Fig 6). There is a caveat for these endopeptidases since they are potentially inhibited by ceftriaxone. To address this issue, the complementation test was repeated with ampicillin, cefsulodin, and aztreonam, which were reported to exhibit different selectivities for inhibition of the PBPs (Henderson et al, 1997; Kocaoglu & Carlson, 2015). Plasmids encoding PBP4, PBP7, and AmpH did not restore growth of BW25113(ycbB, relA′) ΔmepM in the presence of ampicillin, cefsulodin, and aztreonam (Fig EV3) confirming that these endopeptidases are unable to compensate for the absence of MepM. PBP4 and PBP7 were purified and shown to only cleave 4→3 cross‐links (Fig 7C and D). Thus, the absence of complementation of the mepM deletion by the plasmids encoding these PBPs can be accounted for by their lack of hydrolytic activities on 3→3 cross‐linked dimers. PBP4 and PBP7 did not display L,D‐carboxypeptidase activity.
Figure EV3. Complementation of the mepM deletion by endopeptidases of the PBP family.
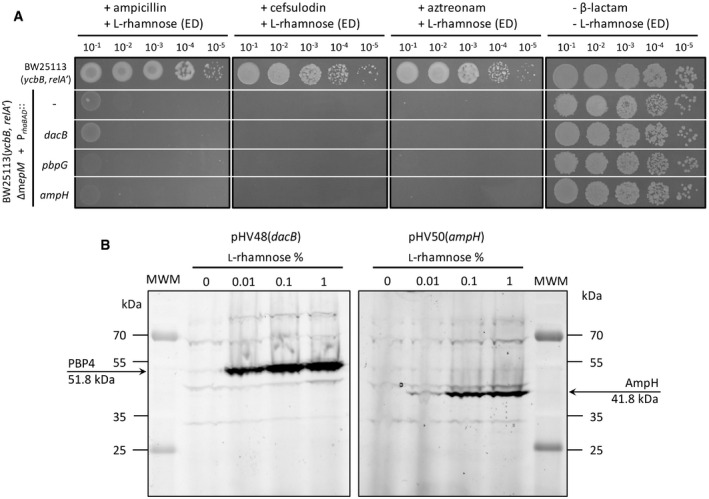
- Functional complementation of the mepM deletion of BW25113(ycbB, relA′) ΔmepM was performed with the pHV30 vector or recombinant plasmids encoding PBP4, PBP7, and AmpH under the control of the PrhaBAD promoter. Induction of endopeptidase (ED) genes was performed with 0.2% L‐rhamnose in the presence or absence of 16 µg/ml ampicillin, 32 µg/ml cefsulodin, or 8 µg/ml aztreonam. BHI agar plates contained 40 µM IPTG and 1% L‐arabinose for induction ycbB and relA′, respectively. It was previously shown that ampicillin, cefsulodin, and aztreonam do not inhibit peptidoglycan cross‐linking in BW25113(ycbB, relA′) due to full bypass of the D,D‐transpeptidase activity of PBPs by the L,D‐transpeptidase activity of YcbB, which is not inactivated by these drugs (Hugonnet et al, 2016).
- Control experiment showing that the PrhaBAD promoter in plasmids pHV48(dacB) and phV50(ampH) enables inducible production PBP4 and AmpH. PBPs were labeled with a fluorescent β‐lactam (BOCILLIN® FL Penicillin) in intact cells and separated by SDS–PAGE. This control experiment was only performed for PBP4 and AmpH since modifications of the resistance or growth phenotypes were observed for the induction of all other endopeptidase genes in at least one of the assays reported in the entire study. MWM, molecular weight marker.
Minimal complement of endopeptidases required for growth in the context of the formation of 4→3 cross‐links by PBPs
Previous analyses based on multiple deletions showed that genes encoding endopeptidases belonging to the PBP family (PBP4, PBP7, and AmpH) are collectively dispensable (Denome et al, 1999). Independently, deletion of the genes encoding MepH, MepM, and MepS in various combinations revealed that at least one of these endopeptidases was essential (Singh et al, 2012). Here, we extend these analyses to the full complement of the eight endopeptidase genes.
Serial deletions of endopeptidase genes were introduced into the chromosome of E. coli BW25113 ΔrelA generating the lineages depicted in Appendix Fig S1. This approach culminated in the construction of a viable derivative of BW25113 ΔrelA, designated Δ7EDs (lineage 5 in Appendix Fig S1), which retained only one of the eight endopeptidase genes (mepM). Thus, MepM alone was necessary and sufficient to support bacterial growth in the context of the 4→3 mode of cross‐linking.
Our next objective was to determine whether deletion of mepM could be complemented by overproduction of other endopeptidases. To address this question, the mepM gene was cloned under the L‐rhamnose‐inducible promoter of vector pHV30 and introduced into the Δ7EDs strain. The chromosomal copy of mepM was deleted from the resulting strain leading to strain Δ8EDs pHV53(mepM), which was dependent upon the presence of L‐rhamnose for growth (Fig 8A). The plasmids enabling L‐arabinose‐inducible expression of the eight endopeptidase genes (above) were introduced in the Δ8EDs pHV53(mepM) strain to determine which endopeptidase could functionally replace MepM (Fig 8B). Induction by L‐arabinose of the genes encoding MepM, MepH, MepS, and PBP7 suppressed the requirement for L‐rhamnose for growth. These results indicate that a single endopeptidase, MepM, MepH, MepS, or PBP7 is potentially sufficient for growth in the context of the 4→3 mode of cross‐linking. Except for MepM, this required overproduction of the enzymes following induction of the ParaBAD promoter of the recombinant plasmids.
Figure 8. Minimal complement of endopeptidases required for growth in the context of the formation of 4→3 cross‐links by PBPs.
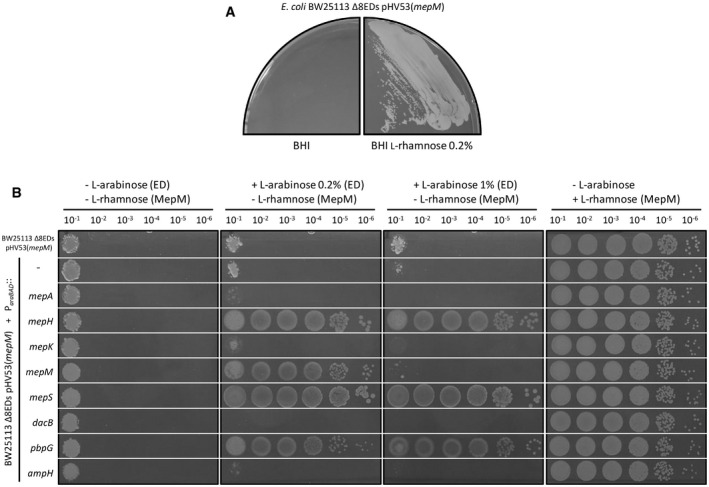
- BW25113 Δ8EDs harboring a plasmid carrying the mepM gene under the control of the PrhaBAD L‐rhamnose‐inducible promoter with the “weak” TIS2 translation initiation signal (plasmid pHV53) was grown on BHI agar in the absence or presence of 0.2% L‐rhamnose. Growth was dependent upon induction of the mepM copy carried by pHV53.
- The plating efficiency assay was performed with derivatives of BW25113 Δ8EDs pHV53(mepM) harboring the vector pHV7 or recombinant plasmids carrying each of the eight endopeptidase genes under the control of the ParaBAD promoter. In this assay, functional replacement of MepM is detected based on growth in media containing 0.2% or 1% L‐arabinose for expression of the endopeptidase gene carried by vector pHV7, thereby by‐passing the requirement for induction of the mepM copy of pHV53 by L‐rhamnose. Complementation was observed with both concentrations of inducer for mepH, mepS, and pbpG. Overproduction of mepM encoded by the pHV7 derivative in the presence of the high dose of L‐arabinose (1%) was lethal. The right panel presents the growth control performed in the presence of 0.2% L‐rhamnose for induction of the mepM copy carried by pHV53.
