

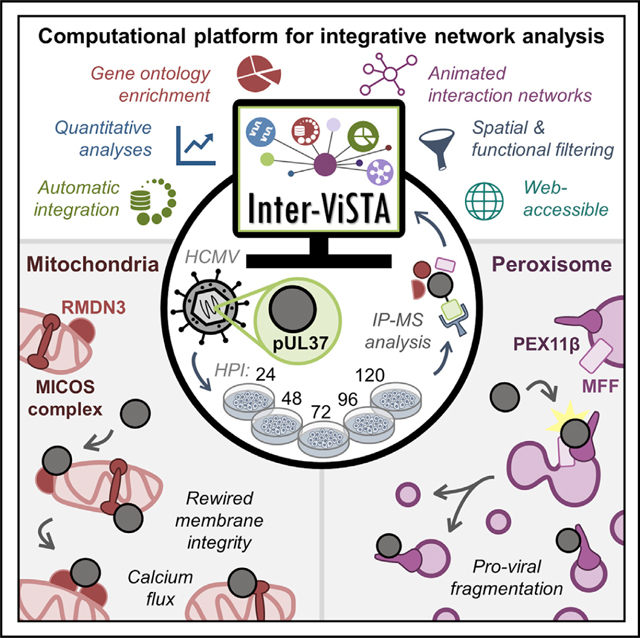
SUMMARY
Nearly all biological processes rely on the finely tuned coordination of protein interactions across cellular space and time. Accordingly, generating protein interactomes has become routine in biological studies, yet interpreting these datasets remains computationally challenging. Here, we introduce Inter-ViSTA (Interaction Visualization in Space and Time Analysis), a web-based platform that quickly builds animated protein interaction networks and automatically synthesizes information on protein abundances, functions, complexes, and subcellular localizations. Using Inter-ViSTA with proteomics and molecular virology, we define virus-host interactions for the human cytomegalovirus (HCMV) anti-apoptotic protein, pUL37×1. We find that spatiotemporal controlled interactions underlie pUL37×1 functions, facilitating the pro-viral remodeling of mitochondria and peroxisomes during infection. Reciprocal isolations, microscopy, and genetic manipulations further characterize these associations, revealing the interplay between pUL37×1 and the MIB complex, which is critical for mitochondrial integrity. At the peroxisome, we show that pUL37×1 activates PEX11β to regulate fission, a key aspect of virus assembly and spread.
In Brief
Inter-ViSTA is a computational platform that accelerates the visualization and analysis of dynamic protein interaction datasets. Using Inter-ViSTA during human cytomegalovirus infection, Federspiel et al. define the temporally and spatially controlled interactome of the viral protein pUL37, revealing virus-host interactions that underlie the pro-viral organelle remodeling of mitochondria and peroxisomes.
Graphical Abstract
INTRODUCTION
Protein interactions, of stable or transient nature, are fundamental to protein functions and govern nearly all biological processes (Wan et al., 2015). Controlling the localization, composition, and frequency of these interactions across both space and time is essential for cellular viability and homeostasis. In addition to regulating protein levels, eukaryotic cells organize their proteomes into organelles, partitioning biological functions and positioning proteins for efficient response to external stimuli. Disrupting or exploiting protein associations is a hallmark of cellular pathologies, well illustrated by virus-host protein interactions during the progression of viral infection. All viruses, despite differences in viral structure, tropism, and replication strategies, rely on protein interactions to infect host cells, produce infectious particles, and spread between organisms (Greco et al., 2014). Similarly, the rapid formation of protein complexes across subcellular space underlies the ability of host cells to induce defense mechanisms, transmitting antiviral signals from the site of pathogen recognition to gene regulation (Crow et al., 2016; McNab et al., 2015; Staring et al., 2018). Protein interactions are thus poised at the interface between a healthy system and infection-induced pathogenesis and are a promising venue for the development of antiviral therapeutics (Greco and Cristea, 2017).
Nearly all viruses have acquired mechanisms to exploit the temporal and spatial sensitivity of host protein interactions (Cook and Cristea, 2019). One striking example of this is during infection with human cytomegalovirus (HCMV), a β-herpesvirus that chronically infects 50%–90% of the population worldwide (Cannon et al., 2010). Upon infection, HCMV orchestrates an array of temporally controlled virus-host protein interactions that remodel organelle structure and proteome organization (Alwine, 2012; Jean Beltran and Cristea, 2014; Jean Beltran et al., 2016). Rewiring begins quickly upon infection with the modulation of apoptotic and immune signaling, including inhibition of the host stress responder TSC2 by the viral protein pUL38 (Moorman et al., 2008) and degradation of the pathogen sensor IFI16 by pUL83 (Li et al., 2013), among others. Later in infection, viral proteins such as pUL99 and pUL32 engage host ESCRTs, clathrin, and the trans-Golgi complex, initiating a global reorganization of secretory organelles into a juxtanuclear viral assembly complex (Das and Pellett, 2011; Moorman et al., 2010). In addition, many other host processes, such as cytoskeletal structure, peroxisomal lipid metabolism, and the unfolded protein response, are hijacked by the virus, yet protein interactions that regulate these changes remain unknown. The continued identification of protein associations, together with their spatiotemporal dynamics and pathogen-driven functions, is critical to understanding the underlying biology of viral infections.
The past decade has seen remarkable growth in the ability to interrogate protein-protein interactions across biological conditions. Mass spectrometry (MS)-based biochemical approaches, such as immunoaffinity purification (IP), chemical crosslinking, and proximity-based labeling (e.g., BioID, APEX, TurboID), have made the generation of large-scale datasets with increasing depth and quality the status quo in proteomic investigations (Greco et al., 2020). However, the interpretation of protein interaction datasets remains an important challenge for biological researchers. A significant barrier is the effective integration and visualization of interaction networks with information related to protein function (e.g., abundance, subcellular localization, gene ontology, and changes across time), which adds depth and understanding to biological investigations. Multiple tools have been developed to facilitate specific aspects of this integration (Keller et al., 2019; Liu et al., 2019; Michalak et al., 2019; Miryala et al., 2018; Rudolph and Cox, 2019; Salamon et al., 2018), which have proved valuable for diverse studies. Yet, these can be manually labor and time intensive and require varying degrees of bioinformatics expertise. There is currently no platform available to the research community that facilitates comprehensive interactome analysis in a user-friendly and interactive manner.
To address this need for integrating spatial, functional, and temporal protein interaction data, we built a web-accessible analysis program called Inter-ViSTA (Interaction Visualization in Space and Time Analysis). Inter-ViSTA enables users to quickly and efficiently visualize their own proteomic data via animated interaction networks, while automatically integrating quantitative and qualitative proteomic information across published databases. Our platform can be used to analyze multiple baits, time points, and experiments, all with minimal manual workup. As proof of concept, we first demonstrate the broad utility of Inter-ViSTA by analyzing several published IP-MS and proximity-labeling interactome datasets. Then, we leverage Inter-ViSTA to examine virus-host protein interactions during HCMV infection, focusing on the spatial-temporal dynamics of the viral immuneevasion protein pUL37 exon 1 (pUL37×1).
Also known as the viral mitochondrial inhibitor of apoptosis (vMIA), pUL37×1 is a multifunctional, transmembrane protein required for HCMV replication (Kouzarides et al., 1988; McCormick et al., 2003; Tenney and Colberg-Poley, 1991). Expressed rapidly upon infection, the majority of pUL37×1 traffics from the endoplasmic reticulum (ER) to the outer mitochondrial membrane (OMM) (Bozidis et al., 2010). Here, pUL37×1 acts quickly to inhibit host immune response, block apoptotic signaling, regulate calcium flux, and induce mitochondrial fragmentation. While it is known pUL37×1 functions rely on interactions with host proteins, only a few have been identified, including inhibition of the apoptotic factor BAX (Zhang et al., 2013) and association with the antiviral protein MAVS (Castanier et al., 2010). Additionally, a subset of pUL37×1 localizes to peroxisomes, causing fragmentation and blocking peroxisomal MAVS signaling (Magalhães et al., 2016), but the mechanisms underlying peroxisome remodeling remain unknown. Moreover, pUL37×1 is an essential viral protein and continually expressed during HCMV infection, yet no late infection functions have been identified and no study has characterized its global protein interactions.
By using Inter-ViSTA to bridge the gap between proteomic discovery and biological application, we define the dynamic interactome of pUL37×1 across subcellular space and infection time. Our work reveals temporally regulated virus-host protein interactions at the mitochondria, with increasing peroxisomal associations as infection progresses, broadening the scope of the prior model that pUL37×1 functions primarily early in infection. Reciprocal isolations, co-localization microscopy, and CRISPR-based genetic perturbations further characterize the functions of these protein interactions, shedding new light on the mechanisms underlying pUL37×1-mediated organelle remodeling during infection. We examine the pro-viral associations between pUL37×1 and integral mitochondrial membrane proteins, including members of the prohibitin and mitochondrial intermembrane space bridging (MIB) complexes. We also uncover a role for pUL37×1 in regulating peroxisome morphology late in HCMV infection, acting in concert with the peroxisomal fission factor PEX11β. Altogether, our results demonstrate that the pro-viral roles of pUL37×1 span the entirety of HCMV infection, rewiring both mitochondria and peroxisomes via an array of previously unrecognized virus-host interactions.
RESULTS
Developing Inter-ViSTA as a Platform for Visualizing Spatiotemporal Protein Interactions
The challenge of integrating experimental interactome datasets with the wealth of information in proteomic databases inspired us to construct Inter-ViSTA, a computational, web-accessible platform that enables users to discover underlying spatial, temporal, and functional patterns in interactome studies. Our tool (available online at https://github.com/cristealab/Inter-ViSTA and intervista.princeton.edu) uses cutting-edge network algorithms and programmatic database access to deliver intuitive data visualization, automatically synthesizing information from multiple experiments (e.g., multiple baits, time points, or conditions) and public databases (Figure 1A). Based on user input, our platform generates interactive protein networks that allow hands-on exploration of quantitative analyses. By simultaneously connecting experimental data with multiple proteomic databases, Inter-ViSTA allows users to readily interpret interaction datasets and reveal their biological significance.
Figure 1. Inter-ViSTA Is a Computational Platform for Real-Time Analysis and Visualization of Interactome Datasets.

(A) A schematic depicting the Inter-ViSTA computational interface, from user input of experimental bait-prey pairs to data visualization and export.
(B) Inter-ViSTA animated network key, with nodes and edges color coded by interaction characteristics.
(C) Single-frame shots of the networks generated by Inter-ViSTA from the cyclin interactions by Pagliuca et al. (2011). The cyclins used as baits are shown adjacent to the indicated cell-cycle stage. Key interaction groups shared between baits are highlighted in blue.
(D) Protein interactions of Us9 during PRV infection (Kramer et al., 2012) clustered by temporal abundance via Inter-ViSTA. Notable GO terms are indicated.
(E) Inter-ViSTA identified organelle-specific abundance profiles of OPRD1 interactors during internalization and trafficking (Lobingier et al., 2017). Experiment temporality is indicated at right.
See also Figures S1 and S2, Videos S1, S2, and S3, and Table S1.
As input, Inter-ViSTA requires specificity-scored bait-prey interaction data. Optionally, this input may include multiple experiments (e.g., time points or conditions), curated subcellular localization information, and/or quantitative information on protein abundance. The user defines a specificity cutoff between zero and one, and the specificity scores can be derived from sources compatible with various biochemical approaches (e.g., IP-MS, BioID, XL-MS). For each pair of proteins with an interaction score that surpasses the cutoff, Inter-ViSTA assigns an “interaction spell” with an onset and terminus. The onset is the first time the score surpasses the cutoff and the terminus is the last time it remains above threshold. To account for small changes between adjacent time points, Inter-ViSTA automatically relaxes the user-provided cutoff by 0.1. If an interaction score drops below the relaxed cutoff and later reappears, it is assigned multiple onsets and termini.
To supplement the user-provided dataset, Inter-ViSTA automatically queries external databases, retrieving known protein interactions from STRING (Szklarczyk et al., 2017), subcellular localizations and gene ontology (GO) terms from UniProt (Consortium, 2019), and protein complexes from CORUM (Giurgiu et al., 2019). Using a pre-loaded or user-defined list of background genes, GO enrichment analysis is performed using topGO (Alexa and Rahnenführer, 2018) and CORUM complexes are extracted if ≥40% of subunits are present. Additionally, if reference proteome data are provided alongside interactions, Inter-ViSTA can normalize interactions to protein abundances, thus testing whether alterations to protein levels drive changes in interactions. Ultimately, Inter-ViSTA synthesizes all this information to build an interactive interface that allows users to identify and explore patterns in their data through quantitative, functional, and network analyses.
