

Abstract
Across a broad range of human cancers, gain-of-function mutations in RAS genes (HRAS, NRAS, and KRAS) lead to constitutive activity of oncoproteins responsible for tumorigenesis and cancer progression. The targeting of RAS with drugs is challenging because RAS lacks classic and tractable drug binding sites. Over the past 30 years, this perception has led to the pursuit of indirect routes for targeting RAS expression, processing, upstream regulators, or downstream effectors. After the discovery that the KRAS-G12C variant contains a druggable pocket below the switch-II loop region, it has become possible to design irreversible covalent inhibitors for the variant with improved potency, selectivity and bioavailability. Two such inhibitors, sotorasib (AMG 510) and adagrasib (MRTX849), were recently evaluated in phase I-III trials for the treatment of non-small cell lung cancer with KRAS-G12C mutations, heralding a new era of precision oncology. In this review, we outline the mutations and functions of KRAS in human tumors and then analyze indirect and direct approaches to shut down the oncogenic KRAS network. Specifically, we discuss the mechanistic principles, clinical features, and strategies for overcoming primary or secondary resistance to KRAS-G12C blockade.
Keywords: Gene mutation, Covalent inhibitor, Drug resistance, KRAS, Targeted therapy
Key points
KRAS is the most frequently mutated oncogene in human cancer and has challenged the development of clinical anticancer therapeutics in the last 30 years.
Mutated KRAS oncoprotein disrupts GAP-mediated GTP hydrolysis and thus remains in a continuous GTP binding activation state.
Small-molecule inhibitors that directly target KRAS-G12C mutants provide new tools for precision oncology.
Clinical trials involving covalent KRAS-G12C inhibitors (adagrasib and sotorasib) have shown promising activity against lung cancers harboring KRAS-G12C mutations.
Secondary KRAS mutations, gain-of-function mutations of the MAPK pathway, loss-of-function mutations in tumor suppressor genes, and other gene alterations are conducive to acquired resistance to KRAS-G12C inhibitors.
The design and implementation of strategies to minimize or overcome drug resistance is an important goal for the further development of KRAS inhibitors.
Introduction
The RAS gene was initially identified as a virus-encoded gene in 1964 [1], and later was found to be a genome-encoded oncogene that is frequently mutated in human cancers [2]. Thus, activating mutations of RAS are found in 19% of neoplasias, corresponding to approximately 3.4 million new diagnoses of malignant disease worldwide each year [2]. The RAS gene family includes HRAS, NRAS, and KRAS, which are encoding proteins that play partially overlapping but specific roles in signaling transduction [3]. For example, the knockout of Kras is embryonically lethal in mice, while the depletion of Nras or Hras does not affect development [4]. The mutation of human KRAS was first detected in non-small cell lung cancer (NSCLC) [5–7]. The earliest evidence that RAS is an oncogene was based on the fact that transfecting mouse Kras causes morphological transformation of NIH-3T3 fibroblasts [8]. Subsequent studies involving transgenic mice confirmed that the mutation of Hras, Nras, or Kras mimicked human oncogenesis by triggering the stochastic transformation of cells [9–12]. Clinical studies revealed the prognostic impact of RAS mutations in certain cancers [2]. However, in spite of decades-long efforts of academia and industry to target RAS protein for cancer therapy [13], the design of direct RAS pharmacological inhibitors has only achieved a major breakthrough during recent years [14]. In 2021, two covalent inhibitors of KRAS-G12C protein (hereafter referred to as G12Ci), sotorasib (AMG 510) [15] and adagrasib (MRTX849) [16], were clinically approved by the U.S. Food and Drug Administration and the European Medicines Agency to treat patients with advanced NSCLC carrying the KRAS-G12C mutation. Monotherapies or combination therapies using G12Ci are being evaluated in clinical trials for advanced or metastatic solid cancer, including NSCLC, colorectal cancer (CRC), and pancreatic ductal adenocarcinoma (PDAC) [17]. This progress is inspiring scientists to continue to design drugs targeting key oncogenic drivers, even those that previously were considered to be “undruggable” like KRAS [18].
Here, we summarize recent therapeutic advances in mutant KRAS blockade that have led to clinical approval or are currently being evaluated in trials (Table 1), while discussing attempts to target upstream regulators, downstream effectors, and mutant KRAS protein itself. We also discuss the challenges associated with G12Ci-based treatments and the future prospects of this evolving topic.
Table 1.
Clinical trials targeting KRAS
| Target | Agent | Combinations | Study phase |
Tumor type | Recruitment status | Trial number |
|---|---|---|---|---|---|---|
| BRAF, CRAF | LXH-254 | None | I | Advanced solid tumors harboring MAPK pathway alterations | Active, not recruiting | NCT02607813 |
| BRAF, CRAF | LXH-254 | Rineterkib (RAF/ERK inhibitor); trametinib (MEK inhibitor); ribociclib (CDK4/6 inhibitor); EGF816 (EGFR inhibitor); dabrafenib (BRAF mutant inhibitor) | I, II | Unresectable or metastatic melanoma; EGFR-mutant NSCLC | Recruiting; active, not recruiting | NCT04417621; NCT02974725; NCT03333343; NCT04294160 |
| ERK | LY3214996; GDC-0994; ulixertinib; MK-8353 | None | I, II | Acute myeloid leukemia; locally advanced or metastatic solid tumors; metastatic uveal melanoma; acute myelogenous leukemia or myelodysplastic syndromes | Recruiting; completed; terminated | NCT04081259; NCT01875705; NCT04488003; NCT03417739; NCT02296242; NCT01358331 |
| ERK | LY3214996 | RMC-4630 (SHP2-inhibitor); abemaciclib (CDK4/6 inhibitor); hydroxychloroquine (autophagy inhibitor) | I, II | Metastatic KRAS mutant cancers; solid tumors harboring pathogenic alterations in BRAF, RAF1, MEK1/2, ERK1/2, and NF1 | Not yet recruiting | NCT04916236; NCT04956640; NCT04534283; NCT04616183; NCT04391595; NCT04386057 |
| ERK | GDC-0994 | Cobimetinib (MEK inhibitor) | I | Locally advanced or metastatic solid tumors | Completed | NCT02457793 |
| ERK | Ulixertinib | Hydroxychloroquine (autophagy inhibitor); palbociclib (CDK4/6 inhibitor) | I, II | Advanced MAPK-mutated gastrointestinal adenocarcinomas; advanced pancreatic and other solid tumors | Recruiting | NCT041452973; NCT03454035 |
| ERK | MK-8353 | Selumetinib (MEK inhibitor); pembrolizumab (anti–PD-1 ab) | I | Advanced malignancies | Completed; active, not recruiting | NCT03745989; NCT02972034 |
| KRAS | AZD4785 | None | I | Advanced solid tumors | Completed | NCT03101839 |
| KRAS-G12C | Sotorasib | None | I, II | KRAS-G12C–mutant advanced/metastatic solid tumors | Recruiting; not yet recruiting | NCT04380753; NCT04625647; NCT04667234; NCT04933695 |
| KRAS-G12C | Sotorasib | Docetaxel (microtubule inhibitor); pembrolizumab (anti–PD-1 ab) | I, II, III | KRAS-G12C–mutant advanced/metastatic solid tumors | Active, not recruiting; recruiting | NCT04303780; NCT03600883; NCT04613596 |
| KRAS-G12C | Adagrasib | Docetaxel (microtubule inhibitor); pembrolizumab (anti–PD-1 ab); cetuximab (anti-EGFR ab); afatinib (pan-EGFR inhibitor); TNO155 (SHP2 inhibitor) | I, II, III | KRAS-G12C–mutant advanced/metastatic solid tumors | Recruiting | NCT04685135; NCT03785249; NCT04330664; NCT04793958 |
| KRAS-G12C | GDC-6036 | Atezolizumab (anti–PD-L1 ab); cetuximab (anti-EGFR ab); bevacizumab (anti-VEGF ab); erlotinib (EGFR inhibitor) | I | KRAS-G12C–mutant advanced/metastatic solid tumors | Recruiting | NCT04449874 |
| KRAS-G12C | D-1553 | Standard treatment | I | KRAS-G12C–mutant advanced/metastatic solid tumors | Recruiting | NCT04585035 |
| KRAS-G12C | JNJ-74699157 | Standard treatment | I | KRAS-G12C–mutant advanced/metastatic solid tumors | Completed | NCT04006301 |
| KRAS-G12C | LY3499446 | Abemaciclib (CDK4/6 inhibitor); cetuximab (anti-EGFR ab); erlotinib (EGFR inhibitor); docetaxel (microtubule inhibitor) | I, II | KRAS-G12C–mutant advanced/metastatic solid tumors | Terminated | NCT04165031 |
| KRAS-G12D | siG12D-LODER | Gemcitabine + nab-paclitaxel; FOLFIRINOX | II | Advanced pancreatic cancer | Recruiting | NCT01676259 |
| MEK | Cobimetinib |
Belvarafenib (RAF inhibitor) |
I | Advanced or metastatic solid tumors | Recruiting | NCT03284502 |
| MEK | Trametinib | LXH254 (RAF inhibitor) | I | NSCLC or melanoma | Recruiting | NCT02974725 |
| MEK | Pimasertib | None | I | N-RAS–mutated locally advanced or metastasis malignant cutaneous melanoma | Recruiting | NCT01693068, NCT00982865 |
| MEK | Pimasertib | SAR405838 (MDM2 antagonist) | I | Advanced solid tumors | Completed | NCT01985191 |
| MEK | Mirdametinib | Lifirfenib | I | Advanced or refractory solid tumors | Recruiting | NCT03905148 |
| p110α | Alpelisib | Capecitabine (nucleoside metabolic inhibitor); paclitaxel (microtubule inhibitor) | I | Patients with PIK3CA mutant metastatic colorectal cancer; PIK3CA-altered metastatic/recurrent gastric cancer | Not yet recruiting | NCT04753203; NCT04526470 |
| p110α | GDC-0077 | Entrectinib (pan-TRK inhibitor) | I | Breast cancer and advanced solid tumors | Recruiting | NCT04632992 |
| RAF | PLX8394; TAK-580 | None | I, II | Advanced unresectable solid tumors; low-grade glioma | Recruiting | NCT02428712; NCT03429803 |
| RAF | Belvarafenib | None | I | Solid tumors | Completed | NCT02405065 |
| RAF | Belvarafenib | Cobimetinib (MEK inhibitor); cetuximab (anti-EGFR ab); atezolizumab (anti–PD-L1 ab) | I | Advanced or metastatic solid tumors; NRAS-mutant advanced melanoma | Recruiting | NCT03284502; NCT04835805 |
| RAF, EGFR | Lifirfenib | None | I | Solid tumors | Completed | NCT02610361; NCT03641586 |
| RAF, EGFR | Lifirfenib | Mirdametinib (MEK inhibitor) | I | NSCLC with confirmed KRAS mutations | Recruiting | NCT04294160 |
| SHP2 | BBP-398; JAB-3068; RMC-4630; RLY-1971; JAB-3312; SH3809 | None | I, II | Advanced or metastatic solid tumors | Recruiting | NCT04528836; NCT03565003; NCT03518554; NCT03634982; NCT04252339; NCT04121286; NCT04045496; NCT04843033 |
| SHP2 | RMC-4630 | LY3214996 (ERK inhibitor); cobimetinib (MEK inhibitor); osimertinib (EGFR inhibitor) | I, II | Advanced or metastatic solid tumors | Not yet recruiting; recruiting | NCT04916236; NCT03989115 |
| SHP2 | ERAS-601 | Cobimetinib (MEK inhibitor) | I | Advanced or metastatic solid tumors | Recruiting | NCT04670679 |
| SHP2 | TNO155 | Nazartinib (EGFR inhibitor); spartalizumab (anti–IL-1β antibody); ribociclib (CDK4/6 inhibitor); adagrasib (KRAS-G12C inhibitor); JDQ443 (KRAS-G12C inhibitor) | I, II | Advanced solid tumors | Recruiting | NCT03114319; NCT04000529; NCT04330664; NCT04699188 |
| SOS1 | BI 1701963 | Trametinib (MEK inhibitor); BI 3011441 (MEK inhibitor); irinotecan (topoisomerase I inhibitor) | I | Advanced or metastatic solid tumors | Recruiting | NCT04111458; NCT04835714; NCT04627142 |
Type and frequency of KRAS mutation
RAS genes are mutated at different prevalence rates in human cancers (Fig. 1a) [19]. Elucidating similarities and differences among these RAS mutations from a developmental or evolutionary perspective remains a challenge [20, 21]. Some RAS gene mutations are innocuous, but others cause cancer by producing oncoproteins. For example, three amino acid residues (G12, G13, and Q61) in HRAS, KRAS, and NRAS are mutational hot spots, though with distinct frequencies in different human tumor types (Fig. 1b) [14]. A KRAS-G12 mutation is a common event in pancreatic (91%), colorectal (68%), and lung adenocarcinoma (85%; a subtype of NSCLC) [14]. Among these, KRAS-G12D is the leading mutation in pancreatic (45%) and colorectal adenocarcinoma (45%), while KRAS-G12C mainly occurs in lung adenocarcinoma (46%) (Fig. 1c) [14]. In contrast, 85% of human melanomas have KRAS-Q61 mutations, especially Q61R (46%) [14]. Whole-exome sequencing of human PDAC or NSCLC tumors shows that, despite the genetic heterogeneity within the tumor, KRAS is mutated in different regions [22, 23]. Consistent with this, the co-mutational interactions between each KRAS allele and other unrelated genes are highly tissue-specific, emphasizing the complexity of cell type-specific oncogenesis [24]. KRAS mutations usually co-occur with mutations in tumor protein p53 (TP53) and cyclin-dependent kinase inhibitor 2A (CDKN2A) in PDAC, Kelch-like ECH-associated protein 1 (KEAP1) and/or serine/threonine kinase 11 (STK11) in NSCLC, or APC regulator of WNT signaling pathway (APC) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) in CRC. While mutations in exon 2 of KRAS are the most common, it is important to note that they affect the differential use of exon 4, giving rise to two splice variants, KRAS4A and KRAS4B (Fig. 1b) [25]. These KRAS variants differ in their C-terminal membrane targeting region, posttranslational modifications, and interactomes, thus exhibiting different signal behaviors in development, metabolism, and proliferation [26–28]. Therefore, to target KRAS, investigators must consider changes in the protein structure caused by point mutations, but also isoform-specific properties [29].