MepA and MepS compensate for the absence of MepK when ycbB is induced
The basal production of the YcbB L,D‐transpeptidase encoded by plasmid pKT2(ycbB) in the absence of induction was found to be lethal in a derivative of BW25113 lacking mepK (above). To investigate the possibility that MepK might be replaced by another endopeptidase, the ycbB gene was cloned under the control of the ParaBAD promoter of vector pHV7 to obtain a lower level of expression of the L,D‐transpeptidase gene. The resulting plasmid, pHV63(ycbB), was successfully introduced into the BW25113 ΔmepK strain indicating that the basal level of expression of ycbB in the absence of induction was compatible with the absence of mepK. The disk diffusion assay revealed a clear zone around the disk containing L‐arabinose indicating that induction of ycbB in the ΔmepK background prevented bacterial growth (Fig 9A). Plasmids for expression of each of the eight endopeptidases under the control of the PrhaBAD promoter (above) were introduced in this strain (Fig 9B). Bacterial growth was observed in conditions of induction of ycbB by L‐arabinose and of genes encoding MepA or MepS by L‐rhamnose. This result indicates that the essential role of MepK for PG polymerization mediated by the YcbB L,D‐transpeptidase was bypassed by overproduction of MepA or MepS.
Figure 9. Complementation of the mepK deletion by plasmids encoding L‐rhamnose‐inducible copies of the eight endopeptidase genes.
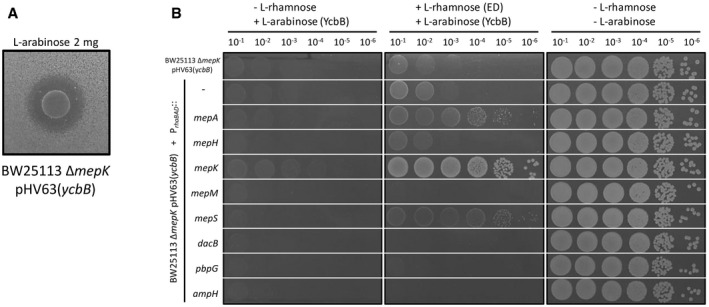
- Induction of ycbB under the control of the ParaBAD promoter in BW25113 ΔmepK pHV63(ycbB) was studied by the disk diffusion assay. The clear zone around the disk containing L‐arabinose indicates that production of YcbB inhibited growth.
- Functional complementation of the mepK deletion of BW25113 ΔmepK pHV63(ycbB) was performed with the pHV30 vector or recombinant plasmids encoding each of the eight endopeptidases under the control of the PrhaBAD promoter. Induction of ycbB and of endopeptidase (ED) genes was performed with 0.2% L‐arabinose and 1% L‐rhamnose, respectively. BHI agar plates contained chloramphenicol (20 µg/ml) to counter‐select loss of pHV63(ycbB).
Complementation of the mepK deletion by catalytically inactivated endopeptidases
Since overproduction of MepK, MepS, and MepA was found to complement the chromosomal deletion of the mepK gene (above, Fig 9), we focused on these three endopeptidases. Plasmids encoding catalytically inactive MepK H133A, MepS C94A, and MepA H113A were used to determine whether the endopeptidase activity of MepK, MepA, and MepS was required to compensate for the chromosomal deletion of mepK. Overproduction of the endopeptidases was tested in the ΔmepK background (single‐deletion mutant retaining all chromosomal endopeptidase genes except mepK, Fig 10A) and in the Δ7EDs background (seven‐deletion mutant retaining only mepM, Fig 10B). Overproduction of MepK but not MepK H133A was essential for growth in both backgrounds indicating that the catalytic activity of the endopeptidase was essential. Overproduction of MepS restored growth in both backgrounds but complementation by MepS C94A was only observed in the ΔmepK single‐deletion background. Since the periplasmic protease Prc hydrolyzes MepS (Singh et al, 2015), overproduction of MepS C94A may saturate the protease enabling sufficient chromosomally‐encoded MepS to escape hydrolysis and support growth. Likewise, saturation of the Prc protease is likely to be responsible for the apparent complementation mediated by overproduction of MepA and MepA H113A since overproduction of these enzymes restored growth in the ΔmepK single‐deletion background but not in the Δ7EDs background. Together, these results indicate that MepS is the only endopeptidase that can compensate for the absence of MepK. This required overproduction of MepS. Alternatively, saturation of the Prc protease by overproduction of MepA, MepA H113A, or MepS C94A prevented hydrolysis of MepS produced at a lower level from the native chromosomal mepS gene.
Figure 10. Complementation of mepK deletion with catalytically inactivated endopeptidases.
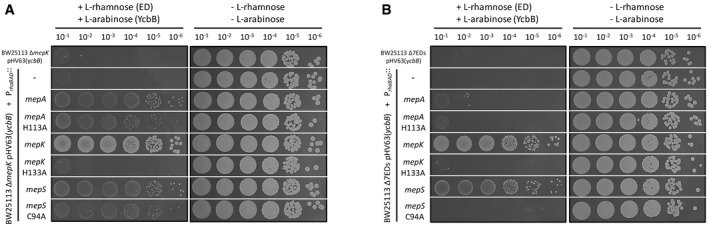
- Functional complementation of the mepK deletion of BW25113 ΔmepK pHV63(ycbB) was performed with the pHV30 vector or recombinant plasmids encoding mepA, mepK, mepS, or derivatives encoding catalytically inactive endopeptidases under the control of the PrhaBAD promoter.
- The complementation assay was repeated for BW25113 Δ7EDs pHV63(ycbB), which was obtained by deletion of all chromosomal endopeptidase genes except mepM. Induction of ycbB and of endopeptidase (ED) genes was performed with 0.2% L‐arabinose and 1% L‐rhamnose, respectively. BHI agar plates contained chloramphenicol (20 µg/ml) to counter‐select loss of pHV63(ycbB).
Minimal complement of endopeptidases required for growth in the presence of β‐lactams in the context of the exclusive formation of 3→3 cross‐links by the YcbB L,D‐transpeptidase
As previously described (Hugonnet et al, 2016), induction of relA′ led to mecillinam resistance in BW25113(ycbB, relA′), whereas induction of both ycbB and relA′ was required for ampicillin and ceftriaxone resistance (Table 1 and Fig EV4). Strain BW25113(ycbB, relA′) Δ6EDs (harboring only mepM and mepK) was also resistant to the three β‐lactams upon induction of ycbB and relA′ indicating that 6 of the 8 endopeptidase genes were dispensable for expression of β‐lactam resistance. In contrast to BW25113(ycbB, relA′), the Δ6EDs derivative was resistant to ampicillin and ceftriaxone in the absence of induction of ycbB by IPTG. The basal level of ycbB expression in the absence of induction was required for resistance since susceptibility to ampicillin and ceftriaxone was observed in the absence of pKT2(ycbB). These observations indicate that deletion of 6 of the 8 endopeptidase genes was associated with a decrease in the level of expression of ycbB required for β‐lactam resistance. In combination with the analysis based on single‐gene deletions (Fig 2B), these results show that MepM and MepK are necessary and sufficient for bacterial growth in conditions in which YcbB is the only functional transpeptidase.
Table 1.
YcbB‐mediated β‐lactam resistance in BW25113 derivatives harboring all endopeptidase genes or only mepM and mepK (Δ6EDs).
| Host | Plasmid | Inducerb | Inhibition zones (mm)a | ||
|---|---|---|---|---|---|
| Mec | Amp | Cro | |||
| BW25113 ΔrelA | |||||
| pKT2(ycbB) pKT8(relA′) | None | 20 | 18 | 34 | |
| IPTG (ycbB) | 21 | 16 | 32 | ||
| Ara (relA′) | 9 | 18 | 35 | ||
| IPTG + Ara | < 8 | < 8 | 15 | ||
| BW25113 ΔrelA Δ6EDs | |||||
| pKT2(ycbB) pKT8(relA′) | None | 19 | 22 | 37 | |
| IPTG (ycbB) | 20 | 19 | 35 | ||
| Ara (relA′) | < 8 | 10 | 18 | ||
| IPTG + Ara | < 8 | < 8 | 16 | ||
| BW25113 ΔrelA Δ6EDs | |||||
| pHV6c pKT8(relA′) | None | 22 | 18 | 35 | |
| IPTG | 22 | 18 | 35 | ||
| Ara (relA′) | 10 | 18 | 37 | ||
| IPTG + Ara | 9 | 17 | 37 | ||
The diameter of inhibition zones was determined by the disk diffusion assay around disks containing 10 µg mecillinam (Mec), 10 µg ampicillin (Amp), or 30 µg ceftriaxone (Cro). Data are the medians from three experiments. Examples of the original results are presented in Fig EV4.
The ycbB and relA′ genes carried by plasmid pKT2 and pKT8 were induced with 40 µM IPTG and 1% L‐arabinose (Ara), respectively.
pHV6 is the vector used for construction of pKT2(ycbB).
