

Abstract
The epigenome of macrophages can be reprogrammed by extracellular cues, but the extent to which different stimuli achieve this is unclear. NFκB is a transcription factor that is activated by all pathogen-associated stimuli and can reprogram the epigenome by activating latent enhancers. However, we show that NFκB does so only in response to a subset of stimuli. This stimulus-specificity depends on the temporal dynamics of NFκB activity, in particular whether it is oscillatory or non-oscillatory. Non-oscillatory NFκB opens chromatin by sustained disruption of nucleosomal histone-DNA interactions, enabling activation of latent enhancers that modulate expression of immune response genes. Thus, temporal dynamics can determine a transcription factor’s capacity to reprogram the epigenome in a stimulus-specific manner.
One Sentence Summary:
Nucleosomal DNA interactions decode NFκB dynamics to regulate activation of latent enhancers of immune response genes.
The cellular epigenome, a regulatory network involving transcription factors, chromatin architecture and histone modifications, contains stable, heritable information that determines cell type-specific programs of gene expression (1, 2). Nevertheless, the epigenome of differentiated cells remains highly plastic, particularly in immune cells like macrophages (3, 4). These immune sentinel cells detect microenvironmental immune threats, mount appropriate gene expression responses, and reprogram their epigenomes to tailor subsequent immune responses (5). At a molecular level, this reprogramming is initiated by the activity of signal-dependent transcription factors (TFs) such as nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (6). In cooperation with chromatin modifiers and pioneering TFs, signal-dependent TFs increase chromatin accessibility and modify histones at previously silent regions of the genome, thus converting latent enhancers to poised or active states (7–9). NFκB activated by bacterial lipopolysaccharide (LPS) has been a model TF in this field. However, the degree to which NFκB or other TFs can alter the epigenome in response to different stimuli is unknown.
To investigate the stimulus-specificity of epigenomic reprogramming, we stimulated bone marrow-derived macrophages (BMDMs) with five well-characterized ligands: tumor necrosis factor (TNF), and the Toll-like receptor agonists Pam3CSK, CpG, LPS, and Poly(I:C). We performed chromatin immunoprecipitation sequencing (ChIP-seq) using antibodies recognizing the mono-methylation of lysine 4 on histone H3 (H3K4me1) to identify latent enhancers that were activated upon stimulation. We found 3978 enhancer regions that segregated into two clusters by the k-means algorithm (Fig. 1A, Fig. S1). The latent enhancers in Cluster 1 were most strongly activated in response to LPS and Poly(I:C) and were enriched for interferon response factor (IRF) motifs (Fig. 1B, top), consistent with the fact that these stimuli activate IRF3 and type I interferon (10); in Irf3−/−Ifnar−/− BMDMs these regions no longer acquired H3K4me1 (Fig. 1C, top). Weak H3K4me1 signal was preserved in response to TNF, which does not activate IRF3 but IRF1 (11).
