

Abstract
As far as comorbidity is concerned, cardiovascular diseases (CVD) appear to be accounted for the highest prevalence, severity, and fatality among COVID 19 patients. A wide array of causal links connecting CVD and COVID-19 baffle the overall prognosis as well as the efficacy of the given therapeutic interventions. At the centre of this puzzle lies ACE2 that works as a receptor for the SARS-CoV-2, and functional expression of which is also needed to minimize vasoconstriction otherwise would lead to high blood pressure. Furthermore, SARS-CoV-2 infection seems to reduce the functional expression of ACE2. Given these circumstances, it might be advisable to consider a treatment plan for COVID-19 patients with CVD in an approach that would neither aggravate the vasodeleterious arm of the renin-angiotensinogen-aldosterone system (RAAS) nor compromise the vasoprotective arm of RAAS but is effective to minimize or if possible, inhibit the viral replication. Given the immune modulatory role of Zn in both CVD and COVID-19 pathogenesis, zinc supplement to the selective treatment plan for CVD and COVID-19 comorbid conditions, to be decided by the clinicians depending on the cardiovascular conditions of the patients, might greatly improve the therapeutic outcome. Notably, ACE2 is a zinc metalloenzyme and zinc is also known to inhibit viral replication.
Keywords: Angiotensinogen converting enzyme, CVD and COVID 19 Comorbidity, High blood pressure, Vasoconstriction, SARS-CoV-2
Introduction
The 2019 Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) responsible for novel coronavirus disease 2019 (COVID -19) has been declared as a pandemic on Mar 11, 2020, by World Health Organization (WHO) and driven the global health care system at bay. While the COVID-19 case fatality rate in children is nil and very low in adults up to 50 years, it is more than 20% for patients aged 80 years or more, and this was well evidenced in Europe [1].
As far as comorbidity is concerned, cardiovascular diseases (CVD) appear to be accounted for the highest prevalence, severity, and fatality among COVID 19 patients. Within the spectrum of CVD, its coexistence has been reported highest for hypertension (35-57%), followed by coronary artery disease (10-17%), and congestive heart failure (6-7%) [2,3]. Based on other reports, 30-35% of COVID-19 related deaths have underlying CVD, while the comorbid patients with CVD have an increased risk of severe manifestations of COVID-19 [[4], [5], [6], [7]]. However, using retrospective analyses of 50 COVID-19 patients (mean age 64.80 ± 14.51 years) who were admitted to ICU, Aladag and Atabey demonstrated that pre-existing CVD is not a significant contributor in mortality with severe COVID-19 [8].
Nevertheless, cardiac injury has been reported in patients with COVID-19 having no history of cardiovascular issues [7]. National Health Commission of China reported that around 11.8% of patients died due to COVID-19-mediated onset of heart dysfunction during the course of illness [9]. Brit Long et al. evaluated 45 recent reports regarding COVID-19 and heart complications and confirmed the fatal outcome of COVID-19 on heart disablement even in patients with no pre-existing CVD record [7]. Notably, a SARS-CoV-2 positive 16-year-old boy developed acute myocarditis without showing any sign of COVID-19, except fever [10].
All these pieces of evidence prompted the establishment of the causal link of fatal co-morbidity of CVD and COVID-19, which might have caused the baffling overall prognosis of COVID-19 patients with or without CVD. Given this perplexing situation, it is important to analyse two basic assumptions whether: (1) the CVD pathologies favour SARS-CoV-2 and vice versa, and (2) the therapies meant for CVD are counterproductive to the treatment for COVID-19 and vice versa. Currently, the molecular evidence might indicate supporting both assumptions – hence clinicians and scientists are facing challenges to offer an effective treatment plan for comorbid patients with CVD and COVID-19.
However, the current knowledge on the CVD pathologies and pathogenesis of SARS-CoV-2 might shed a light on how Zinc (Zn) supplements might offer clinical benefits to this struggle.
Outline of the narrative review
To explain the potential benefits of the Zn supplement, the current review will first demonstrate that angiotensinogen converting enzyme 2 (ACE2) forms the central pathogenic link between CVD and COVID-19. This will then suggest a selective treatment plan to minimize the risk of cardiovascular pathology with COVID-19 comorbidity. The review will finally highlight possible beneficial role of Zn to inhibit SARS-CoV-2 replication as well as its role in the functional expression of ACE2- the receptor of SARS-CoV-2. Scientific rationale of the arguments established in the current review is based on the published papers that have described pathogenesis of CVD and COVID-19 as well as how these two diseases were reported during the ongoing pandemic.
Pathology of CVD and angiotensinogen converting enzyme
Cardiovascular diseases are a group of disorders of the heart and blood vessels resulting in heart attacks or strokes mainly caused by a blockage that prevents blood from flowing to the heart or brain. The cause of heart attacks and strokes is usually the presence of combined risk factors, including hypertension or high (elevated) blood pressure, diabetes, and hyperlipidaemia.
Physiological control of blood pressure is primarily a hormone-mediated system that maintains an equilibrium of fluid and electrolytes. The system starts with renin that carries out the conversion of angiotensinogen (ANGen) to angiotensin I (ANG I) [11]. Angiotensinogen converting enzyme (ACE) then hydrolyzes inactive decapeptide ANG I to the octapeptide ANG II by removing His-Leu residues from the C-terminal end [12]. The binding of ANG II to the type 1 ANG II receptor (ANG 1aR) results in vasoconstriction and therefore increased blood pressure. This cascade of events often is referred to as the vasodeleterious arm of RAAS (renin-angiotensinogen-aldosterone system) (Fig. 1 ). Angiotensin-converting enzyme 2 (ACE2) on the other hand, can lower blood pressure by catalyzing the hydrolysis of ANG II into vasodilator angiotensin (1-7). Thus, the activity of the ACE is balanced by reducing the amount of ANG-II and increasing ANG (1-7) which subsequently binds to Mas receptor (MAS R) causing vasodilation [13]. This cascade of events is referred to as the vasoprotective arm of RAAS (Fig. 1).
