

Abstract
Type III secretion systems (T3SSs) are bacterial membrane-embedded nanomachines that allow a number of humans, plant and animal pathogens to inject virulence factors directly into the cytoplasm of eukaryotic cells. Export of effectors through T3SSs is critical for motility and virulence of most Gram-negative pathogens. Current computational methods can predict type III secreted effectors (T3SEs) from amino acid sequences, but due to algorithmic constraints, reliable and large-scale prediction of T3SEs in Gram-negative bacteria remains a challenge. Here, we present DeepT3 2.0 (http://advintbioinforlab.com/deept3/), a novel web server that integrates different deep learning models for genome-wide predicting T3SEs from a bacterium of interest. DeepT3 2.0 combines various deep learning architectures including convolutional, recurrent, convolutional-recurrent and multilayer neural networks to learn N-terminal representations of proteins specifically for T3SE prediction. Outcomes from the different models are processed and integrated for discriminating T3SEs and non-T3SEs. Because it leverages diverse models and an integrative deep learning framework, DeepT3 2.0 outperforms existing methods in validation datasets. In addition, the features learned from networks are analyzed and visualized to explain how models make their predictions. We propose DeepT3 2.0 as an integrated and accurate tool for the discovery of T3SEs.
INTRODUCTION
Microbial pathogens secrete a wide range of substrates that disrupt host homeostasis and immune defenses, thus resulting in the establishment of infection (1). However, transporting substrates across cellular membranes is a challenging biochemical feat, and to achieve this, bacteria have evolved nine dedicated secretion systems (type I to type IX) (2,3). Of these, the type III secretion system (T3SS) is one of the most sophisticated and best-characterized systems (4,5) and has been widespread in many Gram-negative bacteria, including symbionts, such as Rhizobium, and pathogens that are responsible for a range of severe diseases, such as gastroenteritis (Shigella flexneri), plague (Yersinia pestis), typhoid fever (Salmonella typhi) and infantile bacterial diarrhea (enteropathogenic Escherichia coli; EPEC) (6,7). T3SSs are assembled from three main ‘parts’ including a cytoplasmic ring (C-ring) and sorting platform; a basal body, which is a multi-ring system that embedded in both bacterial membranes, and a translocation pore that is inserted into host cell membranes (8). The overall structure and organization of the T3SS spans three cellular membranes, the bacterial inner membrane, the bacterial outer membrane and the eukaryotic host cell membrane, which uses a one-step secretion mechanism to transport substrates directly from the bacterial cytoplasm into host cells (9,10).
The T3SSs are involved in the manipulation of a vast array of key cellular processes such as the cell cytoskeleton, trafficking, cell death or survival, and the NF-κB and MAPK signaling pathways (3) and these functions are enabled by T3SEs. Unlike other bacterial effectors that exert their function by introducing covalent, non-reversible modifications of their target host cell proteins, T3SEs may act by mimicking the functions of host cell proteins (11,12). This strategy seems appropriate to have been adapted by bacteria which have T3SSs as a central element for the establishment of a close functional interface that is often symbiotic in nature. Since T3SEs often mimic or override the functions of host cell proteins, the relevant studies have also provided remarkable examples of convergent evolution, tools for research and clinical applications, as well as deep insights into host cell processes (13).
Owing to the high cost and technical challenges of testing all possible effector candidates experimentally, researchers have attempted to identify T3SEs by computational methods. Nearly all current T3SE prediction methods rely on well-established machine-learning algorithms such as Naïve Bayes (NB) (14,15), artificial neural network (ANN) (16), support vector machine (SVM) (17–23), random forest (RF) (24), Markov Model (MM) (25), gradient boosting machine (LightGBM) and so on (26). In addition to the quality and quantity of the data set, another important challenge for these methods is to define suitable features or feature sets from data that led to better separation between different classes. Therefore, the feature extraction can be crucial for the process of T3SE classification and the accuracy of the predictions. Several properties of amino acid sequences are found to be important for distinguishing T3SEs from non-T3SEs and can be separated into two broad classes. The first class comprises few but specific features of either N-terminal 30 or 100 amino acids and includes short peptides (14), N-terminal instability (27), solvent accessibility information (23,24), secondary structure (14,23,24) and position-specific composition (14,17,20,23,24) or entropy (24) of amino acids. The second class comprises comprehensive and general features of entire protein sequence, e.g. amino acid composition, dipeptide composition, sequence-order descriptors, physicochemical properties and evolutionary conservation (26,28). Currently, the integrated model based on ensemble learning has shown better performance than training a single machine-learning algorithm when combined with various features for the T3SE detection (26,28). For example, Bastion3, one of the latest predictors, reports a remarkable accuracy value of 0.959 for the two-layer ensemble models, when validated on the test data (26).
The recent revolution in deep learning techniques for biology suggests that convolutional neural networks (CNNs) can serve as an effective tool for ‘sequence-based’ modeling of a broad range of biological questions (29–34). Based on these ideas, several algorithms have been developed to predict secreted effectors and their types from amino acid sequences (35–37). DeepT3 was among the first publicly available methods that used a convolutional neural network with N-terminal sequence data to train a model for predicting T3SEs (35). In this study, we introduce DeepT3 2.0, which is an updated version combines various deep learning architectures and an integrated framework for improving T3SE prediction. The major contributions of our work are fourfold: we systematically and comprehensively explored the effects of model architectures, hyperparameters, encoding methods and sequence lengths on model performance, which provides recommendations for future method development; we analyzed features extracted from the hidden layers of the deep learning models to investigate their ability to distinguish between T3SEs and non-T3SEs; we created an integrative prediction framework for identifying T3SEs in whole-genome scale; we evaluated the performance of our tool on both the aggregate dataset and specific secretion system subsets (e.g. T1SE, T2SE and so on).
MATERIALS AND METHODS
Data collection
We generated a benchmark dataset for building, testing the model and making comparison with other deep learning models. We relied on a combination of the positive training set of the Bastion3 method (26) (which had experimentally verified and database annotated type III secreted effectors for Gram-negative bacteria) together with the negative set collected from the previous work of Dong et al (17), which was compiled from eight well-studied Gram-negative bacterial proteomes, including Escherichia coli O157:H7, Salmonella enterica serovar Typhimurium, Pseudomonas syringae DC3000, Yesinia pestis bv. Antiqua, Chlamydia trachomatics, Shigella flexneri, Yesinia enteroclitica and Burkholderia pesudomallei. We discarded the protein sequences which are <50 amino acids or with noncanonical amino-acid symbols (B, Z, J) contained in the records. To avoid duplicate or homologous proteins, the CD-HIT program (38) was used to filter the positive set with a sequence identity >70% (26) and the negative set >30%. This way, 379 effectors and 755 non-effectors were collected, respectively.
