

Abstract
The intricate process of human mtDNA replication requires the coordinated action of both transcription and replication machineries. Transcription and replication events at the lagging strand of mtDNA prompt the formation of a stem‐loop structure (OriL) and the synthesis of a ∼25 nt RNA primer by mitochondrial RNA polymerase (mtRNAP). The mechanisms by which mtRNAP recognizes OriL, initiates transcription, and transfers the primer to the replisome are poorly understood. We found that transcription initiation at OriL involves slippage of the nascent transcript. The transcript slippage is essential for initiation complex stability and its ability to translocate the mitochondrial DNA polymerase gamma, PolG, which pre‐binds to OriL, downstream of the replication origin thus allowing for the primer synthesis. Our data suggest the primosome assembly at OriL—a complex of mtRNAP and PolG—can efficiently generate the primer, transfer it to the replisome, and protect it from degradation by mitochondrial endonucleases.
Keywords: mitochondrial replication, mitochondrial transcription, PolG, POLRMT, primosome
Subject Categories: Chromatin, Transcription & Genomics; DNA Replication, Recombination & Repair; Organelles
Replication of human mitochondrial DNA involves transcript slippage and RNA primer handover/protection within a primosome formed by mtRNAP and DNA polymerase PolG.
Introduction
Replication and transcription of the multi‐copy human mitochondrial genome require a carefully orchestrated interplay between the corresponding molecular machineries. This involves generation of replication primers by mitochondrial RNA polymerase (mtRNAP or POLRMT), which acts as a primase but is also responsible for the synthesis of all types of mitochondrial RNA (Masters et al, 1987; Wanrooij et al, 2008). Because deletions in mitochondrial DNA are connected to a number of mitochondrial diseases and also accumulate during aging, mechanisms of replication of mitochondrial DNA are the focus of intensive studies (Persson et al, 2019; Falkenberg & Gustafsson, 2020).
Over the past few decades, several models of replication of circular human mitochondrial genome have been put forth; however, the prevailing model postulates that generation of the daughter strands of mtDNA occurs asynchronously (Clayton, 1982; Phillips et al, 2017). First, transcription at the light‐strand promoter, LSP, generates a ∼120 nt RNA product that is transferred to the replisome, which consists of DNA polymerase PolG and the replicative helicase TWINKLE (Jemt et al, 2011; Sen et al, 2012; Milenkovic et al, 2013). Upon replication of the heavy stand through about 2/3 of the length of genome, the region known as an origin of replication of the light strand, or OriL, becomes single‐stranded and forms a hairpin. This hairpin is recognized by mtRNAP, which produces 20–30 nt replication primer (Hixson et al, 1986; Wanrooij et al, 2008; Fuste et al, 2010). The primer is then extended by PolG to generate a copy of the light strand of mtDNA (Fuste et al, 2010; Wanrooij et al, 2012b).
Phylogenetic analysis suggests that OriL appeared early in vertebrate evolution as it is absent in invertebrate chordates but present in vertebrate classes that are distant to Mammalia, such as Chondrichthyes (Wanrooij et al, 2012b). In mammalian mitochondrial genomes, the OriL region is found in a cluster of tRNA‐encoding genes and does not overlap with any genes in either strand of mtDNA. After the hairpin structure is formed, mtRNAP is thought to recognize both its sequence and topology and initiate transcription at a site located in the stretch of dTMP residues in the stem loop (Hixson et al, 1986). The exact location of the transcription start site in OriL has never been determined. The T‐stretch, and the sequence of the stem were both suggested to be important for the OriL transcription (Fuste et al, 2010; Wanrooij et al, 2012b). Although the single‐stranded DNA‐binding protein, mtSSB seems to be dispensable for the process of primer synthesis, it is required to prevent non‐specific transcription initiation at other single‐stranded DNA regions (Miralles Fuste et al, 2014). Because of a lack of RNA displacement, OriL transcription results in the formation of an extended RNA–DNA hybrid, which destabilizes the initial transcription complex and leads to dissociation of mtRNAP (Fuste et al, 2010). In vivo and in vitro experiments show that the region where the transition from RNA to DNA occurs is heterogeneous and is located approximately 16–21 nt downstream from the 5’ end of the T‐stretch region (Tapper & Clayton, 1982; Wong & Clayton, 1985; Kang et al, 1997; Fuste et al, 2010). The exact molecular mechanism of the transfer of the primer from mtRNAP to PolG remains obscure.
Stem loop structures have been previously implicated in replication initiation; however, only few were shown to involve primer synthesis by RNA polymerase. Examples of RNAP‐dependent replication include phage M13, which requires the host polymerase to bind an imperfect hairpin, synthesize an 18–20 nt primer, and then “backtrack” along the RNA–DNA hybrid, leaving the 3’ end of RNA accessible for extension by DNA polymerase III (Zenkin et al, 2006). In bacteria, replication of the Escherichia coli plasmid F is mediated by formation of an extended hairpin structure in the single‐stranded DNA region that is recognized by RNAP (Masai & Arai, 1997). While the N4 coliphage hairpin P2 has not been implicated in DNA replication, it serves as an efficient promoter for a single‐subunit mtRNAP‐like polymerase (Davydova et al, 2007). Unlike the situation with OriL, transcription initiation start sites in these systems are found in the stem of the hairpin, suggesting a different mechanism of promoter recognition and transcription initiation (Tapper & Clayton, 1982; Fuste et al, 2010).
Here, we define the bona fide transcription start site at OriL and demonstrate that the generation of the replication primer by mtRNAP employs a transcript slippage mechanism. This feature is conserved in mammals and is essential for initiation complex stability, replication primer generation, and priming of the light‐strand synthesis. Our data suggest a model for transcription and replication initiation events at OriL that involves a concerted action of RNA and DNA polymerases.
Results
Transcription at OriL starts at position T5751 and involves transcript slippage
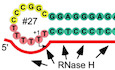
Previous reports have suggested that the OriL transcription start site (TSS) is located at the most upstream thymidine (T5747) in the stretch of six dTMP residues (indicated by the asterisk in Fig 1A; Hixson et al, 1986). To verify this, we used the OriL‐containing DNA template (WT, template #1) and performed in vitro transcription assays in the presence of ATP, CTP, and GTP. Assuming the TSS at position T5747, this template should direct the synthesis of a 27 nt long transcript under these conditions. Instead, the experiment revealed the presence of a set of RNA products of different sizes, with some prominent products exceeding the length of the expected transcript (Fig 1B). This indicated that transcription may have started at a position other than the predicted T5747. To map the exact location of the TSS and to probe the significance of the T‐stretch residues for transcription, we tested a set of OriL variants in which each dTMP residue in the OriL loop was substituted with either dAMP or dCMP (Fig 1B and C, Appendix Table S1). Strikingly, substitution of T5750 to A (template #5, or −1A) resulted in the synthesis of predominantly 23 nt long RNA product (Fig 1B, lane 5). It is apparent that this transcript originated at the base T5751, which we designate here as the TSS (or +1 base). This finding suggests that the heterogeneity of the RNA products observed at OriL is likely due to transcript slippage—repetitive addition of AMP residues to the transcript without translocation of polymerase relative to the DNA template. In agreement with this notion, substitution of the −1T base to C also resulted in the elimination of slippage and generation of predominantly 23 nt long RNA product (Fig 1C, lane 5). Our data show that transcript slippage at OriL requires the presence of an uninterrupted stretch of at least three dTMP residues including the TSS and the flanking T bases (i.e., −1 and +2 bases). Consistently, OriL variants having substitutions that do not disrupt the T‐stretch at the TSS (i.e., −4A, −3A, −2A, −4C, −3C, −2C) were prone to transcript slippage (Fig 1B and C). Notably, while the possibility of slippage on a T‐stretch was eliminated in +2C template, a slippery C‐stretch was created instead (Figs 1C and EV1). Accordingly, transcription of this template could start at +1 with no slippage or it could misstart at +2 and proceed with slippage by repetitive addition GMP residues, albeit with lower efficiency (Figs 1C and EV1).
Figure 1. Transcript slippage occurs during initiation of the replication primer synthesis at OriL.
-
ASchematic representation of the human OriL stem‐loop structure. Human OriL comprises a 12 nt loop and an 11 bp stem. The conserved residues of the OriL stem are shown as green circles and the stretch of the dTMP residues harboring the transcription initiation site are in pink. Non‐conserved sequence in the loop and sequences beyond the stem region are shown in yellow. The OriL transcription initiation start site (+1) is shown as determined in this work (bent arrow) and its location in the human mtDNA (accession NC_012920.1) is indicated.
-
B, CTranscription initiation at OriL involves transcript slippage. The products of transcription by mtRNAP in the presence of a limiting set of substrates (ATP, CTP, and GTP) are shown. Transcription assays were performed using either the WT OriL as DNA template or OriL variants, in which particular dTMP residues in the T‐stretch region were substituted with either dAMP (B) or dCMP (C). The substitutions are specified above each lane and the corresponding template numbers are listed below the images. The labels on the left indicate the length of transcripts.
-
DIdentification of the OriL transcription start site. Transcription assays were performed involving the WT and mutant −1A and +1C OriL templates, as indicated on the top, in the presence of ATP, CTP, and GTP. The transcription products were labeled either at each GMP residue by adding [α‐32P]‐GTP to the substrate mixtures or at their 5’ ends by utilizing [γ‐32P]‐initiating nucleotides, ATP or GTP, as indicated above each lane.