Fig. 1.

COVID-19 is tied to create imbalance between vasoprotective and vasodeleterious arms of RAAS towards high blood pressure. ACE hydrolyzes inactive decapeptide ANG I to the octapeptide ANG II. ANG II binds to either ANG II receptor 1a (ANG 1aR) leading to tissue damage and lung edema, or to ANG II receptor 2 (ANG 2R) reducing tissue damage. Binding to ANG 1aR, ANG II causes vasoconstriction that results in hypertension. ACE2 on the other hand, can lower blood pressure by catalysing the hydrolysis of ANG II into a vasodilator ANG (1-7) thus counters the activity of the ACE by reducing the amount of ANG II and increasing ANG (1-7). While SARS-CoV-2 enters pulmonary cells by binding to ACE2, therefore, in case of SARS-CoV-2 infection, the therapeutic advantage to inhibit functional expression of ACE2 would favour vasodeleterious arm of the RAAS. On the other hand, therapeutic interventions to treat hypertensive patients should favour the vasoprotective arm i.e., towards the functional expression of ACE2. Hence, any individual with both SARS-CoV and hypertension would face a challenge to prioritize the treatment options. [→ = stimulation/activation;  = inhibition/reduction].
= inhibition/reduction].
All these molecules are constitutively expressed in various tissues including the heart and lungs. Hence, a balance in the functional expression of those molecules ensures an optimum blood pressure (Fig. 1).
Pathology of SARS-CoV-2 infection starts with ACE-2
Using spike glycoproteins (S glycoproteins), SARS-CoV-2 particularly binds to ACE2 expressed in various organs of the body including lung alveolar and alveolar monocytes and macrophages [[14], [15], [16]]. It triggers imbalanced T cell activation against the virus hence a massive inflammatory cascade termed ‘cytokine storm’ is ensued eventually [17]. This spurring immune response along with the viral multiplication leads to pulmonary cell destruction [18,19]. Consequently, the blood oxygen level drops, making the heart work harder and faster to pump blood throughout the body.
SARS-CoV-2 binding to ACE2 receptors triggers conformational changes in the S-glycoprotein. The virus is then endocytosed into the cytoplasm. Endosomal pH favours the host protease to cleave the S-glycoprotein resulting in the fusion of the viral envelope. Subsequently, the positive-strand viral genomic RNA is released into the cell cytoplasm. SARS-CoV-2 replication starts with RNA-dependent RNA polymerase (RdRp) which is integrated into a membrane-associated viral enzyme complex to allow the synthesis of negative-strand RNA. The negative RNA strand is used as a template for the synthesis of viral mRNA [20,21].
ACE2: the epicentre of CVD and COVID-19 pathogenesis
ACE2 is mostly bound to cell membranes of various organs of the body including the heart, blood vessels, gut, lung, kidney, testis, brain while only scarcely present in the circulation in a soluble form, as well as in alveolar monocytes and macrophages [[14], [15], [16],22]. The ACE2 receptor, a transmembrane type I glycoprotein, was initially discovered by two independent groups in the year 2000 and has a 40% structural identity to ACE [23,24]. This monocarboxypeptidase has 805 amino acids with one extracellular catalytic domain that catalyses the removal of one amino acid from the C-terminal end of ANG II and converts it to ANG (1-7).
While ACE2 serves as the receptor for SARS-CoV-2 binding and subsequently entering host cells [15], in vivo studies revealed that lung ACE2 expression is markedly decreased upon SARS-CoV infection [22]. Only a handful of molecular evidence, as mentioned below, restricts to conclude convincingly - whether SARS-CoV-2, like its ancestor SARS-CoV, will lead to a decreased expression of ACE2 upon infecting the host cells. However, SARS-CoV and SARS-CoV-2 share more than 70% identity in the amino acid sequence [25], hence is not unlikely to see a reduced expression of ACE2 after SARS-CoV-2 infection.
In vivo experiment involving a rat model of acute respiratory distress syndrome has shown increased ACE activity and ANG II expression, while ACE2 activity and ANG (1–7) levels were reduced [26,27]. Furthermore, a negative correlation was shown between ACE2 expression and COVID‐19 fatality at molecular levels [28]. Again, the SARS-CoV-2 genome was found in 7 out of 20 post-mortem autopsy of heart tissues that were characterized by increased myocardial fibrosis, inflammation, and reduced myocardial ACE2 expression [29].
At the same time, patients with CVDs have increased ACE2 as compared to healthy controls [7]. It is possible then; CVD patients might be more susceptible to SARS-CoV-2 attack due to more viral entry through binding to ACE2. The binding of SARS-CoV-2 to cardiac ACE2 can be assumed to influence two events concurrently: (1) hindering the vasoprotective arm of RAAS by retarding the conversion of ANG II to ANG (1-7) occurring in heart tissues [30], and (2) aggravating the vasodeleterious arm of the RAAS by allowing the conversion of ANG II to ANG 1aR culminating to hypertension. Overall, these lines of evidence place SARS-CoV-2 as a double-edged sword involving the ACE2 at the epicentre of the pathogenesis (Fig. 2 ).