Additionally, to determine how well the DeepT3 2.0 tool performed when predicting T3SEs from a given species, an independent test set which included a plant pathogen P. syringae was created. Proteins experimentally confirmed as type III effectors from P. syringae were collected from the study of Baltrus et al (39). Since it is challenging to define a non-T3SE, we developed a strict filtering criterion to generate the negative set. We first collected the whole UniprotKB reviewed entries of the P. syringae and then removed all possible type III effectors using the Uniprot keyword and QuickGO annotations. We also pruned out proteins with <50 amino acids or containing elements other than the 20 common amino acids. We further excluded protein sequences that were already present in our benchmark dataset and used the CD-HIT to decrease sequence redundancy with a threshold of 30%. Finally, we obtained 32 effectors and 711 non-effectors form P. syringae.
The Bastion3 independent test set consisting of 108 effectors and 108 non-effectors was used as another independent test set to evaluate the performance of several state-of-the-art T3SE predictors.
We collected a set of seven secretion system effectors from T1SS to T8SS after removing the T3SEs and their homologs from our previous study (40) to evaluate the specificity of DeepT3 2.0 in Gram-negative bacterial secretomes. First, we directly extracted a minority of proteins from Uniprot according to the ‘Subcellular location’ comments and various keywords describing their secretory types. Second, we got information about their protein or gene IDs and corresponding secretory types through a careful literature search, and then artificially collected them from the three databases including UniprotKB, TrEMBL and RefSeq. To ensure an accurate collection, the proteins with uncertain secretory types were not selected. With this procedure, we ended-up with 509 proteins. The 161 T5SEs comprised the largest subset. The numbers of T1SE and T2SE were relatively smaller than that of T5SE, including 107 and 106 proteins, respectively. The remaining proteins in this data set were distributed as follows: 65 from T4SEs, 8 from T6SEs, 53 from T7SEs and 3 from T8SEs.
We assessed DeepT3 2.0 running time and whole-genome scale performance using complete genome of Chlamydia trachomatis (strain D/UW-3/Cx). The genome of this Gram-negative bacterial species is 1 042 519 bp long and hosts 936 genes including T3SS (41). Only RefSeq entries were used, and other proteins were not included. From the NCBI, we downloaded all the 1774 proteins. All data sets described herein can be downloaded from http://advintbioinforlab.com/data/dataset.zip.
Amino acid representation
For providing the tensors to the deep learning models appropriately, each amino acid in a protein sequence with N residues is represented by an N × 20 one-hot vector and an N × 256 embedding vector, respectively. One-hot encoding provides a mapping mechanism between the residues and the vectors of zeros with one, for example, alanine can be encoded as [1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]. The embedding vector is a high-dimensional continuous vector that preserve the context of amino-acid symbols in a protein sequence. The benefit of embedding is that it can create a more compact representation of amino acid symbols and can yield semantically similar symbols close to each other in the vector space (42). The embedding process can be implemented by the embedding layer in the Keras framework. Specifically, sequences of amino acids are first dictionary encoded (the amino acids are assigned different numbers from 1 to 20, for example, alanine can be assigned to 1), and then passed through the embedding layer to convert each number into a 256-dimensional vector before being input into other hidden layers in the models.
DeepT3 2.0 model architectures
The DeepT3 2.0 consisted of an integrative framework of six deep learning models, each model can produce a probability score between 0 and 1 (that is interpreted as the predicted likelihood that the protein of interest being a type III secreted effector or non-effector). The final prediction of the framework was the sum of the predictions from the six independent models and determined by an optimized cutoff. Each model is described sequentially going from the encoded protein sequence input to predictions in which the output of a layer is used as the input for the next.
Recurrent neural network with embedding representation—RNN (dictionary)
We selected the bidirectional long short-term memory (BiLSTM) recurrent neural network as RNN representation learner because the recent success of the BiLSTM for the sequence-based deep representation learning in protein engineering (43). Bidirectional LSTM processes sequences in both forward and backward directions, and therefore often captures the context better. Specifically, the architecture selected for training was a single-layer BiLSTM with 64 hidden neurons. Dropout of 0.25 was applied to the BiLSTM layer for preventing overfitting. We connected the BiLSTM layer and a single dense output layer with a sigmoid activation function. The output layer contained one neuron representing effector (T or 1) or non-effector (F or 0) classes.
Convolutional and Recurrent neural network with embedding representation—CNN-RNN(dictionary)
We combined a 1D-CNN that extracts sequence-based features, with an RNN architecture that captures long-term dependency in the sequences. The first layer was a convolutional layer containing 200 filters, each of which had a 1D kernel with width of 9. A Rectified Linear Units (ReLU) activation function was applied to the neuron output. The activation function introduced nonlinearities to avoid the network suffering from the vanishing gradient problem (30). The second layer performed max pooling, which outputted the maximum value over a non-overlapping sliding window of 2. The pooling layer was then fully connected to the 64-neuron BiLSTM layer. The fourth and final layer consisted of only one neuron producing the final probability score using the sigmoid function.
Convolutional neural network with embedding representation—CNN(dictionary)
The general architecture of CNN consisted of four main components: convolutional layer, pooling layer, fully connected layer and an output layer. First, we began by passing the embedding vectors into a convolutional layer consisting of 250 filters with width of 5. Each filter covered all 250-dimensional amino-acid input channels. During training, each filter scanned along the input sequence and computed a score for each five amino acids, followed by a ReLU activation function. These activated scores were then passed through a pooling layer, where the maximum score was computed over a window size of 2. Next, the flattened pooling scores were passed to a single dense layer with 650 hidden neurons. To prevent overfitting, the dense scores proceed through a dropout layer with a dropout rate of 0.25. Finally, the scores were fed into an output layer consisting of a sigmoid function.
Convolutional and Recurrent neural network with one-hot encoding—CNN-RNN(onehot)
For the CNN, we combined a convolutional 2D layer (50 filters with kernel size [20 × 3]) using a ReLU activation function with a pooling 2D layer (pooling size [1 × 2]). We chose max pooling to flatten the output and reduce the number of parameters. Subsequently, the pooling layer was followed by a batch normalization layer and a dropout layer. The batch normalization layer stabilized the output from the pooling layer and dropout layer prevented the network from overfitting. We then passed the output of the dropout layer to a BiLSTM layer. We trained the BiLSTM with 64 hidden neurons. The last layer was a sigmoid activation node.
Convolutional neural network with one-hot encoding—CNN(onehot)
We used a kernel of 20 × 11 with step size 1 for convolutional 2D layer, 1 × 2 aggregation regions for Maxpooling 2D layer, and rectified linear unit nonlinearities for the activation function. The number of filters in the convolution layer was 100, and the number of neurons in the dense layer was 650. We used the dropout with rate of 0.25 for the pooling and dense layers.
Multi-layer perceptron with one-hot encoding—MLP (onehot)
MLP was constructed from two fully connected layers. The number of neurons per hidden layer was 512 and 256, respectively. The dropout layer with a dropout rate of 0.25 followed each layer, which finally connected to a sigmoid activation function that outputted the predicted probability.