Figure EV4. Antibiotic susceptibility testing by the disk diffusion assay.
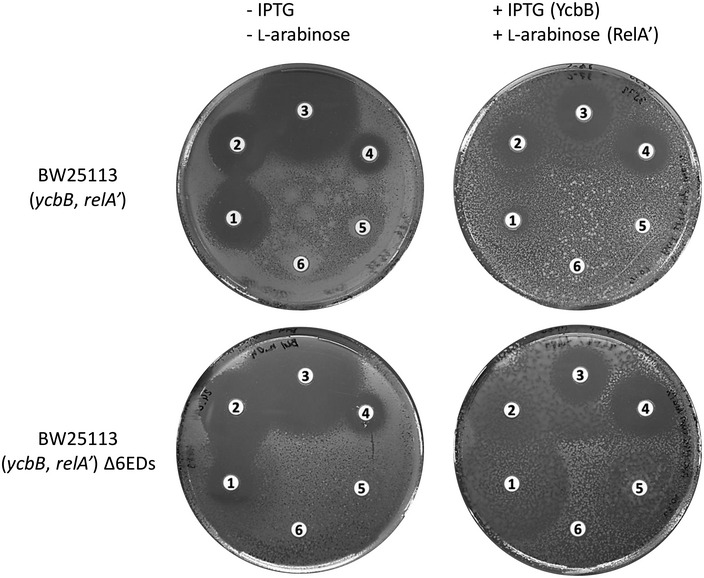
Disks contained 10 µg mecillinam (1), 10 µg ampicillin (2), 30 µg ceftriaxone (3), 30 µg tetracycline (4), 30 µg chloramphenicol (5), and 30 µg kanamycin (6). Plasmids pKT2(ycbB) and pKT8(relA′) confer resistance to tetracycline and chloramphenicol, respectively. Kanamycin resistance is mediated by the KmR cassette inserted in place of relA. Induction of ycbB and relA′ was performed with 40 µM IPTG and 1% L‐arabinose.
Mutations impairing the lytic transglycosylase activity of Slt70 favors YcbB‐mediated PG synthesis
Although BW25113(ycbB, relA′) displays high β‐lactam resistance on BHI agar supplemented with IPTG and L‐arabinose, the strain was found to remain susceptible to β‐lactams in BHI broth supplemented with the same inducers. Mutations leading to expression of β‐lactam resistance in liquid medium were sought by selecting mutants derived from E. coli BW25113 M1 (Hugonnet et al, 2016). The latter strain overexpresses ycbB from plasmid pJEH12(ycbB) in response to induction by IPTG and overproduces the (p)ppGpp alarmone due to impaired expression of the isoleucine tRNA synthetase gene ileS (Hugonnet et al, 2016). Derivatives of BW25113 M1 were selected in BHI broth containing 16 µg/ml ampicillin and 50 µM IPTG. Four independent mutants derived from BW25113 M1 (M1.1 to M1.4) were isolated and shown to grow in liquid medium supplemented with ampicillin and IPTG (Fig 11A). Whole genome sequencing revealed single mutations all located in the sltY gene encoding the lytic transglycosylase Slt70 (Table 2). One of the mutants (M1.3) most probably harbored a null allele of sltY since a 7‐bp deletion introduced a frameshift at the 9th codon of the gene. To confirm this conclusion, the sltY gene was deleted from the chromosome of BW25113(ycbB, relA′) strain. The resulting strain, BW25113(ycbB, relA′) ΔsltY, was also resistant to ceftriaxone in liquid medium in the presence of IPTG and L‐arabinose (Fig 11A).
Figure 11. Growth phenotype of sltY mutants.
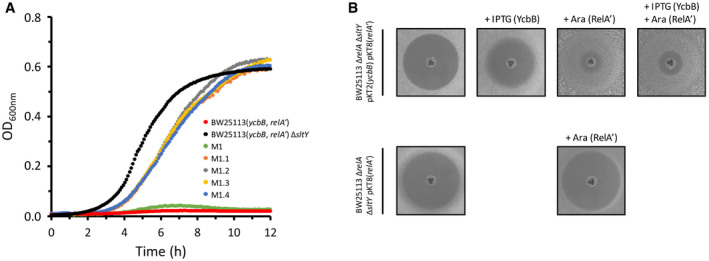
- Growth curves of (i) BW25113(ycbB, relA′) and its ΔsltY derivative; and (ii) strain M1 and its derivatives (M1.1 to 1.4) harboring mutations in sltY. The growth medium contained (i) 40 µM IPTG, 1% L‐arabinose, and 8 µg/ml ceftriaxone for BW25113(ycbB, relA′) and its ΔsltY derivative or (ii) 50 µM IPTG and 16 µg/ml ampicillin for M1 and its derivatives.
- Ceftriaxone‐resistance depends upon ycbB and relA′ expression. The disk diffusion assay was performed with BW25113 ΔrelA ΔsltY harboring pKT2(ycbB) and pKT8(relA′) or pKT8(relA′) only. Induction was performed with 40 µM IPTG and 1% L‐arabinose for ycbB and relA′, respectively. Disks were loaded with 30 µg of ceftriaxone.
Table 2.
Mutations detected in the sltY gene of mutants BW25113 M1.1 to M1.4.
| Mutant | Position | Mutation | Impacta |
|---|---|---|---|
| M1.1 | 4,621,672 | C→T | Gln375stop |
| M1.2 | 4,622,297 | Duplication of AGG: CAGGG→CAGGAGGG | Insertion of Gly position 584 (codon GGN) |
| M1.3 | 4,620,572 | 7‐bp deletion: CCTGGCGGC→CC | Trp9frameshift |
| M1.4 | 4,621,507 | C→T | Arg320stop |
Slt70 comprises 645 amino acid residues.
Comparison of the resistance phenotype of BW25113(ycbB, relA′) and its ΔsltY derivative on BHI agar revealed that induction of ycbB by IPTG is required for ampicillin and ceftriaxone resistance in the former but not in the latter strain (Fig 11B for representative antibiograms and Table 3 for the full set of data). Expression of resistance to ampicillin and ceftriaxone on BHI agar remained dependent upon induction of relA′ since BW25113 ΔrelA ΔsltY pKT2(ycbB) pKT8(relA′) remained susceptible to these drugs in the absence of L‐arabinose. Expression of resistance to ampicillin and ceftriaxone also remained dependent upon the presence of pKT2(ycbB) since replacement of this plasmid by the pHV6 vector abolished resistance [strain BW25113 ΔrelA ΔsltY pHV6 pKT8(relA′); Table 3]. These results indicate that loss of Slt70 enabled expression of YcbB‐mediated β‐lactam resistance in liquid medium and reduced the level of production of the YcbB L,D‐transpeptidase required for expression of resistance to ampicillin and ceftriaxone on solid medium. This observation suggests that accumulation of uncross‐linked glycan chains in the absence of Slt70 may improve the capacity of YcbB to catalyze PG cross‐linking accounting for the lower level of expression of ycbB required for resistance.
Table 3.
YcbB‐mediated β‐lactam resistance in BW25113(ycbB, relA′) and its derivative obtained by deletion of sltY.
| Host | Plasmid | Inducerb | Inhibition zones (mm)a | ||
|---|---|---|---|---|---|
| Mec | Amp | Cro | |||
| BW25113 ΔrelA | |||||
| pKT2(ycbB) pKT8(relA′) | None | 20 | 18 | 34 | |
| IPTG (ycbB) | 21 | 16 | 32 | ||
| Ara (relA′) | 9 | 18 | 35 | ||
| IPTG + Ara | < 8 | < 8 | 15 | ||
| BW25113 ΔrelA ΔsltY | |||||
| pKT2(ycbB) pKT8(relA′) | None | 21 | 21 | 33 | |
| IPTG (ycbB) | 22 | 18 | 26 | ||
| Ara (relA′) | < 8 | < 8 | 9 | ||
| IPTG + Ara | < 8 | < 8 | 12 | ||
| BW25113 ΔrelA ΔsltY | |||||
| pHV6c pKT8(relA′) | None | 20 | 22 | 34 | |
| IPTG | 22 | 22 | 34 | ||
| Ara (relA′) | < 8 | 21 | 36 | ||
| IPTG + Ara | < 8 | 20 | 35 | ||
| BW25113 ΔrelA ΔsltY | |||||
| pKT2(ycbB) pHV7d | None | 23 | 21 | 34 | |
| IPTG (ycbB) | 25 | 18 | 25 | ||
| Ara | 25 | 21 | 34 | ||
| IPTG + Ara | 24 | 17 | 26 | ||
The diameter of inhibition zones was determined by the disk diffusion assay around disks containing 10 µg mecillinam (Mec), 10 µg ampicillin (Amp), or 30 µg ceftriaxone (Cro). Data are the medians from three experiments.
