

Summary
Here, we describe three complementary microscopy-based approaches to quantify morphological changes of chromatin organization in cultured adherent cells: the analysis of the coefficient of variation of DNA, the measurement of DNA-free nuclear areas, and the quantification of chromatin-associated proteins at the nuclear edge. These approaches rely on confocal imaging and stochastic optical reconstruction microscopy and allow a fast and robust quantification of chromatin compaction. These approaches circumvent inter-variability between imaging conditions and apply to every type of adherent cells.
For complete details on the use and execution of this protocol, please refer to Neguembor et al. (2021).
Subject areas: Cell Biology, Microscopy, Molecular Biology
Graphical abstract
Highlights
-
•
Protocol for three complementary techniques to measure chromatin compaction
-
•
Chromatin compaction is quantified from confocal and super-resolution images
-
•
CV and Black Space analyses score for heterogeneity of nuclear DNA distribution
-
•
Edge analysis measures the enrichment of a protein signal around the nuclear edge
Here, we describe three complementary microscopy-based approaches to quantify morphological changes of chromatin organization in cultured adherent cells: the analysis of the coefficient of variation of DNA, the measurement of DNA-free nuclear areas, and the quantification of chromatin-associated proteins at the nuclear edge. These approaches rely on confocal imaging and stochastic optical reconstruction microscopy and allow a fast and robust quantification of chromatin compaction. These approaches circumvent inter-variability between imaging conditions and apply to every type of adherent cells.
Before you begin
The protocol below describes the steps for the confocal- and Stochastic Optical Reconstruction Microscopy (STORM)-based quantifications of morphological changes in chromatin organization in the nucleus of HeLa cells treated with Actinomycin D, an inhibitor of transcription whose action drives major changes in nuclear topology (Neguembor et al., 2021). In addition, we have also successfully used this protocol with other drugs (as α-amanitin, β-Lapachone, which are a transcription inhibitor and a torsional stress inhibitor, respectively) and in different cellular systems (mAID-mClover-POLR2A DLD-1 cells, in which transcriptional inhibition is obtained by degrading the endogenous RNA polymerase II) (Neguembor et al., 2021).
Specifically, the protocol offers a means to (1) quantify the coefficient of variation of DAPI signal; (2) quantify the percentage of the nuclear DNA-free areas; and (3) quantify the percentage of H3K9me3 spatially localizing in the nuclear edge with respect to the total nuclear signal.
Coefficient of variation is a method to assess the degree of heterogeneity of DNA signal across the nucleus. It has been previously used to quantify changes in DNA compaction upon drug treatments or genetic modifications affecting chromatin organization (Grezy et al., 2016; Jeanblanc et al., 2012; Neguembor et al., 2021). The coefficient of variation (CV) of individual nuclei is calculated as , where σ represents the standard deviation of the DAPI intensity values and μ represents the mean value of intensity of the nucleus.
DNA-free area analysis is an alternative method to assess DNA compaction previously reported in Neguembor et al. (2021). Upon DNA compaction, DNA signal becomes redistributed and accumulates in densely packed areas. This drives the appearance of areas with no DNA signal or low-density DNA signal (i.e., DNA free areas). DNA-free nuclear areas are quantified from DNA STORM images by applying a binary threshold based on signal intensity on Gaussian filtered density maps. Regions with DNA signal below the threshold are considered DNA-free areas and the percentage of DNA-free areas over the imaged nuclear area is estimated for each nucleus.
Nuclear edge analysis is a method to quantify the enrichment of specific proteins close to the nuclear rim with respect to the protein signal across the entire nuclear section. The method was previously reported in Neguembor et al. (2021) to assess the redistribution of H3K9me3 and Lamin A/C signals in STORM images. Here, the percentage of H3K9me3 STORM localizations at the edge of nucleus is calculated by first defining an edge region around the border of each nucleus of a width chosen by the user and then calculating the percentage of localizations falling in this area, i.e., the number of localizations inside the edge region divided by the total number of localizations inside the nucleus.
We expect this protocol to be suitable for quantitatively assessing the changes in spatial localization and density of any nuclear protein of interest other than DNA or H3K9me3 with respect to a control condition, provided that such proteins are suitable for confocal or STORM imaging, and that variable parameters of analysis are adjusted and optimized according to the particular experiment.
Solutions are prepared following the recipes in the materials and equipment section. Solutions that can be prepared in advance and stored are indicated. A complete list of materials and equipment required is given in the key resources table.
Installation of software and custom codes for analysis
Timing: 1 h
-
1.Download and install (Fiji Is Just) ImageJ (FIJI) (Rueden et al., 2017) from ImageJ website: download the relevant .zip file depending on the operating system and run installation executable. The codes illustrated in this protocol were tested in ImageJ 2.0.0 and 2.1.0/1.53c versions.
-
a.Upload ImageJ script CV PART 1 IMAGEJ within ChromatinCompactionAnalysis.zip (see key resources table).
-
b.Extract the .zip file in the folder of your choice.
-
a.
-
2.Download and install MATLAB from https://www.mathworks.com/products/matlab.html. The codes illustrated in this protocol were tested in MATLAB R2021a and older versions, such as R2016a, R2016b versions.
- a.
-
b.Extract the .zip file in the folder of your choice.
-
c.In the home section of MATLAB, hit ‘‘Set Path’’.
-
d.Select ‘‘Add with Subfolders…’’ and browse to the folder containing the extracted files.
-
e.Hit ‘‘Save’’. MATLAB can now find and run all the scripts necessary for the analyses.