-
ESchematic illustration of the process of transcript slippage at OriL. A portion of the OriL loop is shown in black in the 3′ to 5′ direction with the transcription start site indicated as +1. Two substrate ATP molecules (red) base‐pair the templating T bases at positions +1 and +2 as shown. Upon formation of the first phosphodiester bond, the resulting AA transcript (red) slides along the DNA template to base‐pair the −1 and +1 T bases. This vacates the substrate‐binding site opposite the +1 T base. Incorporation of another AMP residue results in extension of the nascent transcript, which in turn also “slips” along the DNA template as shown. The nucleotide incorporation and RNA slippage steps then continue. Note that the slippage ultimately results in the formation of a 6 bp long RNA–DNA hybrid because of the presence of six templating T residues in the human OriL.
Figure EV1. Transcript slippage during initiation at OriL.
Left Panel—schematic representation of the mutated OriL templates used in the experiment. Right panel—the products of transcription by mtRNAP in the presence of a limiting set of substrates (ATP, CTP, and GTP) are shown. The 5' ends of the transcripts in lanes 1–3 were labeled by incorporation of [γ‐32P]‐ATP, while transcripts in the reactions shown in lanes 4‐6—internally, by incorporation of [α‐32P]‐GTP. Disruption of the T‐stretch in OriL prevents transcript slippage using AMP (lanes 2,3). Transcript slippage using GMP is observed at +2C OriL due to generation of the C‐stretch in this OriL variant (lane 6).
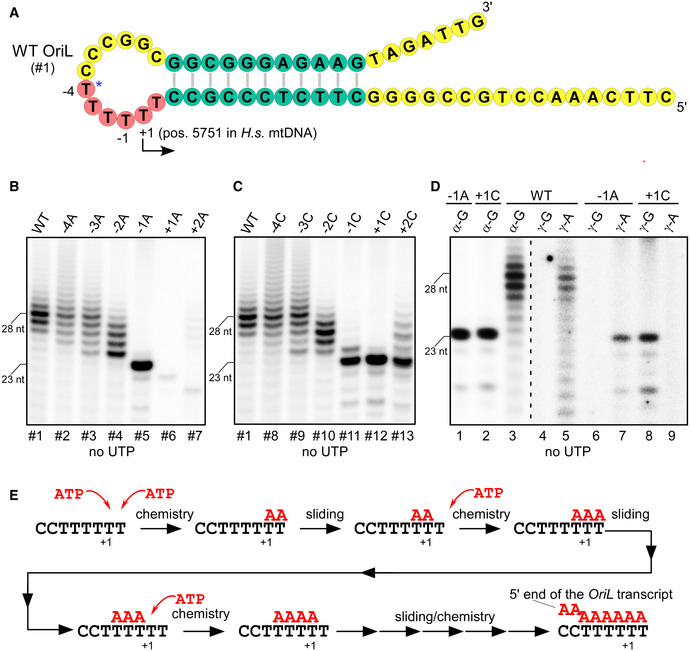
Interestingly, when the +1T base was substituted for C (template #12), a 23 nt long RNA product was also observed, suggesting that this template directs the efficient utilization of GTP as the priming nucleotide (Fig 1C, lane 6). To confirm this, [γ‐32P]‐GTP or [γ‐32P]‐ATP was used to label the 5’ ends of the RNA (Fig 1D). The WT and the −1A OriL template variants produced labeled RNA in the presence of [γ‐32P]‐ATP, in agreement with the TSS being at position T5751 (Fig 1D, lanes 5,7). In the case of the +1C OriL template, transcription products were only observed in the presence of [γ‐32P]‐GTP (Fig 1D, lane 8). This further confirms residue T5751 as the TSS for OriL.
The human OriL contains an uninterrupted stretch of six dTMP residues at the site of transcription initiation. As illustrated in Fig 1E, transcript slippage generates products that have 5–6 AMP residues at their 5’ ends. A major product of the WT OriL transcription generated in the absence of UTP is 28 nt long (Fig 1B). Taking into account the results of the experiments on mapping of the RNA to DNA transition in the OriL region (Hixson et al, 1986; Kang et al, 1997; Fuste et al, 2010), these in vitro RNA products represent the replication primers generated by mtRNAP during replication of the light strand of mtDNA.
The topology but not the sequence of the OriL is critical for transcription initiation
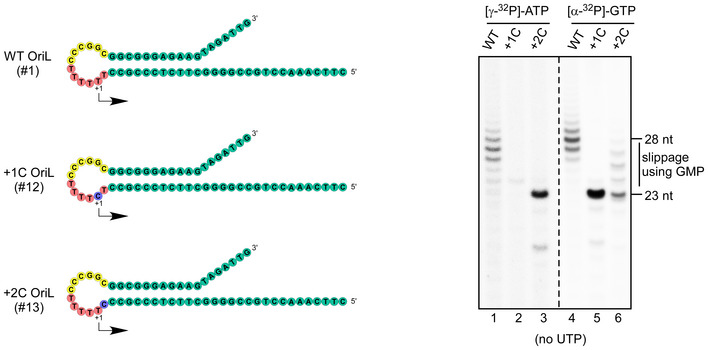
Sequence alignment of the OriL region reveals its high conservation among mammalian species (Fig 2A). The 11–12 bp stem of the OriL hairpin is remarkably well conserved, especially considering how highly divergent the mitochondrial promoters are in various mammals (Gaspari et al, 2004). While the sequences of the 11–14 nt loops vary, all of them contain a conserved stretch of dTMP residues, which can range from 4 to 11 residues in length. We therefore set out to probe the significance of the OriL sequence conservation with respect to primer synthesis by mtRNAP. To do this, we generated several modified OriL templates, in which various conserved sequence elements were altered (Fig 2B) and tested them in transcription assays.
Figure 2. Transcription initiation at OriL is sequence‐independent.
-
AThe OriL sequence is conserved among mammalian species. Sequence alignment of OriL regions of species representing different families of placental mammals. The color code is as follows: green—the conserved bases in the stem; yellow—the conserved bases in the regions beyond the OriL stem; pink: the T‐stretch in the OriL loop.
-
BSchematic representation of the mutated OriL templates used in panels C through G. The templates are grouped by experiments shown below. The color scheme is as indicated in Fig 1A. The purple circles indicate the transcription start site in the templates having +1T to C substitutions and thus initiating with GTP. Other substitutions in the WT sequence are shown in cyan. Partial sequence of the transcribed region is shown. The templates used in the experiments shown in panels C‐G are identified atop of each lane.
-
CSubstitution of the T‐stretch residues upstream of the TSS does not affect transcription initiation. The reaction was performed in the presence of ATP, CTP, and GTP using template #14, in which dTMP residues in the loop (−4 to −1) have been substituted to four dAMP residues.
-
D, EChanges in the OriL sequence do not affect transcription initiation. Transcription assays were performed using WT OriL (D and E, left lanes), the OriL template with substitutions in the loop region (residues −5 to −10, D, middle lane), the OriL template, in which the entire sequence of the stem has been changed to the complementary (D, right lane) and a stem loop found in tomato golden mosaic virus TGMV (E, right lane). Note the difference in mobility of the 35 nt long run‐off transcripts due to purine/pyrimidine composition of the transcribed region of OriL and the other templates (panel D).
-
F, GThe size of the OriL loop is critical for transcription initiation. Products of transcription reactions performed with the indicated templates in the presence of ATP, CTP, and GTP. The size of the OriL loop was decreased by 1, 2, 3, and 4 nt in templates #17 through #20 (F). Substitution of the −10 base with G or A (templates #21 and #23, respectively) results in a smaller loop due to base paring with +2T, which inhibits transcription (panel G).
To simplify the interpretation of the data, we used the OriL templates having +1C mutation, which prevents transcript slippage and enables synthesis of homogeneous RNA transcripts. First, we altered the conserved dTMP residues (−1 to −4) and the sequence upstream of the T‐stretch (−5 to −10) in the OriL loop (templates # 12, 14, and 15). We found that these substitutions had little effect on transcription efficiency (Fig 2C and D). We next made several individual single base‐pair mutations in the stem region (Fig EV2A–D) and substituted the entire stem sequence by swapping the strands of the stem (Fig 2B and D, template #16). Contrary to the previous observation (Wanrooij et al, 2012b), we found that transcription efficiency was not affected by these sequence alterations. To further prove that the sequence of the OriL stem is not required for transcription initiation, we used a stem‐loop of an identical topology (11 bp stem, 12 nt loop) found in the genome of tomato golden mosaic virus (TGMV), which contained a sequence not found in human mtDNA (Orozco & Hanley‐Bowdoin, 1996). We found that it is efficiently transcribed by mtRNAP (Fig 2E).
Figure EV2. Transcription initiation at OriL is sequence‐independent.
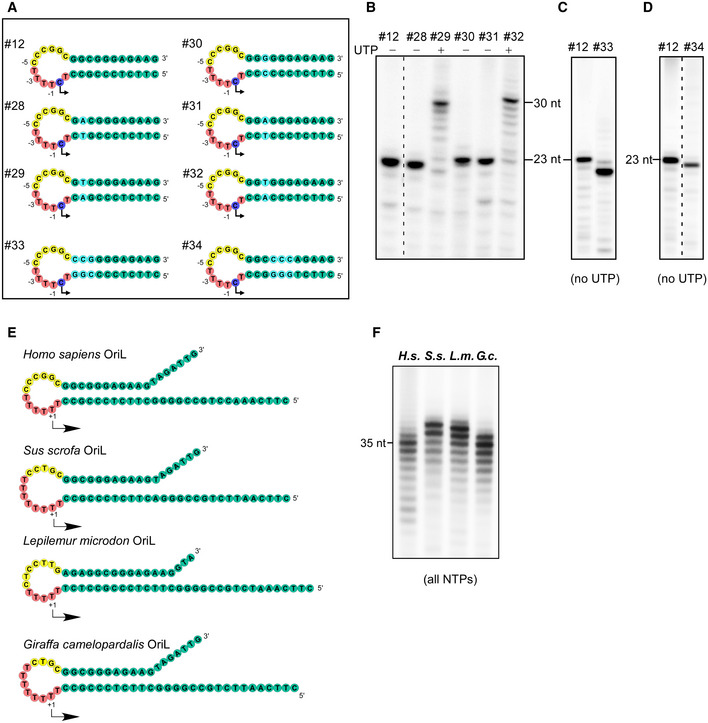
-
ASchematic representation of the mutated OriL templates used in panels B through D. The templates are grouped by experiments shown below. The color scheme is as indicated in Fig 1A. The purple circles indicate the transcription start site in the templates having +1T to C substitutions and thus initiating with GTP. Other substitutions in the WT sequence are shown in cyan. Partial sequence of the transcribed region is shown. The templates used in the experiments shown in panels B–F are identified atop of each lane. Substitutions in the OriL stem have been chosen to match the ones used in the previous study (Wanrooij, 2012b).