Fig. 2.
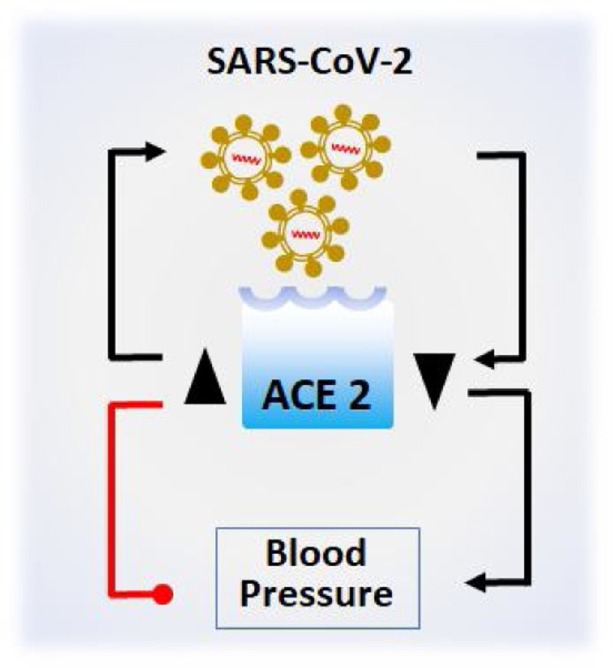
ACE2 is at the centre of COVID-19 and CVD. Increased ACE2 expression (▲) would allow more SARS-CoV-2 to enter host cells by binding to ACE2, while SARS-CoV-2 infection results in the decreased expression of ACE2 (▼). Increased ACE2 expression (▲) on the other hand, reduces blood pressure, while the decreased ACE2 expression (▼) would result in higher blood pressure [→ = stimulation/increase; = inhibition/reduction].
This might be the reason for higher cardiovascular damage during COVID-19 progression [[2], [3], [4], [5], [6], [7],31]. This can be reasonably argued based on the observation that ACE2 becomes depleted due to SARS-CoV-2 infection, which otherwise could convert ANG II into ANG (1-7), but not ANG 1aR, the vasoconstriction consequently results.
Impact of CVD treatment in COVID-19 comorbidity
A range of medications is prescribed for patients with hypertension to prevent heart attacks and strokes. Among them, Angiotensinogen-converting enzyme (ACE) inhibitors, Angiotensin II receptor blockers (ARB), and other renin-angiotensin-aldosterone system (RAAS) inhibitors are the drugs that interrupt different steps in the abnormally activated RAAS system responsible for elevated blood pressure in the human body.
An uncontrolled RAAS activation results in continuous vasoconstriction and hypertension. As a method of restoration, medications like ACE inhibitors are used to relax blood vessels by blocking the formation of ANG II that narrows blood vessels [32] while ARB helps relax blood vessels by blocking the action of ANG II on its receptors [33]. Both ACE inhibitors and ARBs can reduce angiotensin II levels [33]. In general, ACE inhibitors reduce the substrate of ANG II generation, and the ARBs arrest ANG 1aR activity to counter vasoconstriction. RAAS inhibitors on the other hand, slow down the production of renin hormone from the kidney that starts the RAAS [34].
The antihypertensive agent used to treat high blood-pressure patients including ARBs, ACE inhibitors, and RAAS inhibitors can upregulate ACE2 expression in rodent studies, hence is suspected to potentially increase viral entry sites for coronaviruses worsening the outcome in patients with COVID-19 [35].
However, different RAAS inhibitors have different effects on ACE2 levels- the key for SARS-CoV-2 to enter cells for its replication. ACE inhibitor (captopril) caused a significant increase in ACE2 protein expression in rats with acute lung injury [36]. While ACE2 has been reported to have protective effects in acute lung injury [37]. Contrarily, other ACE inhibitors (lisinopril) and ARB (losartan) alone or in combination did not increase cardiac ACE2 activity but caused a significant increase in ACE2 mRNA expression. However, lisinopril caused a 1.8-fold increase in rat plasma ANG-(1–7) and decreased plasma ANG II. Losartan, on the other hand, increased plasma levels of both ANG-(1–7) and ANG II, with increased cardiac ACE2 mRNA and concomitant cardiac ACE2 activity [38]. Therapeutic intervention with the cyclic form of ANG-(1-7) attenuated the inflammatory mediator response, markedly decreased lung injury scores, and increased oxygenation [26].
The above-mentioned evidence demonstrates that therapeutic interventions for CVD involving certain ACE inhibitors would increase the functional expression of ACE2. Continuation of CVD treatment using such inhibitors with COVID-19 comorbidity might have two possible outcomes of opposing spectrum: (1) favouring SARS-CoV-2 to enter host cells resulting in fatal consequences for COVID-19 patients, or (2) improving cardiovascular conditions by prompting functional expression of ACE2 that was reduced as a result of SARS-CoV-2 infection.
The latter outcome could be beneficial based on the observation of a reduced ACE2 expression in the lungs in experimental SARS-CoV infections of wild-type mice. That in turn suggests that a reduced ACE2 expression might have a role in SARS-CoV–mediated severe acute lung pathologies. At the same time, SARS-CoV Spike protein binding to ACE2 in cell lines or SARS-CoV infections in vivo results in reduced ACE2 protein expression [22]. A deficiency of ACE2 in mice results in a dramatic decrease in viral replication and much less severe pathologic alterations in lungs as compared to wild-type mice [22,37].