Note: A digest version of the image analysis steps of this protocol can also be found in the Videos as STEP BY STEP simplified tutorial videos (Methods videos S1, S2, and S3). The protocol does not require an extensive knowledge of MATLAB. However, we recommend looking at the tutorial available at Matlab website, particularly the section about setting up paths and running scripts. We ran this analysis successfully on laptop computers (both Windows and Mac) with ad minima a 1.80 GHz Processor and 16 GB of RAM. Higher specifications can lead to reduced times of analysis.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3K9me3 (dilution 1:50) | Abcam | Cat#ab8898; RRID: AB_306848 |
| Goat anti-Rabbit IgG AF647 (dilution 1:250) | Thermo Fisher Scientific | Cat#A-21244 |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Sigma-Aldrich | Cat#D2650 |
| Actinomycin D (ActD) | Sigma-Aldrich | Cat#A9415 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | Cat#41965039 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat#10270106 |
| Penicillin-Streptomycin (Pen Strep) | Thermo Fisher Scientific | Cat#15140122 |
| GlutaMAX | Thermo Fisher Scientific | Cat#35050038 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Thermo Fisher Scientific | Cat#14190094 |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | Cat#25200056 |
| HEPES | Sigma-Aldrich | Cat#H4034 |
| Hydrochloric acid fuming 37% (HCl) | Millipore | Cat#100317 |
| L-Ascorbic acid | Sigma-Aldrich | Cat#A92902 |
| Amino guanidine | Sigma-Aldrich | Cat#396494 |
| Glucose oxidase | Sigma-Aldrich | Cat#G2133 |
| Catalase | Sigma-Aldrich | Cat#C100 |
| Cysteamine (MEA) | Sigma-Aldrich | Cat#30070 |
| CuSO4 | Sigma-Aldrich | Cat#C1297 |
| Glucose | Sigma-Aldrich | Cat#G8270 |
| 5-Ethynyl-2′-deoxycytidine (EdC) | Sigma-Aldrich | Cat#T511307 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | Cat#A7906 |
| Paraformaldehyde 16% methanol-free (PFA) | Alfa Aesar | Cat#43368 |
| 4′,6-Diamidino-2-phenylindole (DAPI) | Roche | Cat#10236276 |
| Alexa Fluor™ 647 Azide, Triethylammonium Salt (AF647) | Thermo Fisher Scientific | Cat#A10277 |
| Drierite | Sigma-Aldrich | Cat#238961 |
| Deposited data | ||
| Images files (Mendeley data) | This study | DOI: https://doi.org/10.17632/pbnc4zhcwd.1 |
| Experimental models: Cell lines | ||
| HeLa cells (human cervix epithelioid carcinoma, female) | Maria Pia Cosma Lab | N/A |
| Software and algorithms | ||
| ImageJ (2.0.0 and 2.1.0/1.53c) | (Rueden et al., 2017) | https://imagej.nih.gov/ij/download.html |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Data S1. CV analysis, related to steps 14–17, containing CV-Part1-ImageJ (steps 14–16) and CV-Part2-MATLAB (step 17) | This paper |
https://github.com/CosmaLab/CV-Analysis All processing scripts can be found also in Data S1. |
| Data S2. Black Space analysis, related to step 18, used for the quantification of DNA-free areas | This paper |
https://github.com/CosmaLab/Black-Space-Analysis All processing scripts can be found also in Data S2. |
| Data S3. Edge analysis, related to step 19, used for the quantification of the percentage of H3K9me3 signal at nuclear edges | This paper |
https://github.com/CosmaLab/Edge-Analysis All processing scripts can be found also in Data S3. |
| Step-by-step simplified tutorial videos.mp4, a digest version of the protocol | This paper | STEP BY STEP simplified tutorial videos.mp4 |
| LAS X | Leica | https://www.leica-microsystems.com/products/microscope-software/details/product/leica-las-x-ls/ |
| NIS software STORM module | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Graphpad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Insight3 | (Huang et al., 2008) | N/A |
| Other | ||
| Borosilicate glass bottom 8-well chambers (Lab-Tek) | Thermo Fisher Scientific | Cat#155411 |
| N-STORM 4.0 microscope equipped with highly inclined and laminated optical sheet illumination (HILO) | Nikon | N/A |
| LU-NV laser unit with 405 nm (LD, 20 mW output), 488 nm (DPSS, 45 mW output), 560 nm (DPSS, 45 mW output), and 647 nm (fiber, 125 mW output) lasers | Nikon | N/A |
| 405/488/561/647nm Laser Quad Band Set | Chroma | Cat#TRF89902 |
| CFI HP Apochromat TIRF 100XC Oil (NA 1.49) objective | Nikon | N/A |
| iXON Ultra DU-897U EMCCD camera | Andor Technology | N/A |
| Leica TCS SP8 confocal microscopy equipped with laser/filter sets to acquire DAPI signal | Leica | N/A |
| HC Plan-Apochromat CS2 63x/1.40 oil objective | Leica | N/A |
| Type F Immersion liquid (RI 1.518) | Leica | Cat#11944399 |
| Immersion oil (Type F) (RI 1.515) | Nikon | Cat#MXA22168 |
| Stericup Quick Release Millipore Express PLUS 0.22 μm PES, 500 mL | Millipore | Cat#11290 |
| Telstar™ Soporte de mesa para Bio II Advance 4 | Telstar | Cat#15138614 |
| BINDER CO2 incubator CB 160, with O2 control | Binder | Cat# 9040-0092 |
| WHEATON® Dry-Seal vacuum desiccator | Sigma-Aldrich | Cat#Z114375 |
| Sartorius Stedim Biotech SI-215D Analytical Balance | Denver Instrument | Cat#EW-11422-04 |
| Fisherbrand™ accumet™ AE150 Benchtop pH Meter | Thermo Fisher Scientific | Cat#13-636-AE152 |
| Millipore Milli-Q Synthesis A10 Water Purification | Millipore | Cat#15882 |
Materials and equipment
Media for HeLa cell culture
HeLa’s standard cell culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM) | 88% | 44 mL |
| Fetal Bovine Serum (FBS) | 10% | 5 mL |
| Penicillin-Streptomycin | 1% | 0.5 mL |
| GlutaMAX | 1% | 0.5 mL |
| Total | n/a | 50 mL |
HeLa’s resting medium to induce cell cycle arrest (optional)
| Reagent | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM) | 96% | 48 mL |
| Fetal Bovine Serum (FBS) | 2% | 1 mL |
| Penicillin-Streptomycin | 1% | 0.5 mL |
| GlutaMAX | 1% | 0.5 mL |
| Total | n/a | 50 mL |
Note: Hela’s culture media can be stored at 4°C for 1 month.
Note: Optimal medium composition can vary considerably for different cell types.
Note: We recommend filtering FBS under the hood through Stericup 0.22 μm prior to use.
CRITICAL: Penicillin/Streptomycin may cause an allergic skin reaction (H317), may cause allergy or asthma symptoms or breathing difficulties if inhaled (H334), and may damage fertility or the unborn child (H360), and hence it should be manipulated by wearing appropriate recommended PPE.
EdC solution
Ethynil-deoxy-cytidine (EdC) (#T511307, Sigma-Aldrich) is resuspended in DMSO to a concentration of 10 mM and aliquots can be stored at −20°C for 1 year.