Fig. 1.

Type and frequency of RAS mutations in human cancers. a. Somatic mutations of RAS oncogene in the top 10 human cancers. b. The frequency and location of G12, G13, and Q61 mutations in the exons of RAS oncogenes. c. The frequency and type of KRAS mutations in codon 12 in pancreatic cancer, colorectal cancer, and lung adenocarcinoma. The data were derived from recent studies using the COSMIC or cBioPortal database [2, 14, 19]
Prognostic and predictive value of KRAS mutations
Depending on the clinical settings, the prognostic and predictive values of KRAS mutations are variable and even conflicting. For example, an early study of lung adenocarcinoma patients showed that the KRAS mutation in code 12 is an unfavorable prognostic factor [30]. Similarly, a meta-analysis found that KRAS mutations are associated with poor survival in patients with early resectable NSCLC [31]. In contrast, a pooled analysis of NSCLC patients treated with cisplatin-based chemotherapy revealed that a KRAS mutation was not a prognostic factor [32]. Of note, a KRAS mutation is a negative predictor of response to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs; e.g., cetuximab, gefitinib, or erlotinib) (Table 2) [33–36], but a positive predictor of response to immune checkpoint inhibitors (ICIs; including anti–PD-1 and anti–PD-L1) (Table 3) [37–39]. However, response rates are variable and dissenting reports suggest that KRAS mutations cannot guide the therapeutic choice between TKIs and ICIs in NSCLC patients [40, 41].
Table 2.
Tyrosine kinase inhibitors
| Tyrosine kinase inhibitors (TKIs) are a group of drugs that disrupt the tyrosine kinase (TK) signal transduction pathway through a variety of mechanisms. They can compete with adenosine triphosphate (ATP), phosphorylated entities, substrates, or can act in an allosteric manner, that is, bind to sites outside the active site and affect the sites’ activity through conformational changes. TKs can be divided into receptor tyrosine kinases (RTKs), nonreceptor tyrosine kinases (NRTKs), and dual-specific kinases (DSKs). DSKs phosphorylate serine, threonine, and tyrosine residues. Approximately 20 different transmembrane RTK subfamilies have been identified, such the families for vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), insulin receptor (INSR), fibroblast growth factor receptor (FGFR), and epidermal growth factor receptor (EGFR). NRTKs are cytoplasmic proteins and do not have a transmembrane domain. NRTKs are mainly composed of nine families, including those for Abl, Ack, Csk, Fak, Fes/Fer, Jak, Src, Syk/Zap70, and Tec. The most typical example of DSK is mitogen-activated protein kinase kinase (MEK), which is involved in the mitogen-activated protein kinase (MAPK) pathway. More than 50 FDA-approved TKIs (including small-molecule inhibitors and monoclonal antibodies) are used to treat various diseases, including cancer. |
Table 3.
Immune checkpoint inhibitors
| Immune checkpoint inhibitors (ICIs) are a group of drugs that inhibit the activity and function of inhibitory immune checkpoint molecules, such as programmed cell death protein 1 (PD-1), programmed death ligand 1 (PD-L1), cytotoxic T lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG3), and T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3). Under physiological conditions, inhibitory immune checkpoint molecules play an important role in maintaining self-tolerance, preventing autoimmune reactions, and minimizing tissue damage by regulating the duration and intensity of immune responses. However, abnormal expression and excessive activation of immune checkpoint molecules can cause many diseases, including cancer. In particular, inhibitory immune checkpoint molecules are upregulated in various cells within the tumor microenvironment, forming various pairings and limiting the normal antitumor function of immune cells. In contrast, the use of ICIs can restore the function of immune cells hijacked by cancer cells, resulting in an enhanced immunosurveillance with a cytotoxic T lymphocyte (CTL) response. ICIs (e.g., pembrolizumab, nivolumab, cemiplimab, and atezolizumab) have changed the landscape of cancer treatment and become a new hope for cancer patients after the failure of regular chemotherapy or radiotherapy. |
In PDAC, patients with KRAS-G12D mutations (but not total KRAS mutations) have a worse prognosis than patients with wild-type KRAS [42–44]. Other studies suggest that the prognosis of KRAS-G12V is poorer than that for other mutations [45, 46], commensurate to a KRAS-G12V–associated increase in circulating regulatory T cells that most likely limits antitumor immunity [47]. In advanced PDAC, KRAS mutation status is predictive for the efficacy of erlotinib rather than prognostic [48]. This contradicts other studies reporting that KRAS wild-type patients with PDAC have a significant advantage after treatment with gemcitabine/nimotuzumab [49] or gemcitabine/erlotinib [50] with respect to overall survival. Circulating tumor DNA (ctDNA) has recently become a minimally invasive tool used in precision oncology to evaluate genetic alterations. Mutant KRAS in ctDNA might be a more sensitive predictor of survival than the ELISA-based detection of cancer antigen 19-9 (CA 19-9) [51, 52]. Before a clear conclusion can be drawn regarding the impact of KRAS mutations on overall survival in PDAC [53, 54], additional data from analyses of other genetic alterations are needed.
In localized CRC, KRAS mutations usually suggest a poor prognosis [55–57]. Some KRAS mutations, including KRAS-G12V, are more aggressive than others [58]. In contrast to BRAF mutations, KRAS mutations have no major prognostic value in advanced CRC patients [59]. The association between KRAS mutations and poor clinical outcomes from TKI treatment has been confirmed in several independent studies [60–63]. Compared with KRAS-G12V mutations or wild-type tumor groups, CRC patients with KRAS-G13D mutations are insensitive to cetuximab therapy [64, 65]. Nevertheless, the impact of KRAS mutations on cetuximab treatment needs to be further evaluated in prospective randomized trials. Reportedly, KRAS-G12D–mediated inhibition of interferon regulatory factor 2 (IRF2) drives CRC resistance to anti–PD-1 therapy in preclinical models [66]. However, the clinical implications of these findings remain elusive.
In conclusion, the prognostic and predictive values of KRAS mutations are affected by many factors, such as tumor type, stage, patient age, sex, the coexistence of mutations affecting other oncogenes or tumor suppressors, and treatment regimens. For this reason, the clinical utility of detecting KRAS mutations may be over- or underestimated. Careful meta-analyses that avoid nonrandom systematic errors due to variations between trials [67] are needed to clarify this issue.
Activation and modulation of KRAS mutations
Wild-type KRAS protein mostly resides on the cytoplasmic side of the plasma membrane, as well as at membranes of intracellular organelles, and is guided by protein localization signals (such as lipid moieties added to the carboxyl terminus) [68]. The RAS family of proteins belongs to a class of enzymes called small GTPases, which play a central role in cell signal transduction [69]. The functions and activities of RAS protein depend on the transition from an inactive guanosine diphosphate (GDP)-bound state to an active guanosine triphosphate (GTP)-bound state (Fig. 2a) [69]. Normally, this conversion process of RAS status is reversible and is maintained in a balanced manner by GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). GAPs, such as neurofibromin 1 (NF1), accelerate the GTP hydrolysis of RAS, leading RAS to an inactive state [70, 71]. GEFs, including the most universally expressed SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1), are responsible for producing active GTP-bound RAS [72–74]. The balance between hydrolysis and exchange determines the level of activated KRAS in the cell [75]. Oncogenic mutations of RAS disrupt GAP-mediated GTP hydrolysis, allowing these oncoproteins to accumulate in a continuous GTP-binding active state (Fig. 2b) [13]. Among distinct KRAS mutants, the KRAS-G12C protein exhibits the highest intrinsic GTP hydrolysis rate [76].
Fig. 2.

Principle of inhibiting oncogenic KRAS activation. a. The wild-type (WT) KRAS protein maintains a balance between the inactive state of guanosine diphosphate (GDP) binding and the active state of guanosine triphosphate (GTP) binding. This process is mediated by GTPase activating protein (GAP) and guanine nucleotide exchange factor (GEF). b. The KRAS oncoprotein (e.g., KRAS-G12C) disrupts GAP-mediated GTP hydrolysis, allowing these mutants to accumulate in a continuous GTP-binding active state, which is responsible for oncogenic activity. c. The covalent inhibitor of KRAS-G12C protein (G12Ci) achieves allosteric inhibition of mutant cysteine 12 (12C) to prevent GEF-catalyzed nucleotide exchange and block subsequent effector pathways
The upstream regulators of the RAS pathway involve receptor tyrosine kinases (RTKs), which are cell surface receptors for many growth factors, cytokines, and hormones (Fig. 3). One particular RTK subclass, the EGFR family, is composed of four closely related members, EGFR (also known as ERBB1 or HER1), ERBB2/HER2, ERBB3/HER3, and ERBB4/HER4. EGFR/ERBB1, the best characterized activator of RAS signaling, acts through binding to an adaptor protein, namely growth factor receptor bound protein 2 (GRB2) [77, 78]. GRB2 further mediates the recruitment and activation of SOS1- and SH2-containing protein tyrosine phosphatase 2 (SHP2) to activate GTP-bound RAS [79–84]. Activated EGFR mutations are often found in human cancers, especially NSCLC, and lead to the constitutive activation of downstream signals, including RAS [85]. G-protein–coupled receptors (GPCRs), the largest group of membrane receptors, also participate in RAS activation [86, 87]. Thus, the interplay between RTKs and GPCRs may increase the plasticity of RAS activation.
