

Abstract
Cocaine, amphetamine, and methamphetamine abuse disorders are serious worldwide health problems. To date, there are no FDA-approved medications for the treatment of these disorders. Elucidation of the biochemical underpinnings contributing to psychostimulant addiction is critical for the development of effective therapies. Excitatory signaling and glutamate homeostasis are well known pathophysiological substrates underlying addiction-related behaviors spanning multiple types of psychostimulants. To alleviate relapse behavior to psychostimulants, considerable interest has focused on GLT-1, the major glutamate transporter in the brain. While many brain regions are implicated in addiction behavior, this review focuses on two regions well known for their role in mediating the effects of cocaine and amphetamines, namely the nucleus accumbens (NAc) and the ventral tegmental area (VTA). In addition, because many investigators have utilized Cre-driver lines to selectively control gene expression in defined cell populations relevant for psychostimulant addiction, we discuss potential off-target effects of Cre-recombinase that should be considered in the design and interpretation of such experiments.
Keywords: Addiction, psychostimulants, glutamate, dopamine, nucleus accumbens, ventral tegmental area
1. Introduction
Psychostimulants including amphetamine (AMP), methamphetamine (METH), and cocaine are highly addictive drugs. Abuse of these drugs represents a serious health problem that leads to organ damage including the heart, lungs, kidneys, and liver (Lineberry & Bostwick, 2006; Perino, Warren, & Levine, 1987) as well as central nervous system dysregulation (Bramness et al., 2012; Neiman, Haapaniemi, & Hillbom, 2000). While these drugs have medical uses, they exhibit a substantial potential for abuse (George F. Koob, 2006). This potential for abuse occurs in part via their reinforcing effects. Amphetamines and cocaine are reinforcers, that is, they induce rewarding/euphoric effects mediated primarily through activation of the mesocorticolimbic dopamine (DA) system, which increase the probability that they will be taken again (Pierce & Kumaresan, 2006). The ventral tegmental area (VTA) is the origin of DA cell bodies that comprise the mesocortical and mesolimbic circuits (Adinoff, 2004; Ikemoto, 2010). It is well known that a defining characteristic of cocaine and AMP-like psychostimulants is their high affinity for DA transporters (DAT) (Ritz, Lamb, Goldberg, & Kuhar, 1987; Seiden, Sabol, & Ricaurte, 1993) resulting in an increase in the quantity and half-life of synaptic and extrasynaptic DA concentrations (Wayment, Schenk, & Sorg, 2001; J. E. Williams, Wieczorek, Willner, & Kruk, 1995). Traditionally, DAT ligands have been divided into two categories: cocaine-like inhibitors and AMP-like substrates (Schmitt, Rothman, & Reith, 2013). Cocaine-like inhibitors block monoamine uptake but are not translocated across the cell membrane whereas AMP-like substrates are translocated into the cell and induce DAT-mediated release of DA via reversal of DAT (Schmitt et al., 2013).
It has long been known that cocaine acts by elevating extracellular levels of DA via inhibition of the DAT throughout the striatum (Carboni, Imperato, Perezzani, & Di Chiara, 1989; Di Chiara & Imperato, 1988; Giros, Jaber, Jones, Wightman, & Caron, 1996; Rice, Patel, & Cragg, 2011; Sulzer, 2011; Sulzer, Cragg, & Rice, 2016). However, recent work suggests that cocaine also acts independently of DAT to increase DA transmission within the striatum via increasing DA neuronal firing within the VTA which is consistent with the continued reinforcing properties of cocaine in self-administration and cocaine place preference paradigms in mice whereby the DAT gene has been deleted (Buck, Torregrossa, Logan, & Freyberg, 2020; Carboni et al., 2001; Di Chiara et al., 2004). In contrast to cocaine, AMP and METH competitively inhibit DA uptake (Han & Gu, 2006; R. B. Rothman & Baumann, 2003) and increase DA release via reverse transport (Eshleman, Henningsen, Neve, & Janowsky, 1994; J. F. Fischer & Cho, 1976; S. R. Jones, Gainetdinov, Jaber, et al., 1998; S. R. Jones, Gainetdinov, Wightman, & Caron, 1998; Sitte et al., 1998; Wall, Gu, & Rudnick, 1995). DAT is thought to comprise 12 transmembrane segments containing numerous phosphorylation sites in the intracellular domains (Giros & Caron, 1993; Granas, Ferrer, Loland, Javitch, & Gether, 2003; Lin et al., 2003). Reversal of DAT induced by AMP requires phosphorylation of one or more of the first 5 serines in DAT (Khoshbouei et al., 2004). In addition to targeting DAT, amphetamines and cocaine accumulate at acidic, intracellular sites by a “weak base effect” (Sulzer, 2011; Sulzer & Rayport, 1990). More specifically, amphetamines and cocaine are lipophilic weak bases (Beckett & Moffat, 1969; Mack & Bonisch, 1979) that perturb proton gradients in intracellular compartments of DA neurons (Sulzer & Rayport, 1990) resulting in alkalization of vesicles leading to a reduction in the vesicular transmembrane pH gradient (Sulzer, 2011; Sulzer & Rayport, 1990). This reduction in the vesicular transmembrane pH gradient results in the release of DA from vesicles into the cytosol via an unknown mechanism and the release of cytosolic DA into the extracellular space via AMP-induced reversal of DAT (Freyberg et al., 2016). As discussed above, cocaine acts by blocking DAT which differs from AMP in that it does not result in increased extracellular levels of DA via reversal of DAT (Sulzer, 2011; Sulzer & Rayport, 1990). In addition to the accumulation of extracellular DA by blocking DAT, cocaine also results in increased extracellular levels of DA via increasing the firing of midbrain DA neurons (Koulchitsky, De Backer, Quertemont, Charlier, & Seutin, 2012; Sulzer, 2011). Although unexplored, it is potentially the case that the increases in intraneuronal DA induced by cocaine resulting from its “weak base effect” could result in extra DA availability to be released when the DA neurons fire in response to cocaine.
Research focusing on the neurobiology of drug addiction has traditionally focused on the mesocorticolimbic DA system (Pierce & Kumaresan, 2006; Volkow, Fowler, Wang, Swanson, & Telang, 2007; Wise, 2004). However, a growing body of literature has emerged indicating an important role for glutamate (GLU) in mediating the adaptive processes underlying psychostimulant addictions (Ernst & Chang, 2008; Kalivas, 2004; Szumlinski et al., 2017). Accumulating evidence over the years has indicated that disruptions in GLU homeostasis play a critical role (Kalivas, 2009). Additionally, reports over the past decade have shed light on the importance of GLU signaling by DA neurons in mediating responses to psychostimulants (Birgner et al., 2010; Hnasko et al., 2010; Hnasko & Edwards, 2012; Mingote et al., 2017). In contrast to traditional views that each neuron uses a single transmitter, these reports have demonstrated the coexistence of GLU and DA in individual neurons (Bourque & Trudeau, 2000; Dal Bo et al., 2004; Mendez et al., 2008; Stuber, Hnasko, Britt, Edwards, & Bonci, 2010; Sulzer et al., 1998). The main purpose of this review is to highlight literature demonstrating how GLU homeostasis is altered within the mesocorticolimbic DA system during the acute and chronic stages of psychostimulant addiction behaviors.
2. Regulation of Glutamate Homeostasis
2.1. Glutamate Release
GLU homeostasis involves the regulation of extracellular GLU levels in the synaptic and extrasynaptic spaces (Bezzi, Vesce, Panzarasa, & Volterra, 1999; Engeli et al., 2020; Schousboe, 1981; Takahashi et al., 1997). The sinks and sources involved in the regulation of GLU homeostasis are neuronal and glial GLU release and uptake mechanisms. A major source of extracellular GLU is nonvesicular release, as GLU levels are mostly insensitive to blocking voltage-dependent Na+ and Ca2+ channels (Bradford, Young, & Crowder, 1987; Jabaudon et al., 1999; Miele, Boutelle, & Fillenz, 1996; Timmerman & Westerink, 1997). A major source of nonvesicular, extrasynaptic GLU is from the cystine-GLU exchanger (xCT) (Bannai, 1986; Murphy, Schnaar, & Coyle, 1990; Warr, Takahashi, & Attwell, 1999). xCT is plasma membrane bound, Na+-independent, and primarily located on astrocytes (Cho & Bannai, 1990; Danbolt, 2001; Murphy et al., 1990; Ottestad-Hansen et al., 2018; Pow, 2001). This antiporter exchanges one extracellular cystine for one intracellular GLU molecule (Bannai, 1986). Within the NAc a major source of basal GLU levels arises from xCT (Baker, Xi, Shen, Swanson, & Kalivas, 2002). As will be discussed more later, basal levels of GLU are altered by cocaine (Baker, Shen, & Kalivas, 2002), METH (Lominac, Sacramento, Szumlinski, & Kippin, 2012; Parsegian & See, 2014), and other drugs of abuse (Griffin, Haun, Hazelbaker, Ramachandra, & Becker, 2014). Additionally, chronic reductions in extracellular levels of basal GLU contribute to the development of postsynaptic adaptations (Conrad et al., 2008; Massie, Boillee, Hewett, Knackstedt, & Lewerenz, 2015). One example of this is highlighted in Section 6.3, whereby chronic cocaine administration results in chronic reductions of basal GLU levels within the NAc leading to enduring synaptic potentiation within the NAc core (Conrad et al., 2008).
In addition to xCT, Baker and colleagues determined that mGluR2/3 and Na+-dependent GLU transporters also contribute to regulating extracellular GLU levels in the NAc (Baker, Xi, et al., 2002). They found that blocking Na+-dependent GLU transporters results in an increase in extracellular levels of GLU within the NAc that is prevented by blocking xCT. mGluR2/3 belongs to the group II metabotropic receptor family; they are negatively coupled to adenylyl cyclase and normally inhibit GLU neurotransmission (Conn & Pin, 1997). Within the NAc, these receptors are primarily located presynaptically and on glial processes (Robbe, Alonso, Chaumont, Bockaert, & Manzoni, 2002). Pharmacological activation of mGluR2/3 in the NAc inhibits GLU synaptic transmission and alters both the paired pulse ratio as well as the frequency of mEPSCs, thus indicating a presynaptic mechanism by which mGluR2/3s alter release (Robbe et al., 2002). Baker and colleagues demonstrated that the extracellular GLU that arises from xCT binds to mGluR2/3 thus resulting in a decrease in synaptic GLU release (Baker, Xi, et al., 2002). Interestingly, they also demonstrated that mGluR2/3 regulates the release of DA, as had been shown in earlier studies (Hu, Duffy, Swanson, Ghasemzadeh, & Kalivas, 1999). Baker and colleagues determined that xCT was the source for providing endogenous GLU tone on mGluR2/3 that is capable of modulating synaptic activity (Baker, Xi, et al., 2002).
In addition to modulating activity at mGluR2/3, extracellular GLU released via xCT has also been shown to regulate GLU synapse strength by suppressing the number of postsynaptic AMPA receptors in CA3-CA1 synapses (Massie et al., 2015; L. E. Williams & Featherstone, 2014). Using a mouse line in which xCT had been deleted, Williams and Featherstone demonstrated increased EPSC amplitudes at baseline, increased electrically evoked EPCS amplitudes, and increased AMPA receptor accumulation within hippocampus areas C13-CA1(L. E. Williams & Featherstone, 2014). The authors found that this phenotype (increases in baseline and evoked EPSCs and accumulation of AMPA receptors) was reproduced in control mice in which xCT had not been deleted and that the baseline and evoked EPSCs were blocked following application of the AMPA receptor antagonist, NBQX (L. E. Williams & Featherstone, 2014). The authors concluded that xCT in hippocampal astrocytes releases GLU that could potentially trigger loss of postsynaptic AMPA receptors to suppress synapse strength (L. E. Williams & Featherstone, 2014). Taken together, this work demonstrates how GLU homeostasis plays a role in regulating synaptic activity.
2.2. Glutamate Uptake
In addition to GLU release by both synaptic and extrasynaptic sources, another mechanism by which GLU homeostasis is maintained is via its uptake from the extracellular space. Excess levels of extracellular GLU can lead to excitotoxicity (D. W. Choi, 1988; Lipton & Rosenberg, 1994; Meldrum & Garthwaite, 1990). GLU transporters expressed in both glial cells and neurons serve to maintain low extracellular levels of GLU by binding (Wadiche, Amara, & Kavanaugh, 1995) and removing (Danbolt, Storm-Mathisen, & Kanner, 1992; Divac, Fonnum, & Storm-Mathisen, 1977; Levy, Lehre, Rolstad, & Danbolt, 1993; Schousboe, 1981; Storm-Mathisen & Iversen, 1979; Wilkin, Garthwaite, & Balazs, 1982) free GLU from the extracellular space. The removal of GLU from the extracellular space occurs via a family of five Na+-dependent GLU transporters (EAAT1/GLAST, EAAT2/GLT-1, EAAC1, EAAT4, and EAAT5). GLAST is found predominantly in astrocytes, EAAT3 in neurons, EAAT4 in cerebellar Purkinje cells, and EAAT5 is expressed throughout the retina (Danbolt, 2001). GLT-1 is the major GLU transporter in the CNS representing 1% of total brain protein (Lehre & Danbolt, 1998) and is responsible for >90% of synaptosomal GLU uptake (Danbolt, 2001; Petr et al., 2015; Tanaka et al., 1997). GLT-1 is primarily expressed in astrocytes (Rothstein et al., 1994) but also in neurons (Chen et al., 2004; Petr et al., 2015). In the hippocampus, area CA1, neuronal GLT-1 protein expression represents 5–10% of total GLT-1 expression (Furness et al., 2008). GLT-1 is also expressed in neurons within the human cortex (Melone, Bellesi, Ducati, Iacoangeli, & Conti, 2011), rat somatic sensory cortex (Melone, Bellesi, & Conti, 2009), and rat striatum (Petr et al., 2013). Pan knockout of GLT-1, or conditional knockout of GLT-1 restricted to astrocytes results in intractable seizures and premature death (Petr et al., 2015; Tanaka et al., 1997).
As outlined in Table 1, there are two areas of study that have highlighted an important role for GLU homeostasis in addiction behaviors; one involves the acute setting after the initial administration of psychostimulants as exemplified by the work of Wolf and colleagues (Wolf, Xue, Li, & Wavak, 2000) and others (Del Arco, Martinez, & Mora, 1998) and the second is the late post withdrawal symptoms exemplified by the work of Kalivas and colleagues (Kalivas, Lalumiere, Knackstedt, & Shen, 2009; Knackstedt, Melendez, & Kalivas, 2010). As will be discussed more thoroughly in this review, GLT-1 has long been implicated in psychostimulant addiction [see (Reissner & Kalivas, 2010) for review]. Although it has been assumed that the role GLT-1 plays in drug-seeking is due to its location on astrocytes, studies investigating the role of GLT-1 in drug-seeking behaviors were done before the appreciation that GLT-1 is expressed in neurons, with few exceptions (K. D. Fischer et al., 2018; Xu et al., 2003). Thus, there has been no rigorous examination of the role of GLT-1 in neurons in addiction behavior.
Table 1.
Changes in Extracellular Levels of GLU following Acute and Chronic Administration of Psychostimulants
| Acute Administration of Drug | ||||
|---|---|---|---|---|
| Brain Region | Direction and Timepoint of Change in Extracellular Levels | Treatments that Block Drug Effects on Extracellular GLU Levels | Drug Administration Protocol | |
| AMP | NAc (core and shell) | Delayed (~3 hr) Increase (Xue, Ng et al. 1996) | Haloperidol | Single Injection 5 mg/kg i.p. (free base) |
| VTA | Immediate Decrease following by Delayed (~3 hr) Increase (Wolf and Xue 1998; Xue, Ng et al. 1996) | DHK, SCH23390, lesions of PFC (Wolf and Xue 1999) | Single Injection 5 mg/kg i.p. (free base) | |
| Cocaine | NAc (subregion unspecified) | Immediate Increase (Smith, Mo et al. 1995) | TTX (Smith, Mo et al., 1995) | Single Injection 15 and 30 mg/kg i.p. |
| VTA | Immediate Increase (Kalivas and Duffy 1995) | SCH23390 (Kalivas and Duffy 1995) | Single Injection 15 mg/kg i.p. | |
| METH | NAc (subregion unspecified) | Delayed (~2 hr) Increase (Ito et al. 2006) | SCH23390 (Ito et al., 2006) | Single Injection 2.5 mg/kg s.c. (salt form) |
| Striatum | Immediate Increase following 3rd Injection (Stephans and Yamamoto 1994) | Bicuculline (Stephans and Yamamoto 1994) | 10 mg/kg i.p. x 3, every 2 hr | |
| Chronic Administration of Drug | ||||
| Brain Region | Direction of Change in Extracellular Levels | Treatments that Block Drug Effects on Extracellular GLU Levels | Drug Administration Protocol | |
| AMP | NAc (core and shell) | Delayed (~3 hr) Increase (Xue, Ng et al. 1996) | Data Not Shown | 5 mg/kg i.p. x 5 d (challenge 2 d post-withdrawal) (free base) |
| VTA | Delayed (~3 hr) Increase (Wolf and Xue 1998) | MK-801, SCH23390, lesions of PFC (Wolf and Xue 1998, 1999); PBN (Wolf, Xue et al. 2000) | 5 mg/kg i.p. x 5 d (challenge 2 d post-withdrawal) (free base) | |
| Cocaine | NAc core | Decrease after Self-Administration; Increase following Reinstatement or Challenge (McFarland, Lapish et al. 2003; Pierce, Bell et al. 1996) | TTX, muscimol infusions into the Prelimbic Cortex (McFarland, Lapish et al. 2003) | IV Self Administration (2 hr/day)/Extinction Training/Drug-induced Reinstatement |
| METH | NAc core | Decrease after Self-Administration; Increase following Reinstatement (Parsegian and See 2014) | Data Not Shown | IV Self Administration (2 hr/day)/Extinction Training/Drug and Cue-induced Reinstatement |
| NAc (core and shell) | Increase after Self-Administration (Lominac, Sacramento et al. 2012) | Data Not Shown | IV Self Administration (2 hr/day)/Forced Withdrawal | |
| PFC | Decrease after Self-Administration; Increase following Reinstatement (Parsegian and See 2014) | Data Not Shown | IV Self Administration (2 hr/day)/Extinction Training/Drug and Cue-induced Reinstatement | |
In the table. Amphetamine (AMP), Methamphetamine (METH), Haloperidol (D2 antagonist), SCH23390 (D1 antagonist), DHK (GLT-1 blocker), TTX (Sodium channel blocker), Bicuculline (GABA-A antagonist), MK-801 (NMDA antagonist), muscimol (GABA agonist), PBN (spin trapping agent for hydroxyl radicals). Rats were used in all experiments.
3. Pitfalls of Cre/lox technology
Transgenic rodent models utilizing Cre-recombinase driver lines are frequently used to provide genetic access to particular cell populations in the brain (Lammel et al., 2015; Song & Palmiter, 2018). Many reports have used Cre-drivers to target genes expressed in midbrain DA neurons that coexpress other neurotransmitters including GLU (Hnasko et al., 2010; Hnasko & Edwards, 2012; Mingote et al., 2017; Papathanou et al., 2018; Stuber et al., 2010; Wang et al., 2017) and GABA (J. I. Kim et al., 2015; Ntamati & Luscher, 2016; Tritsch, Oh, Gu, & Sabatini, 2014). These studies are generally focused on identifying roles for signaling by these “secondary” transmitters in addiction-related behaviors (Birgner et al., 2010; Hnasko et al., 2010; Hnasko & Edwards, 2012; J. I. Kim et al., 2015; Mingote et al., 2017; Papathanou et al., 2018). Two of the most commonly used Cre-lines drive expression under the tyrosine hydroxylase promoter (TH-Cre; (Lindeberg et al., 2004; Savitt, Jang, Mu, Dawson, & Dawson, 2005) or the DAT promotor (DAT-Cre; (Backman et al., 2006; Zhuang, Masson, Gingrich, Rayport, & Hen, 2005). The Cre-lox method has greatly impacted the field allowing for the understanding of gene functions whose global knockout might otherwise result in lethality (Song & Palmiter, 2018). However, while Cre-recombinase provides control over gene expression, there are limitations that need to be considered when both Cre-recombinase and loxP sites are expressed in animals that will be bred (Song & Palmiter, 2018).
One consideration is that there may be unexpected Cre-recombinase expression in the germline or in the animal during development, referred to as ‘ectopic’ expression (Luo et al., 2020; Rempe et al., 2006; Song & Palmiter, 2018). An example of ectopic Cre-recombinase expression was reported by Lammel and colleagues who showed that TH-Cre expressing mice exhibited profound transgene expression in non-DA neurons (e.g. TH-immunonegative neurons) (Lammel et al., 2015). An example of germline expression of Cre-recombinase was reported by Rempe and colleagues who found germline expression of synapsin-Cre in males related to physiological expression of synapsin 1 in the testes (Rempe et al., 2006). Expression of synapsin 1 in the testes necessitates use of females only for introduction of synapsin 1-Cre into conditional mouse lines. These reports exemplify the need for testing for ectopic or germline expression of Cre-recombinase (K. D. Fischer et al., 2018; Rempe et al., 2006).
The second consideration involves the oversimplification inherent in using single Cre-recombinase driver lines to study neurocircuitry and behavior (Stuber, Stamatakis, & Kantak, 2015). Many reports have demonstrated that midbrain DA neurons are associated with multiple modalities of neurotransmission (Aguilar et al., 2017; Papathanou et al., 2018; Stuber et al., 2010; Tecuapetla et al., 2010; Tritsch, Ding, & Sabatini, 2012). For example, 20% of VTA TH-expressing neurons coexpress a marker for GLU neurons, VGLUT2 (Hnasko et al., 2010; Hnasko & Edwards, 2012; Kawano et al., 2006; X. Li, Qi, Yamaguchi, Wang, & Morales, 2013; Mendez et al., 2008; Root et al., 2014; Yamaguchi, Wang, Li, Ng, & Morales, 2011). Optogenetic experiments have shown that stimulation of DA fibers results in the release of DA, GLU and GABA in striatal subregions (Stuber et al., 2010; Tecuapetla et al., 2010; Tritsch et al., 2012). Additionally, Root and colleagues found that the TH-expressing lateral habenula DA fibers that project to the VTA release both GLU and GABA (Root et al., 2014). These results indicate that single DA, GLU, and GABA related gene markers such as TH, DAT, VGLUT2, or GAD65/67 do not fully define the molecular phenotypes of midbrain DA cell subpopulations (Stuber et al., 2015).
The third consideration involves the potential for Cre-recombinase in itself to induce effects by off-target, “non-specific” actions that may or may not be defined, thus complicating interpretations of the role(s) that a particular gene may play in the phenomenon under investigation (Giusti et al., 2014). One important off target effect is Cre-induced cell toxicity. Cre-recombinase insertion into the genome can result in growth arrest, chromosomal abnormalities, and cell death both in cultured cells and in whole animals (Janbandhu, Moik, & Fassler, 2014; Loonstra et al., 2001; Thanos et al., 2012). In addition. there is the potential for expression of Cre-recombinase in different cells and circuits to produce changes in cell physiology that results in non-lethal biochemical, cellular, electrophysiological, and behavioral alterations that have nothing to do with modulation of expression of the target gene. Giusti and colleagues demonstrated the importance of running relevant experimental groups to control for potential Cre-mediated behavioral effects (Giusti et al., 2014), and this caution applies to all experiments involving the use of Cre-recombinase. One of the most extensively used Cre mouse lines in psychiatric research, referred to as the NestinCre mouse (Tronche et al., 1999) was found to have altered fear conditioning due to the expression of Cre-recombinase per se. Others utilizing this Cre-driver, reported behavioral abnormalities, including diminished fear learning, but these studies used “floxed” mice as their control groups (no Cre) and did not include a control for Cre itself (Gao et al., 2010; Suzuki, Ferris, Chee, Maratos-Flier, & Kahn, 2013). Thus, as pointed out by (Giusti et al., 2014), it was not clear whether the induced mutation in the NestinCre mouse or Cre expression itself was responsible for the diminished fear learning response.
Given the problems inherent in using Cre/lox technology, an important question is how best to design an experiment relying on this approach to target genes for inactivation. Most important is the need to recognize that there are problems, and that appropriate controls need to be included in such experiments. One approach is for all groups to express the floxed allele(s) and for the test group to express Cre-recombinase under the appropriate promoter. Subsequently, if there is an effect of Cre expression, then a second experiment needs to be performed testing the effect of Cre-recombinase itself on a wild type background. The results of the first experiment are uninterpretable without the second experiment. An alternative approach is to design experiments in which all mice express the Cre-recombinase, and for the test mice to have the floxed allele(s). Any effect in this case would be related to modification of the floxed allele(s) and not the expression of Cre-recombinase per se. Obviously additional experiments would need to be done to be sure that the presence of the floxed allele(s) were not responsible for the effects observed. This approach of having all groups express Cre-recombinase would seem to eliminate the problem of confusing Cre-related effects for target specific effects. However, it is also important to note that the physiology of mice expressing Cre is not necessarily the same as the physiology of mice not expressing Cre (Gewin, 2019). Therefore, it is conceivable that effects that appear that are target specific but might occur only on a background of Cre-recombinase expression.
In summary, Cre/lox technology is a critically important approach for determining the role of specific proteins expressed in specific cells in neural circuits (Deisseroth, 2014; Yizhar, Fenno, Davidson, Mogri, & Deisseroth, 2011). However, the aforementioned considerations are crucial for the design and interpretation of any studies involving the expression of Cre-recombinase in mammalian cells.