ActD stock solution
Actinomycin D (ActD) (#A9415, Sigma-Aldrich) is resuspended in DMSO to a concentration of 5 mM and aliquots can be stored foil-wrapped vials at −20°C for up to 1 month. Do not store in glass vials.
Solutions for microscopy
Fixative solution
Fixative solution is prepared by diluting PFA 16% in PBS to obtain a final solution of PFA 4%.
Note: PFA 4% is prepared freshly the same day of the fixation. PFA 16% can be stored at 4°C for 3 months.
Permeabilization solution
Permeabilization solution is prepared by diluting TritonX-100 in PBS to obtain a final solution of 0.4%.
Note: TritonX-100 0.4 % stock solution is stable at 20°C–25°C for 1 month.
Blocking buffer for Immuno-Fluorescence (IF) and STORM
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 10% | 1 g |
| Triton-X 100 0.4 % | 0.01% | 0.25 mL |
| PBS | n/a | 9.75 mL |
| Total | n/a | 10 mL |
Note: Blocking solution is prepared freshly before use.
Washing buffer for IF and STORM
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 2% | 0.2 g |
| Triton-X 100 0.4 % | 0.01% | 0.25 mL |
| PBS | n/a | 9.75 mL |
| Total | n/a | 10 mL |
Note: Washing buffer is prepared freshly before use.
DAPI
DAPI stock is prepared by dissolving DAPI in ultrapure water to 1 mg/mL.
Note: Stock solution is stable for 6 months and repeated use if stored protected from light at −20°C.
Glox solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Glucose Oxidase | 0.1% | 14 mg |
| Catalase | ∼200–500 U/μL | 50 μL |
| PBS | n/a | 200 μL |
| Total | n/a | ∼250 μL |
Note: Glucose Oxidase is stored at −20°C and it is stable for 5 years.
Note: Catalase is prepared as specified in the Product Data Sheet (Sigma-Aldrich) and the aliquots can be stored at 4°C for 12 months.
Note: Thawed Glox aliquots can be stored at 4°C for 1 week, do not re-freeze.
Preparation:
-
•
Weigh 14 mg of Glucose Oxidase.
-
•
Dissolve in 200 μL of PBS. Gently vortex in order to dissolve.
-
•
Add 50 μL of Catalase 1 mg/mL.
-
•
Centrifuge at 20 000 × g for 2 min in order to pellet insolubilities.
-
•
Prepare 200 μL eppendorf tubes. Pipet the supernatant in aliquots of ∼20 μL.
-
•
Store at −20°C or use immediately.
Click Chemistry buffer solution
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES 152 mM pH 8.2 | n/a | 1492 μL |
| CuSO4 solution (100 mM stock) | 1 mM | 20 μL |
| Amino Guanidin (500 mM stock) | 50 mM | 200 μL |
| Ascorbic acid (250 mM) | 25 mM | 200 μL |
| AlexaFluor647-azide (10 mM stock) | 10 μM | 2 μL |
| Glucose 50% | 2% | 80 μL |
| Glox solution (1000X) | 0.1% | 6 μL |
| Total | n/a | 2 mL |
Note: HEPES 152 mM pH 8.2, CuSO4 100 mM solution, and Glucose 50% solution can be prepared in advance and can be stored at 20°C–25°C. Amino Guanidin 500 mM solution, AlexaFluor647-azide and Glox can be prepared in advance and should be stored for 3 months at −20°C in the dark. Ascorbic acid solution should be prepared freshly before use.
Note: To prepare HEPES 152 mM pH 8.2 stock solution, HEPES is resuspended in MilliQ water to a concentration of 152 mM, pH is adjusted to pH 8.2 with HCl using a pH-meter, solution is brought to final desired volume with milliQ water. Stock solution can be prepared in advance and can be stored for 1 year at 20°C–25°C.
Note: To prepare CuSO4 100 mM stock solution, CuSO4 is resuspended in MilliQ water to a concentration of 100 mM. Stock solution can be prepared in advance and can be stored for 1 year at 20°C–25°C, protected from light.
Note: Glucose 50% stock solution is prepared by resuspending Glucose in MilliQ water to a concentration of 50% (w/v). Stock solution can be prepared in advance and can be stored for 1 year at 20°C–25°C.
Note: To prepare Amino Guanidin 500 mM stock solution, Amino Guanidin is resuspended in MilliQ water to a concentration of 500 mM. Stock solution can be prepared in advance and can be stored in aliquots for 1 year at −20°C.
Note: To prepare Ascorbic acid solution, Ascorbic acid is resuspended in MilliQ water to a concentration of 44 mg/mL. Solution should be prepared freshly before use and remaining solution should be discarded.
Note: To prepare AlexaFluor647-azide 10 mM stock solution, Alexa Fluor647-azide is resuspended in DMSO to a concentration of 10 mM. Stock solution can be prepared in advance and can be stored in aliquots for 1 year at −20°C.
MEA 1M solution
MEA 1M is prepared by dissolving 77 mg of MEA in 1 mL of HCl 360 mM.
Note: MEA powder is aliquoted in eppendorfs containing 77 mg each. Eppendorfs should be wrapped in parafilm and stored with Drierite under the vacuum desiccator, at 4°C for 6 months.
Note: MEA solution can be stored at 4°C for 1 month.
Imaging buffer for STORM
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | n/a | 237 μL |
| Glucose 50% | 5% | 30 μL |
| MEA 1M solution | 100 mM | 30 μL |
| Glox solution (100×) | 1% | 3 μL |
| Total | n/a | 300 μL |
Note: Imaging buffer should be prepared freshly right before use using PBS at 20°C–25°C.
Note: Always keep MEA and Glox solution on ice during imaging sessions.
Note: Glucose 50% can be stored at 20°C–25°C or 4°C for 1 year.
Note: MEA solution can be stored at 4°C for 1 month.
Alternatives: Reagents listed for Click chemistry reaction can be substituted with the components of the commercial kit Click-iT EdU Cell proliferation kit for imaging Alexa Fluor 647 dye (Cat#C10340). However, we recommend the use of EdC instead of EdU.
Step-by-step method details
We begin by describing how to culture and treat HeLa cells with Actinomycin D. We then illustrate the protocols for staining DNA with DAPI for confocal microscopy, for labeling DNA through Click Chemistry for STORM, and for immunostaining of H3K9me3 for STORM. Next, we describe the protocol for imaging acquisition, both for confocal imaging and STORM imaging. Finally, we explain the workflows for the analysis and quantification of acquired images through customized codes for ImageJ and MatLab.
