

Abstract
Glioblastoma (GBM) is the most common primary tumor of the central nervous system. Arising from neuroepithelial glial cells, GBM is characterized by invasive behavior, extensive angiogenesis, and genetic heterogeneity that contributes to poor prognosis and treatment failure. Currently, there are several molecular biomarkers available to aid in diagnosis, prognosis, and predicting treatment outcomes; however, all require the biopsy of tumor tissue. Nevertheless, a tissue sample from a single location has its own limitations, including the risk related to the procedure and the difficulty of obtaining longitudinal samples to monitor treatment response and to fully capture the intratumoral heterogeneity of GBM. To date, there are no biomarkers in blood or cerebrospinal fluid for detection, follow‐up, or prognostication of GBM. Liquid biopsy offers an attractive and minimally invasive solution to support different stages of GBM management, assess the molecular biology of the tumor, identify early recurrence and longitudinal genomic evolution, predict both prognosis and potential resistance to chemotherapy or radiotherapy, and allow patient selection for targeted therapies. The aim of this review is to describe the current knowledge regarding the application of liquid biopsy in glioblastoma, highlighting both benefits and obstacles to translation into clinical care.
Implications for Practice
To translate liquid biopsy into clinical practice, further prospective studies are required with larger cohorts to increase specificity and sensitivity. With the ever‐growing interest in RNA nanotechnology, microRNAs may have a therapeutic role in brain tumors.
Keywords: Liquid biopsy, Glioblastoma, Biomarker, Circulating tumor cells, Circulating tumor DNA, Microvescicles, Circulating microRNA, Prognostic, Predictive
Short abstract
This review describes the current knowledge regarding the application of liquid biopsy in glioblastoma, highlighting both the benefits and the obstacles to translation into clinical care.
Introduction
Glioblastoma (GBM) is the most common malignant tumor of the central nervous system, characterized by dismal prognosis, marked genetic heterogeneity, and unpredictable clinical behavior [1, 2].
Diagnosis of GBM remains a clinical challenge. Magnetic resonance imaging (MRI) and advanced MRI techniques, such as diffusion magnetic resonance imaging, perfusion‐weighted imaging, and magnetic resonance spectroscopy, are the optimal neuroradiological technique for the study of GBM, which typically appears as an infiltrative, heterogeneous, ring‐enhancing lesion with central necrosis and surrounding peritumoral edema. Nevertheless, diagnostic errors occur in up to 30% of the cases when clinical decisions are based only upon MRI [3]: this implies that the final pathologic diagnosis of GBM inevitably relies on tissue biopsy. However, this procedure also has several limitations: it is highly invasive and burdened with side effects, such as hemorrhage and neurological function impairment. In addition, samples may be small and not representative of the entire tumor, capturing only a static snapshot of an ever‐changing tumor.
The standard approach for GBM treatment relies on maximal surgical resection followed by concurrent chemoradiation using the alkylating agent temozolomide (TMZ) and a further adjuvant treatment with TMZ for a total of six cycles of maintenance according to the pivotal phase III trial published in 2005 by Stupp et al. [4]. The addition of a tumor treating field to the maintenance therapy with temozolomide, although challenging given low compliance rates and high costs, has shown improved overall survival (OS) in patients with glioblastoma. It is a device that works by delivering low‐intensity alternating electric fields that disrupt the microtubules in the mitotic spindle leading to tumor cell death [5].
Despite this multimodality therapy incorporating surgery, radiotherapy, and chemotherapy, the prognosis remains poor, and the majority of patients with GBM experience recurrence within 6 months. Treatment options in the relapsed or recurrent setting are less well defined, with no established standard of care and little evidence for any interventions that prolong OS; such options include further surgical resection, reirradiation, systemic therapies, or supportive care alone, given the significant proportion of patients not even eligible for second‐line therapy. Lomustine, carmustine, and rechallenge with temozolomide are all potential options, although the benefits are modest, and only patients with O6‐alkylguanine DNA alkyltransferase (MGMT) promoter methylation are likely to benefit [6, 7].
The need for additional therapeutic avenues and evolving insights into the pathophysiology and molecular biology of GBM, have allowed the development of new therapeutic approaches targeting “neovascularity”: bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor (VEGF), received accelerated approval from the U.S. Food and Drug Administration for treatment of recurrent glioblastomas in the U.S. based on the success of two phase II clinical trials [8] showing an improvement in progression‐free survival compared with nitrosourea‐based treatment, but it failed to show a positive effect on overall survival. As a result, bevacizumab is approved in the U.S. but not in Europe, and its role in the treatment of patients with recurrent GBM remains unclear.
Regorafenib is a tyrosine and serine–threonine kinase inhibitor targeting angiogenic (VEGF), stromal (platelet‐derived growth factor receptor, fibroblast growth factor receptors), and oncogenic (KIT, RAF, RET) tyrosine kinase receptors. The REGOMA phase II trial demonstrated the superiority of regorafenib compared with lomustine in the treatment of recurrent GBM, with significant improvement of the median overall survival from 5.6 to 7.4 months [9].
Significant obstacles prevent the development of effective treatments in GBM [10, 11], including genetic instability, intratumoral histopathological variability, and the persistence of a subpopulation of cancer cells with stemlike properties that are believed to be the main causes of cancer recurrence and resistance to radiotherapy and chemotherapy [12].
The interpatient and intratumoral heterogeneity of GBM poses several major challenges: molecular characterization, definition of the prognosis, monitoring of tumor progression, and definition of the response to the treatment. These challenges are further complicated by the invasive nature of brain surgery and tissue biopsy, which requires high economic expenditure and specialized surgical expertise, at the cost of a high morbidity rate, making the procedure of rebiopsy difficult to perform.
It must also be considered that the value of biopsy in a single point is limited because it may not reflect the entire tumor, which is constantly evolving in response to clonal selection, hypoxia, and treatments [13].
Liquid biopsy started to be considered a new standard of care for oncological patients mainly after the U.S. Food and Drug Administration (FDA) approval in 2016 of the first blood‐based test for epidermal growth factor receptor (EGFR) mutational status [14].
In this context, liquid biopsy appears as a minimally invasive, safe, and sensitive alternative approach for tissue biopsies in patients with GBM.
A range of prognostic or predictive molecular biomarkers have been identified in GBM, including IDH mutation, MGMT methylation, phosphatase and tensin homolog (PTEN) mutations, EGFR variant III (EGFRvIII) mutation and/or EGFR amplification, glial fibrillary acidic protein (GFAP) mutation, telomerase reverse transcriptase (TERT) promoter alterations, and loss of heterozygosity in chromosome 10 [15].
In addition, the pivotal role of epigenetics (DNA methylation, histone modification, and microRNA) is becoming increasingly evident in the regulation of the molecular processes and metabolic alterations underlying the aggressiveness and recurrence of GBM [16, 17].
Cell metabolism is mediated by a series of enzymes, whose regulation is important for tumor progression. DNA methylation in the promoter or intron regions of metabolism‐related genes influences cell metabolism by silencing gene expression.
In GBM, in particular, DNA methylation and microRNA affect several metabolic processes, such as glycolysis, oxidative phosphorylation, and lipid metabolism. For example, hypermethylation in the promoter or intron regions is commonly found in IDH1‐mutant GBM, whereas hypomethylation is commonly found in mesenchymal GBM, where the transcriptional activities of these glycolytic genes are activated [18].
The lack of standardized technologies, the small cohorts of patients in all studies, and the lack of confidence in these new methods compared with tissue biopsy are the main limitations in the application of liquid biopsy in GBM.
Rationale for Liquid Biopsy Development in GBM
Genomic characterization is crucial for optimal diagnosis and treatment of GBM, characterized by a complex genomic landscape that changes over time in response to disease progression or treatment effects.
GBM can harbor IDH mutations, present in only about 5% of patients, that beyond their diagnostic role represent the most powerful positive prognostic factor. In addition, GBM is characterized by overexpression and mutations of EGFR (especially EGFRvIII mutation), rearrangements of chromosomes 7 and 10, and TERT mutations [19]. Currently, MGMT promoter methylation is the only established predictive factor of response to alkylating agent temozolomide [20]. Distinct mutations are used for the molecular classification of glioblastoma (EGFR amplification is associated with classical subtype, NF1 and TP53 mutations with the mesenchymal subtype, and IDH1 mutation with the proneural subtype).
Since the 2016 World Health Organization classification, the routine diagnostic workup for GBM requires molecular genetic analysis to guide patients’ prognosis stratification and treatment decisions: among a number of clinically significant biomarkers, the most representative are the mutational status of IDH1/2, MGMT promoter methylation, and TERT mutation [19].
Plasma, serum, and cerebrospinal fluid (CSF) are biosources of tumor‐associated biomarkers [21]. The ideal source of circulating biomarkers in GBM has been much debated over the last years. Peripheral blood has become the most commonly investigated liquid biopsy source, given the advantages of noninvasive collection and dynamic reflection of tumor progress [22]. CSF, on the other hand, continuously circulating around the central nervous system (CNS), can fully contact tumor tissue, transporting tumor metabolites and exfoliated tumor cells.
A tumor‐related biomarker, to be usefully introduced into clinical practice, should be easily detected by an analytical method with high sensitivity and specificity, be economically acceptable, and provide accurate information about tumor burden.
Liquid biopsy allows tumor monitoring by detection in peripheral blood or CSF of diverse classes of biomarkers through the analysis of biological samples by different approaches, including circulating tumor cells (CTCs), plasmatic concentration of circulating free DNA (cfDNA) that contains circulating tumor DNA (ctDNA), circulating cell‐free tumor RNA, extracellular vesicles (EVs), proteins, metabolites, and tumor‐educated platelets. This circulating material can derive from tumor tissue and thus can provide real and representative information for molecular profiling and tumor monitoring in clinical oncology. Integration of these technologies and their role in the different stages of GBM management are presented in Figure 1.