4. Glutamate Signaling by Dopamine Neurons
Evidence demonstrating the release of GLU by midbrain DA neurons was obtained initially in microcultures of isolated VTA neurons (Bourque & Trudeau, 2000; Sulzer et al., 1998). Consistent with the use of GLU as a neurotransmitter in DA neurons was the discovery that VGLUT2 is coexpressed with TH and VMAT2 in DA neurons of adult rodents (Dal Bo et al., 2004; Hnasko et al., 2010; Mendez et al., 2008; Silm et al., 2019). In addition to mice and rats, the coexpression of VGLUT2/TH has been identified within the VTA of non-human primates and humans (Root et al., 2016). The primary question currently is what is the biochemical, cellular, and behavioral significance of GLU co-transmission in DA neurons? Using a VTA-NAc slice preparation, Chuhma and colleagues demonstrated that direct stimulation of VTA DA neurons elicits a monosynaptic EPSC in NAc shell medium spiny neurons (Chuhma, Choi, Mingote, & Rayport, 2009; Chuhma et al., 2004). Using this same preparation, Hnasko and colleagues demonstrated that conditional deletion of VGLUT2 from DA neurons by expression of Cre-recombinase under the influence of the DA transporter promoter (datVGLUT2 KO) results in reduced EPSC amplitude in NAc shell neurons in response to VTA stimulation suggesting that dual GLU-DA neurons in the VTA form excitatory synapses in the NAc (Hnasko et al., 2010). They also found reduced DA tissue content and electrically evoked DA release in the NAc shell of datVGLUT2 KO mice. These findings were explained by the phenomenon of vesicular synergy whereby the transport of GLU into presynaptic vesicles by VGLUT2 facilitates the loading of DA into the same vesicles (Hnasko & Edwards, 2012; Trudeau et al., 2014). It was recently shown in both a Drosophila and mouse model that DA vesicles hyperacidify in response to neuronal depolarization and that GLU transport across VGLUT2 into the synaptic vesicle is necessary to potentiate the depolarization-induced changes in pH (Aguilar et al., 2017). Furthermore, Stuber and colleagues utilized the datVGLUT2 KO combined with optogenetics to selectively stimulate DA terminals while recording postsynaptic currents in NAc medium spiny neurons (Stuber et al., 2010). They found that VGLUT2 in DA neurons is required for GLU release in NAc shell DA terminals of adult mice. While co-release has been detected from VTA DA terminals in the shell, it was not detected in dorsal striatal DA terminals (Stuber et al., 2010). It was recently shown that while DA neurons within the SNc that project to the medial dorsal striatum do not coexpress GLU, SNc DA neurons that project to the lateral dorsal striatum and to the tail of the striatum coexpress GLU (Cai & Ford, 2018; Chuhma et al., 2018; Poulin et al., 2018). Thus, within the striatum, GLU corelease from DA neurons appears to be highly regionally specific.
An important concern to address is the impact that DAT-Cre driven conditional knockout of VGLUT2 could have on the development of DA neurons. Fortin and colleagues found that the abrogation of GLU transmission in the DAT-Cre driven conditional knockout of VGLUT2 results in impairment of DA neuron survival and axonal arborization in vitro, and compromises DA neuron development resulting in a 20% decrease in DA neuron number (Fortin et al., 2012). These findings by Fortin and colleagues (Fortin et al., 2012) as well as by others discussed above (Fortin et al., 2012; Hnasko et al., 2010; Stuber et al., 2010) utilized the DAT-Cre mouse developed by Zhuang and colleagues (JAX Stock No: 020080) that express Cre-recombinase through the 5’UTR, immediately upstream of the DAT translation start codon (Zhuang et al., 2005). The control groups by which comparisons were made to the datVGLUT2 KO mice in these reports were all DAT-Cre expressing. A problem with the 5’ untranslated region (UTR) knockin approach is that it disrupts one copy of the DAT gene, and DAT heterozygous KO mice show decreases in DAT protein expression, D1/D2 receptor mRNA and protein expression and a decrease in DA and TH levels (Backman et al., 2006; Beeler et al., 2020; Giros et al., 1996; S.R. Jones, Gainetdinov, Jaber, et al., 1998). In addition to targeting the 5’UTR of the DAT gene, another DAT-Cre mouse model was developed by Backman (JAX Stock No: 006660) whereby the Cre gene insertion is preceded by an internal ribosomal entry site (IRES) in the 3’UTR of the DAT gene (Backman et al., 2006). Compared to targeting the 5’UTR of the DAT gene, DA transmission and mRNA levels of D1/D2 receptors are significantly less affected by targeting the 3’UTR (Backman et al., 2006). However, recently it was shown that DAT protein expression was downregulated and DA release dynamics were altered in the DAT-Ires-Cre mouse (Beeler et al., 2020). Evoked DA release assessed via fast-scan cyclic voltammetry showed decreased clearance and increased peak amplitude (Beeler et al., 2020). Although DA receptor loss or DA cell loss was not found in the DAT-IRES-Cre mouse (Backman et al., 2006; Steinkellner et al., 2018), it is important to note that the altered DA release dynamics associated with this mutation may complicate the interpretation of behavioral or molecular results derived from lines created by using this Cre-driver. As discussed, utilizing the proper Cre control groups whereby comparisons are made with groups that also express Cre-recombinase could alleviate this concern.
While evidence indicates that VGLUT2 is expressed more broadly in DA neurons during development, it is important to note that VGLUT2 expression is downregulated in vivo over the course of development (Berube-Carriere et al., 2009; Mendez et al., 2008). Though downregulated in the adult, coexpression of VGLUT2 and DA markers in the adult mouse is most pronounced in the medial VTA (Hnasko, Hjelmstad, Fields, & Edwards, 2012; Mendez et al., 2008; Steinkellner et al., 2018; Yamaguchi, Qi, Wang, Zhang, & Morales, 2015). Because it is these medial VTA DA neurons that are generally spared in Parkinson’s Disease (PD) animal models (Berthet et al., 2014; Damier, Hirsch, Agid, & Graybiel, 1999; Hirsch, Graybiel, & Agid, 1988; Jackson-Lewis, Jakowec, Burke, & Przedborski, 1995; Schneider, Yuwiler, & Markham, 1987), Steinkellner wanted to determine the relationship between VGLUT2 expression and vulnerability to DA neuron degeneration (Steinkellner et al., 2018). They reported that VGLUT2 coexpression in adult SNc and VTA DA neurons is low but that the vast majority of these DA neurons expressed VGLUT2 during development (Steinkellner et al., 2018). Interestingly, compared to DAT-Cre expressing control mice, conditional VGLUT2 KO mice driven by the DAT-Ires-Cre promoter were found to have disrupted GLU transmission in DA neurons in the adult conditional VGLUT2 KO observed via a lack of optogenetic-evoked EPSCs in striatal MSNs (Steinkellner et al., 2018). Thus, in contrast to what was originally reported by Stuber and colleagues (Stuber et al., 2010), VGLUT2 expression in SNc DA neurons appears to be important for GLU corelease (Steinkellner et al., 2018). Furthermore, VGLUT2 coexpression specifically within SNc DA neurons reemerges in adult mice following DA neuron insult (6-OHDA or MPTP lesions) and deletion of VGLUT2 from DA neurons resulted in increased susceptibility of SNc DA neurons to Parkinsonian-related injuries such as 6-OHDA and MPTP lesions (Steinkellner et al., 2018). Similarly, over-expression of VGLUT2 from DA neurons induced a Parkinsonian-related phenotype including death of SNc DA neurons and increased rotational behavior (Steinkellner et al., 2018). This group also investigated whether other neuronal populations were affected by over-expression of VGLUT2 and found that GABA neurons in the VTA, GLU neurons in the subthalamic nucleus, striatal cholinergic interneurons, and serotonin neurons in the dorsal raphe were spared (Steinkellner et al., 2018). These data suggest that the homeostatic balance of VGLUT2 levels in SNc DA neurons is a crucial determinant of SNc DA neuron survival and that either too high or too low VGLTU2 levels in SNc DA neurons may contribute to a Parkinsonian-phenotype (Steinkellner et al., 2018).
Mingote and colleagues took an alternate approach to investigating a role for GLU signaling in DA neurons by downregulating GLU signaling from DA neurons using GLU as a neurotransmitter (Mingote et al., 2017). They targeted phosphate-activated glutaminase (PAG; GLS1) in DA neurons with the idea that they would be reducing presynaptic GLU content modestly without having an effect on DA neuron vesicular dynamics or DA neuron development (Mingote et al., 2017). The majority of presynaptic GLU arises from the actions of PAG; once presynaptic GLU is release, it is taken up by neighboring astrocytes and converted to glutamine and then transferred back to the presynaptic terminal where it is converted back to GLU by PAG (Marx, Billups, & Billups, 2015; Mingote et al., 2017). Mingote and colleagues created a DAT-Ires-Cre-driven conditional HET GLS1 mouse (cHET) in order to determine whether GLU cotransmission in DA neurons was altered compared to DAT-Cre expressing control mice (Mingote et al., 2017). Since heterozygous reduction in GLS1 is sufficient to attenuate GLU transmission (Gaisler-Salomon et al., 2009), and in order to minimize compensatory mechanisms observed in the homozygous GLS1 KO mice (Bae, Wang, Li, Rayport, & Lubec, 2013), Mingote and colleagues used DAT GLS1 (cHET) mice and DAT-Ires-Cre mice as controls (Mingote et al., 2017). They measured DA neuron GLU cotransmission within the NAc shell utilizing single pulse photostimulation and burst photostimulation (thought to mimic in vivo phasic firing of DA neurons) (Paladini & Roeper, 2014) in DA nerve terminals (Mingote et al., 2017). Compared to DAT-Cre control mice, the frequency of single-evoked EPSCs in the NAc shell was decreased in cHET mice while the amplitude of EPSCs did not differ between genotypes (Mingote et al., 2017). Interestingly, DA neuron control of NAc shell cholinergic interneurons was attenuated in cHET mice which was quantified using the firing ratio defined as the firing frequency during train photostimulation divided by the preceding 2 s of baseline firing (Mingote et al., 2017). While they found no genotype differences in baseline firing frequencies, the firing ratio of NAc shell cholinergic interneurons was significantly reduced in cHET mice (Mingote et al., 2017). While they showed greater burst firing in DAT-Cre controls compared to cHETs, there was no difference in the post-burst period (Mingote et al., 2017). The authors concluded that PAG plays a role in mediating DA neuron GLU cotransmission at higher firing frequencies and controls their ability to drive NAc shell cholinergic interneurons to fire in bursts (Mingote et al., 2017). Finally, the authors wanted to determine whether DA transmission was altered in the cHET mice. They performed fast-scan cyclic voltammetry within the NAc shell of cHET and DAT-Cre control mice and found no differences in DA release (Mingote et al., 2017). The decay time constant of DA responses did not differ between genotypes following single or burst photostimulation (Mingote et al., 2017). Thus, conditional GLS1 reduction does not appear to affect DA release within the NAc shell. Importantly, these authors also noted behavioral effects of this genetic manipulation that will be discussed in Section 5.5.
All of the gene targeting studies mentioned above that utilized promoters of both VGLUT2 and DAT genes have embryonic onset and thus suffer from the uncertainty that any of the phenotypes observed might be due to developmental circuitry adaptations to the knockout (Papathanou et al., 2018). Additionally, the apparent age-dependent decrease in VGLUT2 expression within VTA DA neurons make it challenging to dissociate a role for VGLUT2 in mature DA neurons from developmental compensatory adaptations (Papathanou et al., 2018). Papathanou and colleagues conducted a thorough series of experiments to address the role of VGLUT2 in fully matured DA neurons by utilizing a tamoxifen-inducible DAT-Cre transgene (DAT-CreERT2) (Engblom et al., 2008; Papathanou et al., 2018). This allowed them to fully control for the temporal aspects of DAT-Cre recombination (Papathanou et al., 2018). They compared the biochemical and behavioral phenotypes of two separate VGLUT2 mouse lines whereby VGLUT2 was deleted either during development that contained embryonal onset of the transgene or in mature DA neurons with a tamoxifen inducible DAT-Cre (Papathanou et al., 2018). The two DAT-Cre drivers that were used in this study were: 1 – DAT-Cre transgenic mouse line with embryonal onset of the transgene (Ekstrand et al., 2007) abbreviated ‘eDAT-Cre’ and 2 – Tamoxifen-inducible DAT-CreERT2 mice (Engblom et al., 2008) abbreviated ‘txDAT-Cre’. To achieve selective gene deletion in DA neurons, the Cre construct was based on a bacterial artificial chromosome harboring the DAT gene at the 5’UTR locus (Ekstrand et al., 2007). The advantage of this approach is that the endogenous DAT gene is not compromised (Papathanou et al., 2018; Parlato, Rieker, Turiault, Tronche, & Schutz, 2006) although it is still possible that the expression of DAT and other DA related genes might be affected. Similar to what they and others had shown with embryonic targeting of VGLUT2 (Wang et al., 2017), light-evoked EPCSs were significantly dampened in the txDAT-Cre VGLUT2 KO mouse thus confirming that GLU transmission is reduced when VGLUT2 is targeted in mature DA neurons (Papathanou et al., 2018). This group further analyzed the effects of cocaine on EPCS in these lines and these results will be discussed in more detail below in Section 5.5. Furthermore, in the following sections we will discuss how GLU signaling by DA neurons plays a role in the underlying adaptations associated with psychostimulant administration and addiction behaviors (Birgner et al., 2010; Fortin et al., 2012; Hnasko et al., 2010; Mingote et al., 2017; Paladini, Fiorillo, Morikawa, & Williams, 2001; Papathanou et al., 2018; Underhill et al., 2014; Ungless, Whistler, Malenka, & Bonci, 2001).
5. Psychostimulant-Induced Alterations in Excitatory Signaling and Glutamate Homeostasis
5.1. Acute AMP-induced Alterations of Excitatory Signaling
Acute administration of cocaine and AMP-related psychostimulants results in alterations in excitatory transmission and in GLU homeostasis (Adrover, Shin, & Alvarez, 2014; S. Jones & Kauer, 1999; M. H. Li et al., 2017; Padgett et al., 2012; Paladini et al., 2001; Schilstrom et al., 2006; Uchimura & North, 1991; Underhill et al., 2014; Ungless et al., 2001). Using whole-cell patch-clamp recordings in VTA DA neurons, Jones and Kauer demonstrated that acute AMP administration results in an immediate depression of excitatory signaling in DA neurons by an unknown mechanism (S. Jones & Kauer, 1999). Paladini and colleagues conducted a thorough set of experiments to determine in what way acute AMP alters excitatory transmission. The authors proposed that AMP alters excitatory signaling in VTA DA neurons via two ways: 1 – inhibition of excitatory signaling via AMP-stimulated DA release and activating D2 autoreceptors on midbrain DA neurons (as had been previously reported) (Groves, Wilson, Young, & Rebec, 1975; Mercuri, Calabresi, & Bernardi, 1989), and 2 – excitation by desensitizing the mGluR-mediated hyperpolarization induced by AMP (Paladini et al., 2001). More specifically, they performed intracellular recordings of VTA DA neurons and found that with repetitive stimulation, the first 3–4 stimuli resulted in AMPA- and NMDA-mediated EPSPs (Paladini et al., 2001). After the first few stimuli, the membrane potential hyperpolarized (assessed via an increase in IPSPs) and this hyperpolarization resulted from mGluR1 activation (Paladini et al., 2001). Superinfusion of AMP in these DA cells attenuated the mGluR1-mediated hyperpolarization. Interestingly, the mechanism underlying the AMP-induced inhibition of IPSPs was found to involve activation α1 adrenergic receptors on VTA DA neurons, presumably via AMP-stimulated DA release (Paladini et al., 2001). Activation of these α1 receptors was found to suppress the release of Ca2+ by activation of mGluR1 or by intracellular application of inositol 1,4,5-triphosphate (InsP3) (Paladini et al., 2001). Production of InsP3 mediates the release of Ca2+ by mGluRs (Morikawa, Imani, Khodakhah, & Williams, 2000). To summarize, Paladini and colleagues propose that the AMP-induced inhibition of mGluR1 IPSPs involves the release of DA stimulated by AMP, DA-mediated activation of α1 receptors on VTA DA neurons, and inhibition of InsP3-mediated Ca2+ release from internal stores (Paladini et al., 2001).
5.2. Acute AMP-induced Alterations of Glutamate Homeostasis
Since changes in excitatory transmission have been demonstrated following AMP then one might speculate that changes in GLU homeostasis, if they occur, might be important. Using in vivo microdialysis, Xue and colleagues demonstrated that acute AMP i.p. resulted in a delayed increase in GLU efflux in VTA and NAc (Xue, Ng, Li, & Wolf, 1996). However, this same group later reported that when AMP was administered directly into the VTA, there was an immediate decrease in VTA GLU efflux followed by a delayed increase in VTA GLU efflux (Wolf & Xue, 1998). The immediate decrease in VTA GLU accumulation reported by Wolf and colleagues is in agreement with Jones and Kauer’s finding that acute AMP attenuates GLU synaptic transmission (S. Jones & Kauer, 1999). Furthermore, Xue and colleagues reported that the delayed increase in extracellular GLU becomes significant at ~3 hrs post-AMP injection and is not Ca2+-dependent (Xue et al., 1996) suggesting that synaptic release is not responsible. Rather, they speculated that the delayed nature of AMP-stimulated GLU release involved inhibition of GLU transporters, allowing build up of GLU from other sources, or reversal of GLU transporters, recognizing that GLU transporters themselves might be the source (Wolf et al., 2000). In fact, they found that the sustained increase in AMP-induced GLU release was blocked by the GLT-1 specific inhibitor DHK (Wolf et al., 2000). This result argues for reversal of transport being the underlying mechanism of the AMP-induced increase in GLU release. In Figure 1, we have depicted potential signaling pathways implicated in producing reversal of GLU transport following AMP administration.
Figure 1. Signaling pathways implicated in the perturbation of glutamate homeostasis in the VTA induced by AMP.
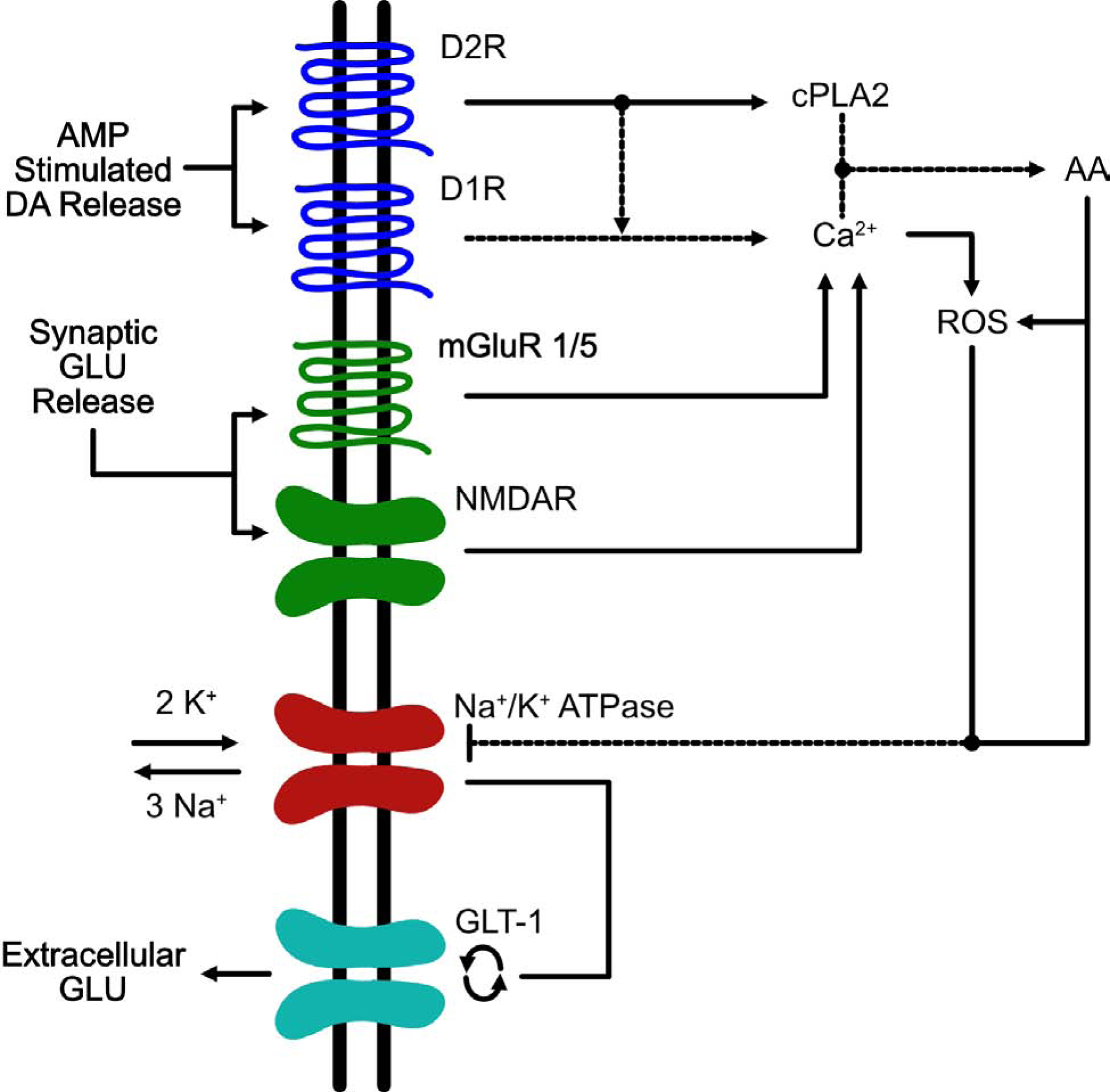
This is an example of a VTA astrocyte in which AMP/METH activates a signaling cascade that ultimately results in the reversal of EAATs. Administration of amphetamines results in the reversal of the dopamine transporter DAT and a decrease in vesicular uptake via VMAT which ultimately results in an accumulation of extracellular levels of DA within multiple brain regions including the VTA (Sulzer and Rayport 1990, Giorgetti, Hotsenpiller et al. 2001). DA released by DAT reversal stimulates D1- and D2-like receptors on VTA astrocytes (Liu, Wang et al. 2009, Zhang, Zhou et al. 2009). Activation of D2 receptors results in activation of the effector enzyme, cytoplasmic phospholipase 2 (cPLA2) (Vial and Piomelli 1995, Bhattacharjee, Chang et al. 2006). The combined stimulation of D1/D2R increases the release of intracellular Ca2+ which along with cPLA2 helps to free arachidonic acid (AA) from phospholipid membranes (Lee, So et al. 2004). AA release results in increased levels of reactive oxygen species (ROS) (Chan and Fishman 1980, Chan, Chen et al. 1988, Sakuma, Kitamura et al. 2012). Both AA and ROS inhibit the Na+/K+ pump (Na+/K+ -ATPase) leading to an increase in cytosolic potassium and a decrease in cytoplasmic sodium (Hexum and Fried 1979, Chan, Kerlan et al. 1983, Volterra, Trotti et al. 1994). The inhibition of Na+/K+ -ATPase results in two events: 1 – depolarization of the cell membrane and 2 – disruption of the Na+/K+ gradients; both of these events can result in the reversal of EAATs (Nicholls and Attwell 1990, Volterra, Trotti et al. 1994, Anderson, Huguenard et al. 2010). The increase in extracellular GLU following AMP administration is most likely due to reversal of GLT-1, as the increases in GLU are completely blocked by the GLT-1 blocker, DHK (Wolf, Xue et al. 2000). Another source of increased extracellular GLU within the VTA arises from GLU input from the PFC. DA projections from the VTA to the PFC play a critical role in modulating PFC GLU output back to the VTA (Sesack and Pickel 1992). By increasing the activity of VTA neurons, AMP/METH could activate this VTA-PFC-VTA circuit resulting in increased extracellular GLU levels within the VTA and in fact it has been shown that acute AMP results in increased activation of this circuit following AMP administration in rats (Colussi-Mas, Geisler et al. 2007). Furthermore, increased levels of extracellular GLU in VTA can activate mGluR1/5 and NMDARs located on the astrocyte resulting in a further increase in intracellular Ca2+ and AA (Biber, Laurie et al. 1999, Daniels and Brown 2001, Lalo, Pankratov et al. 2006, Lee, Ting et al. 2010) which continues the cycle of increases in ROS, decreases in Na+/K+ -ATPase function, and increases in extracellular levels of GLU via reversal of GLT-1.
The delayed nature of AMP-stimulated GLU release found by Wolf and Xue (Wolf & Xue, 1999) suggests that the rise in extracellular GLU is not responsible for the acute locomotor activating effects induced by AMP. In support of this conclusion, these authors reported that while ibotenic lesions of the PFC and administration of either a NMDA or D1 receptor antagonist blocked the delayed increase in AMP-induced GLU efflux, only the D1 receptor antagonist blocked AMP-stimulated locomotor activation (Wolf & Xue, 1999), suggesting a disconnect between the delayed increase in VTA GLU efflux and the increased locomotor activation induced by acute AMP, and also that the increase in locomotion was dependent upon D1R activation. Interestingly, all three of these treatments that were found to block the increase in the delayed AMP-stimulated GLU efflux prevented the development of sensitization suggesting an important role for repeated increases in AMP-stimulated GLU efflux within VTA in mediating longer term addiction behaviors. This is discussed in more detail in Section 6.2.