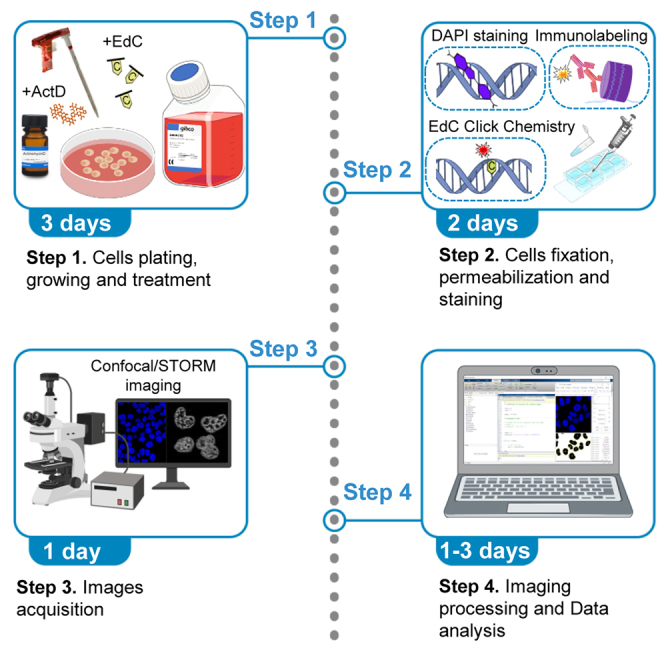
For a global overview of the step-by-step workflow see Figure 1.
Figure 1.

Diagram of the workflow for chromatin compaction quantification
Cell plating, synchronization, and treatment
Note: Pre-warming the complete cell culture medium and Trypsin-EDTA is recommended to avoid exposing culture cells to excessive thermal shift and minimizing detachment time, respectively.
Cells are seeded in 8-well chambers and allowed to attach for 24 h before challenging them with drug treatments or other experimental conditions that may induce changes in the degree of chromatin compaction.
-
1.Detach HeLa cells from maintenance cultures in T75 flasks or p10 dishes.
-
a.Discard exhausted culture medium.
-
b.Wash gently with 5 mL of PBS, and discard.
-
c.Add 1 mL Trypsin-EDTA (0.25%), phenol red to each flask.
-
d.Incubate flasks for 3 min at 37°C in cell incubator.Note: As detachment time can vary considerably with cell type, the plate should be visually inspected for complete cell detachment before proceeding.Note: Tapping gently on the sides of the flask/dish can favour the detachment.Alternatives: Trypsin without Phenol Red/Versene can be employed as an alternative agent for cell detachment.
-
e.Add 9 mL of complete culture medium and pipette gently up and down do disaggregate cells.
-
f.Collect detached cells in 15 mL Falcon tubes.
-
a.
-
2.HeLa cells counting.
-
a.Count cells with a standard or automated cell counter.
-
b.Prepare a cell suspension to achieve 1 × 105 cells/mL.
-
i.Put in a 15 mL Falcon the volume of cell suspension containing 800 000 cells.
-
ii.Dilute it with complete culture medium until reaching the volume of 8 mL.
-
i.
-
a.
Note: The suggested protocol is sufficient to plate 3 chamberslides at the density of 37 500 cells/cm2. In the case of using different cell lines and biological treatments, we recommend scaling up/down the cell density as well as the volumes for culturing cells into higher/lower number of wells. Particular attention should be taken in modifying density plating, since this protocol requires cells to be in a monolayer when imaged and superimposition of nuclei would compromise the analysis.
Note: To perform DAPI confocal imaging or H3K9me3 STORM imaging proceed to step 3, then to steps 5a and 6a. To perform EdC incorporation for DNA STORM imaging proceed directly to step 4, then to steps 5b and 6b.
-
3.HeLa seeding for DAPI confocal imaging or H3K9me3 STORM imaging
-
a.Seed 350 μL of this suspension in 8-well chambers and return plates to the incubator.
-
a.
-
4.HeLa seeding for DNA STORM imaging
-
a.Add 4 μL of EdC 10 mM solution to the 8 mL of cells suspension (1:2000 dilution, final concentration 5 μM).
-
b.Seed 350 μL of this suspension in 8-well chambers and return plates to the incubator.
-
a.
Alternatives: For steps 3 and 4, Borosilicate glass bottom 8-well chambers (Ibidi, #80827) can be used alternatively to Borosilicate glass bottom 8-well chambers (Lab-Tek, #155411), but volumes per well should be adjusted to maximum 250 μL to avoid spill-over. Adjust concentration of cell suspension accordingly.
-
5.Serum-starvation of HeLa cells
-
a.After 24 h from seeding, replace culture medium in all wells with 350 μL of pre-warmed resting medium for DAPI confocal imaging or H3K9me3 STORM imaging.
-
b.For wells designated to DNA STORM imaging, replace culture medium with 350 μL of pre-warmed resting medium plus EdC in the dilution of 1:2000, as the day before.
-
a.
Note: Step 5 can be skipped if cell cycle arrest is not required for your experimental design.
-
6.HeLa cells treatment with ActD (5 h)
-
a.After 43 h from seeding, replace resting medium in all wells with 350 μL of fresh resting medium plus ActD at 1:1000 dilution (final concentration 5 μg/mL).
-
b.For wells designated to DNA STORM imaging, also add EdC in the dilution of 1:2000, as the days before.
-
a.
Alternatives: Different treatment lengths and concentrations or alternative drug treatments can be performed at this stage. Adjust timing and drug concentration accordingly.
Alternatives: If you wish to compare experimental conditions other than drug treatments (for example, genetically modified cell lines), steps 5 and 6 can be skipped.
Cell fixation, permeabilization, and staining
After 5 h of ActD incubation, or corresponding alternative treatment, HeLa cells are subjected to fixation, permeabilization and labeling for IF microscopy-based assessment of DNA compaction and STORM microscopy-based assessment of DNA-free areas and H3K9me3 redistribution.
Note: Optimal fixation and permeabilization conditions may vary with cell type.
Note: Fixative solution should be at 20°C–25°C before being added to the cells, as cell fixation should be carried out at 20°C–25°C.
-
7.Fixation.
-
a.Discard medium.
-
b.Wash 3 × 5 minutes with PBS.