Hyperparameter tuning
The hyperparameters we considered in the DeepT3 2.0 included dropout rate, batch size, embedding dimension, pooling size, pooling type, convolution kernel size, number of filters and number of neurons in BiLSTM (44).
We generated eight sets of hyperparameters from the following: dropout rate = (0, 0.25, 0.5), batch size = (20, 40, 60, 80), embedding dimension = (50, 100, 150, 200, 250), pooling type = (maximum, average), pooling size = (2, 4, 6), convolution kernel size = (3, 5, 7, 9, 11), number of filter = (50, 100, 150, 200, 250) and number of neurons in BiLSTM = (32, 64, 128, 256). We compared the average performance of each of these parameter sets by Matthew’s correlation coefficient (MCC) score for the test data.
We took the sampled parameter set with best performance (mean MCC score) and varied each parameter individually while keeping the rest constant. We measured the performance change with respect to the change in each parameter, again by average performance on test data measured by MCC score for each model. Based on this analysis, the final hyperparameters that gave the best average performance were dropout rate = 0.25, batch size = 60, embedding dimension = 250, pooling type = maximum, pooling size = 2 and number of neurons in BiLSTM = 64. In addition, we observed that the performance of deep learning models depends critically on the choice of kernel size and the number of filters. Both hyperparameters control how and what the convolutional network model can learn. In the result section, we discussed how different models select their optimal kernel sizes and the number of filters.
Model training and integrating predictions
We integrated the predictions to improve performance, where each model was trained on a different random split of the training data and their predictions were summed. We began by evaluating our models on the benchmark dataset of 1134 proteins using the hyper parameter optimization step described in the section above to determine the best performance of a single model. Here, we first split the benchmark dataset into mutually exclusive sets for training and validation (80%) and for testing (20%). Then we made a 90.0–10.0% train-validation split in Python using the numpy package and a fixed random seed. The test set was never used for training so that it could be used to estimate generalization performance when conducting experiments and building models. We trained each model on the training data for 25 epochs, where an epoch is defined as a single pass through all the training data (45), and we evaluated the trained model with updated parameters on the validation data at the end of each epoch. After training was complete, we used the model parameters that performed best on the validation data and tested the model with those parameters on the test data. We repeated this procedure with 10 different random splits of the training and validation data and averaged the results. After the model performance was optimized, we integrated all models to conduct predictions on new datasets. All models were trained using the Adam optimizer. Neural networks were implemented with Keras 2.2.4 (https://keras.io/) using Tensorflow backend and Python 3.5. For training, we utilized an NVIDIA GTX 1060 GPU with CUDA 10.2.95 on a Windows 10 workstation to speed up the gradient descent. A single training run over 25 epochs took only around 1–2 min.
Exploratory analysis and data visualization
We extracted one-hot vectors and embedding vectors of each amino acid from the trained DeepT3 2.0 model. We used Uniform Manifold Approximation and Projection (UMAP) (46)—a common technique for visualizing high-dimensional data (as implemented in the UMAP-learn R package uwot (47), with parameters n_neighbors: 4 and mim_dist: 0.1), to project the 20 common amino acids in a 2D space. We also used this tool to visualize the internal features learned by each deep learning model. Additionally, the intermediate output from the hidden layers was extracted for UMAP projection as well. The intermediate outputs from the hidden layer can represent how the encoded proteins in the training data were processed in the 2D-projection. The related parameter set for the intermediate output’s projection is (n_neighbors: 15, mim_dist: 0.001). Here, all UMAP plots were produced using the R and ggplot2 package (48).
Comparison on prediction sets
We trained the six deep learning models separately and compared their prediction outputs using an independent test set containing 108 effectors and 108 non-effectors. To visualize the differences and similarities among six prediction sets, we used the Venn diagrams to display their unions and intersections. A list of set unions in Venn diagrams was built and analyzed by the UpSetR (49).
Comparison with existing methods
In addition to the previous version of DeepT3 (35), 5 other methods were selected for comparison of predictive performances, including Bastion3 (26), BEAN2 (18), pEffect (19), Effective T3 (14) and BPBAac (20). Most of the methods were run through their respective websites. For the method BPBAac, we downloaded the Perl and R scripts and run locally on our computers using the default parameters.
Bacterial genome visualization
We applied DeepT3 2.0 to the whole-genome of Gram-negative bacteria Chlamydia trachomatis. All RefSeq proteins and predicted effectors were mapped to their corresponding positions on the circular genome using the blastx searches. All known coding sequence, tRNA and rRNA were also mapped on the corresponding DNA strand of the genome. We generated the graphical map of circular genome that shows sequence features, base composition plots and predicted results using the CGView Server (50).
Predictive performance metric calculation
Prediction performance of all T3SE detection algorithms was measured using recall, precision (PRE), accuracy (ACC) and the Matthew's correlation coefficient (MCC). These performance metrics are calculated as:
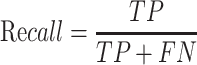 |
(1) |
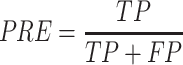 |
(2) |
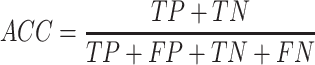 |
(3) |
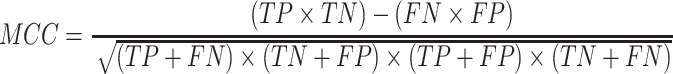 |
(4) |
In the above equations, TP, FP, TN and FN represent true positives, false positives, true negatives and false negatives, respectively. Each of these metrics has different properties (51). Recall measures the proportion of true positives among all truly positives, where precision measures the proportion of true positives among all positively predicted results. The accuracy denotes the proportion of true results (true positives and true negative) among the total number of outcomes. The Matthew’s correlation coefficient is the discrete case for Pearson Correlation Coefficient, which refers to the quality of the binary classification by considering true and false positives and true and false negatives (52).
To further evaluate the performance of deep learning models, receiver operator characteristic (ROC) curves and the areas under ROC curves (AUC) were utilized. The ROC curves evaluate the change in true positive rate with respect to the false positive rate of a predicted class label in accordance with all possible thresholds of a classification score that can be interpreted as probabilities. The AUC scores are calculated based on the area under ROC curves using trapezoidal rule. The value of the AUC falls in the interval of [0, 1], where a perfect classification would read a score of 1 and a random guessing would reach a sore of 0.5. We plotted the ROC curves and calculated the AUC metrics using R pROC package (53).