Because GLU is released by DA neurons, it is a reasonable assumption that DA neurons contain the machinery to remove GLU from the extracellular space. EAAT3 is one of five GLU transporter subtypes and is primarily expressed in neurons (Danbolt, 2001; Holmseth et al., 2012; Rothstein et al., 1994). Underhill and colleagues demonstrated that EAAT3 is coexpressed with DAT in VTA DA neurons that project to NAc (Underhill et al., 2014). They also found that uptake of AMP by DAT induces internalization of EAAT3; using acute brain slices they demonstrated that cocaine, which blocks the function of DAT, blocks the AMP-mediated endocytosis of EAAT3. More recently, they found that AMP is also transported by NET and DAT located in noradrenergic neurons and induces endocytosis of both NET and EAAT3 (Underhill, Colt, & Amara, 2020) thus suggesting that this phenomenon is not specific for DA neurons. Furthermore, Underhill et al. assessed the effects of AMP on evoked GLU synaptic currents (eEPSCs) in DA neurons within the VTA and substantia nigra (M. H. Li et al., 2017; Underhill et al., 2014). They found that acute AMP administration potentiates NMDA-mediated synaptic currents and decreases AMPA/NMDAR ratios (M. H. Li et al., 2017). Inhibition of the NMDA-GluN2B receptor subunit inhibits the potentiation induced by AMP (M. H. Li et al., 2017) thus indicating that NMDAR-GluN2B are activated by AMP. Additionally, inhibition of EAAT3 also blocks the NMDAR potentiation suggesting that the potentiation requires the transport of AMP into the cell by DAT (M. H. Li et al., 2017). A similar effect on GLU transmission in the midbrain has been observed with METH which is discussed in Section 5.6.
5.3. Acute Cocaine-induced Alterations of Excitatory Signaling
Umara and colleagues demonstrated that a single in vivo exposure to cocaine results in long-term potentiation of AMPA-receptor mediated currents at excitatory synapses onto VTA DA neurons (Ungless et al., 2001). Interestingly, when the GluA1 subunit of the AMPA receptor is deleted from VTA DA neurons, the cocaine-induced increase in EPSCs is abolished (Engblom et al., 2008). These results are in line with others showing that acute cocaine exposure results in the insertion of GluA2-lacking AMPA receptors onto VTA DA neurons (Bellone & Luscher, 2006). In addition to enhancing signaling at GluA2-lacking AMPA receptors, acute cocaine exposure increases NMDA receptor function in the VTA that is mediated by DA D5-like receptors leading to the insertion of NR2B-containing NMDARs in the membrane (Schilstrom et al., 2006).
Regarding cocaine-induced adaptations on excitatory signaling within the accumbens, following repeated exposure to cocaine, there is an increase in the amplitude of AMPAR-mediated EPSCs due to an increased insertion of GluA2 lacking (calcium permeable) AMPA receptors into NAc shell D1-expressing MSNs (Engblom et al., 2008; Mameli et al., 2009; Pascoli, Turiault, & Luscher, 2011). In summary, acute cocaine exposure induces a plasticity in both VTA DA neurons and NAc MSNs that is expressed by insertion of GluA2-lacking AMPA receptors and within the VTA induced through enhanced NMDA receptor signaling.
5.4. Acute Cocaine-induced Alterations of Glutamate Homeostasis
Acute cocaine administration was shown to increase extracellular levels of GLU within the VTA 20 mins after injection and this increase in GLU occurred in parallel with the increase in locomotor activity (Kalivas & Duffy, 1995). This effect of cocaine is in contrast with AMP-induced increases in extracellular levels of VTA GLU which does not occur over the same time course as the increase in locomotor activity (Wolf & Xue, 1999). However, similar to what has been observed with AMP, the D1 antagonist SCH-23390 blocked the increase in extracellular levels of VTA GLU induced by cocaine and delayed the increase in locomotor activation suggesting an important role for D1R-mediated GLU accumulation in the VTA. Whether the acute cocaine-induced increases in VTA GLU accumulation arise from synaptic or non-synaptic sources is unknown. It is conceivable that the GLU accumulation following acute cocaine exposure arises from reversal of GLU transport. It is known that D1 and D2 receptor activation inhibits activity of the sodium potassium ATP pump (Na/K-ATPase) (Bertorello, Hopfield, Aperia, & Greengard, 1990). The Na/K-ATPase resides on neurons and astrocytes and is responsible for maintaining ion gradients that underlie resting and action potentials in neurons (Bertorello et al., 1990; Larsen, Stoica, & MacAulay, 2016; Skou, 1965). One proposed mechanism by which exposure to both cocaine and AMP would result in increased accumulation of extracellular GLU is DA receptor-induced inhibition of the Na/K-ATPase leading to disruption of ion gradients necessary for the inward transport of GLU by GLU transporters (Bertorello et al., 1990; Pierce, Bell, Duffy, & Kalivas, 1996; Wolf et al., 2000).
Regarding the effects of acute cocaine on extracellular levels of GLU within the accumbens, in vivo microdialysis reports have shown that cocaine administration results in a dose dependent increase in extracellular levels of NAc GLU that is Ca2+-dependent and coincides with the time course of behavioral activation (Smith, Mo, Guo, Kunko, & Robinson, 1995). The coinciding time course of cocaine-induced increases in locomotor activation and extracellular levels of VTA and NAc GLU are in contrast with the effects observed with AMP, whereby the increase in extracellular levels of GLU is observed 90 ins post-AMP injection, well after the increase in locomotor stimulation (Wolf & Xue, 1999). Furthermore, it is known that intra-NAc infusions of GLU agonists result in an increase in both locomotor activity and extracellular accumulation of DA within the NAc (Donzanti & Uretsky, 1983; Wu, Brudzynski, & Mogenson, 1993; Youngren, Daly, & Moghaddam, 1993) and that the increase in locomotor activity induced by GLU agonists is abolished when the mesolimbic DA projections are abolished via 6-OHDA lesions of the VTA (Wu et al., 1993). Thus, the acute behavioral activation induced by cocaine likely arises from GLU-stimulated DA accumulation within the NAc.
5.5. Importance of Co-Transmission in Responses to AMP and Cocaine
In light of reports demonstrating the release of GLU from DA terminals, and that VGLUT2 is expressed in DA terminals and facilitates DA signaling by DA neurons, Birgner and colleagues predicted that the absence of VGLUT2 would disrupt behavioral responses mediated by AMP (Birgner et al., 2010). They found that datVGLUT2 KO mice, utilizing the DAT-Cre driver that targets the 5’UTR of the DAT gene (Zhuang et al., 2005), displayed a blunted acute locomotor response to AMP. In order to rule out an effect of DAT-Cre in itself, this group also compared AMP-evoked locomotor responses in DAT-Cre expressing and DAT-Cre lacking mice and found no differences (Birgner et al., 2010). Thus, it is unlikely that the blunted locomotor response to AMP was due to DAT-Cre expression in itself.
As discussed above, Mingote and colleagues targeted PAG in DA neurons, creating a DAT-driven conditional HET GLS1, utilizing the DAT-Cre driver that targets the 3’UTR of the DAT gene (Backman et al., 2006) and found that while the acute locomotor response to AMP was intact in the conditional PAG knockout mouse, locomotor sensitization to AMP was diminished (Mingote et al., 2017). Recently, DAT-Ires-Cre expressing mice were reported to show a blunted acute locomotor response to AMP (Chohan, Esses, Haft, Ahmari, & Veenstra-VanderWeele, 2020). This result potentially confounds interpretations of behavioral data reported using this specific Cre-driver. However, Mingote and colleagues took the approach of comparing groups of mice that all expressed DAT-Cre thus diminishing the problem of confusing Cre-related effects for target specific effects (discussed in Section 3). These reports could be interpreted as 1 - DA signaling by DA/GLU coexpressing neurons determine the acute locomotor responses to AMP (Birgner et al., 2010) and 2 - GLU signaling by DA/GLU coexpressing neurons via GLS1 determines the sensitization but not the acute locomotor response to AMP (Mingote et al., 2017). Collectively, these interpretations are consistent a previously mentioned report showing 1 - the temporal disconnect between the acute locomotor response to AMP and the increase in VTA GLU efflux, 2 - the acute locomotor response to AMP is blocked by a D1 receptor antagonist, and 3 - sensitization is blocked by interventions that prevent the elevation of extracellular glutamate in the VTA (Wolf & Xue, 1999).
A role for GLU signaling in VTA DA neurons in mediating the biochemical and locomotor effects induced by cocaine has been investigated. Although GLU transmission from VTA DA neurons onto medium spiny neurons represents a small percentage of all GLU inputs onto these cells (Adrover et al., 2014), some reports have indicated that these inputs are able to regulate the physiological response to cocaine. Hnasko and colleagues utilized a datVGLUT2 KO mouse, targeting the 5’ UTR of the DAT gene (Zhuang et al., 2005), and demonstrated a reduced acute locomotor response to cocaine (20 mg/kg; ip) (Hnasko et al., 2010) which as discussed above was similarly observed with AMP (Birgner et al., 2010). However, it is important to note that sensitization and the conditioned place preference response to cocaine were unaltered and self-administration was actually enhanced in the datVGLUT2 KO (Alsio et al., 2011; Hnasko et al., 2010). Note that the control mice used in these experiments for comparison to the datVGLUT2 KO mice were DAT-Cre expressing mice. Furthermore, if VGLUT2 expression in DA neurons is important for the loading of DA into vesicles (Hnasko et al., 2010; Hnasko & Edwards, 2012) then one possible explanation for the enhanced cocaine SA in the datVGLUT2 KO is reduced extracellular DA concentrations in response to cocaine due to impaired packaging of DA in synaptic vesicles (Ikemoto, Yang, & Tan, 2015; Wang et al., 2017). Thus, in the datVGLUT2 KO, animals may have to work harder (e.g. increased lever pressing) to achieve a similar reinforcing effect of the drug.
As discussed above in Section 4, these VGLUT2 conditional KO models suffer from potential developmental compensatory adaptations, as VGLUT2 is knocked out during development (Alsio et al., 2011; Birgner et al., 2010; Hnasko et al., 2010). Papathanou and colleagues utilized the eDAT-Cre (Ekstrand et al., 2007) and txDAT-Cre (Engblom et al., 2008) VGLUT2 KO mouse lines discussed above in Section 4 to pinpoint the role of the VGLUT2 gene in mature DA neurons (Papathanou et al., 2018). Note that the Cre control mice utilized for this study were DAT-Cre transgenic mice with embryonal onset of the transgene (eDAT-Cre) (Ekstrand et al., 2007) and txCtrl mice (tamoxifen treated control mice without VGLUT2 gene deletion) (Papathanou et al., 2018). The eDAT-Cre VGLU2 KO mice exhibited a blunted acute locomotor response to AMP (Papathanou et al., 2018), as was expected due similar findings discussed above by Birgner and colleagues that utilized a similar DAT-Cre VGLUT2 mouse with embryonic onset of DAT-Cre-recombinase through the 5’UTR, immediately upstream of the DAT translation start codon (Birgner et al., 2010; Zhuang et al., 2005). An additional finding by Papathanou and colleagues that was not assessed by Birgner and colleagues is that the eDAT-Cre VGLUT2 KO mice showed a blunted sensitized response to AMP (Papathanou et al., 2018). Furthermore, Papathanou and colleagues also assessed acute and sensitized locomotor responses to cocaine in the eDAT-Cre VGLUT2 KO and found there were no overall differences between the KO and eDAT-Cre control mice apart from the last day of cocaine administration whereby the eDAT-Cre VGLUT2 KO mice displayed higher locomotor activation than controls (Papathanou et al., 2018). The finding that eDAT-Cre KO mice and WT controls displayed similar acute responses to cocaine and that the KO mice showed enhanced sensitized locomotor responses to cocaine is in contrast with Hnasko and colleagues report that showed a blunted acute response and no differences in the sensitized locomotor responses to cocaine in the VGLUT2 KO mice (Hnasko et al., 2010). While the DAT-Cre drivers utilized in these reports are similar, it is important to note that the DAT-Cre driver utilized by Birgner and Hnasko is a 5’UTR knock-in (Birgner et al., 2010; Hnasko et al., 2010; Zhuang et al., 2005) whereas the DAT-Cre driver utilized by Papathanou and colleagues is a 5’UTR transgenic (Ekstrand et al., 2007; Papathanou et al., 2018). Thus, the discrepancy in the locomotor response to cocaine between these two reports by Hnasko and Papathanou could be due to different genetic backgrounds between the DAT-Cre mouse lines. Moreover, deletion of VGLUT2 in mature DA neurons did not affect the acute or sensitized response to AMP or cocaine, as both txCtrl and txDAT-Cre mice displayed similar increases in acute and sensitized locomotor responses (Papathanou et al., 2018). These findings demonstrate that the temporal onset of VGLUT2 targeting in DA neurons determines the addiction-related phenotype. Finally, because repeated exposure to cocaine induces adaptations in synaptic transmission onto D1-receptor MSNs within the NAc shell (discussed in Section 5.3) (Engblom et al., 2008; Mameli et al., 2009; Pascoli et al., 2011), Papathanou and colleagues wanted to determine if GLU release from mature DA neurons affected synaptic plasticity specifically in NAc D1-receptor expressing MSNs (Papathanou et al., 2018). They found that compared to txCtrol mice, the AMPA/NMDA ratio was greatly increased in D1-receptor expressing MSNs in the txDAT-Cre VGLUT2 KO at baseline (Papathanou et al., 2018). Thus, with deletion of VGLUT2 in mature DA neurons, mice show normal sensitization responses to AMP and cocaine while also showing a decrease in NAc GLU transmission (decrease in EPSCs) and an elevated AMPA/NMDA ratio. The authors concluded that the elevated AMPA/NMDA ratio at baseline observed in the txDAT-Cre VGLUT2 KO was strong enough to block the enhanced synaptic plasticity effect induced by cocaine (Papathanou et al., 2018). These data suggest that even though VGLUT2 is down-regulated in DA neurons in adulthood, VGLUT2 in mature DA neurons nonetheless plays a critical role in regulating GLU transmission as well as synaptic plasticity in NAc MSNs.
As discussed earlier, the neuronal GLU transporter EAAT3 has been shown to colocalize with DAT in VTA DA neurons that project to NAc, and AMP administration results in internalization of this EAAT3 (Underhill et al., 2014). Although unexplored, a potential role for GLT-1 in DA neurons should be considered. The effects of AMP on extracellular GLU release are thought to be due to reversal of GLT-1 (Wolf et al., 2000; Xue et al., 1996). As is discussed in Section 7, GLT-1 has been identified as the major GLU transporter associated with excitatory terminals (Berger, DeSilva, Chen, & Rosenberg, 2005; Chen et al., 2004; Furness et al., 2008). It is conceivable that DA neurons that coexpress GLU also contain GLT-1 as a component of the excitatory transmission phenotype in these neurons, perhaps to assist in removing GLU from the extracellular space or for a metabolic role (McNair et al., 2019; McNair et al., 2020a). Careful investigation of the possibility that GLT-1 is localized in some or all DA neurons will aid in our understanding of its potential contributions to signaling by these neurons and role in mediating the effects of psychostimulants and other drugs of abuse (Xu et al., 2003).
5.6. Acute METH-induced Alterations on Excitatory Signaling
Similar to acute AMP administration, acute METH administration has been shown to decrease inhibitory signaling and enhance excitatory signaling (M. H. Li et al., 2017; Padgett et al., 2012). Padgett and colleagues found that acute METH results in a reduction in the inhibitory presynaptic GABABR currents in VTA DA neurons (Padgett et al., 2012). Furthermore, Li and colleagues demonstrated that similar to acute AMP, acute administration of METH potentiates GLU neurotransmission in midbrain DA neurons via increasing NMDA-GluN2B-mediated synaptic currents and decreasing AMPA/NMDAR ratios (M. H. Li et al., 2017). Inhibition of EAAT3 also blocks the NMDAR potentiation suggesting that the potentiation requires the transport of METH into the cell by DAT (M. H. Li et al., 2017). Altogether, these reports indicate that both AMP and METH potentiate GLU transmission in midbrain DA neurons through internalization of EAAT3 and subsequent activation of GluN2B-containing NMDARs (M. H. Li et al., 2017; Underhill et al., 2014). Li and colleagues postulated that internalization of EAAT3 in DA neurons by amphetamines could result in 1 - increased extracellular levels of GLU in the synapse and subsequent spillover to GLU receptors or alternatively, 2 - decreased GLU uptake by EAAT3 outside the synapse could potentiate GLU receptor activation (M. H. Li et al., 2017).
5.7. Acute METH-induced Alterations of Glutamate Homeostasis
While the effects of acute METH administration on extracellular levels of GLU within the VTA are unknown, as discussed above and postulated by Li and colleagues, it is conceivable that METH-induced internalization of EAAT3 in midbrain VTA DA neurons results in increased extracellular levels of GLU (M. H. Li et al., 2017). Furthermore, similar to AMP, acute METH administration results in a delayed (~2 h post METH injection) and sustained increase in extracellular NAc GLU accumulation (K. Ito, Abekawa, & Koyama, 2006). Similar to AMP, the delayed increase in extracellular GLU accumulation induced by METH is blocked by the NMDA antagonist MK-801 (K. Ito et al., 2006). Similar to both AMP and cocaine, the delayed increase in extracellular GLU accumulation induced by METH is blocked by the D1 antagonist SCH-23390 (K. Ito et al., 2006). Additionally, and consistent with the reports mentioned above on acute AMP and cocaine administration, only the D1 antagonist was found to block the acute locomotor response induced by METH (K. Ito et al., 2006).
Acute METH administration has also been shown to increase extracellular levels of GLU within the dorsal striatum (Mark, Soghomonian, & Yamamoto, 2004; Stephans & Yamamoto, 1994). However, acutely, this effect that METH has on dorsal striatal GLU levels has only been shown following a single day of binge METH treatment (10 mg/kg given 4 times i.p. at 2 h intervals) (Halpin, Northrop, & Yamamoto, 2014; Mark et al., 2004; Pu, Broening, & Vorhees, 1996; Simoes et al., 2008; Stephans & Yamamoto, 1994). The majority of reports that have assessed METH-induced alterations in dorsal striatal GLU levels have utilized binge METH treatment paradigms in order to assess effects on DA terminal depletion (which occurs at higher, more toxic doses of METH) as well as on striatal inflammatory responses (Halpin et al., 2014; Mark et al., 2004; Pereira et al., 2006; Pu et al., 1996; Stephans & Yamamoto, 1994). This is discussed more below in Section 6.4. As discussed above, a single, low dose of METH (2.5 mg/kg) and AMP (5.0 mg/kg) results in an increase in extracellular GLU levels within the ventral striatum (NAc) (K. Ito et al., 2006; Xue et al., 1996). This raises the question whether a single, lower dose of METH alters extracellular levels of dorsal striatal GLU levels.
6. Chronic Administration of Psychostimulants
6.1. Modeling Drug Addiction Behavior
The validity of an animal model in representing a human disorder is established by demonstrating face, predictive, and construct validity (Spanagel, 2017). Face validity is based on the presence of characteristic behavioral features that are seen both in laboratory animals and in humans. Construct validity is based on identity of underlying biological mechanisms in the animal model and in humans (Sarter, 2002). Finally, predictive validity is assessed by success in discovering drugs that are useful in human patients based on performance in the animal model and is arguably the most relevant test of validity for developing potential therapeutics (Haney, 2009). An example of an animal model that has been proposed to be relevant for the understanding of relapse in human drug addicts is chronic exposure to psychostimulants and withdrawal. In this model, following a prolonged withdrawal period, exposure to drug associated stimuli usually results in reinstatement of self-administration, which appears to be due to a disruption in GLU homeostasis within the mesocorticolimbic circuit (Kalivas, 2009).
Models of drug addiction behavior can be broken down into contingent and non-contingent paradigms (Kuhn, Kalivas, & Bobadilla, 2019). A contingent model refers to the animal having to perform a task (i.e. lever press or nose poke) in order to receive the reward (i.e. cocaine) (Kuhn et al., 2019). The reinstatement model of drug self-administration behavior is arguably the most valid contingent animal model for studying addiction and manifests excellent face, predictive, and construct validity (for review see (Spanagel, 2017). The drug reinstatement model is widely used as a model of relapse in human addicts (Bossert, Marchant, Calu, & Shaham, 2013; Kalivas & McFarland, 2003). In this paradigm, animals are first trained to self-administer cocaine by pressing a lever or nose poking for an iv drug infusion in an operant conditioning chamber (David, Polis, McDonald, & Gold, 2001; Kalivas & McFarland, 2003). After this behavior is well learned, the animal is then placed in extinction training where the learned behavior becomes abolished (e.g. lever pressing does not result in delivery of the drug) or through a forced withdrawal period whereby the animal is left in the home cage without access to the drug (Spanagel, 2017). Following extinction training or withdrawal, the animals are re-exposed to a priming stimulus: the cue previously paired with the drug, a stressor, or the drug itself (reinstatement). Exposure to a drug-paired cue, exposure to a stressor, and re-exposure to the previously self-administered drug have all been demonstrated to result in reinstatement in rats (Ahmed & Koob, 1997; Childress et al., 1993; Kufahl & Olive, 2011; McFarland & Kalivas, 2001; Taslimi, Komaki, Haghparast, & Sarihi, 2018; Weiss et al., 2000). Two commonly used models based on non-contingent drug exposure are the conditioned place preference (CPP) and behavioral sensitization paradigms. CPP has been proposed as an alternative to drug self-administration for studying drug-seeking behavior (Spanagel, 2017). In this paradigm, animals are injected daily with the drug paired with a distinguishable compartment in a conditioning box while a second compartment is paired with a vehicle injection; animals will achieve drug-CPP, i.e. spending more time in the compartment paired with drug, after several days of conditioning (Spanagel, 2017). The CPP model can also be utilized to study drug-primed reinstatement after a period of extinction training whereby the drug-paired chamber is paired with a vehicle injection. While some investigators claim that CPP is a model of drug-seeking behavior, CPP is dependent entirely on Pavlovian associations. Therefore, CPP in itself cannot account for the instrumental nature of drug-seeking/drug-taking behavior that is perhaps bettered modeled by drug self-administration (Belin-Rauscent A, 2012). Furthermore, the repeated administration of psychostimulants leads to augmented behavioral effects, termed behavioral sensitization. This phenomenon is well characterized in both experimental animals and humans (Kalivas & Stewart, 1991; T. E. Robinson, 1984, 2010) Sensitization of locomotor activity has been proposed as a model of addiction in humans (T. E. Robinson & Becker, 1986; T. E. Robinson & Berridge, 1993). That sensitization affects addiction behavior is illustrated by studies in which rats that develop locomotor sensitization in response to psychostimulants will subsequently work harder for the drug during self-administration than naïve animals (Lorrain, Arnold, & Vezina, 2000). Non-contingent models such as CPP and behavioral sensitization are easy to run and quick to set up and because of these advantages many investigators have used them to identify how drug exposure alters key reward-related neurobiological substrates (Kuhn et al., 2019). However, it is important to also understand the limitations of these non-contingent models (Kuhn et al., 2019).
6.2. AMP: Alterations in Glutamate Homeostasis Following Chronic Administration
A majority of the reports on the chronic effects of AMP on extracellular levels of GLU have used experimenter administered AMP sensitization paradigms. As mentioned above, acute systemic injection of AMP as well as intra-VTA administration of AMP or D1 agonists results in a delayed and sustained increase in VTA GLU levels (Wei et al., 2016). AMP sensitization can be produced by either repeated systemic or repeated intra-VTA infusions of AMP (Cador, Bjijou, & Stinus, 1995; Kalivas & Weber, 1988; Perugini & Vezina, 1994; Vezina & Stewart, 1990; Wolf, White, & Hu, 1994). The delayed increase in extracellular levels of GLU within the VTA also occurs in sensitized rats [e.g. rats treated with AMP (5 mg/kg) for 5 d and challenged 2 d following a withdrawal period with AMP] (Wolf et al., 2000; Xue et al., 1996). The magnitude of the increase in extracellular levels of GLU within the VTA is the same in acute and chronic AMP treated animals and occurs after each injection in a chronic AMP regimen (Wolf et al., 2000; Xue et al., 1996). Additionally, this increase in VTA GLU following chronic AMP treatment can be produced by intra-VTA infusion of AMPA (Giorgetti, Hotsenpiller, Ward, Teppen, & Wolf, 2001) and is attenuated following intra-VTA infusion with the D1 receptor antagonist SCH-23390 (Wolf & Xue, 1998) thus suggesting a causal role for AMPA and D1 receptor activation involvement in the delayed increase in AMP-induced GLU release in the VTA. Thus, both the acute and chronic effects of AMP on extracellular levels of VTA GLU are blocked by D1 receptor antagonists.