Figure 1.
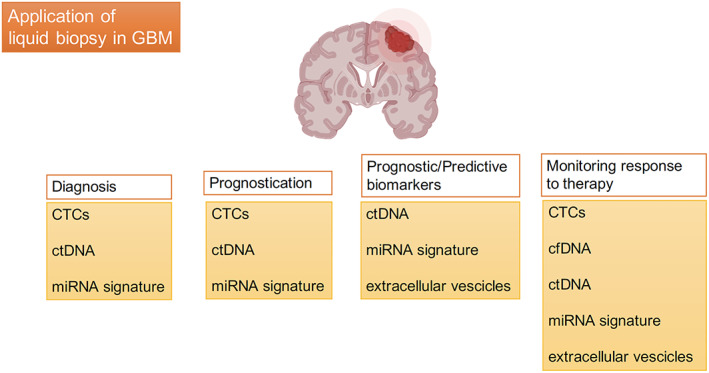
Most promising biomarkers in the different stages of GBM management. Abbreviations: cfDNA, circulating free DNA; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; GBM, glioblastoma; miRNA, microRNA.
The procedure of liquid biopsy offers several advantages in the management of GBM:
early detection of cancer (screening);
discrimination between neoplastic transformation from other processes, such as inflammation or neurodegeneration;
early diagnosis of minimal residual disease after neurosurgery;
prognostication by providing information about stage and spread of the disease;
identification of new targets for personalized treatment;
early detection of tumor progression or pseudoprogression;
assessment of tumor burden;
assessment of prognostic and predictive biomarkers to stratify patients into tumor subgroups;
prediction of tumor sensitivity or resistance to chemotherapy and radiotherapy;
monitoring of tumor response to treatments; and
overcoming the limits of multiple biopsies and, given high repeatability over time, guaranteeing the capture of tumor heterogeneity and clonal evolution of the disease and the ability to monitor “real‐time” pharmacological responses.
Each biosource and biomolecule has its potential advantages and disadvantages for cancer diagnostics, prognostics, and therapy monitoring. For example, ctDNA may reflect the entire tumor genome, and it has gained traction for its potential clinical utility not only of detection of point mutations or structural variants but also of copy number aberrations and methylation status. However, genomic analysis of ctDNA has limited sensitivity, primarily because of its limited abundance in plasma and because of the limited number of somatic mutations that distinguish cancerous ctDNA from normal circulating DNA.
CTCs provide the possibility of a comprehensive molecular characterization of the entire cell, including RNA and proteins, and represent an effective biomarker for predicting the prognosis of various cancers, but searching for CTCs in the serum of patients with GBM is difficult because they are rare and difficult to detect; reliable tumor‐specific cell surface markers are not yet established.
Many studies have shown that both ctDNA and CTCs are present in advanced tumors, although only a few studies have compared the amounts of CTCs and ctDNA templates in the same patients [23, 24, 25, 26]. Comparison of the two approaches has reached opposing conclusions, likely because of technical limitations. EVs have been shown to harbor proteins and RNA biomarker molecules of variable length, but also surface membrane proteins that are correlated to organ tropism for cancer metastasis. Tumor‐educated platelets have emerged as central players acting as local and systemic responders to tumor growth, capable of sequestering EV‐derived RNAs and proteins as well as altering their spliced RNA profile [23, 27, 28].
Both benefits and pitfalls of different techniques are summarized in Table 1.
Table 1.
Pros and cons of different approaches employed for liquid biopsy in glioblastoma
| Approach | Pros | Cons |
|---|---|---|
| CTCs | Information can be provided at the protein, DNA, and RNA levels |
Only a few studies have been carried out by using brain tumor–derived CTCs |
| ctDNA |
Might provide a comprehensive view of the glioblastoma genome [40, 41, 42, 43, 44, 45, 46, 47] Higher ctDNA levels compared with CTC |
Blood ctDNA of patients with primary brain tumor is low compared with other tumors that are able to transfer ctDNA fragments into blood [50, 51, 52, 53, 54] Short half‐life, <1.5 hour Improvement in sequencing technologies is necessary [50, 51, 52, 53, 54] |
| miRNAs |
Extremely stable in biological fluids [90] Using sophisticated software (e.g., TargetScan) it is possible to correlate miRNAs with potential target genes [91] |
Development of panels of validated miRNAS is necessary because of the uncertainties of the current findings [100, 101] |
| Extracellular vesicles |
Easy detection Better source of nucleic acids for tumor molecular profiling as compared with cell‐free nucleic acids [121] Promising data, such as detection of the EGFRvIII deletion variant [129] |
Possible presence of contaminants by current isolation methods |
Abbreviations: CTC, circulating tumor cell; ctDNA, circulating tumor DNA; EGFRvIII, EGFR variant III; miRNA, microRNA.
Interestingly, a combined readout of multiple biosources and biomolecules in parallel could be a promising strategy to make the liquid biopsy a more usable and exploitable tool for brain tumors.
Circulating Tumor Cells
CTCs are nucleated tumor cells released into the bloodstream and CSF from a primary or metastatic tumor; they are believed capable of reflecting the molecular biology of the original tumor [29]. Only CTCs resistant to microenvironment stress agents and to host immune surveillance can be detected in biofluids.
CTCs are extremely rare in the circulation, compared with normal blood cells (one cell in 109 blood cells), which makes their capture challenging and their isolation difficult because of the complexity of the required techniques. CTC analysis provides the possibility of analyzing the entire cell, including RNA and proteins, and can complement liquid biopsies through comprehensive molecular characterization of the cells [30]. Available methods for CTC enrichment and isolation from the peripheral blood rely on antibody‐mediated methods: “positive selection,” which can be achieved by targeting specific tumor markers that are commonly expressed on the surface of these cells, such as epithelial cell adhesion molecules (EpCAMs), and “negative selection methods” based on CTC size (CTCs are larger than normal blood cells) like the EPISPOT assay and the ISET, Parsortix, and DEPArray technologies [23, 31, 32].
The CellSearch system is a circulating tumor cell kit for the enumeration of CTCs of epithelial origin (CD45−, EpCAM+, and cytokeratins 8, 18+, and/or 19+) in whole blood and is the only platform approved by the FDA for monitoring patients with metastatic breast, prostate, and colorectal cancer. The presence of CTCs in the peripheral blood, as detected by the CellSearch test, is associated with decreased progression‐free survival and decreased overall survival in patients treated for metastatic breast, colorectal, or prostate cancer.
CTCs can be used for molecular characterization, using techniques such as reverse‐transcription polymerase chain reaction (PCR), fluorescence in situ hybridization (FISH), and next‐generation sequencing (NGS), to study tumor heterogeneity and clonal evolution.
Despite being one of the most important weapons in the field of liquid biopsy in diverse types of solid tumors, including melanoma, lung cancer, osteosarcoma, and pheochromocytoma [24, 25], in the past years the application of CTCs in GBM was considered a failure because of the special nature of the blood‐brain barrier (BBB) and brain microenvironment; this hypothesis was further supported by the low rate of extracranial metastases despite the high aggression of this malignancy.
Moreover, glioma cells do not express epithelial biomarkers or EpCAM; thus, they are not detectable using conventional methods like the CellSearch system [30, 33].
Therefore, searching for CTCs in the serum of patients with GBM is difficult given that reliable tumor‐specific cell surface markers have yet to be established. Thus, investigators have targeted panels of membrane proteins that may identify GBM‐specific CTCs [33, 34].
Recently, a few studies have reported that CTCs can be detected in high‐grade glioma and, in particular, in GBM, especially in the more aggressive, mesenchymal subtypes and in progressing disease, providing the possibility of their application in brain tumor management [35].
Sullivan et al. [26] detected CTCs in the serum of 13 of 33 patients with GBM using a trio of antibodies (anti‐CD14, anti‐CD16, and anti‐CD45) through CTC‐iChip technology, showing that the majority of the CTCs had a predominant mesenchymal molecular signature typical of the aggressive mesenchymal GBM subtype, with high expression of SERPINE1, TGFB1, TGFBR2, and VIM. FISH analysis highlighted the overexpression of wild‐type EGFR, GFAP, and nestin and the absence of EpCAM.
Macarthur et al. also isolated CTCs using telomerase activity and, in particular, an adenoviral probe to TERT, which is elevated in this malignancy. CTCs were detectable from the serum of 8 of 11 patients with GBM (71% of patients) before the radiotherapy and in one of eight patients postradiotherapy, demonstrating that CTC detection is predictive of disease progression [36].
Similarly, Müller et al. isolated CTCs in 20% of patients with GBM, employing differential centrifugation with Ficoll‐Paque gradients followed by fluorescence immunocytochemistry to marker CTCs, confirming that GBM CTCs have a low proneural signature with elevated EGFR copy number [37].
Krol et al. found the presence of clusters of CTCs in the blood of patients with GBM, ranging from 2 to 23 cells, expressing EGFR, Ki67, and EB1 markers but negative for CD45 [38].
Moreover, CTC detection may be useful to differentiate between real disease progression and pseudoprogression. In fact, there is growing evidence that CTC count reflects tumor burden, increasing with progression disease and drastically shrinking as a result of response to chemoradiotherapy [39].
In conclusion, the studies so far conducted have shown a detection rate of CTCs varying from 20% to 70%; this is due to the heterogeneity of the detection methods, the lack of standardized tumor‐specific cell surface markers, and the absence of a procedural uniformity that can make the various studies comparable.