-
c.Fix cells with 300 μL of PFA 4% for 10 min at 20°C–25°C.
-
d.Wash 3 × 5 minutes with PBS.
-
a.
Pause point: Fixed cells can be stored (before staining) for up to 1 month at 4°C, provided that chamber lids are covered with 400 μL PBS and that plates are sealed with parafilm to avoid PBS evaporation. In this case, wells should be visually inspected to ensure normal cellular morphology and absence of contamination before resuming the protocol. Also, bring cells to 20°C–25°C before proceeding with permeabilization.
-
8.Permeabilization.
-
a.Permeabilize with Triton 0.4% for 15 min.
-
b.Wash 3 × 5 minutes with PBS.
-
a.
Note: For Click-chemistry reaction for DNA STORM imaging of EdC labelled cells proceed to step 9, then to step 13 (STORM imaging).
Note: For immunostaining of H3K9me3 and STORM imaging, proceed to step 10, then to step 13 (STORM imaging).
Note: For DAPI labeling for confocal imaging proceed directly to step 11, then to step 12 (confocal imaging).
-
9.Click Chemistry reaction for DNA STORM imaging.
-
a.Incubate cells with Click Chemistry solution for 30 min protecting samples from light.
-
b.Wash with PBS three times for 5 min each protecting samples from light.
-
c.Proceed directly with STORM imaging (step 13) or store at 4°C until imaging.
-
a.
-
10.Immunostaining for H3K9me3 STORM imaging.
-
a.Incubate wells with blocking buffer for 1 h at 20°C–25°C.
-
b.Incubate wells with Rabbit polyclonal anti-H3K9me3 diluted in blocking buffer 1:50 for 12–16 h at 4°C.
-
c.Wash with Washing buffer three times for 5 min each.
-
d.Incubate cells with 1:250 Goat anti-Rabbit IgG AF647 in blocking buffer for 1 h at 20°C–25°C in the dark.Note: From now on, keep the chamberslides in the dark to avoid bleaching of the AF647 fluorophore.
-
e.Wash secondary antibody with Washing buffer twice for 5 min each.
-
f.Wash once in PBS.
-
g.Proceed directly with STORM imaging (step 13) or store at 4°C until imaging.
-
a.
-
11.DAPI staining for DNA confocal imaging
-
a.Incubate wells with Blocking buffer plus DAPI diluted 1:1000 in PBS (final concentration 1 μg/mL) for 15 min at 20°C–25°C in the dark.
-
b.Wash three times with PBS.
-
a.
Alternatives: Hoechst 33342 at a 1:1000 dilution can be employed as an alternative agent for DNA staining instead of DAPI. Also, DNA labeled with EdC-AF647 can be imaged with confocal imaging.
Slight changes in the concentration of the dye (in the range of 1:500 -1:2000) are not crucial for the positive outcome of the protocol as far as the adjusted concentration is kept constant across all conditions and biological replicates.
Image acquisition
Stained chambers are mounted onto the microscopes (Confocal and STORM respectively) and images are acquired.
-
12.Confocal microscopy imaging.
-
a.Set parameters of image acquisition for the corresponding image size and zoom. We suggest to use an image size of 1024 × 1024, pinhole 1, scan speed 400 Hz, bidirectional scan, and a zoom of 1, in order to have around 50 nuclei per field at a high resolution.
-
b.Adjust laser power and gain for the DAPI channel to satisfy the dynamic range of the experiment by imaging at least 10 fields per condition. We normally use a laser power in a range between 10%–20% and a gain between 300–600V, and we suggest adjusting these parameters if needed, as far as they are kept constant across the whole experiment.
-
c.Visually inspect the objective for proper state. Place one drop of Type F oil (Leica) (RI 1.518) in the center of the objective lens.
-
d.Quickly but carefully, mount the chamber on the stage-holder.
-
e.Adjust the focus on the central stack of the cells and proceed with image acquisition. Repeat the step for at least other 9 fields per condition, in order to have enough nuclei to analyze.
-
a.
-
13.STORM imaging
-
a.Visually inspect the objective for proper state. Place one drop of Type F oil (Nikon) (RI 1.515) in the center of the objective lens.
-
b.Quickly but carefully, mount the chamber on the stage-holder.
-
c.Replace PBS with freshly prepared STORM imaging buffer in the well to image.
-
d.Set camera to 17 MHz, EM Gain 100V and image size to 256 × 256 pixels.
-
e.Detect the AF647 fluorescence in the sample by using a laser compatible with AF647 fluorophore excitation (peak at 647 nm) and starting at a low power coupled with a 100 ms exposure, to avoid blinking and sample bleaching. Adjust focus and illumination angle for optimal signal on the central stack of the cells.
-
f.Take a single snapshot of the nucleus/nuclei to be acquired. It can be used for further detection of nuclear structures (e.g., nucleoli), as well as for assaying the intensity of fluorescence prior to single-molecule acquisition.
-
g.Set the camera exposure time at 10 ms and increase the 647 nm laser power to 2 kW/cm2 approx., to visualize single molecule blinking.
-
h.Launch image acquisition with simultaneous illumination of the sample with 647 nm and 405 nm lasers. Specifically, 647 nm laser was used at constant ∼2 kW/cm2 power density and 405 nm laser power was gradually increased over the imaging, from 0.005 kW/cm2 to 0.5 kW/cm2. Typically, 60 000 frames were acquired and at least 10 fields were imaged per condition.
-
i.Generate localization lists from STORM images and perform drift correction with Insight3 (Huang et al., 2008) or Nikon NIS elements by selecting the command “Automatic drift correction”.In Insight3, select “Save Molecule List”, and a .bin file will be generated.To generate .bin files in NIS elements, go to Input & Output in the Molecule analysis section. Select Export and choose “Export to BIN file (M425)”.
-
j.To generate .bin files of individual selected nuclei in Nikon NIS elements go to Molecule analysis, Turn ON ROI button, open ROI Menu and select “Draw Bezier ROI…”. Check “Autofit ROI” option. Click “Process All Frames”. Export .bin file as described in point i).
-
a.
Note: We recommend the use of 405-nm laser activation during acquisition to maintain a good rate of single molecule blinking per frame throughout the entire image acquisition (approx. 30–50 localizations per frame). Nevertheless, EdC-AF647 signal typically does not require 405 nm laser activation during acquisition as enough single molecules are detected per frame.
Coefficient of variation analysis
For step-by step visual guide of the protocol please refer to Methods videos S1.