The critical question is whether or not the delayed increase in GLU is required for the induction of sensitization. Evidence for a role for elevated extracellular GLU is that intra-VTA infusions of AMP produce sensitization (Cador et al., 1995; Kalivas & Weber, 1988; Perugini & Vezina, 1994; Vezina & Stewart, 1990) and result in the delayed, long-lasting increase in VTA extracellular GLU (Wolf & Xue, 1998). Both sensitization (Bjijou, Stinus, Le Moal, & Cador, 1996; Stewart & Vezina, 1989; Vezina, 1996) and the increase in VTA GLU are blocked by intra-VTA infusion of D1 receptor antagonists (Wolf & Xue, 1998). However, a lower dose of AMP (2.5 mg/kg) is sufficient to induce sensitization but does not result in a delayed increase in extracellular VTA GLU (Xue et al., 1996). Xue and colleagues argue that 2.5 mg/kg AMP could induce small increases in GLU efflux that are not detectable by microdialysis due to efficiency of GLU clearance mechanisms (Xue et al., 1996). Additional evidence that VTA GLU efflux is required for the development of behavioral sensitization is that other treatments that prevent the AMP-induced increase in VTA GLU also prevent the induction to AMP sensitization. Alongside treatment with AMP, treatment with PBN, an agent that blocks the formation of hydroxyl free radicals, blocks both the delayed increase in VTA GLU and the development of sensitization (Wolf et al., 2000). The exact mechanism underlying how PBN prevents the AMP-induced increases in extracellular GLU levels is not known however it is known that PBN also attenuates METH-induced toxicity of DA terminals (Cappon, Broening, Pu, Morford, & Vorhees, 1996). Thus, PBN is acting in some way to reduce increases in GLU levels and DA terminal damage associated with AMP/METH administration. Moreover, administration of the NMDA antagonist, MK-801, as well as ibotenic acid lesions of the PFC, prevent AMP from increasing VTA GLU efflux and also prevent sensitization (Wolf & Xue, 1999). One important question is, why does blocking NMDA receptors with MK-801 prevent AMP-stimulated GLU accumulation? One possible mechanism is that the increase in extracellular DA levels induced by AMP results in D1 receptor activation that evokes glutamate release by inhibition of the Na+/K+ -ATPase, or some other mechanism (see Section 5.4), which results in a disruption of ion gradients necessary for the inward transport of GLU by GLU transporters (Bertorello et al., 1990; Pierce et al., 1996; Wolf et al., 2000). In Fig. 1, we propose a mechanism underlying the GLU-mediated inhibition of the Na+/K+ -ATPase following AMP administration. In brief, AMP results in increased extracellular levels of VTA DA via reversal of DAT. DA released by DAT reversal stimulates DA receptors on VTA astrocytes (Liu et al., 2009; Zhang et al., 2009) resulting in increases in intracellular levels of Ca2+ that then causes the release of arachidonic acid (AA) from the phospholipid membrane (S. P. Lee et al., 2004). The release of AA results in increased levels of reactive oxygen species (ROS) (Chan, Chen, & Yu, 1988; Chan & Fishman, 1980; Sakuma et al., 2012). Both AA and ROS inhibit the Na+/K+ -ATPase (Chan, Kerlan, & Fishman, 1983; Hexum & Fried, 1979; Volterra, Trotti, Tromba, Floridi, & Racagni, 1994) that ultimately leads to a depolarization of the cell membrane and disruption of the Na+/K+ gradients that results in the reversal of EAATs in general and GLT-1 in particular (Anderson, Huguenard, & Prince, 2010; Nicholls & Attwell, 1990; Volterra et al., 1994). Increased levels of extracellular GLU in VTA can activate NMDARs located on the astrocyte resulting in further increases in intracellular Ca2+ and AA and sustained reversal of GLT-1 (Biber et al., 1999; Daniels & Brown, 2001; Lalo, Pankratov, Kirchhoff, North, & Verkhratsky, 2006; M. C. Lee et al., 2010). Thus, the increase in AMP-mediated GLU accumulation could activate NMDA receptors in a feed-forward manner to increase extracellular GLU accumulation further (Wolf & Xue, 1999). Collectively, these data strongly support a direct and necessary role for VTA GLU efflux in the induction of behavioral sensitization.
Repeated intra-VTA, but not intra-NAc, injections of AMP produce enhanced locomotor responses to subsequent systemic administration of AMP (Kalivas & Weber, 1988) suggesting an important role for the VTA but not the NAc in the induction of sensitization. The NAc is referred to as the brain region responsible for the expression of sensitization (Kalivas & Weber, 1988; Paulson & Robinson, 1991). Following induction of AMP sensitization, injection of AMP into the NAc enhances locomotor responses to AMP during expression (Paulson & Robinson, 1991) suggesting that the NAc is important in the expression of sensitization. In line with this, there is a delayed and sustained increase in extracellular levels of NAc GLU in rats pretreated with AMP (5 mg/kg) for 5 d and challenged 2 d following a withdrawal period with AMP (Xue et al., 1996). Moreover, intra-VTA infusions of the non-NMDA receptor GLU agonist AMPA increased extracellular levels of NAc GLU in rats 3 d after the last day of AMP injections. Additionally, intra-VTA preadministration of the competitive GLU reuptake inhibitor, PDC (Bridges, Stanley, Anderson, Cotman, & Chamberlin, 1991), produces sensitization to a systemic AMP challenge (Aked, Coizet, Clark, & Overton, 2005). Collectively, these data suggest that alterations in GLU homeostasis, specifically increases in extracellular GLU within the VTA, are necessary and sufficient for the induction of sensitization. Alterations in GLU homeostasis within the NAc are associated with the expression of sensitization, but a causal relationship has not been demonstrated, unlike for VTA. Potentiation of AMPA receptor signaling in the VTA has been shown to occur following repeated AMP administration and appears to be important for the induction of AMP sensitization (Argilli, Sibley, Malenka, England, & Bonci, 2008; Giorgetti et al., 2001). A major question, then, is the mechanistic relationship between the alteration in GLU homeostasis and the potentiation of AMPA receptor signaling in the VTA that occurs following AMP administration.
Similar to cocaine and METH addictions (discussed below), treatment strategies for reducing the rewarding effects of AMP have focused on targeting GLU homeostasis. As outlined in Table 2, targeting GLT-1 has demonstrated promising results in preclinical animal models. If increases in NAc GLU efflux are responsible for the expression of AMP sensitization, one might expect that reducing the levels of extracellular GLU in this region would attenuate this process. One class of drugs known to increase GLT-1 expression in injured brain, or to prevent downregulation of expression with injury is β-lactam antibiotics (Jagadapillai, Mellen, Sachleben, & Gozal, 2014; Lipski et al., 2007; Miller et al., 2008; Rothstein et al., 2005). Of these, ceftriaxone is the most widely studied for its ability to attenuate relapse and sensitization behavior related to many classes of drugs of abuse (Roberts-Wolfe & Kalivas, 2015; Smaga, Fierro, Mesa, Filip, & Knackstedt, 2020). Repeated administration of ceftriaxone induces a reduction in extracellular levels of GLU within the NAc in healthy control rodents (Rasmussen, Unterwald, & Rawls, 2011). Treatment with ceftriaxone has been shown to attenuate AMP sensitization behavior in rats (Rasmussen et al., 2011). Riluzole, a neuroprotective drug currently marketed for the treatment of amyotropic lateral sclerosis, is another compound that increases GLT-1 protein expression measured via western blot (Carbone, Duty, & Rattray, 2012). Riluzole has demonstrated efficacy in attenuating AMP conditioned place preference in rats, a model of drug-seeking behavior (Tzschentke & Schmidt, 1998). However, this preclinical study with Riluzole was not supported in human studies whereby the subjective reporting of craving by human patients was not reduced following Riluzole treatment (Sofuoglu, Waters, Mooney, & Kosten, 2008). Importantly, there were several limitations discussed in the report by Sofuoglu and colleagues. These limitations included: 1 - a lack of examining the dose-effect relationship for Riluzole’s effects on d-AMP responses, 2 - a lack of examining d-AMP plasma levels in human patients, and 3 - their sample consisted of healthy volunteers and thus the generalizability of these findings to stimulant users would need to be determined in future studies (Sofuoglu et al., 2008).
Table 2.
Treatments Targeting GLT-1 or xCT for Chronic Psychostimulant Addiction Behaviors in Preclinical Animal Models
| Addiction Behavioral Protocol | Psychostimulant-induced effect on GLT-1 or xCT | Treatment Approach | Behavioral Outcome | Treatment Effect on GLU Homeostasis | |
|---|---|---|---|---|---|
| AMP | AMP Sensitization | Data Not Shown | Ceftriaxone (200 mg/kg i.p.) for 8 days during induction (Rasmussen, Baron et al. 2011) | Reduction in Sensitization Expression | Reduced extracellular levels of NAc GLU (Rasmussen, Baron et al. 2011) |
| AMP CPP | Data Not Shown | Riluzole (4 mg/kg i.p.) for 3 days during CPP training (Tzschentke and Schmidt 1998) | Blocked CPP | Data Not Shown | |
| Cocaine | Cocaine Reinstatement following Extinction | Data Not Shown | Ceftriaxone (200 mg/kg i.p.) for 5 days during extinction (Sari, Smith et al. 2009) | Blocked Cue-Induced Reinstatment | Data Not Shown |
| Cocaine Reinstatement following Long Withdrawal Periods (45 d) | Greater reduction in GLT-1 protein expression in NAc core following longer withdrawal periods (Fischer et al. 2012) | Ceftriaxone (200 mg/kg i.p.) for last 5 days of withdrawal (Fischer et al. 2012) | Blocked Cue-Induced Reinstatment | Increased GLT-1 protein expression in NAc (Fischer et al. 2012) | |
| Cocaine Reinstatement following Extinction | Reduced GLU uptake and expression of GLT-1 and xCT in the NAc core following extinction (Knackstedt et al. 2010) | Ceftriaxone (200 mg/kg i.p.) for 7 days during extinction (Knackstedt et al. 2010) | Blocked Cue- and Cocaine-Induced Reinstatment | Increased expression of GLT-1 and xCT in NAc core (Knackstedt et al., 2010) | |
| Cocaine Reinstatement following Extinction | Reduced GLU uptake and expression of GLT-1 and xCT in the NAc core following extinction (Knackstedt et al. 2010) | N-acetylcysteine (100 mg/kg i.p.) for 7 days during extinction (Knackstedt et al. 2010) | Blocked Cue- and Cocaine-Induced Reinstatment | Increased expression of GLT-1 and xCT in NAc core (Knackstedt et al., 2010) | |
| Cocaine Reinstatement following Extinction | Reduced NAc core basal GLU levels | Ceftriaxone (200 mg/kg i.p.) for last 5 days of extinction training) (Trantham-Davidson et al. 2012) | Blocked Cocaine-Induced Reinstatment | Restores basal GLU levels and increases GLU uptake in NAc core (Trantham-Davidson et al. 2012) | |
| Cocaine Self-Administration in Mice | Data Not Shown | Clavulanic acid (1 mg/kg i.p.) for 3 days prior to self-administration (Kim et al. 2015) | Reduced Self-Administration Behavior in Mice | Increased GLT-1 protein expression in NAc (Kim et al. 2015) | |
| METH | METH Sensitization in Mice | Data Not Shown | Riluzole (20 mg/kg i.p.) for 5 days during induction (Itzhak and Martin 2000) | Reduction in Sensitization Expression in Mice | Data Not Shown |
| METH CPP | Data Not Shown | Ceftriaxone (200 mg/kg i.p.) for 7 days during CPP extinction (Abulseoud, Miller et al. 2012) | Blocked METH-induced CPP Reinstatement | Increased GLT-1 protein and mRNA expression in PFC (Abulseoud, Miller et al. 2012) |
In the table. Amphetamine (AMP), Methamphetamine (METH), Conditioned Place Preference (CPP), Unless specified otherwise, rats were used.
6.3. Cocaine: Alterations in Glutamate Homeostasis Following Chronic Administration
A defining characteristic of cocaine addiction is repeated cycles of drug use followed by abstinence (Spencer & Kalivas, 2017). Prevention of craving and relapse has been a continuing focus in the study of cocaine abuse. Many reports have demonstrated that chronic cocaine administration, whether it is self-administered by the animal itself or experimenter administered, alters extracellular levels of GLU within the NAc (Baker, Shen, et al., 2002; Pierce et al., 1996; Schmidt, Anderson, Famous, Kumaresan, & Pierce, 2005). It is important to note that the NAc comprises two subregions, core and shell, which have differential functionality in cocaine-seeking behavior (Ambroggi, Ghazizadeh, Nicola, & Fields, 2011; Everitt & Robbins, 2005; R. Ito & Hayen, 2011; McFarland & Kalivas, 2001). Within the core, basal non-synaptic (TTX-independent) GLU levels are decreased following a history of cocaine self-administration (assessed before reinstatement testing that followed extinction training) (McFarland, Lapish, & Kalivas, 2003) and following experimenter administered cocaine for 7 days followed by a 3-week withdrawal period (Baker, Shen, et al., 2002; Pierce et al., 1996). When cocaine is self-administered or experimenter administered followed by extinction training or a forced withdrawal period (3 weeks in withdrawal length), there is a significant increase in extracellular synaptic (TTX-dependent) levels of GLU in NAc core (but not NAc shell) (McFarland et al., 2003; Pierce et al., 1996). The PFC-NAc core GLU projection has been found to play a role in both the relapse response and the increases in extracellular levels of NAc GLU (McFarland et al., 2003). However, it is important to note that PFC outputs to the NAc are not homogeneously distributed. The prelimbic region of the PFC sends GLU output selectively to the NAc core whereas the ventral PFC (infralimbic PFC) sends GLU output to the NAc shell (Vertes, 2004). Specifically, it has been shown that pharmacological inhibition of the prelimbic PFC projection to the core attenuates both relapse behavior and the rise in extracellular GLU (McFarland et al., 2003) whereas relapse behavior is unaffected by inactivation of the PFC (infralimbic) projection to shell (Capriles, Rodaros, Sorge, & Stewart, 2003). Furthermore, the decrease in non-synaptic levels of NAc core extracellular GLU that is observed 3 weeks following chronic cocaine exposure results from decreased expression and activity of xCT (Baker, McFarland, Lake, Shen, Toda, et al., 2003; Knackstedt et al., 2010) the GLU antiporter that is a major source of extrasynaptic GLU in the NAc (Baker, Xi, et al., 2002). Chronic cocaine administration also alters the regulation of GLU transmission by mGluR2/3 in the core. Following withdrawal from chronic cocaine, there is a decreased mGluR2/3-mediated inhibition of AMPA-mediated EPSCs (Moussawi et al., 2009) and decreased surface expression of mGlu2 in the core (Logan et al., 2020). The increase in extracellular synaptic NAc core GLU levels associated with relapse following withdrawal periods is thought to result from decreased capacity of mGluR2/3 to regulate the presynaptic release of GLU (Xi et al., 2002) as well as decreased expression and function of GLT-1 (Knackstedt et al., 2010). Collectively, these reports demonstrate that chronic cocaine administration reduces the basal extracellular level of non-synaptic NAc GLU and the capacity of mGluR2/3 and GLT-1 to regulate the synaptic release of NAc GLU in response to a cocaine challenge or relapse session.
The animal model of cocaine self-administration and reinstatement has been altered over the past 20 years to more accurately reflect human drug consumption patterns. The majority of cocaine self-administration reports have utilized extinction/reinstatement models in animals exposed to limited-access conditions (~2 h/day in the self-administration chamber). However, increasing access during drug self-administration to 6 h/day and introducing long withdrawal periods (3 weeks or longer) induce behavioral and morphological alterations that more adequately capture key features of human addiction (Ahmed, 2012; Ahmed & Koob, 1998; Ferrario et al., 2005; K. D. Fischer, Houston, & Rebec, 2013). Extending access during self-administration to >6 h/d elicits behaviors that more closely mimic human compulsive drug seeking. Introducing long withdrawal periods following drug self-administration results in an increase in drug relapse behavior in animal models, referred to as the incubation of cocaine craving (Conrad et al., 2008; Grimm, Hope, Wise, & Shaham, 2001; Lu, Grimm, Hope, & Shaham, 2004; Zavala, Biswas, Harlan, & Neisewander, 2007). This effect is associated with increases in GluA2 lacking AMPA receptors in the NAc (Conrad et al., 2008) thus implicating GLU signaling within the NAc in relapse (but also see (See, Elliott, & Feltenstein, 2007)). Interestingly, it was determined that increasing access during cocaine self-administration from 2 to 6 h/d results in a greater NAc core and shell GLT-1 down-regulation and that introducing long withdrawal periods from 1 to 45 d results in greater GLT-1 down-regulation in the core (Fischer-Smith, Houston, & Rebec, 2012). However, these cocaine self-administration and withdrawal parameters that result in downregulation of GLT-1 expression are not accompanied by greater decreases in basal non-synaptic GLU or greater increases in synaptic GLU release during a reinstatement test (Lutgen et al., 2014). Nonetheless, interventions targeting up-regulation of GLT-1 have shown preclinical efficacy in the treatment of cocaine relapse behavior (K. D. Fischer et al., 2013; Knackstedt et al., 2010; Sari, Smith, Ali, & Rebec, 2009; Sondheimer & Knackstedt, 2011; Ward et al., 2011). It was also shown that up-regulation of GLT-1 expression following treatment with ceftriaxone was associated with attenuation of cue-induced cocaine-seeking behavior with a greater effect in rats exposed to both extended-access (6 h/d) and long-withdrawal (45 d) conditions (K. D. Fischer et al., 2013). Critically, in these studies GLT-1 blockade with DHK and DL-TBOA infusions in core, but not shell, reversed the ceftriaxone-induced attenuation of relapse behavior. Collectively these data indicate a critical role for GLT-1 within the NAc core in mediating cocaine relapse behavior. The effects that varying withdrawal periods from cocaine self-administration have on glutamate homeostasis are displayed in Figure 2.
Figure 2. Alterations in GLU homeostasis following cocaine withdrawal within the NAc.
A simplified illustration of how GLU homeostasis is altered within the NAc core in an animal experiencing varying withdrawal lengths from cocaine administration. A. Drug naïve conditions: Under normal, drug naïve, conditions GLU is packaged into vesicles and released from the presynaptic terminal and is taken up by GLT-1 on the astrocyte. The cystine-GLU exchanger (xCT) located on the astrocyte exports GLU out into the extrasynaptic space (Baker, Shen et al. 2002); this GLU binds to mGluR2/3 located on the presynaptic terminal which decreases the synaptic GLU release probability (Moran, McFarland et al. 2005). By removing GLU from the extracellular space and maintaining tone at mGluR2/3, GLT-1 and xCT, respectively, both work to maintain GLU homeostasis (Baker, Xi et al. 2002). B. Short withdrawal following cocaine self-administration: The alterations illustrated in panel B are compared to the drug-naïve synapse presented in panel A. As shown, cocaine self-administration followed by short withdrawal periods (1–3 days) or extinction training results in reduced basal levels of extracellular GLU, most likely due to the decreased expression of xCT (Knackstedt, Melendez et al. 2010, Trantham-Davidson, LaLumiere et al. 2012). Decreased expression of xCT reduces GLU tone at mGluR2/3 (Baker, Xi et al. 2002). Following short withdrawal periods from cocaine self-administration there is also a decrease in mGluR2/3 receptor expression in the NAc (Logan, Bechard et al. 2020). Additionally, GLT-1 protein expression is decreased following short withdrawal periods from cocaine self-administration (Fischer-Smith, Houston et al. 2012). Because mGluR2/3s serve to inhibit neurotransmission, the decreased tone at mGluR2/3s results in an increase in GLU release when the PFC-NAc projection is activated during drug-seeking behavior (Kalivas 2009, Reissner and Kalivas 2010). Decreased expression of GLT-1 results in reduced uptake of GLU from the extracellular space during drug-seeking behavior (Trantham-Davidson, LaLumiere et al. 2012). Furthermore, following short withdrawal from cocaine self-administration there is increased expression of the GluR2 lacking Ca2+ permeable AMPA receptors, more specifically the GluA3 subunit expressing AMPA receptors (Conrad, Tseng et al. 2008), suggesting that cocaine self-administration followed by short withdrawal increases signaling at the Ca2+ permeable GluR2 lacking AMPA receptors. C. Long withdrawal following cocaine self-administration: The alterations illustrated in panel C are compared to animals experiencing short withdrawal from cocaine self-administration in panel B. Long withdrawal periods (40–47 days) following cocaine self-administration yields an even greater reduction in GLT-1 expression compared to short withdrawal conditions (Fischer-Smith, Houston et al. 2012). Additionally, compared to short withdrawal conditions, there is an even greater increase in the expression of the GluR2 lacking Ca2+ permeable AMPA receptors (GluR1/3 subunit expressing) (Conrad, Tseng et al. 2008). There is also an increase in evoked excitatory postsynaptic currents (EPSCs) in NAc core slices that is blocked by Naspm, a selective blocker of GluR2-lacking AMPA receptors (Conrad, Tseng et al. 2008). Naspm has no effect on EPSCs in animals exposed to short withdrawal conditions from cocaine self-administration suggesting that these GluR2-lacking AMPA receptors contribute to enhanced NAc synaptic transmission only after long withdrawal periods (Conrad, Tseng et al. 2008, Wolf 2010).
N-acetylcysteine (NAC) is a cysteine prodrug and antioxidant precursor that has been used in humans for many years as a treatment for acetaminophen overdose (Prescott, Park, Ballantyne, Adriaenssens, & Proudfoot, 1977; Scalley & Conner, 1978). NAC is membrane-permeable and does not require active transport in order to deliver cysteine to the cell (Sen, 1997), although it has limited penetration across the blood brain barrier (Borgstrom & Kagedal, 1990; Sjodin, Nilsson, Hallberg, & Tunek, 1989; Tardiolo, Bramanti, & Mazzon, 2018). NAC is also a precursor of the antioxidant glutathione (GSH); the synthesis of GSH depends on the rate-limiting activity of xCT (Dringen & Hirrlinger, 2003). Treatment with NAC or ceftriaxone normalizes abnormal GLU levels in the NAc via increasing xCT protein expression and function (Baker, McFarland, Lake, Shen, Tang, et al., 2003) and, by mechanisms unknown, GLT-1 (K. D. Fischer et al., 2013; Knackstedt et al., 2010; Sari et al., 2009) expression and function in animals previously exposed to cocaine. While treatment with either ceftriaxone or NAC has been shown to up-regulate GLT-1 and xCT expression and function in the NAc in animals that have been exposed to cocaine, the expression and function of both of these proteins in the NAc is unaltered in control animals (e.g. animals not exposed to cocaine but exposed to ceftriaxone or NAC) (K. D. Fischer et al., 2013; Knackstedt et al., 2010; LaCrosse et al., 2017), although see (Smaga et al., 2020). Furthermore, as outlined in Table 2, both of these treatments attenuate cocaine relapse behavior (Reissner et al., 2015; Sari et al., 2009) and the sensitized response to cocaine (Rasmussen et al., 2011; Sondheimer & Knackstedt, 2011). NAC-induced restoration of abnormal GLU levels was shown to be mGlu2/3 dependent (Kupchik et al., 2012), however, more recently it was shown that the restoration of GLT-1 is also critical for the more enduring protection from cocaine relapse (Reissner et al., 2015). Furthermore, NAC has demonstrated partial success in the translation from preclinical to clinical models of cocaine relapse. In a double-blind placebo-controlled study, NAC did not differ from placebo in ongoing cocaine use, however, this treatment did increase the number of days to relapse in a subset of abstinent patients (LaRowe et al., 2013). Recently, acute NAC was found to attenuate cocaine-primed cocaine-seeking in abstinent cocaine users (Woodcock, Lundahl, Khatib, Stanley, & Greenwald, 2020). These data are consistent with rodent reports indicating this treatment to be more effective at reducing reinstatement following extinction periods than at reducing ongoing cocaine self-administration (Baker, McFarland, Lake, Shen, Toda, et al., 2003; Madayag et al., 2007; Reissner et al., 2015).
In addition to ceftriaxone and NAC, other compounds targeting GLT-1 have shown promising preclinical results in the treatment of cocaine relapse in animal models (J. Kim et al., 2016; Reissner et al., 2014). Reissner and colleagues tested the effects of the glial modulator and neuroprotective agent propentofylline on cocaine relapse behavior and found that this treatment not only attenuates both cue- and cocaine-induced reinstatement but also restores GLT-1 expression to normal levels (similar to levels in a cocaine naïve rat) in the NAc core (Reissner et al., 2014). Additionally, this group found that restoring GLT-1 expression was necessary for propentofylline to inhibit reinstatement, as infusion of GLT-1 antisense into the NAc core reversed the effects of propentofylline on relapse behavior (Reissner et al., 2014). The hypothesized mechanism whereby propentofylline inhibits reinstatement is restoration of astrocytic-mediated clearance of synaptic NAc GLU (Reissner et al., 2014). Furthermore, clavulanic acid is a structural analog of ceftriaxone but has greater brain penetrability, increased oral availability, with negligible antibiotic activity (J. Kim et al., 2016). Clavulanic acid increases GLT-1 expression in the NAc and reduces cocaine self-administration responding on a progressive-ratio (PR) schedule of reinforcement learning (J. Kim et al., 2016). The PR schedule of reinforcement requires the animal to continuously work harder in order to achieve the drug (e.g. increasing the amount of lever presses it takes to receive a reward) and is useful for studying treatments that might affect the reinforcing strength of the drug (e.g. cocaine) (J. Kim et al., 2016; Negus, 2003; Roberts, Bennett, & Vickers, 1989; Roberts, Loh, & Vickers, 1989; Ward, Morgan, & Roberts, 2005). Interestingly, Kim and colleagues showed that Clavulanic acid reduced self-administration of cocaine under a PR schedule at lower doses than ceftriaxone (J. Kim et al., 2016). They also determined that the reinforcing efficacy of cocaine was reduced following treatment with clavulanic acid (J. Kim et al., 2016). However, clavulanic acid did not attenuate cue-primed reinstatement of cocaine-seeking in rats (Bechard, Hamor, Wu, Schwendt, & Knackstedt, 2019). Whether or not this treatment is effective in human patients is currently unknown, however there is an on-going clinical study recruiting cocaine-dependent individuals to assess the tolerability and drug interactions between clavulanic acid and cocaine (NCT02563769).