Fig. 3.

Indirect KRAS suppression strategy. The activation of receptor tyrosine kinases, such as members of the epidermal growth factor receptor (EGFR) family, activate KRAS through the growth factor receptor-bound protein 2 (GRB2)-SH2–containing protein tyrosine phosphatase 2 (SHP2)-SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1) pathway. The mutant KRAS protein accumulates in the guanosine triphosphate (GTP)-bound state, leading to the activation of downstream effector pathways, especially the RAF-MEK-extracellular signal regulated kinase (ERK) and the phosphatidylinositol 3-kinase (PI3K)-AKT-mechanistic target of rapamycin (mTOR) pathways. The localization of KRAS on the cell membrane is the first step in subsequent KRAS activation, which is mediated by enzymes, including but not limited to farnesyltransferase (FT), geranylgeranyltransferase 1 (GGT1), and isoprenylcysteine carboxyl methyltransferase (ICMT). In addition to directly inhibiting KRAS (exemplified by covalent allele-specific inhibitors that bind to KRAS-G12C), multiple approaches can indirectly inhibit the oncogenic pathway of KRAS by targeting upstream regulators, downstream effectors, and KRAS expression and processing. The main drugs or reagents used for indirect KRAS inhibition are shown in red (for clinical trials or approved for use in patients) or green (for preclinical research)
The downstream effectors of the RAS pathway are mainly involved in the activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways, which favor anabolic processes including cell growth, protein translation, and proliferation (Fig. 3) [88, 89]. Thus, the constitutive activation of membrane RAS-dependent signal pathways favors oncogenesis [90]. The aggregation of mitochondrial KRAS-G12V protein also favors tumor cell growth through metabolic effects [91]. Another distinctive feature of KRAS-G12D protein is that it can be released by PDAC cells into the tumor microenvironment and then mediates the polarization of pro-tumor macrophages (Fig. 4) [92]. Indeed, PDAC patients with KRAS-G12D–positive macrophages exhibit low survival rates [92]. There is also strong preclinical evidence that mutated KRAS requires additional factor (such as chronic inflammation or a high-fat or high-iron diet) to deploy its full carcinogenic activity [93–95]. Further work is needed to elucidate the likely complex cell-autonomous and non-autonomous effects of mutated and unmutated KRAS protein on the tumor microenvironment [21].
Fig. 4.

The immunosuppressive function of extracellular KRAS-G12D protein in the tumor microenvironment. KRAS-G12D protein can be released during ferroptosis, which is a regulated cell death caused by reactive oxygen species (ROS) and subsequent lipid peroxidation. The release of KRAS-G12D protein is mediated by exosomes, which are cargo extracellular vesicles produced by multivesicular bodies derived from endosomes. The small GTPase RAB27A regulates exocytosis of multivesicular endosomes, which leads to exosome secretion. This process is further enhanced by autophagy-related 5 (ATG5)-dependent autophagosome formation and autophagy-meditated secretion. Once released, the extracellular KRAS-G12D protein from exosomes is taken up by advanced glycosylation end product-specific receptor (AGER) on macrophages, leading to phosphorylation and activation of signal transducer and activator of transcription 3 (STAT3). Nuclear STAT3 acts as a transcription factor to produce cytokines, such as transforming growth factor beta 1 (TGFB1), interleukin 10 (IL10), and arginase 1 (ARG1), for polarization of M2 macrophages, which limits antitumor immunity
Indirect KRAS suppression strategies
The most successful way to inhibit oncogenic kinases is to develop inhibitors that compete with ATP to bind to the kinase domain. However, KRAS uses GTP instead of ATP as a phosphate donor for signal transmission. Attempts to directly enzymatically inhibit KRAS function have been largely frustrated, leading to the development of indirect methods for KRAS inhibition. Below, we highlight some representative drugs that exemplify the main strategies for indirectly targeting mutant KRAS (Table 1 and Fig. 3).
Inhibition of KRAS expression
AZD4785 is a constrained ethyl-containing antisense oligonucleotide that is complementary to the sequence of the 3'UTR of KRAS mRNA, leading to the downregulation of wild-type and mutant KRAS protein [96]. AZD4785 selectively inhibits the proliferation of mutant KRAS-driven tumor cells in vitro or in xenograft models [96, 97]. However, intravenously infused AZD4785 failed to completely reduce KRAS mRNA in patients with NSCLC (NCT03101839) [14], calling for adjustments of the dose and method of administration. Another approach for transcriptionally inhibiting KRAS expression involves the use of a specific small interfering RNA (siRNA) named siG12D-LODER that specifically targets G12D but not wild-type KRAS. This agent showed promise in a phase I study (NCT01188785) in combination with chemotherapy (gemcitabine or FOLFIRINOX, i.e., a combination of 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) in 12 patients with advanced PDAC [98]. A phase II study (NCT01676259) evaluating this therapeutic strategy is now underway.
Inhibition of KRAS processing
The location of RAS on the cell membrane is the initial step of RAS activation and requires multiple posttranslational processing steps, especially lipid-related prenylation [99]. The two enzymes involved in KRAS prenylation are farnesyltransferase (FT) and geranylgeranyltransferase 1 (GGT1). Although preclinical studies suggest anticancer activity for FT inhibitors (for example, tipifarnib/R115777 and lonafarnib/SCH 66336) against RAS-mutant tumors, clinical studies have been disappointing [100–103]. One possible explanation for this failure is functional redundancy among FT and GGT1 [104]. A single molecule with dual inhibitory activity on FT and GGT1, such as FGTI-2734 [105], might have the potential to eliminate RAS-mutant tumors. Alternatively, targeting downstream RAS processing enzymes, such as isoprenylcysteine carboxyl methyltransferase (ICMT), may be attempted. Two ICMT inhibitors (cysmethynil and UCM-1336) impair the membrane localization of RAS (including that of KRAS) [106], but their application in vivo remains to be studied. Once RAS is effectively processed, membrane RAS protein undergoes activating self-association, and this process can be blocked by a synthetic binding protein called NS1 [107]. Since NS1 is an alien protein, its possible recognition by the immune system needs to be evaluated before it is introduced into clinical trials.
Inhibition of upstream signaling molecules
Gefitinib, erlotinib, afatinib (pan-EGFR inhibitors), icotinib, and osimertinib are first-line EGFR TKIs for treating NSCLC patients with EGFR mutations [108]. Lapatinib is the first dual inhibitor of EGFR and ERBB2/HER2 for treating ERBB2-positive breast cancer, whereas brigatinib is a mixed inhibitor of ALK and EGFR used for the treatment of metastatic NSCLC. The clinical benefit of small-molecule EGFR inhibitors on KRAS-mutant cancers is context-dependent. For example, gefitinib alone is not effective against KRAS-mutant NSCLC [109], while the combination of erlotinib and gemcitabine provides transient benefit to patients with KRAS-mutant PDAC [110]. Another approach to inhibit EGFR activity consists of the use of monoclonal antibodies [108]. Cetuximab and panitumumab are approved for metastatic CRC, while necitumumab is used for the treatment of squamous NSCLC. However, some studies suggest that antibodies against EGFR have no effect on KRAS-mutant CRC [60, 62], while others report that CRC patients with KRAS-G13D are sensitive to cetuximab treatment [65]. Regardless, acquired KRAS mutations are a common mechanism of resistance to EGFR inhibitors [111]. Recently, rybrevant, a bispecific antibody against EGFR and MET receptors, has been approved for the treatment of NSCLC patients with EGFR exon 20 insertion mutations. Other EGFR-specific monoclonal antibodies in clinical development are zalutumumab, nimotuzumab, and matuzumab. It will be important to understand how preexisting or acquired KRAS mutations will affect the clinical activity of such drugs.
The proteins SOS1 and SOS2 promote RAS activation by binding to GRB2. The SOS1 inhibitor BI-1701963 is used in combination with MEK inhibitors (trametinib and BI-3011441) or a topoisomerase I inhibitor (irinotecan) in clinical trials enrolling patients with advanced or metastatic solid cancer (Table 1). BI-1701963 has been designed to bind to the catalytic domain of SOS1 to prevent its interaction with KRAS-GDP [79]. SOS1 mutations lead to dysregulated enzymatic activities, which may cause drug resistance [112]. Because there is currently no SOS2-specific inhibitor, it is unclear whether targeting SOS2 would have the same effects as SOS1 inhibitors. It can be speculated that pan-SOS inhibitors might be particularly efficient in blocking the activation of RAS.
SHP2 not only mediates the RTK-stimulated activation of RAS [113] but also acts as a promoter of immune checkpoint pathways [114]. Certain SHP2 inhibitors are in early-phase clinical development for treating advanced or metastatic solid cancer (Table 1). TNO155 is an allosteric inhibitor that maintains SHP2 in a self-inhibited conformation [115]. Preclinical studies have shown promising anticancer activity from TNO155 combined with inhibitors of EGFR, MEK, ERK, CDK4/6, or KRAS-G12C and anti–PD-1 antibodies in xenograft models of NSCLC or CRC cells [116]. The efficacy and toxicity of combination regimens involving TNO155 together with TKIs or ICIs remain to be determined in clinical trials.
Inhibition of downstream signaling molecules
Oncogenic transformation mediated by RAS requires the downstream activation of the RAF/MEK/ERK and the PI3K/AKT/mTOR pathways. In theory, inhibition of any of these effectors should block oncogenic KRAS signaling. In fact, these two pathways intersect with each other and even form a feedforward loop to activate KRAS as an upstream signal [14]. Nonetheless, the success of targeting KRAS-mutant tumors by inhibiting single downstream molecules has been limited [18]. Despite these results, certain MAPK and PI3K pathway inhibitors have been approved or are entering clinical trials for combination therapies (Table 1) [88, 89]. In this section, we highlight some of these drugs and their application for KRAS-mutant cancers.
RAF inhibitors
The RAF family consists of ARAF, BRAF, and CRAF, all sharing RAS as a common upstream activator. Belvarafenib (HM95573) is a pan-RAF dimerization inhibitor that demonstrates selective anticancer activity with either cobimetinib or cetuximab in preclinical models, as well as in cancer patients with RAS or RAF mutations, especially melanoma patients (Table 1). ARAF mutations are conducive to resistance to bevacafenib, indicating that the RAF subtype has a compensatory function, and a secondary mutation of a RAF member may reactivate the MAPK pathway to avoid cell death [117].
LXH-254, an ATP-competitive inhibitor of BRAF and CRAF [118], is used in multiple clinical trials for patients with NSCLC or melanoma (Table 1). The anticancer activity of LXH-254 is demonstrated in tumors carrying BRAF/RAS co-mutations, but it has moderate activity against cancers driven by KRAS mutants [119]. ARAF may also mediate LXH-254 resistance in RAS-mutant cancer cell lines [119], supporting the hypothesis that all RAF isoforms need to be suppressed at the same time in order to achieve tangible antineoplastic effects.
Lifirafenib (BGB-283), a dual inhibitor of RAF and EGFR, is being used in clinical studies enrolling patients with BRAF- or KRAS/NRAS-mutated solid tumors [120]. Preclinical study suggests that lifirafenib enhances the antitumor activity of MEK inhibitors (mirdametinib and selumetinib) in KRAS-mutant tumors [121]. A phase I study on the safety and pharmacokinetics of the combination with lifirfenib and mirdametinib in KRAS-mutant NSCLC is ongoing (NCT03905148).