The ycbB and relA′ genes carried by plasmid pKT2 and pKT8 were induced with 40 µM IPTG and 1% L‐arabinose (Ara), respectively.
pHV6 is the vector used for construction of pKT2(ycbB).
pHV7 is the vector used for construction of pKT8(relA′).
For the sake on completeness, we also noticed that BW25113 ΔrelA ΔsltY pKT2(ycbB) pKT8(relA′) expressed intermediate resistance to ceftriaxone in the presence of IPTG (induction of ycbB) and in the absence of L‐arabinose (no induction of relA′) because the size of the inhibition zone (26 mm) was intermediate between those of susceptible and resistant strains (range of 32–36 mm versus 9–15 mm) (Table 3). This phenotype was independent from relA′ since replacement of plasmid pKT8(relA′) of BW25113 ΔrelA ΔsltY pKT2(ycbB) pKT8(relA′) by the vector pHV7 also led to intermediate resistance to ceftriaxone in the presence of IPTG for induction of ycbB (25 mm).
Discussion
Endopeptidases are essential for expansion of sacculi
PG polymerization requires a combination of synthetases, the transpeptidases and the glycosyltransferases, in addition to hydrolases that fulfill two essential roles. Since PG is a net‐like macromolecule completely surrounding the cytoplasmic membrane, it seems clear that insertion of new disaccharide peptide subunits into the growing cell wall requires cleavage of covalent bonds (Höltje & Heidrich, 2001; Vollmer, 2012). The stress‐bearing PG being present during the entire cell cycle, it is also obvious that PG hydrolases are required to split daughter cells following completion of septum synthesis (Heidrich et al, 2002). Some of these hydrolases generate PG fragments that are imported into the cell and recycled, a complex pathway that is not essential for growth in laboratory conditions but bears important roles in (i) minimizing energy costs, (ii) sensing the appropriate balance between synthetic and hydrolytic activities, which may be altered by β‐lactam antibiotics and other toxic agents, and (iii) avoiding the release of proinflammatory molecules recognized by the host immune system (Johnson et al, 2013; Bastos et al, 2021). PG hydrolases specifically acting on each of the ten amide, ether, and glycosidic bonds present in the PG polymer have been described and most cleavage specificities involve multiple enzymes (Fig EV5A). Enzymes of different specificities can at least in part compensate for each other, e.g., lytic glycosyltransferases and amidases both contribute to the separation of daughter cells (Heidrich et al, 2002; van Heijenoort, 2011). In this study, we show that endopeptidases are specifically required for bacterial growth not only in the context of the formation of 4→3 cross‐links by PBPs but also in the context of the formation of 3→3 cross‐links by YcbB (Table 4). We also identify for the first time the minimum sets of endopeptidases for each mode of PG cross‐linking, namely MepM for 4→3 cross‐links and MepM plus MepK for 3→3 cross‐links. Endopeptidase overproduction resulting from expression of the genes under the control of heterologous promoters revealed potential functional redundancies in the endopeptidase families. In particular, overproduction of MepH, MepS, or PBP7 compensated for the absence of MepM in the context of a 4→3 cross‐linked PG (Fig 12A). Overproduction of MepS compensated for the absence of MepM or MepK for growth with a 3→3 cross‐linked PG. Overproduction of MepM prevented growth probably due to unbalanced synthesis and hydrolysis of PG cross‐links (observed for both 4→3 and 3→3 cross‐linked PG). Production of catalytically inactive endopeptidases suggested that MepA and MepS are negatively regulated by Prc‐mediated proteolysis, as previously established for MepS (Singh et al, 2015; Lai et al, 2017). In conclusion, MepK is specifically required for growth if the peptidoglycan contains a large proportion of 3→3 cross‐links, while MepM is additionally required for β‐lactam resistance, unless it is the only endopeptidase remaining, in which case it is also required for growth in the absence of the drug.
Figure EV5. Specificity of PG hydrolases.
- Highlight of enzyme stereospecificity. The commonly used endopeptidase designation was employed in the entire manuscript to refer to the cleavage of internal bonds although certain enzymes do not cleave peptide bonds connecting the α amino and carboxyl groups of two consecutive amino acids and should have been more precisely referred to as amidases.
- Recognition of the donor and acceptor stems of dimers by PBP and Mep endopeptidases accounting for the 4→3 versus 4→3 plus 3→3 specificities. According to this model, endopeptidases of the Mep families cleave both 4→3 plus 3→3 cross‐links since they interact with the tripeptide portion of the acceptor stem, which is present in both types of dimers. In contrast, endopeptidases of the PBP family specifically interact with the tetrapeptide donor stem of 4→3 cross‐linked dimers.
Table 4.
Characteristics of the endopeptidases.
| Endopeptidase | In vitro hydrolysis of 4→3 and 3→3 cross‐linksc | Role in the context of the two modes of cross‐linking (revealed by endopeptidase overproductionf) | |
|---|---|---|---|
| 4→3 cross‐links | 3→3 cross‐links | ||
| Acyl‐serine transferase | |||
| PBP4a | 4→3 | Not essential (None) | Not essential (None) |
| PBP7a | 4→3 | Not essential (Compensates for the absence of MepM) | Not essential (None) |
| AmpHb | 4→3d | Not essential (None) | Not essential (None) |
| NlpC/P60 peptidase | |||
| MepHa | 4→3 > 3→3 L,D‐carboxypeptidase | Not essential (Compensates for the absence of MepM) | Not essential (None) |
| MepSa | 4→3 = 3→3 L,D‐carboxypeptidase | Not essential (Compensates for the absence of MepM) | Not essential (Compensates for the absence of MepM or MepK)e |
| Lysostaphin/M23 peptidase | |||
| MepMa | 4→3 = 3→3 | Sufficient (Prevents growth) | Essential (Prevents growth) |
| LAS metallopeptidase | |||
| MepAb | 4→3 = 3→3 | Not essential (None) | Not essential (None) |
| M15 peptidase | |||
| MepKb | 4→3 < 3→3 | Not essential (None) | Essential (None) |
Characterized in this study.
Data from the literature (Engel et al, 1992; Gonzalez‐Leiza et al, 2011; Chodisetti & Reddy, 2019).
MepM, MepS, and MepA cleaved 4→3 and 3→3 cross‐links with similar efficacies (4→3 = 3→3). MepH and MepK displayed a preference for 4→3 cross‐links (4→3 > 3→3) or for 3→3 cross‐links (4→3 < 3→3), respectively. L,D‐carboxypeptidase, hydrolysis of the DAP3‐D‐Ala4 amide bond of tetrapeptide stems.
Hydrolysis of 3→3 cross‐links was not tested.
Replacement of both MepM and MepK by MepS was not tested.
None, no phenotype associated with endopeptidase overproduction.
Figure 12. Proposed model for polymerization of side wall PG by transpeptidases of the D,D or L,D specificity.
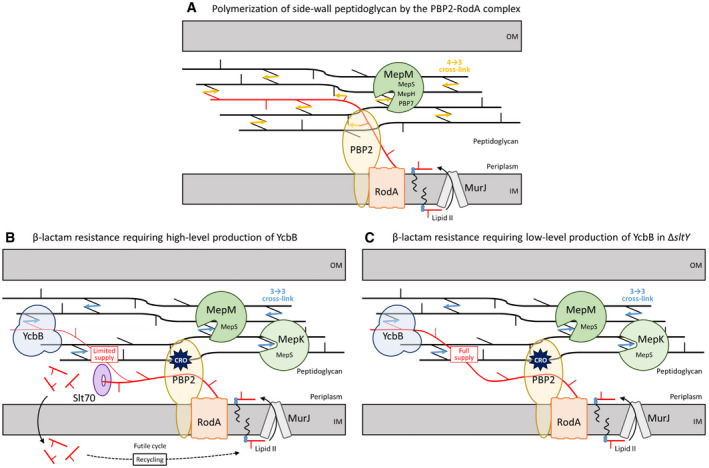
- In wild‐type cells, the disaccharide pentapeptide subunit linked to the undecaprenyl lipid transporter (Lipid II) is translocated to the outer leaflet of the cytoplasmic membrane by MurJ and polymerized by the glycosyltransferase and D,D‐transpeptidase activities of the RodA‐PBP2 complex. MepM is essential and sufficient for insertion of new material in the PG net, although this endopeptidase can be replaced by overproduction of MepH, MepS, or PBP7.