-
B–DChanges in the OriL sequence do not affect transcription initiation. Transcription assays were performed using WT OriL (left lanes in B–D) and the OriL templates with substitutions in the stem region as indicated in panel A. Partial NTP mixture (no UTP) has been used in all assays except for the templates #29 and #32, which required all NTPs to make a run‐off product. Note that the RNA products obtained on templates #33 and #34 appear to be smaller than the RNA made on WT OriL due to difference in their G‐C content.
-
ESchematic representation of the OriL templates used in panel F.
-
FHuman mtRNAP efficiently transcribes OriL from other mammalian species.
Since the sequence context did not appear to play any role in OriL transcription, we supposed that it must be the topology of the hairpin that is critical for mtRNAP to start transcription, as has been suggested earlier (Wanrooij et al, 2012b). To verify this, we designed and tested four OriL variants in which the loop of the hairpin was reduced by 1, 2, 3, and 4 nt (Fig 2B, templates #17–20). Transcription assays showed that deletion of a single nucleotide was well tolerated. Longer deletions caused either a drastic decrease or a complete abolishment of the mtRNAP transcription activity (Fig 2F), consistent with the previous observations (Wanrooij et al, 2012b). Alteration of the −10 base in the loop of the hairpin (−10C to A), which affected the base‐pairing of +2T (Fig 2G, template #23), resulted in dramatic changes in transcription efficiency. Consistently, transcription was significantly decreased in response to the −10C to G substitution (Fig 2G, template #21). This was likely caused by shortening of the loop due to the formation of a wobble −10G/+2T base pair. Noticeably, introduction of a T at position −10, which kept the +2T base mismatched, had little effect on transcription, as expected (Fig 2G, construct #22).
Transcription at OriL is not species‐specific since efficient RNA synthesis was detected when human mtRNAP transcribed lemur, giraffe or porcine stem loops (Fig EV2E and F). Other single‐subunit RNA polymerases of the Pol A superfamily, such as Saccharomyces cerevisiae mtRNAP and T7 RNAP could efficiently utilize the OriL hairpin as a template (Fig 3A). Moreover, a structurally unrelated to mtRNAP multi‐subunit E.coli core RNAP was able to generate RNA transcripts on the WT OriL template (Fig 3B).
Figure 3. Transcription initiation at OriL does not depend on the structural elements important for promoter initiation.
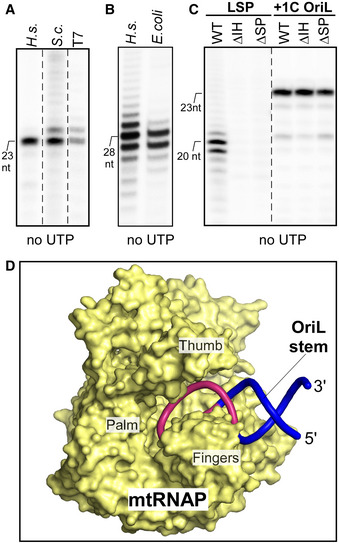
-
A, BSingle‐subunit and multi‐subunit RNA polymerases can transcribe the OriL stem‐loop structure. Transcription assays were run using +1C OriL (template #12) and the human or yeast Saccharomyces cerevisiae mtRNAP and phage T7 RNAP (A), and E. coli core RNAP on WT OriL (B).
-
CThe mtRNAP structural elements required for promoter initiation are dispensable for the OriL transcription. Transcription assays were performed using LSP or +1C OriL (template #12), as indicated, with the WT mtRNAP or its mutants lacking the intercalating hairpin (ΔIH) or the specificity loop (ΔSP).
-
DA structural model of the mtRNAP‐OriL complex. The stem‐loop structure used to represent OriL is shown in pink (13 nt loop region) and blue (8 bp stem region). MtRNAP molecule is depicted as a surface model (yellow) with major subdomains indicated. In this configuration of the complex, the DNA initiating nucleotide is placed in the vicinity of the enzyme’s active center to allow base‐pairing with the priming ATP molecule.
Finally, we probed whether the structural elements responsible for promoter recognition and transcription initiation in mtRNAP also play a role in transcription initiation at OriL (Fig 3C). The specificity loop of the mtRNAP is thought to recognize the promoter sequence, while the “intercalating” hairpin is required for the maintenance of the upstream edge of the transcription bubble (Hillen et al, 2017). Consistently, deletion of these elements results in mtRNAPs that are unable to initiate transcription from LSP or HSP (Fig 3C) (Ringel et al, 2011). In contrast, these mutants were as active in the OriL transcription assays as the WT mtRNAP. These data indicate that transcription initiation at OriL is distinct from transcription at promoter regions in both structural and mechanistic aspects.
A model of the mtRNAP‐OriL transcription initiation complex
Identification of the exact location of the OriL TSS (Fig 1) is significant because it allows for structural modeling of the transcription initiation complex. To build the model, we took into account that the +1T base must occupy a location in the active site of mtRNAP that is similar to that of the priming (or +1) DNA base in the elongation complex. Also, the trajectory of the OriL duplex must be the same as the trajectory of downstream DNA observed in both the promoter initiation and elongation complexes of mtRNAP (Schwinghammer et al, 2013). This would allow for melting of the DNA duplex (i.e., the OriL stem) by helices O and Y of the mtRNAP. To obtain a model of the OriL‐mtRNAP complex, we used the structure of elongation complex of mtRNAP (PDB ID 4BOC). We docked the structure of a stem loop (PDB ID 2M1V), which is similar to OriL in length of the stem and in size of the loop, by superimposing its stem into the downstream DNA duplex of the elongation complex. This resulted in the structure presented in Fig 3D. Analysis of the model suggests that the loop of OriL can be neatly accommodated in the RNA–DNA‐binding cavity of mtRNAP between the "thumb" and the "finger" subdomains (Fig 3D). Also, DNA hairpins having larger loops (> 13–14 nt) would not be able to fit without substantially clashing with mtRNAP. These observations likely explain the conservation of the size of mammalian OriL loops. While the presented model remains tentative, it is consistent with the findings that binding of OriL by mtRNAP is sequence‐independent, and that the specificity loop and the intercalating hairpin do not play a significant role in OriL transcription.
Transcript slippage increases the stability of OriL initiation complex
The apparent conservation of the OriL transcript slippage mechanism prompted us to investigate it further. We first looked at generation of short (2–12 nt) abortive RNA products, which accompanies transcription initiation in various systems (Diaz et al, 1996). Presumably, this occurs because the RNAP complexes are unstable during initiation, thus making it the rate‐limiting stage of the overall process of transcription. We compared the efficiency of abortive initiation by mtRNAP for the WT OriL template and a no‐slippage −1A OriL mutant. Abortive RNA products were hardly detectable when the WT OriL was used (Fig 4A). However, we observed a significant accumulation of 4–5 nt RNA products when transcription slippage was prevented by the −1A substitution (Fig 4A). This is indicative of a much greater transcript release frequency seen with the early −1A OriL initiation complexes. Thus, the ratio between abortive and run‐through products increased dramatically (∼25 fold) when transcript slippage was prevented (Fig 4B). When the transcripts exceed 5 nt in length, the stability of the complexes appeared to be the same for both templates. These data indicate that the transcription slippage events promote the transition of unstable initiation complexes into elongation by enabling incorporation of several AMP residues without translocation along the DNA template.
Figure 4. Transcript slippage at OriL increases efficiency of transcription initiation.
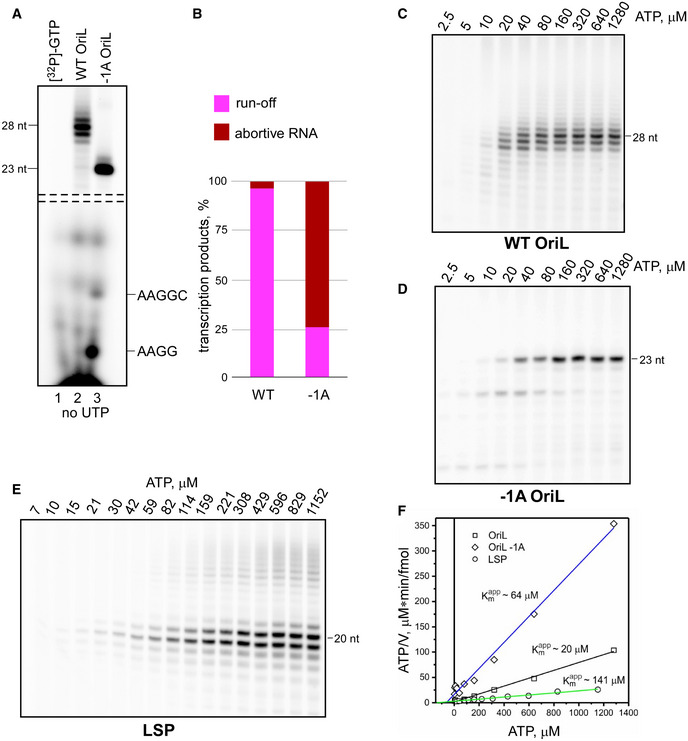
-
A, BTranscript slippage stabilizes the OriL transcription initiation complexes. Transcription products generated on WT and −1A OriL templates in the presence of ATP, CTP, and GTP. The RNA species in the top part of the image correspond to the complexes halted at position +23 relative to the TSS. The RNA length is indicated on the left side. The bottom of the image shows abortive RNA products (identified to the right). A sample of [α‐32P]‐GTP was loaded as a control in lane 1. Note the drastic enhancement of the abortive transcripts when transcript slippage is prevented by the −1A substitution in the template (panel B).