Other RAF inhibitors, such as PLX8394 and TAK-580 (MLN2480), are being evaluated as single-agent therapeutics in patients with advanced unresectable solid tumors (NCT02428712) or low-grade glioma (NCT03429803), respectively. Vemurafenib, dabrafenib, and encorafenib are RAF inhibitors approved for the treatment of tumors with BRAF-V600E/K, but not RAS, mutations. It appears that monotherapy with RAF inhibitors is not efficient against cancers with KRAS mutations, suggesting that combination with other MAPK pathway inhibitors should be attempted.
MEK inhibitors
Three MEK inhibitors, including trametinib (GSK1120212), cobimetinib (XL518), and binimetinib (MEK162), are approved in combination with BRAF inhibitors for the treatment of patients with advanced melanoma harboring BRAF mutations (V600E or V600K). Although the MEK inhibitor selumetinib (AZD6244) is not efficient against melanoma, it has recently been approved for the treatment of neurofibromatosis type 1 in children. Compared with standard treatments, MEK inhibitors alone are not efficient against solid tumors driven by KRAS mutations [122–125]. However, the combination of a MEK inhibitor and RAF inhibitor has shown promising activity against KRAS-mutant (especially KRAS-G13D) cells in vitro [126]. These findings provide the rationale for ongoing clinical trials that combine RAF (belvarafenib or LXH-254) and MEK inhibitors (cobimetinib or trametinib) to treat solid cancer with KRAS mutations (Table 1).
Two MEK inhibitors, pimisertib (MSC1936369B) and mirdametinib (PD0325901), are being evaluated for clinical activity against KRAS-mutant solid cancers (Table 1). Pimasertib alone is better than dacarbazine for improving progression-free survival in patient with BRAF- and NRAS-mutant melanoma [127, 128]. A combination of pimasertib and the MDM2 (a repressor of tumor suppressor TP53) inhibitor SAR405838 has shown preliminary antitumor activity in the treatment of solid cancers with RAS or RAF mutations [129]. Thus, targeting MEK combined with pharmacological TP53 induction may constitute a strategy for combating KRAS-mutant cancers.
ERK inhibitors
Although ERK inhibition is an effective strategy to overcome resistance to upstream MEK or RAF inhibitors [130–132], the clinical development of ERK inhibitors has been retarded when compared to that of MEK and RAF inhibitors. In 2020, the FDA granted an expanded access program for the ERK inhibitor ulixertinib (BVD-523) for the treatment of cancer patients with abnormal MAPK pathways, including but not limited to those involving KRAS, NRAS, HRAS, BRAF, MEK, and ERK mutations [133]. Ulixertinib is currently being evaluated in combination with hydroxychloroquine (an autophagy inhibitor) or palbociclib (a CDK4/6 inhibitor) in patients with advanced pancreatic and other solid tumors (NCT041452973 and NCT03454035).
Certain ERK inhibitors, such as LY3214996, GDC-0994, and MK-8353, alone or in combination with other drugs, are in the early stages of clinical development. LY3214996 is being used with abemaciclib, hydroxychloroquine, or RMC-4630, in solid tumors, including KRAS-mutant cancers (Table 1). Despite strong preclinical data [134], patients with advanced solid tumors cannot tolerate combination therapy with GDC-0994 and cobimetinib [135]. However, GDC-0994 alone has acceptable side effects and showed anticancer activity in two patients with BRAF-mutant CRC [136]. The toxicity of GDC-0994 in combination with other MAPK inhibitors needs to be further investigated.
MK-8353 shows a tolerable safety profile and antitumor activity in melanoma patients with a BRAF-V600 mutation, rather than RAS mutation [137]. MK-8353 in combination with selumetinib or pembrolizumab is being investigated in patients with advanced malignancies, including CRC (Table 1). Although preclinical studies have shown that ERK mutations confer resistance to MAPK inhibitors [138], clinical studies have not yet reported the occurrence of acquired resistance to ERK inhibitors.
PI3K pathway inhibitors
Class I phosphatidylinositol-3-kinase (PI3K) is a lipid kinase that phosphorylates the signaling lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), resulting in the recruitment of protein kinase B (PKB, best known as AKT) to the plasma membrane and subsequent activation of mTOR for cell growth and proliferation [139]. In contrast, PTEN, a tumor suppressor, can convert PIP3 to PIP2, thereby diminishing PI3K activity. PI3K consists of a catalytic subunit (p110, including p110α, p110β, p110γ, and p110δ isoforms) and a regulatory subunit (p85α or p85β). Compared with p110γ and p110δ, which are mainly expressed in immune cells, the expression of p110α and p110β is common in various cells or tissues. PIK3CA, which encodes p110α, is frequently mutated in cancer as an important drug target [139]. PIK3CA mutations can coexist with RAS mutations, while RAS mutations and MAPK pathway mutations are usually mutually exclusive [140, 141]. These co-mutation patterns might guide clinical trials to target different signals from the MAPK and PI3K pathways.
In May 2019, the FDA approved the first drug, alpelisib, as a specific p110α inhibitor for treating breast cancer. The combination of alpelisib with other chemotherapy (capecitabine or paclitaxel) is being tested in patients with PIK3CA-mutant CRC or gastric cancer patients (Table 1). The secondary p110α inhibitor GDC-0077 is under clinical development for the treatment of breast cancer (NCT04632992). Additionally, copanlisib (a pan class I PI3K inhibitor), duvelisib (a p110γ/p110δ inhibitor), and idelalisib (a p110δ inhibitor) are approved to treat adult patients with relapsed or refractory follicular lymphoma, but not solid tumors, with RAS mutations. Despite considerable efforts to combine PI3K and MEK inhibitors in preclinical models [142], such a combination can cause significant toxicity in patients with RAS-mutated cancers [143, 144]. Similarly, the combination of AKT or mTOR inhibitors and MAPK inhibitors is generally poorly tolerated by patients, which may limit their applications [145, 146].
In summary, compared with MAPK pathway inhibitors, monotherapy or combination therapy with PI3K pathway inhibitors has limited benefits for patients with KRAS-mutated cancers, although such PI3K inhibitors may reverse resistance to KRAS-G12C inhibitors (discussed later). Current PI3K inhibitors are still challenged by insufficient selectivity, which results from the close structural resemblance among ATP binding sites of different PI3K isoforms [139].
Others
The MAPK and PI3K pathways activate transcription factors to induce or suppress the expression of genes involved in multiple cellular processes. Targeting related downstream processes (for example, autophagy, glycolysis, and immunosuppression) also may help to mitigate the carcinogenic activity of RAS but will not be discussed in this review. Of note, genomic screenings have enabled the discovery of synthetic lethal partners to inhibit tumor growth in KRAS-mutant cancer cells (Table 4).
Table 4.
Genes involved in synthetic lethality of mutant KRAS-dependent cancers
| Synthetic lethal genes |
Full name | Main function | Tumor type | Reference |
|---|---|---|---|---|
| ANAPC1 | Anaphase-promoting complex subunit 1 | Mediates cell cycle progression | KRAS-mutant colon cancer | [147] |
| ARHGEF2 | Rho/Rac guanine nucleotide exchange factor 2 | Activates Rho-GTPases | KRAS-mutant pancreatic cancer | [148] |
| BCL2L1 (BCL-XL) | BCL2-like 1 | Inhibits apoptosis | KRAS-mutant solid cancer | [149] |
| BIRC5 (survivin) | Baculoviral IAP repeat containing 5 | Inhibits apoptosis | KRAS-mutant colon cancer | [150] |
| CDK1 | Cyclin-dependent kinase 1 | Mediates cell cycle progression | KRAS-mutant colon cancer | [151] |
| CDK4 | Cyclin-dependent kinase 4 | Mediates cell cycle progression | KRAS-mutant lung cancer | [152, 153] |
| DHODH | Dihydroorotate dehydrogenase (quinone) | Inhibits mitochondrial oxidative damage | KRAS-mutant pancreatic cancer | [154] |
| FGFR1 | Fibroblast growth factor receptor 1 | Mediates mitogenesis and differentiation | KRAS-mutant lung cancer | [155] |
| GATA2 | GATA binding protein 2 | Promotes development and survival | KRAS-mutant lung cancer | [156] |
| MAP3K7 (TAK1) | Mitogen-activated protein kinase kinase kinase 7 | Promotes NF-κB activation | KRAS-mutant colon cancer | [157] |
| PLK1 | Polo-like kinase 1 | Promotes centrosome maturation and spindle assembly | KRAS-mutant chronic myelomonocytic leukemia or solid cancer | [147, 158, 159] |
| PRMT5 | Protein arginine methyltransferase 5 | Arginine methyltransferase | KRAS-mutant pancreatic cancer | [160] |
| PSMA5 | Proteasome 20S subunit alpha 5 | Mediates protein degradation | KRAS-mutant colon cancer | [147] |
| SHOC2 | SHOC2 leucine-rich repeat scaffold protein | Promotes RAS signaling | KRAS-mutant leukemia and solid cancer | [161, 162] |
| SHP2 (PTPN11) | SH2 containing protein tyrosine phosphatase 2 | Promotes RAS signaling | KRAS-mutant solid cancer | [113, 163] |
| SNAI2 | Snail family transcriptional repressor 2 | Promotes epithelial-mesenchymal transition | KRAS-mutant colon cancer | [164] |
| STK33 | Serine/threonine kinase 33 | Regulates cell cytoskeleton | KRAS-mutant solid cancer | [165] |
| TBK1 | TANK binding kinase 1 | Promotes NF-κB activation | KRAS-mutant lung cancer | [166] |
| WT1 | WT1 transcription factor | Promotes development and survival | KRAS-mutant lung cancer | [167] |
| XPO1 | Exportin 1 | Mediates nuclear export | KRAS-mutant lung cancer | [168] |
| YAP1 | Yes1-associated transcriptional regulator | Mediates the Hippo signaling pathway | KRAS-mutant pancreatic cancer | [167] |
Direct KRAS inhibition
Covalent KRAS-G12C inhibitors
Historically, KRAS was considered "undruggable" because it does not have a classical pocket suitable for binding small inhibitory molecules [18]. Recent structural studies and drug design efforts to produce G12Ci have changed this view (Table 5). The pioneering work of Shokat and colleagues uncovered a hidden pocket (switch-II) in the KRAS-GDP complex that is located next to the mutant cysteine 12 [172]. The proximity of switch-II to cysteine 12 facilitated the development of covalent inhibitors of switch-II, thereby achieving allosteric inhibition of cysteine 12 to prevent the nucleotide exchange catalyzed by GEF and diminish the subsequent interaction between RAS and RAF (Fig. 2c) [170, 172, 173]. Since wild-type KRAS lacks cysteine in the active site, the covalent inhibition of cysteine 12 is expected to be highly specific. ARS-1620 structurally modified from compound 12 [172], 1_AM [169], and ARS-853 [170] turned out to be the first G12Ci to elicit effective tumor suppression in patient-derived xenograft modes [171].
Table 5.