A major output of Inter-ViSTA is an animated network graph that highlights dynamic interaction properties. Each network describes bait and prey characteristics, indicated by node and edge styles (Figure 1B). Clicking on nodes and edges further reveals attributes such as gene names, confidence scores, complex membership, etc. The network scans across user-provided time points (or conditions) in real time, and the user can filter the network by localization, STRING confidence scores, functional annotations, species, and proteins shared between baits. As an additional layer of analysis, Inter-ViSTA computes and outputs reports describing trends in quantitative data (e.g., conditional protein abundance profiles), including interaction spell plots, temporal clusters, and localization- or function-specific interaction durations. Total analysis time runs on the order of minutes, significantly reducing the time it takes to link proteomic data with meaningful biological insight. Inter-ViSTA annotations and analyses are available for download, allowing the user to import data into other programs if further hands-on analysis is desired. Altogether, Inter-ViSTA facilitates tangible exploratory analyses to uncover the dynamics of specific biological processes or organelles over intervals of network activity.
Proof of Concept: Validating Inter-ViSTA Using Existing IP and Proximity Labeling Interaction Datasets
To validate the performance of Inter-ViSTA, we first analyzed published interaction datasets encompassing cell-cycle progression (Pagliuca et al., 2011), pseudorabies virus (PRV) infection (Kramer et al., 2012), and G-protein-coupled receptor trafficking (Lobingier et al., 2017). These were generated by multi- or single-bait IP-MS and APEX-based proximity labeling, respectively, thus challenging Inter-ViSTA with a range of biochemical approaches. In minutes and with minimal user effort, Inter-ViSTA captured the biological depth presented in these studies, highlighting its robustness and utility (Figures 1C–1E).
Briefly, the study by Pagliuca et al. defined the interactomes of the cyclins E1 (CCNE1), A2 (CCNA2), and B1 (CCNB1) at each phase of the cell cycle (G1, S, G2, M), uncovering distinct, phase-dependent protein interactions in accordance with cyclin function (Pagliuca et al., 2011). Inter-ViSTA built animated networks that emphasized the temporality of each baits’ unique interactome (Figure 1C; Video S1). This included a “hand-off” of key interactors (e.g., the cyclin regulator SKP2) from A2 to B1 as phase G2 cycled to M, the functional redundancy of E1 with A2 interactions in G1 and S phases, and increasing associations between A2 and the cyclin inhibitors CDKN1A-C during the switch to S phase (Figure S1A; Table S1A–S1D).
Next, upon analyzing interactions with the PRV trafficking protein Us9 (Kramer et al., 2012), Inter-ViSTA pointed to functional changes in synaptic vesicle and intracellular transport associations across infection time (Figure 1D; Video S2). Among Us9 interactors, SNARE and endocytic coat complexes were highlighted, confirming the findings of Kramer et al. (2012) (Figure S1B). Additionally, Inter-ViSTA identified previously unreported temporal patterns in functional clusters that support the current model of PRV axonal transport, which relies on endosomal sorting, the motor protein KIF1A, and local protein synthesis (Koyuncu et al., 2013; Kramer et al., 2012). These included enrichment of coatomer subunits following 8 h post-infection (hpi), a sustained increase in calcium-dependent exocytosis proteins, transport and motor proteins (e.g., KIF1A) that peak late in infection, and ribosomal subunits that are maintained throughout infection (Figure 1D; Table S1E and S1F). The Inter-ViSTA temporal analysis pointed to additional players in PRV trafficking, such myosins and dyneins.
Finally, Inter-ViSTA effortlessly recapitulated the spatial-temporal dynamics of the delta-type opioid receptor (OPRD1) degradation pathway reported by Lobingier et al. (2017), demonstrating its ability to visualize localization-specific interactions across biological time (Figure 1E; Video S3; Figure S2). Beyond confirming OPRD1 movement from the plasma membrane to lysosome, we identified distinct categories of protein interaction spells that give context to mechanisms underlying OPRD1 internalization, trafficking, and degradation. This included immediate interactions with proteins involved in membrane curvature and clathrin coat assembly (e.g., PICALM, EPN2), sustained interactions with endosomal sorting and trafficking proteins (e.g., SORT1, WASHC5), and late interactions with ubiquitination (e.g., UBQLN2, WWP2) and lysosomal degradation (e.g., ATP6V1A, TMEM55B) players (Figure S2B; Table S1G–S1I). In each validation described above, Inter-ViSTA rapidly provided the biological context needed to interpret the interactome datasets, reducing manual analysis time from days to minutes and expanding upon the original authors’ findings.
Exploring HCMV-Host Protein Interactions with Inter-ViSTA
Having demonstrated that Inter-ViSTA is an effective tool for analyzing existing datasets, we next used our platform to interrogate virus-host protein interactions during the 5-day cycle of lytic HCMV infection. As described above, HCMV drives extensive changes in cellular organelles and proteome organization (Figure 2A), and this relies on the careful orchestration of protein interactions across both space and time (Alwine, 2012; Jean Beltran et al., 2016). Given its essential roles in immune and apoptotic signaling, we examined the viral protein pUL37×1, which is encoded by the first exon of the HCMV gene UL37 (Kouzarides et al., 1988; Tenney and Colberg-Poley, 1991). Despite the knowledge that pUL37×1 is required for HCMV replication and is dually localized to mitochondria and peroxisomes, its protein interactome has not been elucidated and many of its critical functions remain unexplained.
Figure 2. Investigation of pUL37×1 Spatial and Temporal Dynamics across the Replication Cycle of HCMV.
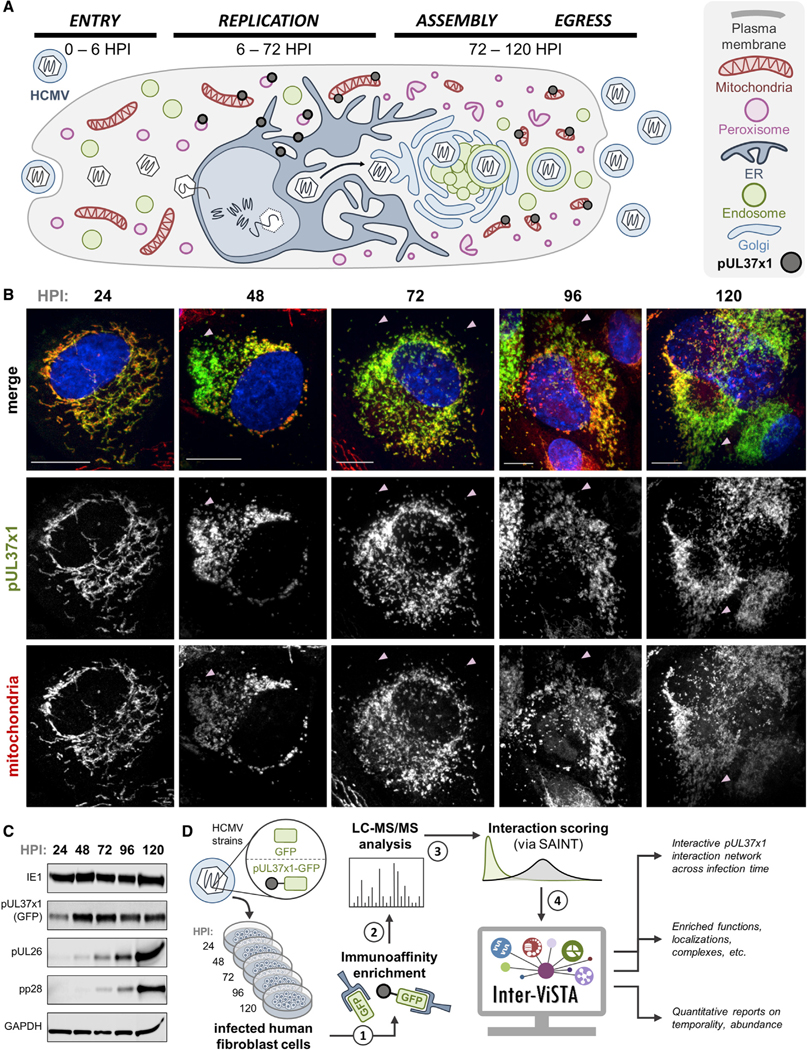
(A) Schematic representing pUL37×1 localization and HCMV biology, highlighting spatial-temporal changes to organelles. pUL37×1 is translated in the ER around6 hpi, quickly localized to mitochondria and peroxisomes, and remains expressed throughout infection.
(B) Fluorescence microscopy images (z stack maximum projections) of human fibroblast cells infected with pUL37×1-GFP virus (green) and labeled for mitochondria (red, MitoTracker). Arrows indicate likely peroxisomal pUL37×1 localization (pUL37×1 puncta not colocalized with mitochondria). Scale bars are 10 μm.
(C) Western blot of pUL37×1-GFP-infected cell lysates across infection time, with antibodies against viral proteins. Consistent protein loading is indicated by GAPDH.
(D) Workflow schematic of the investigation performed in this study. Two biological replicates were performed for each time point.
See also Table S2
Using a GFP-tagged virus strain (BADinUL37GFP [Moorman et al., 2010]) in combination with biochemical, microscopy, and computational approaches, we captured pUL37×1 interaction dynamics across HCMV infection. To first confirm that the GFP virus exhibits the expected temporality of viral protein production, we immunoblotted for different temporal classes of HCMV proteins, including IE1 (immediate-early), pUL26 (delayed-early), and pUL99 (late) (Figure 2C). We also imaged pUL37×1-GFP in tandem with mitochondria throughout infection in human fibroblasts, demonstrating its proper localization to mitochondria, enrichment at the OMM, and induction of mitochondrial fragmentation (Figure 2B). Beginning around 48 hpi, a subset of pUL37×1-GFP appeared distinct from mitochondria, likely representing sites of peroxisomal localization.
Following verification of virus viability, we conducted IP of pUL37×1 using an anti-GFP antibody, at time points spanning HCMV genome replication, virion assembly, and virus egress (24, 48, 72, 96, and 120 hpi, Figure 2D). Parallel control isolations were performed with a virus strain expressing GFP alone (BADinGFP). Captured proteins were then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and interaction specificity was assessed using SAINT (Choi et al., 2011). For Inter-ViSTA analysis, we annotated protein localizations by referencing UniProt and our previously curated dataset that defined protein localizations throughout HCMV infection (Jean Beltran et al., 2016) (Table S2A). A final file containing bait-prey pairs, protein abundances, localizations, and specificity scores was uploaded to Inter-ViSTA (Table S2B and S2C), which generated an animated interaction network replete with functional, protein complex, and quantitative information that captured the temporality of the HCMV infectious cycle (Video S4).
Inter-ViSTA Uncovers Temporally Dynamic pUL37×1 Interactions with Host Proteins Regulating Mitochondrial Integrity, Apoptosis, and Antiviral Response
Given the functions of pUL37×1 in inhibiting apoptosis and immune signaling and inducing mitochondrial fragmentation, we expected most protein interactions to occur early in infection and at the mitochondria. Indeed, we found over 85% of specific interactions were with mitochondrial proteins and only a small fraction was peroxisomal (Figures 3A and 3B). Our data also revealed that the number of pUL37×1 interactors increases approximately 5-fold from 24 to 120 hpi (Figures S3A and S3B). Highlighting its understudied roles late in infection, we find that most pUL37×1 interactions begin after 48 hpi and are temporally dynamic, being specific at three or fewer time points (Figure 3B; Figure S3C). As HCMV broadly upregulates translation (Jean Beltran et al., 2016; Vincent et al., 2016; Weekes et al., 2014), we asked whether the temporal changes in pUL37×1 associations were caused by increased protein abundances. After using Inter-ViSTA to normalize to protein abundance, we determined that pUL37×1 interactions are not primarily driven by changes in protein levels but rather by the specific recruitment or dissociation of interacting partners (Figure S3D; Table S3C).
Figure 3. pUL37×1 Protein and Protein Complex Interactions Are Temporally Controlled during HCMV Infection.