-
C–FTranscript slippage at OriL increases affinity of mtRNAP to the substrate ATP. Products of mtRNAP transcription on WT OriL (C), −1A OriL (D) and LSP (E) performed in the presence of CTP, GTP (OriL) or CTP, GTP, UTP (LSP) and changing concentrations of ATP. To calculate Km app for ATP, Hanes‐Woolf plot analysis (F) was performed for the experiments shown in panels C‐E. The Km values were averaged over three independent experiments and found to be 20 ± 2 μM (WT OriL), 64 ± 9 μM (−1A OriL), and 142 ± 5 μM (LSP).
We next probed the kinetics of transcription initiation as a function of ATP concentration and measured the apparent Km for generation of a 28 nt RNA primer at the WT and a mutant OriL template (−1A), which does not allow for transcript slippage to occur. We found that the apparent Km for the WT OriL was ∼20 μM (Fig 4C and F). In contrast, in the absence of a transcript slippage it increased about threefold (Fig 4D and F), indicating that slippage increases the efficiency of initiation at lower ATP concentrations. The low apparent Km for OriL transcription is in contrast to promoter‐dependent initiation, for which significantly higher levels of ATP (sevenfold) are required to reach a half‐maximum velocity of transcription (Fig 4E and F).
Transcript slippage is required for OriL replication
The observation that transcribing initiation complexes at OriL are greatly stabilized by transcript slippage suggested that this mechanism may have functional importance during early stages of replication of the mtDNA lagging strand. To evaluate this possibility, we used coupled transcription‐replication assays, in which both mtRNAP and PolG were allowed to bind OriL in the presence of their corresponding substrates. To exclude potential effects of a proximal unpaired 3’‐DNA end on PolG binding to OriL, templates with 20 nt long single‐stranded “arms” (#36 and 37) were used (Fig 5A). In these reactions, all four dNTPs were added to support DNA synthesis and a limiting set of rNTPs (no UTP) was present to allow the generation of RNA primers. Thus, the experimental system was expected to produce RNA primers, which are then extended in situ by DNA polymerization (Fig 5A). As with the experiments described in previous sections, we focused on OriL templates that either allowed or prevented transcript slippage during initiation (WT vs. −1A OriL). We found that the ∼28 nt RNA primer generated with the WT OriL was readily extended by PolG to produce ∼38 nt RNA/DNA species (Fig 5B, lanes 1,3). In contrast, no DNA product was observed when the assay was performed using −1A OriL as the template (Fig 5B, lane 4). This suggests that transcript slippage plays a key role in the early stages of mtDNA replication. We therefore set out to investigate this phenomenon further.
Figure 5. Transcript slippage at OriL is required for efficient priming of replication.
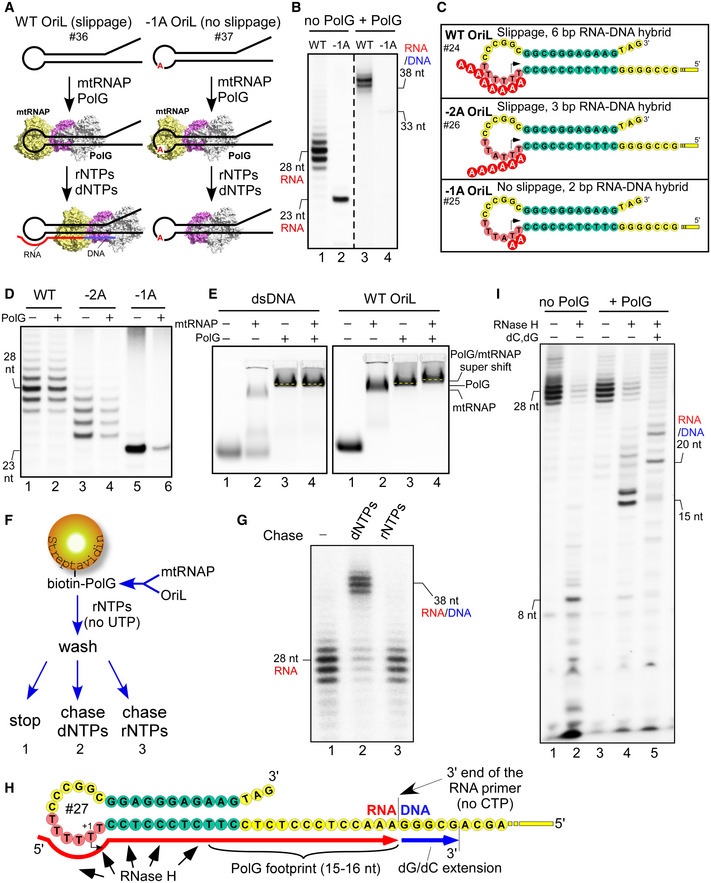
-
ASchematic illustration of the coupled transcription‐replication assay. OriL template, either WT or a no‐slippage −1A, is incubated with mtRNAP and PolG. A mixture of all four dNTPs is then added together with three rNTPs (ATP, CTP, and GTP). The products of transcription and DNA replication are then resolved by denaturing PAGE and visualized.
-
BTranscript slippage at OriL is required for efficient primer extension by PolG. Coupled transcription‐replication assay was performed using WT (lanes 1,3) and −1A OriL (lanes 2,4) as illustrated in panel A. The RNA primer was labeled by incorporation of [α‐32P]‐GTP. PolG was added to the reaction mixtures as specified on the top. The position of the expected RNA/DNA product for −1A OriL (33 nt) is indicated.
- C
-
DBoth slippage and the formation of an extended RNA–DNA hybrid are required for efficient primer generation in the presence of PolG. Transcription assay was performed using templates as illustrated in panel C in the absence of dNTPs.
-
EBoth mtRNAP and PolG bind to the OriL region. EMSA was performed using a random double‐stranded DNA (left) or WT OriL (right). Note the change of mobility of the labeled species in the presence of both mtRNAP and PolG (“super” shift).
-
F, GOriL‐mtRNAP‐PolG complex is able to generate RNA primer and initiate DNA synthesis. Schematic illustration of immobilized transcription‐replication assay (F). Transcription‐replication complex, composed of OriL, mtRNAP, and biotinylated PolG was immobilized on streptavidin‐agarose beads and washed to remove any unbound material. NTP mix (no UTP) was then provided to allow mtRNAP to start transcription. The beads were washed and the transcribing complexes chased with either dNTPs or NTPs. The products of the assay (F) performed in the absence of chase (Panel G, lane 1; note the formation of the set of RNA primers) and after the chase with dNTPs (lane 2) or rNTPs (lane 3) are indicated.
-
GSchematic representation of the mutated OriL template used to probe transcription‐replication in the presence of RNase H. The color scheme is as indicated in Fig 1A. Partial sequence of the transcribed region is shown. The position of the halted mtRNAP in the absence of CTP is indicated. PolG footprint on RNA (red) generated by RNase H cleavage is shown. Extension of the RNA primer by PolG is shown by blue line.
-
HMtRNAP generates products resistant to RNase H degradation only in the presence of PolG. RNase H was added to transcription reaction performed in the absence (lane 2) or in the presence (lanes 4,5) of PolG. Upon RNase H addition, PolG was allowed to extend the RNA primer by 5 nt by incubation with dGTP/dCTP mixture (lane 5).
As both the WT and the −1A OriL transcription appear to generate similar amounts of transcripts in the absence of PolG (Fig 5B, lanes 1, 2), we reason that it is the preloading of PolG onto OriL that causes the observed disruption of RNA synthesis at the −1A OriL template by the transcription initiation complex that is inherently unstable due to the absence of transcript slippage. We therefore compared the transcription efficiency in the presence or absence of PolG on the −1A OriL template; on −2A OriL, which allows transcript slippage but directs the formation of only 3 bp initial A‐T RNA–DNA hybrid; and on the WT OriL (Fig 5 C and D). We found that whereas the primer generation at WT OriL template was only slightly affected by PolG (∼1.3‐fold), transcription of −1A OriL was severely impaired (Fig 5D). A moderate decrease in transcription efficiency (∼twofold) was observed for the −2A OriL variant. These observations suggest that the extended T‐stretch region in human OriL enhances the stability of the initiation complex by facilitating the formation of an extended RNA–DNA hybrid at the 5’ end of the transcript.
Binding of PolG and mtRNAP to OriL and the mechanism of replication primer transfer
Since the transcription‐replication assays suggested binding of PolG to the OriL template, we probed whether a ternary complex between mtRNAP, PolG, and OriL can be assembled. Using an electrophoretic mobility shift assay (EMSA) with a fluorescently labeled OriL template, we found that mtRNAP binds OriL with a sub‐micromolar affinity, while PolG exhibits a slightly better affinity (Fig EV3A–C). When both PolG and mtRNAP were added to the OriL template, a “super‐shifted” band was observed, which migrates above the bands representing the OriL‐mtRNAP or the OriL‐PolG complexes (Fig 5E, right panel). The appearance of the super‐shifted band was not detected when a random double‐stranded DNA was used (Fig 5E, left panel), indicating that formation of the ternary complex was specific to the OriL structure. The −1A substitution in the OriL template did not affect the ternary complex formation, as was evident from binding of mtRNAP to OriL variants and the observed super‐shifted band in EMSA assays (Fig EV3D and E). In addition, we probed the assembly of the ternary complex by analytical gel filtration. Both PolG and mtRNAP bound to OriL and eluted as a single peak from the size‐exclusion column, confirming the formation of a ternary complex (Fig EV3F and G).