6.4. METH: Alterations in Glutamate Homeostasis Following Chronic Administration
While METH and AMP share similar pharmacological and behavioral activating properties in animal models, it is commonly suggested that METH is more potent than AMP (Hall, Stanis, Marquez Avila, & Gulley, 2008). Of the AMP-type stimulants, METH is the most frequently abused (Courtney & Ray, 2014) and has the greatest clinical and societal relevance, particularly given that METH-associated deaths have been on the rise (Hedegaard, Bastian, Trinidad, Spencer, & Warner, 2018). The impact that METH has on mesocorticolimbic circuitry is perhaps more profound than other drugs of abuse due in part to its long half-life, fast uptake and accumulation, and its effects on GLU and DA transmission (Fowler et al., 2008; Parsegian & See, 2014; Stephans & Yamamoto, 1994). In vivo microdialysis reports have demonstrated that METH produces very rapid and large increases in extracellular levels of NAc DA concentrations, an effect believed to underlie the reinforcing effects of the drug (Camp, Browman, & Robinson, 1994; Di Chiara & Imperato, 1988). Additionally, using in vitro electrophysiology in DA neurons, Goodwin and colleagues demonstrated greater DAT-mediated whole-cell currents for METH stimulation than for AMP stimulation and also that METH produced five times greater extracellular levels of DA compared to AMP (Goodwin et al., 2009).
Chronic METH-seeking behaviors in rodents are associated with abnormalities in extracellular levels of GLU. More specifically, extracellular basal GLU levels are significantly reduced in the PFC and NAc core in rats following 10 days of METH self-administration and 10 extinction sessions (Parsegian & See, 2014). When extinguished rats are re-exposed to cues previously paired with the drug and to the drug itself, there is a rise in extracellular GLU within the PFC and NAc core (Parsegian & See, 2014). Notably, this rise in GLU efflux occurs immediately following re-exposure to the drug-paired cues or the drug itself and thus differs from acute exposure to METH which induces a delayed and sustained increase in NAc GLU efflux (K. Ito et al., 2006). Although (Parsegian & See, 2014) did not determine the mechanism underlying the rapid rise in extracellular levels of PFC and NAc core GLU during METH-induced reinstatement, the authors propose that the increase in GLU efflux in both of these regions could derive from multiple neuronal sources of GLU. For example, it is known that the PFC and NAc receive GLU inputs from multiple sources including the VTA, hippocampus, and amygdala (Sesack & Grace, 2010).
Although Parsegian and colleagues demonstrated reduced basal NAc GLU levels in rats following METH self-administration, Lominac and others showed that rats with a history of METH self-administration display increases in basal NAc GLU levels (Lominac et al., 2012). However, there are notable differences in the self-administration models used in these two reports. Parsegian and colleagues measured GLU levels following extinction training whereas Lominac’s measurements occurred after a forced withdrawal paradigm. It is known that extinction vs. forced withdrawal can recruit different neural substrates (Fuchs, Branham, & See, 2006) and can result in differential alterations in GLU receptors (Schwendt, Reichel, & See, 2012). An additional discrepancy between Parsegian’s and Lominac’s reports is the region studied wherein GLU efflux was increased during the METH relapse test. Parsegian and colleagues measured GLU efflux in the NAc core. Lominac and colleagues did not specify what subregion of the NAc they were targeting although according to the microdialysis probe verification figure provided, the NAc core was targeted in some animals and the shell in others (Lominac et al., 2012). The majority of those investigating psychostimulant-induced alterations in GLU efflux following extinction or withdrawal report measurements in the NAc core (Kalivas & Volkow, 2005; McFarland & Kalivas, 2001; McFarland et al., 2003; Parsegian & See, 2014). It is unknown whether NAc shell GLU efflux is altered following chronic METH administration. Furthermore, because increases in extracellular levels of GLU appear to play an important role in METH addiction behaviors, Fugio and colleagues wanted to determine if blocking GLU transport would affect locomotor responses induced by METH. This group found that intracerebroventricular administration of TBOA, a GLU transporter blocker, induced an enhanced locomotor response to a METH challenge injection in rats that were previously treated with METH (Fujio, Nakagawa, Suzuki, Satoh, & Kaneko, 2005). Collectively, these data suggest that preventing the rise in extracellular GLU that occurs during a challenge (or perhaps relapse) test may provide a new strategy to preventing METH addiction behaviors.
In addition to affecting GLU homeostasis within the PFC and NAc, administration of METH (either continuously or acutely in high doses) results in long lasting toxic effects on DA and serotonin neurons (i.e. decreased levels of transmitter, metabolites, and transporters) (Hotchkiss & Gibb, 1980; Morgan & Gibb, 1980; Ricaurte, Guillery, Seiden, Schuster, & Moore, 1982; Volkow, Chang, Wang, Fowler, Ding, et al., 2001; Volkow, Chang, Wang, Fowler, Leonido-Yee, et al., 2001; Wagner et al., 1980; Wilson et al., 1996). These toxic effects are likely due to increases in extracellular levels of GLU; inhibition of corticostriatal GLU release via inhibiting D1 or GABA-A receptors in the SNr which results in a decrease in corticostriatal activation affords protection against METH-induced toxicity to DA terminals (Mark et al., 2004). Additionally, treatment with GLU receptor antagonists (delivered locally within the striatum or systemically) prevents METH-induced damage to DA terminals within the striatum (Finnegan & Taraska, 1996; Halpin & Yamamoto, 2012). Interestingly, acute liver damage and subsequent increases in ammonia have been implicated to play a role in the METH-mediated excitotoxic events including DA and serotonin terminal damage (Halpin & Yamamoto, 2012). Ammonia is metabolized by the liver and has established neurological effects (Halpin & Yamamoto, 2012). Administration of METH acutely in high doses (10 mg/kg, i.p., every 2 h x 4) has been shown to increase plasma and brain ammonia concentrations (Halpin & Yamamoto, 2012). Interestingly, neurotoxicity (cell death) induced by systemic ammonia administration is dampened following systemic administration of GLU receptor antagonists (Kosenko et al., 2003; Saez, Llansola, & Felipo, 1999). Furthermore, intra-striatal infusions of ammonia in combination with METH, but not METH or ammonia alone, recapitulate the DA and serotonin terminal damage (Halpin & Yamamoto, 2012). In order to determine a causative role for ammonia in the release of extracellular levels of GLU and monoamine terminal damage, Halpin and colleagues performed studies whereby they enhanced peripheral ammonia excretion during and after acute binge METH (10 mg/kg, i.p., every 2 h x 4) and examined the long-term effects on monoamine terminal damage in the striatum (Halpin et al., 2014). Following acute binge METH exposure, plasma and brain ammonia levels were increased (Halpin et al., 2014). It was also shown that METH administration in rats pre-treated with lactulose, a compound that lowers plasma and brain ammonia concentrations (Al Sibae & McGuire, 2009; Halpin et al., 2014; Jia & Zhang, 2005; Nicaise et al., 2008), prevents the METH-induced increases in extracellular levels of striatal GLU as well as the monoamine terminal damage (Halpin et al., 2014). In order to determine the mechanism by which ammonia increases extracellular levels of striatal GLU, the authors infused TBOA, a GLU transporter blocker, directly into the striatum of acute binge METH exposed rats (Halpin et al., 2014). This study revealed that intra-striatal infusions of TBOA blocked the ammonia-induced increases in extracellular levels of striatal GLU (Halpin et al., 2014), thus indicating that similar to the actions of AMP, ammonia-induced increases in extracellular levels of striatal GLU may involve the reversal of GLU transporters.
In addition to monoamine terminal damage, METH administration has also been shown to induce inflammatory responses within the CNS. Astrogliosis is a marker of inflammation and is demonstrated by increases in glial fibrillary acidic protein (GFAP) expression; this occurs in the somatosensory cortex 3 days following a single day of binge METH treatment (10 mg/kg given 4 times i.p. at 2 h intervals) (Pu et al., 1996). Microglial activation is elicited by METH in mouse and rat striatum (10–15 mg/kg given 4 times i.p. at 2 h intervals) (Guilarte, Nihei, McGlothan, & Howard, 2003; Thomas et al., 2004). It is conceivable that the increases in GLU in response to METH may activate inflammatory mediators. Indeed, GLU receptor stimulation increases proinflammatory cytokine production of interleukin IL-1β, tumor necrosis factor-α (TNF-α), and IL-6 (Chaparro-Huerta, Rivera-Cervantes, Flores-Soto, Gomez-Pinedo, & Beas-Zarate, 2005; de Bock, Dornand, & Rondouin, 1996; Marini et al., 2004; Vezzani et al., 1999). Conversely, GLU receptor antagonists decrease microglial activation (Taylor, Jones, Kubota, & Pocock, 2005; Thomas & Kuhn, 2005). This interaction between GLU and cytokine production may play a role in promoting dysregulation of glutamate homeostasis and consequent excitotoxicity.
Similar to cocaine, there is currently no FDA approved medication for the treatment of METH addiction. However, investigators have shown promising preclinical results in treating METH addiction behaviors by targeting GLU receptors and GLU transporters. More specifically, targeting mGluR5 has shown promising efficacy for the treatment of METH addiction in preclinical animal models. While mGluR5 receptors are expressed in numerous regions of the brain, they are expressed at relatively high levels within the NAc as well as on DA neurons within the VTA (Ferrada, Sotomayor-Zarate, Abarca, & Gysling, 2017; Mitrano & Smith, 2007; Romano et al., 1995; Shigemoto et al., 1993). MTEP is a selective mGluR5 antagonist that was shown to attenuate reinstatement of METH-seeking behavior induced by cues previously paired with METH or by METH itself (Gass, Osborne, Watson, Brown, & Olive, 2009; Osborne & Olive, 2008). This drug had no effect on food self-administration thus indicating specificity of effect on pathways reinforced by METH. Riluzole and ceftriaxone are other compounds that have been tested for their preclinical efficacy in the treatment of METH addiction; as mentioned above, both of these treatments increase the expression and function of the GLT-1. Riluzole treatment was shown to reduce the expression of locomotor sensitization to METH (Itzhak & Martin, 2000) and ceftriaxone blocked the reinstatement of METH-seeking behavior in a CPP paradigm (Abulseoud, Miller, Wu, Choi, & Holschneider, 2012). Ceftriaxone was shown to increase the expression of GLT-1 in the PFC from METH treated animals compared to METH only treated controls (Abulseoud et al., 2012). One unanswered question from the studies by Abulseoud and colleagues and Itzhak and Martin is whether GLT-1 expression is altered following METH administration in itself (by comparing to expression in naïve animals). While this wasn’t investigated directly, as discussed above in Section 6.3, ceftriaxone treatment seems to affect GLT-1 levels only in injured brain (Jagadapillai et al., 2014; Lipski et al., 2007; Miller et al., 2008; Rothstein et al., 2005). Therefore, it is likely that METH decreases GLT-1 expression, and that the effects of ceftriaxone are dependent on the injury produced by METH.
7. GLT-1 Contributions to Psychostimulant Addiction: Neuronal or Astrocytic?
As has been discussed, GLT-1 plays an important role in psychostimulant addiction behaviors and has served as a therapeutic target in many studies for the treatment of relapse behaviors (Abulseoud et al., 2012; Baker, McFarland, Lake, Shen, Toda, et al., 2003; K. D. Fischer et al., 2013; Itzhak & Martin, 2000; Knackstedt et al., 2010; Rasmussen et al., 2011; Smaga et al., 2020; Spencer & Kalivas, 2017; Tzschentke & Schmidt, 1998). GLT-1 is expressed primarily in astrocytes (Rothstein et al., 1994) and to a lesser extent in neurons (~5–10% of the total, determined in the hippocampus) (Furness et al., 2008). Thus, assay of protein expression by immunoblot would be expected to assess astrocytic GLT-1, because changes in neuronal expression would a priori be expected to be at or below the level of detection. To dissect the contributions of astrocytic and neuronal GLT-1 to GLU homeostasis, a conditional GLT-` knockout was generated and mouse lines produced using Cre/lox technology lacking GLT-1 in astrocytes or in neurons (Petr et al., 2015). The GLT-1 protein expression in the neuronal knockout was not distinguishable by immunoblot analysis from GLT-1 protein expression in wild-type littermates. The efficacy of the neuronal knockout was confirmed by EM immunocytochemistry. In contrast, GLT-1 expression assayed by immunoblot in the astrocytic knockout was reduced by 70–90%. These results establish that immunoblot analysis cannot reliably be used to detect changes in expression of neuronal GLT-1, and that when changes in GLT-1 expression are detected, these changes likely represent changes in astrocytic GLT-1.
A common approach to assessing GLT-1 function is by measuring uptake of radioactive substrates for GLU transporters, typically [3H]L-GLU or [3H]D-aspartate, in crude synaptosomal preparations. These typically are prepared simply by resuspension of the P2 pellet from brain, or brain region homogenates (M. B. Robinson, Sinor, Dowd, & Kerwin, 1993). Because of the predominance of astrocytic GLT-1 over neuronal GLT-1 in the brain, and the erroneous but persistent consensus that GLT-1 is not expressed in neurons, it had long been assumed that uptake of GLU or aspartate into synaptosomal preparations represented astrocytic GLT-1 function (Rimmele & Rosenberg, 2016). However, it was actually found using the conditional GLT-1 knockout that genetic deletion of astrocytic GLT-1 did not significantly decrease uptake of [3H]L-GLU into crude synaptosomes prepared from the forebrain (Petr et al., 2015). In contrast, knockout of neuronal GLT-1 using a synapsin 1-Cre driver reduced GLU uptake significantly by c. 40% in synaptosomes from the forebrain (Petr et al., 2015) and up to 80% in specific regions (Laprairie et al., 2019; McNair et al., 2019; McNair et al., 2020b; Zhou, Hassel, Eid, & Danbolt, 2019). When uptake was assayed into reconstituted liposomes using protein derived from either neuronal GLT-1 knockout or astrocytic GLT-1 knockout brain, then uptake was reduced to a degree expected from the relative amounts of GLT-1 in astrocytes and neurons—that is, a c. 85% decrease in liposomes reconstituted from the astrocytic GLT-1 KO brain, and no significant decrease in liposomes reconstituted from the neuronal GLT1–1 knockout brain (Petr et al., 2015). Synaptosomal uptake measured in crude synaptosomes, therefore, does not reflect the function of GLT-1 in astrocytic membranes, but rather represents the function of GLT-1 expressed in neuronal membranes, even though no effort was made to exclude astrocytic membranes from the preparation.
A powerful and elegant approach that distinguishes neuronal from astrocytic GLU transport sites is electron microscopic D-aspartate uptake autoradiography (Furness et al., 2008; Gundersen, Shupliakov, Brodin, Ottersen, & Storm-Mathisen, 1995). Using this approach in hippocampal slices, it was found that approximately 50% of the immunogold immunocytochemical labeling for D-aspartate was in axon terminals despite the fact that immunogold labeling of terminals for GLT-1 was only 6% of the total (Furness et al., 2008). D-aspartate uptake into terminals was shown to be mediated by GLT-1 because the build-up of D-aspartate immunoreactivity was blocked by an inhibitor of GLT-1 (e.g. DHK) and by GLT-1 knockout (Furness et al., 2008). These results, taken together, establish: 1) that changes in synaptosomal uptake likely reflect changes in function of neuronal GLT-1 (Zhou et al., 2019); and 2) that changes in uptake of GLU transporter substrates into slices cannot be assumed to represent uptake into astrocytes (Furness et al., 2008). Furthermore, these results demonstrate that assessing changes in neuronal GLT-1 function can only be achieved, at this time, by measuring changes in synaptosomal uptake, or using EM autoradiographic or immunocytochemical methods. Given the importance of ceftriaxone as a modulator of GLT-1 expression and function, it is important to note that in a careful study of its effects it was determined that this drug upregulates expression of GLT-1 in axon terminals as well as in astrocytic membranes of the cerebral cortex (Capuani et al., 2016). In addition, recent reports demonstrate that GLT-1 expressed in axon terminals has a significant metabolic function to promote the utilization of GLU by synaptic mitochondria (McNair et al., 2019; McNair et al., 2020b). Therefore, a potential role for neuronal GLT-1 in modulating glutamatergic synaptic transmission in the setting of exposure to drugs of abuse should not be overlooked (K. D. Fischer et al., 2018).
With these recent findings in mind, it is instructive to review work in which attempts have been made to assess changes in GLU transporter activity related to exposure to drugs of abuse by assaying radioactive substrates. The effects of chronic cocaine and heroin on [3H]L-GLU uptake have been assessed in NAc core slices (Knackstedt et al., 2010; Shen, Scofield, Boger, Hensley, & Kalivas, 2014; Trantham-Davidson, LaLumiere, Reissner, Kalivas, & Knackstedt, 2012). Knackstedt et al. found that cocaine self-administration followed by 3 weeks of extinction training resulted in a reduction in NAc core [3H]L-GLU uptake in rats (Knackstedt et al., 2010). This group subsequently demonstrated that treatment with ceftriaxone reversed the cocaine-induced deficits in NAc core [3H]L-GLU uptake (Trantham-Davidson et al., 2012). Shen et al. found that heroin self-administration attenuates NAc core slice [3H]L-GLU uptake and that NAc core slices obtained from heroin-extinguished rats treated with ceftriaxone showed elevated uptake (Shen et al., 2014). Given what has been learned regarding cellular localization of the GLU uptake activity that is measured using radioactive substrates, these demonstrations of changes in GLU uptake activity should not be assumed to be showing changes exclusively localized to astrocytes, and may indicate a contribution of changes in neuronal GLT-1 activity to the alterations in GLU homeostasis produced by exposure to drugs of abuse.
8. Concluding Remarks
The concept of GLU homeostasis first appeared in the scientific literature nearly 40 years ago (Schousboe & Hertz, 1981). When first introduced, the focus was primarily on GLU metabolism and the compartmentalization of different aspects of GLU metabolism in neurons and astrocytes. Subsequently, with the discovery of the phenomenon of excitotoxicity (D.W. Choi, 1988; J.W. Olney, 1969; J.W. Olney & Ho, 1970; J. W. Olney & Sharpe, 1969; S. M. Rothman, 1983; S. M. Rothman & Olney, 1986) and the high sensitivity of CNS neurons to the toxic effects of GLU (M.B. Robinson, Djali, & Buchhalter, 1993; Rosenberg & Aizenman, 1989; Rosenberg, Amin, & Leitner, 1992), GLU homeostasis acquired additional importance because the regulation of extracellular GLU concentration was seen to be critical in determining whether neurons would survive the continual release of GLU from excitatory synapses (Lipton & Rosenberg, 1994; Schousboe, Sonnewald, Civenni, & Gegelashvili, 1997). In the last 20 years, our understanding of GLU homeostasis has been greatly expanded by a large body of evidence that suggests that the control of extracellular GLU concentration around synapses actually determines the changes in behavior that follow exposure to drugs of abuse. This is the subject of the present review. There is not a more compelling body of evidence suggesting that GLU homeostasis regulates behavior, although the evidence is mounting for a similarly important role for GLU homeostasis in other areas of investigation, including mental illness (Cui et al., 2014), pain (Inquimbert et al., 2012; Inquimbert et al., 2018), epilepsy, synaptic plasticity (Levenson, Weeber, Sweatt, & Eskin, 2002; Omrani et al., 2009; Pita-Almenar, Sol Collado, Colbert, & Eskin, 2006), and neurodegeneration (Scimemi et al., 2013; Zott et al., 2019). Collectively, the body of work reviewed here produced by many dedicated investigators support the need for further research focusing on the regulation of GLU homeostasis in the pathophysiology of addiction and as a target for clinical intervention.
Highlights.
METH, AMP, and cocaine alter glutamate homeostasis within the mesolimbic circuit
GLT-1 and xCT are the glutamate transporters primarily responsible for drug-induced changes in glutamate homeostasis
GLT-1 and xCT are therapeutic targets for METH, AMP, and cocaine addiction behaviors
Glutamate co-transmission by dopamine neurons is important for behavioral responses to psychostimulants
Acknowledgments
The authors would like to thank Dr. Stephen Rayport and Dr. Susana Mingote for reviewing this manuscript and providing invaluable suggestions. The authors were partially funded by the following grants: NIH NS066019, MH104318, HD018655. LK was supported by DA045140. KF was supported by T32 NS007473.
Abbreviations:
- GLU
glutamate
- DA
dopamine
- NAc
nucleus accumbens
- VTA
ventral tegmental area
- PFC
prefrontal cortex
- METH
methamphetamine
- AMP
amphetamine
- mGluR
metabotropic glutamate receptor
- NAC
N-acetylcysteine
- SA
Self-Administration
- CPP
Conditioned Place Preference
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest: None
References
- Abulseoud OA, Miller JD, Wu J, Choi DS, & Holschneider DP (2012). Ceftriaxone upregulates the glutamate transporter in medial prefrontal cortex and blocks reinstatement of methamphetamine seeking in a condition place preference paradigm. Brain Res, 1456, 14–21. 10.1016/j.brainres.2012.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinoff B (2004). Neurobiologic processes in drug reward and addiction. Harv Rev Psychiatry, 12(6), 305–320. 10.1080/10673220490910844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrover MF, Shin JH, & Alvarez VA (2014). Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J Neurosci, 34(9), 3183–3192. 10.1523/JNEUROSCI.4958-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar JI, Dunn M, Mingote S, Karam CS, Farino ZJ, Sonders MS, … Freyberg Z(2017). Neuronal Depolarization Drives Increased Dopamine Synaptic Vesicle Loading via VGLUT. Neuron, 95(5), 1074–1088 10.1016/j.neuron.2017.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH (2012). The science of making drug-addicted animals. Neuroscience, 211, 107–125. 10.1016/j.neuroscience.2011.08.014 [DOI] [PubMed] [Google Scholar]
- Ahmed SH, & Koob GF (1997). Cocaine- but not food-seeking behavior is reinstated by stress after extinction. Psychopharmacology (Berl), 132(3), 289–295. 10.1007/s002130050347 [DOI] [PubMed] [Google Scholar]
- Ahmed SH, & Koob GF (1998). Transition from moderate to excessive drug intake: change in hedonic set point. Science, 282(5387), 298–300. 10.1126/science.282.5387.298 [DOI] [PubMed] [Google Scholar]
- Aked J, Coizet V, Clark D, & Overton PG (2005). Local injection of a glutamate uptake inhibitor into the ventral tegmental area produces sensitization to the behavioural effects of d-amphetamine. Neuroscience, 134(2), 361–367. 10.1016/j.neuroscience.2005.04.044 [DOI] [PubMed] [Google Scholar]
- Al Sibae MR, & McGuire BM (2009). Current trends in the treatment of hepatic encephalopathy. Ther Clin Risk Manag, 5(3), 617–626. 10.2147/tcrm.s4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsio J, Nordenankar K, Arvidsson E, Birgner C, Mahmoudi S, Halbout B, … Wallen-Mackenzie A (2011). Enhanced sucrose and cocaine self-administration and cue-induced drug seeking after loss of VGLUT2 in midbrain dopamine neurons in mice. J Neurosci, 31(35), 12593–12603. 10.1523/JNEUROSCI.2397-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggi F, Ghazizadeh A, Nicola SM, & Fields HL (2011). Roles of nucleus accumbens core and shell in incentive-cue responding and behavioral inhibition. J Neurosci, 31(18), 6820–6830. 10.1523/JNEUROSCI.6491-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TR, Huguenard JR, & Prince DA (2010). Differential effects of Na+-K+ ATPase blockade on cortical layer V neurons. J Physiol, 588(Pt 22), 4401–4414. 10.1113/jphysiol.2010.191858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM, & Bonci A (2008). Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci, 28(37), 9092–9100. 10.1523/JNEUROSCI.1001-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, … Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis, 44(8), 383–390. 10.1002/dvg.20228 [DOI] [PubMed] [Google Scholar]
- Bae N, Wang Y, Li L, Rayport S, & Lubec G (2013). Network of brain protein level changes in glutaminase deficient fetal mice. J Proteomics, 80, 236–249. 10.1016/j.jprot.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, & Kalivas PW (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci, 6(7), 743–749. 10.1038/nn1069 [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Toda S, & Kalivas PW (2003). N-acetyl cysteine-induced blockade of cocaine-induced reinstatement. Ann N Y Acad Sci, 1003, 349–351. 10.1196/annals.1300.023 [DOI] [PubMed] [Google Scholar]
- Baker DA, Shen H, & Kalivas PW (2002). Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino Acids, 23(1–3), 161–162. 10.1007/s00726-001-0122-6 [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, & Kalivas PW (2002). The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci, 22(20), 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai S (1986). Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem, 261(5), 2256–2263. [PubMed] [Google Scholar]
- Bechard AR, Hamor PU, Wu L, Schwendt M, & Knackstedt LA (2019). The effects of clavulanic acid and amoxicillin on cue-primed reinstatement of cocaine seeking. Behav Neurosci, 133(2), 247–254. 10.1037/bne0000297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett AH, & Moffat AC (1969). Correlation of partition coefficients in n-heptane-aqueous systems with buccal absorption data for a series of amines and acids. J Pharm Pharmacol, 21, 10.1111/j.2042-7158.1969.tb08365.x [DOI] [PubMed] [Google Scholar]
- Beeler JA, Mourra D, Zanca M, Kalmbach A, Gellman C, Klein BY, … Burghardt NS (2020). VULNERABLE AND RESILIENT PHENOTYPES IN A MOUSE MODEL OFANOREXIA NERVOSA. Biol Psychiatry. 10.1016/j.biopsych.2020.06.030 [DOI] [PMC free article] [PubMed]
- Belin-Rauscent A BD (2012). Animal Models of Drug Addiction Addictions - From Pathophysiology to Treatment (pp. 3–20).