Single-channel confocal microscopy images are processed for the assessment of chromatin compaction through the measurement of the coefficient of variation.
Note: (Fiji Is Just) ImageJ imaging software is employed to perform this automated analysis.
Note: Before to begin, create one folder containing the .tiff files to be analyzed and the script FIJI_MACRO_CV.txt, contained in Data S1.
Output files will be saved automatically in the same folder. The name of the output file will include both the name of the source image and the index of the selected ROI.
Note: We also suggest generating a shortcut for the command “Run” to make the process of analysis even faster. To do this, select from (Fiji Is Just) ImageJ menu: > Plugins > Shortcuts > Add Shortcut…, and associate the Command “Run…” to the Shortcut of your preference.
In that case, step 14b will be substituted by “Click your personalized Shortcut for the command “Run…”.
-
14.Identification of single nuclei and generation of cropped single nuclei .tiff files.
-
a.Open one single-channel .tiff image.
-
b.Plugins > Macros > Run…
-
c.Run FIJI_MACRO_CV.txt.
-
a.
-
15.Check generated masks if they correctly match single nuclei.
-
a.Visually inspect if masks generated are correct.
-
b.If yes, > Close all.
-
c.If not, in FIJI_MACRO_CV.txt change the minimum and maximum threshold values set to generate the masks:
-
i.Open the .txt.
-
ii.In line 10,
- > //setThreshold(12, 255);
- the former parameter (i.e., “12”) is the threshold of the minimum intensity value detected by the mask, while the latter (i.e., “255”) is the threshold of the maximum intensity one. Change value “12” with customized values between 0 and 255 adapted to your particular condition. Note that lowering the minimum intensity value (i.e., <”12”) would allow the detection of weaker signal, while increasing it (i.e., >”12”) would increase the stringency. The maximum intensity value should be kept as “255”, to include the broadest range of intensity in the image.
-
iii.Save modified .txt.
-
iv.Re-analyze images from step 14.
-
i.
-
a.
Note: Although this code has been successfully tested on several different conditions, we are aware that the density of cell seeding and the quality of labeling can both affect the recognition of nuclei performed by FIJI_MACRO_CV.txt script. For this reason, we recommend modifying the ROI analyzed (see troubleshooting 1) and the command “Watershed” if needed (see troubleshooting 2).
Note: If after applying troubleshooting 2 and 3 some nuclei are still incorrectly identified, these can be discarded easily in step 15 by checking the mask numbers associated to them and eliminating the correspondent .tiff files in the folder.
-
16.
Run step 14 and step 15 for every image you want to analyze.
Note: We recommend analyzing at least 5 images per condition for a total of at least 100 nuclei per condition, and multiple biological replicates (at least 3).
-
17.Calculation of the Coefficient of Variation.
-
a.Open MatLab software.
-
b.Open the MATLAB_CV_Confocal.m script, contained in Data S1.
-
c.Change the following parameters according to your experiment:
-
i.>categ =; with the number of experimental conditions.
-
ii.>Folder = ; with the path of the data folder given as a string.
-
iii.>categname = ; with the names of the experimental categories given in cell format.
-
iv.>colors =[] ; with a 3-columns matrix with the color code for each category in each subsequent row. You can use predefined functions as>colors = colormap(jet());to define color series.
-
i.
-
d.Click Run.
-
e.Load the single nuclei .tiff files generated in step 15. One different window will open for each category.
-
f.Click Done.
-
g.Two pop-up windows with graphic representations of the results will appear. Figure 1 represents the distribution of pixel intensities of single cells (“CV_Histplot_individualcells”) (see Methods videos S1, 00:02:34), while Figure 2 plots the mean and standard deviation of the distribution of pixel intensities for each category (“CV_Histplot_smooth”) (see Methods videos S1, 00:02:36).
-
h.In the Command Window, the line
- >Do Boxplot (Y/N)>will appear.
Write “Y” (Yes) to additionally visualize the boxplot of the CV calculated (“Coefficient_Variation_Boxplot”) or “N” (No) to skip the visualization, and press Enter. All the .fig visualized, plus the .xls output file generated and the CV workspace are automatically saved in your folder of choice defined in step 17) c) ii).
-
a.
Figure 2.

Schematic view of the code to crop images in batch (left) of the corresponding subROI selection (right)
DNA-free area analysis
For step-by step visual guide of the protocol please refer to Methods videos S2.
Single-color STORM microscopy images of EdC-AF647 labeled DNA are processed for the assessment of chromatin compaction through the measurement of the percentage of DNA free areas. The same procedure can be applied for STORM images of core histones or histone modifications.
-
18.Calculation of the percentage of DNA-free areas
-
a.Open MatLab software.
-
b.Open Main code: “BlackSpace_MAIN”, contained in Data S2.
-
c.Change the following parameters according to your experiment:
-
i.>categ =; with the number of experimental conditions.
-
ii.>Directorio = ; with the path of the data folder given as a string.
-
iii.>categname = ; with the names of the experimental categories given in cell format.
-
iv.Check and adjust to image dimension by defining>maxX = ;>maxY = ;values (e.g., 256 or 512 pixels).
-
v.>h = fspecial('gaussian',[5 5], 2); (line 44) with “2” being the value of Gaussian sigma recommended. Adjust it if needed depending on your dataset.Increasing the value will increase the level of blurring of the image from which the masks of DNA and nucleus are generated.
-
vi.>MaskDNA = imbinarize(Dens,'adaptive','Sensitivity',0.001); (line 48) with “0.001” being the value of adaptive threshold recommended. Adjust it if needed depending on your dataset.
-
i.
-
d.Click Run.
-
e.Load .bin files for each experimental category.
-
f.Click Done.
-
g.One pop-up window with graphic representations of the results will appear for each analyzed file. Each figure is a panel displaying the density rendering of the nucleus, the DNA mask and the nuclear mask generated.
-
h.Calculated values of percentage of DNA-free areas are automatically saved as the .xls file called “Blackspace_results.xlsx” in your folder of choice defined in step 18) c) ii).
-
a.
Note: Although this code has been successfully tested on several different conditions, we are aware that the quality of labeling and alternative DNA distributions across cell types can affect the generation of nuclear masks and the identification of DNA-free areas performed by this script. For this reason, we recommend to visually verify the generated masks and modify the adaptive threshold value, pixel size and Gaussian sigma if needed (see troubleshooting 3).