Figure EV3. PolG and mtRNAP assemble into a ternary complex on WT and −1A OriL.
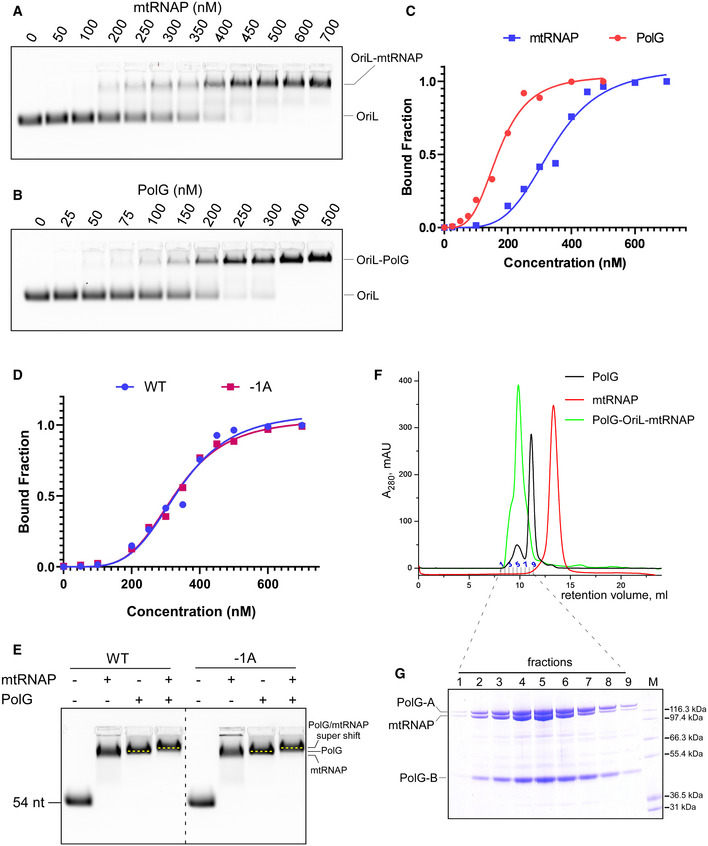
-
A, BEMSA using OriL and mtRNAP (A) and PolG (B).
-
CRelative affinity of mtRNAP and PolG to OriL as observed in A and B.
-
DRelative affinity of mtRNAP to WT and −1A OriL assayed using EMSA.
-
EThe primosome forms on both WT and −1A OriL. EMSA was performed using WT (left) or −1A OriL (right). Note the change of mobility of the labeled species in the presence of both mtRNAP and PolG (“super shift”).
-
FElution profile of PolG‐OriL‐mtRNAP, PolG and mtRNAP during size‐exclusion chromatography on Superdex 200 column.
-
GCoomassie stained SDS–PAGE showing the composition of fractions obtained in size‐exclusion experiment with the primosome complex. Molecular weight markers Mark12 (Invitrogen) are show in lane M.
The mode of binding of mtRNAP to the loop of OriL (Fig 3D) and the length of dsDNA needed for PolG binding (Lee et al, 2009) suggest that these polymerases load onto the hairpin in close proximity to each other. To probe this, we used a bifunctional cross‐linking reagent, DSG, which has a ∼8 Å cross‐linking range. We detected specific cross‐linking products formed between 32P‐labeled mtRNAP and both subunits of PolG in the presence of OriL only (Fig EV4), suggesting proximity of these proteins while bound to OriL and confirming the ternary complex formation.
Figure EV4. Proximity of mtRNAP and PolG in the primosome revealed by DSG cross‐linking.
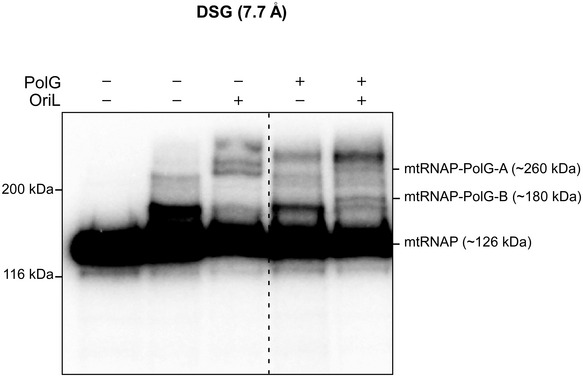
Cross‐linking was performed using 32P‐labeld mtRNAP in the presence or absence of PolG and OriL, as indicated. The leftmost lane represents a control reaction, in which the DSG reagent has not been added. The products of the reaction were resolved using 8% Tris‐glycine SDS–PAGE. The positions of the protein markers are indicated to the left of the gel. The positions of the adducts representing cross‐link between mtRNAP and PolG are indicated to the right.
To test whether this ternary complex represents a functional mitochondrial "primosome", we devised an experiment to probe whether RNA synthesis followed by DNA replication can be made on the same OriL template without dissociation of RNA or DNA polymerase. For this purpose, we generated a biotinylated version of PolG, which was capable of binding to streptavidin beads and retaining the OriL DNA and, via the latter, mtRNAP (Fig 5F and G). The ternary complex (biotinPolG‐OriL‐mtRNAP) was immobilized on streptavidin beads and allowed to generate a 28 nt RNA primer (Fig 5G, lane 1). The resulting complex was chased with either rNTP or dNTP mixture. The rNTP chase was performed to test whether any mtRNAP‐OriL complex has remained after the RNA primer synthesis was halted. We found no extension of the RNA primer by rNTPs, suggesting that, upon synthesis of a ∼28 nt RNA, mtRNAP dissociated from the templates (Fig 5G, lane 3). The chase with dNTPs was carried out to probe whether the RNA primer can be extended by PolG, which we found to be the case (Fig 5F). This suggests that the immobilized PolG‐OriL complex was able to synthesize DNA upon mtRNAP dissociation (Fig 5G, lane 2). We therefore conclude that the assembled ternary complex is functionally competent and likely represents the basic primosome during the replication of the light strand of human mitochondrial DNA.
Finally, we probed whether assembly of the primosome would provide more efficient primer protection against cleavage by the essential mitochondrial nuclease RNase H. OriL transcription complexes, which were allowed to generate a ∼28 nt RNA primer in the absence or presence of PolG, were incubated with RNase H to monitor the extent of the RNA primer protection (Fig 5 H and I). When only mtRNAP was present, the RNA primer was readily digested by RNase H, producing a weak, 8 nt long RNA footprint (Fig 5I, lane 2). In contrast, in the case of the primosome we observed a stable 15–16 nt RNA footprint, suggesting that PolG has readily taken over the RNA primer and protected the 3’ end portion of the RNA hybrid from nuclease degradation (Fig 5I, lane 4). The subsequent chase with dGTP/dCTP mixture revealed extension of the RNA primer by ∼5 nt, confirming the functional activity of the primosome complexes (Fig 5I, lane 5).
Discussion
Transcript slippage as a pre‐requisite for replication
Transcription and replication are inherently conflicting processes in living cells because they occur on the same template and may happen simultaneously. This is particularly exemplified in human mitochondria, where collisions between the two molecular machineries on a small circular genome seem unavoidable. In the absence of a dedicated primase, mtRNAP is recruited to generate RNA primers for the replication machinery. Involvement of the enzyme that is responsible for the synthesis of all RNA species in DNA replication may pose a major problem to mitochondrial biogenesis because initiation of transcription by mtRNAP is a rate‐limiting step of the transcription cycle. An elegant solution to this predicament is the employment of a non‐sequence‐specific (or non‐promoter) region that is only transiently available in the replicating mtDNA—the OriL stem‐loop. Indeed, while OriL transcription is less robust compared to promoter‐driven transcription (Fig 4F), it offers significant benefits. First, it does not depend on transcription initiation and elongation factors and therefore is significantly simplified in its utilization. Second, binding to a secondary structure that only forms transiently limits the possibility that mtRNAP will generate an aberrant transcript. Finally, transcription on a single‐stranded template provides a mechanism of termination (and thus primer generation): the lack of a complementary strand results in extended RNA–DNA hybrid that destabilizes mtRNAP and results in its dissociation from the DNA, making the 3’ end of RNA available for the replication machinery.
The sequence conservation of the OriL region of mtDNA in various mammals is astonishing. For example, the promoter regions in humans and giraffes (Giraffa camelopardalis) share no apparent homology, yet the OriL regions in these species are nearly identical and support efficient transcription (Fig EV2). Previous reports suggested the importance of the conserved sequence in the stem of the OriL hairpin (Wanrooij et al, 2012b). Our results, however, revealed no sequence‐specific interactions (Fig 2). Instead, mtRNAP recognizes only the secondary structure of OriL explaining why the size of the loop and likely the position of the TSS are conserved among mammalian species (Fig 2A). At first sight, transcription initiation based on secondary structure recognition may appear non‐specific and ambiguous. However, scrupulous inspection of human mtDNA reveals few, if any, regions that are capable of folding into a stable hairpin of a similar topology (Fig EV5A and B). In all instances, only OriL contains a homopolymeric T‐stretch in the loop of the hairpin, suggesting that transcription potential of other putative hairpins is low and their involvement in alternative sites of replication is unlikely to be mechanistically similar to that at OriL (Brown et al, 2005).