Additional research is required to more effectively detect CTCs, which have proven to be an intriguing and promising modality for GBM diagnosis.
Circulating Tumor Nucleic Acid
ctDNA is a portion of the overall cfDNA and is a short fragment (usually 130–180 base pairs) of single‐ or double‐stranded DNA released by the tumor cells into the bloodstream harboring the same mutational status of the original tumor and is thus useful, for example, to establish sensitivity or resistance to targeted therapies [40, 41, 42, 43, 44, 45, 46, 47].
Given the abundance of cfDNA released by normal cells, it is challenging to separate, in the blood samples, the low concentrations of ctDNA from the high “noisy” amount of cfDNA.
Boisselier et al. detected small‐size DNA in the plasma of patients with glioma and reported that cfDNA concentration was correlated with the grade and the enhancing tumor burden in these patients [48].
Patients with high‐grade glioma had a significantly higher plasma small‐size DNA concentration than patients with low‐grade glioma and healthy controls. This is probably due to the presence of the BBB. The intact BBB in low‐grade glioma limits the diffusion of small‐size DNA into the bloodstream, whereas high‐grade glioma, characterized by a disrupted BBB, exhibits a large amount of circulating small‐size DNA.
ctDNA analysis might be the best option to obtain the genomic profile of patients with glioblastoma because it provides information on the specific gene mutation and dynamically reflects tumor progression and drug resistance mutations at an early stage [35].
For ctDNA analyses, plasma samples are preferable to serum samples because they represent a good source of ctDNA and lack background levels of cfDNA, which are higher in serum, probably because of contamination with DNA released from immune cells lysed during the clotting process [35].
Initial optimism for the use of plasma ctDNA in GBM has turned into disappointment after the results of studies demonstrating low rates of plasma ctDNA in patients with glioma compared with other solid tumors [46, 48, 49].
Boisselier et al. first [48] demonstrated a glioma‐specific mutation in plasma ctDNA, detecting the IDH1 R132H mutation in plasma from patients with mutated glioma, confirming that small‐size DNA originates from tumor cells. Boisselier et al. used co‐amplification at lower denaturation temperature PCR, which preferentially amplifies mutant DNA, and digital PCR, which is a highly sensitive approach to detect distinct mutations, and found that a total of 15 out of 25 mutated tumors exhibited the IDH1 R132H mutation in plasma (compared with none of the 14 nonmutated tumors), with sensitivity increasing for high‐grade gliomas and enhancing tumor volume. Particularly, they found the IDH1 R132H mutation in 60% of patients with IDH‐mutant GBM. In this study, the specificity of the circulating tumor DNA measurement was low, and the plasma DNA concentration in the patients widely overlapped with that of the healthy subjects. One possible reason is that Boisselier et al. performed a second centrifugation to reduce the contamination of the small‐size DNA delivered by tumor cells from DNA of circulating normal cells, which could explain the lower DNA concentration found here compared with other studies. This makes this study not very reproducible.
In glioblastoma, the presence of tumor‐derived ctDNA in plasma is low compared with other cancers because GBM does not metastasize beyond the central nervous system and because of the presence of the BBB, in contrast to other tumors that are able to transfer ctDNA fragments into blood.
Bettegowda et al. found a substantial reduction in detectability of ctDNA, mutations in localized disease compared with metastatic tumors. In particular, they found that ctDNA was detectable in >75% of patients with advanced colorectal, ovarian pancreatic, bladder, gastroesophageal, breast, melanoma, hepatocellular, and head and neck cancers but in less than 10% of patients with glioma. ctDNA was often present in patients without detectable circulating tumor cells, suggesting that these two biomarkers are distinct entities [49].
A substantial issue regarding the use of ctDNA lies in the importance of researching distinct tumor mutations, as the amount of ctDNA alone is not an applicable as a diagnostic tool.
The “ideal” mutation should be frequent, having diagnostic, prognostic, or predictive value, and for patients with glioma the IDH1/2 mutations meet these conditions.
Because of the challenge involved in identifying ctDNA, technology is refining: the two methods currently used to detect somatic alteration in ctDNA are next‐generation sequencing and digital PCR [50, 51, 52, 53, 54].
Digital droplet PCR (ddPCR) consists in the subdivision of a sample into many subsamples, each undergoing an individual PCR reaction. It is considered the most sensitive and specific method for detection and quantification of ctDNA when compared with Sanger sequencing, quantitative PCR, and next‐generation sequencing because it works with smaller amounts of nucleic acid (i.e., millions of isolated droplets), allowing the detection of rare, low mutation allelic frequencies [55, 56].
Whereas NGS explores a wide range of nucleic acids present in a sample, allowing the detection of unknown genetic mutations, ddPCR allows rare event detection and quantification.
Although ddPCR experiments are easier to set up and faster, present lower costs, and do not need complex bioinformatic analysis, nevertheless, they only enable the study of known mutations; NGS, instead, has the advantage of massive whole‐exome sequencing in a single experiment.
Several studies recently compared the sensitivity of two methods, ddPCR and targeted NGS, for the detection of somatic mutations in ctDNA.
Hattori et al. demonstrated that ddPCR detected MYD88 c.T778C in 93% (13/14) of patients with primary CNS lymphoma (PCNSL), whereas targeted NGS did not (0/14) [57].
In a similar study, the histone 3 p.K27M (H3K27M) mutation was detected by ddPCR in 88% of plasma samples of patients with high‐grade gliomas [55].
Studies performing comprehensive ctDNA analysis and a highly sensitive and specific NGS assay yielded approximately 50% of the ctDNA detection rate in patients with advanced GBM and showed that the ctDNA detection rate in gliomas may increase in high‐grade gliomas and in the presence of a more aggressive histology [35, 58, 59]. The most frequent somatic mutations detected using NGS technology were observed in TP53, NF1, EGFR1, MET, APC, and PDGFRA genes together with amplifications of ERBB2, MET, and EGFR, among others [35, 58, 59].
Examining MGMT promoter methylation or loss of heterozygosity in chromosome 1p, 19q, and 10q through next‐generation sequencing, several authors have detected at least one somatic alteration in the plasma of patients with glioma, with higher sensitivity and specificity in presence of high measurable tumor volume by MRI [60, 61].
Fontanilles et al. [33] provided evidence that somatic mutations can be detected by NGS in the circulating cfDNA released by a subset of patients suffering from PCNSL, characterized by MYD88 and CD79B alterations [15, 18, 62]. Fontanilles et al. used a two‐step approach. First, patient‐specific somatic mutations were identified using a targeted panel. Then, a second sequencing using a restricted panel targeting MYD88 c.T778C was performed and compared with plasma samples from control patients with other cancers. The researchers found that a total of 32% patients (8/25) had detectable somatic mutations in cfDNA.
Nassiri et al. [63] performed a plasma‐based liquid biopsy approach for diagnostics of tumors of the central nervous system and for differential diagnosis between primitive gliomas and brain metastasis, without relying on information obtained from a tumor tissue biopsy. Their approach, named cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing, was based on the detection of the methylome signature of glioma ctDNA in plasma. This approach allowed the researchers not only to effectively detect ctDNA despite low ctDNA abundance but also to noninvasively discriminate among common intracranial tumors that share similar cell‐of‐origin lineages (meningiomas, hemangiopericytomas, low‐grade glioneuronal tumors, IDH‐mutant gliomas, IDH wild‐type gliomas).
Another issue regarding the use of ctDNA concerns the high fragmentation that characterizes tumor‐derived circulating DNA. Underhill et al. have highlighted that ctDNA length is consistently shorter than normal cell‐free DNA. Subsequently, size selection for shorter cell‐free DNA fragments may increase the proportion of ctDNA within a sample [64].
Existing methods to improve detection of ctDNA have focused on sensitivity for detecting genomic alterations but have rarely considered the biological properties of plasma cfDNA. Mouliere et al. hypothesized that differences in fragment lengths of circulating DNA could be exploited to enhance sensitivity for detecting the presence of ctDNA and for noninvasive genomic analysis of cancer. They analyzed 13 CSF samples from patients with primary high‐grade glioma and detected enrichment of ctDNA in fragment sizes between 90 and 150 base pairs and developed methods for in vitro and in silico size selection of these fragments. They demonstrated that a specific fragmentation signature, with enrichment in small fragments between 90 and 150 base pairs, improved detection of tumor DNA in CSF, with more than twofold median enrichment in >95% of cases and more than fourfold enrichment in >10% of cases [65]. This innovative approach may represent an interesting alternative way to detect GBM ctDNA in CSF at acceptable cost.
Another blood‐based liquid biopsy novel approach relies on tumor‐educated platelets (TEPs). Platelets are anucleate cells originating from megakaryocytes and have been recognized for their complex interaction with cancer and for their central function as local and systemic responders to tumor growth [66, 67]. TEPs are platelets that undergo a tumor‐induced biomolecule transfer process called “education” that consists in receiving tumor‐associated molecules from neoplastic cells and that results in altered platelet behavior [68, 69]. Cancer cells transfer mutant RNA into blood platelets; thus, TEPs are characterized by distinct RNA profiles. During tumor growth platelets are “educated” by the tumor environment and exhibit altered spliced RNA profiles by several mechanisms: tumor‐induced alteration of RNA transferred by megakaryocytes into platelets; platelet RNA alternative splicing events derived from tumor, stromal, or immune cells; evolution of differential platelet subpopulations; and altered platelet turnover [70, 71, 72, 73, 74].
Several methods for isolation and analysis of platelet‐spliced RNA profile have been developed, establishing TEPS as a valuable complement of the approaches employed for liquid biopsy.