Fig. 1. NF-kB–activated latent enhancers are stimulus-specific and correlate to dynamic features of NF-kB activity.
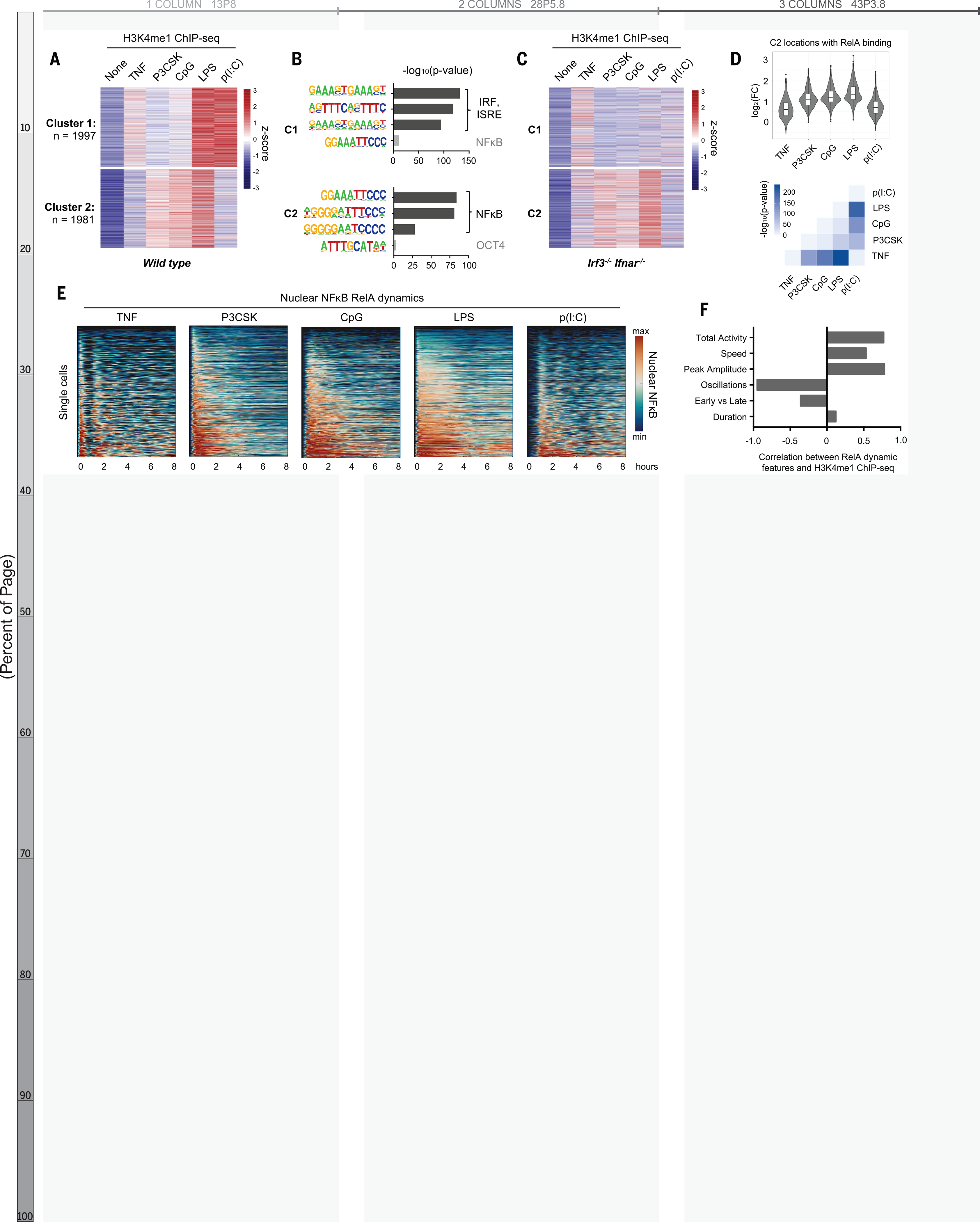
(A) Heatmap of H3K4me1 ChIPseq inducible peaks from BMDMs stimulated with five ligands for 8 hours, unsupervised k-means clustering. Average of two biological replicates. (B) Known transcription factor motifs with greatest enrichment in cluster 1 and cluster 2 peaks. ISRE, interferon-stimulated response element; OCT4, organic cation/carnitine transporter 4. (C) Heatmap of H3K4me1 ChIP-seq in Irf3−/−Ifnar−/−BMDMs, using the same clusters as in (A). (D) Violin and box plots of log2 fold change in H3K4me1 signal of 1071 NF-kB enhancers from cluster 2 that also contain an NF-kB–RelA binding event. Corresponding matrix of P values of H3K4me1 ChIP-seq fold change, by two-tailed t test between pairs of conditions. (E) Heatmaps of NF-kB activity in single cells by live cell microscopy of mVenus-RelA BMDMs, showing nuclear abundance of NF-kB in response to five stimuli over 8 hours. (F) Bar graph of correlation coefficients between mean H3K4me1 ChIP-seq counts of NF-kB enhancers and the six key features of NF-kB dynamics (20); see also fig. S3
In contrast, the regions in Cluster 2 were highly enriched for NFκB motifs (Fig. 1b, bottom), implying that these were latent NFκB enhancers. We examined the contribution of other stimulus-responsive signaling pathways and found that the gain of H3K4me1 was preserved in Irf3−/−Ifnar−/− BMDMs (Fig. 1C, bottom), but disrupted by pharmacologic inhibition of mitogen-activated protein kinase (MAPK) pathways (Fig. S2a–b). MAPK inhibition also blocked activation of latent enhancers in Cluster 1, suggesting that the MAPK pathway is generally critical for epigenomic reprogramming (12) and does not specifically collaborate with NFκB.
We next examined the contribution of NFκB family members. RelA:p50 is the dominant NFκB dimer in macrophages (13), but cRel also plays a role (14). We knocked out cRel (Rel−/−) and found that H3K4me1 ChIP-seq signals were unchanged (Fig. S2c), including at the Il12b promoter (14). Knocking out p50 (nfkb1−/−) only weakly diminished H3K4me1 signals, indicating that partial compensation by RelA:p52 or RelA homodimers was sufficient (15). These data indicated that RelA is the primary activator of latent NFκB enhancers in macrophages.
To focus on latent NFkB enhancers, we used RelA ChIP-seq data (16) to identify 1071 regions in Cluster 2 that contained a RelA binding event. Surprisingly, these regions acquired H3K4me1 in a stimulus-specific manner, even though all five stimuli tested activate NFκB (17). Within these regions, the H3K4me1 signal was strongly induced by Pam3CSK, CpG, and LPS, with median log2 fold-changes of 1.07, 1.16, and 1.33, respectively. TNF and Poly(I:C) produced less H3K4me1, with median log2 fold-changes of 0.60 and 0.70, respectively (Fig. 1D, top). A pairwise comparison of samples quantitatively confirmed the stimulus-specificity of these NFκB enhancers (Fig. 1D, bottom).