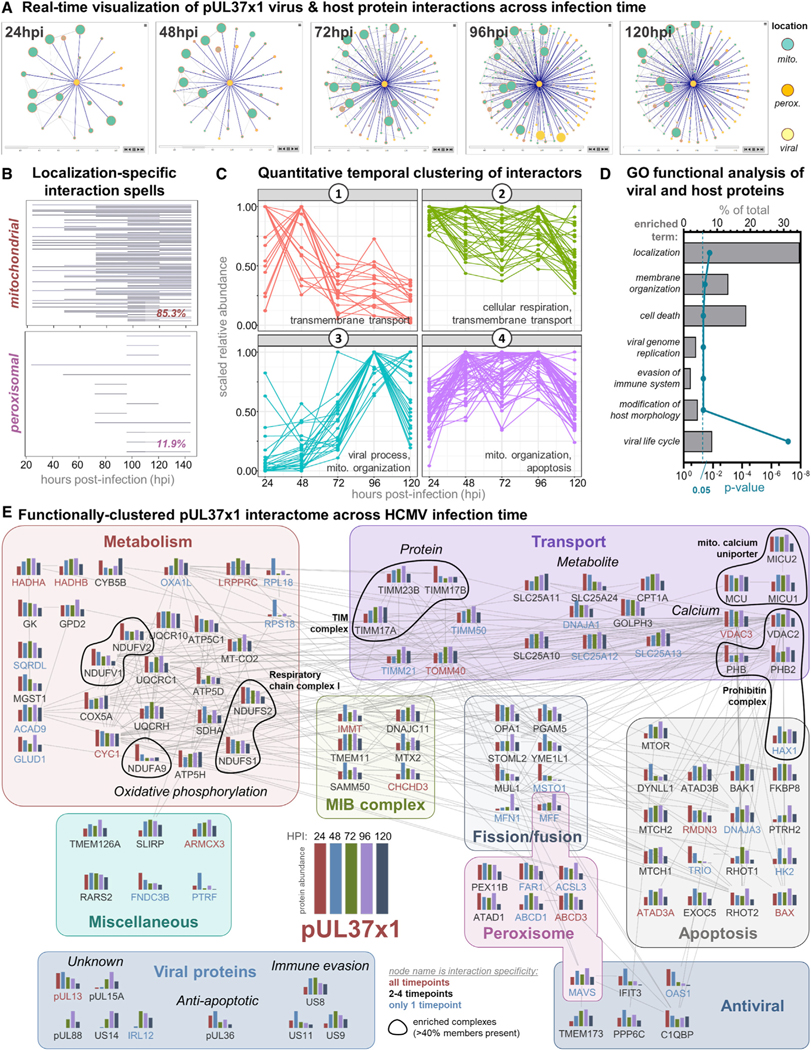
(A) Single time point shots of the animated pUL37×1 interaction network generated by Inter-ViSTA, with a specificity cutoff of 0.9 (see Video S4 for animation).
(B) Graphical representation of pUL37×1 prey protein interaction spells organized by organelle localization. Each line represents an interacting protein, and the line width represents the duration of interaction with pUL37×1. The percentage of specific pUL37×1 interactors localized to either the mitochondria or peroxisome is indicated at the lower right.
(C) Enriched GO terms of pUL37×1 interactors across all time points, plotted as the percentage of proteins out of the total interactome (upper x axis). The corrected p value for each term is plotted on the lower x axis. A complete list of GO terms is in Table S3D.
(D) pUL37×1 interactors cluster into four temporal groups by quantitative abundance profiles. Select GO terms enriched in each cluster are indicated.
(E) The pUL37×1 interactome as annotated by Inter-ViSTA and assembled in Cytoscape. Proteins are clustered by function, and edges represent STRING-based protein interactions. Bar graphs depict the association abundance at each time point. Protein names indicate the duration of passing the specificity cutoff: all time points (red), 2–4 (black), or only one time point (blue). Black circles denote complex members.
Inter-ViSTA highlighted four temporal clusters of protein interactions: decreasing (cluster 1), maintained (cluster 2), increasing (cluster 3), and specifically enriched late in infection (cluster 4) (Figure 3C; Table S3A and S3B). GO analyses of each cluster revealed functions in accordance with the known roles of pUL37×1 in modulating mitochondrial calcium flux, integrity, and metabolism (Figures 3C and 3D; Table S3D), suggesting these associations contribute to pUL37×1 functions. For example, cluster 2 includes proteins that regulate calcium flux (e.g., VDACs, MCU complex) and metabolism (e.g., respiratory chain complex I, ATP synthases ATP5C1/5H). Among those enriched during virus replication and assembly (cluster 4), we find proteins involved in mitochondrial integrity (e.g., TIM/TOM complex, RHOT1) and apoptotic and immune signaling (discussed below). Perhaps expected, cluster 3 includes viral proteins known to increase in abundance throughout infection. However, several of these are functionally uncharacterized, such as pUL15a, pUL85, and pUL13, which also localizes to mitochondria (Jean Beltran et al., 2016), pointing to the presence of untapped processes at the core of HCMV replication. In line with this, the peroxisomal functions of pUL37×1 had not been investigated past 24 hpi, and we find most peroxisome protein interactions in clusters 2 and 4, suggesting pUL37×1 participates in previously unrecognized peroxisomal processes late in infection.
Mitochondrial complexes and their associated proteins are prominent within the pUL37×1 interactome (Table S3E). These likely contribute to two major pUL37×1 functions: (1) control of mitochondrial metabolism, such as the mitochondrial calcium uniporter (MCU) mentioned above and the respiratory chain complex I, which controls cellular respiration and oxidative stress (Sharma et al., 2009), and (2) manipulation of membrane integrity and fragmentation. The latter category includes the TIM complex, which facilitates the ER-to-mitochondria transfer of pUL37×1 (Bozidis et al., 2008), and the evolutionarily conserved MIB complex. The MIB, composed of both the MICOS and SAMM50 complexes, bridges the inner and OMMs to maintain mitochondrial organization, integrity, and cristae structure (Kozjak-Pavlovic, 2017). MIB proteins were among the most continuous and specific pUL37×1 interactors, pointing to a function at the core of HCMV-induced mitochondrial manipulation. In addition, pUL37×1 associated with the mitochondrial prohibitin complex, an essential player in cristae morphogenesis, membrane scaffolding, and mitochondrial fusion (Merkwirth and Langer, 2009). Notably, we found that pUL37×1 associates with 80% of prohibitin complex members, including the core components PHB/2, the PHB interactor OPA1, and MFN1, all key players in mitochondrial fusion. As these interactions are maintained throughout infection (cluster 2), it is possible that pUL37×1 antagonizes prohibitin proteins to compromise the balance between fission and fusion, resulting in the fragmentation caused by infection (Figure 2B). Further supporting this, we identified interactions with mitochondrial fission proteins, including the OPA1 protease YME1L1 and essential fission factor, MFF.
Combining the functional, localization-specific, and complex information provided by Inter-ViSTA, we further visualized the pUL37×1 interactome by constructing a static, functionally clustered network in Cytoscape (Shannon et al., 2003) (Figure 3E). Evident in our network are associations reflective of pUL37×1 suppression of apoptotic signaling, including the previously reported interactor BAX (Zhang et al., 2013). In addition, we identified other apoptotic proteins as highly specific pUL37×1 interactors, including the anti-apoptotic ATPase ATAD3A (Fang et al., 2010), the Rho GTPase RHOT1 (MIRO1) (Fransson et al., 2003), and the calcium-regulatory protein RMDN3 (PTPIP51) (Lv et al., 2006). Like BAX, ATAD3A and RMDN3 passed specificity criteria at all infection time points, while RHOT1 peaks midway through infection. These stable interactions suggest that mechanisms other than BAX inhibition also support pUL37×1 regulation of apoptosis and calcium flux.
Our network also revealed interactions reflective of the roles of pUL37×1 in inhibiting host immune response. Among these were the host antiviral proteins MAVS and its downstream component STING (TMEM173). pUL37×1 is known to inhibit the MAVS antiviral response at both mitochondria (Castanier et al., 2010) and peroxisomes (Magalhães et al., 2016), but an interaction with the ER-localized STING has not been reported. Interestingly, pUL37×1 also interacted with another HCMV protein inhibitor of MAVS and STING, US9 (Choi et al., 2018). Considering that pUL37×1 interactions with US9, MAVS, and STING have similar temporal profiles, continuing throughout virus replication and assembly (Figure 3E), these four proteins may interact in concert to suppress host defense across subcellular space. We also found an association between pUL37×1 and the stress response kinase mTOR, which is inhibited during infection by pUL38 (Moorman et al., 2008). As pUL38 is not among the viral proteins we detected, the pUL37×1-mTOR interaction may represent an additional pro-viral mechanism to subvert host homeostasis. Overall, the interactome we report here reveals a striking range of interactions across both space and time, highlighting the multifaceted roles of pUL37×1 in suppressing host defense and rewiring mitochondrial biology to benefit HCMV infection.
Validation and Analysis of Mitochondrial pUL37×1 Interactions
Our finding that pUL37×1 forms associations with a broad range of host proteins involved in apoptosis, immune signaling, and mitochondrial biology led us to further examine the temporality and spatial specificity of these interactions. Using parallel reaction monitoring (PRM) targeted MS, we built a peptide library for pUL37×1 interactors of interest, monitoring three to five unique signature peptides per protein (Figure 4A). By coupling PRM to IP of pUL37×1-GFP, we measured the abundance of host and viral pUL37×1 interactors across infection time, spanning the mitochondrial functions described above. Indeed, this quantitative approach not only confirmed these interactions, but also their temporality (Figure 4A; Table S4). Proteins involved in mitochondrial structure and integrity were most enriched at 48–72 hpi, corresponding to the time of mitochondrial fragmentation. The pUL37×1 association with BAX was retained throughout infection, which agrees with both our interactome data and prior reports (Zhang et al., 2013). Proteins that modulate host calcium flux, immune signaling, and apoptosis were also maintained during infection.
Figure 4. Validation of Temporal pUL37×1 Mitochondrial Interactions.
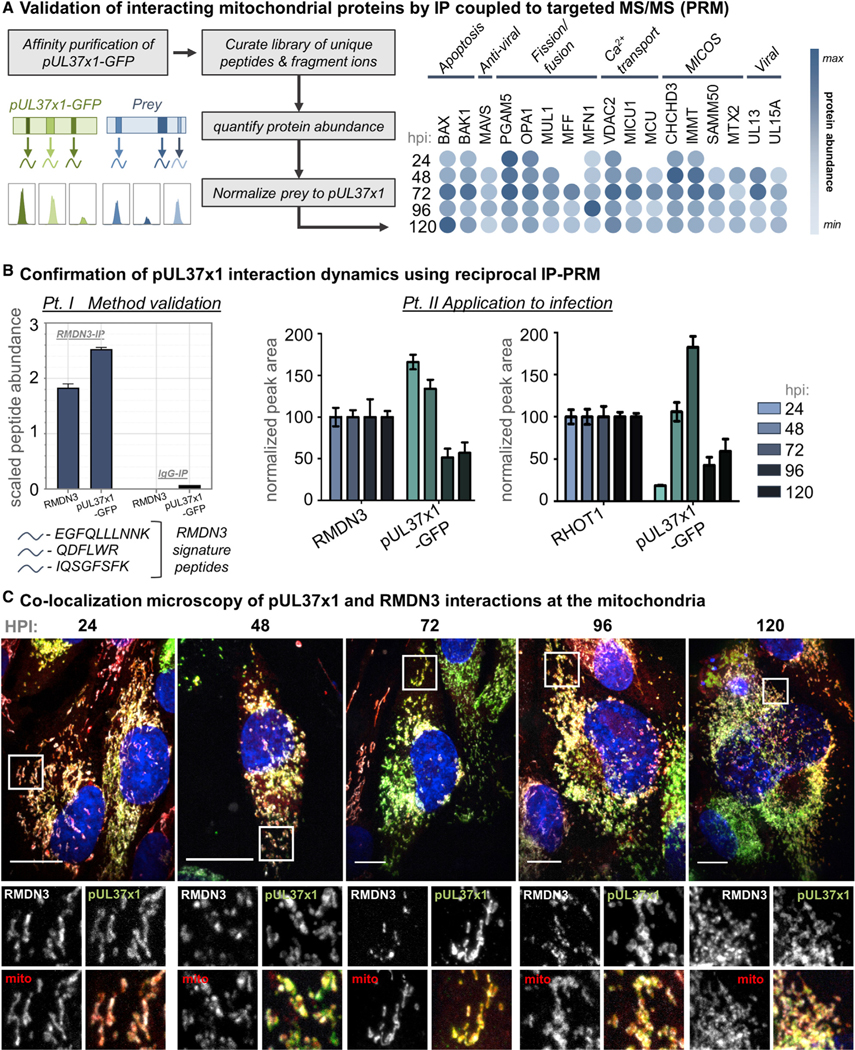
(A) Left: design of a parallel reaction monitoring (PRM) assay for the sensitive validation of pUL37×1 interactions. Right: heatmap depicting the minimum to maximum association with pUL37×1 (bait) for each prey protein, determined by PRM. White space indicates time points when the protein was not detected as interacting with pUL37×1.
(B) Reciprocal isolations of endogenous pUL37×1 interactors, RMDN3 and RHOT1, followed by PRM analyses to validate temporal interactions with pUL37×1. Left: an IgG control was used for each IP, confirming the specificity of endogenous IP. Right: isolation of RMDN3 and RHOT1 was performed across infection and pUL37×1 co-isolation was monitored by PRM. Error bars denote SEM from measuring three unique peptides per protein.