Note: We recommend analyzing at least 10 images per condition.
Edge analysis
For step-by step visual guide of the protocol please refer to Methods videos S3.
Single-color STORM microscopy images of H3K9me3 are processed for the assessment of chromatin compaction through the measurement of the percentage of localizations found at the nuclear edge versus the total number of nuclear localizations. The same procedure can be applied to STORM images of any nuclear staining (for example: core histones, lamins, DNA) to quantify differences in signal enrichment at nuclear edge.
-
19.Calculation of the percentage of localizations at nuclear edge and nuclear interior
-
a.Open MatLab software.
-
b.Open Main script: "edgeLocDensity_main.m", contained in Data S3.
-
c.Change the following parameters according to your experiment:
-
i.>categ =; with the number of experimental conditions.
-
ii.>Folder = ; with the path of the data folder given as a string.
-
iii.>CategLabels =; with the names of the experimental categories given in cell format.
-
iv.>colors =[]; with a 3-columns matrix with the color code for each category in each subsequent row. You can use predefined functions as>colors = colormap(jet());to define color series.
-
v.>SR_px = ; with the size of super resolved pixels. We recommend starting with the value “20” and change it if needed.
-
vi.>NPx_edge = ; with the width in pixels of the edge mask desired. We recommend starting with the value “50”and adjust it if needed depending on your dataset. Note that very small values could cause a partial loss of the enrichment while too high values would cause the inclusion of also the inner part of the nucleus, not-being specific for the edge area anymore. Both conditions would cause an error of underestimation of the percentage of enrichment.
-
vii.>nmperpx = ; with the original pixel size in nm. Adjust this value (e.g., “160”) depending on the microscope model.
-
viii.>sigma = ; with the desired Gaussian Sigma value. We recommend starting with the value “1.5”. This parameter regulates the degree of blurring of the signal from which the nuclear mask is generated (the lower the value, the higher the stringency). If the signal of the protein labeled is not present across the whole nuclear area, we suggest to increase the value of blurring (lowering the stringency) in order to avoid the generation of artifactual holes.
-
i.
-
d.Click Run.
-
e.Load .bin files for each experimental category.
- f.
-
g.In the Command Window, the line>Do Density rendering (Y/N)>will appear.Write “ Y” (Yes) to visualize additionally the density renderings or “N” (No) to skip the visualization and press Enter.
-
h.Calculated values of percentage of localizations at nuclear edge and nuclear interior, as well as average and median density values, are automatically saved in a single file called ‘EDGE_analysis_results.xlsx’ in your folder of choice defined in step 19) c) ii).
-
a.
Note: Although this code has been successfully tested on several different conditions, we are aware that the quality of labeling can affect the generation of nuclear masks performed by this script. For this reason, we recommend to visually verify the generated masks and change the Gaussian sigma if needed (see troubleshooting 4).
Note: We recommend analyzing at least 10 images per condition.
Expected outcomes
Confocal and STORM images of cells exposed to transcription inhibition treatment processed as described above reveal the compaction of chromatin preferentially around the nuclear and nucleolar rims.
See Figures S2B, S2D, S2F and S2I in Neguembor et al. (2021), for comparison of coefficient of variation analysis between control and treated cells. See Figures 2B and 6C in Neguembor et al. (2021), for comparison of DNA-free areas between control and treated cells. See Figures 4D and 4F in Neguembor et al. (2021), for both comparison of percentage of Lamin A/C and of H3K9me3 at the nuclear rim between control and WAPL mutant conditions.
Quantification and statistical analysis
Quantitative results obtained upon image analysis as detailed above are processed with GraphPad Prism software as follows.
Coefficient of variation data generated through the MATLAB_CV_Confocal.m script were automatically saved as .xls file (step 17). The .xls was imported in a new GraphPad Prism file and plotted as column scatter plot with mean and standard deviation per conditions.
DNA-free areas data generated through the BlackSpace_MAIN.m script were automatically saved as .xls file (step 18). The .xls was imported in a new GraphPad Prism file and plotted as column scatter plot with mean and standard deviation per conditions.
Edge quantification data generated through the edgeLocDensity_main.m script were automatically saved as .xls file (step 19). The .xls was imported in a new GraphPad Prism file and plotted as column scatter plot with mean and standard deviation per conditions.
For all the above-mentioned quantification approaches, statistical analysis has been performed in Graphpad Prism. For every dataset, normality tests (D’Agostino-Pearson and Shapiro-Wilk) were run to assess normal distribution. For datasets with Gaussian distribution of values, parametric tests were applied. In all cases, two-tailed tests were run and multiple comparison corrections were applied for datasets with more than two groups and multiple comparisons. For datasets with no Gaussian distribution, non-parametric tests were applied. Statistical significance was represented in the following manner: ns p > 0.05, ∗ p ≤ 0.05, ∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001.
Limitations
Our protocol has been carried out only on adherent cells. Non-adherent cells pose a challenge for imaging because of their constant movements in the medium, even when sedimented. That can be addressed by coating the support with components allowing immobilization of cells. Since, EdC incorporation requires DNA replication, Click chemistry labeling can be performed only on replicating cells. If cell cycle arrest is desired, perform EdC incorporation before cell cycle arrest treatment.
Moreover, the staining of cells described in the protocol requires a step of permeabilization; thus, it is not compatible with live imaging. Nevertheless, in the case of coefficient of variation analysis this limitation can be overcome by skipping the step of fixation and performing nuclear staining with Hoechst instead of DAPI.
Troubleshooting
Problem 1
Not homogeneous cell density (steps 2, 3, and 4):
Homogeneous cell seeding and attachment is critical to ensure robust conditions for labeling, imaging and downstream analysis. Cell clumps and overlaps should be avoided as well as cell detachment from the well.
Potential solution
To ensure homogeneous cell seeding, it is important to disaggregate cell clumps by pipetting well and to prepare cell dilutions at the final concentration desired for plating, as described in the protocol, instead of plating cells at higher concentrations and adjusting volumes with medium in the wells. Make sure the cells are homogeneously seeded by gentle agitation of the chamberslide before placing them in the incubator. Avoid circular movements that tend to bring cells to the center of the well. Steps of pipetting and aspiration should be done carefully to avoid cell detachment. We recommend inclining the wells and placing the tip at a bottom corner of the wells.
Problem 2
Not homogeneous illumination (step 15):
The fields of confocal imaging could present regions more illuminated than others. Even though our method of analysis is independent on absolute values of pixel intensities belonging to distinct nuclei, the CV quantification can be affected if one single nucleus shows inhomogeneous illumination.