- Inhibition of PBP2 by β‐lactams leads to the accumulation of uncross‐linked glycan chains that are cleaved by the Slt70 lytic transglycosylase. This limits the supply of glycan chains for cross‐linking by the YcbB L,D‐transpeptidase. Under this condition, MepM or overproduction of MepS is required for insertion of new glycan strands. Production of YcbB leads to a requirement for an additional endopeptidase, MepK. This function of MepK can be bypassed by overproduction of MepS.
- Deletion of the sltY gene encoding lytic transglycosylase Slt70 prevents digestion of uncross‐linked glycan chains leading to a full supply of neo‐synthesized glycan chains to YcbB and improved expression of β‐lactam resistance. IM, inner membrane; OM, outer membrane; CRO, ceftriaxone.
Specificity of purified endopeptidases for 4→3 and 3→3 cross‐links
The cleavage specificity of the endopeptidases was determined by mass spectrometry (Figs 5 and 7, Appendix Fig S2). Endopeptidases of the PBP family were specific to 4→3 cross‐links. Endopeptidases belonging to other families cleaved both 4→3 and 3→3 cross‐links with similar efficacies (MepM, MepS, MepA) or with a preference for 4→3 (MepH) or 3→3 (MepK) cross‐links (Figs 5 and 7) (Engel et al, 1992; Chodisetti & Reddy, 2019). This is unexpected since 4→3 and 3→3 cross‐links contain amide bonds connecting two R stereocenters (D‐Ala4→DAP3) or an S to an R stereocenter (DAP3→DAP3), respectively (Fig EV5A). This could imply that endopeptidases of the Mep families mainly interact with the acceptor stems of cross‐linked muropeptides, which are the same for both types of cross‐links, whereas endopeptidases of the PBP family interact with a donor tetrapeptide stem only present in 4→3 cross‐linked muropeptides (Fig EV5B).
Integration of endopeptidases into the global regulation of 4→3 and 3→3 PG cross‐linking
D,D‐carboxypeptidases, which cleave off the terminal residue (D‐Ala5) of pentapeptide stems, are thought to negatively control the transpeptidase activity of PBPs since these enzymes require a pentapeptide donor (Fig 1). A less studied impact of D,D‐carboxypeptidases is the formation of the essential tetrapeptide donor substrate of the LDTs, except for two publications reporting that PBP5 and PBP6a are essential for YcbB‐mediated β‐lactam resistance and for rescue of a defect in lipopolysaccharide synthesis, respectively (Hugonnet et al, 2016; Morè et al, 2019). Thus, D,D‐carboxypeptidases have crucial roles in controlling the relative contributions of transpeptidases of the D,D and L,D specificities to PG cross‐linking by both decreasing access of PBPs to pentapeptide stems and increasing access of LDTs to tetrapeptide stems.
Endopeptidases participate in the metabolism of PG cross‐links in several ways. MepH and MepS display L,D‐carboxypeptidase activity leaving tripeptides as the main (> 80%) end product of in vitro PG hydrolysis (Fig 7). This activity may negatively control the L,D‐transpeptidase activity of YcbB by hydrolysis of D‐Ala4, thereby preventing access to its tetrapeptide donor. In addition, hydrolysis of 4→3 cross‐linked Tetra→Tetra and Tetra→Tri dimers by the endopeptidases generates free tetrapeptide stems (Figs 5 and 7). These tetrapeptide stems can be used as donor by YcbB for formation of 3→3 cross‐links, as demonstrated for the LDTs of Mycobacterium smegmatis (Baranowski et al, 2018). In contrast, the D,D‐transpeptidase activity of PBPs exclusively requires pentapeptide‐containing donors. Analyses of peptidoglycan structure indicate that D‐Ala5 is rapidly cleaved off from pentapeptide stems by D,D‐carboxypeptidases if they are not used for formation of 4→3 cross‐links (Glauner et al, 1988). These observations suggest that the transpeptidase activity of PBPs exclusively relies on de novo synthesis and translocation of pentapeptide‐containing subunits. Thus, YcbB is expected to function as a rescue enzyme to restore cross‐linking in regions of the PG that are compromised by 4→3 or 3→3 endopeptidases. This mechanism was proposed for PG repair following disassembly of the lipopolysaccharide export machinery that crosses the PG layer (Morè et al, 2019). The combined activities of endopeptidases cleaving 4→3 cross‐links and of L,D‐transpeptidases could contribute to the enrichment in 3→3 cross‐links in stationary phase cultures (Pisabarro et al, 1985). In turn, this enrichment may protect cells from hydrolases active on 4→3 cross‐linked PG. Previous analyses proposed that two L,D‐transpeptidases may contribute to the enrichment of PG in 3→3 cross‐links, namely YcbB (LdtD), which is induced by the cell envelope Cpx stress system, and YnhG (LdtE), which is expressed under the control of sigma S and induced in stationary phase (Weber et al, 2005; Delhaye et al, 2016).
Participation of YcbB to PG polymerization complexes
PG polymerization is generally thought to be performed by two multiprotein complexes involved in the expansion of the lateral cell wall (elongasome) and in the formation of the septum (divisome) (Pazos et al, 2017). Replacement of the D,D‐transpeptidase activity of all PBPs by the L,D‐transpeptidase activity of YcbB raises several questions regarding the identity of the partners of YcbB for the assembly of lateral wall and septum PG, and whether YcbB physically replaces PBPs in the PG polymerization complexes. Our data support a model in which YcbB functions with two different sets of partners for lateral wall and septum PG assembly as follows.
For the assembly of lateral wall PG, inactivation of the transpeptidase domain of PBP2 by β‐lactams leads to uncoupling of the glycosyltransfer and transpeptidation reactions, the former being most probably catalyzed by RodA (Uehara & Park, 2008; Cho et al, 2014) (Fig 12B). Uncross‐linked glycan chains accumulate in the periplasm and are eventually cleaved by the Slt70 lytic transglycosylase and recycled. According to the model presented in Fig 12, and in agreement with a previous study (Cho et al, 2014), the reactions catalyzed by Slt70 and YcbB occur in competition implying that YcbB‐mediated cross‐linking is not coupled to glycan chain polymerization by RodA. This also implies that YcbB could function in the PG layer in combination with the MepM endopeptidase known to participate in cell elongation (Uehara et al, 2009; Singh et al, 2012; Banzhaf et al, 2020; Truong et al, 2020). In agreement with this model, impaired Slt70 activity had a positive impact on β‐lactam resistance mediated by YcbB (Fig 12C). This was established both by the selection of mutations enabling expression of β‐lactam resistance in liquid medium, which mapped in the sltY gene encoding Slt70 (Table 2) and by the deletion of sltY, which lowered the level of ycbB expression required for resistance (Fig 11).
For the assembly of septum PG, YcbB was proposed to cooperate with the glycosyltransferase activity of Class A PBP1b (Hugonnet et al, 2016; Caveney et al, 2019). In support of this hypothesis, microscale thermophoresis experiment revealed that purified YcbB interacts with PBP1b and PBP5 (D,D‐carboxypeptidase). Furthermore, the glycosyltransferase activity of PBP1b is essential for YcbB‐mediated β‐lactam resistance, whereas the combined deletion of class A PBP1a and PBP1c had no impact. We cannot rule out the possibility that the glycosyltransferase activity of FtsW also contributes to septum PG polymerization in the presence of β‐lactams but this would require that the inactivation of PBP3 by β‐lactams lead to the uncoupling of glycan chain polymerization and PG cross‐linking, as proposed for the RodA‐PBP2 complex (see above). This is not supported by the analyses of PG recycling in conditions of selective inhibition of PBP3 by aztreonam (Uehara & Park, 2008). The endopeptidases involved in septum formation have not been identified, except for a contribution of PBP4, which has an effect on the timing of septation (preprint: Verheul et al, 2020). MepK is a candidate for this function in 3→3 cross‐linked PG although this is currently not supported by experimental evidence, and it remains to be seen if endopeptidases are needed for septum PG synthesis.