(C) Immunofluorescence microscopy images (z stack maximum projections) of infected human fibroblast cells co-labeled with four fluorescent probes: an antibody against endogenous RMDN3 (white), pUL37×1-GFP (green), MitoTracker (red), and DAPI (blue). Individual channels from a region of interest (white box) are shown below each merged image. Scale bars represent 10 μm.
See also Table S4.
Given the reported roles for RHOT1 in regulating mitochondrial dynamics (Fransson et al., 2003; Modi et al., 2019; Safiulina et al., 2019) and RMDN3 in calcium homeostasis and apoptosis (Lv et al., 2006; De vos et al., 2012), these proteins may be key to pUL37×1 functions during infection. We aimed to further validate their associations using reciprocal IP-PRM analyses, isolating endogenous RHOT1 or RMDN3 and using our PRM peptide library to quantify co-isolated pUL37×1 (Figure 4B). Despite repeated attempts, we were not able to isolate endogenous RMDN3 at 24 hpi. As we previously noted that RMDN3 levels increase during infection (Murray et al., 2018), this is likely because the protein levels of RMDN3 were too low early in infection for the antibody to effectively bind its antigen. In agreement with this, from 48 hpi onward we were able to IP RMDN3 and detect its association with pUL37×1. Supporting the temporal interactions indicated by Inter-ViSTA, we find that RMDN3 and RHOT1 associations with pUL37×1 peak at 48 and 72 hpi, though pUL37×1 is detected at every time point (Figure 4B). We further monitored the RMDN3-pUL37×1 association by microscopy across infection time (Figure 4C). Our proteomic data suggested that, although RMDN3 was a specific pUL37×1 interactor at all time points, this association was more pronounced early in infection. Accordingly, RMDN3 and pUL37×1 co-stained mitochondrial membranes early in infection but, beginning at 72 hpi, RMDN3 localization became restricted while pUL37×1 continually coats the outer membrane. Considering the multifunctionality of both pUL37×1 and RMDN3, this temporal interaction may link the manipulation of calcium flux to host apoptotic signaling early in infection, controlling mitochondrial dynamics to support HCMV infection.
pUL37×1 Engages the MIB Complex to Facilitate HCMV Production
Our IP-PRM assay also confirmed the temporality of the interactions between pUL37×1 and MIB complex members, showing a sharp increase in association at 48 hpi (Figure 5A). Notably, we found associations with nearly all MIB proteins known to localize near the OMM, including the MICOS (IMMT, CHCHD3, DNAJC11, TMEM11) and SAMM50 (MTX2, SAMM50) components. The single exception was MTX1, which was detected but did not pass our stringent specificity filtering. Moreover, pUL37×1-MIB interactions coincide with HCMV-induced mitochondrial fragmentation, which peaks at 48 hpi (Figure 2B), and the MIB complex is central to regulating mitochondrial structure (Kozjak-Pavlovic, 2017; Tang et al., 2020).
Figure 5. The Mitochondrial Intermembrane Space Bridging Complex Associates with pUL37×1 and Is Required for HCMV Infection.

(A) Left: schematic of MICOS and SAMM50 complex proteins detected as specific pUL37×1 interactors. Right: relative temporal abundances of protein interactions between MIB complex members and pUL37×1, all assigned to cluster 4 by Inter-ViSTA. Protein colors correspond with the schematic at left.
(B) Immunoblot of the IMMT reciprocal isolation at 96 hpi stained for GFP and IMMT, confirming pUL37×1 association. Arrowhead denotes pUL37×1 band; the asterisk denotes IgG heavy chain bands.
(C) IP-PRM of the IMMT reciprocal isolation further confirmed the temporality of IMMT-pUL37×1 interactions. Left: an IgG control was used to confirm the specificity of the endogenous IP. Right: isolation of IMMT was done at five time points of infection, and the abundance of co-isolated pUL37×1 was monitored by PRM. Error bars denote SEM from three unique peptides per protein.
(D) CRISPR-mediated knockout of CHCHD3 was validated by PRM (n = 6 unique peptides), leading to a mean ∼55% decrease in protein abundance (left). Viral titers from the CHCHD3 CRISPR and control cells (right: ***p ≤ 0.001, n = 3 biological replicates). Error bars represent SEM.
(E) Interactions between pUL37×1 and integral mitochondrial membrane proteins are poised to regulate changes to mitochondrial structure during HCMV infection.
See also Table S4.
Among the MIB proteins, both our untargeted (Figures 3E and 5A) and targeted (Figure 4A) MS analyses identified IMMT (also known as Mic60) and CHCHD3 (also known as Mic19) as the most specific, enriched, and continuous pUL37×1 interactors. These proteins are the core scaffolding components of MICOS, with CHCHD3 connecting MICOS to SAMM50 (Tang et al., 2020) and IMMT thought to be the most ancient unit of the entire complex (Kaurov et al., 2018). Indeed, IP of endogenous IMMT did co-isolate pUL37×1-GFP in infected fibroblasts (Figure 5B). As the pUL37×1-GFP band runs just above a non-specific immunoglobulin G (IgG) heavy chain band on a western blot, we further confirmed the specific co-isolation of pUL37×1 by monitoring three signature peptides for either IMMT or pUL37×1-GFP by PRM (Figure 5C; Table S4). We then examined the temporality of this interaction by reciprocal IP-PRM, isolating endogenous IMMT and quantifying co-isolated pUL37×1 throughout infection. Again, we find that pUL37×1 interacts most abundantly with IMMT at 48 and 72 hpi (Figure 5C), in tandem with mitochondrial fragmentation.
To gain further functional insight into these interactions, we turned to CRISPR-based genetic knockouts of MIB proteins in fibroblasts. Given that IMMT knockdown severely compromises cell viability and mitochondrial function (Ott et al., 2015; Tarasenko et al., 2017), we chose to generate CRISPR knockouts of CHCHD3. We achieved an approximately 50% knockout of CHCHD3, as determined by PRM quantification of six unique CHCHD3 peptides (Figure 5D; Table S4). When we infected these cells and measured HCMV production, we found a more than 9-fold decrease in viral titers, indicating that CHCHD3 is necessary for effective HCMV replication (Figure 5D). Altogether, our results lead us to propose a model where pUL37×1 is positioned to compromise MIB-based mitochondrial integrity by interacting with both the MICOS and SAMM50 complexes (Figure 5E). Additional associations with the prohibitin complex and other fission/fusion factors, such as OPA1, MFN1, and MFF, demonstrate that pUL37×1 engages cellular machinery at the core of mitochondrial structure, poised to toggle the balance between fission and fusion.
Peroxisome Morphology Is Controlled by pUL37×1-Mediated Activation of the Fission Factor PEX11β
While our findings thus far focused largely on the roles of pUL37×1 at mitochondria, we next turned our attention to its functions at the peroxisome. Peroxisomes have been increasingly recognized as key organelles during a variety of human pathogen infections, regulating antiviral signaling, cytotoxicity, and cellular metabolism (Cook et al., 2019). During HCMV infection, pUL37×1 localizes to peroxisomes, causes their fragmentation, and inhibits downstream antiviral signaling from the peroxisome-localized pool of MAVS (Magalhães et al., 2016). This is similar to what occurs at mitochondria, especially considering that peroxisomes share MAVS and nearly all fission proteins with mitochondria, including MFF, FIS1, and DRP1 (Koch and Brocard, 2012). Recently, a pro-viral function for peroxisomes during HCMV infection was identified, whereby increased peroxisome numbers and altered membrane morphology enhance lipid metabolism for construction of the viral envelope (Jean Beltran et al., 2018). However, the protein-protein interactions driving pUL37×1-induced peroxisomal fragmentation remain undiscovered.
Our Inter-ViSTA analysis of pUL37×1 interactions demonstrated that only 12% are peroxisomal. Most of these were detected at every time point but were specific only late (72–120 hpi) in infection (Figures 3B–3E; Table S3A and S3B). We also found that peroxisomal interactions sharply decrease in abundance at 120 hpi, although they remain specific (Figure 6A). This suggests that pUL37×1 either leaves peroxisomes or is isolated to a subset distinct from the general population. Given our previous findings that HCMV induces the formation of two peroxisome populations, enlarged versus fragmented (Jean Beltran et al., 2018), we aimed to further investigate this by microscopy. We found that the majority of pUL37×1 localizes to mitochondria at all time points, in agreement with previous studies and our proteomic datasets, while a small subset co-localized with peroxisomes (Figures 6B and 6C). This peroxisome-specific pool of pUL37×1 was often difficult to distinguish from the abundance of pUL37×1-labeled mitochondria, especially as both organelles increase in number throughout infection (Video S5). However, as populations of fragmented and enlarged peroxisomes became readily distinguishable (72 hpi), we observed that pUL37×1 localized nearly exclusively to small peroxisomes, and consequently absent from enlarged peroxisomes (Figure 6D; Video S6). We also noted that peroxisome fission factors are among the most enriched pUL37×1 interactors identified in our study, including MFF and the peroxisome-specific factor PEX11β, which recruits MFF and FIS1 to initiate the division process (Koch and Brocard, 2012).
Figure 6. pUL37×1 Preferentially Localizes to Fragmented, Rather Than Enlarged, Peroxisomes during HCMV Infection.
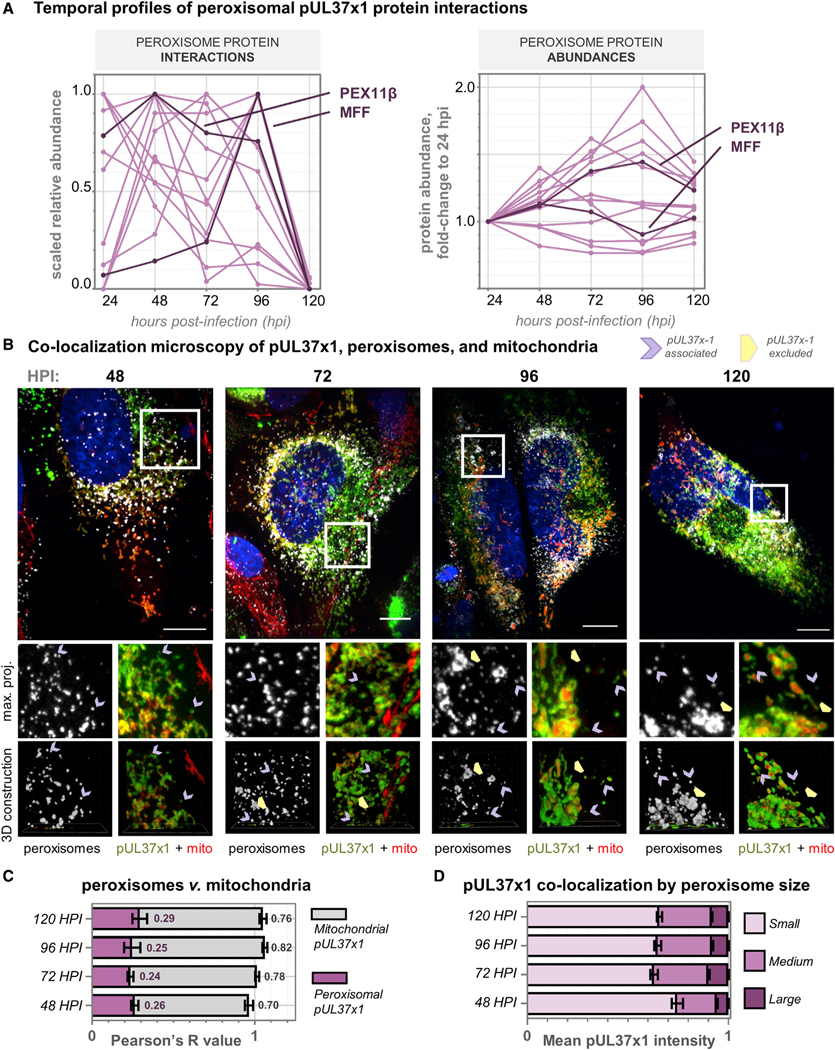
(A) Left: relative abundances of pUL37×1 peroxisomal protein interactions, plotted across infection time. Right: protein levels of pUL37×1 peroxisomal interactors during infection, displayed as a fold change to the abundance at 24 hpi. In contrast to the changes in interaction levels, peroxisome proteins do not decrease in abundance at 120 hpi. PEX11β and MFF profiles are highlighted in darker color.
(B) Immunofluorescence images (z stack maximum projections) across HCMV infection, showing pUL37×1-GFP (green), mitochondria (red, MitoTracker), peroxisomes (white, PEX14 antibody), and DAPI (blue). Channels from a region of interest (white box) are shown below each merged image with a 3D reconstruction. Purple arrows indicate peroxisomes co-localized with pUL37×1 puncta, distinct from mitochondria. Yellow arrowheads indicate enlarged peroxisomes devoid of pUL37×1. Scale bars represent 10 μm.
(C) Colocalization quantification of data in (B) (n = 9 cells per time point), showing Pearson’s R values for pUL37×1 with peroxisomes (pink) or mitochondria (gray). Error bars denote SEM, and averages are indicated as text.