In 2015, Best et al. [27] first developed the ThromboSeq platform to perform an extensive RNA sequencing from 283 platelet samples aimed at determining differentially spliced RNA profiles in platelets from patients with cancer and healthy controls. Best et al. correctly distinguished 228 patients with localized and metastasized tumors (across six different tumor types including non‐small cell lung carcinoma, colorectal cancer, glioblastoma, pancreatic cancer, hepatobiliary cancer, and breast cancer) from 55 healthy individuals with 84%–96% accuracy, glioblastoma cases being among the most accurately distinguished. The location of the primary tumor was correctly identified with 71% accuracy.
Additionally, microarray analysis revealed that TEPs from patients with glioblastoma present a distinct RNA profile, and, in particular, they capture tumor‐derived EVs containing EGFRvIII mutant RNA. Specifically, EGFRvIII mutation was detected in 80% of glioblastomas but not in healthy controls [27].
Sol et al. showed that patients with glioblastoma have markedly altered TEP‐spliced RNA profiles that enable high‐accuracy differential diagnosis between GBM and other neurological diseases, like multiple sclerosis or brain metastasis. Moreover, Sol et al. developed the digitalSWARM algorithm to improve monitoring of glioblastoma progression and demonstrate that the TEP tumor scores of individual patients with glioblastoma could be used to distinguish pseudoprogression from true progression [75].
TEPs offer certain advantages over other blood‐based approaches, including their abundance and easy isolation and their high‐quality RNA. Combinatorial analysis of TEPs with complementary biosources such as EVs, ctDNA, and CTCs, but possibly also imaging and protein markers, may provide optimal diagnostic synergy and warrants consideration as a next‐generation biomarker option [76].
In addition to circulating nucleic acid, analytical methods to capture the “metabolome,” the complex of tumor‐associated proteins whose abundance drastically increases as a consequence of cancer‐associated somatic mutations in specific enzymes, have also been revealed as a promising approach [77].
The most commonly known oncometabolite is D‐2‐hydroxyglutarate (2HG), which accumulates in glioma cells with IDH1 mutation and is responsible for gliomagenesis via multiple processes [78].
Because 2HG is a small molecule, it can reach the systemic circulation; thus, altered 2HG serum or urine levels, detected by mass spectrometry or liquid chromatography, might help to identify patients harboring IDH1‐mutated gliomas [79].
Lombardi et al. [80] measured and compared the 2HG levels in plasma and urine of 84 patients with glioma using liquid chromatography tandem mass spectrometry. Lombardi et al. found a significant difference in the 2HG plasma and urine levels between patients with and without an IDH1 mutation. Furthermore, they observed that plasma and urine levels of 2HG could serve as a surrogate biomarker of the treatment response and disease recurrence in patients with glioma and could be used to predict response to treatment [80].
Picca et al. reported that 2HG can be used as an “imaging biomarker,” and noninvasive diagnostic approaches have been developed for the detection of 2HG through magnetic resonance spectroscopy of the brain; thus, the IDH mutation is a candidate to become a possibly theranostic marker in the near future [81].
CSF Tumor Nucleic Acid
Given that the brain is immersed in CSF, this biofluid lies in close proximity to tumor tissue and is an appealing and accessible biosource for GBM liquid biopsy. CSF continuously circulating in the ventricles and cisterns is called “cisternal” CSF, whereas we term “lumbar” the CSF located in the spinal cord. Cisternal and lumbar CSF have different chemical compositions [82, 83], suggesting limited exchange between these two anatomic compartments. Whether these differences affect their diagnostic value for glioblastoma remains an open question.
The collection of CSF, however, involves a moderately invasive procedure; thus, identifying a biomarker in the serum would be preferable.
CSF has a higher amount of nucleic acid than CTCs [22, 34]. Although detection of ctDNA in blood remains challenging, CSF from surgical procedures or lumbar puncture seems to be the best source of ctDNA in patients with GBM and would allow monitoring of every step of glioma management, from diagnosis to tumor response [84, 85].
Bettegowda et al. confirmed that CSF is better than serum as a biosource for detecting ctDNAs derived from primary brain tumors [49].
Pan et al. analyzed ctDNA from seven patients with solid brain tumors and detected tumor‐related mutations in CSF from six of the seven patients. Interestingly, concentration of ctDNA was higher in the CSF than in plasma when the systemic disease burden was low [84].
In 2015, Wang et al. [85] investigated the presence of ctDNA in the CSF of 35 patients with different brain tumors (low‐grade and high‐grade gliomas, medulloblastomas, and ependymomas). Most of the CSF samples were collected directly from the CNS cavities at the time of the initial surgery, and using PCR and whole‐exome sequencing, Wang et al. found that 74% of 35 CSF samples contained detectable levels of tumor DNA. Notably, they observed significant association between the location and type of the tumor and the presence of ctDNA. They detected ctDNA in all World Health Organization grade III or IV gliomas and all medulloblastomas and ependymomas (100% of 21 cases) from samples from lesions adjacent to a CSF reservoir in the brain or spinal cord, whereas no ctDNA was present in the CSF of tumors embedded into deep brain tissue (p < .0001). These findings suggest that ctDNA, especially from a CSF reservoir and from high‐grade gliomas, represents a valuable source for liquid biopsy [85].
Although several studies have confirmed that tumor DNA can be detected in CSF from patients with CNS cancers, most often low numbers of patients with different types of brain tumors were included and the CSF samples used were collected mainly during surgery [86, 87].
The study of Miller and colleagues distinguishes itself from prior studies by its exclusive focus on diffuse glioma, the larger number of patients examined, the detailed clinical and radiographic annotation of each patient, the comprehensive nature of the NGS test, and the method of CSF collection (spinal tap) [86]. Miller et al. collected CSF samples from 85 patients affected by diffuse glioma and searched for mutations, copy number alterations, and chromosomal aberrations using the FDA‐authorized MSK‐IMPACT assay. Tumor DNA was detected in the CSF of about 50% of patients and was associated with disease burden and adverse outcome. Miller et al. observed high concordance of the genomic profile between CSF and matched tumor samples, the former harboring a broad spectrum of genetic alterations that commonly occur during gliomagenesis, including 1p/19q co‐deletions and IDH1, TP53, ATRX, and/or TERT promoter mutations [86].
Another potential avenue for CSF biomarkers lies in microRNA (miRNA). In 2017, Akers et al. [87] identified a unique cerebrospinal fluid miRNA signature in patients with GBM. Akers et al. performed miRNA profiling with a TaqMan OpenArray Human MicroRNA Panel. Comparison of CSF miRNA profiles between patients with GBM and those without brain tumors yielded a tumor “signature” consisting of nine miRNAs, correlated with tumor volume. Akers et al. demonstrated acceptable accuracy in the sensitivity and specificity of the signature, respectively, 28% and 95%, for lumbar CSF.
Teplyuk et al. [88] reported that microRNA (miR)‐10b and miR‐21 levels in the CSF are significantly increased in patients with GBM, whereas miR‐200 levels are elevated in brain metastases, enabling clinicians to differentiate CNS metastasis from primary malignancies with an accuracy of 91%–99% [88]. Similarly, Drusco et al. demonstrated that elevated levels of miR‐223, miR‐451, and miR‐711 in the CSF can be used for differential diagnosis between GBM and other types of brain tumors [89].
Circulating miRNAs
In addition to CTCs and circulating nucleic acid, microRNAs have been recognized as a promising diagnostic, prognostic, and therapeutic opportunity, although transferability of miRNA signature into clinical practice is still far away and limited by the uncertainties of the current findings.
MicroRNAs belong to the family of short noncoding RNAs that are about 22 nucleotides in size, playing an important regulatory role in transcription processes and intercellular communication, powerfully controlling the expression of approximately 65% of the human protein‐coding genes.
miRNAs regulate a very wide spectrum of cellular functions, including neuron differentiation and maturation, metabolism, tumorigenesis, invasiveness, angiogenesis, resistance to treatment, modulation of the immune system, and apoptosis, through the capacity of controlling gene expression at the post‐transcriptional level. Furthermore, miRNA can also function as a tumor suppressor gene, oncogene, or both, depending on the cell context.
To prevent degradation in the circulation, miRNAs are released by cells in both exosomes (lipid vesicles) and miRNA/protein complexes, such as Ago‐2, or high‐density lipoproteins [90], resulting in forms remarkably stable in plasma and serum, also resistant to various storage conditions such as extreme pH and multiple freeze‐thaw cycles.
Using sophisticated software (e.g., TargetScan) it is possible to correlate miRNAs with potential target genes [91].
Several recent studies have examined miRNA signatures in tissue, CSF, and serum samples of patients with glioblastoma [92].
A variety of circulating miRNAs have a critical role in GBM pathogenesis, becoming ideal candidates for noninvasive diagnosis and monitoring of GBM. The role of circulating miRNAs in GBM is reported in Table 2.
Table 2.
Role of circulating microRNAs in glioblastoma
| Diagnostic value | Prognostic value | Drug resistance | Radioresistance |
|---|---|---|---|
| miR‐21, miR‐128, and miR‐342‐3p [98] | miR‐21 [105] | miR‐21, miR‐15b, miR‐181, miR‐30b, and miR‐93 [112, 116] | miR‐128 and miR‐301 [119, 120] |
| miR‐15b, miR‐23a, miR‐133a, miR‐150, miR‐197, miR‐497, and miR‐548b‐5p [102] | miR‐182 [106] | miR‐223 [117] | |
| miR‐128 and miR‐342‐3p [99] | miR‐145‐5p [106] | miR‐125b‐2 [118] | |
| miR‐185 [103] | miR‐301 and miR‐205 [107] | miR‐221/miR‐222 [113] | |
| miR‐210 [104] | miR‐485‐3p [109] | miR‐1238 [114] | |
| miR‐21 [45] | miR‐20a‐5p, miR‐106a‐5p, miR‐222‐3p, miR‐182, and miR‐145‐5p [109] | ||
| miR‐17‐5p, miR‐20a, and miR‐106a [110] |
Abbreviation: miR, microRNA.