Materials and Methods
Strains, plasmids, and growth conditions
All strains were derived from E. coli BW25113 (Baba et al, 2006). The origin and characteristics of plasmids are listed in Appendix Table S1. The origin and genotype of strains are listed in Appendix Table S2. Bacteria were grown in brain heart infusion (BHI; Difco) broth or agar at 37°C unless otherwise specified. Liquid cultures were performed with aeration (180 rpm). The growth media were systemically supplemented with drugs to counter‐select plasmid loss (Appendix Table S1). The same drugs at the same concentrations were used to select transformants. Kanamycin at 50 µg/ml was used for the KmR cassette. Induction of the lacZYA, araBAD, and rhaBAD promoters was performed with isopropyl β‐D‐1‐thiogalactopyranoside (IPTG, 40 or 50 µM), L‐arabinose (0.2 or 1%), and L‐rhamnose (0.2 or 1%), respectively. Plasmids constructed in this study were obtained by using NEBuilder HiFi DNA assembly (New England Biolabs) method, unless otherwise specified.
Growth curves were obtained in a 96‐well plate using an Infinite 200 PRO microplate reader (TECAN). Briefly, bacteria were grown to the late exponential phase, i.e., to an optical density at 600 nm (OD600) greater than 1.0 (ca. 6 h at 37°C under agitation). The OD600 was adjusted to 1.0, and 5 µl were inoculated in 195 µl of BHI broth supplemented with drugs and inducers, as specified in the legends to figures. Growth was monitored at 600 nm every 5 min for 12 h at 37°C with vigorous shaking.
Construction of E. coli strains carrying gene deletions
The Keio collection comprises 3,985 mutants obtained by replacement of non‐essential genes by a kanamycin resistance (KmR) cassette (Baba et al, 2006). P1 transduction of the KmR cassette from selected mutants was used to introduce deletions of specific genes involved in PG synthesis (Datsenko & Wanner, 2000). For multiple gene deletions, the KmR cassette was removed by the FLT recombinase encoded by plasmid pCP20. The presence of the expected deletions was confirmed by PCR amplification at each deletion step. Appendix Fig S1 shows lineages that have been obtained by parallel serial deletions.
To study the 3→3 mode of cross‐linking, plasmid pKT2(ycbB) and pKT8(relA′) were introduced into the derivatives of E. coli BW25113 ΔrelA obtained by deletion of various combinations of endopeptidase genes. For the sake of simplicity, the latter strains were referred to as BW25113(ycbB, relA′) derivatives even though gene deletions preceded the introduction of pKT2(ycbB) and pKT8(relA′).
Construction of E. coli BW25113 ΔrelA Δ8EDs
The plasmid pHV53(PrhaBAD‐TIS2‐mepM) was introduced into BW25113 ΔrelA Δ7EDs harboring mepM as the only chromosomal endopeptidase‐encoding gene. The mepM deletion was introduced into BW25113 ΔrelA Δ7EDs pHV53(PrhaBAD‐TIS2‐mepM) in the presence of 0.2% L‐rhamnose by P1 transduction as described above. Growth of the resulting BW25113 ΔrelA Δ8EDs pHV53(PrhaBAD‐TIS2‐mepM) strain was dependent on the induction of the plasmid copy of mepM mediated by L‐rhamnose.
Mutant selection and whole‐genome sequencing
E. coli BW25113 M1 was streaked for isolated colonies on agar plates containing 10 µg/ml tetracycline to counter‐select loss of plasmid pJEH12(ycbB). The selection procedure was independently carried out starting with four independent colonies. Briefly, 5 ml of BHI broth supplemented with 10 µg/ml tetracycline and 50 µM IPTG were inoculated with a colony. Bacteria were grown overnight with shaking (180 rpm). A fraction of 1 ml of the culture was inoculated in 250 ml of BHI broth supplemented with 16 µg/ml ampicillin and 50 µM IPTG. Bacteria were grown overnight with shaking and streaked on BHI agar containing 16 µg/ml ampicillin and 50 µM IPTG. A colony was inoculated in 250 ml of BHI broth supplemented with 16 µg/ml ampicillin and 50 µM IPTG. Bacteria were grown overnight with shaking and streaked on BHI agar containing 16 µg/ml ampicillin and 50 µM IPTG. Five ml of BHI broth containing 10 µg/ml tetracycline was inoculated with a single colony and genomic DNA was extracted (Wizard DNA extraction kit, Promega). Genomic DNA was sequenced by paired‐end joining Illumina (Biomics Platform of the Institut Pasteur, Paris, France). Identification of the mutations was performed with the breseq pipeline (Deatherage & Barrick, 2014).
Plating efficiency assay
Bacteria were grown to the late exponential phase, i.e., to an optical density at 600 nm (OD600) greater than 1.0 (ca. 6 h at 37°C under agitation). The OD600 was adjusted to 1.0‐ and 10‐fold dilutions (10−1 to 10−6) were prepared in BHI broth. Ten µl of the resulting bacterial suspensions was spotted on BHI agar supplemented with inducers and drugs as indicated in the legend to figures. For the disk diffusion assay, 5 µl of the bacterial suspension adjusted to an OD600 of 1.0 was inoculated in 5 ml of water. BHI agar plates were flooded with the latter suspension, excess liquid was removed, and the plates were kept at room temperature for 15 min prior to the addition of paper disks containing antibiotics or inducers. Plates were imaged after 16 h (or 24 h for plates containing ceftriaxone) of incubation at 37°C.
Detection of the production of PBP4 and AmpH with a fluorescent penicillin
Derivatives of BW25113(ycbB, relA′) ΔmepM harboring plasmids pHV48(dacB) or pHV50(ampH) were grown in the presence of L‐rhamnose (0, 0.01, 0.1, and 1%), 10 µg/ml tetracycline, 20 µg/ml chloramphenicol, and 25 µg/ml zeocin to the early stationary phase (OD600 = 3.0). The antibiotics were used to counter‐select loss of plasmids pKT2, pKT8, and pHV48 or pHV50, respectively. Bacteria (1.5 ml) were washed in PBS, concentrated 10‐fold (150 µl), and incubated with 200 µM BOCILLIN® FL Penicillin (Invitrogen) for 10 min at 20°C. Bacteria were washed with PBS, resuspended in 100 µl of PBS, and 100 µl of 2× Laemmli loading buffer was added. The mixture was incubated at 92°C for 3 min, and 30 µl was analyzed by 12% SDS–PAGE. Images were acquired using the IBright FL 1500 (Thermo Fisher) imager in the protein fluorescence mode.
Purification of endopeptidases
The mepM gene was amplified by PCR and cloned into pET‐TEV between the NdeI and XhoI restriction sites. The fusion protein comprised a 6× histidine tag, a TEV protease cleavage site, and residues 41–440 of MepM. The mepH and mepS genes were independently amplified by PCR and cloned in frame with dsbC into pETMM82 using NEBuilder HiFi DNA assembly (New England Biolabs). The fusion proteins comprised the DsbC chaperone (Firczuk & Bochtler, 2007), a 6× histidine tag, a TEV protease cleavage site, and residues 28–271 of MepH or 25–188 of MepS. The enzymes were produced in E. coli BL21(DE3) following induction by 0.5 mM IPTG for 18 h at 16°C. The endopeptidases were purified in 50 mM Tris–HCl pH 8.0 from a clarified lysate by nickel affinity chromatography (elution with 0.5 mM imidazole). The endopeptidases were dialyzed overnight at 4°C against 50 mM Tris–HCl pH 8.0, 0.5 mM EDTA. N‐terminal tags were cleaved overnight at room temperature following addition of 10 µg of TEV protease for every mg of protein and DTT at a final concentration of 0.5 mM. MepM, MepH, and MepS were further purified by size‐exclusion chromatography (Superdex 75 HiLoad 26/60, GE Healthcare) in 50 mM Tris–HCl pH 7.5, 200 mM NaCl.