On the other hand, ACE inhibitors that have no impact on the functional expression of ACE2 which could favour the vasoprotective arm of RAAS to reduce high blood pressure might be futile for the patients with COVID-19, since SARS-CoV-2 infection reduces ACE2 expression.
Notably, ACE2 is greatly expressed in epithelial cells of alveoli, trachea, bronchi, bronchial serous glands [16], and alveolar monocytes and macrophages, as well as in coronary vessels along with cardiac myocytes and fibroblasts [39].
Alternative treatment plans for CVD and COVID-19 comorbidity
Given the above discussion, it might be advisable to consider a treatment plan for COVID-19 patients with CVD in an approach that would neither aggravate the vasodeleterious arm of RAAS nor compromise the vasoprotective arm of RAAS which at the same time would minimize or if possible inhibit viral (SARS-CoV-2) replication.
First, this would require activation of ACE2. Because ACE2 can convert ANG II to ANG (1-7) and eventually can cause vasodilation. However, SARS-CoV-2 infection results in the downregulation of ACE2, hence an additional Zn supplement might aid to activate or upregulate functional ACE2 expression. This can be reasonably argued based on the fact that ACE2 is a zinc-containing metalloenzyme [40] (Fig. 3 , indicated with the letter ‘a’).
Fig. 3.

Potential sites of drug target and Zn action to favour both CVD and COVID-19 treatments. Zn supplement might aid to activate or upregulate functional ACE2 expression countering SARS-CoV-2 mediated ACE2 downregulation (indicated with a). Zn can also inhibit SARS-CoV-2 replication by inhibiting RdRp. The second possible target might aid to increase the conversion of ANGII to ANG (1-7) to favour vasodilatory impact (indicated with b). The third possible therapeutic target might focus on the antagonist to ANG II to bind ANG 1aR (indicated with c). [→ = stimulation/activation; = inhibition or reduction].
The second possible target might aid to increase the conversion of ANGII to ANG (1-7) as this has vasodilatory impact hence would reduce the blood pressure (Fig. 3, indicated with the letter ‘b’). In the same line of direction, RAAS inhibitors were suggested to be beneficial in COVID-19 treatment [41].
The third possible therapeutic target might focus on the reduction of the binding of the ANGII with angiotensin II receptor 1a (ANG1aR) as this binding aggravates the tissue damage. This could be achieved by designing any antagonist to ANG II to bind ANG 1aR. (Fig. 3, indicated with the letter ‘c’). In favour of this notion, it can be highlighted that COVID-19 patients once given angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II type 1 receptor blockers (ARBs) showed reduced severity and a lower level of IL-6 in peripheral blood which in turn also increased CD3 and CD8 T cell counts in peripheral blood and decreased the peak viral load compared to other antihypertensive drugs [42].
How Zn2+ supplement would offer a beneficial role in CVD and COVID-19 comorbidity?
In biological fluids, zinc ion (Zn2+) works as a Lewis acid hence acts as a stable cofactor in redox reactions occurring in cellular and extracellular environment [43]. Zinc is directly involved in cell-mediated immunity against infectious bacteria and viruses [[44], [45], [46], [47]]. For example, cell specific impact of Zn deficiency may result in a decreased activation of NF-κB that in turn cause reduced IL-2 and IL-2 receptor-α in T helper cell line. Thus, a potential therapeutic roles of Zn was suggested in acute infantile diarrhea, acrodermatitis enteropathica, blindness in patients with age-related macular degeneration, and the treatment of common cold [44]. In addition, in vivo, zinc deficiency negatively impact immunological functions of granulocytes, monocytes, NK-, T-, and B-cells. Regulatory role of free Zn2+ in signal transduction in cells of the immune system involves several molecular targets, such as phosphatases, phosphodiesterases, caspases, and kinases [45].
Again, increased Zn at the site of infection causing upregulated expression of Zn importing proteins (i.e., ZIP) by the circulating leukocytes or infiltrated inflammatory cells, induces expression of metallothionein (MT) which not only protect leukocytes from the increased influx of Zn at the site of infections but also helps those cells to fight against the infection [47].
Furthermore, Zn is involved in the proliferation of neutrophils, NK cells, macrophages, and T and B lymphocytes as well as their immune functions such as cytokine production. Zn also mediates protection against reactive oxygen species (ROS) that are generally produced during inflammatory processes. Free intracellular Zn2+ is essential in extravasation to the site of the infection and uptake and killing of microorganisms by neutrophils [48].
Activation of immune responses such as NF-κB signalling requires Zn2+. Notably, NF-κB regulates the expression of pro-inflammatory cytokines such as IL-6, IL-8, and TNF-α, chemokines, acute phase proteins, matrix metalloproteinases, adhesion molecules, growth factors, and other factors involved in the inflammatory response, such as COX-2 and iNOS [49,50]. Zinc administration in mixed lymphocyte cultures induces and stabilizes subsets of CD4+ T cells [51], while CD4+ T-cells, commonly known as helper T-cells, play critical roles in antiviral immunity [52,53]. Zn2+ also serves as an intracellular second messenger and triggers apoptosis or a decrease in protein synthesis at elevated concentrations [[54], [55], [56]].