RESULTS
Overview of the DeepT3 2.0
The DeepT3 2.0 integration framework is shown in Figure 1A and described in the Materials and methods section. Briefly, we aimed to improve upon previous T3SE prediction methods by developing a tool by integrating different deep learning models instead of using a single model. Consequently, we constructed six models involving two types of data encodings: one-hot and embedding encodings, and four types of neural networks: convolutional, recurrent, convolutional-recurrent and multilayer neural networks. For each protein, we encoded the amino acids using the one-hot and embedding matrices so that each position in the sequence became two vectors of length 20 and 256, respectively, containing the information about the co-occurrences of amino acids (Figure 1B). The architectural components of four types of neural networks were mainly consisting of the following types of layers: convolution, recurrent, maxpooling and fully connected layers (Figure 1C). We designed different model architectures that were of varying layers and connections, and further examined whether network hyperparameters influenced predicting T3SEs. Several published studies have shown that the N-terminal sequences are adequate for the identification of T3SEs (20,24,35), we therefore tested whether T3SE prediction could be refined by learning directly from first 100 N-terminal residues of proteins, instead of full-length sequences. To prevent information leaking, the sequences were clustered to remove redundancy from the training, validation and test sets by using CD-HIT (38). We evaluated the performance of individual models and of their integration into DeepT3 2.0.
Figure 1.

Overview of the DeepT3 2.0 framework. (A) Detailed description of the RNN(dictionary), CNN-RNN(dictionary), CNN(dictionary), CNN(onehot), CNN-RNN(onehot) and MLP(onehot) models. The six different models are integrated using a unified framework that uses amino acid sequence of interest as input, and output the prediction of the protein being a T3SE or non-T3SE. (B) One-hot and embedding encoded amino acid sequence matrices. (C) Illustration of different network layers.
Effect of hyperparameters on performance
We first explored the influence of two hyperparameters on prediction performance of four convolution-based models. Then a grid hyperparameter search for each model was performed on the validation set, and the top-performing tuned models were generated and evaluated on the test set afterward. Specifically, we tried a range of kernel size from 3 to 11 in steps of 2, and a range of filter number from 50 to 250 in steps of 50. Optimization results based on MCC for each of the four models are shown in Supplementary Figure S1.
For these models, hyperparameters tuning on the filter number and kernel size offered significant performance increases. The maximum increase on MCC across models ranged from 0.060 for CNN(onehot) to 0.322 for CNN-RNN(onehot). When utilizing the best hyperparameters, CNN-RNN(dictionary), CNN(dictionary), CNN(onehot) and CNN-RNN(onehot) achieved the highest MCC values of 0.830, 0.798, 0.743 and 0.662, respectively. In addition, the performance of CNN-RNN(onehot) was found to be highly sensitive to the choice of filter number. We observed no statistically significant difference in performance between the kernels (pairwise P values for MCC ranged between 0.66 and 0.98), but the performance dropped sharply as the filter number increased. We also evaluated the effect of the number of convolutional layers on performance (Supplementary Figure S2). The CNN-RNN(dictionary) with single-layer architecture showed better prediction performance, indicating that adding more convolutional layers would not improve performance for this dataset.
Performance comparison of various models
We evaluated and compared six deep learning models, which were optimized and represented the best from each proposed architecture, using the held-out test set. Prediction performance was measured using the accuracy, precision, recall and Matthew’s correlation coefficient metrics. We summarize the benchmark results in the Figure 2A, where the asterisk on the right of the bars shows the best performance among the models for each metric. Overall, for models trained with N-terminal sequences, RNN(dictionary) showed superior performance for all the metrics, with an accuracy of 0.920. The CNN-RNN(onehot) showed the lowest performance, obtaining an accuracy of 0.849. The accuracy of the remaining four models, including CNN-RNN(dictionary), CNN(dictionary), CNN(onehot) and MLP (onehot), was 0.916, 0.905, 0.879 and 0.864, respectively. The RNN(dictionary) shows the best performance because the N-terminal residues appear to provide targeting information for T3SE translocation or secretion, while bidirectional approach of LSTM is the best candidate for protein sequences containing secretion signals due to its previous memory recalling capability. To assess the effect of sequence length on predictive power, we repeated the training and testing experiment using the full-length sequence. The new models achieved no statistically significant difference in all metrics compared to using N-terminal sequences (P-values for accuracy, recall, precision and MCC were 0.879, 0.967, 0.930 and 0.941, respectively). The RNN(dictionary) was also the best one across all metrics with the exception of recall, where MLP(onehot) showed the highest value (Figure 2A). The second to fifth ranked performance was CNN-RNN(dictionary), CNN(dictionary), CNN(onehot) and MLP (onehot), respectively. The CNN-RNN(onehot) had the lowest performance in all the metrics, but with a high precision of 0.943.
Figure 2.

Comparison of models. (A) Performance of six deep learning models trained using two different sequence lengths. (B) ROC curves are shown for each model and separately for N-terminal sequence (left) and full-length sequence (right).
The receiver operating characteristic (ROC) curves were exploited to evaluate the overall performance of models, which indicate how effectively the probabilities of T3SEs are differentiated from non-T3SEs (Figure 2B). Regarding area under ROC curves (AUC), the performance of all models varied between 0.887 and 0.954, with a median score of 0.934. Collectively, the above results provide the first comprehensive comparison of deep learning models for the classification between T3SEs and non-T3SEs.
Visualizing the hidden feature representations of deep learning models
To interpret a deep learning model and explain its predictions, we visualized the representations of residues and proteins learned by the models in the two-dimensional uniform manifold approximation and projection (UMAP) space (46). First, we ran UMAP on the trained amino acid embedding and one-hot vectors simultaneously (Figure 3A). UMAP learned to embed similar activation pattern residues close to each other, while dissimilar residues were embedded far. On the embedding vector projection, UMAP visually revealed three main amino acid clusters. One was composed of eight residues (close to the top left), including alanine (A), glutamic acid (E), arginine (R), aspartic acid (D), methionine (M), lysine (K), histidine (H) and glutamine (Q). Five residues clustered in the center corresponding respectively to serine (S), asparagine (N), proline (P), threonine (T) and glycine (G). The last cluster contained remaining seven residues and appeared in the bottom right of embedding space. In contrast, UMAP identified no cluster on the one-hot vector projection, notably splitting residues into discrete points. This is consistent with the previous observation that one-hot vector is much sparser than embedding vector (Figure 1B). To further elucidate model’s inner workings or shed light on its internal representations, we visualized the T3SEs and non-T3SEs based on the features learned at different network layers of CNN-RNN (onehot). As shown in Figure 3B, the features become more and more discriminative along the layer hierarchy, with T3SEs and non-T3SEs mixed without a clear decision boundary at the input layer, culminating with a clear visual separation between classes in the BiLSTM layer. These results reveal that deep learning models have gradually learned sequence determinants in the inter-layer evolution. We further showed the projections of the last hidden layers representations of all models. By color-coding the protein representations, we observed that UMAP clearly separated T3SEs and non-T3SEs into two distinct clusters on the projections of training set (Figure 3C) and test set (Supplementary Figure S3), indicating the robustness and good generalization of models.