This stimulus-specificity would be difficult to explain if NFκB acted as a binary on-off switch, but NFκB is in fact activated with complex, stimulus-specific temporal dynamics (17–19). In response to various stimuli, NFκB enters the nucleus with distinct speeds, amplitudes, and durations, and may oscillate between the nucleus and cytoplasm. To determine whether stimulus-specific NFκB dynamics play a role in stimulus-specific activation of latent enhancers, we used live-cell microscopy of BMDMs expressing NFκB-RelA fused with the mVenus fluorophore (mVenus-RelA) (20) to measure the single-cell dynamics of NFκB-RelA in response to each of the five ligands (Fig. 1E). We quantified the six NFκB dynamic features that function to encode ligand identity and dose (20) and correlated them to mean H3K4me1 counts in the NFκB- activated latent enhancers (Fig. S3). Oscillatory power (r = −0.95), total activity (r = 0.77), and peak amplitude (r = 0.78) were highly correlated with the capacity of a given stimulus to activate latent enhancers (Fig. 1F).
We hypothesized that temporal dynamics of NFκB activity might affect its interaction with chromatin. Crystallographic studies imply that stable NFκB-DNA binding requires the DNA to be nucleosome-free because NFκB dimers embrace the DNA double helix circumferentially (21, 22) (Fig. 2A). However, NFκB can interact with nucleosomal DNA, particularly when its binding site is distal to the nucleosome dyad (23). Indeed, the DNA-histone interface is composed of low-affinity interactions that allow spontaneous disassociation or “breathing” (24). Thus, successive disruptions of DNA-histone contacts by NFκB, in collaboration with remodeling complexes such as SWI/SNF (25), chaperone proteins such as FACT (26, 27), and/or pioneer factors such as Pu.1 or CEB/Pα (28), may displace the nucleosome (Fig. 2B). This may be followed by the deposition of histone modifications on neighboring nucleosomes, resulting in a poised or active enhancer (7).
Fig. 2. Mathematical model predicts epigenetic response to distinct dynamic features of NF-kB.
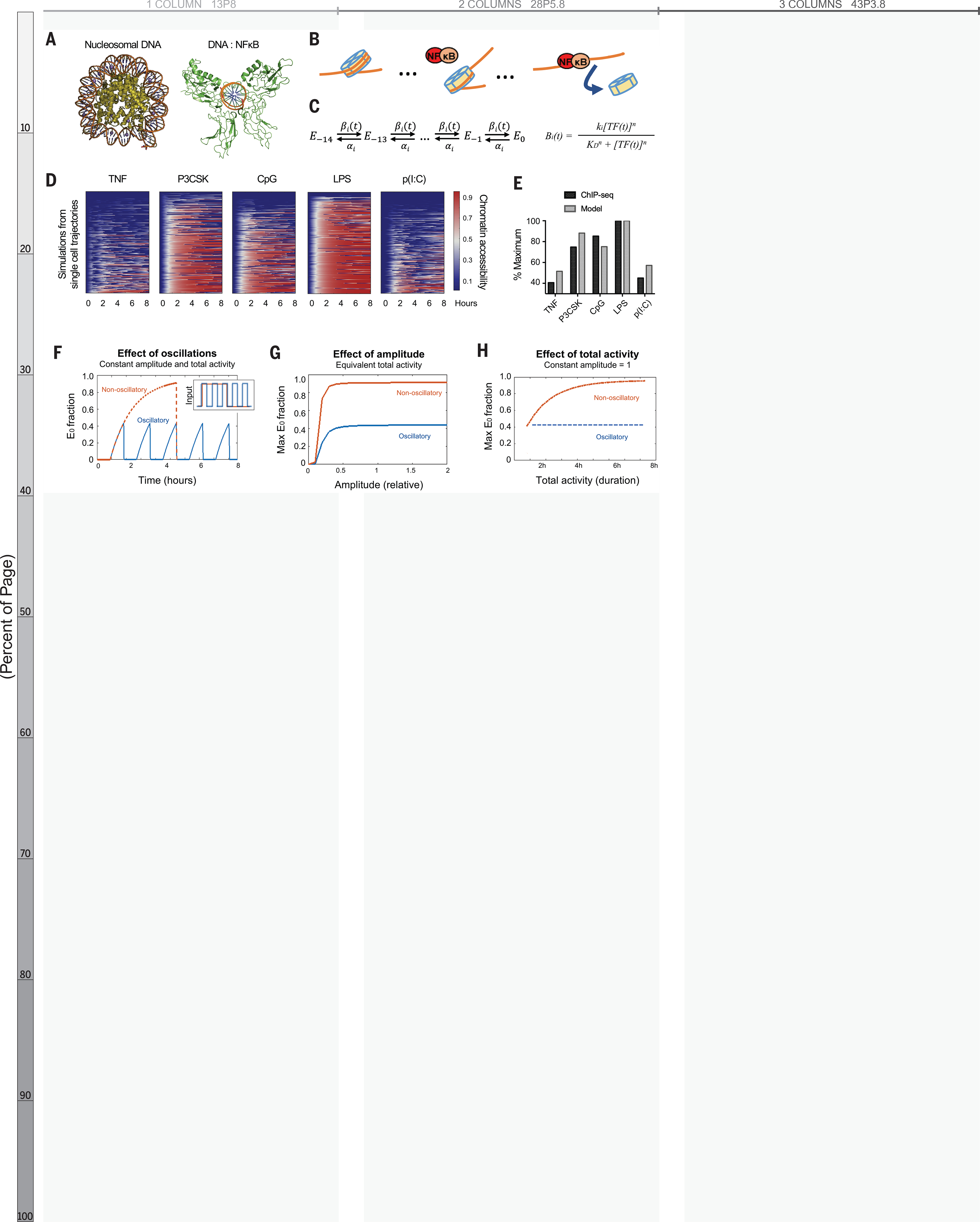
(A) Crystal structures of nucleosomal DNA [Protein Data Bank (PDB) ID 1F66] versus NF-kB–bound DNA (PDB ID 1VKX), where the p65:p50 NF-kB dimer is in green. (B) Schematic of model illustrating NF-kB–driven nucleosome displacement. (C) Multistep model with 14 steps to complete nucleosome unwrapping, each expressed as a Hill function (for further explanation, see supplementary materials). (D) Heatmaps of simulations of chromatin opening in response to different stimuli, using single-cell trajectories from NF-kB microscopy data as input. (E) Model simulation versus ChIP-seq data. Mean ChIP-seq counts from 1071 latent NF-kB enhancers (Fig. 1D), background-subtracted and scaled to maximum signal (LPS stimulation). Model simulations are mean of maximum E0 fraction per cell (compare with fig. S3A), scaled to LPS condition. (F) Model simulation of predicted chromatin accessibility comparing oscillatory versus non-oscillatory input activities. (G and H) Model simulation of predicted chromatin opening across a range of amplitudes and durations.