Zn also plays a critical role in autophagy under basal conditions - a natural, controlled mechanism to remove unnecessary or dysfunctional components from within the cells [57]. This notion was supported by an in vitro experiment using human hepatoma cells, VL-17A where Zn depletion caused a significant suppression of autophagy; on the contrary, an addition of the element to the culture enhanced autophagy [57]. Notably, in autophagy, the intracellular components such as protein aggregates and damaged organelles are degraded by lysosomal enzymes [58,59] including proteases, peptidases, phosphatases, nucleases, glycosidases, sulfatases, and lipases [60] — while structural and functional integrity of many of these enzymes depends on Zn [61]. In fact, during viral infections, autophagy provides protection as a defence mechanism by auto-degradation of the infected cells [[62], [63], [64]].
As a Zn-metalloenzyme, the functional expression of ACE2 [40] is expected to be increased by Zn2+ supplement. At the same time, Zn2+ is expected to inhibit SARS-CoV-2 replication as Zn2+ was reported to inhibit the in vitro RNA-dependent RNA polymerase (RdRp) activity by inhibiting the SARS-CoV RdRp elongation and template binding [65].
Earlier it was also shown that Zn2+ inhibited the proteolytic processing of replicase polyproteins [66]. To allow Zn2+ to exert its inhibitory effect on SARS-CoV-2 viral replication, Zn2+ entry inside the cell might be enhanced by ionophores such as dithiocarbamates [67], pyrithione [65,68], zincophorin [69], and hydroxychloroquine [65,70,71]. It can be noted that a meta-analysis involving 19 reported studies suggested that chloroquine/hydroxychloroquine was associated with a reduced risk of CVD in patients with rheumatic diseases [72].
The ability of Zn2+ to inhibit the replication of various RNA viruses has been demonstrated in a good number of in vitro studies. For example, in the presence of its cellular import stimulatory compounds such as hinokitol (HK), pyrrolidine dithiocarbamate (PDTC), and pyrithione (PT), the added Zn2+ inhibited the replication of influenza virus [73], respiratory syncytial virus [74], and several picornaviruses [71,75,76]. Their interference with polyprotein processing in cells infected with human rhinovirus and coxsackievirus B3 is well evidenced [71].
The other modes of action that Zn salts exhibit to inhibit SARS-CoV-2 as well as other viruses, viz. HIV, HSV, and vaccinia virus are inhibition of the viral entry, blocking of polyprotein processing, and inhibition of viral RdRp activity [65,77,78]. Zn salts namely Zn-sulfate and Zn-acetate were also shown to inhibit viral sense and antisense RNA levels by approximately 50%, thus inhibiting viral replication [79].
A retrospective case series study confirmed that triple therapy involving Zn, low-dose HCQ, and azithromycin as early as possible after symptom onset results in significantly fewer hospitalisations [80]. In the same line of direction, a multi-centre cohort study revealed an association between Zn supplement with an ionophore, and both increased rates of discharge home and a 24% reduced risk of in-hospital mortality among COVID-19 patients [81]. Again, adequate intake of Zn, vitamins C, and D was earlier suggested to combat the SARS-CoV-2 infection [82]. However, in a randomized clinical trial involving 214 adult COVID-19 patients, a high-dose of Zn with vitamin C (ascorbic acid) supplement was found ineffective to alleviate symptoms of the disease [83].
In addition to those direct effects on the virus as well as to improve the clinical outcome of CVD treatment, several immunome pathways such as the NF-κB signalling pathway are activated by Zn2+ [84]. This might control cytokine storm in COVID-19 patients by regulating the expression of pro-inflammatory cytokines namely IL-1b, IL-6, IL-8, TNF-α, and MCP-1; chemokines, acute phase proteins, matrix metalloproteinases, adhesion molecules, growth factors, as well as COX-2 and iNOS [49,50].
Conclusion
COVID positive patients’ cardiac health, with or without respiratory symptoms could be evaluated for resolving primary complications of COVID-19 and to reduce mortality from potential cardiovascular conditions. Based on the patients’ condition, a selective treatment plan might be required to ensure minimal lung injury or cardiovascular pathologies. Special attention might be required to influence the functional expression of ACE2. Given the above discussion on the involvement of Zn2+, its use as a supplement with an appropriate ionophore would offer multiple benefits to CVD and COVID-19 comorbid patients: (1) preventing viral replication by inhibiting the RdRP of the SARS-CoV-2, (2) enhance protective immune responses, and (3) restoring the functional balance of ACE2.
Conflict of interest statement
All authors declare that there is no conflict of interest.
Acknowledgement
No funds were available for this review work.