Development history and application status of KRAS-G12C inhibitors
| Name | Application date | Institutions | Structure | Status | Reference/ trial number |
|---|---|---|---|---|---|
| 1_AM | August 2017 | Dana-Farber Cancer Institute | 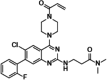 |
Preclinical | [169] |
| Adagrasib | October 2019 | Mirati | 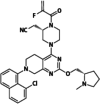 |
Clinical (approved) |
[16] |
| ARS-853 | January 2016 | Memorial Sloan Kettering Cancer Center | 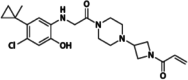 |
Preclinical | [170] |
| ARS-1620 | January 2018 | Wellspring Biosciences | 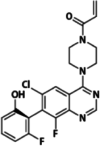 |
Preclinical | [171] |
| Compound 12 | November 2013 | University of California | 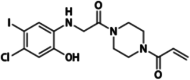 |
Preclinical | [172] |
| D-1553 | October 2020 | InventisBio | Structure not disclosed | Clinical (recruiting) | NCT04585035 |
| GDC-6036 | June 2020 | Genentech | Structure not disclosed | Clinical (recruiting) | NCT04449874 |
| JNJ-74699157 | July 2019 | Araxes/J&J | Structure not disclosed | Clinical (terminated) | NCT04006301 |
| LY3499446 | November 2019 | Eli Lilly | Structure not disclosed | Clinical (terminated) | NCT04165031 |
| Sotorasib | October 2019 | Amgen | 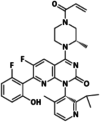 |
Clinical (approved) |
[15] |
Amgen and Mirati Therapeutics developed two structure-optimized covalent G12Ci formulations, sotorasib [15] and adagrasib [16]. Compared with ARS-1620, sotorasib and adagrasib have larger surface grooves, which enhance the effectiveness of irreversible interactions with the H95 residue in the 3 helix of KRAS-G12C protein [15, 16]. Sotorasib and adagrasib mediate selective tumor suppression activity across a panel of cancer cell lines harboring the KRAS-G12C mutation [14, 16, 174]. Durable responses to sotorasib have been observed in immunocompetent rather than immunodeficient tumor-bearing mice [175]. This may be explained by the fact that sotorasib induces the production of chemokines (CXCL10 and CXCL11) and potential damage-associated molecular patterns (DAMPs), leading to an immune response mediated by cytotoxic lymphocytes (Fig. 5) [175]. Accordingly, the combined use of anti–PD-1 antibodies further enhances sotorasib-induced tumor suppression in mouse models [175]. Whether G12Ci can be used to produce cancer-preventive or therapeutic immune responses is an open question. Interestingly, patient-derived xenograft models indicate that individual genetic alteration (such as in KRAS, TP53, STK11, or CDKN2A) cannot predict the anticancer activity of adagrasib [176]. CRISPR screens have identified negative (MYC, SHP2, mTOR, RPS6, CDK1, CDK2, CDK4/6, and RB1) and positive (KEAP1 and CBL) regulators of adagrasib sensitivity in NSCLC cells [176]. Drug screening further revealed that a pan-EGFR family inhibitor (afatinib), a SHP2 inhibitor (RMC-4550), mTOR inhibitors (vistusertib and everolimus), and a CDK4/6 inhibitor (palbociclib) increase the response rate to adagrasib in cell cultures and mouse models [176], providing potential optimization strategies for translational research.
Fig. 5.

Immunostimulation by sotorasib acting on the tumor microenvironment. Sotorasib is a highly selective inhibitor of KRAS-G12C that reacts with mutant cysteine at position 12 by connecting to a structural feature called the switch II pocket. Sotorasib can induce the production of chemokines, such as C-X-C motif chemokine ligand 10 (CXCL10) and CXCL11, as well as the release of damage-associated molecular patterns (DAMPs), leading to dendritic cell (DC) maturation and activation. The priming of naive T cells to generate cytotoxic T lymphocytes (CTLs) requires mature DC-mediated antigen presentation. The number and function of tumor-targeted CTLs is a prerequisite for the immune system to attack cancer cells. However, the expression of immune checkpoint substances (such as programmed cell death protein 1 [PD-1]) limit the anticancer activity of CTLs, and the administration of anti–PD-1 antibodies reverses this process
Clinical studies have shown some promising antitumor activity of sotorasib or adagrasib in patients with KRAS-G12C-mutated NSCLC that previously had been treated with platinum-based chemotherapy and/or PD-1/PD-L1 blockade. In a phase II trial, 37.1% (46/124) of such NSCLC patients responded to single-agent sotorasib (900 mg/kg, once daily) with a median duration of response of 11.1 months across all PD-L1 expression level subgroups (Table 6) [178]. The activity of sotorasib has been observed in patients with mutations in TP53, STK11, and KEAP1 [178], which are associated with a poor prognosis in NSCLC [179]. Another phase I/II trial involved 129 KRAS-G12C–mutant cancer patients, in which 32.2% of NSCLC, 7.1% of CRC, and 14.3% of other tumor patients showed an objective response to sotorasib (Table 6) [177]. In May 2021, the FDA granted accelerated approval to sotorasib for the treatment of KRAS-G12C–mutated NSCLC. In June 2021, the FDA awarded breakthrough therapy designation to adagrasib for KRAS-G12C–mutated NSCLC based on an unpublished phase I/II study showing that 45% (23/51) of participants responded and 51% (26/51) of them were in stable conditions after using adagrasib (600 mg/kg, twice daily). Although the elimination half-lives of sotorasib (6 hours) and adagrasib (25 hours) are different, they have similar treatment-related adverse events (e.g., nausea, diarrhea, and vomiting). A number of clinical trials are underway to evaluate the antitumor activity of sotorasib or adagrasib alone or in combination with target drugs (docetaxel, pembrolizumab, cetuximab, afatinib, or TNO155) in solid cancers carrying KRAS-G12C mutations (Table 1). Specifically, two phase III trials will test the combination of sotorasib or adagrasib with docetaxel or cetuximab to treat KRAS-G12C–mutant NSCLC or CRC, respectively (NCT04685135 and NCT04793958).
Table 6.
Clinical results of sotorasib therapy in advanced cancer with KRAS-G12C
| Hong et al., 2020 (n = 129) [177] |
Skoulidis et al., 2021 (n = 126) [178] |
|
|---|---|---|
| Characteristics | ||
| Median age (range, year) | 62 (33–83) | 63.5 (37–80) |
| NSCLC (n) | 59 | 126 |
| CRC (n) | 42 | 0 |
| Other solid cancer (n) | 28 | 0 |
| Treatment | Sotorasib (orally 180-960 mg/kg, once daily) | Sotorasib (orally 960 mg/kg, once daily) |
| Efficacy | ||
| Objective response (%) | NSCLC: 32.2; CRC: 7.1; Other: 14.3 | 37.1 |
| Disease control (%) | NSCLC: 88.1; CRC: 73.8; Other: 75.0 | 80.6 |
| Complete response (%) | NSCLC: 0; CRC: 0; Other: 0 | 3.2 |
| Partial response (%) | NSCLC: 32.2; CRC: 7.1; Other: 14.3 | 33.9 |
| Stable disease (%) | NSCLC: 55.9; CRC: 66.7; Other: 60.7 | 43.5 |
| Progressive disease (%) | NSCLC: 8.5; CRC: 23.8; Other: 14.3 | 16.1 |
| Could not be evaluated (%) | NSCLC: 1.7; CRC: 2.4; Other: 7.1 | 1.6 |
| Adverse events | ||
| Any grade (%) | 96.9 | 99.2 |
| Serious (%) | 45.0 | 45.3 |
| Resulting in discontinuation of treatment (%) | 7.0 | 7.1 |
The clinical development of other G12Ci compounds, such as GDC-6036 and D-1553, might provide additional opportunities for selectively targeting advanced solid tumors with KRAS-G12C mutations (Table 1). Notably, the clinical trial of two G12Ci formulations, LY3499446 and JNJ-74699157, has been terminated due to significant toxicity (NCT04165031 and NCT04006301). It remains to be seen whether these toxicities are caused by covalent or noncovalent off-targets.
Pan-KRAS inhibitors
BI-2852 is a pan-KRAS inhibitor that binds between the switch-I and switch-II pockets, thereby blocking the interaction of KRAS protein with GEF, GAP, and its downstream effectors [180]. Early preclinical studies confirmed its activity in blocking the KRAS pathway in NSCLC cells [180]. Using FR054 to inhibit glycosylation reactions further enhances the anticancer activity of BI-2852 against PDAC cells [181], supporting that the hexosamine biosynthesis pathway is a potential target for the treatment of KRAS-mutant cancers [182].
Revolution Medicines utilized a tri-complex technology platform to design a type of RAS(ON) inhibitor. RAS(ON) inhibitors (for example, RM-007 and RM-008) act as molecular glues to mediate protein-protein interactions between different mutant KRAS proteins and an endogenous protein (cyclophilin), thereby inhibiting the binding of mutant KRAS to SOS1 and effector proteins. Thus, the mode of action of RAS(ON) inhibitors is different from that of RAS(OFF) inhibitors, including G12Ci.
More recently, a small-molecule compound called Pen-cRaf-v1 has been identified as a pan-RAS inhibitor capable of targeting G12C and non-G12C RAS mutants to inhibit RAS-effector interaction [183]. Further animal studies are needed to determine the activity, metabolism, and toxicity of pan-KRAS inhibitors before their translational application into clinical medicine.
Others
An interesting trend in recent drug discovery is the selective induction of protein degradation through the proteasome. Proteolysis-targeting chimera (PROTAC) technology can be used to design new drugs that bridge the target protein to E3 ligases, hoping to achieve the target’s degradation to nonfunctional fragments. Whether this approach may be successful for the destruction of oncogenic RAS remains to be explored [184, 185].
Mechanisms of adaptation or resistance to KRAS-G12C inhibitors
Producing new KRAS-G12C protein
The activation of the EGFR-SHP2 pathway maintains newly synthesized KRAS-G12C protein in an active GTP-binding form, thereby leading to the adaptation of KRAS-G12C–mutated cancer cells to ARS-1620 (Fig. 6a) [186]. The cell cycle regulator aurora kinase A (AURKA) binds newly produced KRAS-G12C, which in turn stabilizes the interaction between CRAF and KRAS and mediates subsequent ERK effector signals for cell cycle progression [186]. Consequently, the inhibition of EGFR or AURKA reverses the adaptation of cancer cells to ARS-1620. These preclinical studies provide clues for the development of combined strategies that target EGFR or the cell cycle regulator to delay the development of resistance to KRAS-G12C inhibitors [186]. In addition to AURKA inhibitors (e.g., alisertib), many drugs that are already in clinical use or under development target various cell cycle regulators, especially CDKs. AURKA inhibitors and CDK inhibitors both have shown promise in the treatment of various types of cancer, including KRAS-mutant cancers [187–192].
Fig. 6.

Mechanisms of adaptation or resistance to KRAS-G12C inhibitors. a. Production of new KRAS-G12C protein. Activation of the pathway involving epidermal growth factor receptor (EGFR)–SH2-containing protein tyrosine phosphatase 2 (SHP2)–SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1) is necessary to maintain the newly produced KRAS-G12C protein in an active GTP-bound form, which leads to the adaptation of ARS-1620 through the RAF-MEK-extracellular signal regulated kinase (ERK) pathway. The cell cycle regulator aurora kinase A (AURKA) can further enhance KRAS-G12C–mediated activation of mitogen-activated protein kinase (MAPK) effector pathways. b. Activating wild-type NRAS and HRAS. Multiple receptor tyrosine kinases (RTKs), rather than a single RTK, activate wild-type NRAS and HRAS, leading to acquired resistance to ARS-1620 and sotorasib by the RAF-MEK-ERK and the phosphatidylinositol 3-kinase (PI3K)-AKT-mechanistic target of rapamycin (mTOR) pathways. c. Inducing epithelial-to-mesenchymal transition (EMT). The insulin-like growth factor receptor (IGFR)-insulin receptor substrate 1 (IRS1) pathway mediates PI3K activation in a SHP2-independent manner, leading to acquired resistance to sotorasib or ARS-1620 through snail family transcriptional repressor 1 (SNAI1)-mediated EMT. d. Inducting secondary genetic alterations. An analysis of the genetic alterations of patients with acquired adagrasib resistance showed that 45% of the cases had a putative genetic mechanism of drug resistance. In short, acquired KRAS mutations in drug binding sites or oncogenic hotspots, gain-of-function mutations in the MAPK pathway, and loss-of-function mutations in tumor suppressor genes favor the acquisition of resistance to KRAS-G12C inhibitors
Activating wild-type RAS
Wild-type and mutant RAS subtypes co-exist in the same cell, thus providing a feedback mechanism to reactivate RAS signaling if one of the two RAS pathways is blocked. As such, even if KRAS-G12C is effectively and completely inhibited, residual wild-type RAS (NRAS and HRAS) activity may confer resistance to G12Ci. Multiple RTKs (EGFR, HER2, FGFR, and c-MET), instead of a single RTK, activate wild-type RAS, resulting in acquired resistance to ARS-1620 and sotoracide (Fig. 6b) [193]. This feedback reactivation of wild-type RAS occurs in parallel to the neosynthesis of KRAS-G12C protein, resulting in drug resistance. Since SHP2 and SOS1 are the common nodes of RTK signals, SHP2 inhibitors (e.g., TNO155, SHP099, and RMC-4550) or SOS1 inhibitors (e.g., BAY-293) may either enhance the activity of G12Ci or reverse adaptive resistance. This hypothesis has been confirmed in preclinical models [83, 116, 186, 193–197], and such inhibitors are now entering clinical evaluation (NCT04330664).