We created a multi-step model describing how dynamical NFκB activity might interact with nucleosomal DNA. A series of 14 Hill equations described the competition between NFκB and histone for interacting with DNA (Fig. 2C), reflecting the number of contact points in the histone octamer-DNA crystal structure (29). Relative rates of nucleosome wrapping and unwrapping were based on available biophysical data (30). With measured single-cell NFκB activities (Fig. 1E) as inputs, the model simulations reproduced the differences in experimental H3K4me1 ChIP-seq data (Fig. 2D–E, S4A–B).
We used the model to investigate which features of NFκB dynamics affect chromatin accessibility. We examined the three features most highly correlated with the H3K4me1 ChIP-seq data (Fig. 1F): non-oscillatory, amplitude, and total activity. The model indicated that a non-oscillatory dynamic produces a two-fold greater chromatin accessibility than an oscillatory dynamic (Fig. 2F). The model also indicated that NFκB activity must have a minimal amplitude (Fig. 2G, S4C) and extend for a minimal duration (Fig. 2H, S4D) to open chromatin; but above these thresholds, non-oscillatory NFκB always has greater capacity to open chromatin than oscillatory NFκB. This was consistent across a range of parameter values (Fig. S5). These simulations led to the striking prediction that the presence or absence of oscillations, not the maximum amplitude or duration of activity, is the key determinant of whether NFκB preserves or alters the chromatin state.
To test this prediction, we generated a mouse in which NFκB dynamics are perturbed. When activated, NFκB rapidly induces expression of Nfkbia, whose gene product is the negative regulator IκBα (Fig. 3A) (31). IκBα knockout alone is perinatal lethal due to persistent inflammation (32), but we rescued this lethality by genetically ablating basal TNF expression (33). We then crossed the composite knockout strain with mVenus-RelA knock-in mice to examine the dynamics of NFκB by live-cell microscopy. IκBα−/− BMDMs responded to TNF with altered NFκB dynamics compared to WT controls (Fig. 3B). We quantified the differences in the distribution of single cell dynamic features by the nonparametric Kolmogorov-Smirnov test and found that the greatest dynamic difference between IκBα−/− and WT was a loss of oscillatory content, with a test statistic (D) of 0.85, corresponding to a p-value < 10−16 (Fig. 3C, Fig. S6A). Other dynamic features were either unaffected or favored WT cells in the case of activation speed (D = 0.66) and early-vs-late activity (D = 0.52). The area under the NFκB activity curve slightly favored WT cells at all time points (Fig. 3C, Fig. S6B). We concluded that loss of IκBα abolished NFκB oscillations without increasing its total activity.
Fig. 3. IkBa knockout abolishes NF-kB oscillations, increases chromatin accessibility, and activates latent enhancers.