- Bellone C, & Luscher C (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci, 9(5), 636–641. 10.1038/nn1682 [DOI] [PubMed] [Google Scholar]
- Berger UV, DeSilva TM, Chen W, & Rosenberg PA (2005). Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol, 492(1), 78–89. 10.1002/cne.20737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, … Nakamura K (2014). Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci, 34(43), 14304–14317. 10.1523/JNEUROSCI.0930-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorello AM, Hopfield JF, Aperia A, & Greengard P (1990). Inhibition by dopamine of (Na(+)+K+)ATPase activity in neostriatal neurons through D1 and D2 dopamine receptor synergism. Nature, 347(6291), 386–388. 10.1038/347386a0 [DOI] [PubMed] [Google Scholar]
- Berube-Carriere N, Riad M, Dal Bo G, Levesque D, Trudeau LE, & Descarries L (2009). The dual dopamine-glutamate phenotype of growing mesencephalic neurons regresses in mature rat brain. J Comp Neurol, 517(6), 873–891. 10.1002/cne.22194 [DOI] [PubMed] [Google Scholar]
- Bezzi P, Vesce S, Panzarasa P, & Volterra A (1999). Astrocytes as active participants of glutamatergic function and regulators of its homeostasis. Adv Exp Med Biol, 468, 69–80. 10.1007/978-1-4615-4685-6_6 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee AK, Chang L, White L, Bazinet RP, & Rapoport SI (2006). D-Amphetamine stimulates D2 dopamine receptor-mediated brain signaling involving arachidonic acid in unanesthetized rats. J Cereb Blood Flow Metab, 26(11), 1378–1388. 10.1038/sj.jcbfm.9600290 [DOI] [PubMed] [Google Scholar]
- Biber K, Laurie DJ, Berthele A, Sommer B, Tolle TR, Gebicke-Harter PJ, … Boddeke HW (1999). Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem, 72(4), 1671–1680. 10.1046/j.1471-4159.1999.721671.x [DOI] [PubMed] [Google Scholar]
- Birgner C, Nordenankar K, Lundblad M, Mendez JA, Smith C, le Greves M, … Wallen-Mackenzie A (2010). VGLUT2 in dopamine neurons is required for psychostimulant-induced behavioral activation. Proc Natl Acad Sci U S A, 107(1), 389–394. 10.1073/pnas.0910986107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjijou Y, Stinus L, Le Moal M, & Cador M (1996). Evidence for selective involvement of dopamine D1 receptors of the ventral tegmental area in the behavioral sensitization induced by intra-ventral tegmental area injections of D-amphetamine. J Pharmacol Exp Ther, 277(2), 1177–1187. [PubMed] [Google Scholar]
- Borgstrom L, & Kagedal B (1990). Dose dependent pharmacokinetics of N-acetylcysteine after oral dosing to man. Biopharm Drug Dispos, 11(2), 131–136. 10.1002/bdd.2510110205 [DOI] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, & Shaham Y (2013). The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl), 229(3), 453–476. 10.1007/s00213-013-3120-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque MJ, & Trudeau LE (2000). GDNF enhances the synaptic efficacy of dopaminergic neurons in culture. Eur J Neurosci, 12(9), 3172–3180. 10.1046/j.1460-9568.2000.00219.x [DOI] [PubMed] [Google Scholar]
- Bradford HF, Young AM, & Crowder JM (1987). Continuous glutamate leakage from brain cells is balanced by compensatory high-affinity reuptake transport. Neurosci Lett, 81(3), 296–302. 10.1016/0304-3940(87)90399-5 [DOI] [PubMed] [Google Scholar]
- Bramness JG, Gundersen OH, Guterstam J, Rognli EB, Konstenius M, Loberg EM, … Franck J (2012). Amphetamine-induced psychosis--a separate diagnostic entity or primary psychosis triggered in the vulnerable? BMC Psychiatry, 12, 221. 10.1186/1471-244X-12-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges RJ, Stanley MS, Anderson MW, Cotman CW, & Chamberlin AR (1991). Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem, 34(2), 717–725. 10.1021/jm00106a037 [DOI] [PubMed] [Google Scholar]
- Buck SA, Torregrossa MM, Logan RW, & Freyberg Z (2020). Roles of dopamine and glutamate co-release in the nucleus accumbens in mediating the actions of drugs of abuse. FEBS J. 10.1111/febs.15496 [DOI] [PMC free article] [PubMed]
- Cador M, Bjijou Y, & Stinus L (1995). Evidence of a complete independence of the neurobiological substrates for the induction and expression of behavioral sensitization to amphetamine. Neuroscience, 65(2), 385–395. 10.1016/0306-4522(94)00524-9 [DOI] [PubMed] [Google Scholar]
- Cai Y, & Ford CP (2018). Dopamine Cells Differentially Regulate Striatal Cholinergic Transmission across Regions through Corelease of Dopamine and Glutamate. Cell Rep, 25(11), 3148–3157 10.1016/j.celrep.2018.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp DM, Browman KE, & Robinson TE (1994). The effects of methamphetamine and cocaine on motor behavior and extracellular dopamine in the ventral striatum of Lewis versus Fischer 344 rats. Brain Res, 668(1–2), 180–193. 10.1016/0006-8993(94)90523-1 [DOI] [PubMed] [Google Scholar]
- Cappon GD, Broening HW, Pu C, Morford L, & Vorhees CV (1996). alpha-Phenyl-N-tert-butyl nitrone attenuates methamphetamine-induced depletion of striatal dopamine without altering hyperthermia. Synapse, 24(2), 173–181. [DOI] [PubMed] [Google Scholar]
- Capriles N, Rodaros D, Sorge RE, & Stewart J (2003). A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl), 168(1–2), 66–74. 10.1007/s00213-002-1283-z [DOI] [PubMed] [Google Scholar]
- Capuani C, Melone M, Tottene A, Bragina L, Crivellaro G, Santello M, … Pietrobon D(2016). Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol Med, 8(8), 967–986. 10.15252/emmm.201505944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, Duty S, & Rattray M (2012). Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem Int, 60(1), 31–38. 10.1016/j.neuint.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni E, Imperato A, Perezzani L, & Di Chiara G (1989). Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience, 28(3), 653–661. 10.1016/0306-4522(89)90012-2 [DOI] [PubMed] [Google Scholar]
- Carboni E, Spielewoy C, Vacca C, Nosten-Bertrand M, Giros B, & Di Chiara G (2001). Cocaine and amphetamine increase extracellular dopamine in the nucleus accumbens of mice lacking the dopamine transporter gene. J Neurosci, 21(9), RC141: 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, Chen SF, & Yu AC (1988). Induction of intracellular superoxide radical formation by arachidonic acid and by polyunsaturated fatty acids in primary astrocytic cultures. J Neurochem, 50(4), 1185–1193. 10.1111/j.1471-4159.1988.tb10591.x [DOI] [PubMed] [Google Scholar]
- Chan PH, & Fishman RA (1980). Transient formation of superoxide radicals in polyunsaturated fatty acid-induced brain swelling. J Neurochem, 35(4), 1004–1007. 10.1111/j.1471-4159.1980.tb07100.x [DOI] [PubMed] [Google Scholar]
- Chan PH, Kerlan R, & Fishman RA (1983). Reductions of gamma-aminobutyric acid and glutamate uptake and (Na+ + K+)-ATPase activity in brain slices and synaptosomes by arachidonic acid. J Neurochem, 40(2), 309–316. 10.1111/j.1471-4159.1983.tb11284.x [DOI] [PubMed] [Google Scholar]
- Chaparro-Huerta V, Rivera-Cervantes MC, Flores-Soto ME, Gomez-Pinedo U, & Beas-Zarate C (2005). Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J Neuroimmunol, 165(1–2), 53–62. 10.1016/j.jneuroim.2005.04.025 [DOI] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, … Rosenberg PA (2004). The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci, 24(5), 1136–1148. 10.1523/JNEUROSCI.1586-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress AR, Hole AV, Ehrman RN, Robbins SJ, McLellan AT, & O’Brien CP (1993). Cue reactivity and cue reactivity interventions in drug dependence. NIDA Res Monogr, 137, 73–95. [PubMed] [Google Scholar]
- Cho Y, & Bannai S (1990). Uptake of glutamate and cysteine in C-6 glioma cells and in cultured astrocytes. J Neurochem, 55(6), 2091–2097. 10.1111/j.1471-4159.1990.tb05800.x [DOI] [PubMed] [Google Scholar]
- Chohan MO, Esses S, Haft J, Ahmari S, & Veenstra-VanderWeele J (2020). Altered baseline and amphetamine-mediated behavioral profiles in dopamine transporter Cre (DAT-Ires-Cre) mice compared to tyrosine hydroxylase Cre (TH-Cre) mice. Psychopharmacology (Berl). 10.1007/s00213-020-05635-4 [DOI] [PMC free article] [PubMed]
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1(8), 623–634. 10.1016/0896-6273(88)90162-6 [DOI] [PubMed] [Google Scholar]
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1, 623–634. [DOI] [PubMed] [Google Scholar]
- Chuhma N, Choi WY, Mingote S, & Rayport S (2009). Dopamine neuron glutamate cotransmission: frequency-dependent modulation in the mesoventromedial projection. Neuroscience, 164(3), 1068–1083. 10.1016/j.neuroscience.2009.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Yetnikoff L, Kalmbach A, Ma T, Ztaou S, … Rayport S (2018). Dopamine neuron glutamate cotransmission evokes a delayed excitation in lateral dorsal striatal cholinergic interneurons. Elife, 7. 10.7554/eLife.39786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, & Rayport S (2004). Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci, 24(4), 972–981. 10.1523/JNEUROSCI.4317-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi-Mas J, Geisler S, Zimmer L, Zahm DS, & Berod A (2007). Activation of afferents to the ventral tegmental area in response to acute amphetamine: a double-labelling study. Eur J Neurosci, 26(4), 1011–1025. 10.1111/j.1460-9568.2007.05738.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, & Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol, 37, 205–237. 10.1146/annurev.pharmtox.37.1.205 [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, … Wolf ME (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature, 454(7200), 118–121. 10.1038/nature06995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KE, & Ray LA (2014). Methamphetamine: an update on epidemiology, pharmacology, clinical phenomenology, and treatment literature. Drug Alcohol Depend, 143, 11–21. 10.1016/j.drugalcdep.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Mizukami H, Yanagisawa M, Aida T, Nomura M, Isomura Y, … Aizawa H (2014). Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. J Neurosci, 34(49), 16273–16285. 10.1523/JNEUROSCI.1465-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Bo G, St-Gelais F, Danik M, Williams S, Cotton M, & Trudeau LE (2004). Dopamine neurons in culture express VGLUT2 explaining their capacity to release glutamate at synapses in addition to dopamine. J Neurochem, 88(6), 1398–1405. 10.1046/j.1471-4159.2003.02277.x [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, & Graybiel AM (1999). The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain, 122 (Pt 8), 1437–1448. 10.1093/brain/122.8.1437 [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol, 65(1), 1–105. 10.1016/s0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, & Kanner BI (1992). An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience, 51(2), 295–310. 10.1016/0306-4522(92)90316-t [DOI] [PubMed] [Google Scholar]
- Daniels M, & Brown DR (2001). Astrocytes regulate N-methyl-D-aspartate receptor subunit composition increasing neuronal sensitivity to excitotoxicity. J Biol Chem, 276(25), 22446–22452. 10.1074/jbc.M101740200 [DOI] [PubMed] [Google Scholar]
- David V, Polis I, McDonald J, & Gold LH (2001). Intravenous self-administration of heroin/cocaine combinations (speedball) using nose-poke or lever-press operant responding in mice. Behav Pharmacol, 12(1), 25–34. 10.1097/00008877-200102000-00003 [DOI] [PubMed] [Google Scholar]
- de Bock F, Dornand J, & Rondouin G (1996). Release of TNF alpha in the rat hippocampus following epileptic seizures and excitotoxic neuronal damage. Neuroreport, 7(6), 1125–1129. 10.1097/00001756-199604260-00004 [DOI] [PubMed] [Google Scholar]
- Deisseroth K (2014). Circuit dynamics of adaptive and maladaptive behaviour. Nature, 505(7483), 309–317. 10.1038/nature12982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Martinez R, & Mora F (1998). Amphetamine increases extracellular concentrations of glutamate in the prefrontal cortex of the awake rat: a microdialysis study. Neurochem Res, 23(9), 1153–1158. 10.1023/a:1020769816332 [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, … Lecca D (2004). Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology, 47 Suppl 1, 227–241. 10.1016/j.neuropharm.2004.06.032 [DOI] [PubMed] [Google Scholar]
- Di Chiara G, & Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A, 85(14), 5274–5278. 10.1073/pnas.85.14.5274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divac I, Fonnum F, & Storm-Mathisen J (1977). High affinity uptake of glutamate in terminals of corticostriatal axons. Nature, 266(5600), 377–378. 10.1038/266377a0 [DOI] [PubMed] [Google Scholar]
- Donzanti BA, & Uretsky NJ (1983). Effects of excitatory amino acids on locomotor activity after bilateral microinjection into the rat nucleus accumbens: possible dependence on dopaminergic mechanisms. Neuropharmacology, 22(8), 971–981. 10.1016/0028-3908(83)90213-7 [DOI] [PubMed] [Google Scholar]
- Dringen R, & Hirrlinger J (2003). Glutathione pathways in the brain. Biol Chem, 384(4), 505–516. 10.1515/BC.2003.059 [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, … Larsson NG (2007). Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A, 104(4), 1325–1330. 10.1073/pnas.0605208103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, … Spanagel R (2008). Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron, 59(3), 497–508. 10.1016/j.neuron.2008.07.010 [DOI] [PubMed] [Google Scholar]
- Engeli EJE, Zoelch N, Hock A, Nordt C, Hulka LM, Kirschner M, … Herdener M (2020). Impaired glutamate homeostasis in the nucleus accumbens in human cocaine addiction. Mol Psychiatry. 10.1038/s41380-020-0828-z [DOI] [PubMed]
- Ernst T, & Chang L (2008). Adaptation of brain glutamate plus glutamine during abstinence from chronic methamphetamine use. J Neuroimmune Pharmacol, 3(3), 165–172. 10.1007/s11481-008-9108-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Henningsen RA, Neve KA, & Janowsky A (1994). Release of dopamine via the human transporter. Mol Pharmacol, 45(2), 312–316. [PubMed] [Google Scholar]
- Everitt BJ, & Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci, 8(11), 1481–1489. 10.1038/nn1579 [DOI] [PubMed] [Google Scholar]
- Ferrada C, Sotomayor-Zarate R, Abarca J, & Gysling K (2017). The activation of metabotropic glutamate 5 receptors in the rat ventral tegmental area increases dopamine extracellular levels. Neuroreport, 28(1), 28–34. 10.1097/WNR.0000000000000708 [DOI] [PubMed] [Google Scholar]
- Ferrario CR, Gorny G, Crombag HS, Li Y, Kolb B, & Robinson TE (2005). Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry, 58(9), 751–759. 10.1016/j.biopsych.2005.04.046 [DOI] [PubMed] [Google Scholar]
- Finnegan KT, & Taraska T (1996). Effects of glutamate antagonists on methamphetamine and 3,4-methylenedioxymethamphetamine-induced striatal dopamine release in vivo. J Neurochem, 66(5), 1949–1958. 10.1046/j.1471-4159.1996.66051949.x [DOI] [PubMed] [Google Scholar]
- Fischer JF, & Cho AK (1976). Properties of dopamine efflux from rat striatal tissue caused by amphetamine and p-hydroxyamphetamine. Proc West Pharmacol Soc, 19, 179–182. [PubMed] [Google Scholar]
- Fischer KD, Houston AC, & Rebec GV (2013). Role of the major glutamate transporter GLT1 in nucleus accumbens core versus shell in cue-induced cocaine-seeking behavior. J Neurosci, 33(22), 9319–9327. 10.1523/JNEUROSCI.3278-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KD, Houston ACW, Desai RI, Doyle MR, Bergman J, Mian M, … Rosenberg PA (2018). Behavioral phenotyping and dopamine dynamics in mice with conditional deletion of the glutamate transporter GLT-1 in neurons: resistance to the acute locomotor effects of amphetamine. Psychopharmacology (Berl), 235(5), 1371–1387. 10.1007/s00213-018-4848-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith KD, Houston AC, & Rebec GV (2012). Differential effects of cocaine access and withdrawal on glutamate type 1 transporter expression in rat nucleus accumbens core and shell. Neuroscience, 210, 333–339. 10.1016/j.neuroscience.2012.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin GM, Bourque MJ, Mendez JA, Leo D, Nordenankar K, Birgner C, … Trudeau LE (2012). Glutamate corelease promotes growth and survival of midbrain dopamine neurons. J Neurosci, 32(48), 17477–17491. 10.1523/JNEUROSCI.1939-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Logan J, Alexoff D, Telang F, Wang GJ, … Apelskog K (2008). Fast uptake and long-lasting binding of methamphetamine in the human brain: comparison with cocaine. Neuroimage, 43(4), 756–763. 10.1016/j.neuroimage.2008.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Sonders MS, Aguilar JI, Hiranita T, Karam CS, Flores J, … Javitch JA (2016). Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat Commun, 7, 10652. 10.1038/ncomms10652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Branham RK, & See RE (2006). Different neural substrates mediate cocaine seeking after abstinence versus extinction training: a critical role for the dorsolateral caudate-putamen. J Neurosci, 26(13), 3584–3588. 10.1523/JNEUROSCI.5146-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio M, Nakagawa T, Suzuki Y, Satoh M, & Kaneko S (2005). Facilitative effect of a glutamate transporter inhibitor (2S,3S)-3-{3-[4-(trifluoromethyl)benzoylamino]benzyloxy}aspartate on the expression of methamphetamine-induced behavioral sensitization in rats. J Pharmacol Sci, 99(4), 415–418. 10.1254/jphs.sc0050144 [DOI] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, … Danbolt NC (2008). A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience, 157(1), 80–94. 10.1016/j.neuroscience.2008.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, … Rayport S (2009). Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacology, 34(10), 2305–2322. 10.1038/npp.2009.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, … Tsai LH (2010). A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature, 466(7310), 1105–1109. 10.1038/nature09271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JT, Osborne MP, Watson NL, Brown JL, & Olive MF (2009). mGluR5 antagonism attenuates methamphetamine reinforcement and prevents reinstatement of methamphetamine-seeking behavior in rats. Neuropsychopharmacology, 34(4), 820–833. 10.1038/npp.2008.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George F Koob MLM (2006). Psychostimulants Neurobiology of Addiction (pp. 69–120): Science Direct. [Google Scholar]
- Gewin LS (2019). The Cre/lox system: Cre-ating unintended damage. Am J Physiol Renal Physiol, 316(5), F873–F874. 10.1152/ajprenal.00428.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti M, Hotsenpiller G, Ward P, Teppen T, & Wolf ME (2001). Amphetamine-induced plasticity of AMPA receptors in the ventral tegmental area: effects on extracellular levels of dopamine and glutamate in freely moving rats. J Neurosci, 21(16), 6362–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, & Caron MG (1993). Molecular characterization of the dopamine transporter. Trends Pharmacol Sci, 14(2), 43–49. 10.1016/0165-6147(93)90029-j [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, & Caron MG (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature, 379(6566), 606–612. 10.1038/379606a0 [DOI] [PubMed] [Google Scholar]
- Giusti SA, Vercelli CA, Vogl AM, Kolarz AW, Pino NS, Deussing JM, & Refojo D (2014). Behavioral phenotyping of Nestin-Cre mice: implications for genetic mouse models of psychiatric disorders. J Psychiatr Res, 55, 87–95. 10.1016/j.jpsychires.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Goodwin JS, Larson GA, Swant J, Sen N, Javitch JA, Zahniser NR, … Khoshbouei H (2009). Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J Biol Chem, 284(5), 2978–2989. 10.1074/jbc.M805298200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granas C, Ferrer J, Loland CJ, Javitch JA, & Gether U (2003). N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J Biol Chem, 278(7), 4990–5000. 10.1074/jbc.M205058200 [DOI] [PubMed] [Google Scholar]
- Griffin WC 3rd, Haun HL, Hazelbaker CL, Ramachandra VS, & Becker HC (2014). Increased extracellular glutamate in the nucleus accumbens promotes excessive ethanol drinking in ethanol dependent mice. Neuropsychopharmacology, 39(3), 707–717. 10.1038/npp.2013.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, & Shaham Y (2001). Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature, 412(6843), 141–142. 10.1038/35084134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves PM, Wilson CJ, Young SJ, & Rebec GV (1975). Self-inhibition by dopaminergic neurons. Science, 190(4214), 522–528. 10.1126/science.242074 [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Nihei MK, McGlothan JL, & Howard AS (2003). Methamphetamine-induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience, 122(2), 499–513. 10.1016/s0306-4522(03)00476-7 [DOI] [PubMed] [Google Scholar]
- Gundersen V, Shupliakov O, Brodin L, Ottersen OP, & Storm-Mathisen J (1995). Quantification of excitatory amino acid uptake at intact glutamatergic synapses by immunocytochemistry of exogenous D-aspartate. Journal Of Neuroscience, 15, 4417–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Stanis JJ, Marquez Avila H, & Gulley JM (2008). A comparison of amphetamine- and methamphetamine-induced locomotor activity in rats: evidence for qualitative differences in behavior. Psychopharmacology (Berl), 195(4), 469–478. 10.1007/s00213-007-0923-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Northrop NA, & Yamamoto BK (2014). Ammonia mediates methamphetamine-induced increases in glutamate and excitotoxicity. Neuropsychopharmacology, 39(4), 1031–1038. 10.1038/npp.2013.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, & Yamamoto BK (2012). Peripheral ammonia as a mediator of methamphetamine neurotoxicity. J Neurosci, 32(38), 13155–13163. 10.1523/JNEUROSCI.2530-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DD, & Gu HH (2006). Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol, 6, 6. 10.1186/1471-2210-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney M (2009). Self-administration of cocaine, cannabis and heroin in the human laboratory: benefits and pitfalls. Addict Biol, 14(1), 9–21. 10.1111/j.1369-1600.2008.00121.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard H, Bastian BA, Trinidad JP, Spencer M, & Warner M (2018). Drugs Most Frequently Involved in Drug Overdose Deaths: United States, 2011–2016. Natl Vital Stat Rep, 67(9), 1–14. [PubMed] [Google Scholar]
- Hexum TD, & Fried R (1979). Effects of superoxide radicals on transport (Na + K) adenosine triphosphatase and protection by superoxide dismutase. Neurochem Res, 4(1), 73–82. 10.1007/BF00963833 [DOI] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, & Agid YA (1988). Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature, 334(6180), 345–348. 10.1038/334345a0 [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, … Edwards RH (2010). Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron, 65(5), 643–656. 10.1016/j.neuron.2010.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, & Edwards RH (2012). Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol, 74, 225–243. 10.1146/annurev-physiol-020911-153315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Hjelmstad GO, Fields HL, & Edwards RH (2012). Ventral tegmental area glutamate neurons: electrophysiological properties and projections. J Neurosci, 32(43), 15076–15085. 10.1523/JNEUROSCI.3128-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, … Danbolt NC (2012). The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci, 32(17), 6000–6013. 10.1523/JNEUROSCI.5347-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss AJ, & Gibb JW (1980). Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther, 214(2), 257–262. [PubMed] [Google Scholar]
- Hu G, Duffy P, Swanson C, Ghasemzadeh MB, & Kalivas PW (1999). The regulation of dopamine transmission by metabotropic glutamate receptors. J Pharmacol Exp Ther, 289(1), 412–416. [PubMed] [Google Scholar]
- Ikemoto S (2010). Brain reward circuitry beyond the mesolimbic dopamine system: a neurobiological theory. Neurosci Biobehav Rev, 35(2), 129–150. 10.1016/j.neubiorev.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S, Yang C, & Tan A (2015). Basal ganglia circuit loops, dopamine and motivation: A review and enquiry. Behav Brain Res, 290, 17–31. 10.1016/j.bbr.2015.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inquimbert P, Bartels K, Babaniyi OB, Barrett LB, Tegeder I, & Scholz J (2012). Peripheral nerve injury produces a sustained shift in the balance between glutamate release and uptake in the dorsal horn of the spinal cord. Pain, 153(12), 2422–2431. 10.1016/j.pain.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inquimbert P, Moll M, Latremoliere A, Tong CK, Whang J, Sheehan GF, … Scholz J (2018). NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep, 23(9), 2678–2689. 10.1016/j.celrep.2018.04.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Abekawa T, & Koyama T (2006). Relationship between development of cross-sensitization to MK-801 and delayed increases in glutamate levels in the nucleus accumbens induced by a high dose of methamphetamine. Psychopharmacology, 187(3), 293–302. 10.1007/s00213-006-0423-2 [DOI] [PubMed] [Google Scholar]
- Ito R, & Hayen A (2011). Opposing roles of nucleus accumbens core and shell dopamine in the modulation of limbic information processing. J Neurosci, 31(16), 6001–6007. 10.1523/JNEUROSCI.6588-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak Y, & Martin JL (2000). Effect of riluzole and gabapentin on cocaine- and methamphetamine-induced behavioral sensitization in mice. Psychopharmacology (Berl), 151(2–3), 226–233. 10.1007/s002130000394 [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, & Gerber U (1999). Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proceedings of the National Academy of Sciences of the United States of America, 96(15), 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, & Przedborski S (1995). Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration, 4(3), 257–269. 10.1016/1055-8330(95)90015-2 [DOI] [PubMed] [Google Scholar]
- Jagadapillai R, Mellen NM, Sachleben LR Jr., & Gozal E (2014). Ceftriaxone preserves glutamate transporters and prevents intermittent hypoxia-induced vulnerability to brain excitotoxic injury. PLoS One, 9(7), e100230. 10.1371/journal.pone.0100230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janbandhu VC, Moik D, & Fassler R (2014). Cre recombinase induces DNA damage and tetraploidy in the absence of loxP sites. Cell Cycle, 13(3), 462–470. 10.4161/cc.27271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, & Zhang MH (2005). Comparison of probiotics and lactulose in the treatment of minimal hepatic encephalopathy in rats. World J Gastroenterol, 11(6), 908–911. 10.3748/wjg.v11.i6.908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, & Kauer JA (1999). Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci, 19(22), 9780–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, & Caron MG (1998). Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A, 95(7), 4029–4034. 10.1073/pnas.95.7.4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, & Caron MG (1998). Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci, 18(6), 1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol, 4(1), 23–29. 10.1016/j.coph.2003.11.002 [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci, 10(8), 561–572. 10.1038/nrn2515 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, & Duffy P (1995). D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci, 15(7 Pt 2), 5379–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Lalumiere RT, Knackstedt L, & Shen H (2009). Glutamate transmission in addiction. Neuropharmacology, 56 Suppl 1, 169–173. 10.1016/j.neuropharm.2008.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, & McFarland K (2003). Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology (Berl), 168(1–2), 44–56. 10.1007/s00213-003-1393-2 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, & Stewart J (1991). Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev, 16(3), 223–244. 10.1016/0165-0173(91)90007-u [DOI] [PubMed] [Google Scholar]