Inducing epithelial-to-mesenchymal transition
Epithelial-to-mesenchymal transition (EMT), the process whereby epithelial cells are transformed into mesenchymal cells, is one of the acquired resistance mechanisms to antineoplastic therapies, including TKIs [198]. The induction of EMT in sotorasib-sensitive NSCLC cells by adding TGFβ or using transfection with SNAIL leads to acquired resistance to sotorasib through the activation of the PI3K pathway, which is not associated with significant AKT activation [199]. This suggests that the classical KRAS-PI3K-AKT pathway is not essential for acquired resistance to sotorasib, whereas KRAS-independent PI3K activation favors such resistance in lung cancer cells [200]. In the latter, the insulin-like growth factor receptor (IGFR)-IRS1 pathway, as a key upstream signal, mediates PI3K activation in a SHP2-independent manner, leading to acquired resistance to sotorasib or ARS-1620 in NSCLC cells (Fig. 6c) [199]. Therefore, the clinical optimization of G12Ci may profit from patient stratification based on EMT status.
In other cases, the activation of the PI3K-AKT-mTOR pathway clearly limits the efficacy of G12Ci, such as sotoracide or ARS1620, against NSCLC and PDAC cells [176, 201, 202]. Hence, the mechanisms of PI3K pathway-mediated resistance to G12Ci may depend on the tumor type and the degree of cellular (de)differentiation.
Inducing secondary genetic alterations
Specific secondary genetic alterations may provide additional information to predict G12Ci responses (Fig. 6d). Among 38 patients with KRAS-G12C-mutated solid cancers who received adagrasib monotherapy, 45% displayed a putative genetic mechanism for resistance [203]. The reactivation of RAS-MAPK signaling by 10 genetic alterations affecting the RAS-RAF-MEK-ERK pathway has been described in an NSCLC patient with acquired adagrasib resistance [204]. Secondary KRAS mutations, gain-of-function mutations of the MAPK pathway, loss-of-function mutations in tumor suppressor genes, gene fusion, and gene amplification are conducive to acquired resistance to G12Ci (Fig. 6d).
In vitro and in vivo, KRAS mutation studies further confirmed that the expression of clinically observed switch-II pocket mutations conferred resistance to adagrasib in KRAS-G12C–mutant BaF3 cells (a murine interleukin-3–dependent pro-B cell line) [203, 204]. Thus, the mutations Y96C, Y96D, or Y96S led to resistance to both adagrasib and sotorasib [203, 204]. In contrast, H95D, H95Q, and H95R mutants remained sensitive to sotorasib [203]. Interestingly, a RAS(ON) inhibitor retained its therapeutic index against cells harboring dual G12C/Y96D mutations [204], supporting the notion that RAS(ON) inhibitors mediate the inhibition of oncogenic RAS by a completely different mechanism.
Collectively, these studies provide proof of concept and mechanistic support for a combination therapy that suppresses adaptive genetic alterations. In fact, clinical trials evaluating adagrasib or sotorasib in combination with inhibitors of RTKs, MAPK, SOS1, or SHP2 are underway (NCT04330664 and NCT04185883) to explore novel strategies for overcoming cancer drug resistance [79, 205].
Conclusions and future perspectives
With the development of G12Ci, we now have a tool to directly and irreversibly inhibit KRAS-G12C oncoprotein in patients [17]. However, the excitement spurred by this discovery has been dampened by the fact that the vast majority of patients fail to respond to G12Ci treatment due to primary or acquired resistance [177, 203, 204, 206]. Thus, there are still many outstanding problems to be solved.
First, how can we develop next-generation inhibitors?
Different G12Ci compounds exhibit distinct activities and toxicities. For example, adagrasib was found to bind to KRAS-G12C equally in a series of different cell lines, despite the major variability in downstream signals [176]. The main risk of covalent inhibitors is the possibility of nonspecific reactions with off-target proteins containing cysteine residues [173]. Thus, the molecular identification of proteins accounting for off-target effects of G12Ci may improve structure-based drug design [203]. In addition to organ-mediated drug metabolism, tumor-resident microorganisms can directly decompose chemotherapeutic drugs to cause drug resistance [207]. Hence, optimizing the in vivo pharmacokinetic properties of KRAS inhibitors and evaluating the (in)activity of their metabolites is still an important area for examination. Finally, it remains to be seen whether it is possible to develop additional mutation-specific inhibitors or pan-mutant KRAS inhibitors [208].
Second, how can we design combination therapies?
The design and implementation of strategies to minimize or overcome drug resistance is an important goal of clinical oncology in order to achieve complete and durable clinical responses [209]. The observed tumor heterogeneity and the extensive feedback between RAS and other tumor-related signals may promote drug resistance. The combination of several drugs intercepting different signaling pathways (e.g., upstream signaling, downstream signaling, parallel signaling, cell cycle processes, or immune checkpoints) may prevent or delay the development of therapy resistance, but usually at the cost of increased toxicity [14, 17]. A number of clinical trials combining G12Ci with other established agents (including TKIs and ICIs) are being launched. The design of such trials should avoid random combinations and follow a rationale based on the genetic, metabolic, and immune mechanisms of drug resistance.
Third, how can we develop predictive cancer biomarkers?
Predictive biomarkers can guide treatment decisions by indicating the likely impact of a particular therapy on a patient. Some genetic alterations, especially secondary gene mutation, are associated with the development of adagrasib resistance in patients. In addition to using endoscopic ultrasound-guided fine needle aspiration, the analysis of circulating tumor DNA combined with next-generation sequencing technologies provides a way for examining the genetic characteristics of tumors. Despite these obvious technological advances, circulating metabolites or proteins should not be neglected as potential biomarkers, given that their quantitation would be much more convenient.
In summary, the development of ever more efficient and specific drugs blocking oncogenic RAS must be in parallel with major efforts to avoid and overcome therapeutic resistance. Given the continued efforts of industry, academia, and health care providers, as well as the latest breakthroughs in basic, clinical, and translational research on G12Ci, the prospects look bright.
Acknowledgements
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
Authors’ contributions
All authors contributed to, and read, the manuscript.
Funding
The authors declare no funding support was received for this study.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to publication.
Competing interests
The authors declare no competing interests. GK is a co-founder of EverImmune, Samsara Therapeutics, and Therafast Bio.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Daolin Tang, Email: daolin.tang@utsouthwestern.edu.
Guido Kroemer, Email: kroemer@orange.fr.
Rui Kang, Email: rui.kang@utsouthwestern.edu.
References
- 1.Harvey JJ. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature. 1964;204:1104–1105. doi: 10.1038/2041104b0. [DOI] [PubMed] [Google Scholar]
- 2.Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020;80(14):2969–2974. doi: 10.1158/0008-5472.CAN-19-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 4.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11(19):2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci U S A. 1983;80(8):2112–2116. doi: 10.1073/pnas.80.8.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu K, Birnbaum D, Ruley MA, Fasano O, Suard Y, Edlund L, et al. Structure of the Ki-ras gene of the human lung carcinoma cell line Calu-1. Nature. 1983;304(5926):497–500. doi: 10.1038/304497a0. [DOI] [PubMed] [Google Scholar]
- 7.Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223(4637):661–664. doi: 10.1126/science.6695174. [DOI] [PubMed] [Google Scholar]
- 8.Parada LF, Weinberg RA. Presence of a Kirsten murine sarcoma virus ras oncogene in cells transformed by 3-methylcholanthrene. Mol Cell Biol. 1983;3(12):2298–2301. doi: 10.1128/mcb.3.12.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 10.Sukumar S, Notario V, Martin-Zanca D, Barbacid M. Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature. 1983;306(5944):658–661. doi: 10.1038/306658a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, et al. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A. 2009;106(19):7979–7984. doi: 10.1073/pnas.0900343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014;4(12):1418–1429. doi: 10.1158/2159-8290.CD-14-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J Med Chem. 2020;63(1):52–65. doi: 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 16.Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem. 2020;63(13):6679–6693. doi: 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- 17.Kim D, Xue JY, Lito P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell. 2020;183(4):850–859. doi: 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murugan AK, Grieco M, Tsuchida N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol. 2019;59:23–35. doi: 10.1016/j.semcancer.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer. 2018;18(12):767–777. doi: 10.1038/s41568-018-0076-6. [DOI] [PubMed] [Google Scholar]
- 22.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346(6206):256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook JH, Melloni GEM, Gulhan DC, Park PJ, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun. 2021;12(1):1808. doi: 10.1038/s41467-021-22125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Cao J, Miller SP, Jing H, Lin H. Comparative Nucleotide-Dependent Interactome Analysis Reveals Shared and Differential Properties of KRas4a and KRas4b. ACS Cent Sci. 2018;4(1):71–80. doi: 10.1021/acscentsci.7b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112(3):779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, et al. KRAS4A directly regulates hexokinase 1. Nature. 2019;576(7787):482–486. doi: 10.1038/s41586-019-1832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plowman SJ, Williamson DJ, O'Sullivan MJ, Doig J, Ritchie AM, Harrison DJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23(24):9245–9250. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer. 2017;3(10):686–697. doi: 10.1016/j.trecan.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slebos RJ, Kibbelaar RE, Dalesio O, Kooistra A, Stam J, Meijer CJ, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med. 1990;323(9):561–565. doi: 10.1056/NEJM199008303230902. [DOI] [PubMed] [Google Scholar]
- 31.Pan W, Yang Y, Zhu H, Zhang Y, Zhou R, Sun X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget. 2016;7(7):8373–8388. doi: 10.18632/oncotarget.7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepherd FA, Domerg C, Hainaut P, Janne PA, Pignon JP, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31(17):2173–2181. doi: 10.1200/JCO.2012.48.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douillard JY, Shepherd FA, Hirsh V, Mok T, Socinski MA, Gervais R, et al. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol. 2010;28(5):744–752. doi: 10.1200/JCO.2009.24.3030. [DOI] [PubMed] [Google Scholar]
- 34.Zhu CQ, da Cunha SG, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26(26):4268–4275. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 35.Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13(10):2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 36.Hames ML, Chen H, Iams W, Aston J, Lovly CM, Horn L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer. 2016;92:29–34. doi: 10.1016/j.lungcan.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: A meta-analysis and review. Oncotarget. 2017;8(29):48248–48252. doi: 10.18632/oncotarget.17594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeanson A, Tomasini P, Souquet-Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC) J Thorac Oncol. 2019;14(6):1095–1101. doi: 10.1016/j.jtho.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 39.Sun L, Hsu M, Cohen RB, Langer CJ, Mamtani R, Aggarwal C. Association Between KRAS Variant Status and Outcomes With First-line Immune Checkpoint Inhibitor-Based Therapy in Patients With Advanced Non-Small-Cell Lung Cancer. JAMA Oncol. 2021;7(6):937–939. doi: 10.1001/jamaoncol.2021.0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kartolo A, Feilotter H, Hopman W, Fung AS, Robinson A. A single institution study evaluating outcomes of PD-L1 high KRAS-mutant advanced non-small cell lung cancer (NSCLC) patients treated with first line immune checkpoint inhibitors. Cancer Treat Res Commun. 2021;27:100330. doi: 10.1016/j.ctarc.2021.100330. [DOI] [PubMed] [Google Scholar]