(A) Schematic of IκBα as inducible negative regulator of NF-κB. IKK, IκB kinase. (B) Heatmap of single-cell NF-kB activity by microscopy comparing TNF response in WT versus IkBa−/− macrophages. A.U., arbitrary units. (C) Bar graph of K-S test statistic for difference in distribution of six key signaling features and areas under NF-kB activity curve (AUC), comparing IkBa−/− and WT. (D) Heatmap of ATACseq signal after TNF stimulation at 322 genomic regions that are TNF-inducible and differential between IkBa−/− and control. Average of two biological replicates. The term “wash” indicates 8 hours with and 16 hours without TNF stimulation. (E) Known transcription factor motifs with greatest enrichment in differentially inducible ATAC-seq regions. (F) Genome browser tracks for representative differentially inducible ATAC-seq regions, two replicates per time point. (G) Percentage of cells with non-oscillatory NF-kB trajectories by microscopy, compared with relative percentage of cells with accessible chromatin by ATAC-seq at Chr15 intergenic peak (F). (H) Heatmap of H3K4me1 ChIP-seq signal after TNF stimulation over the 322 regions defined as differentially inducible by ATAC-seq. Average of two biological replicates. The term “wash” indicates 8 hours with and 16 hour without TNF stimulation.
We examined the chromatin state by stimulating BMDMs from IκBα−/− and littermate control mice with TNF and performed the assay for transposase-accessible chromatin (ATAC-seq) at two, four, and eight hours. This was followed by a 16-hour period without TNF after which a final time point was collected (Fig. S7A). We identified 1443 genomic regions that demonstrated TNF-inducible chromatin accessibility in either genotype. Of these, 332 were differentially inducible between control and IκBα−/−. Of these regions, 97% (n=322) had greater chromatin accessibility in the knockout than control (Fig. 3D), despite the slight reduction in total NFκB activity (Fig. S6B). These differentially inducible regions were enriched for NFκB motifs (Fig. 3E), and 311 of 322 regions showed RelA binding by ChIP-seq (Fig. S7B). Ninety-six percent were located in intergenic or intronic portions of the genome (Fig. S7C), suggesting that they function as cis-acting enhancers of immune genes such as Ccl5 (Fig. 3F), which requires chromatin remodeling for maximal expression (16).
Our model predicted that chromatin accessibility is primarily determined by whether NFκB is oscillatory or non-oscillatory at the single cell level. We therefore considered that the magnitude of ATAC-seq signal can be interpreted as the proportion of cells in a sample in which a particular region of DNA is accessible. By microscopy, 87% of IκBα−/− cells had non-oscillatory NFκB, compared to 25% in WT cells. This was similar to the magnitude of ATAC-seq differences between IκBα−/− and control. For example, at an intergenic peak on chromosome 15, 67% of IκBα−/− cells showed accessible chromatin, compared to 22% of control cells (Fig. 3G).
To confirm that the negative feedback function of IκBα was indeed critical for the observed effects, we used an IκBακB/κB mutant in which NFκB-binding sites in the promoter of the Nfkbia gene are disrupted (34) (Fig. S8A). In this model, basal IκBα expression is preserved, and the mice live into adulthood without requiring compound suppressor mutations. We found that upon TNF stimulation IκBακB/κB BMDMs activated NFκB in a non-oscillatory manner with minimal disruption of other dynamic features (Fig. S8B–E). ATAC-seq analysis of TNF-stimulated WT vs. IκBακB/κB BMDMs recapitulated our findings in the IκBα−/− system, with 131 genomic regions demonstrating greater gain of chromatin accessibility in the mutant compared to WT (Fig. S8F). These regions were enriched for NFκB motifs, and 90% showed RelA binding by ChIP-seq (Fig. S8G–H). Taken together, the ATAC-seq data from both IκBα−/− and IκBακB/κB experimental models indicated that loss of NFκB oscillations results in greater chromatin accessibility at NFκB binding sites.
We examined whether regions with differentially inducible chromatin accessibility acquired the corresponding histone mark of enhancers. H3K4me1 ChIP-seq in TNF-stimulated BMDMs showed that in the 322 differentially inducible ATAC-seq regions there was also a greater gain of H3K4me1 signal in IκBα−/− than in littermate controls (Fig. 3H). These histone marks persisted for 16 hours after TNF was removed. This suggested that chromatin opening facilitated by NFκB may be transient but leads to durable H3K4 methylation even after the stimulus is removed, thus activating a latent enhancer and reprogramming the epigenome.
Because histone methylation is more durable and more indicative of enhancer function, we analyzed the H3K4me1 ChIP-seq data independently and identified 2081 regions that acquired more H3K4 methylation in IκBα−/− cells than controls (Fig. 4A). These differentially induced, dynamics-dependent enhancers persisted after the TNF stimulus was removed, were enriched for NFκB motifs (Fig. 4B), and showed significant overlap with the set of stimulus-specific NFκB enhancers identified previously (Fig. 1D, 211 genomic regions, p = 3.0 e-135). The inducible ChIP-seq signal was consistently greater when NFκB dynamics were non-oscillatory rather than oscillatory, whether by genetic perturbation or by stimulus-specific signaling mechanisms (Fig. 4C).
Fig. 4. NF-kB dynamics-dependent enhancers are associated with dynamics-dependent gene expression.