- Kalivas PW, & Volkow ND (2005). The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry, 162(8), 1403–1413. 10.1176/appi.ajp.162.8.1403 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, & Weber B (1988). Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther, 245(3), 1095–1102. [PubMed] [Google Scholar]
- Kawano M, Kawasaki A, Sakata-Haga H, Fukui Y, Kawano H, Nogami H, & Hisano S (2006). Particular subpopulations of midbrain and hypothalamic dopamine neurons express vesicular glutamate transporter 2 in the rat brain. J Comp Neurol, 498(5), 581–592. 10.1002/cne.21054 [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, … Javitch JA(2004). N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol, 2(3), E78. 10.1371/journal.pbio.0020078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, John J, Langford D, Walker E, Ward S, & Rawls SM (2016). Clavulanic acid enhances glutamate transporter subtype I (GLT-1) expression and decreases reinforcing efficacy of cocaine in mice. Amino Acids, 48(3), 689–696. 10.1007/s00726-015-2117-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Ganesan S, Luo SX, Wu YW, Park E, Huang EJ, … Ding JB (2015). Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science, 350(6256), 102–106. 10.1126/science.aac4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI, & Kalivas PW (2010). Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry, 67(1), 81–84. 10.1016/j.biopsych.2009.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko E, Llansola M, Montoliu C, Monfort P, Rodrigo R, Hernandez-Viadel M, … Felipo V (2003). Glutamine synthetase activity and glutamine content in brain: modulation by NMDA receptors and nitric oxide. Neurochem Int, 43(4–5), 493–499. 10.1016/s0197-0186(03)00039-1 [DOI] [PubMed] [Google Scholar]
- Koulchitsky S, De Backer B, Quertemont E, Charlier C, & Seutin V (2012). Differential effects of cocaine on dopamine neuron firing in awake and anesthetized rats. Neuropsychopharmacology, 37(7), 1559–1571. 10.1038/npp.2011.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufahl PR, & Olive MF (2011). Investigating Methamphetamine Craving Using the Extinction-Reinstatement Model in the Rat. J Addict Res Ther, S1(3). 10.4172/2155-6105.s1-003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn BN, Kalivas PW, & Bobadilla AC (2019). Understanding Addiction Using Animal Models. Front Behav Neurosci, 13, 262. 10.3389/fnbeh.2019.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Moussawi K, Tang XC, Wang X, Kalivas BC, Kolokithas R, … Kalivas PW (2012). The effect of N-acetylcysteine in the nucleus accumbens on neurotransmission and relapse to cocaine. Biol Psychiatry, 71(11), 978–986. 10.1016/j.biopsych.2011.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCrosse AL, O’Donovan SM, Sepulveda-Orengo MT, McCullumsmith RE, Reissner KJ, Schwendt M, & Knackstedt LA (2017). Contrasting the Role of xCT and GLT-1 Upregulation in the Ability of Ceftriaxone to Attenuate the Cue-Induced Reinstatement of Cocaine Seeking and Normalize AMPA Receptor Subunit Expression. J Neurosci, 37(24), 5809–5821. 10.1523/JNEUROSCI.3717-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, & Verkhratsky A (2006). NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci, 26(10), 2673–2683. 10.1523/JNEUROSCI.4689-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Steinberg EE, Foldy C, Wall NR, Beier K, Luo L, & Malenka RC (2015). Diversity of transgenic mouse models for selective targeting of midbrain dopamine neurons. Neuron, 85(2), 429–438. 10.1016/j.neuron.2014.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Petr GT, Sun Y, Fischer KD, Denovan-Wright EM, & Rosenberg PA (2019). Huntington’s disease pattern of transcriptional dysregulation in the absence of mutant huntingtin is produced by knockout of neuronal GLT-1. Neurochem Int, 123, 85–94. 10.1016/j.neuint.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRowe SD, Kalivas PW, Nicholas JS, Randall PK, Mardikian PN, & Malcolm RJ (2013). A double-blind placebo-controlled trial of N-acetylcysteine in the treatment of cocaine dependence. Am J Addict, 22(5), 443–452. 10.1111/j.1521-0391.2013.12034.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BR, Stoica A, & MacAulay N (2016). Managing Brain Extracellular K(+) during Neuronal Activity: The Physiological Role of the Na(+)/K(+)-ATPase Subunit Isoforms. Front Physiol, 7, 141. 10.3389/fphys.2016.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Ting KK, Adams S, Brew BJ, Chung R, & Guillemin GJ (2010). Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One, 5(11), e14123. 10.1371/journal.pone.0014123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, … George SR (2004). Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem, 279(34), 35671–35678. 10.1074/jbc.M401923200 [DOI] [PubMed] [Google Scholar]
- Lehre KP, & Danbolt NC (1998). The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci, 18(21), 8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Weeber EJ, Sweatt JD, & Eskin A (2002). Glutamate uptake in synaptic plasticity: from mollusc to mammal. Curr Mol Med, 2(7), 593–603. [DOI] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Rolstad B, & Danbolt NC (1993). A monoclonal antibody raised against an [Na(+)+K+]coupled L-glutamate transporter purified from rat brain confirms glial cell localization. FEBS Lett, 317(1–2), 79–84. 10.1016/0014-5793(93)81495-l [DOI] [PubMed] [Google Scholar]
- Li MH, Underhill SM, Reed C, Phillips TJ, Amara SG, & Ingram SL (2017). Amphetamine and Methamphetamine Increase NMDAR-GluN2B Synaptic Currents in Midbrain Dopamine Neurons. Neuropsychopharmacology, 42(7), 1539–1547. 10.1038/npp.2016.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Qi J, Yamaguchi T, Wang HL, & Morales M (2013). Heterogeneous composition of dopamine neurons of the rat A10 region: molecular evidence for diverse signaling properties. Brain Struct Funct, 218(5), 1159–1176. 10.1007/s00429-012-0452-z [DOI] [PubMed] [Google Scholar]
- Lin Z, Zhang PW, Zhu X, Melgari JM, Huff R, Spieldoch RL, & Uhl GR (2003). Phosphatidylinositol 3-kinase, protein kinase C, and MEK1/2 kinase regulation of dopamine transporters (DAT) require N-terminal DAT phosphoacceptor sites. J Biol Chem, 278(22), 20162–20170. 10.1074/jbc.M209584200 [DOI] [PubMed] [Google Scholar]
- Lindeberg J, Usoskin D, Bengtsson H, Gustafsson A, Kylberg A, Soderstrom S, & Ebendal T (2004). Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis, 40(2), 67–73. 10.1002/gene.20065 [DOI] [PubMed] [Google Scholar]
- Lineberry TW, & Bostwick JM (2006). Methamphetamine abuse: a perfect storm of complications. Mayo Clin Proc, 81(1), 77–84. 10.4065/81.1.77 [DOI] [PubMed] [Google Scholar]
- Lipski J, Wan CK, Bai JZ, Pi R, Li D, & Donnelly D (2007). Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience, 146(2), 617–629. 10.1016/j.neuroscience.2007.02.003 [DOI] [PubMed] [Google Scholar]
- Lipton SA, & Rosenberg PA (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med, 330(9), 613–622. 10.1056/NEJM199403033300907 [DOI] [PubMed] [Google Scholar]
- Liu J, Wang F, Huang C, Long LH, Wu WN, Cai F, … Chen JG (2009). Activation of phosphatidylinositol-linked novel D1 dopamine receptor contributes to the calcium mobilization in cultured rat prefrontal cortical astrocytes. Cell Mol Neurobiol, 29(3), 317–328. 10.1007/s10571-008-9323-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CN, Bechard AR, Hamor PU, Wu L, Schwendt M, & Knackstedt LA (2020). Ceftriaxone and mGlu2/3 interactions in the nucleus accumbens core affect the reinstatement of cocaine-seeking in male and female rats. Psychopharmacology (Berl), 237(7), 2007–2018. 10.1007/s00213-020-05514-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominac KD, Sacramento AD, Szumlinski KK, & Kippin TE (2012). Distinct neurochemical adaptations within the nucleus accumbens produced by a history of self-administered vs non-contingently administered intravenous methamphetamine. Neuropsychopharmacology, 37(3), 707–722. 10.1038/npp.2011.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, … Jonkers J (2001). Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci U S A, 98(16), 9209–9214. 10.1073/pnas.161269798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorrain DS, Arnold GM, & Vezina P (2000). Previous exposure to amphetamine increases incentive to obtain the drug: long-lasting effects revealed by the progressive ratio schedule. Behav Brain Res, 107(1–2), 9–19. 10.1016/s0166-4328(99)00109-6 [DOI] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Hope BT, & Shaham Y (2004). Incubation of cocaine craving after withdrawal: a review of preclinical data. Neuropharmacology, 47 Suppl 1, 214–226. 10.1016/j.neuropharm.2004.06.027 [DOI] [PubMed] [Google Scholar]
- Luo L, Ambrozkiewicz MC, Benseler F, Chen C, Dumontier E, Falkner S, … Craig AM (2020). Optimizing Nervous System-Specific Gene Targeting with Cre Driver Lines: Prevalence of Germline Recombination and Influencing Factors. Neuron, 106(1), 37–65 10.1016/j.neuron.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutgen V, Kong L, Kau KS, Madayag A, Mantsch JR, & Baker DA (2014). Time course of cocaine-induced behavioral and neurochemical plasticity. Addict Biol, 19(4), 529–538. 10.1111/j.1369-1600.2012.00493.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack F, & Bonisch H (1979). Dissociation constants and lipophilicity of catecholamines and related compounds. Naunyn Schmiedebergs Arch Pharmacol, 310(1), 1–9. 10.1007/BF00499868 [DOI] [PubMed] [Google Scholar]
- Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, … Baker DA (2007). Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci, 27(51), 13968–13976. 10.1523/JNEUROSCI.2808-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, & Luscher C (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci, 12(8), 1036–1041. 10.1038/nn.2367 [DOI] [PubMed] [Google Scholar]
- Marini H, Altavilla D, Bellomo M, Adamo EB, Marini R, Laureanti F, … Squadrito F (2004). Modulation of IL-1 beta gene expression by lipid peroxidation inhibition after kainic acid-induced rat brain injury. Exp Neurol, 188(1), 178–186. 10.1016/j.expneurol.2004.03.023 [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, & Yamamoto BK (2004). High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci, 24(50), 11449–11456. 10.1523/JNEUROSCI.3597-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx MC, Billups D, & Billups B (2015). Maintaining the presynaptic glutamate supply for excitatory neurotransmission. J Neurosci Res, 93(7), 1031–1044. 10.1002/jnr.23561 [DOI] [PubMed] [Google Scholar]
- Massie A, Boillee S, Hewett S, Knackstedt L, & Lewerenz J (2015). Main path and byways: non-vesicular glutamate release by system xc(−) as an important modifier of glutamatergic neurotransmission. J Neurochem, 135(6), 1062–1079. 10.1111/jnc.13348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, & Kalivas PW (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci, 21(21), 8655–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, & Kalivas PW (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci, 23(8), 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair LF, Andersen JV, Aldana BI, Hohnholt MC, Nissen JD, Sun Y, … Waagepetersen HS (2019). Deletion of Neuronal GLT-1 in Mice Reveals Its Role in Synaptic Glutamate Homeostasis and Mitochondrial Function. J Neurosci, 39(25), 4847–4863. 10.1523/JNEUROSCI.0894-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair LF, Andersen JV, Nissen JD, Sun Y, Fischer KD, Hodgson NW, … Aldana BI (2020a). Conditional Knockout of GLT-1 in Neurons Leads to Alterations in Aspartate Homeostasis and Synaptic Mitochondrial Metabolism in Striatum and Hippocampus. Neurochem Res, 45(6), 1420–1437. 10.1007/s11064-020-03000-7 [DOI] [PubMed] [Google Scholar]
- McNair LF, Andersen JV, Nissen JD, Sun Y, Fischer KD, Hodgson NW, … Aldana BI (2020b). Conditional Knockout of GLT-1 in Neurons Leads to Alterations in Aspartate Homeostasis and Synaptic Mitochondrial Metabolism in Striatum and Hippocampus. Neurochem Res. 10.1007/s11064-020-03000-7 [DOI] [PubMed]
- Meldrum B, & Garthwaite J (1990). Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci, 11(9), 379–387. 10.1016/0165-6147(90)90184-a [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, & Conti F (2009). Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia, 57(1), 108–117. 10.1002/glia.20744 [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Ducati A, Iacoangeli M, & Conti F (2011). Cellular and Synaptic Localization of EAAT2a in Human Cerebral Cortex. Front Neuroanat, 4, 151. 10.3389/fnana.2010.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez JA, Bourque MJ, Dal Bo G, Bourdeau ML, Danik M, Williams S, … Trudeau LE (2008). Developmental and target-dependent regulation of vesicular glutamate transporter expression by dopamine neurons. J Neurosci, 28(25), 6309–6318. 10.1523/JNEUROSCI.1331-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, & Bernardi G (1989). The mechanism of amphetamine-induced inhibition of rat substantia nigra compacta neurones investigated with intracellular recording in vitro. Br J Pharmacol, 98(1), 127–134. 10.1111/j.1476-5381.1989.tb16872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele M, Boutelle MG, & Fillenz M (1996). The source of physiologically stimulated glutamate efflux from the striatum of conscious rats. J Physiol, 497 (Pt 3), 745–751. 10.1113/jphysiol.1996.sp021805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, … Rebec GV (2008). Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience, 153(1), 329–337. 10.1016/j.neuroscience.2008.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingote S, Chuhma N, Kalmbach A, Thomsen GM, Wang Y, Mihali A, … Rayport S (2017). Dopamine neuron dependent behaviors mediated by glutamate cotransmission. Elife, 6. 10.7554/eLife.27566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrano DA, & Smith Y (2007). Comparative analysis of the subcellular and subsynaptic localization of mGluR1a and mGluR5 metabotropic glutamate receptors in the shell and core of the nucleus accumbens in rat and monkey. J Comp Neurol, 500(4), 788–806. 10.1002/cne.21214 [DOI] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, & Seamans JK (2005). Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci, 25(27), 6389–6393. 10.1523/JNEUROSCI.1007-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan ME, & Gibb JW (1980). Short-term and long-term effects of methamphetamine on biogenic amine metabolism in extra-striatal dopaminergic nuclei. Neuropharmacology, 19(10), 989–995. 10.1016/0028-3908(80)90010-6 [DOI] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, & Williams JT (2000). Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci, 20(20), RC103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, & Kalivas PW (2009). N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci, 12(2), 182–189. 10.1038/nn.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, & Coyle JT (1990). Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J, 4(6), 1624–1633. [PubMed] [Google Scholar]
- Negus SS (2003). Rapid assessment of choice between cocaine and food in rhesus monkeys: effects of environmental manipulations and treatment with d-amphetamine and flupenthixol. Neuropsychopharmacology, 28(5), 919–931. 10.1038/sj.npp.1300096 [DOI] [PubMed] [Google Scholar]
- Neiman J, Haapaniemi HM, & Hillbom M (2000). Neurological complications of drug abuse: pathophysiological mechanisms. Eur J Neurol, 7(6), 595–606. 10.1046/j.1468-1331.2000.00045.x [DOI] [PubMed] [Google Scholar]
- Nicaise C, Prozzi D, Viaene E, Moreno C, Gustot T, Quertinmont E, … Hols P (2008). Control of acute, chronic, and constitutive hyperammonemia by wild-type and genetically engineered Lactobacillus plantarum in rodents. Hepatology, 48(4), 1184–1192. 10.1002/hep.22445 [DOI] [PubMed] [Google Scholar]
- Nicholls D, & Attwell D (1990). The release and uptake of excitatory amino acids. Trends Pharmacol Sci, 11(11), 462–468. 10.1016/0165-6147(90)90129-v [DOI] [PubMed] [Google Scholar]
- Ntamati NR, & Luscher C (2016). VTA Projection Neurons Releasing GABA and Glutamate in the Dentate Gyrus. eNeuro, 3(4). 10.1523/ENEURO.0137-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science, 164, 719–721. [DOI] [PubMed] [Google Scholar]
- Olney JW, & Ho O-L (1970). Brain Damage in Infant Mice following Oral Intake of Glutamate, Aspartate or Cystine. Nature, 227, 609–610. [DOI] [PubMed] [Google Scholar]
- Olney JW, & Sharpe LG (1969). Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science, 166(903), 386–388. [DOI] [PubMed] [Google Scholar]
- Omrani A, Melone M, Bellesi M, Safiulina V, Aida T, Tanaka K, … Conti F (2009). Up-regulation of GLT-1 severely impairs LTD at mossy fibre--CA3 synapses. J Physiol, 587(Pt 19), 4575–4588. 10.1113/jphysiol.2009.177881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MP, & Olive MF (2008). A role for mGluR5 receptors in intravenous methamphetamine self-administration. Ann N Y Acad Sci, 1139, 206–211. 10.1196/annals.1432.034 [DOI] [PubMed] [Google Scholar]
- Ottestad-Hansen S, Hu QX, Follin-Arbelet VV, Bentea E, Sato H, Massie A, … Danbolt NC (2018). The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia, 66(5), 951–970. 10.1002/glia.23294 [DOI] [PubMed] [Google Scholar]
- Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, … Slesinger PA (2012). Methamphetamine-evoked depression of GABA(B) receptor signaling in GABA neurons of the VTA. Neuron, 73(5), 978–989. 10.1016/j.neuron.2011.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, Fiorillo CD, Morikawa H, & Williams JT (2001). Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci, 4(3), 275–281. 10.1038/85124 [DOI] [PubMed] [Google Scholar]
- Paladini CA, & Roeper J (2014). Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience, 282, 109–121. 10.1016/j.neuroscience.2014.07.032 [DOI] [PubMed] [Google Scholar]
- Papathanou M, Creed M, Dorst MC, Bimpisidis Z, Dumas S, Pettersson H, … Wallen-Mackenzie A (2018). Targeting VGLUT2 in Mature Dopamine Neurons Decreases Mesoaccumbal Glutamatergic Transmission and Identifies a Role for Glutamate Co-release in Synaptic Plasticity by Increasing Baseline AMPA/NMDA Ratio. Front Neural Circuits, 12, 64. 10.3389/fncir.2018.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato R, Rieker C, Turiault M, Tronche F, & Schutz G (2006). Survival of DA neurons is independent of CREM upregulation in absence of CREB. Genesis, 44(10), 454–464. 10.1002/dvg.20236 [DOI] [PubMed] [Google Scholar]
- Parsegian A, & See RE (2014). Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology, 39(4), 811–822. 10.1038/npp.2013.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, & Luscher C (2011). Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature, 481(7379), 71–75. 10.1038/nature10709 [DOI] [PubMed] [Google Scholar]
- Paulson PE, & Robinson TE (1991). Sensitization to systemic amphetamine produces an enhanced locomotor response to a subsequent intra-accumbens amphetamine challenge in rats. Psychopharmacology (Berl), 104(1), 140–141. 10.1007/bf02244569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FC, Lourenco ES, Borges F, Morgadinho T, Ribeiro CF, Macedo TR, & Ali SF (2006). Single or multiple injections of methamphetamine increased dopamine turnover but did not decrease tyrosine hydroxylase levels or cleave caspase-3 in caudate-putamen. Synapse, 60(3), 185–193. 10.1002/syn.20285 [DOI] [PubMed] [Google Scholar]
- Perino LE, Warren GH, & Levine JS (1987). Cocaine-induced hepatotoxicity in humans. Gastroenterology, 93(1), 176–180. 10.1016/0016-5085(87)90331-3 [DOI] [PubMed] [Google Scholar]
- Perugini M, & Vezina P (1994). Amphetamine administered to the ventral tegmental area sensitizes rats to the locomotor effects of nucleus accumbens amphetamine. J Pharmacol Exp Ther, 270(2), 690–696. [PubMed] [Google Scholar]
- Petr GT, Schultheis LA, Hussey KC, Sun Y, Dubinsky JM, Aoki C, & Rosenberg PA (2013). Decreased expression of GLT-1 in the R6/2 model of Huntington’s disease does not worsen disease progression. Eur J Neurosci, 38(3), 2477–2490. 10.1111/ejn.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, … Rosenberg PA (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci, 35(13), 5187–5201. 10.1523/JNEUROSCI.4255-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, & Kalivas PW (1996). Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci, 16(4), 1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, & Kumaresan V (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev, 30(2), 215–238. 10.1016/j.neubiorev.2005.04.016 [DOI] [PubMed] [Google Scholar]
- Pita-Almenar JD, Sol Collado M, Colbert CM, & Eskin A (2006). Different mechanisms exist for the plasticity of glutamate reuptake during early long-term potentiation (LTP) and late LTP. J Neurosci, 26(41), 10461–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, … Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci, 21(9), 1260–1271. 10.1038/s41593-018-0203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV (2001). Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia, 34(1), 27–38. 10.1002/glia.1037 [DOI] [PubMed] [Google Scholar]
- Prescott LF, Park J, Ballantyne A, Adriaenssens P, & Proudfoot AT (1977). Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine. Lancet, 2(8035), 432–434. 10.1016/s0140-6736(77)90612-2 [DOI] [PubMed] [Google Scholar]
- Pu C, Broening HW, & Vorhees CV (1996). Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse, 23(4), 328–334. [DOI] [PubMed] [Google Scholar]
- Rasmussen B, Unterwald EM, & Rawls SM (2011). Glutamate transporter subtype 1 (GLT-1) activator ceftriaxone attenuates amphetamine-induced hyperactivity and behavioral sensitization in rats. Drug Alcohol Depend, 118(2–3), 484–488. 10.1016/j.drugalcdep.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Brown RM, Spencer S, Tran PK, Thomas CA, & Kalivas PW (2014). Chronic administration of the methylxanthine propentofylline impairs reinstatement to cocaine by a GLT-1-dependent mechanism. Neuropsychopharmacology, 39(2), 499–506. 10.1038/npp.2013.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Gipson CD, Tran PK, Knackstedt LA, Scofield MD, & Kalivas PW (2015). Glutamate transporter GLT-1 mediates N-acetylcysteine inhibition of cocaine reinstatement. Addict Biol, 20(2), 316–323. 10.1111/adb.12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, & Kalivas PW (2010). Using glutamate homeostasis as a target for treating addictive disorders. Behav Pharmacol, 21(5–6), 514–522. 10.1097/FBP.0b013e32833d41b2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, & Federoff HJ (2006). Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis, 44(1), 44–49. 10.1002/gene.20183 [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, & Moore RY (1982). Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res, 235(1), 93–103. 10.1016/0006-8993(82)90198-6 [DOI] [PubMed] [Google Scholar]
- Rice ME, Patel JC, & Cragg SJ (2011). Dopamine release in the basal ganglia. Neuroscience, 198, 112–137. 10.1016/j.neuroscience.2011.08.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmele TS, & Rosenberg PA (2016). GLT-1: The elusive presynaptic glutamate transporter. Neurochem Int, 98, 19–28. 10.1016/j.neuint.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, & Kuhar MJ (1987). Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science, 237(4819), 1219–1223. 10.1126/science.2820058 [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J, & Manzoni OJ (2002). Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci, 22(11), 4346–4356. doi:20026420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DC, Bennett SA, & Vickers GJ (1989). The estrous cycle affects cocaine self-administration on a progressive ratio schedule in rats. Psychopharmacology (Berl), 98(3), 408–411. 10.1007/BF00451696 [DOI] [PubMed] [Google Scholar]
- Roberts DC, Loh EA, & Vickers G (1989). Self-administration of cocaine on a progressive ratio schedule in rats: dose-response relationship and effect of haloperidol pretreatment. Psychopharmacology (Berl), 97(4), 535–538. 10.1007/BF00439560 [DOI] [PubMed] [Google Scholar]
- Roberts-Wolfe DJ, & Kalivas PW (2015). Glutamate Transporter GLT-1 as a Therapeutic Target for Substance Use Disorders. CNS Neurol Disord Drug Targets, 14(6), 745–756. 10.2174/1871527314666150529144655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB, Djali S, & Buchhalter JR (1993). Inhibition of glutamate uptake with L-trans-pyrrolidine-2,4-dicarboxylate potentiates glutamate toxicity in primary hippocampal cultures. J.Neurochem., 61, 2099–2103. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Sinor JD, Dowd LA, & Kerwin JF Jr. (1993). Subtypes of sodium-dependent high-affinity L-[3H]glutamate transport activity: pharmacologic specificity and regulation by sodium and potassium. J Neurochem, 60(1), 167–179. [DOI] [PubMed] [Google Scholar]
- Robinson TE (1984). Behavioral sensitization: characterization of enduring changes in rotational behavior produced by intermittent injections of amphetamine in male and female rats. Psychopharmacology (Berl), 84(4), 466–475. 10.1007/BF00431451 [DOI] [PubMed] [Google Scholar]
- Robinson TE (2010). Sensitization to Drugs. Encyclopedia of Psychopharmacology (Stolerman IP ed., pp. 1203–1208): Springer, Berlin, Heidelberg. [Google Scholar]
- Robinson TE, & Becker JB (1986). Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res, 396(2), 157–198. doi:S0006–8993(86)80193–7 [pii] [DOI] [PubMed] [Google Scholar]
- Robinson TE, & Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev, 18(3), 247–291. 10.1016/0165-0173(93)90013-p [DOI] [PubMed] [Google Scholar]
- Romano C, Sesma MA, McDonald CT, O’Malley K, Van den Pol AN, & Olney JW (1995). Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol, 355(3), 455–469. 10.1002/cne.903550310 [DOI] [PubMed] [Google Scholar]
- Root DH, Mejias-Aponte CA, Zhang S, Wang HL, Hoffman AF, Lupica CR, & Morales M (2014). Single rodent mesohabenular axons release glutamate and GABA. Nat Neurosci, 17(11), 1543–1551. 10.1038/nn.3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Wang HL, Liu B, Barker DJ, Mod L, Szocsics P, … Morales M (2016). Glutamate neurons are intermixed with midbrain dopamine neurons in nonhuman primates and humans. Sci Rep, 6, 30615. 10.1038/srep30615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, & Aizenman E (1989). Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett, 103(2), 162–168. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, & Leitner M (1992). Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci, 12(1), 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, & Baumann MH (2003). Monoamine transporters and psychostimulant drugs. Eur J Pharmacol, 479(1–3), 23–40. 10.1016/j.ejphar.2003.08.054 [DOI] [PubMed] [Google Scholar]
- Rothman SM (1983). Synaptic activity mediated death of hypoxic neurons. Science, 220, 526–527. [DOI] [PubMed] [Google Scholar]
- Rothman SM, & Olney JW (1986). Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann. Neurol., 19, 105–111. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, … Kuncl RW (1994). Localization of neuronal and glial glutamate transporters. Neuron, 13(3), 713–725. 10.1016/0896-6273(94)90038-8 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, … Fisher PB (2005). Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature, 433(7021), 73–77. [DOI] [PubMed] [Google Scholar]
- Saez R, Llansola M, & Felipo V (1999). Chronic exposure to ammonia alters pathways modulating phosphorylation of microtubule-associated protein 2 in cerebellar neurons in culture. J Neurochem, 73(6), 2555–2562. 10.1046/j.1471-4159.1999.0732555.x [DOI] [PubMed] [Google Scholar]
- Sakuma S, Kitamura T, Kuroda C, Takeda K, Nakano S, Hamashima T, … Fujimoto Y (2012). All-trans Arachidonic acid generates reactive oxygen species via xanthine dehydrogenase/xanthine oxidase interconversion in the rat liver cytosol in vitro. J Clin Biochem Nutr, 51(1), 55–60. 10.3164/jcbn.11-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Smith KD, Ali PK, & Rebec GV (2009). Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci, 29(29), 9239–9243. 10.1523/JNEUROSCI.1746-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter B (2002). Animal models in biological psychiatry. Biological Psychiatry.