(D) pUL37×1 localization as a function of peroxisome size, measured from data in (B) (n > 6,000 peroxisomes per time point). Mean pUL37×1 intensity perperoxisome is plotted as a fraction of the total for each time point. Error bars denote SEM.
Given the knowledge that pUL37×1 drives peroxisome fragmentation early in infection and peroxisome sizes become increasingly polarized late in infection (Jean Beltran et al., 2018), our findings prompted us to examine whether pUL37×1 is involved in generating the dual population of peroxisome sizes observed late in HCMV infection. We hypothesized that pUL37×1 activates fission via PEX11β throughout infection, thus uncoupling peroxisomal and mitochondrial fragmentation. To address this question, we generated a CRISPR-based knockout of PEX11β and used a UL37 deletion virus (ΔUL37), assessing each impact on peroxisome morphology during HCMV infection with microscopy. In comparison to wild-type HCMV, infection with ΔUL37 resulted in tubular, elongated peroxisomes and reduced fragmentation (Figure 7A). This phenotype was reminiscent of the elongated peroxisomes that persist throughout infection in PEX11β KO cells (Figure 7A). We next quantified peroxisome morphology in each of these conditions by measuring surface area (SA), volume (V), and calculating their ratio (SA:V), which represents the relative geometry of a three-dimensional object in terms of irregular shape. We found that ΔUL37-infected and uninfected PEX11β KO conditions nearly phenocopied each other, increasing the median, range, and average peroxisome SA compared to wild-type conditions (Figure 7B). This suggests that pUL37 and PEX11β have similar functions in generating fragmented peroxisomes during wild-type HCMV infection. However, all infected cells, including wild type, PEX11β, and ΔUL37, exhibited a similar increase in SA:V compared to uninfected control cells, signifying peroxisome flattening late in infection. The ΔUL37 peroxisomes exhibited slightly higher SA:V than other infected conditions, likely due to the lack of fragmentation (Figure 7A).
Figure 7. Peroxisome Fragmentation during HCMV Infection Is Regulated by pUL37×1-Mediated Activation of PEX11β.

(A) Images of peroxisomes (white, antibody against PEX14) from control cells (left), PEX11β KO CRISPR cells (middle), and control cells infected with ΔUL37 virus (right), before and after infection with HCMV. Infection is confirmed with an antibody against pUL99 (red). The peroxisome channel from three regions of interest is below each merged image. Scale bars represent 10 μm.
(B) For each condition in (A), peroxisome SA and SA:V was quantified (n > 20,000 peroxisomes in each condition). The SA:V schematic is shown to right, demonstrating the shift from spherical to flattened as the ratio increases. Box-and-whisker plots show whiskers at 10–90 percentiles, midline at median, and numerical value representing the mean. ***p < 0.001 compared to the wild-type mock-infected condition.
(C) Proposed model for the function of pUL37×1-PEX11β and MFF interactions, whereby pUL37×1 binds and activates PEX11β to promote peroxisome fission, generating fragmented peroxisomes (1). Alternatively, pUL37×1 is excluded from peroxisome membranes, likely by a currently unidentified host or viral factor, to form the enlarged and irregular peroxisomes (2).
Our findings lead us to propose a model whereby pUL37×1 activates PEX11β, which in turn activates MFF to upregulate peroxisome fission during infection (Figure 7C). This begins early in infection and causes an accumulation of increasingly fragmented peroxisomes as infection progresses. Peroxisomes that are excluded from pUL37×1 become enlarged and irregular in shape, possibly through the participation of an unidentified host or viral protein. As we have previously shown that increased peroxisome numbers are beneficial to HCMV, enhancing virus assembly (Jean Beltran et al., 2018), it is likely that the pUL37×1-PEX11β interaction represents a pro-viral mechanism beyond its classic role as an early immune-modulating viral protein.
DISCUSSION
The continued exploration of protein-protein interactions is necessary to further our understanding of biological processes. Computational tools that integrate spatial, temporal, and functional information from large-scale proteomic studies are needed to effectively interpret experimental datasets. Here, we report the development and implementation of Inter-ViSTA, a computational platform for the visualization and analysis of dynamic protein-protein interaction datasets. Inter-ViSTA acts as a research accelerator, allowing users to quickly interpret protein functions, localizations, multiprotein complex membership, and temporal or conditional trends from their experimental data, with minimal manual input. Our tool is amenable to many biochemical techniques, biological conditions, and model organisms, offering a robust platform for future scientific investigations.
We applied Inter-ViSTA to uncover dynamic virus-host protein interactions during infection with HCMV, a critical human pathogen and masterful manipulator of host cell organization. HCMV relies on the finely tuned regulation of protein interactions to rewire cellular organelles for virus replication and spread. We focused on characterizing the interactome of the multifunctional viral protein pUL37×1, which is instrumental in regulating mitochondrial biology to control metabolism, apoptosis, calcium flux, mitochondrial structure, and immune signaling (McCormick et al., 2003; Sharon-Friling et al., 2006; Zhang et al., 2013). pUL37×1 has also been reported to localize to peroxisomes, disrupting peroxisome-mediated antiviral signaling (Magalhães et al., 2016). Despite its range of functions, our knowledge of pUL37×1 interactions with cellular and viral proteins throughout infection remained limited. By performing a temporal interaction study, we aimed to offer additional insights into the mechanistic underpinnings of pUL37×1 activity.
The pUL37×1 protein interactions, validations, and functional investigations reported here present two major points of discussion: (1) pUL37×1-mediated regulation of mitochondrial processes is more complex than previously thought, linking functions across subcellular space and infection time, and (2) the peroxisome-centric roles of pUL37×1 may represent a pro-viral function later in infection, challenging the idea that it participates only in early infection processes.
pUL37×1 is well known for inhibiting apoptosis, which is closely linked to disruption of mitochondrial calcium homeostasis early in HCMV infection. Previous work has predominantly focused on interactions with BAX (Zhang et al., 2013), which we observe as a robust pUL37×1 association throughout infection. However, we additionally discovered and validated its interactions with other apoptotic cellular proteins, including RMDN3 and RHOT1, which also regulate mitochondrial calcium levels (Fransson et al., 2003; Lv et al., 2006; Safiulina et al., 2019; De vos et al., 2012). These associations suggest that pUL37×1-mediated regulation of apoptosis is complex, especially considering that RMDN3 and RHOT1 are important players in regulating mitochondrial dynamics like motility, calcium flux, mitophagy, and cristae organization. Moreover, they participate in functions at the ER-mitochondria contact site, which pUL37×1 engages to increase mitochondrial calcium levels, promote BAX degradation, and insert itself into the OMM (Bozidis et al., 2010). Multifunctional proteins like RMDN3 and RHOT1, among others detected in our dataset, may underlie the ability of pUL37×1 to manipulate diverse aspects of mitochondrial biology. This is further highlighted by our finding that pUL37×1 interacts with the MIB, an ancient component of mitochondrial structure and thus a likely player in viral mitochondrial manipulation. Indeed, we find that the core MIB proteins IMMT and CHCHD3 are among the most abundant and specific pUL37×1 interactors, peaking at the onset of mitochondrial fragmentation (48 hpi). We also demonstrate that CHCHD3 itself is required for productive HCMV replication, reducing virus titers by nearly 9-fold. Importantly, a mechanism for pUL37×1-induced mitochondrial fission has not been fully defined, and the protein interactions we describe here are promising candidates, including the MIB and prohibitin complexes, RHOT1, and the fission/fusion factors MFF and OPA1.
In addition to pUL37×1 mitochondrial functions, our interactome provided insights into its peroxisomal activity. pUL37×1 has been shown to cause peroxisomal fragmentation early in infection (Magalhães et al., 2016), yet how this occurs was not known. Here, we show that pUL37×1 localizes to peroxisomes throughout HCMV infection and interacts with the primary regulator of peroxisomal division, PEX11β, thus activating the peroxisome fission pathway to form fragmented peroxisomes. In agreement, our previous study established that HCMV infection preferentially induces peroxisome fission to increase numbers, which coincides with the translocation of MFF to peroxisome membranes at 72 hpi (Jean Beltran et al., 2018). Here, we observe increased pUL37×1 associations with MFF at this time, agreeing with the knowledge that MFF acts downstream of PEX11β during peroxisome fission.
Peroxisome-localized pUL37×1 was not a pervasive phenotype, however, as we found pUL37×1 to be excluded from the enlarged population of peroxisomes late in HCMV infection. This morphology was recently shown to promote the synthesis of plasmalogen lipids, supporting the envelopment of newly replicated virions (Jean Beltran et al., 2018). Among peroxisomal pUL37×1 interactors we identified FAR1, one of the two upstream enzymes in the plasmalogen synthesis pathway and the only one localized on the cytosolic face of peroxisome membranes (Braverman and Moser, 2012; Lodhi and Semenkovich, 2014). This association was specific at only one time point and decreased in abundance as infection progressed, possibly due to the increasing isolation of pUL37×1 to small peroxisomes. A next key step will be determining the host or viral factors that prevent pUL37×1 from localizing to these enlarged, irregular peroxisomes. Given the multifunctionality of peroxisomes and the temporality of the interactions we describe here, pUL37×1 peroxisomal functions could reflect a balance between inhibiting immune signaling early in infection and increasing metabolic output late in infection.
Altogether, the dynamic interactome of pUL37×1 represents an elegant example of co-evolution, whereby HCMV uses a single protein to regulate two organelles that are quite similar, considering their fission pathways and functions in cellular metabolism and antiviral signaling, yet this pro-viral remodeling plays out differently across infection time. By using Inter-ViSTA to uncover these spatiotemporal interactions, our study demonstrates its value in adding biological depth and understanding to the study of protein-protein interactions, pinpointing promising avenues for follow-up functional analyses. We anticipate that Inter-ViSTA will be useful for specialist and non-specialist researchers alike and used in a broad range of contexts to bridge the gap between proteomic discovery and biological application.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
For further information and requests for resources or reagents, contact Dr. Ileana M. Cristea, Princeton University (icristea@princeton.edu).
Materials Availability
Virus strains and CRISPR cell lines used in this study are available on request from the Lead Contact.
Data and Code Availability
All data-dependent MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019). Access at PRIDE: PXD017163.
The targeted (PRM) MS data have been deposited to Panorama Public (Sharma et al., 2018): https://panoramaweb.org/InterViSTA.url and the RAW data to ProteomeXchange, which can be accessed at PRIDE: PXD013848.
Inter-ViSTA is freely available for download at https://github.com/cristealab/Inter-ViSTA and at intervista.princeton.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Viral strains, cell culture, and viral infection
HCMV virus strains BADinGFP, BADinUL37GFP, and BADin ΔUL37×1 that express free GFP, pUL37×1-GFP, or no pUL37×1, respectively, were used for the IP-MS and microscopy experiments where indicated. These virus strains are derivatives of BADwt (Yu et al., 2002), which was cloned from the HCMV laboratory-strain AD169. The BADinUL37GFP and BADinΔUL37×1 virus strains were gifts from Dr. Thomas Shenk at Princeton University, Princeton, NJ. Viral propagation and titering were performed as previously described (Britt, 2010). Each virus was used at a multiplicity of infection (MOI) of 3.
All experiments were carried out in HFF-1 cells (SCRC-1041, ATCC, Manassas, VA), which were cultured in Dulbecco’s modified Eagle medium (high-glucose) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and grown at 37°C in a 5% CO2 atmosphere.
CRISPR cell lines
CHCHD3 CRISPR cells were generated via transfection using TrueCut Cas9 Protein V2 (A36499, ThermoFisher Scientific) and CRISPRmax (CMAX0001, ThermoFisher Scientific) with a synthetic guide RNA sequence (ACAUAUCCUCUCCCGAAGGA, ThermoFisher Scientific) against CHCHD3 according to the manufacturer’s instructions. Control CRISPR cells were generated via the same protocol using a non-targeting synthetic guide RNA sequence (AAAUGUGAGAUCAGAGUAAU, ThermoFisher Scientific). CRISPR KO efficiency was determined via targeted mass spectrometry, which showed an approximately 55% knockout of CHCHD3, likely due to the long half-life of this protein. Viral titers of CRISPR cells were determined via immunofluorescent detection of viral infection five days post-infection as previously described (Jean Beltran et al., 2016).
METHOD DETAILS
Uploading protein interaction data to Inter-ViSTA
Inter-ViSTA is freely available for download at https://github.com/cristealab/Inter-ViSTA and at intervista.princeton.edu and can be used on nearly all interactome proteomic datasets. Given the name similarity, we consider Inter-ViSTA the friend of the computationally distinct, seemingly inactive but not forgotten, platform “Vista” previously developed by the Gygi group for the high-throughput quantification of mass spectrometry data (Bakalarski et al., 2008).