References
- 1.Vrints C.J.M., Krychtiuk K.A., Van Craenenbroeck E.M., Segers V.F., Price S., Heidbuchel H. Endothelialitis plays a central role in the pathophysiology of severe COVID-19 and its cardiovascular complications. Acta Cardiol. 2020:1–16. doi: 10.1080/00015385.2020.1846921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goyal P., Choi J.J., Pinheiro L.C., Schenck E.J., Chen R., Jabri A., et al. Clinical Characteristics of Covid-19 in New York City. N Engl J Med. 2020;100:1–3. doi: 10.1056/NEJMc2005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson S., Hirsch J.S., Narasimhan M., Crawford J.M., McGinn T., Davidson K.W., et al. Presenting Characteristics, Comorbidities, and Outcomes among 5700 Patients Hospitalized with COVID-19 in the New York City Area. J Am Med Assoc. 2020;323:2052–2059. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onder G., Rezza G., Brusaferro S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. J Am Med Assoc. 2020;323:1775–1776. doi: 10.1001/jama.2020.4683. [DOI] [PubMed] [Google Scholar]
- 5.Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., et al. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;2019:811–818. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., He W., Yu X., Hu D., Bao M., Liu H., et al. Coronavirus Disease 2019 in elderly patients: characteristics and prognostic factors based on 4-week follow-up. J Infect. 2020 doi: 10.1016/j.jinf.2020.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long B., Brady W.J., Koyfman A., Gottlieb M. Cardiovascular complications in COVID-19. Am J Emerg Med. 2020;38:1504–1507. doi: 10.1016/j.ajem.2020.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aladağ N., Atabey R.D. The role of concomitant cardiovascular diseases and cardiac biomarkers for predicting mortality in critical COVID-19 patients. Acta Cardiol. 2020:1–8. doi: 10.1080/00015385.2020.1810914. [DOI] [PubMed] [Google Scholar]
- 9.Clerkin K.J., Fried J.A., Raikhelkar J., Sayer G., Griffin J.M., Masoumi A., et al. COVID-19 and Cardiovascular Disease. Circulation. 2020;2019:1648–1655. doi: 10.1161/CIRCULATIONAHA.120.046941. [DOI] [PubMed] [Google Scholar]
- 10.Gnecchi M., Moretti F., Bassi E.M., Leonardi S., Totaro R., Perotti L., et al. Myocarditis in a 16-year-old boy positive for SARS-CoV-2. Lancet. 2020;395:e116. doi: 10.1016/S0140-6736(20)31307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar R., Singh V.P., Baker K.M. The intracellular renin-angiotensin system in the heart. Curr Hypertens Rep. 2009;11:104. doi: 10.1007/s11906-009-0020-y. [DOI] [PubMed] [Google Scholar]
- 12.Coates D. The angiotensin converting enzyme (ACE) Int J Biochem Cell Biol. 2003;35:769–773. doi: 10.1016/S1357-2725(02)00309-6. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues Prestes T.R., Rocha N.P., Miranda A.S., Teixeira A.L., Simoes-E-Silva A.C. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: Evidence from basic and clinical research. Curr Drug Targets. 2017;18:1301–1313. doi: 10.2174/1389450117666160727142401. [DOI] [PubMed] [Google Scholar]
- 14.Hamming I., Timens W., Bulthuis M.L.C., Lely A.T., Navis G.J., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L., Wei Q., Alvarez X., Wang H., Du Y., Zhu H., et al. Epithelial cells lining salivary gland ducts are early target cells of severe acute respiratory syndrome coronavirus infection in the upper respiratory tracts of rhesus macaques. J Virol. 2011;85:4025–4030. doi: 10.1128/JVI.02292-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L., Zhang Y., Zhang S. Cardiovascular impairment in COVID-19: Learning from current options for cardiovascular anti-inflammatory therapy. Front Cardiovasc Med. 2020;7:78. doi: 10.3389/fcvm.2020.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz M.D., Emerson S.G., Punt J., Goff W.D. Decreased Naïve T-cell Production Leading to Cytokine Storm as Cause of Increased COVID-19 Severity with Comorbidities. Aging Dis. 2020;11:742–745. doi: 10.14336/AD.2020.0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 24.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 25.Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wösten-van Asperen R.M., Lutter R., Specht P.A., Moll G.N., van Woensel J.B., van der Loos C.M., et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–627. doi: 10.1002/path.2987. [DOI] [PubMed] [Google Scholar]
- 27.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J., Jiang Q., Xia X., Liu K., Yu Z., Tao W., et al. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell. 2020;19 doi: 10.1111/acel.13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oudit G.Y., Kassiri Z., Jiang C., Liu P.P., Poutanen S.M., Penninger J.M., et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39:618–625. doi: 10.1111/j.1365-2362.2009.02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y., Roever L., Tse G., Liu T. 2019-Novel Coronavirus-Related Acute Cardiac Injury Cannot Be Ignored. Curr Atheroscler Rep. 2020;22:2019–2020. doi: 10.1007/s11883-020-00842-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.South A.M., Diz D.I., Chappell M.C. COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol. 2020;318:H1084–90. doi: 10.1152/ajpheart.00217.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzau V.J. Mechanism of Action of Angiotensin-Converting Enzyme (ACE) Inhibitors in Hypertension and Heart Failure. Drugs. 1990;39:11–16. doi: 10.2165/00003495-199000392-00004. [DOI] [PubMed] [Google Scholar]