Figure 3.

Visualization of learned features. (A) Two-dimensional UMAP maps of 256-dimensional embedding vectors and 20-dimensional onehot vectors, colored according to amino acid type. (B) UMAP visualization of inter-layer evolution. Four CNN-RNN(onehot) hidden layers after training. Data points are colored based on their true class label, where T3SEs and non-T3SEs are represented by red and blue points, respectively. (C) Comparison of the last hidden layer representations of the training sequences for the six deep learning models.
DeepT3 2.0 improves performance by integrating predictions
We asked whether T3SE prediction across models could be used to create a meta-predictor with even higher accuracy and coverage. We hypothesized that a protein supported by most models would be more likely to be a T3SE than a protein supported by only a few or no models. To confirm this speculation, we selected the Bastion3 independent set of 108 T3SEs and 108 non-T3SEs for initial analysis (26), as it has the most abundant and balanced test data for existing prediction methods. We assessed the performance of six models on the new test set (Supplementary Figures S4 and S5) and visualized all positive prediction sets and their intersections quantitatively (Figure 4A). We found that 69.4% of the predictions were consistent across all models, while 20 of the 108 T3SEs were correctly identified by only one particular model. These observations imply that different deep learning models learn different sets of features that can complement each other for the task of identifying T3SEs. Therefore, we derived a voting-based score for a protein based on its predictions by the different models. This score represents the minimum number of models that support a protein as a T3SE. We optimized this score and tested the influence of its values on the performance of the integrating predictions. From the Figure 4B, we saw that the ACC and MCC values were highest when the score was equal to 4. So we considered T3SEs as those with score ≥ 4 and the non-T3SEs as those having a score <4. Using the new score, integrating predictions gave improvements over each model taken individually, with improved ACC and MCC values ranging from 0.009 to 0.069 and from 0.003 to 0.131, respectively.
Figure 4.

Performance of the integrated model. (A) The intersection diagram of T3SEs for the six deep learning models. The horizontal histogram shows the number of T3SEs of each model, while the vertical histogram shows the size of different intersections of these six T3SEs sets. (B) Effect of the voting-based score in the Bastion3 independent set on integrated model's performance. Performance is measured using the MCC detection and accuracy metric. (C and D) Benchmarking DeepT3 2.0 performance against existing T3SEs prediction algorithms with independent Bastion3 and P. syringae test sets, respectively.
To detect the latent overfitting of the model, we have generated the learning curve by resampling the data and refitting the untrained models. The training dataset was resampled to 20%, 40%, 60%, 80% and 100% of the original scale. The independent test dataset was not changed. At every scale, a 5-fold cross validation on training dataset and independent test on independent test dataset were performed for comparing the ACCs and MCCs. To gain the robustness of the comparison, we repeated 10 times of the training/testing comparison of every scale. As the result in Supplementary Figure S6, the learning curve increased gradually after the scale >0.6, and the standard variance (reflected by the semi-transparent shape) reduced to nearly 0.01 finally. The difference between cross validation and independent test shown that there is not a significant overfitting of this model since the difference is only about 5% and the tendency of the curves are the same.
Based on the finding above, we developed a new tool to better predict T3SEs called DeepT3 2.0. We compared DeepT3 2.0 with six existing methods that can also apply to this task: Bastion3 (26), BEAN2 (18), pEffect (19), Effective T3 (14), BPBAac (20) and DeepT3 (35). Benchmarking on the Bastion3 independent test set, the Bastion3 method was proved to be the best predictor with an accuracy equal to 0.977 (Figure 4B). DeepT3 2.0 fell behind Bastion3 but performed better than other predictors with an accuracy of 0.903. The performance of Bastion3 and DeepT3 2.0 was followed by BEAN2 (0.879), pEffect (0.875), DeepT3 (0.815), Effective T3 (0.759) and BPBAac (0.634). Next, all methods were evaluated and compared using the P. syringae independent set. The ratio of T3SE to non-T3SE in P. syringae independent set is approximately 1: 22, which is a clear class imbalance. DeepT3 2.0 had the best T3SEs and non-T3SEs discrimination across all metrics in the P. syringae benchmark, except for recall, for which it ranked third after Bastion3 and BEAN2 (Figure 4C). Bastion3 had the highest recall, and the second highest accuracy and MCC values. Of all the methods tested, Effective T3 achieved the lowest accuracy, precision and MCC metrics. Collectively, the above results demonstrate the utility of integrative strategy in improving predictive accuracy and establish DeepT3 2.0 as an effective method to identify T3SEs.
DeepT3 2.0 outperforms other methods for Gram-negative bacterial secretome prediction
We benchmarked the performance of five methods across seven secretion system datasets used for the specificity evaluation. These datasets used in this study vary in the sample size and secretion type, to represent different levels of challenges in the classification task and to evaluate how each method performs in each case. The specificity was calculated as: Specificity = TN/N, where ‘TN (true negative)’ is the number of correctly predicted non-T3SEs, and ‘N (negative)’ is the number of non-T3SEs shown. Figure 5 shows the performance of all methods on seven selected classes of secreted proteins. DeepT3 2.0 demonstrated superior specificity compared with other methods across all types of secreted proteins, although Effective T3 achieved the highest specificity in T2SP. Specifically, DeepT3 2.0 achieved a specificity of 83.2%, 87.7%, 76.9%, 82.6%, 87.5%, 96.2% and 66.7% for predicting non-T3SE for T1SP, T2SP, T4SP, T5SP, T6SP, T7SP and T8SP, respectively. The performance of BEAN2 and pEffect on T4SP was worse than other methods, with a specificity of 21.5% and 16.9%, respectively. Baston3 performed poorest among all methods on T5SP, below the average value with a specificity of 34.8%. In addition, pEffect and Bastion3 failed to correctly predict the three T8SPs. The otherwise successful methods BEAN2, pEffect and Bastion3 performed relatively poor for T4SP, T5SP and T8SP, probably because some of these effectors share similar evolutionary conserved profiles and sequence motifs with T3SP, which poses a challenge to distinguish them accurately. Collectively, these results indicate that DeepT3 2.0 can predict secreted proteins across all secretion systems and classify them as a negative class with a high accuracy.
Figure 5.

Performance comparison of T3SE prediction methods on seven secretion system effectors in Gram-negative bacteria using the specificity detection as metric. The number of proteins tested is shown in parentheses.