Figure EV5. OriL is the only stem loop that can drive efficient primer generation by mtRNAP.
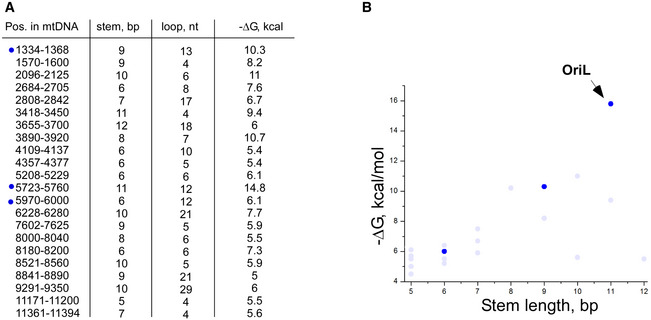
- Putative stem loops found in the light strand of human mtDNA. The size of the stem and the loop of the hairpins, and the stability of the hairpins (ΔG) are indicated. Dark blue dots indicate stem loops that satisfy the topological requirements to serve as a potential template for transcription.
- Stability of the stem loops found in human mtDNA as a function of the stem length. Stability of stem loops (−ΔG) found in human mtDNA as a function of the stem length. Light blue dots—stem loops that have a “disallowed” number of nucleotides in the loop (< 10 or more than 16). Note that neither of these stem loops, except OriL, encodes a stretch of the dTMP residues required for efficient transcription and primer generation.
Another interesting feature of OriL is the presence of a conserved stretch of dTMP residues. As we demonstrated in our experiments, the presence of the T‐stretch results in transcript slippage, which allows mtRNAP to build a homopolymeric transcript by sequential incorporation of AMP residues without having to translocate along the DNA template. The process of slippage continues until at least a 5–6 bp RNA–DNA hybrid is generated involving the positions of the template from +2 to −4 (or −3). Moreover, quite frequently even longer oligo‐A RNA species are produced, which have several unpaired AMP residues at the 5’ end besides those engaged in the RNA–DNA hybrid. The addition of just two such extra residues to the 6 bp hybrid would fill the RNA–DNA‐binding cavity of the mtRNAP and allow the transcription complex to assume a stable, elongation‐like conformation (Schwinghammer et al, 2013). While the 6 bp AT‐rich RNA–DNA hybrid is expected to be relatively weak, its contribution to the stability of the mtRNAP‐OriL complex seems significant. This stabilizing effect is reflected in an almost complete lack of abortive products during OriL initiation, as compared to the mutant OriL, which does not allow transcript slippage to occur (Fig 4A). In comparison with the case of double‐stranded DNA promoters, an additional factor stabilizing the early OriL transcription initiation complex is the lack of a complementary non‐template DNA strand in the region of TSS. Thus, the nascent OriL transcripts are not displaced by the re‐annealing of the non‐template strand of DNA. According to our in vitro data, 3 out of 4 attempts of mtRNAP to initiate at OriL in the absence of transcript slippage are not successful and results in abortive transcription (Fig 4B). Thus, the ability of mtRNAP to “slip” during initiation at OriL appears to be a major contributor to the efficiency of generation of the RNA primers necessary to replicate lagging strand of the mtDNA.
OriL as a molecular device to uncouple transcription and replication in human mitochondria
An increased stability of initiation complexes due to transcript slippage may also explain the lower apparent Km value for the initiating substrate when OriL is compared to a double‐stranded promoter (Fig 4). It is therefore tempting to speculate that low ATP requirement for OriL may represent a fail‐safe mechanism, which would allow replication but not transcription under unfavorable cellular conditions. How would this be possible considering that transcription initiation at LSP is required to generate a replication primer for the leading strand of mtDNA at the origin of replication OriH (Chang & Clayton, 1985)? There, the RNA primer is first transferred to the replisome formed by PolG and the replicative helicase, TWINKLE, and then converted into a ∼ 650 nt replication intermediate, known as 7S DNA (Wanrooij et al, 2010; Wanrooij et al, 2012a). Synthesis of the 7S DNA is responsible for the formation of the D‐loop containing mtDNA, which represents a significant (up to 50‐90%) pool of mtDNA molecules in human cells (Nicholls & Minczuk, 2014). In these mtDNA molecules, the occupancy of both PolG and TWINKLE was enriched in the region corresponding to the location of the 3’ end of the 7S DNA (Jemt et al, 2015). We suggest that the replication of both strands of mtDNA can be completely independent of promoter‐initiated transcription because utilization of the 7S DNA as the de facto replication primer circumvents the need for RNA primer synthesis at LSP to initiate replication. Therefore, replication of both mtDNA strands may require significantly lower ATP levels than transcription. Uncoupling of mitochondrial transcription from replication has been previously suggested via a molecular switch mechanism, which involves transcription elongation and anti‐termination factor, TEFM (Agaronyan et al, 2015).
A model of the replication primer generation at OriL
Perhaps the most surprising finding of this study is that binding of PolG affects primer synthesis on OriL templates. As discussed above, transcript slippage is a major contributing factor to the effectiveness of the process of primer generation. Transcript slippage helps to overcome an obstacle, be it a low concentration of ATP, high salt concentration or the presence of another protein, which loads in the proximity of the TSS in OriL. Our experiments using a coupled transcription‐replication assay and no‐slippage −1A OriL template demonstrate that PolG can, in fact, load onto OriL and, together with mtRNAP, form a ternary complex in vitro (Fig 5A and B). What would be the benefits of having PolG pre‐loaded onto the OriL region during replication? As mtRNAP is making an extended RNA–DNA hybrid that reaches a length of 20–30 nt, the transcription complex becomes destabilized and would readily dissociate. The nascent RNA–DNA hybrid is a substrate for the mitochondrial endoribonuclease RNase H. This nuclease is highly abundant and active in human mitochondria explaining why the activity of mtRNAP on OriL template (or promoter‐containing template) is not detectable in mitochondrial lysates (Chang et al, 1985). The preloading of PolG onto OriL serves to protect the 3' end of the RNA in RNA–DNA hybrid from degradation and also decreases the time required for the RNA primer transfer to the replisome.
According to the model (Fig 6), PolG can preload onto OriL near the TSS and bind to the stem of the OriL. MtRNAP lacks extensive contacts with downstream DNA, where the leading edge of its footprint is just 3–4 bp from the active site (Schwinghammer et al, 2013). To position the active site over the TSS and to engage into early transcription steps, mtRNAP must accommodate the loop of the OriL hairpin in the RNA–DNA‐binding cavity (Fig 3D) and compete with the pre‐loaded PolG for the downstream region. Only transcription complexes engaged in transcript slippage may build an RNA–DNA hybrid sufficient to overcome this barrier and push forward against PolG, forcing the latter to slide downstream along the DNA template. Transcription by mtRNAP continues until the size of the nascent RNA–DNA hybrid plays a major destabilization role (Temiakov et al, 2000; Kent et al, 2009) and results in mtRNAP dissociation. As mtRNAP dissociates, PolG slides backward and takes over the 3’ end of the RNA primer to initiate DNA replication (Fig 6). As implied by the model, the nascent RNA primer is protected by mtRNAP until it is transferred to PolG, enabling efficient and timely priming of replication.
Figure 6.
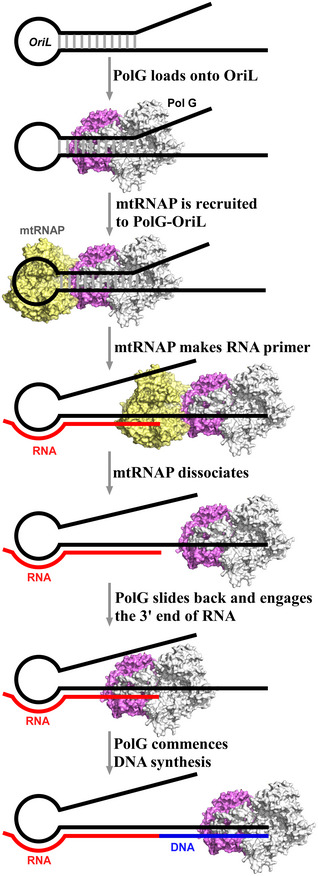
A model of transcription‐replication initiation at OriL.
To conclude, transcription at OriL represents an efficient mechanism of primer generation, which critically depends upon a concerted action of RNA and DNA polymerases. This mechanism may not be exclusive to mitochondria, as a multi‐subunit bacterial RNAP can efficiently initiate transcription at OriL‐like hairpin (Fig 3B) and a remarkably similar stem loop has been implicated in replication of a genome of TGMV virus (Orozco & Hanley‐Bowdoin, 1996).
Materials and Methods
Protein expression and purification
Human Δ43 mtRNAP (mature mtRNAP), Δ119, and Δ150 mtRNAP variants, were expressed and purified as described previously (Sologub et al, 2009). A variant of Δ119 mtRNAP (Δ119PKA mtRNAP) containing an engineered site for the protein kinase PKA (NEB) was purified as described previously (Morozov et al, 2014). MtRNAP variants—specificity loop deletion (residues 1,086–1,106) and intercalating hairpin deletion (residues 611–618)—were expressed and purified as previously described (Schwinghammer et al, 2013). WT TFAM and TFB2 M (Δ20) were expressed as described previously (Morozov et al, 2015).