- 33.Mirabito Colafella K.M., Bovée D.M., Danser A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res. 2019;186 doi: 10.1016/j.exer.2019.05.020. [DOI] [PubMed] [Google Scholar]
- 34.Gradman A.H., Schmieder R.E., Lins R.L., Nussberger J., Chiang Y., Bedigian M.P. Aliskiren, a novel orally effective renin inhibitor, provides dose-dependent antihypertensive efficacy and placebo-like tolerability in hypertensive patients. Circulation. 2005;111:1012–1018. doi: 10.1161/01.CIR.0000156466.02908.ED. [DOI] [PubMed] [Google Scholar]
- 35.Nicholls J., Peiris M. Good ACE, bad ACE do battle in lung injury, SARS. Nat Med. 2005;11:821–822. doi: 10.1038/nm0805-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y., Zeng Z., Li Y., Huang W., Zhou M., Zhang X., et al. Angiotensin-converting enzyme inhibition attenuates lipopolysaccharide-induced lung injury by regulating the balance between angiotensin-converting enzyme and angiotensin-converting enzyme 2 and inhibiting mitogen-activated protein kinase activation. Shock. 2015;43:395–404. doi: 10.1097/SHK.0000000000000302. [DOI] [PubMed] [Google Scholar]
- 37.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrario C.M., Jessup J., Chappell M.C., Averill D.B., Brosnihan K.B., Tallant E.A., et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 39.Varagic J., Ahmad S., Nagata S., Ferrario C.M. ACE2: angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr Hypertens Rep. 2014;16:420. doi: 10.1007/s11906-014-0420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner A.J. Prot. Arm Renin Angiotensin Syst. Funct. Asp. Ther. Implic. Elsevier Inc.; 2015. ACE2 Cell Biology, Regulation, and Physiological Functions; pp. 185–189. [DOI] [Google Scholar]
- 41.Guzik T.J., Mohiddin S.A., Dimarco A., Patel V., Savvatis K., Marelli-Berg F.M., et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116:1666–1687. doi: 10.1093/cvr/cvaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng J., Xiao G., Zhang J., He X., Ou M., Bi J., et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. 2020;9:757–760. doi: 10.1080/22221751.2020.1746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baś S., Szewczyk Marcin, Mlynarski J. In: Chiral Lewis Acids Org. Synth. Mlynarski J., editor. Wiley Online Library; 2017. Zinc‐based Chiral Lewis Acids. [DOI] [Google Scholar]
- 44.Prasad A.S. Zinc in human health: effect of zinc on immune cells. Mol Med. 2008;14:353–357. doi: 10.2119/2008-00033.Prasad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haase H., Rink L. Zinc signals and immune function. Biofactors. 2014;40:27–40. doi: 10.1002/biof.1114. [DOI] [PubMed] [Google Scholar]
- 46.Bonaventura P., Benedetti G., Albarede F., Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev. 2015;14:277–285. doi: 10.1016/j.autrev.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Rahman M.T., Karim M.M. Metallothionein: a Potential Link in the Regulation of Zinc in Nutritional Immunity. Biol Trace Elem Res. 2018;182:1–13. doi: 10.1007/s12011-017-1061-8. [DOI] [PubMed] [Google Scholar]
- 48.Hasan R., Rink L., Haase H. Chelation of Free Zn(2)(+) Impairs Chemotaxis, Phagocytosis, Oxidative Burst, Degranulation, and Cytokine Production by Neutrophil Granulocytes. Biol Trace Elem Res. 2016;171:79–88. doi: 10.1007/s12011-015-0515-0. [DOI] [PubMed] [Google Scholar]
- 49.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1 doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayden M.S., Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Semin Immunol. 2014;26:253–266. doi: 10.1016/j.smim.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maywald M., Rink L. Zinc supplementation induces CD4(+)CD25(+)Foxp3(+) antigen-specific regulatory T cells and suppresses IFN-gamma production by upregulation of Foxp3 and KLF-10 and downregulation of IRF-1. Eur J Nutr. 2017;56:1859–1869. doi: 10.1007/s00394-016-1228-7. [DOI] [PubMed] [Google Scholar]
- 52.Whitmire J.K., Ahmed R. Costimulation in antiviral immunity: differential requirements for CD4(+) and CD8(+) T cell responses. Curr Opin Immunol. 2000;12:448–455. doi: 10.1016/s0952-7915(00)00119-9. [DOI] [PubMed] [Google Scholar]
- 53.Jansen J.M., Gerlach T., Elbahesh H., Rimmelzwaan G.F., Saletti G. Influenza virus-specific CD4+ and CD8+ T cell-mediated immunity induced by infection and vaccination. J Clin Virol. 2019;119:44–52. doi: 10.1016/j.jcv.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 54.Alirezaei M., Nairn A.C., Glowinski J., Premont J., Marin P. Zinc inhibits protein synthesis in neurons. Potential role of phosphorylation of translation initiation factor-2alpha. J Biol Chem. 1999;274:32433–32438. doi: 10.1074/jbc.274.45.32433. [DOI] [PubMed] [Google Scholar]
- 55.Frederickson C.J., Koh J.-Y., Bush A.I. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 56.Lazarczyk M., Favre M. Role of Zn2+ ions in host-virus interactions. J Virol. 2008;82:11486–11494. doi: 10.1128/JVI.01314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liuzzi J.P., Yoo C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol Trace Elem Res. 2013;156:350–356. doi: 10.1007/s12011-013-9816-3. [DOI] [PubMed] [Google Scholar]
- 58.Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018;20:521–527. doi: 10.1038/s41556-018-0092-5. [DOI] [PubMed] [Google Scholar]