It emerged that, among different miRNAs, miR‐21 plays a critical role in GBM pathogenesis and progression and is actually the only circulating miRNA on which abundant and congruent data have been accumulated, making it the ideal candidate diagnostic and therapeutic biomarker for patients with GBM [93]. miR‐21, located on chromosome 17, is one of the most frequently overexpressed miRNAs in many types of human cancers (colorectal, lung, breast), with a predominant role in GBM, where it acts as an oncogene, inhibiting the expression of proapoptotic genes: tropomyosin, programmed cell death protein 4 (PDCD4), and PTEN. It regulates the main mechanisms of tumorigenesis: cell proliferation, migration, and cell invasion. miR‐21 exerts its effects by modulating several genes involved in neoangiogenesis, such as VEGF, PTEN, and hypoxia‐inducible factor‐1α [94], and by modulating apoptosis pathways [95].
Yang et al. indicated that insulin‐like growth factor–binding protein‐3 (IGFBP3), which acts as an oncosuppressor, inhibiting cell proliferation of glioma xenografts in vivo, is a target gene of miR‐21, which possesses the ability to downregulate IGFBP3, inducing tumor growth in patients with GBM [96].
In a meta‐analysis study, miR‐21 emerged as a reliable diagnostic biomarker in blood and as a diagnostic and prognostic biomarker if detected in CSF in patients with glioma [97].
Currently, miR‐21 is the only miRNA whose dysregulation has a clear and defined diagnostic, prognostic, and predictive value.
To understand the diagnostic value of circulating miRNAs, consider that specific miRNA signatures could differentiate between the blood samples of patients with GBM and healthy controls.
Herein we report the main miRNAs whose dysregulation has proved to be useful in discriminating between patients affected by GBM and healthy controls:
miR‐21, having a pivotal role in GBM, has been detected upregulated in plasma of patients with glioblastoma, and its levels decrease after tumor resection [98]. Qu et al. conducted a meta‐analysis to evaluate the accuracy of miRNAs in the diagnosis of glioma, analyzing 28 studies from 11 articles. They confirmed the pivotal role of miR‐21 as a single miRNA exhibiting high sensitivity and specificity. They showed that extracellular miR‐21 exhibited an outstanding diagnostic accuracy in detecting glioma. Moreover, at subgroup analysis, they demonstrated that panels of multiple miRNAs could largely improve the diagnostic accuracy [97].
miR‐128 and miR‐342‐3p have been found significantly downregulated, with an accuracy of 81%, sensitivity of 79%, and specificity of 81% in an interesting study conducted by Roth et al., performed using machine learning analysis of 1,158 circulating miRNAs of 20 patients with GBM [99].
A peculiar miRNA signature with 115 miRNAs significantly upregulated and 24 miRNAs downregulated has been identified in the serum samples of patients with GBM. This observation suggests the possibility of considering not a single miRNA but entire panels of miRNAs to improve the accuracy of GBM diagnosis [100]. Other putative miRNA signatures in patients with GBM have been identified in tumoral brain tissue [101].
miR‐15b, miR‐23a, miR‐133a, miR‐150, miR‐197, miR‐497, and miR‐548b‐5p downregulation predicts malignant astrocytoma with high sensitivity (88.00%) and specificity (97.87%). The levels of these miRNAs dramatically fall after tumor resection, demonstrating that circulating miRNAs reflect brain tumor burden [102].
miR‐185 and miR‐210 are significantly upregulated in patients with glioma compared with normal controls. Interestingly, circulating levels of miR‐185 levels dramatically decrease after surgery and chemoradiation [103, 104].
Despite the blood‐brain barrier and its restrictive nature, these studies confirm the potential use of circulating miRNAs in GBM diagnosis.
Nevertheless, the different results of these studies, the large amount of miRNAs analyzed, and the heterogeneity of the technologies make the application of circulating mirRNA for the diagnosis of glioblastoma still very far from clinical practice.
Dysregulated miRNAs in the serum of patients with GBM may also correlate with GBM survival, having a potential prognostic value, particularly miR‐21.
Upregulation of miR‐21 in patients with GBM may affect survival; in particular, miR‐21 achieves its protumorigenic action by downregulating IGFBP3, a GBM tumor suppressor [96].
Higher levels of expression of miR‐182 in the plasma are associated with poor prognosis and shorter overall survival [105].
miR‐145‐5p, miR‐301, and miR‐205 are strongly associated with Karnofsky performance status [106, 107].
Other circulating miRNas, associated with GBM prognosis, are reported in Table 2 [108, 109, 110]. Circulating miRNAs could also play a role in monitoring treatment response; their dysregulation in serum may reflect the antitumor effect of therapy and can predict response to anticancer drugs or radiotherapy [111]. The main players involved in resistance mechanisms and therefore markers of response to treatment are mir‐21, mir‐221, mir‐222, and mir‐1238.
Association between dysregulation of miR‐21 and GBM response to chemotherapy and radiotherapy was first described by Tumilson et al. [112].
miR‐221 and miR‐222 play a key role in both temozolomide resistance and radiation therapy resistance mechanisms. In particular, Li et al. found that miR‐221 and miR‐222 induce radioresistance in glioblastoma cells, modulating radioinduced DNA damage repair mechanisms through activation of the AKT kinase pathway. Activated AKT mediates several of the pro‐survival PI3K responses: cell growth, metabolism activation, and proliferation [113].
miR‐1238 levels are higher in TMZ‐resistant GBM cells than in sensitive cells. The loss of miR‐1238 may sensitize resistant GBM cells by directly targeting the CAV1/EGFR pathway. These results, obtained by Yin et al. measuring the expression levels of miR‐1238 in GBM cell lines and their exosomes, clinical tissues, and serum by quantitative reverse‐transcription PCR, suggest that circulating miR‐1238 could serve as a clinical biomarker and a promising therapeutic target for TMZ resistance in GBM [114].
Higher blood levels of miR‐10b and miR‐21 have been described in patients with GBM undergoing treatment with bevacizumab. In this study it was also observed that lower levels of expressions of miR‐10b and miR‐21 are associated with higher tumor volume in patients treated with bevacizumab. This correlation has not been found in patients treated with temozolomide [115].
Other relevant miRNAs affecting sensitivity to temozolomide are reported in Table 2 [116, 117, 118, 119, 120].
Extracellular Vesicles
EVs are small particles formed with a lipid bilayer, typically released into the extracellular space, and classified into two categories: exosomes, which are generated intracellularly and fuse with the plasma membrane upon release and which range from 30 to 100 nm in size, and microvesicles, which are produced directly from the extracellular membrane via budding and range from 100 to 1000 nm in size. EVs carry components from the cell membrane and cytoplasm, including DNA, RNA, and proteins, which can be transferred cell by cell through the cell trafficking or gap junctions, raising the possibility of malignant transformation by horizontal transmission. EVs represent a better source of nucleic acids for tumor molecular profiling as compared with cell‐free nucleic acids. EVs can be isolated from body fluids with different methods, including differential centrifugation gradients and immunoaffinity capture [121].
Accumulating evidence indicates that exosomes play a pivotal role in cell‐to‐cell communication. Because contents carried by EVs are protected from the surrounding environment, both exosomes and microvesicles reflect the molecular identity of their cell of origin [122].
Considering that neoplastic cells secrete more EVs than healthy cells, circulating EVs may be a valuable source of information regarding the heterogenetic biological landscape of GBM, the state of the tumor, and the disease progression [123, 124].
EVs released by tumor cells may be taken up by neighboring stromal cells, leading to alteration of cell program, or by cells of the immune system, causing immunosuppression [125, 126, 127, 128].
Among potential biomarkers contained within EVs, miRNAs are some of the most promising.
Several studies report that exosomal miRNAs can be used in GBM as predictive biomarkers for diagnosis, monitoring, and predicting treatment response in patients with GBM, the most important possibilities summarized below:
With regard to exosomes in GBM, the most promising data are those relative to EGFRvIII expression in exosomes. Analysis of serum EV‐derived RNA has demonstrated unique signatures, such as detection of EGFRvIII in patients with glioma. Figueroa et al. have published an interesting multicenter study including 71 patients with GBM, analyzing extracellular vesicles derived from CSF for EGFRvIII status (a frequent activating mutation in EGFR in gliomas) and EGFR amplification: they demonstrated that the detection of the EGFRvIII in extracellular vesicular RNA can be achieved with a 60% sensitivity and greater than 98% specificity as compared with the gold standard of PCR from brain tumor tissue [129].
Exosomal miRNA can regulate response to temozolomide. There is evidence that exosomal transfer of miRNAs derived from temozolomide‐resistant GBM cells could confer resistance to temozolomide [130, 131, 132].
EVs from CSF of patients with GBM present significantly increased levels of exosomal miR‐21 versus healthy controls, with a sensitivity of 87% and specificity of 93% [133].
Santangelo et al. reported the accuracy of circulating exosomal miR‐21, miR‐222, and miR‐124‐3p for GBM diagnosis. Notably, the levels of these miRNAs dramatically decrease after tumor resection [134].
miR‐320 and miR‐574‐3p have been found to be increased in exosomes isolated from serum of 75 patients with diagnosis of GBM [135].