N‐terminal His‐tagged human PolG‐B subunit (residues 26–485) was cloned into a pProEx vector using NcoI and XbaI endonuclease sites. A variant of PolG‐B subunit carrying both the N‐terminal 6‐His and the AviTag (GLNDIFEAQKIEWHE, Avidity) was obtained by site‐directed mutagenesis (QuikChange, Agilent). To express human PolG‐B and Avitag‐PolG‐B, BL21‐CodonPlus (DE3)‐RIPL (Agilent) or CVB 101 competent cells (Avidity) were transformed with the respective plasmid and grown at 37°C in LB media until OD600 reached 0.5 units. The proteins were induced by the addition of 0.15 mM IPTG and 50 μM biotin (for Avitag‐PolG‐B) for 18 h at 16°C. Both WT and biotinylated PolG‐B were purified by affinity chromatography using Ni‐NTA beads (Thermo Fisher Scientific), followed by affinity chromatography using a HiTrap heparin HP column (GE Healthcare). The heparin column was equilibrated in buffer A (40 mM Tris, pH 8.0, 300 mM NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol) and the protein was eluted by linear gradient 0‐70% of buffer B (40 mM Tris, pH 8.0, 1.5 M NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol). Peak fractions were pooled, concentrated, and stored at −80°C.
Human PolG‐A subunit (residues 26–1,239) lacking exonuclease activity (D198A/E200A) with N‐terminal 6‐His was cloned into pFastBac1 vector (Thermo Fisher Scientific) using EcoRI and NotI endonuclease sites. PolG‐A was expressed in Spodoptera frugiperda (Sf9) insect cells based on the protocol from the Bac‐to‐Bac® Baculovirus Expression System (Thermo Fisher Scientific). Human PolG‐A was first purified using HisTrap™ HP column (GE Healthcare) followed by anion exchange chromatography on Mono Q 5/50 GL column (GE Healthcare). Mono Q 5/50 GL column was equilibrated in buffer A (50 mM Tris, pH 7.5, 100 mM NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol) and PolG‐A was eluted by 0–70% linear gradient of buffer B (50 mM Tris, pH 7.5, 1 M NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol). PolG was reconstituted by incubating PolG‐A with 2.6‐fold molar excess of PolG‐B on ice for 10 min at 4°C followed by purification using Superdex® 200 Increase 10/300 GL column (GE Healthcare) using buffer containing 50 mM Tris, pH 7.5, 500 mM NaCl, 5% Glycerol, and 5 mM β‐mercaptoethanol.
Human mtSSB (residues 17–148) having C‐terminal 6‐His‐tag was cloned into pET22b (+) vector using NdeI and XhoI endonuclease sites and expressed in RIPL cells. The cells were grown until the OD600 reached 1.0 unit and mtSSB expression was induced by addition of 0.8 mM IPTG for 2 h at 37°C. MtSSB was purified by affinity chromatography using a HisTrap HP column (GE Healthcare) followed by affinity chromatography using a HiTrap heparin HP column (GE Healthcare). The heparin column was equilibrated in buffer A (40 mM Tris, pH 8.0, 100 mM NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol) and mtSSB was eluted by 0‐70% linear gradient of buffer B (40 mM Tris, pH 8.0, 1.5 M NaCl, 5% Glycerol, 5 mM β‐mercaptoethanol). Peak fractions were pooled, concentrated, and stored at −80°C.
RPO41 and T7 RNAPs were purified as previously described (Temiakov et al, 2003; Savkina et al, 2010). Core E. coli RNAP was from NEB.
Transcription and a coupled transcription‐replication assay
To carry out transcription assays, synthetic DNA oligonucleotides containing OriL sequence (positions 5723‐5780 in human mtDNA) were used (IDT DNA). The complete sequence of OriL templates is given in Appendix Table S1. To anneal, the templates were diluted in a “duplex buffer” (100 mM potassium acetate; 30 mM HEPES, pH 7.5), heated for 7 min at 85°C, and cooled down (1°C/min) for 1 h to 25°C in a thermocycler. Transcription reactions were carried out using OriL template (50 nM) and WT mtRNAP (150 nM) in a transcription buffer containing 20 mM Tris, pH 7.9, 30 mM NaCl, 10 mM MgCl2, and 20 mM β‐mercaptoethanol in the presence of BSA (0.1 mg/ml), and a limited set of NTPs (no UTP) as follows: ATP (0.3 mM), CTP (0.3 mM), GTP (0.05 mM), and 0.3 μCi [α‐32P] GTP (3,000 Ci/mmol). Where indicated, a complete mixture of the NTPs was used to generate run‐off products. Reactions were carried out for 30 min at 30°C and stopped by the addition of an equal volume of 95% formamide/0.05 M EDTA. The products of the reaction were resolved by 20% PAGE containing 6 M Urea and visualized by autoradiography using PhosphorImager (GE Healthcare).
In a coupled transcription‐replication assay, PolG (200 nM), mtSSB (300 nM), and dNTPs (0.3 mM) were added to the reaction mix prior to addition of the OriL template. Reactions were carried out for 30 min at 30°C, treated with proteinase K (0.2 μg/μl) for 1 h at 55°C followed by the addition of an equal volume of stop buffer (95% formamide and 0.05 M EDTA). The products of the reaction were resolved by 20% PAGE containing 6 M Urea and visualized by autoradiography using PhosphorImager (GE Healthcare).
RNase H protection assay
In RNase H assay, a modified OriL template (#27, Appendix Table S1) was used to allow the labeling of the 3’ end of the transcripts using [α‐32P] UTP. First, the transcription assay was carried out in the presence or absence of PolG for 15 min at 30°C. RNase H (0.1 U/μl, NEB) was then added to the reaction and incubated for 3 min at RT. To extend the RNA primer after the RNase H treatment, 0.1 mM dCTP/dGTP mix was added and the reaction was incubated for 15 s at RT. Reactions were treated by proteinase K and the products were resolved and visualized as described above.
Electrophoretic mobility shift assay (EMSA)
The 5ʹ‐Cy3‐labeled variant of OriL DNA was obtained from IDT DNA. The 5ʹ‐Cy3‐labeled dsDNA (IDT DNA) was a kind gift from Dr. Cingolani (Thomas Jefferson University). To perform EMSA, complexes of OriL/mtRNAP, OriL/PolG, and OriL/PolG/mtRNAP were assembled in transcription buffer containing 20 mM Tris, pH 7.9, 30 mM NaCl, 10 mM MgCl2, 5% glycerol, 20 mM β‐mercaptoethanol, and 1 mg/ml BSA, and incubated for 10 min at RT. The same protocol was followed to perform EMSA using non‐specific dsDNA. The reactions were resolved in 0.5% agarose gel run in 0.5× TBE buffer for 30 min at 100 V at 4°C. The products of the reactions were visualized using Bio‐Rad ChemiDoc™ imager.
Solid‐phase transcription‐replication assay
To perform a solid phase coupled transcription‐replication assay, the ternary complex (1 μM WT OriL (template #36), 1 μM Δ119 mtRNAP, 1 μM biotinylated PolG) was assembled in the binding buffer containing 50 mM Tris, pH 7.9, 100 mM NaCl, 10 mM MgCl2, 5% glycerol, 0.05% Tween 20, and 5 mM β‐mercaptoethanol. The complex was immobilized on streptavidin beads (Thermo Fisher Scientific) for 5 min at RT. After the unbound material was removed by washing the beads six times with the binding buffer (1 ml), the NTP mixture containing 0.3 mM ATP, 0.3 mM CTP, 0.05 mM GTP, and 0.3 μCi [α‐32P] GTP (3,000 Ci/mmol) was added to allow transcription for 15 min at RT. After washing the beads (6 × 1 ml), dNTPs (0.3 mM) or rNTPs (0.3 mM) were added to the reactions and incubated for 10 min at RT. The reactions were then stopped by the addition of an equal volume of the stop buffer (95% formamide and 0.05 M EDTA). The products were resolved by 20% PAGE containing 6 M Urea and visualized by autoradiography.
Measurement of apparent Km
The apparent Km of ATP for OriL was measured by performing the transcription assay as described above at 0.0025–1.28 mM concentrations of ATP.
To measure the apparent Km of ATP for LSP, transcription assays were carried out using annealed synthetic LSP templates (50 nM) having −60 upstream region (Morozov et al, 2015), Δ119 mtRNAP (150 nM), TFAM (50 nM), and TFB2 M (150 nM) in a transcription buffer containing (20 mM Tris, pH 7.9, 30 mM NaCl, 10 mM MgCl2, and 20 mM β‐mercaptoethanol) in the presence of BSA (0.1 mg/ml), ATP (0.007–1.2 mM), CTP (0.3 mM), GTP (0.3 mM), UTP (0.05 mM) and 0.3 μCi [α‐32P] UTP (800 Ci/mmol). Reactions were carried out for 30 min at 35°C and analyzed as described above for OriL transcription.
The amount of the transcripts produced at different ATP concentrations was quantified by ImageQuantTL software. The apparent Km was calculated using Hanes–Woolf equation. The apparent K m values are an average of three independent experiments, and the standard error of the mean (SEM) was calculated for each set of data.
Analytical size‐exclusion chromatography
The OriL‐containing complexes were assembled at 20 μM concentration using Δ119 mtRNAP, PolG, and OriL (template #35) for 10 min at RT. The size‐exclusion chromatography was performed using Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in a buffer containing 50 mM Tris, pH 7.9, 150 mM NaCl, 5% glycerol, 5 mM MgCl2, and 5 mM β‐mercaptoethanol. The 0.4 ml fractions were collected and analyzed using PAGE electrophoresis.
Protein–protein cross‐linking
Protein cross‐linking was performed using a homo‐bifunctional reagent—disuccinimidyl glutarate (DSG, Pierce). A primosome complex containing 32P‐labeled mtRNAP was assembled at 2 μM concentration for 10 min in 40 mM HEPES (pH 7.9), 5 mM MgCl2, 5% glycerol, and 20 mM β‐mercaptoethanol. Freshly prepared DSG (4 mM solution in DMSO) was added to the final concentration of 0.4 mM, and the reactions incubated for 30 min at RT. The reactions were stopped by the addition of 50 mM Tris pH 7.9, and the products were resolved using 8% Tris‐glycine SDS–PAGE. The gel was dried, and the reaction adducts were visualized by autoradiography.