Inter-ViSTA requires two files, one with node (protein) information and another with edge (interaction) information. The files can contain nodes and edges from distinct experiments with distinct bait proteins. A helper executable Python script is provided with Inter-ViSTA to allow users to convert their data into the file format accepted by Inter-ViSTA. The nodes csv file requires the following information for each protein: (1) UniProt Accession Number (ex: Q96RT1), (2) Gene Name (ex: ERBB2IP), (3) Species ID (UniProt Taxonomic Identifier; ex: 9606 for human), and (4, optionally) Localization (with multiple localizations separated by a semi-colon; ex: Cytoplasm; Mitochondria). The edges csv file contains the following information for each interaction: (1) Bait Protein Accession Number (ex: P16755), (2) Prey Protein Accession Number (ex: Q96RT1), (3) Interaction Confidence at Time point 1 (between 0–1; ex: 0.65), (4) Interaction Confidence at Time point 2, 3, etc., (5, optionally) Prey Protein Abundance at Time point 1, 2, 3, etc. The only required information is the specificity-scored bait-prey pairs and the specificity cutoff desired for visualization.
Confidence scoring is required to distinguish biologically and functionally relevant interactions from false positives. There are several algorithms to assign confidence scores to PPIs depending on how the data was collected – here, we use Significance Analysis of INTeractome (SAINT) to score PPIs based on label-free quantitative proteomics data (Choi et al., 2011). Confidence scores must be normalized on a 0–1 scale. Finally, the user must specify an edge confidence threshold. Only interactions with confidence scores over the threshold in at least one condition are retained for further analysis. The threshold can either be provided in the helper executable script or incorporated into the name of the edges file as “edges_x.csv,” where ‘x’ is the confidence threshold (ex: “edges_0.7.csv”).
Calculating protein and interaction activity spells
An activity spell specifies the time interval during which an interaction is valid, i.e., has a confidence score over the threshold. An activity spell begins at interaction “onset,” which is the first time point or condition at which an interaction score is over the confidence threshold, and ends at the “terminus,” which is the last time point or condition at which the score is over the threshold. To prevent interactions from disappearing during short intervals where the confidence score drops slightly below the stringent cutoff, the threshold is relaxed to (provided threshold – 0.1) for the calculation of interaction activity spells. Thus, when a 0.9 cutoff is applied, an interaction with a specificity score of 0.88 in one out of six time points will be kept for all six. An interaction is assigned multiple onsets and termini if there are non-overlapping spells of activity. Protein activity spells are assigned similarly by encompassing the onsets and termini of their associated edges. As a protein may have multiple interactions with different baits, the protein onset is the earliest onset of all its associated interactions, while its terminus is the latest terminus of all its associated interactions. Bait proteins are assigned to remain active throughout the entire time period or across all conditions. Finally, if there are multiple baits in the dataset, proteins shared between baits are annotated with their associated bait proteins at each time point or condition.
Querying external databases for integration with Inter-ViSTA
Additional information on known associations between the provided proteins, functional annotations, and subcellular localization information (if not provided by the user) are obtained by automatically querying external databases via the Inter-ViSTA script. Published PPIs are derived from STRINGdb via the STRING API for Python using the provided species IDs (Szklarczyk et al., 2017). Those that are experimentally verified are retained in the edge dataset along with their experimental confidence score (“e-score” in STRING). Functional annotations and localization information for each protein are automatically queried from UniProt with the UniProt.ws package (Carlson) in R. For each species, the “GO-ID” and (optionally) “SUBCELLULAR-LOCATION” attribute are selected for functional annotation and localization information respectively. Proteins may have multiple localizations and GO-IDs. The localization terms are added as attributes to the protein dataset. For mammalian proteins, the provided proteins are matched against complexes in CO-RUM, the comprehensive resource of mammalian protein complexes (Giurgiu et al., 2019). Complexes with ≥ 40% of their members identified in the provided dataset are listed for export and added as attributes to the protein dataset.
Gene ontology enrichment implementation in Inter-ViSTA
GO enrichment is performed using the topGO package (Alexa and Rahnenführer, 2018) in R. The background gene list provided by the user is used to define the “gene universe,” and the proteins in the nodes file are used to define “interesting” genes on which to calculate enrichment. For the pUL37×1 interactome, we use a gene list from human fibroblast cells as the background. After employing the Fisher test for enrichment in topGO, multiple hypothesis correction is applied on the calculated Fisher p values using the base R “p.adjust” function with the “BH” (or “fdr”) method. GO terms with corrected p value < 0.05 are reported in a table along with their Fisher p value, corrected p value, and proteins associated with the term. If fewer than 5 GO terms pass the stringent multiple-testing corrected p value cutoff, all terms with uncorrected p value < 0.05 are listed. The top 10 enriched GO terms by p value are added as attributes to the protein dataset.
Clustering Proteins by Temporal Abundance Profiles
If prey protein abundances are provided, Inter-ViSTA clusters proteins by their temporal or conditional abundance profiles per bait protein. Here, we normalized prey abundances to bait abundance before further analysis. In Inter-ViSTA, each prey protein abundance is scaled to 1 by dividing the abundance by the maximum abundance of that protein in any condition. Principal component analysis (PCA) is then performed on the scaled abundances with the base R “prcomp” function. The number of components that explain > 90% of variation are used to define the number of clusters for k-means clustering with the base R “kmeans” function. For each bait, the scaled prey protein abundances are plotted over time and colored by cluster. Finally, the cluster numbers are added as prey protein attributes to the edge dataset.
Normalizing Interaction Abundance by Proteome Abundance
To normalize the abundance scores from interaction data by overall proteome abundance, the user may provide an additional quantitative proteomics dataset with proteome abundance across the same time points or conditions in the interaction data. If this is provided, Inter-ViSTA normalizes abundances by dividing interaction abundance at each time point or condition to proteome abundance at the corresponding time point or condition. The abundances before and after normalization are added as attributes to the proteins. Heatmaps comparing abundances prior to and after normalization to the proteome are plotted by scaling abundance data from 0 to 1 at each condition using the formula:
Building and Plotting a Dynamic Network in Inter-ViSTA
The protein and edge datasets with all the above attributes are piped into an R network dynamic object and manipulated with the Network Dynamic Temporal Visualization (ndtv) package (Bender-deMoll, 2016). Edges are assigned weights based on their confidence scores and colored by type – user provided or inferred from STRING. Nodes are automatically colored based on their protein localizations. Those with multiple localizations are assigned a separate color, while those with unspecified locations are colored by species instead. Node outlines are colored on a scale of light to dark red based on their activity duration. Proteins in the top quartile of activity (longest duration active) are outlined in the darkest red, while those in the bottom quartile of activity (shortest duration active) are outlined in the lightest red. Finally, the network layout at each time point is calculated using the Kamada-Kawai force-directed algorithm (Kamada and Kawai, 1989).
Creating interactive output
Users interact with the constructed network using the R Shiny interface. Proteins and interactions in the network can be activated and deactivated based on user-selected options, which include STRING edge confidence threshold, localization, GO term, and species. Moreover, the network can be reconstructed to include only nodes shared between at least ‘n’ baits, where ‘n’ is between 1 and the number of baits in the input. The Shiny output also includes a “Quantitative Plots” tab for abundance plots, where the user can view and download plots of log10-protein abundance over time for input prey proteins and their interactors, as well as the cluster number and cluster profiles for each bait that the prey interacts with. If the user provided proteome abundances, heatmaps comparing abundances prior to and after normalization are displayed and can be downloaded. The “Gene Ontology” tab has an interactive, searchable table with significant GO terms and their annotated proteins, while the “Protein Complexes” tab has an interactive table with detected protein complexes and their annotated proteins. The “Downloads” tab allows users to download various aspects of the network, such as an analysis report with basic network features (plots of protein activity separated by localization and function, edge onset times, and edge durations), as well as tab-separated files with node and edge attributes that can be directly exported to Cytoscape for further analysis.
Inter-ViSTA analysis of published datasets
Cyclin interactions from Pagliuca et al. were input to Inter-ViSTA after being assigned confidence scores from 0 – 1 across the four cell cycle stages. Scores were assigned for all interactions by dividing the provided MaxQuant specificity ratios for each interaction by the highest MaxQuant ratio (39.4) observed for any interaction across the dataset. This linear scaling retained the distribution of ratios. The confidence threshold for visualization was assigned based on the suggested MaxQuant ratio in the original paper (1.8), scaled appropriately to a confidence threshold of 0.05 for Inter-ViSTA. Networks were visualized at a STRING confidence score of 0.1. GO enrichment was performed with a reference gene list for proteins expressed in HeLa cells, the cell line used by Pagliuca et al., derived from Bekker-Jensen et al. (2017).
Specificity-filtered interactions of Us9 from Kramer et al. were input to Inter-ViSTA with provided abundances across five infection time points (3, 8, 12, 18, and 24 hpi). As specificity scores were not provided, we set all edge scores to 1. Networks were visualized at a STRING confidence score of 0.1. For GO enrichment, a background gene list of proteins expressed in differentiated PC12 cells, the cell line used by Kramer et al., was derived from(Mullenbrock et al. (2011).
SAINT specificity filtered interactions from Lobingier et al. were used directly for the APEX data analysis. We used a SAINT cutoff of 0.6, which corresponded to the FDR of 0.05 used in the original publication. Networks were visualized at a STRING confidence score of 0.1. GO enrichment was performed with a reference gene list for proteins expressed in HEK293 cells downloaded from https://www.pax-db.org/ (Wang et al., 2015).
Immunofluorescence microscopy, image acquisition, and analysis
HFF-1 cells were plated on coverslips in 6-well dishes and infected at MOI 3 with the indicated viral strains. For mitochondrial colocalization experiments, cells were stained with MitoTracker CMXROS (M7512, Thermo Fisher Scientific) prior to fixation at a concentration of 250 nM in serum-free media for 15 minutes at 37°C. Cells were then fixed with 4% paraformaldehyde (PFA) in PBS for 15 minutes at room temperature, permeabilized in cold methanol for 20 minutes at −20°C, then blocked with 10% goat serum + 5% human serum in 0.1% Tween in PBS for 30 minutes. Slides were incubated in primary antibody for 2 hours and secondary antibodies with DAPI for 1 hour (see specific antibody concentrations below). Coverslips were mounted onto slides with 10uL Prolong Diamond Antifade Mountant (P36961, Thermo Fisher Scientific).
All images were acquired with a Nikon Ti-E inverted fluorescence confocal microscope (Nikon Instruments, Melville, NY) equipped with a Yokogawa spinning disc (CSU-21), digital CMOS camera (Hamamatsu ORCA-Flash TuCam), and precision microscope stage (Piezo). Z stacks were acquired with 0.2 μm steps throughout the cell depth using a Nikon 100X Plan Apo objective. Microscopy images shown in this manuscript were processed using ImageJ (National Institutes of Health), and are displayed as z stack maximum projections, with a background subtraction (rolling ball radius = 200 pixels) applied to each channel. All scale bars correspond to 10 μm in length. For the peroxisome colocalization in Figure 6, Pearson’s R values were calculated by the Coloc2 ImageJ plugin (Johannes Schindelin). For the morphology quantifications in Figure 6D and Figure 7, peroxisome surface areas and volumes were measured with the 3D Objects Counter ImageJ plugin (Bolte and Cordelières, 2006), with a minimum size defined as 10 consecutive voxels (at a resolution of 0.065 μm/pixel), and 3D manager and Quantif 3D were used to map pUL37×1 channels onto each 3D peroxisome map and measure their corresponding intensity. Reported intensities were normalized to each peroxisome’s surface area and categorized as small (< 0.89 μm2), medium (0.9–3.09 μm2) or large (> 3.1 μm2). Three-dimensional movies were generated using the volume viewer and movie maker in NIS Elements (Nikon) software.
Antibodies used in IF experiments were as follows: RMDN3 (1:750, HPA013144 Sigma), PEX14 (1:500, ab183885 Abcam), UL99 (1:100, gift from Dr. Thomas Shenk, Princeton University, Princeton, NJ), Goat anti-Rabbit Alexa Fluor 633 (1:2000, A-21071, Thermo Fisher Scientific), Goat anti-Rabbit Alexa Fluor 488 (1:2000, A-32731, Thermo Fisher Scientific), and Goat anti-Mouse Alexa Fluor 633 (1:2000, A-21052, Thermo Fisher Scientific). DAPI (62248, Thermo Fisher Scientific) was used at a concentration of 1:1000.