- Savitt JM, Jang SS, Mu W, Dawson VL, & Dawson TM (2005). Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci, 25(29), 6721–6728. 10.1523/JNEUROSCI.0760-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalley RD, & Conner CS (1978). Acetaminophen poisoning: a case report of the use of acetylcysteine. Am J Hosp Pharm, 35(8), 964–967. [PubMed] [Google Scholar]
- Schilstrom B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, … Bonci A (2006). Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci, 26(33), 8549–8558. 10.1523/JNEUROSCI.5179-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Anderson SM, Famous KR, Kumaresan V, & Pierce RC (2005). Anatomy and pharmacology of cocaine priming-induced reinstatement of drug seeking. Eur J Pharmacol, 526(1–3), 65–76. 10.1016/j.ejphar.2005.09.068 [DOI] [PubMed] [Google Scholar]
- Schmitt KC, Rothman RB, & Reith ME (2013). Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther, 346(1), 2–10. 10.1124/jpet.111.191056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JS, Yuwiler A, & Markham CH (1987). Selective loss of subpopulations of ventral mesencephalic dopaminergic neurons in the monkey following exposure to MPTP. Brain Res, 411(1), 144–150. 10.1016/0006-8993(87)90691-3 [DOI] [PubMed] [Google Scholar]
- Schousboe A (1981). Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol, 22, 1–45. 10.1016/s0074-7742(08)60289-5 [DOI] [PubMed] [Google Scholar]
- Schousboe A, & Hertz L (1981). Role of astroglial cells in glutamate homeostasis. Adv Biochem Psychopharmacol, 27, 103–113. [PubMed] [Google Scholar]
- Schousboe A, Sonnewald U, Civenni G, & Gegelashvili G (1997). Role of astrocytes in glutamate homeostasis - Implications for excitotoxicity. Advances in Experimental Medicine and Biology, 429, 195–206. [DOI] [PubMed] [Google Scholar]
- Schwendt M, Reichel CM, & See RE (2012). Extinction-dependent alterations in corticostriatal mGluR2/3 and mGluR7 receptors following chronic methamphetamine self-administration in rats. PLoS One, 7(3), e34299. 10.1371/journal.pone.0034299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, & Cook DG (2013). Amyloid-beta1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci, 33(12), 5312–5318. 10.1523/JNEUROSCI.5274-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RE, Elliott JC, & Feltenstein MW (2007). The role of dorsal vs ventral striatal pathways in cocaine-seeking behavior after prolonged abstinence in rats. Psychopharmacology (Berl), 194(3), 321–331. 10.1007/s00213-007-0850-8 [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, & Ricaurte GA (1993). Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol, 33, 639–677. 10.1146/annurev.pa.33.040193.003231 [DOI] [PubMed] [Google Scholar]
- Sen CK (1997). Nutritional biochemistry of cellular glutathione. J. Nutr. Biochem, 8(12). [Google Scholar]
- Sesack SR, & Grace AA (2010). Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology, 35(1), 27–47. 10.1038/npp.2009.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, & Pickel VM (1992). Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol, 320(2), 145–160. 10.1002/cne.903200202 [DOI] [PubMed] [Google Scholar]
- Shen HW, Scofield MD, Boger H, Hensley M, & Kalivas PW (2014). Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci, 34(16), 5649–5657. 10.1523/JNEUROSCI.4564-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, & Mizuno N (1993). Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett, 163(1), 53–57. 10.1016/0304-3940(93)90227-c [DOI] [PubMed] [Google Scholar]
- Silm K, Yang J, Marcott PF, Asensio CS, Eriksen J, Guthrie DA, … Edwards RH (2019). Synaptic Vesicle Recycling Pathway Determines Neurotransmitter Content and Release Properties. Neuron, 102(4), 786–800 10.1016/j.neuron.2019.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes PF, Silva AP, Pereira FC, Marques E, Milhazes N, Borges F, … Macedo TR (2008). Methamphetamine changes NMDA and AMPA glutamate receptor subunit levels in the rat striatum and frontal cortex. Ann N Y Acad Sci, 1139, 232–241. 10.1196/annals.1432.028 [DOI] [PubMed] [Google Scholar]
- Sitte HH, Huck S, Reither H, Boehm S, Singer EA, & Pifl C (1998). Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem, 71(3), 1289–1297. 10.1046/j.1471-4159.1998.71031289.x [DOI] [PubMed] [Google Scholar]
- Sjodin K, Nilsson E, Hallberg A, & Tunek A (1989). Metabolism of N-acetyl-L-cysteine. Some structural requirements for the deacetylation and consequences for the oral bioavailability. Biochem Pharmacol, 38(22), 3981–3985. 10.1016/0006-2952(89)90677-1 [DOI] [PubMed] [Google Scholar]
- Skou JC (1965). Enzymatic Basis for Active Transport of Na+ and K+ across Cell Membrane. Physiol Rev, 45, 596–617. 10.1152/physrev.1965.45.3.596 [DOI] [PubMed] [Google Scholar]
- Smaga I, Fierro D, Mesa J, Filip M, & Knackstedt LA (2020). Molecular changes evoked by the beta-lactam antibiotic ceftriaxone across rodent models of substance use disorder and neurological disease. Neurosci Biobehav Rev. 10.1016/j.neubiorev.2020.05.016 [DOI] [PMC free article] [PubMed]
- Smith JA, Mo Q, Guo H, Kunko PM, & Robinson SE (1995). Cocaine increases extraneuronal levels of aspartate and glutamate in the nucleus accumbens. Brain Res, 683(2), 264–269. 10.1016/0006-8993(95)00383-2 [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Waters AJ, Mooney M, & Kosten T (2008). Riluzole and D-amphetamine interactions in humans. Prog Neuropsychopharmacol Biol Psychiatry, 32(1), 16–22. 10.1016/j.pnpbp.2007.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer I, & Knackstedt LA (2011). Ceftriaxone prevents the induction of cocaine sensitization and produces enduring attenuation of cue- and cocaine-primed reinstatement of cocaine-seeking. Behav Brain Res, 225(1), 252–258. 10.1016/j.bbr.2011.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AJ, & Palmiter RD (2018). Detecting and Avoiding Problems When Using the Cre-lox System. Trends Genet, 34(5), 333–340. 10.1016/j.tig.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R (2017). Animal models of addiction. Dialogues Clin Neurosci, 19(3), 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S, & Kalivas PW (2017). Glutamate Transport: A New Bench to Bedside Mechanism for Treating Drug Abuse. Int J Neuropsychopharmacol, 20(10), 797–812. 10.1093/ijnp/pyx050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkellner T, Zell V, Farino ZJ, Sonders MS, Villeneuve M, Freyberg RJ, … Hnasko TS (2018). Role for VGLUT2 in selective vulnerability of midbrain dopamine neurons. J Clin Invest, 128(2), 774–788. 10.1172/JCI95795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephans SE, & Yamamoto BK (1994). Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse, 17(3), 203–209. 10.1002/syn.890170310 [DOI] [PubMed] [Google Scholar]
- Stewart J, & Vezina P (1989). Microinjections of Sch-23390 into the ventral tegmental area and substantia nigra pars reticulata attenuate the development of sensitization to the locomotor activating effects of systemic amphetamine. Brain Res, 495(2), 401–406. 10.1016/0006-8993(89)90236-9 [DOI] [PubMed] [Google Scholar]
- Storm-Mathisen J, & Iversen LL (1979). Uptake of [3H]Glutamic acid in excitatory nerve endings: light and electronmicroscopic observations in the hippocampal formation of the rat. Neuroscience, 4(9), 1237–1253. 10.1016/0306-4522(79)90154-4 [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, & Bonci A (2010). Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci, 30(24), 8229–8233. 10.1523/JNEUROSCI.1754-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Stamatakis AM, & Kantak PA (2015). Considerations when using cre-driver rodent lines for studying ventral tegmental area circuitry. Neuron, 85(2), 439–445. 10.1016/j.neuron.2014.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D (2011). How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron, 69(4), 628–649. 10.1016/j.neuron.2011.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Cragg SJ, & Rice ME (2016). Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia, 6(3), 123–148. 10.1016/j.baga.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, & Rayport S (1998). Dopamine neurons make glutamatergic synapses in vitro. J Neurosci, 18(12), 4588–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, & Rayport S (1990). Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron, 5(6), 797–808. 10.1016/0896-6273(90)90339-h [DOI] [PubMed] [Google Scholar]
- Suzuki R, Ferris HA, Chee MJ, Maratos-Flier E, & Kahn CR (2013). Reduction of the cholesterol sensor SCAP in the brains of mice causes impaired synaptic transmission and altered cognitive function. PLoS Biol, 11(4), e1001532. 10.1371/journal.pbio.1001532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumlinski KK, Lominac KD, Campbell RR, Cohen M, Fultz EK, Brown CN, … Kippin TE (2017). Methamphetamine Addiction Vulnerability: The Glutamate, the Bad, and the Ugly. Biol Psychiatry, 81(11), 959–970. 10.1016/j.biopsych.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, & Attwell D (1997). The role of glutamate transporters in glutamate homeostasis in the brain. J Exp Biol, 200(Pt 2), 401–409. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, … Wada K (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science, 276(5319), 1699–1702. 10.1126/science.276.5319.1699 [DOI] [PubMed] [Google Scholar]
- Tardiolo G, Bramanti P, & Mazzon E (2018). Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules, 23(12). 10.3390/molecules23123305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taslimi Z, Komaki A, Haghparast A, & Sarihi A (2018). Effects of Acute and Chronic Restraint Stress on Reinstatement of Extinguished Methamphetamine-induced Conditioned Place Preference in Rats. Basic Clin Neurosci, 9(3), 157–166. 10.29252/nirp.bcn.9.3.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Jones F, Kubota ES, & Pocock JM (2005). Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci, 25(11), 2952–2964. 10.1523/JNEUROSCI.4456-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, … Koos T (2010). Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci, 30(20), 7105–7110. 10.1523/JNEUROSCI.0265-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos A, Morizane Y, Murakami Y, Giani A, Mantopoulos D, Kayama M, … Vavvas DG (2012). Evidence for baseline retinal pigment epithelium pathology in the Trp1-Cre mouse. Am J Pathol, 180(5), 1917–1927. 10.1016/j.ajpath.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, & Kuhn DM (2004). Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett, 367(3), 349–354. 10.1016/j.neulet.2004.06.065 [DOI] [PubMed] [Google Scholar]
- Thomas DM, & Kuhn DM (2005). MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res, 1050(1–2), 190–198. 10.1016/j.brainres.2005.05.049 [DOI] [PubMed] [Google Scholar]
- Timmerman W, & Westerink BH (1997). Brain microdialysis of GABA and glutamate: what does it signify? Synapse, 27(3), 242–261. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, & Knackstedt LA (2012). Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci, 32(36), 12406–12410. 10.1523/JNEUROSCI.1976-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, & Sabatini BL (2012). Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature, 490(7419), 262–266. 10.1038/nature11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Oh WJ, Gu C, & Sabatini BL (2014). Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. Elife, 3, e01936. 10.7554/eLife.01936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, … Schutz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet, 23(1), 99–103. 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Hnasko TS, Wallen-Mackenzie A, Morales M, Rayport S, & Sulzer D (2014). The multilingual nature of dopamine neurons. Prog Brain Res, 211, 141–164. 10.1016/B978-0-444-63425-2.00006-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke TM, & Schmidt WJ (1998). Blockade of morphine- and amphetamine-induced conditioned place preference in the rat by riluzole. Neurosci Lett, 242(2), 114–116. 10.1016/s0304-3940(98)00023-8 [DOI] [PubMed] [Google Scholar]
- Uchimura N, & North RA (1991). Baclofen and adenosine inhibit synaptic potentials mediated by gamma-aminobutyric acid and glutamate release in rat nucleus accumbens. J Pharmacol Exp Ther, 258(2), 663–668. [PubMed] [Google Scholar]
- Underhill SM, Colt MS, & Amara SG (2020). Amphetamine Stimulates Endocytosis of the Norepinephrine and Neuronal Glutamate Transporters in Cultured Locus Coeruleus Neurons. Neurochem Res, 45(6), 1410–1419. 10.1007/s11064-019-02939-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill SM, Wheeler DS, Li M, Watts SD, Ingram SL, & Amara SG (2014). Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron, 83(2), 404–416. 10.1016/j.neuron.2014.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, & Bonci A (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature, 411(6837), 583–587. 10.1038/35079077 [DOI] [PubMed] [Google Scholar]
- Vertes RP (2004). Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse, 51(1), 32–58. 10.1002/syn.10279 [DOI] [PubMed] [Google Scholar]
- Vezina P (1996). D1 dopamine receptor activation is necessary for the induction of sensitization by amphetamine in the ventral tegmental area. J Neurosci, 16(7), 2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P, & Stewart J (1990). Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res, 516(1), 99–106. 10.1016/0006-8993(90)90902-n [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, & De Simoni MG (1999). Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci, 19(12), 5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial D, & Piomelli D (1995). Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J Neurochem, 64(6), 2765–2772. 10.1046/j.1471-4159.1995.64062765.x [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Ding YS, Sedler M, … Pappas N (2001). Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry, 158(12), 2015–2021. 10.1176/appi.ajp.158.12.2015 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, … Miller EN (2001). Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry, 158(3), 377–382. 10.1176/appi.ajp.158.3.377 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM, & Telang F (2007). Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol, 64(11), 1575–1579. 10.1001/archneur.64.11.1575 [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Tromba C, Floridi S, & Racagni G (1994). Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci, 14(5 Pt 1), 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, & Kavanaugh MP (1995). Ion fluxes associated with excitatory amino acid transport. Neuron, 15(3), 721–728. 10.1016/0896-6273(95)90159-0 [DOI] [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, & Westley J (1980). Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res, 181(1), 151–160. 10.1016/0006-8993(80)91265-2 [DOI] [PubMed] [Google Scholar]
- Wall SC, Gu H, & Rudnick G (1995). Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol Pharmacol, 47(3), 544–550. [PubMed] [Google Scholar]
- Wang DV, Viereckel T, Zell V, Konradsson-Geuken A, Broker CJ, Talishinsky A, … Ikemoto S (2017). Disrupting Glutamate Co-transmission Does Not Affect Acquisition of Conditioned Behavior Reinforced by Dopamine Neuron Activation. Cell Rep, 18(11), 2584–2591. 10.1016/j.celrep.2017.02.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SJ, Morgan D, & Roberts DC (2005). Comparison of the reinforcing effects of cocaine and cocaine/heroin combinations under progressive ratio and choice schedules in rats. Neuropsychopharmacology, 30(2), 286–295. 10.1038/sj.npp.1300560 [DOI] [PubMed] [Google Scholar]
- Ward SJ, Rasmussen BA, Corley G, Henry C, Kim JK, Walker EA, & Rawls SM (2011). Beta-lactam antibiotic decreases acquisition of and motivation to respond for cocaine, but not sweet food, in C57Bl/6 mice. Behav Pharmacol, 22(4), 370–373. 10.1097/FBP.0b013e3283473c10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr O, Takahashi M, & Attwell D (1999). Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol, 514 (Pt 3), 783–793. 10.1111/j.1469-7793.1999.783ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayment HK, Schenk JO, & Sorg BA (2001). Characterization of extracellular dopamine clearance in the medial prefrontal cortex: role of monoamine uptake and monoamine oxidase inhibition. J Neurosci, 21(1), 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Chen XC, Song Y, Pan XD, Dai XM, Zhang J, … Zhu YG (2016). Amyloid beta Protein Aggravates Neuronal Senescence and Cognitive Deficits in 5XFAD Mouse Model of Alzheimer’s Disease. Chin Med J (Engl), 129(15), 1835–1844. 10.4103/0366-6999.186646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Maldonado-Vlaar CS, Parsons LH, Kerr TM, Smith DL, & Ben-Shahar O (2000). Control of cocaine-seeking behavior by drug-associated stimuli in rats: effects on recovery of extinguished operant-responding and extracellular dopamine levels in amygdala and nucleus accumbens. Proc Natl Acad Sci U S A, 97(8), 4321–4326. 10.1073/pnas.97.8.4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin GP, Garthwaite J, & Balazs R (1982). Putative acidic amino acid transmitters in the cerebellum. II. Electron microscopic localization of transport sites. Brain Res, 244(1), 69–80. 10.1016/0006-8993(82)90905-2 [DOI] [PubMed] [Google Scholar]
- Williams JE, Wieczorek W, Willner P, & Kruk ZL (1995). Parametric analysis of the effects of cocaine and cocaine pretreatment on dopamine release in the nucleus accumbens measured by fast cyclic voltammetry. Brain Res, 678(1–2), 225–232. 10.1016/0006-8993(95)00188-v [DOI] [PubMed] [Google Scholar]
- Williams LE, & Featherstone DE (2014). Regulation of hippocampal synaptic strength by glial xCT. J Neurosci, 34(48), 16093–16102. 10.1523/JNEUROSCI.1267-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, … Kish SJ (1996). Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med, 2(6), 699–703. 10.1038/nm0696-699 [DOI] [PubMed] [Google Scholar]
- Wise RA (2004). Dopamine, learning and motivation. Nat Rev Neurosci, 5(6), 483–494. 10.1038/nrn1406 [DOI] [PubMed] [Google Scholar]
- Wolf ME (2010). Dysregulation of AMPA receptor transmission in the nucleus accumbens in animal models of cocaine addiction. Neurotox Res., 18(0), 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, White FJ, & Hu XT (1994). MK-801 prevents alterations in the mesoaccumbens dopamine system associated with behavioral sensitization to amphetamine. J Neurosci, 14(3 Pt 2), 1735–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, & Xue CJ (1998). Amphetamine and D1 dopamine receptor agonists produce biphasic effects on glutamate efflux in rat ventral tegmental area: modification by repeated amphetamine administration. J Neurochem, 70(1), 198–209. 10.1046/j.1471-4159.1998.70010198.x [DOI] [PubMed] [Google Scholar]
- Wolf ME, & Xue CJ (1999). Amphetamine-induced glutamate efflux in the rat ventral tegmental area is prevented by MK-801, SCH 23390, and ibotenic acid lesions of the prefrontal cortex. J Neurochem, 73(4), 1529–1538. 10.1046/j.1471-4159.1999.0731529.x [DOI] [PubMed] [Google Scholar]
- Wolf ME, Xue CJ, Li Y, & Wavak D (2000). Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem, 75(4), 1634–1644. 10.1046/j.1471-4159.2000.0751634.x [DOI] [PubMed] [Google Scholar]
- Woodcock EA, Lundahl LH, Khatib D, Stanley JA, & Greenwald MK (2020). N-acetylcysteine reduces cocaine-seeking behavior and anterior cingulate glutamate/glutamine levels among cocaine-dependent individuals. Addict Biol, e12900. 10.1111/adb.12900 [DOI] [PMC free article] [PubMed]
- Wu M, Brudzynski SM, & Mogenson GJ (1993). Functional interaction of dopamine and glutamate in the nucleus accumbens in the regulation of locomotion. Can J Physiol Pharmacol, 71(5–6), 407–413. 10.1139/y93-061 [DOI] [PubMed] [Google Scholar]
- Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, & Kalivas PW (2002). Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther, 303(2), 608–615. 10.1124/jpet.102.039735 [DOI] [PubMed] [Google Scholar]
- Xu NJ, Bao L, Fan HP, Bao GB, Pu L, Lu YJ, … Pei, G. (2003). Morphine withdrawal increases glutamate uptake and surface expression of glutamate transporter GLT1 at hippocampal synapses. J Neurosci, 23(11), 4775–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue CJ, Ng JP, Li Y, & Wolf ME (1996). Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem, 67(1), 352–363. 10.1046/j.1471-4159.1996.67010352.x [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Qi J, Wang HL, Zhang S, & Morales M (2015). Glutamatergic and dopaminergic neurons in the mouse ventral tegmental area. Eur J Neurosci, 41(6), 760–772. 10.1111/ejn.12818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Wang HL, Li X, Ng TH, & Morales M (2011). Mesocorticolimbic glutamatergic pathway. J Neurosci, 31(23), 8476–8490. 10.1523/JNEUROSCI.1598-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, & Deisseroth K (2011). Optogenetics in neural systems. Neuron, 71(1), 9–34. 10.1016/j.neuron.2011.06.004 [DOI] [PubMed] [Google Scholar]
- Youngren KD, Daly DA, & Moghaddam B (1993). Distinct actions of endogenous excitatory amino acids on the outflow of dopamine in the nucleus accumbens. J Pharmacol Exp Ther, 264(1), 289–293. [PubMed] [Google Scholar]
- Zavala AR, Biswas S, Harlan RE, & Neisewander JL (2007). Fos and glutamate AMPA receptor subunit coexpression associated with cue-elicited cocaine-seeking behavior in abstinent rats. Neuroscience, 145(2), 438–452. 10.1016/j.neuroscience.2006.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou Z, Wang D, Li A, Yin Y, Gu X, … Zhou J (2009). Activation of phosphatidylinositol-linked D1-like receptor modulates FGF-2 expression in astrocytes via IP3-dependent Ca2+ signaling. J Neurosci, 29(24), 7766–7775. 10.1523/JNEUROSCI.0389-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Hassel B, Eid T, & Danbolt NC (2019). Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochem Int, 123, 101–113. 10.1016/j.neuint.2018.03.006 [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, & Hen R (2005). Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods, 143(1), 27–32. 10.1016/j.jneumeth.2004.09.020 [DOI] [PubMed] [Google Scholar]
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, … Konnerth A (2019). A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science, 365(6453), 559–565. 10.1126/science.aay0198 [DOI] [PMC free article] [PubMed] [Google Scholar]