Potential solution
A simple solution is to analyze a subROI with homogeneous illumination.
The subROI can be either drawn by hand in individual files or a series of images displaying the same problem can be cropped systematically in batch.
The former method is suggested for single cases and requires the use of the tool Draw.
The latter is preferred when analyzing in batch and is performed by modifying the FIJI_MACRO_CV.txt file as following:
- The commands
>makeRectangle(xA, yA, width, height);
>run(“Crop”);
should be added as line 8 and 9, respectively.
The brackets of >makeRectangle(xA, yA, width, height); should contain the pixel coordinates of the upper left (xA, yA) vertex and the width and height of the rectangular subROI (see Figure 2).
Problem 3
Incorrect nuclei masks generation (step 15):
In rare cases, it can occur that one single nucleus is recognized as two different ones (Figure 3). This occurs most frequently in ROIs with cells at very low confluence or with very irregular shapes.
Figure 3.

Example of nuclei and relative masks displaying erroneous nuclear segmentation
Potential solution
This problem can be circumvented by deleting the line 15
>run("Watershed");
from the script FIJI_MACRO_CV.txt and running this modified script only on the ROI displaying the erroneous nuclei division. Alternatively, if this problem is very infrequent, individual masks presenting problems of segmentation can be discarded from the analysis (see step 15, note 2).
Problem 4
Incorrect nuclear mask generation and associated wrong recognition of DNA-free areas (step 18):
In rare cases, insufficient nuclear labeling or peculiar DNA distributions can lead to incorrect generation of nuclear masks with parts of nuclei being excluded from the analysis.
Potential solution
When setting up the analysis, visually inspect the nuclear masks generated for the different cells. If masks are suboptimal, modify the adaptive threshold value and Gaussian sigma until nuclear area and DNA-free areas are correctly segmented. We suggest starting with values of 0.001 for adaptive threshold and 2 for Gaussian sigma, and adjust them if needed.
Problem 5
Incorrect nuclear mask generation and associated wrong recognition of nuclear edges (step 19):
Potential solution
When setting up the analysis, visually inspect the nuclear masks generated for the different cells. If masks are suboptimal, modify the Gaussian sigma until nuclear areas are correctly segmented. We recommend starting with a Gaussian sigma value of 1.5, and adjust it if needed. We suggest to increase the value of Gaussian sigma if the signal is not homogeneously distributed across the nuclear area, since in this way the increase in blurring and the decrease in stringency would help in generating a correct nuclear mask.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maria Pia Cosma (pia.cosma@crg.es).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
The authors acknowledge the support from European Union's Horizon 2020 Research and Innovation Programme (CellViewer no. 686637 to M.P.C. and M.L.); Ministerio de Ciencia e Innovación, grant (BFU2017-86760-P [AEI/FEDER, UE] to M.P.C.), and an AGAUR grant from Secretaria d’Universitats i Recerca del Departament d’Empresa i Coneixement de la Generalitat de Catalunya ([2017 SGR 689] to M.P.C.); Centro de Excelencia Severo Ochoa (2013–2017 to M.P.C.); CERCA Programme/Generalitat de Catalunya (to M.P.C.); the Spanish Ministry of Science and Innovation to the EMBL partnership (to M.P.C.); National Natural Science Foundation of China (31971177 to M.P.C.); Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110103001 to M.P.C.); People Program (Marie Curie Actions) FP7/2007–2013 under REA grant 608959 (to M.V.N.); Juan de la Cierva-Incorporación 2017 (to M.V.N.); grant for the recruitment of early-stage research staff FI-2020 (Operational Program of Catalonia 2014-2020 CCI 2014ES05SFOP007 of the European Social Fund to L.M.); "La Caixa" Foundation Fellowship (ID 100010434, #LCF/BQ/DR20/11790016) (to L.M.); and "La Caixa-Severo Ochoa" pre-doctoral fellowship (to A.C.-G.). The authors acknowledge the Advanced Light Microscopy Unit from CRG for their excellent technical support.
Author contributions
Methodology, M.V.N., L.M., and C.V.; software, L.M., C.V., and A.C.-G.; formal analysis, M.V.N. and L.M.; investigation, M.V.N., L.M., and A.C.-G; visualization, M.V.N. and L.M.; writing – original draft, L.M., M.V.N., and M.P.C.; writing – review & editing, C.V., A.C.-G., and M.L.; funding acquisition, M.P.C. and M.L.; supervision, M.V.N. and M.P.C.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100865.
Contributor Information
Maria Victoria Neguembor, Email: victoire.neguembor@crg.es.
Maria Pia Cosma, Email: pia.cosma@crg.es.
Supplemental information
Data and code availability
The codes generated during this study are available at Github (https://github.com/CosmaLab/) and are provided as Data S1, S2, S3.
Original/source data for figures in the paper are related to Neguembor et al. and available at Mendeley Data: https://doi.org/10.17632/pbnc4zhcwd.1.
References
- Grezy A., Chevillard-Briet M., Trouche D., Escaffit F. Control of genetic stability by a new heterochromatin compaction pathway involving the Tip60 histone acetyltransferase. Mol. Biol. Cell. 2016;27:599–607. doi: 10.1091/mbc.E15-05-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Wang W., Bates M., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblanc M., Ragu S., Gey C., Contrepois K., Courbeyrette R., Thuret J.Y., Mann C. Parallel pathways in RAF-induced senescence and conditions for its reversion. Oncogene. 2012;31:3072–3085. doi: 10.1038/onc.2011.481. [DOI] [PubMed] [Google Scholar]
- Neguembor M.V., Martin L., Castells-Garcia A., Gomez-Garcia P.A., Vicario C., Carnevali D., AlHaj Abed J., Granados A., Sebastian-Perez R., Sottile F. Transcription-mediated supercoiling regulates genome folding and loop formation. Mol. Cell. 2021;81:3065–3081.e12. doi: 10.1016/j.molcel.2021.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden C.T., Schindelin J., Hiner M.C., DeZonia B.E., Walter A.E., Arena E.T., Eliceiri K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18:529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes generated during this study are available at Github (https://github.com/CosmaLab/) and are provided as Data S1, S2, S3.
Original/source data for figures in the paper are related to Neguembor et al. and available at Mendeley Data: https://doi.org/10.17632/pbnc4zhcwd.1.