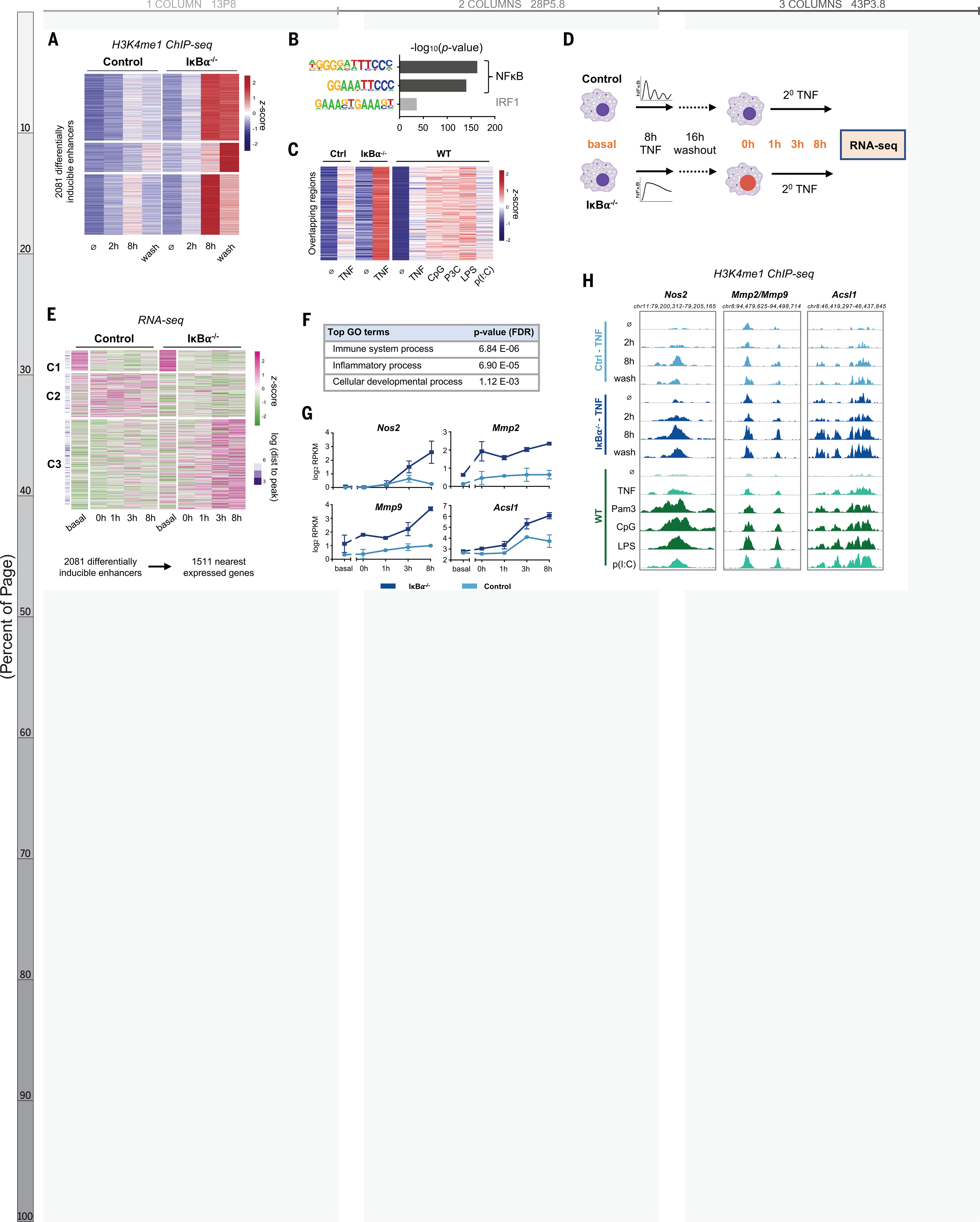
(A) Heatmap of H3K4me1 ChIP-seq signal after TNF stimulation at 2081 regions that are TNF-inducible and differential etween IkBa−/− and control, that is, which are dynamics-dependent enhancers. Average of two biological replicates. The term “wash” indicates 8 hours with and 16 hours without TNF. (B) Known transcription factor motifs with greatest enrichment in dynamics-dependent enhancers. (C) Heatmap of H3K4me1 signal after 8-hour stimulation at regions that overlap between (A) and Fig. 1D (n = 211 regions, P for overlap = 3.0 Å~ 10−135). (D) Schematic of RNA-seq experiment. (E) Heatmap showing expression of genes closest to dynamics-dependent enhancers, where cluster 3 exhibits differential gene expression between IkBa−/− and control. Average of two biological replicates. (F) Top biological process ontology terms for genes in cluster 3 of (E). FDR, false discovery rate. (G) Examples of genes differentially induced between IkBa−/− and control, average and standard deviation of two replicates. RPKM, reads per kilobase per million mapped reads. (H) Genome browser tracks of differentially inducible H3K4me1 peaks near differentially inducible genes, showing TNFstimulated IkBa−/− versus control and stimulus-specific response in WT BMDMs. More darkly shaded tracks indicate non-oscillatory NF-kB conditions. Average of two biological replicates.