Western Blotting
Cell pellets were resuspended in lysis buffer composed of 4% SDS, 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 0.5 mM EDTA. Protein concentration was assessed by BCA assay (23225, Thermo Fisher Scientific) and samples were adjusted to equal concentrations, reduced with 50 mM dithiothreitol, and boiled for 5 minutes at 95°C. Samples were run on 4%–12% Bis-Tris gels (NP0323, Thermo Fisher Scientific) and electrophoresed onto Immobilon-FL PVDF (IPFL00010, Millipore Sigma, Burlington, MA) membranes at 35 V overnight at 4°C. Blots were blocked with 5% milk in tris-buffered saline (TBS) and incubated with primary antibodies diluted in 5% milk in TBS plus 0.2% Tween-20 (TBST) for 1 h at room temperature. Primary antibodies used were: IE1 (1:100), pUL26 (1:100), pUL99 (1:100), and GAPDH (1:1000, Cell Signaling). IE1, pUL26, and pUL99 primary antibodies were gifts from Dr. Thomas Shenk, Princeton University, Princeton, NJ. For reciprocal IPs, antibodies against ATAD3A (H00055210-D01P, Novus Biologicals, Centennial, CO), IMMT (PA1–16918, ThermoFisher Scientific), RMDN3 (NBP1–47293, Novus Biologicals), and GFP (11814460001, Millipore Sigma) were used at a 1:1000 dilution. Secondary antibodies used were anti-mouse and -rabbit Alexafluor 680 conjugates (Thermo Fisher Scientific) at a dilution of 1:10,000 in 5% milk in TBST. Blots were visualized using an Odyssey CLx Imager (LI-COR Biosciences, Lincoln, NE).
Immunoaffinity Purification (IP) and protein digestion
pUL37-GFP IPs, along with the matched GFP-virus control, were performed in biological duplicate at each time point of infection (24, 48, 72, 96, and 120 hpi) for discovery and targeted experiments. For each replicate, one 10cm dish of infected cells was harvested via scraping into cold PBS and centrifuging. Cell pellets were washed twice with cold PBS, spiked with 5 μL of 100X HALT protease and phosphatase inhibitor (78438, Thermo Fisher Scientific, Waltham, MA), snap frozen in liquid nitrogen, and stored at −80°C. Frozen cell pellets were briefly thawed on ice and then suspended in 500 μL of lysis buffer consisting of 20 mM HEPES KOH, pH 7.4, 110 mM potassium acetate, 2 mM MgCl2, 0.1% Tween-20, 1 μM ZnCl2, 1 μM CaCl2, 0.6% Triton, 200 mM NaCl, 1:100 HALT protease and phosphatase inhibitor cocktail, 1:2500 Universal Nuclease (88700, Thermo Fisher Scientific). Samples were incubated at RT with inversion for 10 minutes, and the lysates were subjected to Polytron homogenization for 2 cycles of 15 s at a speed of 22,500 rpm. Samples were then spun at 8,000 x g for 10 minutes to pellet the insoluble fraction.
For anti-GFP IPs, preconjugated GFP-Trap MA beads (gtma-100, Chromotek, Hauppauge, NY) were used. For each IP, 20 μL of bead slurry was washed 3 × 500 μL in wash buffer (lysis buffer without inhibitors and nuclease). Soluble lysates were added to the beads and incubated for 30 min at 4°C with end-over-end rotation. Following the incubation, the beads with bound proteins were collected via a magnetic rack and then suspended in wash buffer and transferred to a new tube. The beads were then washed 3 × 500 μL in wash buffer with magnetic collection in between each wash and then resuspended in 500 μL H2O and transferred to another tube. The beads were washed a final time with H2O and then eluted in 85uL of 106 mM Tris HCl, 141 mM Tris Base, 2% LDS, 0.5 mM EDTA. Eluates were reduced and alkylated with 25 mM TCEP (77720, Thermo Fisher Scientific) and 50 mM chloroacetamide at 70°C for 20 minutes.
For reciprocal IPs, 5 μg of antibodies against IMMT (PA1–16918, ThermoFisher Scientific), RMDN3 (NBP1–47293, Novus Biologicals), and RHOT1 (ab188029, Abcam) were pre-conjugated to magnetic protein A/G beads (88802, Thermo Fisher Scientific) and IPs were performed using the same buffer conditions as the GFP IPs above as previously described (Federspiel et al., 2019). Samples were digested for mass spectrometry using Filter Aided Sample Prep (FASP) and desalted via StageTips as previously described (Greco et al., 2016). Peptides were eluted as a single fraction in 5% ammonium hydroxide, 80% acetonitrile (ACN) and analyzed by LC-MS/MS.
Mass spectrometry analysis by data dependent acquisition
pUL37×1 IPs were analyzed via nano-liquid chromatography tandem mass spectrometry (LC-MS/MS) using a Dionex Ultimate 3000 nanoRSLC (Thermo Fisher Scientific) coupled to an LTQ-Orbitrap Velos ETD mass spectrometer (Thermo Fisher Scientific). Peptides were separated over a 150 min gradient (5% B to 30% B) with a 250 nl/min flow rate by reverse-phase chromatography on a 20 cm column packed in-house with 1.9 μm ReproSil-Pur C18-AQ (Dr. Maisch, GmbH). Mobile phase A consisted of 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 97% ACN. An MS1 scan was performed at a resolution of 120,000 across a mass range of 350–1,700 with an automatic gain control (AGC) of 1 × 106, max injection time of 300 ms, FT preview scan disabled, and waveform injection and dynamic exclusion enabled. Data-dependent MS2 scans of the top 15 ions followed each MS1 scan with collision-induced dissociation fragmentation, an AGC of 1 × 104, max injection time of 125 ms, a minimum signal of 1,000, isolation width of 2.0, normalized collision energy of 30% and activation time of 10 ms.
The MS/MS data were analyzed by Proteome Discoverer 2.2 (Thermo Fisher Scientific) using a FASTA file containing human and herpesvirus protein sequences and common contaminants (22,269 sequences, download 4/2016 from Uniprot). The Spectrum Files RC node and Minora Feature Detector nodes were used to perform offline mass recalibration and label-free MS1 quantitation respectively. MS/MS spectra were analyzed using Sequest HT for forward and reverse searches to determine FDR. Sequest was run with settings for a fully tryptic search with a maximum of two missed cleavages, precursor mass tolerance of 4 ppm, fragment mass tolerance of 0.3 Da, static carbamidomethylation of cysteine, dynamic phosphorylation of serine, threonine, and tyrosine, dynamic deamidation of asparagine, dynamic oxidation of methionine, and dynamic loss of methione plus acetylation of the protein N terminus. The matched spectra were validated by the Percolator node and then PTM site localization was determined in the ptmRS node.
A consensus file was then created in Proteome Discoverer 2.2 for each dataset using the Feature Mapper and Precursor Ions Quantifier nodes. Label-free MS1 signals were mapped across samples to pick up missing values from peptides not selected for sequencing in every sample and then were quantified by taking the max peak intensity for each peptide. Label-free quant data was not normalized or scaled in Proteome Discoverer and protein and peptide FDR was required to be 1%. Two unique peptide sequences were required for protein inference and only unique or razor peptides were used for label-free quantitation.
Bioinformatic analysis of protein-protein interaction data
The assembled datasets were exported to Excel for further analysis. Total spectral count data was analyzed by SAINT (Choi et al., 2011) using the REPRINT (Mellacheruvu et al., 2013) interface to determine interaction specificity compared to control isolations. SAINT was run with LowMode off, MinFold on, and Normalize on. Label-free MS1 abundance values were normalized to the respective bait levels in each sample. Each dataset was analyzed in Inter-ViSTA with a visualization specificity threshold of 0.9, bait-normalized MS1 abundances, and pre-assigned subcellular localizations derived from UniProt and a previous dataset that identified proteins with changed subcellular localizations during infection (Jean Beltran et al., 2016). Network interaction diagrams incorporating relative quantitation and known protein-protein interaction data were made in Cytoscape (Shannon et al., 2003) using information generated by the Inter-ViSTA interface.
Targeted mass spectrometry analysis by parallel reaction monitoring (PRM)
Targeted mass spectrometry was used to specifically examine a subset of identified pUL37×1 interactors for validation during reciprocal IPs and more precise quantification over the course of infection. PRM samples were analyzed via LC-MS/MS using a Dionex Ultimate 3000 nanoRSLC coupled to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Peptides were separated on a reversed phase C18 column by a linear gradient from 0%–35% B over 60 min. The PRM method consisted of targeted MS2 scans recorded in profile mode performed at a resolution of 15,000, with a AGC target of 2e5, maximum inject time of 175 ms, isolation window of 0.8, and normalized collision energy of 27 controlled by a peptide inclusion list containing 2–4 unique peptides for each of the targeted proteins with retention time windows of 5 min. Label-free quantitation of peptides specific for the proteins of interest was designed and analyzed using the Skyline software for targeted proteomics (MacLean et al., 2010). Summed area under the curve of 3 transitions per peptide was used for quantitation.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all MS and microscopy data in this manuscript, the quantification workflows, software, replicates (N values), results, and graphical display keys can be found in the figure legends and/or experiment-specific detailed descriptions are included in the Method Details sections above.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-RMDN3, Rabbit polyclonal (for IF) | Sigma Aldrich | Cat# HPA009975; RRID: AB_1078811 |
| anti-RMDN3, Rabbit polyclonal (for IP, WB) | Novus Biologicals | Cat# NBP1–47293; RRID: AB_10010247 |
| anti-RHOT1 (MIRO1), Mouse monoclonal | Abcam | Cat# ab188029 |
| anti-pUL99, Mouse polyclonal | Gift from Dr. Thomas Shenk | N/A |
| anti-IE1, Mouse polyclonal | Gift from Dr. Thomas Shenk | N/A |
| anti-pUL26, Mouse polyclonal | Gift from Dr. Thomas Shenk | N/A |
| anti-GFP, Mouse monoclonal (for WB) | Millipore Sigma | Cat# 11814460001; RRID: AB_390913 |
| anti-GAPDH, Rabbit polyclonal | Cell Signaling | Cat# D16H11 (5174S); RRID: AB_11129865 |
| anti-IMMT, Rabbit polyclonal | ThermoFisher Scientific | Cat# PA1–16918; RRID: AB_2127191 |
| anti-PEX14, Rabbit polyclonal | Abcam | Cat# ab183885; RRID: AB_2744539 |
| Goat anti-Rabbit Alexa Fluor 633 conjugate | ThermoFisher Scientific | Cat# A-21071; RRID: AB_2535732 |
| Goat anti-Rabbit Alexa Fluor 488 conjugate | ThermoFisher Scientific | Cat# A-21052; RRID: 2535719 |
| Goat anti-Rabbit Alexa Fluor 680 conjugate | ThermoFisher Scientific | Cat# A27042; RRID: 2536103 |
| Goat anti-Mouse Alexa Fluor 680 conjugate | ThermoFisher Scientific | Cat# A-21057; RRID: AB_141436 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| HCMV AD169 | Gift from Dr. Thomas Shenk, Princeton University | N/A |
| BADinGFP | Gift from Dr. Thomas Shenk (Yu et al., 2002) | N/A |
| BADinUL37GFP | Gift from Dr. Thomas Shenk (Yu et al., 2002) | N/A |
| BADinDUL37×1 | Gift from Dr. Thomas Shenk (Yu et al., 2002) | N/A |
|
| ||
| Chemicals Peptides, and Recombinant Proteins | ||
|
| ||
| GFP-Trap MA beads | Chromotek | Cat# gtma-100 |
| magnetic protein A/G beads | ThermoFisher Scientific | Cat# 88802 |
| DAPI | Thermo Fisher Scientific | Cat# 62248 |
| TrueCut Cas9 Protein V2 | ThermoFisher Scientific | Cat# A36499 |
| CRISPRmax | ThermoFisher Scientific | Cat# CMAX0001 |
| Prolong Diamong Antifade Mountant | ThermoFisher Scientific | Cat# P36961 |
| 4–12% Bis-Tris gels | ThermoFisher Scientific | Cat# NP0323 |
| Immobilon-FL PVDF membranes | Millipore Sigma | Cat# IPFL00010 |
| MitoTracker CMXROS | ThermoFisher Scientific | Cat# M7512 |
| HALT protease and phosphatase inhibitor | ThermoFisher Scientific | Cat# 78438 |
| Universal Nuclease | ThermoFisher Scientific | Cat# 88700 |
| TCEP | ThermoFisher Scientific | Cat# 77720 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| BCA Assay | ThermoFisher Scientific | Cat# 23225 |
|
| ||
| Deposited Data | ||
|
| ||
| DDA MS/MS RAW files and PD2.2 analysis files | This study | ProteomeXchange (PXD017163) |
| PRM MS/MS data (Panorama) | This study | https://panoramaweb.org/InterVista.url |
| PRM MS/MS data (RAW files) | This study | ProteomeXchange (PXD013848) |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human foreskin fibroblast 1 (HFF-1) cells | ATCC | Cat# SCRC-1041 |
| CHCHD3 CRISPR Cells | This study | N/A |
| Control CRISPR Cells | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| CHCHD3 CRISPR guide RNA: ACAUAUCCUCUCCCGAAGGA | ThermoFisher Scientific | N/A |
| Control CRISPR guide RNA: AAAUGUGAGAUCAGAGUAAU | ThermoFisher Scientific | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| ProteomeDiscoverer v2.2 | Thermo Fisher Scientific, https://www.thermofisher.com/order/catalog/product/OPTON-30795 | N/A |
| Skyline | (MacLean et al., 2010), https://skyline.ms | N/A |
| GraphPad Prism v8 | GraphPad, https://www.graphpad.com/scientific-software/prism/ | N/A |
| SAINT | (Choi et al., 2011), https://reprint-apms.org/ | N/A |
| STRING | STRING Consortium, https://string-db.org/ | N/A |
| Cytoscape v7.2.1 | Cytoscape, https://cytoscape.org/ | N/A |
| Scaffold v4.7.3 | Proteome Software, http://www.proteomesoftware.com/products/scaffold/ | N/A |
| Pantherdb | (Mi et al., 2019), http://www.pantherdb.org/ | N/A |
| ImageJ | National Institutes of Health | N/A |
| NIS Elements AR Analysis | Nikon | N/A |
| Inter-ViSTA | This study |
intervista.princeton.edu; https://github.com/cristealab/Inter-ViSTA |
Highlights.