Author contributions
Cloning and purifying proteins, and transcription and transcription‐replication assay, EMSA, cross‐linking, and analytical size‐exclusion chromatography: AS; Cloning and purifying proteins: AZ‐O; Data analysis: AS, AZ‐O, MA, and DT; Manuscript writing: DT.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgments
We thank past and present members of the Temiakov laboratory, in particular Dr. Yaroslav Morozov. We thank Dr. William McAllister for critical reading of the manuscript and helpful discussion and Dr. William Copeland for the generous gift of Exo‐ PolG‐A plasmid. NIH R35 GM131832 (D.T.).
The EMBO Journal (2021) 40: e107988.
Data availability
This study includes no data deposited in external repositories.
References
- Agaronyan K, Morozov YI, Anikin M, Temiakov D (2015) Replication‐transcription switch in human mitochondria. Science 347: 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Cecconi C, Tkachuk AN, Bustamante C, Clayton DA (2005) Replication of mitochondrial DNA occurs by strand displacement with alternative light‐strand origins, not via a strand‐coupled mechanism. Genes Dev 19: 2466–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA (1985) Priming of human mitochondrial DNA replication occurs at the light‐strand promoter. Proc Natl Acad Sci USA 82: 351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Hauswirth WW, Clayton DA (1985) Replication priming and transcription initiate from precisely the same site in mouse mitochondrial DNA. Embo J 4: 1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA (1982) Replication of animal mitochondrial DNA. Cell 28: 693–705 [DOI] [PubMed] [Google Scholar]
- Davydova EK, Santangelo TJ, Rothman‐Denes LB (2007) Bacteriophage N4 virion RNA polymerase interaction with its promoter DNA hairpin. Proc Natl Acad Sci USA 104: 7033–7038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz GA, Rong M, McAllister WT, Durbin RK (1996) The stability of abortively cycling T7 RNA polymerase complexes depends upon template conformation. Biochemistry 35: 10837–10843 [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Gustafsson CM (2020) Mammalian mitochondrial DNA replication and mechanisms of deletion formation. Crit Rev Biochem Mol Biol 55: 509–524 [DOI] [PubMed] [Google Scholar]
- Fuste JM, Wanrooij S, Jemt E, Granycome CE, Cluett TJ, Shi Y, Atanassova N, Holt IJ, Gustafsson CM, Falkenberg M (2010) Mitochondrial RNA polymerase is needed for activation of the origin of light‐strand DNA replication. Mol Cell 37: 67–78 [DOI] [PubMed] [Google Scholar]
- Gaspari M, Larsson NG, Gustafsson CM (2004) The transcription machinery in mammalian mitochondria. Biochem Biophys Acta 1659: 148–152 [DOI] [PubMed] [Google Scholar]
- Hillen HS, Morozov YI, Sarfallah A, Temiakov D, Cramer P (2017) Structural basis of mitochondrial transcription initiation. Cell 171: 1072–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixson JE, Wong TW, Clayton DA (1986) Both the conserved stem‐loop and divergent 5'‐flanking sequences are required for initiation at the human mitochondrial origin of light‐strand DNA replication. J Biol Chem 261: 2384–2390 [PubMed] [Google Scholar]
- Jemt E, Farge G, Backstrom S, Holmlund T, Gustafsson CM, Falkenberg M (2011) The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res 39: 9238–9249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemt E, Persson O, Shi Y, Mehmedovic M, Uhler JP, Davila Lopez M, Freyer C, Gustafsson CM, Samuelsson T, Falkenberg M (2015) Regulation of DNA replication at the end of the mitochondrial D‐loop involves the helicase TWINKLE and a conserved sequence element. Nucleic Acids Res 43: 9262–9275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Miyako K, Kai Y, Irie T, Takeshige K (1997) In vivo determination of replication origins of human mitochondrial DNA by ligation‐mediated polymerase chain reaction. J Biol Chem 272: 15275–15279 [DOI] [PubMed] [Google Scholar]
- Kent T, Kashkina E, Anikin M, Temiakov D (2009) Maintenance of RNA‐DNA hybrid length in bacterial RNA polymerases. J Biol Chem 284: 13497–13504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kennedy WD, Yin YW (2009) Structural insight into processive human mitochondrial DNA synthesis and disease‐related polymerase mutations. Cell 139: 312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Arai K (1997) Frpo: a novel single‐stranded DNA promoter for transcription and for primer RNA synthesis of DNA replication. Cell 89: 897–907 [DOI] [PubMed] [Google Scholar]
- Masters BS, Stohl LL, Clayton DA (1987) Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell 51: 89–99 [DOI] [PubMed] [Google Scholar]
- Milenkovic D, Matic S, Kuhl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M, Larsson NG (2013) TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D‐loop strands and complete mtDNA replication. Hum Mol Genet 22: 1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles Fuste J, Shi Y, Wanrooij S, Zhu X, Jemt E, Persson O, Sabouri N, Gustafsson CM, Falkenberg M (2014) In vivo occupancy of mitochondrial single‐stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet 10: e1004832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov YI, Agaronyan K, Cheung AC, Anikin M, Cramer P, Temiakov D (2014) A novel intermediate in transcription initiation by human mitochondrial RNA polymerase. Nucleic Acids Res 42: 3884–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov YI, Parshin AV, Agaronyan K, Cheung AC, Anikin M, Cramer P, Temiakov D (2015) A model for transcription initiation in human mitochondria. Nucleic Acids Res 43: 3726–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls TJ, Minczuk M (2014) In D‐loop: 40 years of mitochondrial 7S DNA. Exp Gerontol 56: 175–181 [DOI] [PubMed] [Google Scholar]
- Orozco BM, Hanley‐Bowdoin L (1996) A DNA structure is required for geminivirus replication origin function. J Virol 70: 148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson O, Muthukumar Y, Basu S, Jenninger L, Uhler JP, Berglund AK, McFarland R, Taylor RW, Gustafsson CM, Larsson Eet al (2019) Copy‐choice recombination during mitochondrial L‐strand synthesis causes DNA deletions. Nat Commun 10: 759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AF, Millet AR, Tigano M, Dubois SM, Crimmins H, Babin L, Charpentier M, Piganeau M, Brunet E, Sfeir A (2017) Single‐Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol Cell 65: 527–538 [DOI] [PubMed] [Google Scholar]
- Ringel R, Sologub M, Morozov YI, Litonin D, Cramer P, Temiakov D (2011) Structure of human mitochondrial RNA polymerase. Nature 478: 269–273 [DOI] [PubMed] [Google Scholar]
- Savkina M, Temiakov D, McAllister WT, Anikin M (2010) Multiple functions of yeast mitochondrial transcription factor Mtf1p during initiation. J Biol Chem 285: 3957–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinghammer K, Cheung AC, Morozov YI, Agaronyan K, Temiakov D, Cramer P (2013) Structure of human mitochondrial RNA polymerase elongation complex. Nat Struct Mol Biol 20: 1298–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Nandakumar D, Tang GQ, Patel SS (2012) Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J Biol Chem 287: 14545–14556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sologub M, Litonin D, Anikin M, Mustaev A, Temiakov D (2009) TFB2 is a transient component of the catalytic site of the human mitochondrial RNA polymerase. Cell 139: 934–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper DP, Clayton DA (1982) Precise nucleotide location of the 5' ends of RNA‐primed nascent light strands of mouse mitochondrial DNA. J Mol Biol 162: 1–16 [DOI] [PubMed] [Google Scholar]
- Temiakov D, Mentesana PE, Ma K, Mustaev A, Borukhov S, McAllister WT (2000) The specificity loop of T7 RNA polymerase interacts first with the promoter and then with the elongating transcript, suggesting a mechanism for promoter clearance. Proc Natl Acad Sci USA 97: 14109–14114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiakov D, Tahirov TH, Anikin M, McAllister WT, Vassylyev DG, Yokoyama S (2003) Crystallization and preliminary crystallographic analysis of T7 RNA polymerase elongation complex. Acta Crystallogr D Biol Crystallogr 59: 185–187 [DOI] [PubMed] [Google Scholar]
- Wanrooij PH, Uhler JP, Simonsson T, Falkenberg M, Gustafsson CM (2010) G‐quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc Natl Acad Sci USA 107: 16072–16077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij PH, Uhler JP, Shi Y, Westerlund F, Falkenberg M, Gustafsson CM (2012a) A hybrid G‐quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R‐loop. Nucleic Acids Res 40: 10334–10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij S, Fuste JM, Farge G, Shi Y, Gustafsson CM, Falkenberg M (2008) Human mitochondrial RNA polymerase primes lagging‐strand DNA synthesis in vitro. Proc Natl Acad Sci USA 105: 11122–11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij S, Miralles Fuste J, Stewart JB, Wanrooij PH, Samuelsson T, Larsson NG, Gustafsson CM, Falkenberg M (2012b) In vivo mutagenesis reveals that OriL is essential for mitochondrial DNA replication. EMBO Rep 13: 1130–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TW, Clayton DA (1985) In vitro replication of human mitochondrial DNA: accurate initiation at the origin of light‐strand synthesis. Cell 42: 951–958 [DOI] [PubMed] [Google Scholar]
- Zenkin N, Naryshkina T, Kuznedelov K, Severinov K (2006) The mechanism of DNA replication primer synthesis by RNA polymerase. Nature 439: 617–620 [DOI] [PubMed] [Google Scholar]