- 41.Nardo G, Carlet J, Marra L, Bonanno L, Boscolo A, Dal Maso A, et al. Detection of Low-Frequency KRAS Mutations in cfDNA From EGFR-Mutated NSCLC Patients After First-Line EGFR Tyrosine Kinase Inhibitors. Front Oncol. 2020;10:607840. doi: 10.3389/fonc.2020.607840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P, et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. doi: 10.1038/ctg.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinemann V, Vehling-Kaiser U, Waldschmidt D, Kettner E, Marten A, Winkelmann C, et al. Gemcitabine plus erlotinib followed by capecitabine versus capecitabine plus erlotinib followed by gemcitabine in advanced pancreatic cancer: final results of a randomised phase 3 trial of the 'Arbeitsgemeinschaft Internistische Onkologie' (AIO-PK0104) Gut. 2013;62(5):751–759. doi: 10.1136/gutjnl-2012-302759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian ZR, Rubinson DA, Nowak JA, Morales-Oyarvide V, Dunne RF, Kozak MM, et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018;4(3):e173420. doi: 10.1001/jamaoncol.2017.3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ako S, Nouso K, Kinugasa H, Dohi C, Matushita H, Mizukawa S, et al. Utility of serum DNA as a marker for KRAS mutations in pancreatic cancer tissue. Pancreatology. 2017;17(2):285–290. doi: 10.1016/j.pan.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 46.Cheng H, Liu C, Jiang J, Luo G, Lu Y, Jin K, et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J Cancer. 2017;140(10):2344–2350. doi: 10.1002/ijc.30650. [DOI] [PubMed] [Google Scholar]
- 47.Cheng H, Luo G, Jin K, Fan Z, Huang Q, Gong Y, et al. Kras mutation correlating with circulating regulatory T cells predicts the prognosis of advanced pancreatic cancer patients. Cancer Med. 2020;9(6):2153–2159. doi: 10.1002/cam4.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haas M, Ormanns S, Baechmann S, Remold A, Kruger S, Westphalen CB, et al. Extended RAS analysis and correlation with overall survival in advanced pancreatic cancer. Br J Cancer. 2017;116(11):1462–1469. doi: 10.1038/bjc.2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schultheis B, Reuter D, Ebert MP, Siveke J, Kerkhoff A, Berdel WE, et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: a multicenter, randomized phase IIb study. Ann Oncol. 2017;28(10):2429–2435. doi: 10.1093/annonc/mdx343. [DOI] [PubMed] [Google Scholar]
- 50.Boeck S, Jung A, Laubender RP, Neumann J, Egg R, Goritschan C, et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48(4):544–548. doi: 10.1007/s00535-013-0767-4. [DOI] [PubMed] [Google Scholar]
- 51.Perets R, Greenberg O, Shentzer T, Semenisty V, Epelbaum R, Bick T, et al. Mutant KRAS Circulating Tumor DNA Is an Accurate Tool for Pancreatic Cancer Monitoring. Oncologist. 2018;23(5):566–572. doi: 10.1634/theoncologist.2017-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Del Re M, Vivaldi C, Rofi E, Vasile E, Miccoli M, Caparello C, et al. Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci Rep. 2017;7(1):7931. doi: 10.1038/s41598-017-08297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe F, Suzuki K, Tamaki S, Abe I, Endo Y, Takayama Y, et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS One. 2019;14(12):e0227366. doi: 10.1371/journal.pone.0227366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48(5):640–646. doi: 10.1007/s00535-012-0664-2. [DOI] [PubMed] [Google Scholar]
- 55.Cerottini JP, Caplin S, Saraga E, Givel JC, Benhattar J. The type of K-ras mutation determines prognosis in colorectal cancer. Am J Surg. 1998;175(3):198–202. doi: 10.1016/s0002-9610(97)00283-3. [DOI] [PubMed] [Google Scholar]
- 56.Yoon HH, Tougeron D, Shi Q, Alberts SR, Mahoney MR, Nelson GD, et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in BRAF-wild-type stage III colon cancers from an adjuvant chemotherapy trial (N0147 alliance) Clin Cancer Res. 2014;20(11):3033–3043. doi: 10.1158/1078-0432.CCR-13-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taieb J, Zaanan A, Le Malicot K, Julie C, Blons H, Mineur L, et al. Prognostic Effect of BRAF and KRAS Mutations in Patients With Stage III Colon Cancer Treated With Leucovorin, Fluorouracil, and Oxaliplatin With or Without Cetuximab: A Post Hoc Analysis of the PETACC-8 Trial. JAMA Oncol. 2016;2(5):643–653. doi: 10.1001/jamaoncol.2015.5225. [DOI] [PubMed] [Google Scholar]
- 58.Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter "RASCAL" study. J Natl Cancer Inst. 1998;90(9):675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 59.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28(3):466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 60.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 61.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19(3):508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 62.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 63.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 64.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30(29):3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 65.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304(16):1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 66.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35(4):559–572. doi: 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahn E, Kang H. Introduction to systematic review and meta-analysis. Korean J Anesthesiol. 2018;71(2):103–112. doi: 10.4097/kjae.2018.71.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 69.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93(1):269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 70.Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62(3):599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 71.Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987;238(4826):542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 72.Bonfini L, Karlovich CA, Dasgupta C, Banerjee U. The Son of sevenless gene product: a putative activator of Ras. Science. 1992;255(5044):603–606. doi: 10.1126/science.1736363. [DOI] [PubMed] [Google Scholar]
- 73.Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 1993;73(3):611–620. doi: 10.1016/0092-8674(93)90146-h. [DOI] [PubMed] [Google Scholar]
- 74.Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280(5366):1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 75.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170(1):17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res. 2015;13(9):1325–1335. doi: 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- 77.Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363(6424):45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 78.Gale NW, Kaplan S, Lowenstein EJ, Schlessinger J, Bar-Sagi D. Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras. Nature. 1993;363(6424):88–92. doi: 10.1038/363088a0. [DOI] [PubMed] [Google Scholar]
- 79.Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021;11(1):142–157. doi: 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24(7):961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 81.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954–960. doi: 10.1038/s41591-018-0024-8. [DOI] [PubMed] [Google Scholar]
- 82.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24(7):968–977. doi: 10.1038/s41591-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A. 2019;116(7):2551–2560. doi: 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin CC, Wieteska L, Suen KM, Kalverda AP, Ahmed Z, Ladbury JE. Grb2 binding induces phosphorylation-independent activation of Shp2. Commun Biol. 2021;4(1):437. doi: 10.1038/s42003-021-01969-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci U S A. 1994;91(26):12706–12710. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wan Y, Kurosaki T, Huang XY. Tyrosine kinases in activation of the MAP kinase cascade by G-protein-coupled receptors. Nature. 1996;380(6574):541–544. doi: 10.1038/380541a0. [DOI] [PubMed] [Google Scholar]
- 88.Vanhaesebroeck B, Perry MWD, Brown JR, Andre F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov. 2021. 10.1038/s41573-021-00209-1. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 89.Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel). 2019;11(10):1618. [DOI] [PMC free article] [PubMed]
- 90.Uprety D, Adjei AA. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat Rev. 2020;89:102070. [DOI] [PubMed]
- 91.Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22(2):399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-Dependent Ferroptosis Drives Tumor-Associated Macrophage Polarization via Release and Uptake of Oncogenic KRAS Protein. Autophagy. 2020;16(11):2069–83. [DOI] [PMC free article] [PubMed]
- 93.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382(3):561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145(6):1449–1458. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. doi: 10.1038/s41467-020-20154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017;9(394):eaal5253. [DOI] [PubMed]
- 97.Sacco A, Federico C, Todoerti K, Ziccheddu B, Palermo V, Giacomini A, et al. Specific targeting of the KRAS mutational landscape in myeloma as a tool to unveil the elicited anti-tumor activity. Blood. 2021. 10.1182/blood.2020010572. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 98.Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560–24570. doi: 10.18632/oncotarget.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11(11):775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cohen SJ, Ho L, Ranganathan S, Abbruzzese JL, Alpaugh RK, Beard M, et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J Clin Oncol. 2003;21(7):1301–1306. doi: 10.1200/JCO.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 101.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22(8):1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 102.Chow LQ, Eckhardt SG, O'Bryant CL, Schultz MK, Morrow M, Grolnic S, et al. A phase I safety, pharmacological, and biological study of the farnesyl protein transferase inhibitor, lonafarnib (SCH 663366), in combination with cisplatin and gemcitabine in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2008;62(4):631–646. doi: 10.1007/s00280-007-0646-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma S, Kemeny N, Kelsen DP, Ilson D, O'Reilly E, Zaknoen S, et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann Oncol. 2002;13(7):1067–1071. doi: 10.1093/annonc/mdf173. [DOI] [PubMed] [Google Scholar]
- 104.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272(22):14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 105.Kazi A, Xiang S, Yang H, Chen L, Kennedy P, Ayaz M, et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin Cancer Res. 2019;25(19):5984–5996. doi: 10.1158/1078-0432.CCR-18-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Manu KA, Chai TF, Teh JT, Zhu WL, Casey PJ, Wang M. Inhibition of Isoprenylcysteine Carboxylmethyltransferase Induces Cell-Cycle Arrest and Apoptosis through p21 and p21-Regulated BNIP3 Induction in Pancreatic Cancer. Mol Cancer Ther. 2017;16(5):914–923. doi: 10.1158/1535-7163.MCT-16-0703. [DOI] [PubMed] [Google Scholar]
- 107.Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 2017;13(1):62–68. doi: 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ayati A, Moghimi S, Salarinejad S, Safavi M, Pouramiri B, Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem. 2020;99:103811. doi: 10.1016/j.bioorg.2020.103811. [DOI] [PubMed] [Google Scholar]
- 109.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 110.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 111.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cai D, Choi PS, Gelbard M, Meyerson M. Identification and Characterization of Oncogenic SOS1 Mutations in Lung Adenocarcinoma. Mol Cancer Res. 2019;17(4):1002–1012. doi: 10.1158/1541-7786.MCR-18-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535(7610):148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 114.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J Med Chem. 2020;63(22):13578–13594. doi: 10.1021/acs.jmedchem.0c01170. [DOI] [PubMed] [Google Scholar]
- 116.Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin Cancer Res. 2021;27(1):342–354. doi: 10.1158/1078-0432.CCR-20-2718. [DOI] [PubMed] [Google Scholar]
- 117.Yen I, Shanahan F, Lee J, Hong YS, Shin SJ, Moore AR, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature. 2021;594(7863):418–423. doi: 10.1038/s41586-021-03515-1. [DOI] [PubMed] [Google Scholar]
- 118.Park S, Kim TM, Cho SY, Kim S, Oh Y, Kim M, et al. Combined blockade of polo-like kinase and pan-RAF is effective against NRAS-mutant non-small cell lung cancer cells. Cancer Lett. 2020;495:135–144. doi: 10.1016/j.canlet.2020.09.018. [DOI] [PubMed] [Google Scholar]
- 119.Monaco KA, Delach S, Yuan J, Mishina Y, Fordjour P, Labrot E, et al. LXH254, a Potent and Selective ARAF-Sparing Inhibitor of BRAF and CRAF for the Treatment of MAPK-Driven Tumors. Clin Cancer Res. 2021;27(7):2061–2073. doi: 10.1158/1078-0432.CCR-20-2563. [DOI] [PubMed] [Google Scholar]