Performance of DeepT3 2.0 in whole-genome prediction
We used DeepT3 2.0 to analyze a well-annotated reference genome of Chlamydia trachomatis, one of the world's most common pathogenic bacteria that has been associated with prevalent bacterial sexually transmitted infection (41). We first measured the execution time of DeepT3 2.0 across data subsamples of sizes ranging from 100 to 1774 proteins (Figure 6A). Across all ranges of subsample sizes, DeepT3 2.0 appeared consistently fast, taking about 5 s for subsamples of size 100 and 16 s for all 1774 proteins. The running time of DeepT3 2.0 for genome-wide prediction is short because the processing of individual protein sequences by DeepT3 2.0 is not dependent on the length of the protein sequence. In fact, DeepT3 2.0 completed the entire prediction process with only the N-terminal 100 amino acids. Moreover, DeepT3 2.0 detected 14 of the 21 experimentally verified effectors. DeepT3 2.0 also found new 186 effectors with high probability, and these may be interesting candidates for verification (Figure 6B and Supplementary Table S1). To compare DeepT3 2.0 with other methods for large-scale prediction, we applied another tool, Effective T3, to search for putative effectors using the optimal probability threshold. The application of Effective T3 to this genome resulted in more predicted effectors (322 of the 1774 proteins), including the 14 known effectors (Figure 6B). We further conducted Gene Ontology (GO) term enrichment analysis (54) to functionally characterize the predicted effectors and to analyze the functional prevalence among the effectors. The GOanna tool, developed as a part of AgBase resource (55) was used for GO enrichment analysis. The GO terms of predicted effectors are mainly associated with enzyme regulator activity, such as ‘thiol-dependent ubiquitin-specific protease activity’, ‘peptidase activity’, ‘cysteine-type peptidase activity’, ‘hydrolase activity’ and ‘NEDD8-specific protease activity’ (Supplementary Table S2). We finally visualized the relationships between genome features and the distributions of predicted effectors in the Chlamydia trachomatis genome (Figure 6C).
Figure 6.

The application of DeepT3 2.0 to whole-genome prediction in the Chlamydia trachomatis genome. (A) Run times of DeepT3 2.0 for inputs of varying sizes. (B) Detailed proportions of predicted effector for two T3SE prediction methods. (C) Visualizing features, ORFs, start and stop codons of Chlamydia trachomatis genome and comparing all RefSeq proteins and DeepT3 2.0 predicted effectors. The proteins are mapped according to their corresponding positions on the circular bacterial genome.
DISCUSSION
Bacteria have evolved a variety of highly specialized protein transport nanomachines, also known as secretion systems, which can export numerous effector proteins into the target eukaryotic cell cytoplasm or the plasma membrane (3). These secreted effectors modulate or subvert specific host cell functions, thereby promoting bacterial adhesion, adaptation and survival (16). Understanding the link between effector sequences and secretory origins is a key challenge for understanding the complex mechanisms of protein secretion and its role in the interaction between bacteria and their environment and other organisms. Many methods have been developed to elucidate the relationship between secreted effectors and secretion systems (14–28,56,57). Using the XGBoost algorithm to extract the features solely from PSSM profiles, Ding et al. introduced an SVM-based classifier-iT3SE-PX, to improve the prediction performance on T3SEs with only protein sequences (58). By integrating the advantages of multiple homology-based biological features and various machine learning algorithms, Hui et al. recently suggested a unified prediction pipeline-T3SEpp, to more accurately identify T3SEs in novel and existing bacterial whole-genome sequences (59). More recently, Wang et al. proposed a universal platform of Gram-negative secreted substrates-BastionHub, which is not only the most comprehensive online database but also helpful for in-depth analysis of five major types of secreted substrates such as the type I, II, III, IV and VI secreted proteins (60). Furthermore, there have been several excellent reviews on machine learning algorithms and protein characterization methods for T3SE predictions (61–64).
In this study, we developed DeepT3 2.0, a new deep learning framework using various neural network models trained on protein sequences to accurately predict Gram-negative bacterial type III secreted effector in a genome-wide unbiased manner (Supplementary Figure S7). We have performed a comprehensive assessment of four types of neural networks in their ability to distinguish T3SEs from non-T3SEs using the same benchmark dataset. Our results suggest that the RNN model with BiLSTM units is the best performing deep learning approach on this classification task. The performance of CNN-RNN varies when using different encoding methods. The CNN performs better than MLP but shows worse overall performance than RNN. We have also explored the influences of two encoding methods (one-hot encoding and embedding encoding) and two sequence lengths (N-terminal sequence and full-length sequence) on model performance. In our analysis, we found that the embedding encoding is a better feature representation, preserving the primitive sequences in comparison with the one-hot format. The most obvious example is the CNN-RNN model, where CNN-RNN(dictionary) performs the second best, while CNN-RNN(onehot) performs the worst. In addition, we observed that the top three best-performing models are all embedding-based models. Various model architectures with different sequence lengths have been evaluated; we set the lengths of N-terminal and full-length sequences to 100 and 2500, where two numbers were chosen to fit the maximal secretion or translocation signal and the longest sequence in the dataset, respectively. If the length of sequences exceeds 100 or 2500 amino acids, the excess will be ignored; otherwise the ‘X’ character (unknown residue, encoded as a zero vector) will be padded at the tail of the sequence to fit the set length. Despite the differences in parameter scale and run time, we did not observe a statistically significant difference in performance among the six different deep learning models trained using different sequence lengths. However, we noted that training time is a limiting factor for models trained on long sequences. In practice, using full-length sequences to train models requires more running time and consumes more computational power. Considering these factors, we chose the N-terminal sequence to develop the subsequent integration tool. We hope the summary of these deep learning methods, the detailed comparison results, and the recommendations and guidelines for model construction can assist researchers in the development of their own new methods.
Despite the superior performance of deep learning methods, there are limitations including interpretability and visual analysis. Usually, a deep learning model is treated as a ‘black-box’ model, which only maps a given input to a classification output. Without a clear understanding of how and why these neural networks work, the development of high-quality deep learning models typically relies on a substantial amount of trial-and-error. To tackle these challenges, in the present study we have applied a dimensionality reduction technique (UMAP) to interpret and visualize what the model has learned. We show in detail how to visually track the inter-layer evolution of learned representations to understand the model behavior. We also show how visualizations can be used to explain the model's predictions and provide insightful feedback for model design and diagnosis. Our proposed visualization strategy can be extended for any types of networks.
In conclusion, we have demonstrated that DeepT3 2.0 has the potential to be a powerful tool for a large-scale prediction of T3SEs. We expect DeepT3 2.0 (http://advintbioinforlab.com/deept3/) will make better utilization of the vast amount of existing well-annotated bacterial genomes and enable researchers to accurately identify and annotate T3SEs in their studies.
DATA AVAILABILITY
All data sets used in this study can be freely available at http://advintbioinforlab.com/data/dataset.zip.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
Supplementary Material
ACKNOWLEDGEMENTS
J.L. and L.Y. conceived the study and wrote the paper. R.J. contributed to the design, implementation and testing of the model and performed the data analysis. All authors have read and agreed to the published version of the manuscript. The authors would like to thank the anonymous reviewers for their comments and suggestions, which improved the manuscript and the software.
Notes
Present address: Jiesi Luo, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou 646000, China.