PBP4 was purified from strain BL21(DE3) pET21bΩPBP4Δ1‐60 as previously reported (Banzhaf et al, 2020). Briefly, cells were grown in the presence of 1 mM IPTG for 8 h at 20°C and then harvested by centrifugation at 7,500 × g, 4°C, 15 min. Cell pellets were resuspended in 50 mM Tris–HCl pH 8.0, 300 mM NaCl and lysed by sonication. Following centrifugation at 14,000 × g, 1 h, 4°C, the NaCl concentration was reduced by stepwise dialysis in a Spectra/Por dialysis membrane (MWCO 12–14 kDa) against 50 mM Tris–HCl pH 8.5 containing (i) 200 mM NaCl, (ii) 100 mM NaCl, and (iii) 30 mM NaCl. The sample was centrifuged at 7,500 × g, 4°C, 10 min, and the supernatant was applied to a 5 ml HiTrap Q HP IEX column in 25 mM Tris–HCl pH 8.5, 30 mM NaCl. Protein was eluted from the column with a linear gradient from 50 mM Tris–HCl pH 8.5, 100 mM NaCl, to 25 mM Tris–HCl pH 8.0, 1 M NaCl, over a 100 ml volume. Fractions containing PBP4 were combined and dialyzed against 10 mM potassium phosphate pH 6.8, 300 mM NaCl. Protein was applied at 1 ml/min to a 5 ml ceramic hydroxyapatite column (Bio‐Rad BioscaleTM) in the dialysis buffer, and a 50 ml linear gradient to 500 mM potassium phosphate pH 6.8, 300 mM NaCl was applied. Fractions with PBP4 were dialyzed overnight against 25 mM HEPES‐NaOH pH 7.5, 300 mM NaCl, 10% glycerol, and concentrated to ca. 5 ml using a Vivaspin concentrator spin column (Sartorius). The protein sample was applied to a HiLoad 16/600 Superdex 200 column (GE healthcare) at 1 ml/min and eluted in a linear gradient to 25 mM HEPES‐NaOH pH 7.5, 300 mM NaCl, 10% glycerol. The collected fractions containing PBP4 were combined.
PBP7 was purified from strain BL21(DE3) pET28aΩpbpGΔ1‐75 as previously reported (Banzhaf et al, 2020). Briefly, cells were grown in the presence of 1 mM IPTG for 3 h at 30°C before being harvested by centrifugation and resuspended in 25 mM Tris–HCl pH 7.5, 500 mM NaCl, 20 mM imidazole. Following sonication and subsequent centrifugation, the lysate was applied to a 5 ml HisTrap HP column (GE healthcare) and washed with 4 column volumes of 25 mM Tris–HCl pH 7.5, 500 mM NaCl, 20 mM imidazole. Bound protein was eluted with 25 mM Tris–HCl pH 7.5, 300 mM NaCl, 400 mM Imidazole. Elution fractions containing PBP7 were combined and dialyzed overnight against 25 mM HEPES‐NaOH pH 7.5, 300 mM NaCl, 10% glycerol, in the presence of 1 unit/ml of restriction grade thrombin (Novagen) to remove the oligohistidine tag. The sample was then concentrated to ca. 5 ml using a Vivaspin concentrator spin column (Sartorius) at 4,500 × g, 4°C. The protein sample was applied to a HiLoad 16/600 Superdex 200 column (GE healthcare) at 1 ml/min and eluted in 25 mM HEPES‐NaOH pH 7.5, 300 mM NaCl, 10% glycerol. Elution fractions containing PBP7 were combined.
Protein concentrations were determined by the Bio‐Rad protein assay using bovine serum albumin as a standard. Endopeptidases were stored at −80°C.
Preparation of sacculi
Bacteria were grown in M9 minimal medium supplemented with 0.1% glucose at 37°C for 48 h. Bacteria were harvested by centrifugation and boiled in 4% sodium dodecyl sulfate (SDS) for 1 h. Sacculi were harvested by centrifugation (20,000 × g for 20 min at 20°C), washed five times with water, and incubated with 100 µg/ml pronase overnight at 37°C in 20 mM Tris–HCl pH 7.5. Sacculi were washed five times with water and incubated overnight at 37°C with 100 µg/ml trypsin in 20 mM sodium phosphate pH 8.0. Sacculi were washed five times with water, boiled for 5 min, collected by centrifugation, resuspended in water, and stored at −20°C.
Digestion of sacculi
Sacculi were digested overnight at 37°C with 120 µM lysozyme alone or in association with an endopeptidase in 40 mM Tris–HCl pH 8.0. Insoluble material was removed by centrifugation, and the soluble fraction containing muropeptides was reduced with sodium borohydride for 1 h in 125 mM borate buffer pH 9.0. The pH of the solution containing the reduced muropeptides was adjusted to 4.0 with phosphoric acid. Muropeptides were separated by rpHPLC in a C18 column (Hypersil GOLD aQ; 250 × 4.6 mm; 3 µm, Thermo Scientific) at a flow rate of 1 ml/min with a linear gradient (0–20%) applied between 10 and 60 min (buffer A, TFA 0.1%; buffer B, acetonitrile 20% TFA 0.1%). Absorbance was monitored at 205 nm and fractions were collected, lyophilized, and analyzed by mass spectrometry. Mass spectra were obtained on a Bruker Daltonics maXis high‐resolution mass spectrometer (Bremen, Germany) operating in the positive mode (Analytical Platform of the Muséum National d’Histoire Naturelle, Paris, France). Theoretical masses were predicted using massXpert, and mass spectral data were explored using mineXpert2 (Rusconi, 2009; Langella & Rusconi, 2021).
Author contributions
HV: conception and design, acquisition, analysis, and interpretation of data, drafting, and revising the article. DD and AL: acquisition of data. WV: interpretation of data, drafting and revising the article. MA and JEH: conception and design, analysis and interpretation of data, drafting and revising the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
This work was supported by the French National Research Agency ANR “RegOPeps” (grant ANR‐19‐CE44‐0007 to JEH). HV is the recipient of a doctoral fellowship from Sorbonne‐Université (ED 515, complexité du vivant). We thank L. Dubost and A. Marie for technical assistance in the collection of mass spectra at the Bio‐organic Mass Spectrometry Plateform. We also thank L. Ma and R. Legendre for technical assistance in genome sequencing. The Biomics Platform is a member of the “France Génomique” consortium supported by the French National Research Agency ANR (grant ANR‐10‐INBS‐0009) and IBISA. We also thank Z. Edoo for proofreading the manuscript.
The EMBO Journal (2021) 40: e108126.
Contributor Information
Michel Arthur, Email: michel.arthur@crc.jussieu.fr.
Jean‐Emmanuel Hugonnet, Email: jean-emmanuel.hugonnet@crc.jussieu.fr.
Data availability
Whole genome sequencing raw data of sltY mutants (M1.1 to M1.4) are available at the Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA748867).