- 120.Desai J, Gan H, Barrow C, Jameson M, Atkinson V, Haydon A, et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. J Clin Oncol. 2020;38(19):2140–2150. doi: 10.1200/JCO.19.02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yuan X, Tang Z, Du R, Yao Z, Cheung SH, Zhang X, et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K-RAS mutant tumors. Mol Oncol. 2020;14(8):1833–1849. doi: 10.1002/1878-0261.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA. 2017;317(18):1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blumenschein GR, Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol. 2015;26(5):894–901. doi: 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig New Drugs. 2012;30(3):1216–1223. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- 125.Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig New Drugs. 2011;29(5):1021–1028. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- 126.Yen I, Shanahan F, Merchant M, Orr C, Hunsaker T, Durk M, et al. Pharmacological Induction of RAS-GTP Confers RAF Inhibitor Sensitivity in KRAS Mutant Tumors. Cancer Cell. 2018;34(4):611–625. doi: 10.1016/j.ccell.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 127.Lebbe C, Italiano A, Houede N, Awada A, Aftimos P, Lesimple T, et al. Selective Oral MEK1/2 Inhibitor Pimasertib in Metastatic Melanoma: Antitumor Activity in a Phase I, Dose-Escalation Trial. Target Oncol. 2021;16(1):47–57. doi: 10.1007/s11523-020-00767-1. [DOI] [PubMed] [Google Scholar]
- 128.Lebbe C, Dutriaux C, Lesimple T, Kruit W, Kerger J, Thomas L, et al. Pimasertib Versus Dacarbazine in Patients With Unresectable NRAS-Mutated Cutaneous Melanoma: Phase II, Randomized, Controlled Trial with Crossover. Cancers (Basel). 2020;12(7):1727. [DOI] [PMC free article] [PubMed]
- 129.de Weger VA, de Jonge M, Langenberg MHG, Schellens JHM, Lolkema M, Varga A, et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer. 2019;120(3):286–293. doi: 10.1038/s41416-018-0355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3(7):742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 131.Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 132.Ma Y, Xu R, Liu X, Zhang Y, Song L, Cai S, et al. LY3214996 relieves acquired resistance to sorafenib in hepatocellular carcinoma cells. Int J Med Sci. 2021;18(6):1456–1464. doi: 10.7150/ijms.51256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018;8(2):184–195. doi: 10.1158/2159-8290.CD-17-1119. [DOI] [PubMed] [Google Scholar]
- 134.Blake JF, Burkard M, Chan J, Chen H, Chou KJ, Diaz D, et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-y l)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J Med Chem. 2016;59(12):5650–5660. doi: 10.1021/acs.jmedchem.6b00389. [DOI] [PubMed] [Google Scholar]
- 135.Weekes C, Lockhart A, LoRusso P, Murray E, Park E, Tagen M, et al. A Phase Ib Study to Evaluate the MEK Inhibitor Cobimetinib in Combination with the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Oncologist. 2020;25(10):833–e1438. doi: 10.1634/theoncologist.2020-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Varga A, Soria JC, Hollebecque A, LoRusso P, Bendell J, Huang SA, et al. A First-in-Human Phase I Study to Evaluate the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2020;26(6):1229–1236. doi: 10.1158/1078-0432.CCR-19-2574. [DOI] [PubMed] [Google Scholar]
- 137.Moschos SJ, Sullivan RJ, Hwu WJ, Ramanathan RK, Adjei AA, Fong PC, et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight. 2018;3(4):e92352. [DOI] [PMC free article] [PubMed]
- 138.Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74(23):7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang M, Jang H, Nussinov R. PI3K inhibitors: review and new strategies. Chem Sci. 2020;11(23):5855–5865. doi: 10.1039/d0sc01676d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6(7):e22769. doi: 10.1371/journal.pone.0022769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cisowski J, Bergo MO. What makes oncogenes mutually exclusive? Small GTPases. 2017;8(3):187–192. doi: 10.1080/21541248.2016.1212689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shapiro GI, LoRusso P, Kwak E, Pandya S, Rudin CM, Kurkjian C, et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Investig New Drugs. 2020;38(2):419–432. doi: 10.1007/s10637-019-00776-6. [DOI] [PubMed] [Google Scholar]
- 144.Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21(4):730–738. doi: 10.1158/1078-0432.CCR-14-1814. [DOI] [PubMed] [Google Scholar]
- 145.Tolcher AW, Kurzrock R, Valero V, Gonzalez R, Heist RS, Tan AR, et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother Pharmacol. 2020;85(4):673–683. doi: 10.1007/s00280-020-04038-8. [DOI] [PubMed] [Google Scholar]
- 146.Mita M, Fu S, Piha-Paul SA, Janku F, Mita A, Natale R, et al. Phase I trial of MEK 1/2 inhibitor pimasertib combined with mTOR inhibitor temsirolimus in patients with advanced solid tumors. Investig New Drugs. 2017;35(5):616–626. doi: 10.1007/s10637-017-0442-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell. 2014;25(2):181–195. doi: 10.1016/j.ccr.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 149.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sarthy AV, Morgan-Lappe SE, Zakula D, Vernetti L, Schurdak M, Packer JC, et al. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007;6(1):269–276. doi: 10.1158/1535-7163.MCT-06-0560. [DOI] [PubMed] [Google Scholar]
- 151.Costa-Cabral S, Brough R, Konde A, Aarts M, Campbell J, Marinari E, et al. CDK1 Is a Synthetic Lethal Target for KRAS Mutant Tumours. PLoS One. 2016;11(2):e0149099. doi: 10.1371/journal.pone.0149099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 153.Mao CQ, Xiong MH, Liu Y, Shen S, Du XJ, Yang XZ, et al. Synthetic lethal therapy for KRAS mutant non-small-cell lung carcinoma with nanoparticle-mediated CDK4 siRNA delivery. Mol Ther. 2014;22(5):964–973. doi: 10.1038/mt.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell Chem Biol. 2018;25(6):705–717. doi: 10.1016/j.chembiol.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 155.Manchado E, Weissmueller S, Morris JP, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534(7609):647–651. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149(3):642–655. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 157.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148(4):639–650. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Carr RM, Vorobyev D, Lasho T, Marks DL, Tolosa EJ, Vedder A, et al. RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat Commun. 2021;12(1):2901. doi: 10.1038/s41467-021-23186-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, et al. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun. 2016;7:11363. doi: 10.1038/ncomms11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wei X, Yang J, Adair SJ, Ozturk H, Kuscu C, Lee KY, et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc Natl Acad Sci U S A. 2020;117(45):28068–28079. doi: 10.1073/pnas.2009899117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168(5):890–903. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, et al. Synthetic Lethal Interaction of SHOC2 Depletion with MEK Inhibition in RAS-Driven Cancers. Cell Rep. 2019;29(1):118–134. doi: 10.1016/j.celrep.2019.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20(9):1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29(33):4658–4670. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 166.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4(4):452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158(1):185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature. 2016;538(7623):114–117. doi: 10.1038/nature19771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem Biol. 2017;24(8):1005–1016. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 170.Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172(3):578–589. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 172.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6(3):316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 174.Saleh K, Kordahi M, Felefly T, Kourie HR, Khalife N. KRAS-targeted therapies in advanced solid cancers: drug the undruggable? Pharmacogenomics. 2021;22(10):587–590. doi: 10.2217/pgs-2021-0045. [DOI] [PubMed] [Google Scholar]
- 175.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 176.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10(1):54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383(13):1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384(25):2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2018;24(2):334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116(32):15823–15829. doi: 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ricciardiello F, Bergamaschi L, De Vitto H, Gang Y, Zhang T, Palorini R, et al. Suppression of the HBP Function Increases Pancreatic Cancer Cell Sensitivity to a Pan-RAS Inhibitor. Cells. 2021;10(2):431. [DOI] [PMC free article] [PubMed]
- 182.Kim J, Lee HM, Cai F, Ko B, Yang C, Lieu EL, et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat Metab. 2020;2(12):1401–12. [DOI] [PMC free article] [PubMed]
- 183.Nomura TK, Heishima K, Sugito N, Sugawara R, Ueda H, Yukihiro A, et al. Specific inhibition of oncogenic RAS using cell-permeable RAS-binding domains. Cell Chem Biol. 2021. 10.1016/j.chembiol.2021.04.013. Online ahead of print. [DOI] [PubMed]
- 184.Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C) Cell Chem Biol. 2020;27(1):19–31. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 185.Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent Sci. 2020;6(8):1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577(7790):421–425. doi: 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang J, Chen P, Liu K, Liu J, Zhou B, Wu R, et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut. 2021;70(5):890–9. [DOI] [PubMed]
- 188.Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu J, et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology. 2017;153(5):1429–1443. doi: 10.1053/j.gastro.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018;8(2):216–233. doi: 10.1158/2159-8290.CD-17-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell. 2018;175(5):1244–1258. doi: 10.1016/j.cell.2018.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353(6297):399–403. doi: 10.1126/science.aae0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ryan MB. Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res. 2020;26(7):1633–1643. doi: 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal. 2019;12(583):eaaw9450. [DOI] [PMC free article] [PubMed]
- 195.Santana-Codina N, Chandhoke AS, Yu Q, Malachowska B, Kuljanin M, Gikandi A, et al. Defining and Targeting Adaptations to Oncogenic KRAS(G12C) Inhibition Using Quantitative Temporal Proteomics. Cell Rep. 2020;30(13):4584–4599. doi: 10.1016/j.celrep.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 196.Solanki HS, Welsh EA, Fang B, Izumi V, Darville L, Stone B, et al. Cell Type-specific Adaptive Signaling Responses to KRAS(G12C) Inhibition. Clin Cancer Res. 2021;27(9):2533–2548. doi: 10.1158/1078-0432.CCR-20-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021;218(1):e20201414. [DOI] [PMC free article] [PubMed]
- 198.Suda K, Murakami I, Yu H, Kim J, Tan AC, Mizuuchi H, et al. CD44 Facilitates Epithelial-to-Mesenchymal Transition Phenotypic Change at Acquisition of Resistance to EGFR Kinase Inhibitors in Lung Cancer. Mol Cancer Ther. 2018;17(10):2257–2265. doi: 10.1158/1535-7163.MCT-17-1279. [DOI] [PubMed] [Google Scholar]
- 199.Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, et al. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26(22):5962–5973. doi: 10.1158/1078-0432.CCR-20-2077. [DOI] [PubMed] [Google Scholar]
- 200.Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011;2(3):261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF, et al. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep Med. 2020;1(8):100131. doi: 10.1016/j.xcrm.2020.100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin Cancer Res. 2019;25(2):796–807. doi: 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
- 203.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. 2021;384(25):2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021;11(8):1913–22. [DOI] [PMC free article] [PubMed]
- 205.Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From the In Vitro Experiments. J Thorac Oncol. 2021;16(8):1321–32. [DOI] [PubMed]
- 206.Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of Resistance to KRAS(G12C) Inhibitors. Cancers (Basel). 2021;13(1):151. [DOI] [PMC free article] [PubMed]
- 207.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357(6356):1156–1160. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Erlanson DA, Webster KR. Targeting mutant KRAS. Curr Opin Chem Biol. 2021;62:101–108. doi: 10.1016/j.cbpa.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 209.Thein KZ, Biter AB, Hong DS. Therapeutics Targeting Mutant KRAS. Annu Rev Med. 2021;72:349–364. doi: 10.1146/annurev-med-080819-033145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.