Tzaridis et al. have studied a panel of microRNAs in EVs from the serum of glioblastoma, evaluating the correlation with the prognosis. They have identified four microRNAs (miR‐15b‐3p, miR‐106a‐5p, miR‐21‐3p, and miR‐328‐3p) in patients with GBM whose levels, in combination, can predict the prognosis for these patients [136].
miRNA Targeted Therapies in Gbm
Galunisertib is a small molecule, an inhibitor of the transforming growth factor‐β (TGF‐β) type I receptor kinase, that can reduce the secretion of miR‐21 from glioma cells. TGF‐β is a major driver of glioma progression that participates in mediating the release of miR‐21 from glioma cells. Targeting TGF‐β signaling using galunisertib can reduce the extracellular levels of miR‐21. Qu et al. [97] showed that miR‐21 exerts its effects via the TGF‐β/Smad3 signaling pathway and that the use of galunisertib, which is an inhibitor of the TGF‐β type I receptor, could decrease expression of miR‐21 in GBM cells.
Nevertheless, the phase Ib/IIa trial published by Wick et al. [137] investigating the clinical benefit of combining galunisertib with temozolomide‐based radiochemotherapy showed that the combination of galunisertib with standard radiochemotherapy did not improve OS and PFS. The authors only observed a higher disease control rate in the galunisertib plus radiochemotherapy arm when compared with radiochemotherapy treatment alone. Also, the phase II study published by Brandes et al. [138], investigating the antitumor activity of galunisertib plus lomustine, failed to demonstrate improved OS of the experimental arm compared with placebo plus lomustine.
Interestingly, Belter and colleagues indicated that the inhibition of miR‐21 employing anti‐miRNA catalytic nucleic could decrease expression of miR‐21 and lead to silencing of miR‐21 functions. These findings indicated that anti‐miRNA catalytic nucleic acids are a new promising therapeutic option for GBM [139].
Tan et al. have reported the antitumor effect of the combination of curcumin and miR21ASO, an antisense oligonucleotide against miR‐21, in GBM cells. Curcumin is a phytochemical product endowed with anti‐inflammatory, antibacterial, and anticancer properties and is an efficient carrier of miR21ASO. The combined delivery of miR21ASO and curcumin was found to be able to reduce miR‐21 levels and thus may be a useful therapy for GBM [140].
RNA nanotechnology is a rapidly evolving field for a gene‐based targeted treatment of human cancers, including GBM. It is still at a stage of embryonic development for GBM treatment, but the first results are encouraging [141].
A number of nanoparticles mainly based on lipids, polymers, and inorganic and organic materials are being developed for targeting glioma.
For effective glioma therapy and to avoid toxicity and side effects, RNA nanoparticles must reach and target intracranial tumors with minimal accumulation in adjacent healthy brain tissues or in major healthy internal organs.
Construction of RNA nanoparticles requires several steps and involves RNA packaging (pRNA). The pRNA of the bacteriophage phi29 DNA packaging motor is a molecule with great plasticity that allows the construction of various RNA nanoparticles, incorporating into the pRNA scaffold therapeutic modules including small interfering RNA, miRNA, or ribozymes, with precise control of shape, size, and stoichiometry [141].
Given their great versatility and stability, the RNA nanoparticles fabricated using the ultrastable three‐way junction (3WJ)–based RNA nanoparticles (RNP) motif as a scaffold, artificially derived from pRNA of bacteriophage phi29 DNA packaging motor, have emerged as a novel vector system for targeted gene therapy in glioblastoma.
Lee et al. reported that multivalent folate–conjugated 3WJ RNPs constructed to harbor anti–miR‐21 sequences specifically targeted miR‐21 and enabled effectively knocked down of miR‐21 expression in glioblastoma cells, in vitro and in vivo, by rescuing tumor suppressors PTEN and PDCD4. This resulted in glioblastoma cell apoptosis, tumor growth regression, and increased overall survival rate [142].
Conclusion
Despite serious efforts to improve diagnostics and therapeutic strategies [88, 89], GBM remains a lethal disease, extremely difficult to treat.
Finding new therapeutic approaches is necessary: thus, identification of new diagnostic and therapeutic circulating biomarkers could contribute to the development of new personalized treatments.
A significant number of potential tumor‐specific liquid biomarkers for GBM have been identified; however, until now they have not found application in clinical practice, and, to date, no validated circulating biomarkers exist for managing patients with GBM .
Whether liquid biopsy will overtake tissue biopsies remains uncertain today but seems possible.
The main difficulty with all studies on liquid biopsy so far conducted in gliomas is the lack of standardized technologies, leading to increasingly different results and to the impossibility of biomarker validation.
The development of more sensitive techniques, such as improved targeted deep NGS, droplet digital PCR [106], DNA methylation profiling [110], and specific PCR, has improved the utility and applicability of liquid biopsies. Nevertheless, we must admit that we have not yet sufficient evidence to transfer liquid biopsy into clinical practice.
Further prospective studies for large‐scale validation of the available liquid biopsy biosources and biomolecules, standardization of detection approaches, and more cost‐effective methods are needed in the near future. New developments, to improve the performance of liquid biopsy in GBM, should include the use of different combined approaches, integrating the analysis of multiple biomarkers, to significantly increase sensitivity and specificity.
Author Contributions
Conception/design: Lidia Gatto, Enrico Franceschi
Collection and/or assembly of data: Lidia Gatto, Enrico Franceschi, Vincenzo Di Nunno, Alicia Tosoni, Raffaele Lodi, Alba Ariela Brandes
Data analysis and interpretation: Lidia Gatto, Enrico Franceschi
Manuscript writing: Lidia Gatto, Enrico Franceschi
Final approval of manuscript: Lidia Gatto, Enrico Franceschi, Vincenzo Di Nunno, Alicia Tosoni, Raffaele Lodi, Alba Ariela Brandes
Disclosures
The authors indicated no financial relationships.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1.Wick W, Platten M. Understanding and treating glioblastoma. Neurol Clin 2018;36:485–499. [DOI] [PubMed] [Google Scholar]
- 2.Soeda A, Hara A, Kunisada T et al. The evidence of glioblastoma heterogeneity. Sci Rep 2015;5:7979. Erratum in: Sci Rep 2015;5:9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vuorinen V, Hinkka S, Färkkilä M et al. Debulking or biopsy of malignant glioma in elderly people ‐ a randomised study. Acta Neurochir (Wien) 2003;145:5– 10. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–996. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Taillibert S, Kanner A et al. Effect of tumor‐treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017;318:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013;310:1842–1850. [DOI] [PubMed] [Google Scholar]
- 7.Brandes AA, Brandes AA, Bartolotti M et al. Nitrosoureas in the management of malignant gliomas. Curr Neurol Neurosci Rep 2016;16:13. [DOI] [PubMed] [Google Scholar]
- 8.Friedman HS, Prados MD, Wen PY et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009;27:4733–4740. [DOI] [PubMed] [Google Scholar]
- 9.Lombardi G, De Salvo GL, Brandes AA et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open‐label, randomised, controlled, phase 2 trial. Lancet Oncol 2019;20:110–119. [DOI] [PubMed] [Google Scholar]
- 10.Franceschi E, Minichillo S, Brandes AA. Pharmacotherapy of glioblastoma: Established treatments and emerging concepts. CNS Drugs 2017;31:675–684. [DOI] [PubMed] [Google Scholar]
- 11.Franceschi E, Stupp R, van den Bent MJ et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol 2012;14:1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couturier CP, Ayyadhury S, Le PU et al. Single‐cell RNA‐seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun 2020;11:3406. Erratum in: Nat Commun 2020;11:4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zachariah MA, Oliveira‐Costa JP, Carter BS et al. Blood‐based biomarkers for the diagnosis and monitoring of gliomas. Neuro Oncol 2018;20:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres S, González Á, Cunquero Tomas AJ et al. A profile on cobas EGFR Mutation Test v2 as companion diagnostic for first‐line treatment of patients with non‐small cell lung cancer. Expert Rev Mol Diagn 2020;20:575–582. [DOI] [PubMed] [Google Scholar]
- 15.Weller M, Stupp R, Reifenberger G et al. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat Rev Neurol 2010;6:39–51. [DOI] [PubMed] [Google Scholar]
- 16.Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell 2012;150:12–27. [DOI] [PubMed] [Google Scholar]
- 17.Johnson C, Warmoes MO, Shen X et al. Epigenetics and cancer metabolism. Cancer Lett 2015;356:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong Z, Cui H. Epigenetic modulation of metabolism in glioblastoma. Semin Cancer Biol 2019;57:45–51. [DOI] [PubMed] [Google Scholar]
- 19.Louis DN, Perry A, Reifenberger G et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol 2016;131:803–820. [DOI] [PubMed] [Google Scholar]
- 20.Mansouri A, Hachem LD, Mansouri S et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro Oncol 2019;21:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell 2017;31:172–179. [DOI] [PubMed] [Google Scholar]
- 22.Siravegna G, Marsoni S, Siena S and Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 2017;14:531–548. [DOI] [PubMed] [Google Scholar]
- 23.Alix‐Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu Rev Med 2012;63:199–215. [DOI] [PubMed] [Google Scholar]
- 24.Best MG, Sol N, Zijl S et al. Liquid biopsies in patients with diffuse glioma. Acta Neuropathol 2015;129:849–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge F, Zhang H, Wang DD et al. Enhanced detection and comprehensive in situ phenotypic characterization of circulating and disseminated heteroploid epithelial and glioma tumor cells. Oncotarget 2015;6:27049–27064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan JP, Nahed BV, Madden MW et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov 2014;4:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Best MG, Sol N, Kooi I et al. RNA‐seq of tumor‐educated platelets enables blood‐based pan‐cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 2015;28:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Best MG, Wesseling P, Wurdinger T. Tumor‐educated platelets as a noninvasive biomarker source for cancer detection and progression monitoring. Cancer Res 2018;78:3407–3412. [DOI] [PubMed] [Google Scholar]