Contributor Information
Runyu Jing, School of Cyber Science and Engineering, Sichuan University, Chengdu 610065, China; Medical Big Data Center, Sichuan University, Chengdu 610065, China.
Tingke Wen, School of Cyber Science and Engineering, Sichuan University, Chengdu 610065, China.
Chengxiang Liao, School of Cyber Science and Engineering, Sichuan University, Chengdu 610065, China.
Li Xue, School of Public Health, Southwest Medical University, Luzhou 646000, China.
Fengjuan Liu, School of Geography and Resources, Guizhou Education University, Guiyang 550018, China.
Lezheng Yu, School of Chemistry and Materials Science, Guizhou Education University, Guiyang 550018, China.
Jiesi Luo, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou 646000, China; Department of Pharmacy, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, China; Sichuan Key Medical Laboratory of New Drug Discovery and Druggability Evaluation, Luzhou Key Laboratory of Activity Screening and Druggability Evaluation for Chinese Materia Medica, Southwest Medical University, Luzhou 646000, China.
FUNDING
National Natural Science Foundation of China [21803045]; Joint project of Luzhou Municipal People's Government and Southwest Medical University [2020LZXNYDJ39].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pinaud L., Sansonetti P.J., Phalipon A.. Host cell targeting by enteropathogenic bacteria T3SS effectors. Trends Microbiol. 2018; 26:266–283. [DOI] [PubMed] [Google Scholar]
- 2.Lasica A.M., Ksiazek M., Madej M., Potempa J.. The type IX secretion system (T9SS): highlights and recent insights into its structure and function. Front. Cell. Infect. Microbiol. 2017; 7:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng W., Marshall N.C., Rowland J.L., McCoy J.M., Worrall L.J., Santos A.S., Strynadka N.C.J., Finlay B.B.. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017; 15:323–337. [DOI] [PubMed] [Google Scholar]
- 4.Portaliou A.G., Tsolis K.C., Loos M.S., Zorzini V., Economou A.. Type III secretion: building and operating a remarkable nanomachine. Trends Biochem. Sci. 2016; 41:175–189. [DOI] [PubMed] [Google Scholar]
- 5.Abrusci P., Vergara-Irigaray M., Johnson S., Beeby M.D., Hendrixson D.R., Roversi P., Friede M.E., Deane J.E., Jensen G.J., Tang C.M.et al.. Architecture of the major component of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 2013; 20:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Büttner D.Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2012; 76:262–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hueck C.J.Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 1998; 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhlen L., Abrusci P., Johnson S., Gault J., Deme J., Caesar J., Dietsche T., Mebrhatu M.T., Ganief T., Macek B.et al.. Structure of the core of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 2018; 25:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stebbins C.E., Galán J.E.. Structural mimicry in bacterial virulence. Nature. 2001; 412:701–705. [DOI] [PubMed] [Google Scholar]
- 10.Büttner D.Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2012; 76:262–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jennings E., Thurston T.L.M., Holden D.W.. Salmonella SPI-2 type III secretion system effectors: molecular mechanisms and physiological consequences. Cell Host Microbe. 2017; 22:217–231. [DOI] [PubMed] [Google Scholar]
- 12.Rêgo A.T., Chandran V., Waksman G.. Two-step and one-step secretion mechanisms in Gram-negative bacteria: contrasting the type IV secretion system and the chaperone-usher pathway of pilus biogenesis. Biochem. J. 2010; 425:475–488. [DOI] [PubMed] [Google Scholar]
- 13.Costa T.R.D., Felisberto-Rodrigues C., Meir A., Prevost M.S., Redzej A., Trokter M., Waksman G.. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 2015; 13:343–359. [DOI] [PubMed] [Google Scholar]
- 14.Arnold R., Brandmaier S., Kleine F., Tischler P., Heinz E., Behrens S., Niinikoski A., Mewes H.W., Horn M., Rattei T.. Sequence-based prediction of type III secreted proteins. PLoS Pathog. 2009; 5:e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tay D.M.M., Govindarajan K.R., Khan A.M., Ong T.Y.R., Samad H.M., Soh W.W., Tong M., Zhang F., Tan T.W.. T3SEdb: data warehousing of virulence effectors secreted by the bacterial type III secretion system. BMC Bioinform. 2010; 11:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Löwer M., Schneider G.. Prediction of type III secretion signals in genomes of gram-negative bacteria. PLoS One. 2009; 4:e5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong X.B., Zhang Y.J., Zhang Z.D.. Using weakly conserved motifs hidden in secretion signals to identify type-III effectors from bacterial pathogen genomes. PLoS One. 2013; 8:e56632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong X.B., Lu X.T., Zhang Z.D.. BEAN 2.0: an integrated web resource for the identification and functional analysis of type III secreted effectors. Database. 2015; 2015:bav064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg T., Rost B., Bromberg Y.. Computational prediction shines light on type III secretion origins. Sci. Rep. 2016; 6:34516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y., Zhang Q., Sun M.A., Guo D.. High-accuracy prediction of bacterial type III secreted effectors based on position-specific amino acid composition profiles. Bioinformatics. 2011; 27:777–784. [DOI] [PubMed] [Google Scholar]
- 21.Samudrala R., Heffron F., McDermott J.E.. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009; 5:e1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Sun M., Bao H., Zhang Q., Guo D.. Effective identification of bacterial type III secretion signals using joint element features. PLoS One. 2013; 8:e59754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y., Zhao J., Morgan R.L., Ma W., Jiang T.. Computational prediction of type III secreted proteins from gram-negative bacteria. BMC Bioinform. 2010; 11:S47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X., Guo Y., Luo J., Pu X., Li M.. Effective identification of Gram-negative bacterial type III secreted effectors using position-specific residue conservation profiles. PLoS One. 2013; 8:e84439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Sun M., Bao H., White A.P.. T3_MM: a markov model effectively classifies bacterial type III secretion signals. PLoS One. 2013; 8:e58173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J., Li J., Yang B., Xie R., Marquez-Lago T.T., Leier A., Hayashida M., Akutsu T., Zhang Y., Chou K.C.et al.. Bastion3: a two-layer ensemble predictor of type III secreted effectors. Bioinformatics. 2019; 35:2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato Y., Takaya A., Yamamoto T.. Meta-analytic approach to the accurate prediction of secreted virulence effectors in gram-negative bacteria. BMC Bioinform. 2011; 12:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J., Wei L., Guo F., Zou Q.. EP3: an ensemble predictor that accurately identifies type III secreted effectors. Brief. Bioinform. 2021; 22:1918–1928. [DOI] [PubMed] [Google Scholar]