We tested whether NFκB dynamics-dependent enhancers alter macrophage transcriptional responses to subsequent stimulation. We treated IκBα−/− and littermate control BMDMs with TNF for eight hours, let cells rest for 16 hours, and then re-stimulated with TNF for up to eight hours and collected samples for mRNA-seq (Fig. 4D). We explored the relation between differentially inducible enhancers and gene expression with two approaches. First, we identified the nearest expressed genes to the 2081 enhancers, removed duplicates, and found three distinct patterns of expression for the resulting 1511 genes (Fig. 4E). Cluster 1 and 2 genes were not TNF-responsive in either condition, reflecting an intrinsic limitation of this approach in which enhancers often do not regulate their nearest genes (35). Despite this limitation, Cluster 3 genes (58% of total) were both TNF-responsive and more strongly induced in IκBα−/− BMDMs. Of these genes, 88% were not induced in controls at all (using a two-fold threshold). These differentially regulated genes were enriched for ontology terms “Immune system process” and “Inflammatory process” (Fig. 4F), indicating that non-oscillatory NFκB epigenetically reprograms macrophages to enhance their immune response.
We also examined our data with a gene-centric approach. From the RNA-seq dataset we identified 1958 TNF-inducible genes, 482 of which were differentially induced in IκBα−/− versus control (Fig. S9A–B). For each gene, we annotated the genomic distance to the nearest differentially inducible H3K4me1 ChIP-seq peak. Differentially induced genes were closer to differentially induced ChIP-seq peaks (p = 1.13 e-9) than genes that were not differentially induced (Fig. S9C–D). Thus, both analyses indicated that NFκB dynamics-dependent enhancers regulate gene expression responses to a subsequent stimulus.
The dynamics-dependent gene expression program included Nos2, Mmp2, and Mmp9, which are well-defined markers of classical macrophage activation (36), as well as Acsl1, which plays a role in the pathogenesis of atherosclerosis (37) (Fig. 4G). Each of these genes had a nearby enhancer that acquired more H3K4me1 signal in the presence of non-oscillatory NFκB, whether in the IκBα−/− system or in WT BMDMs stimulated with various ligands (Fig. 4H). These specific examples further suggested that de novo enhancers formed by non-oscillatory NFκB regulate genes involved in macrophage activation.
In summary, our results indicate that the dynamics of NFκB activity, particularly whether it is oscillatory or non-oscillatory, determine NFκB’s capacity to reprogram the macrophage epigenome. We show with a mathematical model how biophysical principles governing nucleosome dynamics might decode stimulus-specific NFκB dynamical features. The role of temporal dynamics may thus complement the structure-function model in which distance from the nucleosome core determines accessibility to partially exposed DNA motifs (38). Together, TF dynamics and motif accessibility may regulate the sensitivity of a particular nucleosome to eviction. To date, the function of NFκB oscillations has been unclear given that there is little difference in the expression of poised inflammatory-response genes induced by oscillatory vs. non-oscillatory NFκB (39, 40). We propose that in response to some stimuli, the role of oscillations is to maintain the epigenomic state while exploiting existing poised enhancers for inflammatory gene activation. However, in response to other immune threats, non-oscillatory NFκB induces a comparable gene expression program while also activating latent enhancers, thus changing the epigenomic state of the cell and its response to subsequent stimuli. While further work will be needed to determine the physiological implications of NFκB dynamics-dependent enhancers and to identify the proteins that collaborate with NFκB to evict nucleosomes, our study establishes TF temporal dynamics as a key mechanistic determinant of epigenomic reprogramming.
Supplementary Material
Acknowledgements:
We would like to thank Kensei Kishimoto, Diane Lefaudeux, Xiao-Fei Lin, and Anup Mazumber for their experimental and analytical contributions, and Siavash Kurdistani, Michael Carey, Eason Lin and Ying Tang for their insights and critical reading of the manuscript.
Funding: This work was supported by NIH grants R01-AI127864, R01-GM117134, F31-AI138450, T32-GM008042, T32-HL069766, and T32-AI089398, as well as the Specialty Training and Advanced Research (STAR) program of the UCLA Department of Medicine. Sequencing was performed at the UCLA Broad Stem Cell Center Sequencing Core.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Raw data and count tables for ChIP-seq, ATAC-seq, and RNA-seq data have been submitted to the NCBI GEO repository under accession number GSE146068. Code is available at https://github.com/signalingsystemslab/NFkB_enhancer_dynamics (DOI: 10.5281/zenodo.4698447).