- 29.Haber DA, Velculescu VE. Blood‐based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov 2014;4:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poudineh M, Sargent EH, Pantel K et al. Profiling circulating tumour cells and other biomarkers of invasive cancers. Nat Biomed Eng 2018;2:72–84. [DOI] [PubMed] [Google Scholar]
- 31.Talasaz AH, Powell AA, Huber DE et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA 2009;106:3970–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laget S, Broncy L, Hormigos K et al. Technical insights into highly sensitive isolation and molecular characterization of fixed and live circulating tumor cells for early detection of tumor invasion. PLoS One 2017;12:e0169427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fontanilles M, Marguet F, Bohers É et al. Non‐invasive detection of somatic mutations using next‐generation sequencing in primary central nervous system lymphoma. Oncotarget 2017;8:48157–48168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fontanilles M, Duran‐Peña A, Idbaih A. Liquid biopsy in primary brain tumors: Looking for stardust! Curr Neurol Neurosci Rep 2018;18:13. [DOI] [PubMed] [Google Scholar]
- 35.Saenz‐Antoñanzas A, Auzmendi‐Iriarte J, Carrasco‐Garcia E et al. Liquid biopsy in glioblastoma: Opportunities, applications and challenges. Cancers (Basel) 2019;11:950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macarthur KM, Kao GD, Chandrasekaran S et al. Detection of brain tumor cells in the peripheral blood by a telomerase promoter‐based assay. Cancer Res 2014;74:2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller C, Holtschmidt J, Auer M et al. Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med 2014;6:247ra101. [DOI] [PubMed] [Google Scholar]
- 38.Krol I, Castro‐Giner F, Maurer M et al. Detection of circulating tumour cell clusters in human glioblastoma. Br J Cancer 2018;119:487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao F, Cui Y, Jiang H et al. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget 2016;7:71330–71340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diehl F, Schmidt K, Choti MA et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leon SA, Shapiro B, Sklaroff DM et al. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977;37:646–650. [PubMed] [Google Scholar]
- 42.Diaz LA Jr, Williams RT, Wu J et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aggarwal C, Thompson JC, Black TA et al. Clinical implications of plasma‐based genotyping with the delivery of personalized therapy in metastatic non‐small cell lung cancer. JAMA Oncol 2019;5:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sacher AG, Paweletz C, Dahlberg SE et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol 2016;2:1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin KK, Harrell MI, Oza AM et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high‐grade ovarian carcinoma. Cancer Discov 2019;9:210–219. [DOI] [PubMed] [Google Scholar]
- 46.Bagley SJ, Nabavizadeh SA, Mays JJ et al. Clinical utility of plasma cell‐free DNA in adult patients with newly diagnosed glioblastoma: A pilot prospective study. Clin Cancer Res 2020;26:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fontanilles M, Marguet F, Beaussire L et al. Cell‐free DNA and circulating TERT promoter mutation for disease monitoring in newly‐diagnosed glioblastoma. Acta Neuropathol Commun 2020;8:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boisselier B, Gállego Pérez‐Larraya J, Rossetto M et al. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology 2012;79:1693–1698. [DOI] [PubMed] [Google Scholar]
- 49.Bettegowda C, Sausen M, Leary RJ et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014;6:224ra24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diaz LA Jr, Bardelli A. Liquid biopsies: Genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Müllauer L.Next generation sequencing: Clinical applications in solid tumours. Memo 2017;10:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malapelle U, Pisapia P, Rocco D et al. Next generation sequencing techniques in liquid biopsy: Focus on non‐small cell lung cancer patients. Transl Lung Cancer Res 2016;5:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olmedillas‐López S, García‐Arranz M, García‐Olmo D. Current and emerging applications of droplet digital PCR in oncology. Mol Diagn Ther 2017;21:493–510. [DOI] [PubMed] [Google Scholar]
- 54.Pentsova EI, Shah RH, Tang J et al. Evaluating cancer of the central nervous system through next‐generation sequencing of cerebrospinal fluid. J Clin Oncol 2016;34:2404–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panditharatna E, Kilburn LB, Aboian MS et al. Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient‐derived liquid biopsy. Clin Cancer Res 2018;24:5850–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang W, Song Z, Zhang Y. A Comparison of ddPCR and ARMS for detecting EGFR T790M status in ctDNA from advanced NSCLC patients with acquired EGFR‐TKI resistance. Cancer Med 2017;6:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hattori K, Hattori K, Sakata‐Yanagimoto M et al. Clinical significance of disease‐specific MYD88 mutations in circulating DNA in primary central nervous system lymphoma. Cancer Sci 2018;109:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piccioni DE, Achrol AS, Kiedrowski LA et al. Analysis of cell‐free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol 2019;8:CNS34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zill OA, Banks KC, Fairclough SR et al. The landscape of actionable genomic alterations in cell‐free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res 2018;24:3528–3538. [DOI] [PubMed] [Google Scholar]
- 60.Lavon I, Refael M, Zelikovitch B et al. Serum DNA can define tumor‐specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol 2010;12:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majchrzak‐Celińska A, Paluszczak J, Kleszcz R et al. Detection of MGMT, RASSF1A, p15INK4B, and p14ARF promoter methylation in circulating tumor‐derived DNA of central nervous system cancer patients. J Appl Genet 2013;54:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sareen H, Garrett C, Lynch D et al. The role of liquid biopsies in detecting molecular tumor biomarkers in brain cancer patients. Cancers (Basel) 2020;12:1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nassiri F, Chakravarthy A, Feng S et al. Detection and discrimination of intracranial tumors using plasma cell‐free DNA methylomes. Nat Med 2020;26:1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Underhill HR, Kitzman JO, Hellwig S et al. Fragment length of circulating tumor DNA. PLoS Genet 2016;12:e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mouliere F, Chandrananda D, Piskorz AM et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10:eaat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nilsson RJA, Balaj L, Hulleman E et al. Blood platelets contain tumor‐derived RNA biomarkers. Blood 2011;118:3680–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janowska‐Wieczorek A, Wysoczynski M, Kijowski J et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 2005;113:752–760. [DOI] [PubMed] [Google Scholar]
- 69.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial‐mesenchymal‐like transition and promotes metastasis. Cancer Cell 2011;20:576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alhasan AA, Izuogu OG, Al‐Balool HH et al. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood 2016;127:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Angénieux C, Maître B, Eckly A et al. Time‐dependent decay of mRNA and ribosomal RNA during platelet aging and its correlation with translation activity. PLoS One 2016;11:e0148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rondina MT, Schwertz H, Harris ES et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost 2011;9:748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagalla S, Shaw C, Kong X et al. Platelet microRNA‐mRNA coexpression profiles correlate with platelet reactivity. Blood 2011;117:5189–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rondina MT, Weyrich AS. Regulation of the genetic code in megakaryocytes and platelets. J Thromb Haemost 2015;13(suppl 1):S26–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sol N, In't Veld SGJG, Vancura A et al. Tumor‐educated platelet RNA for the detection and (pseudo)progression monitoring of glioblastoma. Cell Rep Med 2020;1:100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.In't Veld SGJG, Wurdinger T. Tumor‐educated platelets. Blood 2019;133:2359–2364. [DOI] [PubMed] [Google Scholar]
- 77.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dang L, White DW, Gross S et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 2009;462:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Janin M, Mylonas E, Saada V et al. Serum 2‐hydroxyglutarate production in IDH1‐ and IDH2‐mutated de novo acute myeloid leukemia: A study by the Acute Leukemia French Association group. J Clin Oncol 2014;32:297–305. [DOI] [PubMed] [Google Scholar]
- 80.Lombardi G, Corona G, Bellu L et al. Diagnostic value of plasma and urinary 2‐hydroxyglutarate to identify patients with isocitrate dehydrogenase‐mutated glioma. The Oncologist 2015;20:562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Picca A, Di Stefano AL, Sanson M. Current and future tools for determination and monitoring of isocitrate dehydrogenase status in gliomas. Curr Opin Neurol 2018;31:727–732. [DOI] [PubMed] [Google Scholar]
- 82.Rubalcava MA, Sotelo J. Differences between ventricular and lumbar cerebrospinal fluid in hydrocephalus secondary to cysticercosis. Neurosurgery 1995;37:668–671. [DOI] [PubMed] [Google Scholar]
- 83.Weisner B, Bernhardt W. Protein fractions of lumbar, cisternal, and ventricular cerebrospinal fluid. Separate areas of reference. J Neurol Sci 1978;37:205–214. [DOI] [PubMed] [Google Scholar]