Inter-ViSTA is a web-based platform for analyzing dynamic protein interactions
Inter-ViSTA provides biological depth by automatically integrating protein features
The HCMV protein pUL37 rewires mitochondria by engaging the conserved MICOS complex
pUL37 activates the fission factor PEX11 to achieve pro-viral peroxisome remodeling
ACKNOWLEDGMENTS
We are grateful for funding from the NIH (GM114141) and a Mallinckrodt Scholar Award to I.M.C., an NIH Predoctoral Individual National Research Service Award (F31AI147637) and Princeton Centennial Award to K.C.C., a National Science Foundation Graduate Research Fellowship to M.A.K., a B1G Post-Graduate Scholarship (Northwestern) to W.A.H., and the National Institutes of Health NIGMS (T32GM007388).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107943.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alexa A, and Rahnenführer J. (2018). Gene set enrichment analysis with topGO. [Google Scholar]
- Alwine JC (2012). The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog. 8, e1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakalarski CE, Elias JE, Villén J, Haas W, Gerber SA, Everley PA, and Gygi SP (2008). The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. Journal of Proteome Research 7, 4756–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB, Sørensen KD, Høyer S, Ørntoft TF, Andersen CL, et al. (2017). An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 4, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender-deMoll S. (2016). Temporal network tools in statnet: networkDynamic, ndtv and tsna. https://statnet.org/Workshops/ndtv_workshop.html.
- Bolte S, and Cordelières FP (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Bozidis P, Williamson CD, and Colberg-Poley AM (2008). Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J. Virol 82, 2715–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozidis P, Williamson CD, Wong DS, and Colberg-Poley AM (2010). Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. J. Virol 84, 7898–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman NE, and Moser AB (2012). Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Britt WJ (2010). Human cytomegalovirus: propagation, quantification, and storage. Curr. Protoc. Microbiol. Chapter 14, 3. [DOI] [PubMed] [Google Scholar]
- Cannon MJ, Schmid DS, and Hyde TB (2010). Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol 20, 202–213. [DOI] [PubMed] [Google Scholar]
- Castanier C, Garcin D, Vazquez A, and Arnoult D. (2010). Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 11, 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, and Nesvizhskii AI (2011). SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Park A, Kang S, Lee E, Lee TA, Ra EA, Lee J, Lee S, and Park B. (2018). Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun 9, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, T.U.; UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47 (D1), D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KC, and Cristea IM (2019). Location is everything: protein translocations as a viral infection strategy. Curr. Opin. Chem. Biol 48, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KC, Moreno JA, Jean Beltran PM, and Cristea IM (2019). Peroxisome Plasticity at the Virus-Host Interface. Trends Microbiol. 27, 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MS, Lum KK, Sheng X, Song B, and Cristea IM (2016). Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit. Rev. Biochem. Mol. Biol 51, 452–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, and Pellett PE (2011). Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol 85, 5864–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, and Miller CCJ (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet 21, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang HY, Chang CL, Hsu SH, Huang CY, Chiang SF, Chiou SH, Huang CH, Hsiao YT, Lin TY, Chiang IP, et al. (2010). ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci 123, 1171–1180. [DOI] [PubMed] [Google Scholar]
- Federspiel JD, Greco TM, Lum KK, and Cristea IM (2019). Hdac4 Interactions in Huntington’s Disease Viewed Through the Prism of Multiomics. Mol. Cell. Proteomics 18 (8, suppl 1), S92–S113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, and Aspenström P. (2003). Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem 278, 6495–6502. [DOI] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A. (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47 (D1), D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, and Cristea IM (2017). Proteomics tracing the footsteps of infectious disease. Mol. Cell. Proteomics 16 (4, suppl 1), S5–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Diner BA, and Cristea IM (2014). The Impact of Mass Spectrometry-Based Proteomics on Fundamental Discoveries in Virology. Annu. Rev. Virol 1, 581–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Guise AJ, and Cristea IM (2016). Determining the composition and stability of protein complexes using an integrated label-free and stable isotope labeling strategy. Methods Mol. Biol 1410, 39–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Kennedy MA, and Cristea IM (2020). Proteomic Technologies for Deciphering Local and Global Protein Interactions. Trends Biochem. Sci 45, 454–455. [DOI] [PubMed] [Google Scholar]
- Jean Beltran PM, and Cristea IM (2014). The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev. Proteomics 11, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean Beltran PM, Mathias RA, Cristea IM, Beltran PMJ, Mathias RA, and Cristea IM (2016). A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst. 3, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean Beltran PM, Cook KC, Hashimoto Y, Galitzine C, Murray LA, Vitek O, and Cristea IM (2018). Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell Host Microbe 24, 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada T, and Kawai S. (1989). An algorithm for drawing general undirected graphs. Inf. Process. Lett 31, 7–15. [Google Scholar]
- Kaurov I, Vancová M, Schimanski B, Cadena LR, Heller J, Bílý T, Potěšil D, Eichenberger C, Bruce H, Oeljeklaus S, et al. (2018). The Diverged Trypanosome MICOS Complex as a Hub for Mitochondrial Cristae Shaping and Protein Import. Curr. Biol 28, 3393–3407. [DOI] [PubMed] [Google Scholar]
- Keller A, Chavez JD, Eng JK, Thornton Z, and Bruce JE (2019). Tools for 3D Interactome Visualization. J. Proteome Res 18, 753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J, and Brocard C. (2012). PEX11 proteins attract Mff and human Fis1 to coordinate peroxisomal fission. J. Cell Sci 125, 3813–3826. [DOI] [PubMed] [Google Scholar]
- Kouzarides T, Bankier AT, Satchwell SC, Preddy E, and Barrell BG (1988). An immediate early gene of human cytomegalovirus encodes a potential membrane glycoprotein. Virology 165, 151–164. [DOI] [PubMed] [Google Scholar]
- Koyuncu OO, Perlman DH, and Enquist LW (2013). Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe 13, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozjak-Pavlovic V. (2017). The MICOS complex of human mitochondria. Cell Tissue Res. 367, 83–93. [DOI] [PubMed] [Google Scholar]
- Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, and Enquist LW (2012). Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12, 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen J, and Cristea IM (2013). Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chang C, Han M, Yin R, Zhan Y, Li C, Ge C, Yu M, and Yang X. (2019). PPIExp: A Web-Based Platform for Integration and Visualization of Protein-Protein Interaction Data and Spatiotemporal Proteomics Data. J. Proteome Res 18, 633–641. [DOI] [PubMed] [Google Scholar]
- Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, and Krogan NJ (2017). An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 169, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, and Semenkovich CF (2014). Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 19, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv BF, Yu CF, Chen YY, Lu Y, Guo JH, Song QS, Ma DL, Shi TP, and Wang L. (2006). Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N-terminal mitochondrial targeting sequence and induces apoptosis. Apoptosis 11, 1489–1501. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães AC, Ferreira AR, Gomes S, Vieira M, Gouveia A, Valença I, Islinger M, Nascimento R, Schrader M, Kagan JC, and Ribeiro D. (2016). Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep 6, 26028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick AL, Smith VL, Chow D, and Mocarski ES (2003). Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol 77, 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, Mayer-Barber K, Sher A, Wack A, and O’Garra A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low T, et al. (2013). The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, and Langer T. (2009). Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 1793, 27–32. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak W, Tsiamis V, Schwämmle V, and Rogowska-Wrzesińska A. (2019). ComplexBrowser: A Tool for Identification and Quantification of Protein Complexes in Large-scale Proteomics Datasets. Mol. Cell. Proteomics 18, 2324–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miryala SK, Anbarasu A, and Ramaiah S. (2018). Discerning molecular interactions: A comprehensive review on biomolecular interaction databases and network analysis tools. Gene 642, 84–94. [DOI] [PubMed] [Google Scholar]
- Modi S, López-Doménech G, Halff EF, Covill-Cooke C, Ivankovic D, Melandri D, Arancibia-Cárcamo IL, Burden JJ, Lowe AR, and Kittler JT (2019). Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun 10, 4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, and Shenk T. (2008). Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Sharon-Friling R, Shenk T, and Cristea IM (2010). A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullenbrock S, Shah J, and Cooper GM (2011). Global expression analysis identified a preferentially nerve growth factor-induced transcriptional program regulated by sustained mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) and AP-1 protein activation during PC12 cell differentiation. J. Biol. Chem 286, 45131–45145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LA, Sheng X, and Cristea IM (2018). Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun 9, 4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott C, Dorsch E, Fraunholz M, Straub S, and Kozjak-Pavlovic V. (2015). Detailed analysis of the human mitochondrial contact site complex indicate a hierarchy of subunits. PLoS ONE 10, e0120213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Collins MO, Lichawska A, Zegerman P, Choudhary JS, and Pines J. (2011). Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Mol. Cell 43, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph JD, and Cox J. (2019). A Network Module for the Perseus Software for Computational Proteomics Facilitates Proteome Interaction Graph Analysis. J. Proteome Res 18, 2052–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina D, Kuum M, Choubey V, Hickey MA, and Kaasik A. (2019). Mitochondrial transport proteins RHOT1 and RHOT2 serve as docking sites for PRKN-mediated mitophagy. Autophagy 15, 930–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon J, Goenawan IH, and Lynn DJ (2018). Analysis and Visualization of Dynamic Networks Using the DyNet App for Cytoscape. Curr. Protoc. Bioinformatics 63, e55. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma LK, Lu J, and Bai Y. (2009). Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr. Med. Chem 16, 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Schilling B, Ludwig C, Jaffe JD, MacCoss MJ, and MacLean B. (2018). Panorama public: A public repository for quantitative data sets processed in skyline. Mol. Cell. Proteomics 17, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon-Friling R, Goodhouse J, Colberg-Poley AM, and Shenk T. (2006). Human cytomegalovirus pUL37×1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA 103, 19117–19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staring J, Raaben M, and Brummelkamp TR (2018). Viral escape from endosomes and host detection at a glance. J. Cell Sci Published online August 3, 2018. 10.1242/jcs.216259. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45 (D1), D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Zhang K, Dong J, Yan C, Hu C, Ji H, Chen L, Chen S, Zhao H, and Song Z. (2020). Sam50–Mic19–Mic60 axis determines mitochondrial cristae architecture by mediating mitochondrial outer and inner membrane contact. Cell Death Differ. 27, 146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasenko D, Barbot M, Jans DC, Kroppen B, Sadowski B, Heim G, Möbius W, Jakobs S, and Meinecke M. (2017). The MICOS component Mic60 displays a conserved membrane-bending activity that is necessary for normal cristae morphology. J. Cell Biol 216, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney DJ, and Colberg-Poley AM (1991). Human cytomegalovirus UL36–38 and US3 immediate-early genes: temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J. Virol 65, 6724–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent HA, Ziehr B, and Moorman NJ (2016). Human cytomegalovirus strategies to maintain and promote mRNA translation. Viruses 8, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, and von Mering C. (2015). Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15, 3163–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang ECY, Aicheler R, Murrell I, Wilkinson GWG, et al. (2014). Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Smith GA, Enquist LW, and Shenk T. (2002). Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol 76, 2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Hildreth RL, and Colberg-Poley AM (2013). Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J. Virol 87, 5657–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data-dependent MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019). Access at PRIDE: PXD017163.
The targeted (PRM) MS data have been deposited to Panorama Public (Sharma et al., 2018): https://panoramaweb.org/InterViSTA.url and the RAW data to ProteomeXchange, which can be accessed at PRIDE: PXD013848.
Inter-ViSTA is freely available for download at https://github.com/cristealab/Inter-ViSTA and at intervista.princeton.edu.