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzhaf M, Yau HC, Verheul J, Lodge A, Kritikos G, Mateus A, Cordier B, Hov AK, Stein F, Wartel Met al (2020) Outer membrane lipoprotein NlpI scaffolds peptidoglycan hydrolases within multi‐enzyme complexes in Escherichia coli . EMBO J 39: e102246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranowski C, Welsh MA, Sham L‐T, Eskandarian HA, Lim HC, Kieser KJ, Wagner JC, McKinney JD, Fantner GE, Ioerger TRet al (2018) Maturing Mycobacterium smegmatis peptidoglycan requires non‐canonical crosslinks to maintain shape. Elife 7: e37516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos PAD, Wheeler R, Boneca IG (2021) Uptake, recognition and responses to peptidoglycan in the mammalian host. FEMS Microbiol Rev 45: fuaa044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonis M, Ecobichon C, Guadagnini S, Prévost MC, Boneca IG (2010) A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol Microbiol 78: 809–819 [DOI] [PubMed] [Google Scholar]
- Caveney NA, Caballero G, Voedts H, Niciforovic A, Worrall LJ, Vuckovic M, Fonvielle M, Hugonnet J‐E, Arthur M, Strynadka NCJ (2019) Structural insight into YcbB‐mediated beta‐lactam resistance in Escherichia coli . Nat Commun 10: 1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Uehara T, Bernhardt TG (2014) Beta‐lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159: 1300–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodisetti PK, Reddy M (2019) Peptidoglycan hydrolase of an unusual cross‐link cleavage specificity contributes to bacterial cell wall synthesis. Proc Natl Acad Sci USA 116: 7825–7830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonge BLM, Wientjes FB, Jurida I, Driehuis F, Wouters JTM, Nanninga N (1989) Peptidoglycan synthesis during the cell cycle of Escherichia coli: composition and mode of insertion. J Bacteriol 171: 5783–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE (2014) Identification of mutations in laboratory‐evolved microbes from next‐generation sequencing data using breseq. Methods Mol Biol 1151: 165–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye A, Collet JF, Laloux G (2016) Fine‐tuning of the Cpx envelope stress response is required for cell wall homeostasis in Escherichia coli . MBio 7: 47–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD (1999) Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol 181: 3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Leibler S (2000) A synthetic oscillatory network of transcriptional regulators. Nature 403: 335–338 [DOI] [PubMed] [Google Scholar]
- Engel H, van Leeuwen AM, Dijkstra A, Keck W (1992) Enzymatic preparation of 1,6‐anhydro‐muropeptides by immobilized murein hydrolases from Escherichia coli fused to staphylococcal protein A. Appl Microbiol Biotechnol 37: 772–783 [DOI] [PubMed] [Google Scholar]
- Firczuk M, Bochtler M (2007) Mutational analysis of peptidoglycan amidase MepA. Biochemistry 46: 120–128 [DOI] [PubMed] [Google Scholar]
- Glauner B, Holtje JV, Schwarz U (1988) The composition of the murein of Escherichia coli . J Biol Chem 263: 10088–10095 [PubMed] [Google Scholar]
- Gonzalez‐Leiza SM, de Pedro MA, Ayala JA (2011) AmpH, a bifunctional DD‐endopeptidase and DD‐carboxypeptidase of Escherichia coli . J Bacteriol 193: 6887–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy C, Gouzy A, Ehrt S (2020) Peptidoglycan hydrolases RipA and Ami1 are critical for replication and persistence of Mycobacterium tuberculosis in the host. MBio 11: e03315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Glasgow J, Jaschke PR, Bawazer LA, Munson MS, Cochran JR, Endy D, Salit M (2017) Measurements of translation initiation from all 64 codons in E. coli . Nucleic Acids Res 45: 3615–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Höltje JV (2002) Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli . J Bacteriol 184: 6093–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heijenoort J (2011) Peptidoglycan Hydrolases of Escherichia coli . Microbiol Mol Biol Rev 75: 636–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson TA, Young KD, Denome SA, Elf PK (1997) AmpC and AmpH, proteins related to the class C beta‐lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli . J Bacteriol 179: 6112–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltje JV, Heidrich C (2001) Enzymology of elongation and constriction of the murein sacculus of Escherichia coli . Biochimie 83: 103–108 [DOI] [PubMed] [Google Scholar]
- Hugonnet JE, Mengin‐Lecreulx D, Monton A, den Blaauwen T, Carbonnelle E, Veckerlé C, Yves VB, van Nieuwenhze M, Bouchier C, Tu Ket al (2016) Factors essential for L, D‐transpeptidase‐mediated peptidoglycan cross‐linking and β‐lactam resistance in Escherichia coli . Elife 5: e19469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Fisher JF, Mobashery S (2013) Bacterial cell‐wall recycling. Ann N Y Acad Sci 1277: 54–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck W, Schwarz U (1979) Escherichia coli murein‐DD‐endopeptidase insensitive to beta‐lactam antibiotics. J Bacteriol 139: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaoglu O, Carlson EE (2015) Profiling of β‐lactam selectivity for penicillin‐binding proteins in Escherichia coli strain DC2. Antimicrob Agents Chemother 59: 2785–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korat B, Mottl H, Keck W (1991) Penicillin‐binding protein 4 of Escherichia coli: molecular cloning of the dacB gene, controlled overexpression, and alterations in murein composition. Mol Microbiol 5: 675–684 [DOI] [PubMed] [Google Scholar]
- Lai GC, Cho H, Bernhardt TG (2017) The mecillinam resistome reveals a role for peptidoglycan endopeptidases in stimulating cell wall synthesis in Escherichia coli . PLOS Genet 13: e1006934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langella O, Rusconi F (2021) mineXpert2: full‐depth visualization and exploration of MSnMass spectrometry data. J Am Soc Mass Spectrom 32: 1138–1141 [DOI] [PubMed] [Google Scholar]
- Lenz JD, Hackett KT, Dillard JP (2017) A single dual‐function enzyme controls the production of inflammatory NOD agonist peptidoglycan fragments by neisseria gonorrhoeae. MBio 8: e01464‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnet S, Dubost L, Marie A, Arthur M, Gutmann L (2008) Identification of the L, D‐transpeptidases for peptidoglycan cross‐linking in Escherichia coli . J Bacteriol 190: 4782–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, Arthur M (2005) A novel peptidoglycan cross‐linking enzyme for a β‐lactam‐resistant transpeptidation pathway. J Biol Chem 280: 38146–38152 [DOI] [PubMed] [Google Scholar]
- Mainardi JL, Villet R, Bugg TD, Mayer C, Arthur M (2008) Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram‐positive bacteria. FEMS Microbiol Rev 32: 386–408 [DOI] [PubMed] [Google Scholar]
- Morè N, Martorana AM, Biboy J, Otten C, Winkle M, Serrano CKG, Montón Silva A, Atkinson L, Yau H, Breukink Eet al (2019) Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. MBio 10: e02729‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos M, Peters K, Vollmer W (2017) Robust peptidoglycan growth by dynamic and variable multi‐protein complexes. Curr Opin Microbiol 36: 55–61 [DOI] [PubMed] [Google Scholar]
- Pazos M, Peters K (2019) Peptidoglycan subcellular biochemistry. New York: Springer; 127–168 [DOI] [PubMed] [Google Scholar]
- Pisabarro AG, De Pedro MA, Vazquez D (1985) Structural modifications in the peptidoglycan of Escherichia coli associated with changes in the state of growth of the culture. J Bacteriol 161: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo GD, Gold L (1992) Translation initiation in Escherichia coli: sequences within the ribosome‐binding site. Mol Microbiol 6: 1219–1229 [DOI] [PubMed] [Google Scholar]
- Romeis T, Holtje J‐V (1994) Penicillin‐binding protein 7/8 of Escherichia coli is a dd‐endopeptidase. Eur J Biochem 224: 597–604 [DOI] [PubMed] [Google Scholar]
- Rusconi F (2009) massXpert 2: A cross‐platform software environment for polymer chemistry modelling and simulation/analysis of mass spectrometric data. Bioinformatics 25: 2741–2742 [DOI] [PubMed] [Google Scholar]
- Sanders AN, Pavelka MS (2013) Phenotypic analysis of Eschericia coli mutants lacking l, d‐transpeptidases. Microbiology 159: 1842–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P (2008) The penicillin‐binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32: 234–258 [DOI] [PubMed] [Google Scholar]
- Schreiber G, Metzger S, Aizenman E, Roza S, Cashel M, Glaser G (1991) Overexpression of the relA gene in Escherichia coli . J Biol Chem 266: 3760–3767 [PubMed] [Google Scholar]
- Singh SK, Parveen S, SaiSree L, Reddy M (2015) Regulated proteolysis of a cross‐link–specific peptidoglycan hydrolase contributes to bacterial morphogenesis. Proc Natl Acad Sci USA 112: 10956–10961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, SaiSree L, Amrutha RN, Reddy M (2012) Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Mol Microbiol 86: 1036–1051 [DOI] [PubMed] [Google Scholar]
- Truong TT, Vettiger A, Bernhardt TG (2020) Cell division is antagonized by the activity of peptidoglycan endopeptidases that promote cell elongation. Mol Microbiol 114: 966–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RD, Vollmer W, Foster SJ (2014) Different walls for rods and balls: the diversity of peptidoglycan. Mol Microbiol 91: 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Dinh T, Bernhardt TG (2009) LytM‐domain factors are required for daughter cell separation and rapid ampicillin‐induced lysis in Escherichia coli . J Bacteriol 191: 5094–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Park JT (2008) Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J Bacteriol 190: 3914–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellanoweth RL, Rabinowitz JC (1992) The influence of ribosome‐binding‐site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo . Mol Microbiol 6: 1105–1114 [DOI] [PubMed] [Google Scholar]
- Verheul J, Lodge A, Yau HC, Liu X, Typas A, Banzhaf M, Vollmer W & Den T (2020) Midcell localization of PBP4 of Escherichia coli is essential for the timing of divisome assembly. bioRxiv 10.1101/2020.07.30.230052 [PREPRINT] [DOI]
- Vollmer W (2012) Bacterial growth does require peptidoglycan hydrolases. Mol Microbiol 86: 1031–1035 [DOI] [PubMed] [Google Scholar]
- Weber H, Polen T, Heuveling J, Wendisch VF, Hengge R (2005) Genome‐wide analysis of the general stress response network in Escherichia coli: σS‐dependent genes, promoters, and sigma factor selectivity. J Bacteriol 187: 1591–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]