- 84.Pan W, Gu W, Nagpal S et al. Brain tumor mutations detected in cerebral spinal fluid. Clin Chem 2015;61:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y, Springer S, Zhang M et al. Detection of tumor‐derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA 2015;112:9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller AM, Shah RH, Pentsova EI et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019;565:654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akers JC, Hua W, Li H et al. A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget 2017;8:68769–68779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teplyuk NM, Mollenhauer B, Gabriely G et al. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro Oncol 2012;14:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Drusco A, Bottoni A, Laganà A et al. A differentially expressed set of microRNAs in cerebrospinal fluid (CSF) can diagnose CNS malignancies. Oncotarget 2015;6:20829–20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang CS, Rana TM. Learning the molecular mechanisms of the reprogramming factors: Let's start from microRNAs. Mol Biosyst 2013;9:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peterson SM, Thompson JA, Ufkin ML et al. Common features of microRNA target prediction tools. Front Genet 2014;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hua D, Mo F, Ding D et al. A catalogue of glioblastoma and brain microRNAs identified by deep sequencing. OMICS 2012;16:690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saadatpour L, Fadaee E, Fadaei S et al. Glioblastoma: Exosome and microRNA as novel diagnosis biomarkers. Cancer Gene Ther 2016;23:415–418. [DOI] [PubMed] [Google Scholar]
- 94.Hermansen SK, Nielsen BS, Aaberg‐Jessen C et al. miR‐21 is linked to glioma angiogenesis: A co‐localization study. J Histochem Cytochem 2016;64:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chan JA, Krichevsky AM, Kosik KS. MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005;65:6029–6033. [DOI] [PubMed] [Google Scholar]
- 96.Yang CH, Yue J, Pfeffer SR et al. MicroRNA‐21 promotes glioblastoma tumorigenesis by down‐regulating insulin‐like growth factor‐binding protein‐3 (IGFBP3). J Biol Chem 2014;289:25079–25087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qu S, Guan J, Liu Y. Identification of microRNAs as novel biomarkers for glioma detection: A meta‐analysis based on 11 articles. J Neurol Sci 2015;348:181–187. [DOI] [PubMed] [Google Scholar]
- 98.Kalkan R, Atli Eİ. The impacts of miRNAs in glioblastoma progression. Crit Rev Eukaryot Gene Expr 2016;26:137–142. [DOI] [PubMed] [Google Scholar]
- 99.Roth P, Wischhusen J, Happold C et al. A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem 2011;118:449–457. [DOI] [PubMed] [Google Scholar]
- 100.Dong L, Li Y, Han C et al. miRNA microarray reveals specific expression in the peripheral blood of glioblastoma patients. Int J Oncol 2014;45:746–756. [DOI] [PubMed] [Google Scholar]
- 101.Visani M, de Biase D, Marucci G et al. Expression of 19 microRNAs in glioblastoma and comparison with other brain neoplasia of grades I‐III. Mol Oncol 2014;8:417–430. Erratum in: Mol Oncol 2015;9:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang Q, Wang R, Wei B et al. Gene and microRNA signatures are associated with the development and survival of glioblastoma patients. DNA Cell Biol 2019;38:688–699. [DOI] [PubMed] [Google Scholar]
- 103.Tang H, Liu Q, Liu X et al. Plasma miR‐185 as a predictive biomarker for prognosis of malignant glioma. J Cancer Res Ther 2015;11:630–634. [DOI] [PubMed] [Google Scholar]
- 104.Lai NS, Wu DG, Fang XG et al. Serum microRNA‐210 as a potential noninvasive biomarker for the diagnosis and prognosis of glioma. Br J Cancer 2015;112:1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xiao Y, Zhang L, Song Z et al. Potential diagnostic and prognostic value of plasma circulating microRNA‐182 in human glioma. Med Sci Monit 2016;22:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Y, Ta WW, Sun PF et al. Diagnostic and prognostic significance of serum miR‐145‐5p expression in glioblastoma. Int J Clin Exp Pathol 2019;12:2536–2543. [PMC free article] [PubMed] [Google Scholar]
- 107.Yue X, Lan F, Hu M et al. Downregulation of serum microRNA‐205 as a potential diagnostic and prognostic biomarker for human glioma. J Neurosurg 2016;124:122–128. [DOI] [PubMed] [Google Scholar]
- 108.Wang ZQ, Zhang MY, Deng ML et al. Low serum level of miR‐485‐3p predicts poor survival in patients with glioblastoma. PLoS One 2017;12:e0184969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao H, Shen J, Hodges TR et al. Serum microRNA profiling in patients with glioblastoma: A survival analysis. Mol Cancer 2017;16:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Srinivasan S, Patric IR, Somasundaram K. A ten‐microRNA expression signature predicts survival in glioblastoma. PLoS One 2011;6:e17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang SW, Ali ND, Zhong L et al. MicroRNAs as biomarkers for human glioblastoma: Progress and potential. Acta Pharmacol Sin 2018;39:1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tumilson CA, Lea RW, Alder JE et al. Circulating microRNA biomarkers for glioma and predicting response to therapy. Mol Neurobiol 2014;50:545–558. [DOI] [PubMed] [Google Scholar]
- 113.Li W, Guo F, Wang P et al. miR‐221/222 confers radioresistance in glioblastoma cells through activating Akt independent of PTEN status. Curr Mol Med 2014;14:185–195. [DOI] [PubMed] [Google Scholar]
- 114.Yin J, Zeng A, Zhang Z et al. Exosomal transfer of miR‐1238 contributes to temozolomide‐resistance in glioblastoma. EBioMedicine 2019;42:238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Siegal T, Charbit H, Paldor I et al. Dynamics of circulating hypoxia‐mediated miRNAs and tumor response in patients with high‐grade glioma treated with bevacizumab. J Neurosurg 2016;125:1008–1015. [DOI] [PubMed] [Google Scholar]
- 116.Gwak HS, Kim TH, Jo GH et al. Silencing of microRNA‐21 confers radio‐sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS One 2012;7:e47449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang BS, Luo QZ, Han Y et al. MiR‐223/PAX6 axis regulates glioblastoma stem cell proliferation and the chemo resistance to TMZ via regulating PI3K/Akt pathway. J Cell Biochem 2017;118:3452–3461. [DOI] [PubMed] [Google Scholar]
- 118.Shi L, Zhang S, Feng K et al. MicroRNA‐125b‐2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol 2012;40:119–129. [DOI] [PubMed] [Google Scholar]
- 119.Sun J, Ye L, Wang C et al. MicroRNA‐128 increases glioma cell radio‐sensitivity by suppressing senescent evasion through oncogene Bmi‐1. Int J Clin Exp Pathol 2018;11:1423–1430. [PMC free article] [PubMed] [Google Scholar]
- 120.Yue X, Lan F, Xia T. Hypoxic glioma cell‐secreted exosomal miR‐301a activates Wnt/β‐catenin signaling and promotes radiation resistance by targeting TCEAL7. Mol Ther 2019;27:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 2015;40:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li CC, Eaton SA, Young PE et al. Glioma microvesicles carry selectively packaged coding and non‐coding RNAs which alter gene expression in recipient cells. RNA Biol 2013;10:1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim HK, Song KS, Park YS et al. Elevated levels of circulating platelet microparticles, VEGF, IL‐6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur J Cancer 2003;39:184–191. [DOI] [PubMed] [Google Scholar]
- 124.Zwicker JI, Liebman HA, Neuberg D et al. Tumor‐derived tissue factor‐bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res 2009;15:6830–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hoshino A, Costa‐Silva B, Shen TL et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Costa‐Silva B, Aiello NM, Ocean AJ et al. Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat Cell Biol 2015;17:816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu Y, Gu Y, Han Y et al. Tumor exosomal RNAs promote lung pre‐metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 2016;30:243–256. [DOI] [PubMed] [Google Scholar]
- 128.Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013;91:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Figueroa JM, Skog J, Akers J et al. Detection of wild‐type EGFR amplification and EGFRvIII mutation in CSF‐derived extracellular vesicles of glioblastoma patients. Neuro Oncol 2017;19:1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shao H, Chung J, Lee K et al. Chip‐based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat Commun 2015;6:6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zeng A, Wei Z, Yan W et al. Exosomal transfer of miR‐151a enhances chemosensitivity to temozolomide in drug‐resistant glioblastoma. Cancer Lett 2018;436:10–21. [DOI] [PubMed] [Google Scholar]
- 132.Lucero R, Zappulli V, Sammarco A et al. Glioma‐derived miRNA‐containing extracellular vesicles induce angiogenesis by reprogramming brain endothelial cells. Cell Rep 2020;30:2065–2074.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Akers JC, Ramakrishnan V, Kim R et al. MiR‐21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): A platform for glioblastoma biomarker development. PLoS One 2013;8:e78115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Santangelo A, Imbrucè P, Gardenghi B et al. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J Neurooncol 2018;136:51–62. [DOI] [PubMed] [Google Scholar]
- 135.Manterola L, Guruceaga E, Gallego Perez‐Larraya J et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol 2014;16:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tzaridis T, Reiners KS, Weller J et al. Analysis of serum miRNA in glioblastoma patients: CD44‐based enrichment of extracellular vesicles enhances specificity for the prognostic signature. Int J Mol Sci 2020;21:7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wick A, Desjardins A, Suarez C et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor‐beta receptor I, in combination with standard temozolomide‐based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest New Drugs 2020;38:1570–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brandes AA, Carpentier AF, Kesari S et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol 2016;18:1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Belter A, Rolle K, Piwecka M et al. Inhibition of miR‐21 in glioma cells using catalytic nucleic acids. Sci Rep 2016;6:24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tan X, Kim G, Lee D et al. A curcumin‐loaded polymeric micelle as a carrier of a microRNA‐21 antisense‐oligonucleotide for enhanced anti‐tumor effects in a glioblastoma animal model. Biomater Sci 2018;6:407–417. [DOI] [PubMed] [Google Scholar]
- 141.Lee TJ, Haque F, Vieweger M et al. Functional assays for specific targeting and delivery of RNA nanoparticles to brain tumor. In: Guo P, Haque F, eds. RNA Nanotechnology and Therapeutics. New York: Humana Press, 2015:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee TJ, Haque F, Shu D et al. RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget 2015;6:14766–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]