- 59.Levine B., Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lubke T., Lobel P., Sleat D.E. Proteomics of the lysosome. Biochim Biophys Acta. 2009;1793:625–635. doi: 10.1016/j.bbamcr.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maret W. Zinc biochemistry: from a single zinc enzyme to a key element of life. Adv Nutr. 2013;4:82–91. doi: 10.3945/an.112.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang P., Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi A.M.K., Ryter S.W., Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 64.Meijer A.J., Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci. 2009;46:210–240. doi: 10.1080/10408360903044068. [DOI] [PubMed] [Google Scholar]
- 65.te Velthuis A.J.W., van den Worm S.H.E., Sims A.C., Baric R.S., Snijder E.J., van Hemert M.J. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denison M.R., Zoltick P.W., Hughes S.A., Giangreco B., Olson A.L., Perlman S., et al. Intracellular processing of the N-terminal ORF 1a proteins of the coronavirus MHV-A59 requires multiple proteolytic events. Virology. 1992;189:274–284. doi: 10.1016/0042-6822(92)90703-r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim C.H., Kim J.H., Hsu C.Y., Ahn Y.S. Zinc is required in pyrrolidine dithiocarbamate inhibition of NF-κB activation. FEBS Lett. 1999;449:28–32. doi: 10.1016/S0014-5793(99)00390-7. [DOI] [PubMed] [Google Scholar]
- 68.Kim C.H., Kim J.H., Moon S.J., Chung K.C., Hsu C.Y., Seo J.T., et al. Pyrithione, a Zinc Ionophore, Inhibits NF-κB Activation. Biochem Biophys Res Commun. 1999;259:505–509. doi: 10.1006/bbrc.1999.0814. [DOI] [PubMed] [Google Scholar]
- 69.Brooks H.A., Gardner D., Poyser J.P., King T.J. The structure and absolute stereochemistry of zincophorin (antibiotic M144255): a monobasic carboxylic acid ionophore having a remarkable specificity for divalent cations. J Antibiot (Tokyo) 1984;37:1501–1504. doi: 10.7164/antibiotics.37.1501. [DOI] [PubMed] [Google Scholar]
- 70.Xue J., Moyer A., Peng B., Wu J., Hannafon B.N., Ding W.-Q. Chloroquine is a zinc ionophore. PLoS One. 2014;9 doi: 10.1371/journal.pone.0109180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krenn B.M., Gaudernak E., Holzer B., Lanke K., Van Kuppeveld F.J.M., Seipelt J. Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J Virol. 2009;83:58–64. doi: 10.1128/JVI.01543-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu D., Li X., Zhang Y., Kwong J.S.-W., Li L., Zhang Y., et al. Chloroquine and hydroxych loroquine are associated with reduced cardiovascular risk: a systematic review and meta-analysis. Drug Des Devel Ther. 2018;12:1685–1695. doi: 10.2147/DDDT.S166893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uchide N., Ohyama K., Bessho T., Yuan B., Yamakawa T. Effect of antioxidants on apoptosis induced by influenza virus infection: inhibition of viral gene replication and transcription with pyrrolidine dithiocarbamate. Antiviral Res. 2002;56:207–217. doi: 10.1016/s0166-3542(02)00109-2. [DOI] [PubMed] [Google Scholar]
- 74.Suara R.O., Crowe J.E.J. Effect of zinc salts on respiratory syncytial virus replication. Antimicrob Agents Chemother. 2004;48:783–790. doi: 10.1128/aac.48.3.783-790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Korant B.D., Kauer J.C., Butterworth B.E. Zinc ions inhibit replication of rhinoviruses. Nature. 1974;248:588–590. doi: 10.1038/248588a0. [DOI] [PubMed] [Google Scholar]
- 76.Lanke K., Krenn B.M., Melchers W.J.G., Seipelt J., van Kuppeveld F.J.M. PDTC inhibits picornavirus polyprotein processing and RNA replication by transporting zinc ions into cells. J Gen Virol. 2007;88:1206–1217. doi: 10.1099/vir.0.82634-0. [DOI] [PubMed] [Google Scholar]
- 77.Katz E., Margalith E. Inhibition of vaccinia virus maturation by zinc chloride. Antimicrob Agents Chemother. 1981;19:213–217. doi: 10.1128/aac.19.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haraguchi Y., Sakurai H., Hussain S., Anner B.M., Hoshino H. Inhibition of HIV-1 infection by zinc group metal compounds. Antiviral Res. 1999;43:123–133. doi: 10.1016/s0166-3542(99)00040-6. [DOI] [PubMed] [Google Scholar]
- 79.Kaushik N., Subramani C., Anang S., Muthumohan R., Shalimar, Nayak B., et al. Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase. J Virol. 2017;91 doi: 10.1128/JVI.00754-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Derwand R., Scholz M., Zelenko V. COVID-19 outpatients: early risk-stratified treatment with zinc plus low-dose hydroxychloroquine and azithromycin: a retrospective case series study. Int J Antimicrob Agents. 2020;56 doi: 10.1016/j.ijantimicag.2020.106214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frontera J.A., Rahimian J.O., Yaghi S., Liu M., Lewis A., de Havenon A., et al. Treatment with Zinc is Associated with Reduced In-Hospital Mortality Among COVID-19 Patients: A Multi-Center Cohort Study. Res Sq. 2020 doi: 10.21203/rs.3.rs-94509/v1. rs.3.rs-94509. [DOI] [Google Scholar]
- 82.Name J.J., Souza A.C.R., Vasconcelos A.R., Prado P.S., Pereira C.P.M. Zinc, Vitamin D and Vitamin C: Perspectives for COVID-19 With a Focus on Physical Tissue Barrier Integrity. Front Nutr. 2020;7 doi: 10.3389/fnut.2020.606398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thomas S., Patel D., Bittel B., Wolski K., Wang Q., Kumar A., et al. 2021. Effect of High-Dose Zinc and Ascorbic Acid Supplementation vs Usual Care on Symptom Length and Reduction Among Ambulatory Patients With SARS-CoV-2 Infection The COVID A to Z Randomized Clinical Trial; pp. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim Y.M., Cao D., Reed W., Wu W., Jaspers I., Tal T., et al. Zn2+-induced NF-κB-dependent transcriptional activity involves site-specific p65/RelA phosphorylation. Cell Signal. 2007;19:538–546. doi: 10.1016/j.cellsig.2006.08.003. [DOI] [PubMed] [Google Scholar]