- 29.Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K.M., Cofer E.M., Zhou J., Troyanskaya O.G.. Selene: a pytorch-based deep learning library for sequence data. Nat. Methods. 2019; 16:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almagro Armenteros J.J., Sønderby C.K., Sønderby S.K., Nielsen H., Winther O.. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017; 33:3387–3395. [DOI] [PubMed] [Google Scholar]
- 33.Kelley D.R., Snoek J., Rinn J.L.. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szalkai B., Grolmusz V.. Near perfect protein multi-label classification with deep neural networks. Methods. 2018; 132:50–56. [DOI] [PubMed] [Google Scholar]
- 35.Xue L., Tang B., Chen W., Luo J.. DeepT3: deep convolutional neural networks accurately identify Gram-negative bacterial type III secreted effectors using the N-terminal sequence. Bioinformatics. 2019; 35:2051–2057. [DOI] [PubMed] [Google Scholar]
- 36.Li J., Li Z., Luo J., Yao Y.. ACNNT3: Attention-CNN framework for prediction of sequence-based bacterial type III secreted effectors. Comput. Math. Methods Med. 2020; 2020:3974598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong J., Luo Y., Mou M., Fu J., Zhang Y., Xue W., Xie T., Tao L., Lou Y., Zhu F.. Convolutional neural network-based annotation of bacterial type IV secretion system effectors with enhanced accuracy and reduced false discovery. Brief. Bioinform. 2020; 21:1825–1836. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y., Niu B., Gao Y., Fu L., Li W.. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010; 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baltrus D.A., Nishimura M.T., Romanchuk A., Chang J.H., Mukhtar M.S., Cherkis K., Roach J., Grant S.R., Jones C.D., Dangl J.L.. Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 pseudomonas syringae isolates. PLoS Pathog. 2011; 7:e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L., Luo J., Guo Y., Li Y., Pu X., Li M.. In silico identification of Gram-negative bacterial secreted proteins from primary sequence. Comput. Biol. Med. 2013; 43:1177–1181. [DOI] [PubMed] [Google Scholar]
- 41.Dhroso A., Eidson S., Korkin d. Genome-wide prediction of bacterial effector candidates across six secretion system types using a feature-based statistical framework. Sci. Rep. 2018; 8:17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veltri D., Kamath U., Shehu A.. Deep learning improves antimicrobial peptide recognition. Bioinformatics. 2018; 34:2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alley E.C., Khimulya G., Biswas S., AlQuraishi M., Church G.M.. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods. 2019; 16:1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan H., Kshirsagar M., Zamparo L., Lu Y, Leslie C.S.. BindSpace decodes transcription factor binding signals by large-scale sequence embedding. Nat. Methods. 2019; 16:858–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stokes J.M., Yang K., Swanson K., Jin W., Cubillos-Ruiz A., Donghia N.M., MacNair C.R., French S., Carfrae L.A., Bloom-Ackermann Z.et al.. A deep learning approach to antibiotic discovery. Cell. 2020; 180:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McInnes L., Healy J.. UMAP: uniform manifold approximation and projection for dimension reduction. J. Open Source Softw. 2018; 3:861. [Google Scholar]
- 47.Melville J.uwot: the uniform manifold approximation and projection (UMAP) method for dimensionality reduction. 2019; https://github.com/jlmelville/uwot.
- 48.Hadley W.ggplot2: Elegant Graphics for Data Analysis. 2016; Switzerland: Springer. [Google Scholar]
- 49.Conway J.R., Lex A., Gehlenborg N.. UpSetR: an r package for the visualization of intersecting sets and their properties. Bioinformatics. 2017; 33:2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grant J.R., Stothard P.. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008; 36:W181–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rifaioglu A.S., Atas H., Martin M.J., Cetin-Atalay R., Atalay V., Doğan T.. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief. Bioinform. 2019; 20:1878–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lombardi E.P., Londoño-Vallejo A.. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020; 48:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.C., Müller M.. pROC: an open-source package for r and S+ to analyze and compare ROC curves. BMC Bioinform. 2011; 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J., Aronow B.J., Jegga A.G.. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinform. 2009; 10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCarthy F.M., Wang N., Magee G.B., Nanduri B., Lawrence M.L., Camon E.B., Barrell D.G., Hill D.P., Dolan M.E., Williams W.P.et al.. AgBase: a functional genomics resource for agriculture. BMC Genomics. 2006; 7:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu Y., Huang H., Cheng X., Shu X., White A.P., Stavrinides J., Köster W., Zhu G., Zhao Z., Wang Y.. A global survey of bacterial type III secretion systems and their effectors. Environ. Microbiol. 2017; 19:3879–3895. [DOI] [PubMed] [Google Scholar]
- 57.Guo Z., Cheng X., Hui X., Shu X., White A.P., Hu Y., Wang Y.. Prediction of new bacterial type III secreted effectors with a recursive hidden markov model profile-alignment strategy. Curr. Bioinform. 2018; 13:280–289. [Google Scholar]
- 58.Ding C., Han H., Li Q., Yang X., Liu T.. iT3SE-PX: identification of bacterial type III secreted effectors using PSSM profiles and XGBoost feature selection. Comput. Math. Methods Med. 2021; 2021:6690299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hui X., Chen Z., Lin M., Zhang J., Hu Y., Zeng Y., Cheng X., Ou-Yang L., Sun M.A., White A.P.et al.. T3SEpp: an integrated prediction pipeline for bacterial type III secreted effectors. mSystems. 2020; 5:e00288–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J., Li J., Hou Y., Dai W., Xie R., Marquez-Lago T.T., Leier A., Zhou T., Torres V., Hay I.et al.. BastionHub: a universal platform for integrating and analyzing substrates secreted by Gram-negative bacteria. Nucleic Acids Res. 2021; 49:D651–D659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDermott J.E., Corrigan A., Peterson E., Oehmen C., Niemann G., Cambronne E.D., Sharp D., Adkins J.N., Samudrala R., Heffron F.. Computational prediction of type III and IV secreted effectors in gram-negative bacteria. Infect. Immun. 2011; 79:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.An Y., Wang J., Li C., Leier A., Marquez-Lago T., Wilksch J., Zhang Y., Webb G.I., Song J., Lithgow T.. Comprehensive assessment and performance improvement of effector protein predictors for bacterial secretion systems III, IV and vI. Brief. Bioinform. 2018; 19:148–161. [DOI] [PubMed] [Google Scholar]
- 63.Zalguizuri A., Caetano-Anollés G., Lepek V.C.. Phylogenetic profiling, an untapped resource for the prediction of secreted proteins and its complementation with sequence-based classifiers in bacterial type III, IV and VI secretion systems. Brief. Bioinform. 2019; 20:1395–1402. [DOI] [PubMed] [Google Scholar]
- 64.Hui X., Chen Z., Zhang J., Lu M., Cai X., Deng Y., Hu Y., Wang Y.. Computational prediction of secreted proteins in gram-negative bacteria. Comput. Struct. Biotechnol. J. 2021; 19:1806–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data sets used in this study can be freely available at http://advintbioinforlab.com/data/dataset.zip.