References and Notes:
- 1.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allis CD, Jenuwein T, The molecular hallmarks of epigenetic control. Nat Rev Genet. 17, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, Natoli G, Molecular control of activation and priming in macrophages. Nature Immunology. 17, 26–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivashkiv LB, Epigenetic regulation of macrophage polarization and function. Trends Immunol. 34, 216–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray PJ, Wynn TA, Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 11, 723–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence T, The Nuclear Factor NF- B Pathway in Inflammation. Cold Spring Harbor Perspectives in Biology. 1, a001651–a001651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G, Latent enhancers activated by stimulation in differentiated cells. Cell. 152, 157–171 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, Glass CK, Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 51, 310–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinz S, Romanoski CE, Benner C, Glass CK, The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol 16, 144–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda K, Takaoka A, Taniguchi T, Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity. 25, 349–360 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Yarilina A, Park-Min K-H, Antoniv T, Hu X, Ivashkiv LB, TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol 9, 378–387 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Klein AM, Zaganjor E, Cobb MH, Chromatin-tethered MAPKs. Curr Opin Cell Biol. 25, 272–277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell S, Vargas J, Hoffmann A, Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med. 8, 227–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanjabi S, Hoffmann A, Liou H-C, Baltimore D, Smale ST, Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proceedings of the National Academy of Sciences. 97, 12705–12710 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann A, Genetic analysis of NF-kB/Rel transcription factors defines functional specificities. The EMBO Journal. 22, 5530–5539 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong A-J, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST, A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell. 165, 165–179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werner SL, Barken D, Hoffmann A, Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 309, 1857–1861 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Behar M, Hoffmann A, Understanding the temporal codes of intra-cellular signals. Curr. Opin. Genet. Dev 20, 684–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Covert MW, Leung TH, Gaston JE, Baltimore D, Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 309, 1854–1857 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Taylor B, Adelaja A, Liu Y, Luecke S, Hoffmann A, Macrophages classify immune threats using at least six codewords of the temporal NFkB code. BioRxiv (2020). [Google Scholar]
- 21.Chen FE, Huang DB, Chen YQ, Ghosh G, Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 391, 410–413 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Luger K, Suto RK, Clarkson MJ, Tremethick DJ, [No title found]. Nat. Struct Biol 7, 1121–1124 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Lone IN, Shukla MS, Charles Richard JL, Peshev ZY, Dimitrov S, Angelov D, Binding of NF-κB to Nucleosomes: Effect of Translational Positioning, Nucleosome Remodeling and Linker Histone H1. PLoS Genet. 9, e1003830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li G, Levitus M, Bustamante C, Widom J, Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol. 12, 46–53 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi K, Hiramatsu H, Nakamura S, Kobayashi K, Haraguchi T, Iba H, Tumor suppression via inhibition of SWI/SNF complex-dependent NF-κB activation. Sci Rep. 7, 11772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A, Sviridov S, Koman IE, Veith J, Komar AA, Gudkov AV, Gurova KV, Curaxins: Anticancer Compounds That Simultaneously Suppress NF- B and Activate p53 by Targeting FACT. Science Translational Medicine. 3, 95ra74–95ra74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkler DD, Luger K, The Histone Chaperone FACT: Structural Insights and Mechanisms for Nucleosome Reorganization. J. Biol. Chem 286, 18369–18374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin F, Li Y, Ren B, Natarajan R, PU.1 and C/EBP(alpha) synergistically program distinct response to NF-kappaB activation through establishing monocyte specific enhancers. Proc Natl Acad Sci U S A. 108, 5290–5295 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ, Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol 319, 1097–1113 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Tims HS, Gurunathan K, Levitus M, Widom J, Dynamics of Nucleosome Invasion by DNA Binding Proteins. Journal of Molecular Biology. 411, 430–448 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann A, Levchenko A, Scott ML, Baltimore D, The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 298, 1241–1245 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Beg AA, Sha WC, Bronson RT, Baltimore D, Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 9, 2736–2746 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Shih VF-S, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A, Kinetic control of negative feedback regulators of NF- B/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proceedings of the National Academy of Sciences. 106, 9619–9624 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng B, Ling J, Lee AJ, Wang Z, Chang Z, Jin W, Kang Y, Zhang R, Shim D, Wang H, Fleming JB, Zheng H, Sun S-C, Chiao PJ, Defective feedback regulation of NF-kappaB underlies Sjogren’s syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc. Natl. Acad. Sci. U.S.A 107, 15193–15198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, Satpathy AT, Mumbach MR, Hoadley KA, Robertson AG, Sheffield NC, Felau I, Castro MAA, Berman BP, Staudt LM, Zenklusen JC, Laird PW, Curtis C, The Cancer Genome Atlas Analysis Network†, W. J. Greenleaf, H. Y. Chang, The chromatin accessibility landscape of primary human cancers. Science. 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA, Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, Li LO, Becker L, Yuan W, Chait A, Braun KR, Potter-Perigo S, Sanda S, Wight TN, Pennathur S, Serhan CN, Heinecke JW, Coleman RA, Bornfeldt KE, Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. U.S.A 109, E715–724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS, Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 161, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barken D, Wang CJ, Kearns J, Cheong R, Hoffmann A, Levchenko A, Comment on “Oscillations in NF-kappaB signaling control the dynamics of gene expression.” Science. 308, 52; author reply 52 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, Hoffmann A, Iterative Modeling Reveals Evidence of Sequential Transcriptional Control Mechanisms. Cell Syst. 4, 330–343 e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.